
Submitted:
20 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
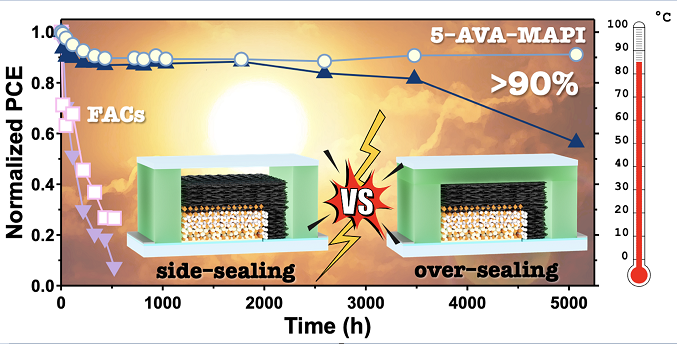
The key to the practical application of organometal–halide–crystals perovskite solar cells (PSCs) is to achieve thermal stability through robust encapsulation. This paper presents a method to significantly extend the thermal stability lifetime of perovskite solar cells to over 5,000 h at 85 °C by demonstrating an optimal combination of encapsulation methods and perovskite composition for carbon–based multiporous–layered–electrode (MPLE)–PSCs. We fabricated the four types of MPLE-PSCs using two encapsulation structures (over– and side–sealing with thermoplastic resin films) and two perovskite compositions ((5–AVA)x(methylammonium (MA))1–xPbI3 and (formamidinium (FA))0.9Cs0.1PbI3), and analyzed the 85 ºC thermal stability followed by ISOS–D–2 protocol. Without encapsulation, FA0.9Cs0.1PbI3 exhibited higher thermal stability than (5–AVA)x(MA)1–xPbI3. However, encapsulation reversed the phenomenon (that of (5–AVA)x(MA)1–xPbI3 became stronger). The combination of (5–AVA)x(MA)1–xPbI3 perovskite absorber and over–sealing encapsulation effectively suppressed the thermal degradation, resulting in the PCE value at 91.2% of the initial value after 5,072 h. On the other hand, another combination (side–sealing on (5–AVA)x(MA)1–xPbI3, over– and side–sealing on FA0.9Cs0.1PbI3) resulted in decreased stability. The FA–based perovskite was decomposed for these degradation mechanisms by the condensation reaction between FA and carbon. For side–sealing, the space between the cell and the encapsulant was estimated to contain approximately 1,260,000 times more H2O than in over–sealing, which catalyzed the degradation of the perovskite crystal. Our results demonstrate that MA–based PSCs, which are generally considered to be thermally sensitive, can significantly extend their thermal stability after the proper encapsulation. Therefore, we emphasize that finding the appropriate combination of encapsulation technique and perovskite composition is quite important to achieve further device stability.
Keywords:
carbon electrode
; fully printable
; thermal stability
; long-term stability
1. Introduction
Society is demanding renewable energy sources to replace CO2–emitting energy. As an alternative, perovskite solar cells (PSCs) have attracted much attention. PSCs were invented by Prof. T. Miyasaka and his group in 2009.[1] As of May 2024, a power conversion efficiency (PCE) of 26.07% (certified 25.8%) for PSCs has been achieved by Prof. H. Zhou and his group.[2] This is close to the world's highest efficiency of 26.81% for widely used Si solar cells.[3] In addition, the PSCs can be fabricated using only low–cost processes such as coating and printing. Therefore, it has the potential to be a cheaper source of energy than existing solar cells.
However, to make PSCs more viable than existing solar cells, it is necessary to reduce material and manufacturing costs and improve durability.[4] Typical PSCs use expensive organic charge transport materials (e.g., Spiro–MeOTAD, fullerene C60, and PEDOT: PSS) and precious metals as back contact (e.g., Au and Ag). These electrodes are formed by vacuum evaporation, and the equipment cost can be high. In addition, it has been reported that Au and Ag electrodes are easily degraded by metal migration under operating conditions.[5,6] It has also been reported that perovskite crystals are easily decomposed by moisture and oxygen in the air.[7]
To solve these problems of PSCs, important points are (1) to replace unstable materials with stable materials and (2) to establish an appropriate encapsulation technology.
Regarding points (1), to utilize stable materials, carbon–based fully printable mesoscopic PSCs were developed by Prof. H. Han and his group in 2013, replacing expensive and unstable organic materials and metal electrodes with carbon electrodes.[8,9] These PSCs used a three–layer mesoscopic structure of mesoporous TiO2 (m–TiO2) as the electron transport layer (0.5 μm), m–ZrO2 as the insulating layer (1–3 μm), and porous carbon as both the hole transport layer and back contact (>10 μm). This structure is based on the prototype monolithic dye–sensitized solar cells (DSSCs) developed by Prof. M Grätzel and his group in 1996.[10] This monolithic structure will be referred to as “multiporous–layered–electrode (MPLE) structure” in this present manuscript. The PCE of MPLE–PSCs is lower than thin film (TF)–PSCs with metal electrodes. In the published work on MPLE–PSCs, the PCE is mostly 10~19%,[11,12,13] with a maximum of 22.2%.[14] On the other hand, in the published work on TF–PSCs, the PCE can be over 20%,[15] with a maximum of 26.07%.[2] The low PCE of MPLE–PSCs can be improved by strategies such as enhanced hole transport capability, energy level matching, and dense packing of larger perovskite crystals within the porous layer. For more information, see the excellent review articles.[16,17,18]
However, the most important features of MPLE–PSCs are their low manufacturing cost and excellent stability.[19] This is because the thick carbon electrodes of MPLE–PSCs effectively prevent water and oxygen from entering the cell.[20,21] In fact, many of the published papers reported results of high stability to light, maximum power point tracking (MPPT), heat, and moisture. G. Grancini et al. reported light stability of one year (>10,000 h) for MPLE–PSCs with 2D/3D perovskite with AVAI as an additive.[22] In addition, A. Mei et al. reported that MAPbI3–based MPLE–PSCs with (5–AVA) iodide as an additive passed the MPPT test for >9,000 h.[23] Furthermore, the authors reported that encapsulated MPLE–PSCs with similar (5–AVA)xMA1–xPbI3 passed the dump–heat test (85 °C/85 %RH, International Summit on Organic Photovoltaic Stability (ISOS)–D–3 protocol) for >3,000 h.[20]
Regarding point (2), to commercialize PSCs, a proper encapsulation technology is required to withstand harsh outdoor conditions.[24] L. Shi et al. reported that encapsulated TF–PSCs using Cs0.05FA0.8MA0.15Pb(I0.85Br0.15)3 perovskite with a polyisobutylene–based polymer blanket and cover glass sheets passed a dump–heat test of >1,800 h and a humidity freeze test of >75 cycles.[25] Their results show that mixed cation cells containing Cs have the highest stability. In addition, the encapsulation method is also examined, showing that a wide blanket is superior to an edge seal. Therefore, the combination of perovskite crystal composition, encapsulant, and encapsulation method must be optimized to effectively prevent perovskite crystal degradation.[26]
Similarly, encapsulants and sealing methods have been also investigated for MPLE–PSCs. Z. Fu et al. reported that MPLE–PSC submodules (10×10 cm) encapsulated with polyurethane and glass sheets maintained 97.52% of their initial efficiency after 2,136 h under outdoor conditions (–10 to 35 °C).[27] They used an over–sealing method that covered the entire surface of the solar cells. In a previous study by the authors, over– and side–sealing methods were compared for MPLE–PSCs using UV–curing glue.[28,29] When UV–curing glue was used as the encapsulant, the side–sealing showed better stability than the over–sealing. Therefore, based on the results of these studies, appropriate sealing methods should be used depending on the type of encapsulant.
Based on these previous studies, we have attempted here to optimize the appropriate combination of encapsulation method and perovskite crystal compositions for the commercialization of MPLE–PSCs. The authors recently reported that MPLE–PSC modules encapsulated with hot–melt ionomer films have a stability equivalent to 20 years of outdoor use.[20] In the previous report with hot–melt ionomer films, only the over–sealing method was used, and the side–sealing method was not investigated. Therefore, this study compared the effectiveness of over– and side–sealing methods using hot–melt ionomer films (Figure 1a).
This work also compared the perovskite crystal composition of MPLE–PSCs was compared between methylammonium (MA)–based and formamidinium (FA)–based perovskites (Figure 1b). Due to differences in molecular size and bonding strength, MA perovskite has lower thermal stability than FA perovskite.[30] However, MA–based perovskites are widely used in MPLE–PSCs and have demonstrated high stability, making them the mainstream. On the other hand, high–efficiency FA–based MPLE–PSCs have been successful in only a few groups, and their potential is still unknown.[31,32,33,34] Therefore, we also addressed the comparison between MA and FA perovskites.
In this study, in order to properly evaluate the thermal stability of MPLE–PSCs with MA and FACs perovskites, thermal stability tests were performed at 85°C according to the ISOS–D–2 protocol.[35] The standard thermal stability test for solar cells is typically performed in the range of –40 to +85 °C. In this study, the only high–temperature side (85 °C) was focused on first because perovskite crystals, especially MA perovskite, are known to decompose when exposed to high temperatures (Figure 1c).
2. Results and Discussion
The compositions of the perovskite crystals used for MPLE–PSCs in this study were (5–AVA)0.05(MA)0.95PbI3 and FA0.9Cs0.1PbI3. Herein, these perovskite compositions are simplified and referred to as MA perovskite and FACs perovskite, respectively.
Morphology of PSCs Used in Thermal Stability Test
The MA and FACs perovskites compared in this study have different perovskite crystal states due to the presence of additives and different crystallization conditions. Figures S1a and b show the SEM images of the perovskite crystals deposited on the carbon electrode surface of the MPLE–PSCs with MA and FACs perovskites. The crystal grain size showed an obvious difference between MA and FACs perovskites, with MA perovskite having a grain size of about 20 μm, while FACs perovskite had larger grains at about 500 μm. The reason for the smaller MA perovskite grains may be due to the addition of the 5–AVAI as an additive. A. Mei et al. reported that the 5–AVA cations have a large effective ion radius exceeding the tolerance factor to replace the MA site, 5–AVA could grow on the surfaces of MAPbI3 grains and adjust the grain size of MAPbI3.[23] For this reason, the grain size of MAPbI3 with 5–AVAI may be limited below a certain size. They have also reported that the addition of 5–AVAI to MAPbI3 increases the stability of the perovskite crystals by strengthening the grain boundaries and forming strong bonds with TiO2 and ZrO2 nanoparticles.[23] On the other hand, it is believed that the larger crystal grain size observed in FACs perovskite can be attributed to the use of the solvent evaporation controlled crystallization (SECC) method, as proposed by Q. Wang et al.[31] This method allows for a relatively slower crystallization process to occur, which may contribute to the formation of larger crystal grains. These grain boundaries can be the starting point for degradation. The 25–fold difference in crystal size between the two perovskite absorbers is due to the difference in the method used to densely pack the perovskite crystals within the multiporous layered structure. In MA perovskite, this is due to the effect of the 5–AVAI cation's effect on grain size, while in FACs perovskite, it is due to the SECC method, which allows slow crystal growth without the use of additive molecules. The SECC method is based on the Ostwald ripening phenomenon, in which small particles shrink and disappear and larger particles grow in a system of particles of different sizes dispersed in the matrix phase.[31] This allows the formation of such large perovskite crystals. In addition, in FACs perovskite, grain boundaries between grains are large and prominent (Figure S1b).
The presence of multiple voids in both MPLE–PSCs was confirmed by cross–sectional SEM images (Figures S1c and d, the areas indicated by arrow). Thus, both MPLE–PSCs with MA and FACs perovskites had an incomplete filling of the perovskite crystals in the MPLE. These voids, as defects, can degrade device performance. The formation of these defects can be effectively suppressed by controlling the perovskite composition and the crystal growth process.[31,36]
Initial Device Performance
Then, the J–V, IPCE, and EIS results were carried out to compare and analyze the performances of MPLE–PSCs with MA and FACs perovskites for the thermal stability tests. Figure 2a,b show the J–V curves at forward and reverse scans for MPLE–PSCs with MA and FACs perovskites, respectively. The parameters at reverse scan for the champion devices were MA and FACs perovskites with short circuit current density (JSC) of 20.25 mA cm–2 and 17.74 mA cm–2, open circuit voltage (VOC) of 0.899 V and 0.912 V, fill factor (FF) of 66.71% and 76.52%, and PCE of 12.14% and 12.38%, respectively. Figure 2c shows the box–and–whisker plots of the J–V parameters in 15 cells. The JSC tended to be higher for MA perovskite than for FACs perovskite. On the contrary, the VOC and FF tended to be higher for FACs perovskite than for MA perovskite. However, both devices had similar average PCE values of 10–11%.
From the IPCE spectra, FACs perovskite shows lower efficiencies than MA perovskite over the entire wavelength range (Figure 2d). The integrated JSC was 19.02 mA cm–2 and 16.46 mA cm–2 for MA and FACs perovskite, respectively. The lower JSC value for FACs perovskite can be attributed to the reduced active area due to grain boundaries, as seen in the SEM image. As supporting information, the slight difference between integrated JSC and J–V JSC is due to the leakage of scattered light from the glass substrate to the outside of the device without absorption by the photoactive area. This is due to the different optical diffusion (or parallel) characteristics of the two measurements.[37]
In addition, the resistance component of the devices with MA and FACs perovskites was analyzed by EIS at 0 V bias under the light. The Nyquist plots are shown in Figure 2e and the resistivity parameters obtained from the fitting results are shown in Table S1. The EIS analysis was performed using the equivalent circuit model (Figure 2e), which was modeled as a mixed conductor system proposed by M. Bag et al..[38] The model circuit consists of the following elements. The high–frequency component consists of the charge and ion transport resistance (Rtr) coupled with the interfacial charge transfer resistance (RCT). The ion accumulation at the interface and the charge stored in the bulk perovskite layer are modeled as the interfacial Debye–layer capacitance (Cdl) and the chemical capacitance (Cg), respectively. Free carrier recombination/transport (both electrons and holes) is modeled by another resistance term (Relectr). The series resistance (RS) represents the electrical resistance present on the carrier transport path in the device. The linear region in the low–frequency range is modeled by the Warburg diffusion (WS). The RS and Relectr were smaller in MA perovskite than in FACs perovskite. On the other hand, Rtr and RCT were smaller in FACs perovskite than in MA perovskite. Therefore, MA perovskite has higher charge/ion transport resistance and interfacial charge transfer resistance, and lower free carrier recombination/transport resistance than FACs perovskite. This can be attributed to the addition of 5–AVAI to MAPbI3, which limited the size of the perovskite crystals and increased defects.[23] Despite the differences in device parameters, the PCEs of the MPLE–PSCs using MA and FACs perovskites were found to be almost similar. As a result, the MPLE–PSCs with similar PCEs worked for the comparisons of thermal stability tests.
Thermal Stability of Unencapsulated Devices
The thermal stability at 85 °C of MPLE–PSCs with and without encapsulation was then investigated. First, the variation of properties with heat on unencapsulated devices was compared by XRD and J–V analysis. The results confirmed that, as is well known, MA perovskite crystals are more sensitive to the thermal environment than FACs perovskite crystals.[39,40] Figure 3a,b show the XRD patterns of MPLE–PSCs with MA and FACs perovskites in the 10–18° range. Each peak near 14° shows MAPbI3 (110) and FACsPbI3 (001). The peaks near 13° are attributed to PbI2 (001), and the peaks near 12° seen in FACs perovskite are associated with Bragg reflections of FAI.[41] The XRD peaks showing each perovskite crystal decreased with aging time, and the PbI2 peaks appeared simultaneously. The aging variation in these MA and FACs perovskites was compared (Figure 3c). The XRD intensity of MAPbI3 (110) decreased rapidly to about 20% of its initial value after 100 h. On the other hand, the XRD intensity of FACsPbI3 (001) remained at 90% after 100 h and decreased to 40% after 300 h. In addition, Figure 3d shows the aging variation of the PCE, which continued to be acquired simultaneously with the XRD measurements during the thermal stability test. The PCE of MPLE–PSC with MA perovskite crystal continued to decrease with aging time and reached almost 0% after 300 h. On the other hand, the PCE of MPLE–PSC with FACs perovskite decreased to 80% of its initial value in the first 10 h and maintained 50% of its initial value after 300 h. The initial degradation in the thermal stability test can be attributed to changes in the state of the perovskite crystals due to the transition from the device fabrication environment in a vapor solvent atmosphere to the thermal stability test in a dry atmosphere. Therefore, without encapsulation, both perovskites were degraded by heat, but it was more pronounced for the MA perovskite.
Thermal Stability of Encapsulated Devices
Surprisingly, the trend of stability was reversed in the encapsulated device, contrary, with MA perovskite showing higher thermal stability than FACs perovskite. Figure 1, Figures S2, and S3 show the schematic diagram, sealing procedure, and photographs of the over– and side–sealing methods. In the over–sealing structure, the top of the cell contacts the encapsulant, which covers the entire cell. In contrast, as a side–sealing structure, the top of the cell does not contact the encapsulant, which covers only the surrounding area of the cell. The difference between the two encapsulation structures is the presence or absence of space above the cell.
Thermal stability tests were performed by placing the MPLE-PSCs on a hot plate at 85 °C in an ambient atmosphere (Figure S4). Figure 4a shows the PCE variation from the initial value during thermal stability tests of encapsulated MPLE–PSCs with MA and FACs perovskites. In addition, Figures S5 and S6 show the variations of JSC, VOC, FF and Rs, Rsh. In FACs perovskite, after 500 h, the PCE decreased to about 30% and 10% of the initial value for over– and side–sealing, respectively. These degradations were due to the overall degradation of device performance, with the JSC degradation rate being particularly significant. Therefore, the FACs perovskite did not show any improvement in thermal stability after the encapsulation. It is known that the amine groups in FA can react with carbon via a condensation reaction, which may have accelerated the degradation of the perovskite crystals.[42] In addition, large grain boundaries exist between the large crystals of FACs perovskite. These grain boundaries are the area where the perovskite crystals are not packed. H2O in the encapsulated device could easily reach the perovskite crystals in the ZrO2 and TiO2 layers, and degradation could have proceeded immediately from these starting points.
On the other hand, the MA perovskite maintained about 90% of its initial PCE up to 1800 h for both encapsulation methods. The side–sealing devices showed a gradual decrease in PCE, down to about 60% of the initial value after more than 5000 h. However, the over–sealing device surprisingly maintained 90% of its initial PCE more over 5000 h. The lifetime of each device is then compared when its performance drops to 80% of its initial value (T80 lifetime). The T80 lifetimes were 16, >5000, 3580 h for without encapsulation, over–, and side–sealing for MA perovskite, and 125, 11, and 35 h for without encapsulation, over–, and side–sealing for FA perovskite, respectively (Table S2). Initial degradation was observed for MA perovskite. This is explained by the burn–in effects observed in the early stages of the thermal stability test.[35] The ability to achieve high stability despite the presence of several defects in the perovskite crystals of the device, as seen in the cross–sectional SEM, indicates the high potential for commercialization of MPLE–PSCs. The reason for the improved thermal stability of MA perovskite can be attributed to the effective suppression of the MAPbI3 decomposition reaction by encapsulation.
The main degradation factors in these MA– and FACs–based MPLE–PSCs were the decrease in JSC and FF. Therefore, the changes in IPCE and EIS during the thermal stability test were also investigated (Figure S7–10). For FACs perovskite, the total IPCE continued to decrease and the resistive component continued to increase regardless of the encapsulation method. On the other hand, MA perovskite showed changes in IPCE and EIS after 5000 h of thermal stability test depending on the encapsulation method. No significant changes were observed in over–sealing, while side–sealing showed a decrease in IPCE and an increase in the resistive component in after 5000 h. Thus, the thermal degradation rate of perovskite varies depending on the perovskite crystal and the encapsulation method.
In addition, changes in the appearance of the active area of the device during the thermal stability test were compared (Figure 4b). In the MA perovskite, there was no noticeable change after 5000 h in the over–sealing devices. In the side–sealing device, several yellow dots appeared in the active area after 5000 h. This is attributed to the decomposition of the perovskite crystal into PbI2. Interestingly, the yellow dots associated with PbI2 precipitation did not appear at the edges of the cell, but rather throughout the active area. This can be attributed to the presence of space above the cell, even though the area around the cell is completely sealed by the encapsulant. On the other hand, for the FACs perovskite, the perovskite decomposed and turned yellow during the thermal test regardless of the encapsulation method. The degree of discoloration was more pronounced on the side–sealing device. Therefore, regardless of the perovskite composition, side–sealing would have a more negative effect on the device than over–sealing.
As supporting information, a similar thermal stability test was performed at 100 °C which is 15 °C higher than the 85 °C specified in the ISOS–D–2 protocol to confirm thermal stability at even higher temperatures (Figures S11 and 12). The difference in thermal stability between the perovskite composition and encapsulation method combinations was the same as for the 85 °C case. However, for all combinations, the thermal stability was lower than at 85 °C. The T80 lifetime was 432, 22, 48, and 15 h for over– and side–sealing for MA perovskite and for over– and side–sealing for FA perovskite, respectively (Table S3). At 100 °C, the combination of MA perovskite and over–sealing showed the highest stability, with a change from the initial value of about 70% at 3500 h and about 40% at 5000 h. Thus, it should be noted that for MA–based perovskites, there is a temperature limit at which the cell can be protected by proper encapsulation.
Degradation Mechanisms for Over– and Side–Sealing Devices
Based on these results, we propose a thermal degradation model for the effect of over– and side–sealing using thermoplastic resin films.
As a prerequisite, all processes including the encapsulation of the MPLE–PSCs fabricated in this study were performed under an ambient atmosphere, which is not a special environment. Therefore, the encapsulated device contains a certain amount of water (H2O) depending on the encapsulation method. The amount of H2O molecules contained in the device with over- and side-sealing was estimated by calculation (the details of the calculation are described in Supplementary Text S1). The estimated number of H2O molecules sealed in the over–sealing and side–sealing devices are 1.47×1010 and 1.85×1016, respectively (Figure 5a). This amount of H2O in the side–sealing is 1,263,160 times the amount in the over–sealing. The large difference is due to the space formed by the side–sealing method. The H2O has a catalytic effect and reacts with MAPbI3 to decompose the perovskite crystals.[43] The decomposition reaction equation for MAPbI3 under the influence of water is shown below.
CH3NH3PbI3 ⇄ CH3NH3PbI3・H2O, (CH3NH3)4PbI6・2H2O
→ CH3NH2 (gas) + PbI2 + HI (gas) + other compositions
Here, the number of perovskite units in the devices is estimated to be 2.04×1012 (Figure 5b, the details of the calculation are described in Supplementary Text S2). The number of perovskite crystal units is approximately 13,000,000 and 11 times the number of H2O molecules in the over– and side–sealing devices, respectively. Thus, the number of H2O molecules in the side-sealing device will be sufficient to fracture the perovskite crystals.
Furthermore, it has been reported that in MPLE–PSCs, MAPbI3 perovskite crystals in the multiporous layered electrode decompose in a thermal environment. Z. Fu et al. showed that after approximately 200 h of thermal stability testing at 85 °C in an unencapsulated (5–AVA)x(MA)1–xPbI3–based MPLE–PSC device, holes formed in the TiO2/ZrO2 layer and the perovskite absorber decomposed.[27] In addition, A. K. Baranwal et al. disassembled side–sealing MAPbI3–based MPLE–PSC devices after 7000 h of thermal stability testing at 100 °C and analyzed the internal perovskite state. [29] They found that MA+ species in MA–based perovskites readily react with carbon under applied thermal stress, resulting in perovskite crystal decomposition.
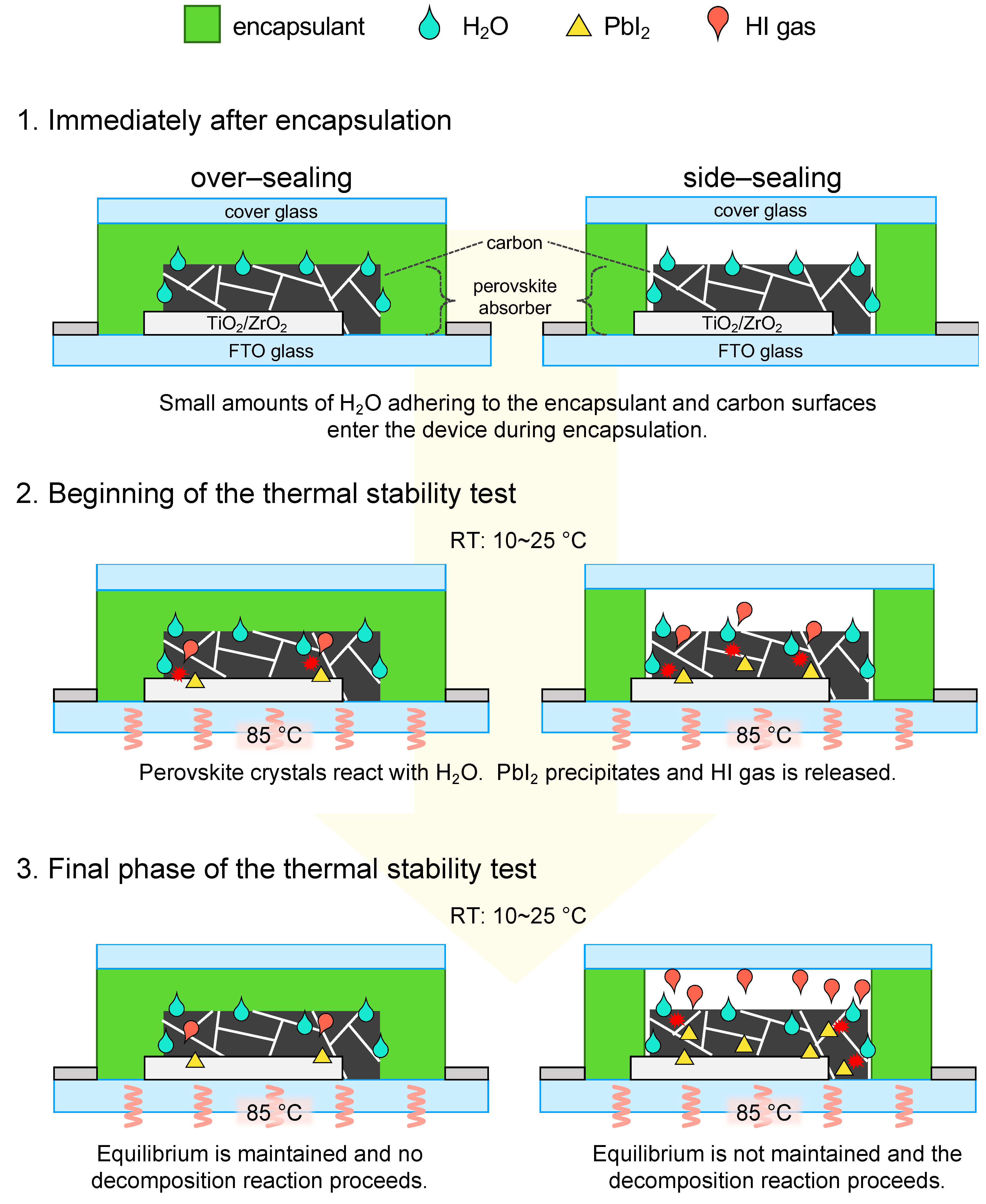
Based on these results and assumptions, when encapsulated cells are placed in a thermal environment, the following phenomena are expected to occur, depending on the encapsulation method, as shown in Figure 6.
Over–sealing: 1. Immediately after encapsulation in an ambient atmosphere, the device is completely covered by the encapsulant and contains a small amount of H2O between the encapsulant and the device. The estimated number of H2O molecules sealed in the over–sealing device is 1.47×1010. 2. When the thermal stability test starts, the heated H2O becomes water vapor and permeates enters the cell. The equilibrium reaction is shown in Eq. (1) proceeds, some perovskite crystals are decomposed, PbI2 is precipitated, and HI gas is released. 3. However, the amount of H2O sealed in the over–seal device is relatively small. Therefore, the equilibrium condition in Eq. (1) is expected to be maintained and the precipitation of PbI2 and the release of HI gas will not exceed a certain amount. Therefore, the degradation of perovskite crystals is not expected to progress beyond a certain level that has reacted at the beginning of the thermal stability test.
Side–sealing: 1. Immediately after encapsulation under an ambient atmosphere, an enclosed space is formed between the device and the cover glass, trapping air containing H2O. In addition, H2O adheres to the encapsulant and device. The estimated number of H2O molecules sealed in the side–sealing device is 1.85×1016. This amount of H2O is approximately 1,260,000 times the amount in the over–sealing device. 2. At the beginning of the thermal stability test, the perovskite crystals in the carbon layer react with H2O to precipitate PbI2 and release HI gas. This HI gas is released toward the space between the device and the encapsulating components. Therefore, the equilibrium reaction Eq. (1) of the device is no longer valid, and only the positive reactions will continue to proceed. In addition, the top of the cover glass was in contact with the surrounding atmosphere (RT: 10~25 °C), which constantly cooled the space between the device and the cover glass. This may have cooled the H2O and volatile gases in the space and reduced the gas pressure in the space, which further promoted the HI gas volatilization of the device. 3. Once the equilibrium is lost, the perovskite crystals continue to decompose. In addition, humid air containing H2O from outside the device can enter the device through a small gap between the encapsulant of the side–sealing and the adhesive surface of the cover glass. In the final phase of the thermal stability test, the decomposition reaction will eventually progress to the perovskite crystals in the exposed m–ZrO2 and m–TiO2 layers, and the device performance is expected to deteriorate as a result.
3. Conclusions
Solving the problems of cost and stability is the main key to the practical application of PSCs. In this study, the thermal stability (at 85 °C) of low–cost carbon–based MPLE–PSCs was investigated. The light absorbers (MA and FACs perovskites), as well as the encapsulation methods, over– and side–sealing with thermoplastic resin films, were compared. As a result, without encapsulation, FACs perovskite showed higher thermal stability than MA perovskite. However, with encapsulation, the phenomenon of thermal stability was reversed as the MA perovskite showed higher thermal stability than the FACs perovskite. It is believed that the thermal decomposition reaction of MAPbI3 was effectively suppressed by encapsulating the device. On the other hand, the thermal stability of MPLE–PSCs with FACs perovskites was not improved by encapsulation. This was an issue of crystal quality and material of the FACs perovskite. In particular, the condensation reaction between the amine groups in the FACs and carbon can occur in the encapsulated cells, resulting in the decomposition of the perovskite crystals starting from the grain boundaries.
Using the thermoplastic ionomer sealant, the over–sealing devices in this study showed higher thermal stability than the side–sealing devices. For MA perovskite, the normalized PCE at the initial value after >5,000 h was approximately 90% and 60% for over– and side–sealing devices, respectively. It was estimated that approximately 1,260,000 times more H2O is trapped in the cell in side–sealing devices than in over–sealing devices due to the space at the top of the cell. Because of this relatively large amount of H2O, the device degraded faster in side–sealing. In addition, because the upper part of the space is always cooled by cold air from outside, the gas pressure in the space decreases and equilibrium reactions are not established. Once equilibrium was lost, the decomposition of the perovskite crystals continued to progress. On the other hand, in the over–sealing device, the top of the cell is completely covered by the encapsulant, which suppresses the release of these volatiles to the outside of the cell. These results indicate that over–sealing encapsulation can significantly extend the thermal stability of devices, even when using MA perovskites with low thermal stability. However, it should be noted that different encapsulant materials may change the optimal encapsulation method. Therefore, this study demonstrates the high reliability of carbon–based MPLE–PSCs for commercialization, with the thermoplastic ionomer sealant.
4. Experimental
4.1. Materials
All of the commercial materials were used as received without any purification including titanium diisopropoxide bis (acetylacetonate) (75 wt.% in isopropanol, Sigma–Aldrich, United States); titanium dioxide (TiO2) paste (PST–30NRD, JGC Catalysts and Chemicals, Japan); zirconium dioxide (ZrO2) paste (Zr–Nanoxide ZT/SP, Solaronix SA, Switzerland); α–terpineol (Kanto Chemical, Japan); lead iodide (PbI2, 99.99%, Tokyo Chemical Industry, Japan); methylammonium iodide (MAI, 98.0%, Tokyo Chemical Industry, Japan); 5–ammonium valeric acid iodide (5–AVAI, Greatcell Solar, Australia); γ–butyrolactone (GBL, Electrochemical grade, Kanto Chemical, Japan); formamidine hydroiodide (FAI, 99.99%, Tokyo Chemical Industry, Japan); cesium iodide (CsI, 99.0%, Tokyo Chemical Industry, Japan); N,N–dimethylformamide (DMF, FUJIFILM Wako Pure Chemical Corporation); dimethyl sulfoxide (DMSO, FUJIFILM Wako Pure Chemical Corporation); fluorine–doped tin oxide (FTO) glass (TEC–15, Nippon Sheet Glass–Pilkington, Japan); thermoplastic resin films consisting of an ionomer–based material (KuranSeal–ES, Kurabo Industries Ltd., Japan); and cover–glass (ASLAB Slide Glass, AS ONE Corporation, Japan). TiO2 paste for electron transport layer (ETL) was obtained by 2 g TiO2 paste was diluted with 3 g α–terpineol (1:1.5 weight ratio) and ultrasonically stirred for 30 minutes using an ultrasonic bath, and then, stirred for 24 h at room temperature using a magnetic stirrer (500 r/min). Carbon paste for back contact was obtained by 0.5 g of carbon black, 2 g of ultra–fine artificial graphite, 0.3 g of ZrO2 nanoparticles, 0.4 g of the polymer binder thickeners, 10 g of α–terpineol, 1 g of ethanol, and three sizes ZrO2 beads (10, 5, and 2 mm) were placed in a zirconia ceramic bowl and crushed and stirred in a planetary ball mill (Planet M2–3F, Nagao System, Japan) for 5 h (700 r/min), then milled at low speed (300 r/min) for 5 min to remove air bubbles in the paste. Preparation of the perovskite precursor solution was performed in a glove box filled with N2 gas (the dew point adjusted to –30 to –20 °C). The 1.2 mol/L (5–AVA)0.05(MA)0.95PbI3 perovskite precursor solution was obtained by dissolving 0.2776 g PbI2, 0.0906 g MAI and 0.0074 g 5–AVAI in 500 μL GBL, and then stirred at 70 °C and 800 r/min for overnight. The 1.0 mol/L FA0.9Cs0.1PbI3 perovskite precursor solution was obtained by dissolving 0.2305 g PbI2, 0.0774 g FAI, and 0.0130 g CsI in 500 μL DMF/DMSO (=4:1, v/v), and then stirred at 50 °C and 800 r/min for overnight.
4.2. Device Fabrication
The device used in this work was fabricated according to procedures previously reported in the literature.[37,44,45,46,47,48] All device fabrication processes, including the perovskite crystal filling process, were performed under ambient air conditions (room temperature was 15 to 25 °C and the relative humidity was 40% to 80% RH). The FTO conductive layer on the glass substrate (100×60 mm) was separated to be small solar cells using a laser etching instrument. The electrical insulation between the FTO layers of solar cells was confirmed using an electrical multimeter, and then, the etched substrate was ultrasonically cleaned with a detergent solution (1 wt.%, white 7–AL, Yuai Kasei, Japan) and ethanol for 15 min, respectively. A blocking layer of compact TiO2 (c–TiO2) was deposited on patterned substrates by spray pyrolysis deposition (SPD) of a 0.66 mL titanium diisopropoxide bis (acetylacetonate) solution diluted in 22.5 mL ethanol (1:34 volume ratio) evenly on the hot plate at 500 °C. The SPD was performed on the entire hot plate with 10–sec spray intervals with glass plate masks on electrical contacting parts. The 0.5 μm thick mesoporous TiO2 (m–TiO2) layer was screen printed using the TiO2 paste, kept in a container with a lid at room temperature for 10 min for leveling, and then dried on a hot plate at 50 °C for 5 min and 140 °C for 5 min. The 1.0 to 1.5 μm thick mesoporous ZrO2 (m–ZrO2) spacer layer was screen printed using the ZrO2 paste, kept in a container with a lid at room temperature for 5 min for leveling, and then dried on a hot plate at 50 °C for 5 min and 140 °C for 5 min. The TiO2 and ZrO2 layers were annealed at 500 °C for 1 h in a hot plate with the lid (temperature rising time was 20 min from room temperature to 500 °C and holding time at 500 °C was 40 min). After annealing of the TiO2 and ZrO2 layers, the 10 to 20 μm thick carbon layer was screen printed using carbon paste on the porous ZrO2 layer, kept in a leveling container with a lid at room temperature for 10 min, and then dried on a hot plate at 125 °C for 10 min. The printed carbon layer was annealed at 400 °C for 1 h on a hot plate with the metal lid closed (temperature rising time was 15 min from room temperature to 400 °C and holding time at 400 °C was 45 min). Then, the substrates were cooled to room temperature and separated into single cells. The multiporous–layered–electrodes were sintered at 400 °C for 50 min on a hot plate with the metal lid closed (temperature rising time was 15 min, holding time at 400 °C was 35 min) before applying the perovskite precursor solution. Then, the substrates were cooled to room temperature, and ultrasonic soldering to attach electrical metal contacts onto both external contacts on the cell substrate. The area around the porous electrodes was masked with heat–resistant polyimide tape so that the two types of perovskite solutions ((5–AVA)0.05(MA)0.95PbI3 or FA0.9Cs0.1PbI3) filled in the porous electrodes, as below;
(5–AVA)0.05(MA)0.95PbI3 perovskite: 4.0 μL of the precursor solution was supplied to the porous electrodes (area: 0.7 cm2) by drop–casting. The filled devices were kept with a petri dish cover at room temperature for 30 min, and then transferred to a 50 °C hot plate with a petri dish cover and kept for 30 min. Then, the petri dish cover was removed and the device was dried at 50 °C for 1 h. This procedure was performed under ambient conditions (without a glove box).
FA0.9Cs0.1PbI3 perovskite: 4.0 μL of the precursor solution was supplied to the porous electrodes by drop–casting in a dry–air–filled glove box. The filled devices were kept with a petri dish cover at room temperature for 5 min, and then transferred to a 70 °C hot plate with a petri dish cover and kept for 2 h.
4.3. Encapsulation
Thermoplastic ionomer films were used as the encapsulant. The films were placed between the cells and cover glass. The difference between over– and side–sealing methods is as follows; over–sealing: Encapsulant covers the entire edge of the cell and the top of the cell. side–sealing: Encapsulant seals only the edge of the cell. The samples were laminated at 110 °C for 5 min using a commercial vacuum laminator (PVL0202S, Nisshinbo Mechatronics, Japan).[20]
4.4. Characterization
The surface of the carbon electrode with perovskite crystal and the cross–section of MPLE–PSC devices were observed using a scanning electron microscope (SEM, JSM–6510, JEOL, Japan). The photocurrent density–voltage (J–V) curves were measured with a DC voltage current source (B2901A, Agilent, USA) under a solar simulator (AM1.5G, 100 mW cm–2) equipped with a 500 W xenon lamp (YSS–100A, Yamashita Denso, Japan). The irradiation intensity of the AM1.5G solar simulator was calibrated using a reference silicon photodiode (Bunkoukeiki Co., Ltd., Japan). The light–irradiation area was 0.3×0.3 cm (the active area of the device was 0.7 cm2 and the mask opening area was 0.09 cm2). The measurement voltage ranged from –0.05 to 1.05 V with forward and reverse scans, the step was set to 0.01 V, the integration time was set to 16.7 ms, and the scan delay time was 10 ms (the measured scan rate was 250 mV s–1). The device was irradiated in AM1.5G light for 1 min before the J–V measurement. The J–V measurements were performed three times, with an interval of 1 min between each measurement, and the device was illuminated continuously with light between measurements for the photoactivation under open–circuit conditions. The incident photon–to–current efficiency (IPCE) spectra were measured using a 150 W Xe lamp (TSM–K1, BSO–X150) equipped with a monochromator (MHM–K1) as a monochromatic light source. Calibration with the silicon photodiode (Bunkoukeiki Co., Ltd., Japan) was carried out before the IPCE measurements. The IPCE measurement was performed three times, and the interval was set to 3 min for the weak–photoactivation. For more information on IPCE measurement methods for MPLE–PSCs, please refer to previous papers by the authors.[37] At J–V and IPCE measurements, the stabilized results of the 3rd measurement were used and compared. Electrochemical impedance spectroscopy (EIS) of the devices was performed on electrochemical workstations (Bio–Logic, France) in the frequency range from 1 Hz to 100 kHz at 0 V bias under the light. The thermal stability of perovskite crystals was characterized by x–ray diffraction (XRD, MiniFlex II, Rigaku, Japan).
4.5. Stability Test
The thermal stability test was performed by keeping the device on a hot plate at 85 °C in the ambient atmosphere (RT, 20–60 %RH). During measurement and analysis, the devices were taken down from the hot plate and returned to room temperature before measurement and analysis were performed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
S.I. supervised the project. R.T. and S.I. conceived the concept. R.T., Y.N., and K.O. performed the experiments. R.T. wrote the original manuscript. R.T. and S.I. revised and edited of the paper. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
This work was supported by the Japan Science and Technology Agency (JST), EIG CONCERT–Japan, Strategic International Collaborative Research Program (SICORP), the project name “Printable Fully Inorganic Porous Metal Oxide Based Perovskite Solar Cells: Defining Charge Selective Oxides for High–efficient and Low–cost Device Structure (PROPER)” (19–191029644) , Japan Aerospace Exploration Agency (JAXA), Space Exploration Innovation Hub 3rd Request for Proposal (RFP), the project name “Development for Enhancing the Durability of Highly Efficient, Low–cost, and Lightweight Thin–film Perovskite Solar Cell Devices”, Japan Society for the Promotion of Science (JSPS) KAKENHI Grant number 22KJ2631, Grant–in–Aid for JSPS Fellows, DC2, the project name of “Development of Ultra–Low–Cost Perovskite Solar Cells based on Printed Inorganic Nanoporous Electrodes”, and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant number 23KJ1175, Grant–in–Aid for JSPS Fellows, PD, the project name of “Development of Ultra–Low–Cost, High–Durability Perovskite Solar Cells Based on Inorganic–Carbon Materials”. A part of this research was supported by the Azbil Yamatake General Foundation and The Electrochemical Society of Japan (Special Scholarship Donations). R.T. acknowledges the scholarship support of the Takigawa Scholarship Foundation, Tokyo Electric Power Company Holdings, Incorporated (TEPCO) Memorial Foundation, and Miyake Shotaro Scholarship Society.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bai, Y.; Huang, X.; Li, J.; Wu, Y.; Chen, Y.; Li, K.; Niu, X.; Li, N.; Liu, G.; Zhang, Y.; Zai, H.; Chen, Q.; Lei, T.; Wang, L.; Zhou, H. Anion–π Interactions Suppress Phase Impurities in FAPbI3 Solar Cells. Nature 2023, 623, 531–537. [Google Scholar] [CrossRef]
- Lin, H.; Yang, M.; Ru, X.; Wang, G.; Yin, S.; Peng, F.; Hong, C.; Qu, M.; Lu, J.; Fang, L.; Han, C.; Procel, P.; Isabella, O.; Gao, P.; Li, Z.; Xu. X. Silicon Heterojunction Solar Cells with up to 26.81% Efficiency Achieved by Electrically Optimized Nanocrystalline-Silicon Hole Contact Layers. Nat. Energy 2023, 8, 789–799. [Google Scholar] [CrossRef]
- Meng, L.; You, J.; Yang, Y. Addressing the Stability Issue of Perovskite Solar Cells for Commercial Applications. Nat. Commun. 2018, 9, 5265. [Google Scholar] [CrossRef]
- Domanski, K.; Correa-Baena, J.P.; Mine, N.; Nazeeruddin, M.K.; Abate, A.; Saliba, M.; Tress, W.; Hagfeldt, A.; Grätzel, M. Not All That Glitters Is Gold: Metal-Migration-Induced Degradation in Perovskite Solar Cells. ACS Nano 2016, 10, 6306–6314. [Google Scholar] [CrossRef] [PubMed]
- Svanström, S.; Jacobsson, T.J.; Boschloo, G.; Johansson, E.M.J.; Rensmo, H.; Cappel, U.B. Degradation Mechanism of Silver Metal Deposited on Lead Halide Perovskites. ACS Appl. Mater. Interfaces 2020, 12, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Im, H.-S.; Seo, J.-Y.; Park, N.-G. Material and Device Stability in Perovskite Solar Cells. ChemSusChem 2016, 9, 2528–2540. [Google Scholar] [CrossRef]
- Ku, Z.; Rong, Y.; Xu, M.; Liu, T.; Han, H. Full Printable Processed Mesoscopic CH3NH3PbI3 /TiO2 Heterojunction Solar Cells with Carbon Counter Electrode. Sci. Rep. 2013, 3, 3132. [Google Scholar] [CrossRef]
- Mei, A.; Li, X.; Liu, L.; Ku, Z.; Liu, T.; Rong, Y.; Xu, M.; Hu, M.; Chen, J.; Yang, Y.; et al. A Hole-Conductor-Free, Fully Printable Mesoscopic Perovskite Solar Cell with High Stability. Science 2014, 345, 295–298. [Google Scholar] [CrossRef]
- Kay, A.; Gräitzel, M. Low Cost Photovoltaic Modules Based on Dye Sensitized Nanocrystalline Titanium Dioxide and Carbon Powder. Sol. Energy Mater. Sol. Cells 1996, 44, 99–117. [Google Scholar] [CrossRef]
- Meroni, S.M.P.; Worsley, C.; Raptis, D.; Watson, T.M. Triple-Mesoscopic Carbon Perovskite Solar Cells: Materials, Processing and Applications. Energies 2021, 14, 386. [Google Scholar] [CrossRef]
- Fagiolari, L.; Bella, F. Carbon-Based Materials for Stable, Cheaper and Large-Scale Processable Perovskite Solar Cells. Energy Environ. Sci. 2019, 12, 3437–3472. [Google Scholar] [CrossRef]
- Duan, M.; Hu, Y.; Mei, A.; Rong, Y.; Han, H. Printable Carbon-Based Hole-Conductor-Free Mesoscopic Perovskite Solar Cells: From Lab to Market. Mater. Today Energy 2018, 7, 221–231. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Chen, K.; Tian, W.; Sheng, Y.; Jiang, Y.; Zhang, D.; Liu, Y.; Qi, J.; Chen, K.; Ma, Q.; Qiu, Z.; Wang, C.; Yin, Y.; Zhao, S.; Leng, J.; Jin, S.; Zhao, W.; Qin, Y.; Su, Y.; Li, X.; Li, X.; Zhou, Y.; Zhou, Y.; Ling, F.; Mei, A.; Han, H. Electron Injection and Defect Passivation for High-Efficiency Mesoporous Perovskite Solar Cells. Science 2024, 383, 1198–1204. [Google Scholar] [CrossRef]
- Jacobsson, T. J.; Hultqvist, A.; García-Fernández, A.; Anand, A.; Al-Ashouri, A.; Hagfeldt, A.; Crovetto, A.; Abate, A.; Ricciardulli, A. G.; Vijayan, A.; Kulkarni, A.; Anderson, A. Y.; Darwich, B. P.; Yang, B.; Coles, B. L.; Perini, C. a. R.; Rehermann, C.; Ramirez, D.; Fairen-Jimenez, D.; Di Girolamo, D.; Jia, D.; Avila, E.; Juarez-Perez, E. J.; Baumann, F.; Mathies, F.; González, G. S. A.; Boschloo, G.; Nasti, G.; Paramasivam, G.; Martínez-Denegri, G.; Näsström, H.; Michaels, H.; Köbler, H.; Wu, H.; Benesperi, I.; Dar, M. I.; Pehlivan, I. B.; Gould, I. E.; Vagott, J. N.; Dagar, J.; Kettle, J.; Yang, J.; Li, J.; Smith, J. A.; Pascual, J.; Jerónimo-Rendón, J. J.; Montoya, J. F.; Correa-Baena, J.-P.; Qiu, J.; Wang, J.; Sveinbjörnsson, K.; Hirselandt, K.; Dey, K.; Frohna, K.; Mathies, L.; Castriotta, L. A. ; Aldamasy, Mahmoud. H.; Vasquez-Montoya, M.; Ruiz-Preciado, M. A.; Flatken, M. A.; Khenkin, M. V.; Grischek, M.; Kedia, M.; Saliba, M.; Anaya, M.; Veldhoen, M.; Arora, N.; Shargaieva, O.; Maus, O.; Game, O. S.; Yudilevich, O.; Fassl, P.; Zhou, Q.; Betancur, R.; Munir, R.; Patidar, R.; Stranks, S. D.; Alam, S.; Kar, S.; Unold, T.; Abzieher, T.; Edvinsson, T.; David, T. W.; Paetzold, U. W.; Zia, W.; Fu, W.; Zuo, W.; Schröder, V. R. F.; Tress, W.; Zhang, X.; Chiang, Y.-H.; Iqbal, Z.; Xie, Z.; Unger, E. An open-access database and analysis tool for perovskite solar cells based on the FAIR data principles. Nat. Energy. [CrossRef]
- Bogachuk, D.; Zouhair, S.; Wojciechowski, K.; Yang, B.; Babu, V.; Wagner, L.; Xu, B.; Lim, J.; Mastroianni, S.; Pettersson, H.; Hagfeldt, A.; Hinsch, A. Low-temperature carbon-based electrodes in perovskite solar cells. Energy Environ. Sci. 2020, 13, 3880–3916. [Google Scholar] [CrossRef]
- Chen, H.; Yang, S. Carbon-Based Perovskite Solar Cells without Hole Transport Materials: The Front Runner to the Market? Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, S. Methods and Strategies for Achieving High-Performance Carbon-Based Perovskite Solar Cells without Hole Transport Materials. J. Mater. Chem. A 2019, 7, 15476–15490. [Google Scholar] [CrossRef]
- Wagner, L.; Qiu, C.; Unmüssig, M.; Bogachuk, D.; Mastroianni, S.; Würfel, U.; Hu, Y.; Han, H.; Hinsch, A. A 2D Model for Interfacial Recombination in Mesoscopic Perovskite Solar Cells with Printed Back Contact. Solar RRL 2021, 5, 2000595. [Google Scholar] [CrossRef]
- Kobayashi, E.; Tsuji, R.; Martineau, D.; Hinsch, A.; Ito, S. Light-Induced Performance Increase of Carbon-Based Perovskite Solar Module for 20-Year Stability. Cell Rep. Phys. Sci. 2021, 2, 100648. [Google Scholar] [CrossRef]
- Emami, S.; Martins, J.; Ivanou, D.; Mendes, A. Advanced Hermetic Encapsulation of Perovskite Solar Cells: The Route to Commercialization. J. Mater. Chem. A 2020, 8, 2654–2662. [Google Scholar] [CrossRef]
- Grancini, G.; Roldán-Carmona, C.; Zimmermann, I.; Mosconi, E.; Lee, X.; Martineau, D.; Narbey, S.; Oswald, F.; De Angelis, F.; Graetzel, M.; Nazeeruddin, M. K. One-Year stable perovskite solar cells by 2D/3D interface engineering. Nat. Commun. 2017, 8, 15684. [Google Scholar] [CrossRef] [PubMed]
- Mei, A.; Sheng, Y.; Ming, Y.; Hu, Y.; Rong, Y.; Zhang, W.; Luo, S.; Na, G.; Tian, C.; Hou, X.; Xiong, Y.; Zhang, Z.; Liu, S.; Uchida, S.; Kim, T.-W.; Yuan, Y.; Zhang, L.; Zhou, Y.; Han, H. Stabilizing Perovskite Solar Cells to IEC61215:2016 Standards with over 9,000-h Operational Tracking. Joule 2020, 4, 2646–2660. [Google Scholar] [CrossRef]
- Ma, S.; Yuan, G.; Zhang, Y.; Yang, N.; Li, Y.; Chen, Q. Development of Encapsulation Strategies towards the Commercialization of Perovskite Solar Cells. Energy Environ. Sci. 2022, 15, 13–55. [Google Scholar] [CrossRef]
- Shi, L.; Bucknall, M. P.; Young, T. L.; Zhang, M.; Hu, L.; Bing, J.; Lee, D. S.; Kim, J.; Wu, T.; Takamure, N.; McKenzie, D. R.; Huang, S.; Green, M. A.; Ho-Baillie, A. W. Y. Gas Chromatography-Mass Spectrometry Analyses of Encapsulated Stable Perovskite Solar Cells. Science 2020, 368, eaba2412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ahmad, I.; Leung, T.; Lin, J.; Chen, W.; Liu, F.; Ng, A.M.C.; Zhang, Y.; Djurišić, A.B. Encapsulation and Stability Testing of Perovskite Solar Cells for Real Life Applications. ACS Mater. Au 2022, 2, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xu, M.; Sheng, Y.; Yan, Z.; Meng, J.; Tong, C.; Li, D.; Wan, Z.; Ming, Y.; Mei, A.; Hu, Y.; Rong, Y.; Han, H. Encapsulation of printable mesoscopic perovskite solar cells enables high temperature and Long-Term outdoor stability. Adv. Funct. Mater. 2019, 29, 1809129. [Google Scholar] [CrossRef]
- Baranwal, A.K.; Kanaya, S.; Peiris, T.A.N.; Mizuta, G.; Nishina, T.; Kanda, H.; Miyasaka, T.; Segawa, H.; Ito, S. 100 °C Thermal Stability of Printable Perovskite Solar Cells Using Porous Carbon Counter Electrodes. ChemSusChem 2016, 9, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, A.K.; Kanda, H.; Shibayama, N.; Masutani, H.; Peiris, T.A.N.; Kanaya, S.; Segawa, H.; Miyasaka, T.; Ito, S. Thermal Degradation Analysis of Sealed Perovskite Solar Cell with Porous Carbon Electrode at 100 °C for 7000 h. Energy Technol. 2019, 7, 245–252. [Google Scholar] [CrossRef]
- Ava, T.T.; Al Mamun, A.; Marsillac, S.; Namkoong, G. A Review: Thermal Stability of Methylammonium Lead Halide Based Perovskite Solar Cells. Appl. Sci. 2019, 9, 188. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, W.; Zhang, Z.; Liu, S.; Wu, J.; Guan, Y.; Mei, A.; Rong, Y.; Hu, Y.; Han, H. Crystallization Control of Ternary-Cation Perovskite Absorber in Triple-Mesoscopic Layer for Efficient Solar Cells. Adv. Energy Mater. 2020, 10, 1903092. [Google Scholar] [CrossRef]
- Keremane, K.S.; Prathapani, S.; Haur, L.J.; Bahulayan, D.; Adhikari, A.V.; Priyadarshi, A.; Mhaisalkar, S.G. Solvent Selection for Highly Reproducible Carbon-Based Mixed-Cation Hybrid Lead Halide Perovskite Solar Cells via Adduct Approach. Sol. Energy 2020, 199, 761–771. [Google Scholar] [CrossRef]
- Bidikoudi, M.; Dracopoulos, V.; Stathatos, E. Low-Temperature Annealed Methylammonium-Free Perovskites Prepared under Ambient Conditions in C Electrode-Based Perovskite Solar Cells. Energy Adv. 2022, 1, 76–86. [Google Scholar] [CrossRef]
- Zheng, Z.; Xia, M.; Chen, X.; Xiao, X.; Gong, J.; Liu, J.; Du, J.; Tao, Y.; Hu, Y.; Mei, A.; Lu, X.; Han, H. Enhancing the performance of FA-Based printable mesoscopic perovskite solar cells via the polymer additive. Adv. Energy Mater. 2023, 13, 2204335. [Google Scholar] [CrossRef]
- Khenkin, M. V.; Katz, E. A.; Abate, A.; Bardizza, G.; Berry, J. J.; Brabec, C.; Brunetti, F.; Bulović, V.; Burlingame, Q.; Di Carlo, A.; Cheacharoen, R.; Cheng, Y.-B.; Colsmann, A.; Cros, S.; Domanski, K.; Dusza, M.; Fell, C. J.; Forrest, S. R.; Galagan, Y.; Di Girolamo, D.; Grätzel, M.; Hagfeldt, A.; Von Hauff, E.; Hoppe, H.; Kettle, J.; Köbler, H.; Leite, M. S.; Liu, S. F.; Loo, Y.-L.; Luther, J. M.; Ma, C.-Q.; Madsen, M.; Manceau, M.; Matheron, M.; McGehee, M.; Meitzner, R.; Nazeeruddin, M. K.; Nogueira, A. F.; Odabaşı, Ç.; Osherov, A.; Park, N.-G.; Reese, M. O.; De Rossi, F.; Saliba, M.; Schubert, U. S.; Snaith, H. J.; Stranks, S. D.; Tress, W.; Troshin, P. A.; Turkovic, V.; Veenstra, S.; Visoly-Fisher, I.; Walsh, A.; Watson, T.; Xie, H.; Yıldırım, R.; Zakeeruddin, S. M.; Zhu, K.; Lira-Cantu, M. Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures. Nat. Energy 2020, 5, 35–49. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, D.; Sheng, Y.; Zhang, W.; Qin, Z.; Qin, M.; Li, S.; Wang, Y.; Gao, C.; Wang, Q.; Ming, Y.; Liu, C.; Yang, K.; Huang, Q.; Qi, J.; Gao, Q.; Chen, K.; Hu, Y.; Rong, Y.; Lu, X.; Mei, A.; Han, H. Highly oriented MAPbI3 crystals for efficient hole-conductor-free printable mesoscopic perovskite solar cells. Fundam. Res. 2022, 2, 276–283. [Google Scholar] [CrossRef]
- Tsuji, R.; Bogachuk, D.; Luo, B.; Martineau, D.; Kobayashi, E.; Funayama, R.; Mastroianni, S.; Hinsch, A.; Ito, S. Activation of Weak Monochromic Photocurrents by White Light Irradiation for Accurate IPCE Measurements of Carbon-Based Multi-Porous-Layered-Electrode Perovskite Solar Cells. Electrochemistry 2020, 88, 418–422. [Google Scholar] [CrossRef]
- Bag, M.; Renna, L.A.; Adhikari, R.Y.; Karak, S.; Liu, F.; Lahti, P.M.; Russell, T.P.; Tuominen, M.T.; Venkataraman, D. Kinetics of Ion Transport in Perovskite Active Layers and Its Implications for Active Layer Stability. J. Am. Chem. Soc. 2015, 137, 13130–13137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Meng, X.; Bai, Y.; Xiao, S.; Hu, C.; Yang, Y.; Chen, H.; Yang, S. Profiling the Organic Cation-Dependent Degradation of Organolead Halide Perovskite Solar Cells. J. Mater. Chem. A 2017, 5, 1103–1111. [Google Scholar] [CrossRef]
- Kundu, S.; Kelly, T. L. In situ studies of the degradation mechanisms of perovskite solar cells. EcoMat 2020, 2, e12025. [Google Scholar] [CrossRef]
- Haris, M.P.U.; Kazim, S.; Ahmad, S. Low-Temperature-Processed Perovskite Solar Cells Fabricated from Presynthesized CsFAPbI3 Powder. ACS Appl. Energy Mater. 2021, 4, 2600–2606. [Google Scholar] [CrossRef]
- Xue, B.; Zhu, J.; Liu, N.; Li, Y. Facile Functionalization of Graphene Oxide with Ethylenediamine as a Solid Base Catalyst for Knoevenagel Condensation Reaction. Catal. Commun. 2015, 64, 105–109. [Google Scholar] [CrossRef]
- Huang, J.; Tan, S.; Lund, P.D.; Zhou, H. Impact of H2O on Organic-Inorganic Hybrid Perovskite Solar Cells. Energy Environ. Sci. 2017, 10, 2284–2311. [Google Scholar] [CrossRef]
- Tsuji, R.; Bogachuk, D.; Martineau, D.; Wagner, L.; Kobayashi, E.; Funayama, R.; Matsuo, Y.; Mastroianni, S.; Hinsch, A.; Ito, S. Function of Porous Carbon Electrode during the Fabrication of Multiporous-Layered-Electrode Perovskite Solar Cells. Photonics 2020, 7, 133. [Google Scholar] [CrossRef]
- Tsuji, R.; Tanaka, K.; Oishi, K.; Shioki, T.; Satone, H.; Ito, S. Role and Function of Polymer Binder Thickeners in Carbon Pastes for Multiporous-Layered-Electrode Perovskite Solar Cells. Chem. Mater. 2023, 35, 8574–8589. [Google Scholar] [CrossRef]
- Bogachuk, D.; Tsuji, R.; Martineau, D.; Narbey, S.; Herterich, J.P.; Wagner, L.; Suginuma, K.; Ito, S.; Hinsch, A. Comparison of Highly Conductive Natural and Synthetic Graphites for Electrodes in Perovskite Solar Cells. Carbon 2021, 178, 10–18. [Google Scholar] [CrossRef]
- Ito, S.; Tsuji, R. Mesoscopic Anodes and Cathodes for Printable Perovskite Solar Cells. In Printable Mesoscopic Perovskite Solar Cells; Wiley-VCH GmbH, 2023; pp. 89–104.
- Shioki, T.; Tsuji, R.; Oishi, K.; Fukumuro, N.; Ito, S. Designed Mesoporous Architecture by 10-100 nm TiO2 as Electron Transport Materials in Carbon-Based Multiporous-Layered-Electrode Perovskite Solar Cells. Photonics 2024, 11, 236. [Google Scholar] [CrossRef]
Figure 1.
(a) Schematic encapsulation diagram of over– and side–sealing multiporous–layered–electrode perovskite solar cells (MPLE–PSCs). (b) Chemical structures of methylammonium (MA) and formamidinium (FA). (c) Schematic illustration of thermal stability test of encapsulated MPLE–PSCs.
Figure 1.
(a) Schematic encapsulation diagram of over– and side–sealing multiporous–layered–electrode perovskite solar cells (MPLE–PSCs). (b) Chemical structures of methylammonium (MA) and formamidinium (FA). (c) Schematic illustration of thermal stability test of encapsulated MPLE–PSCs.
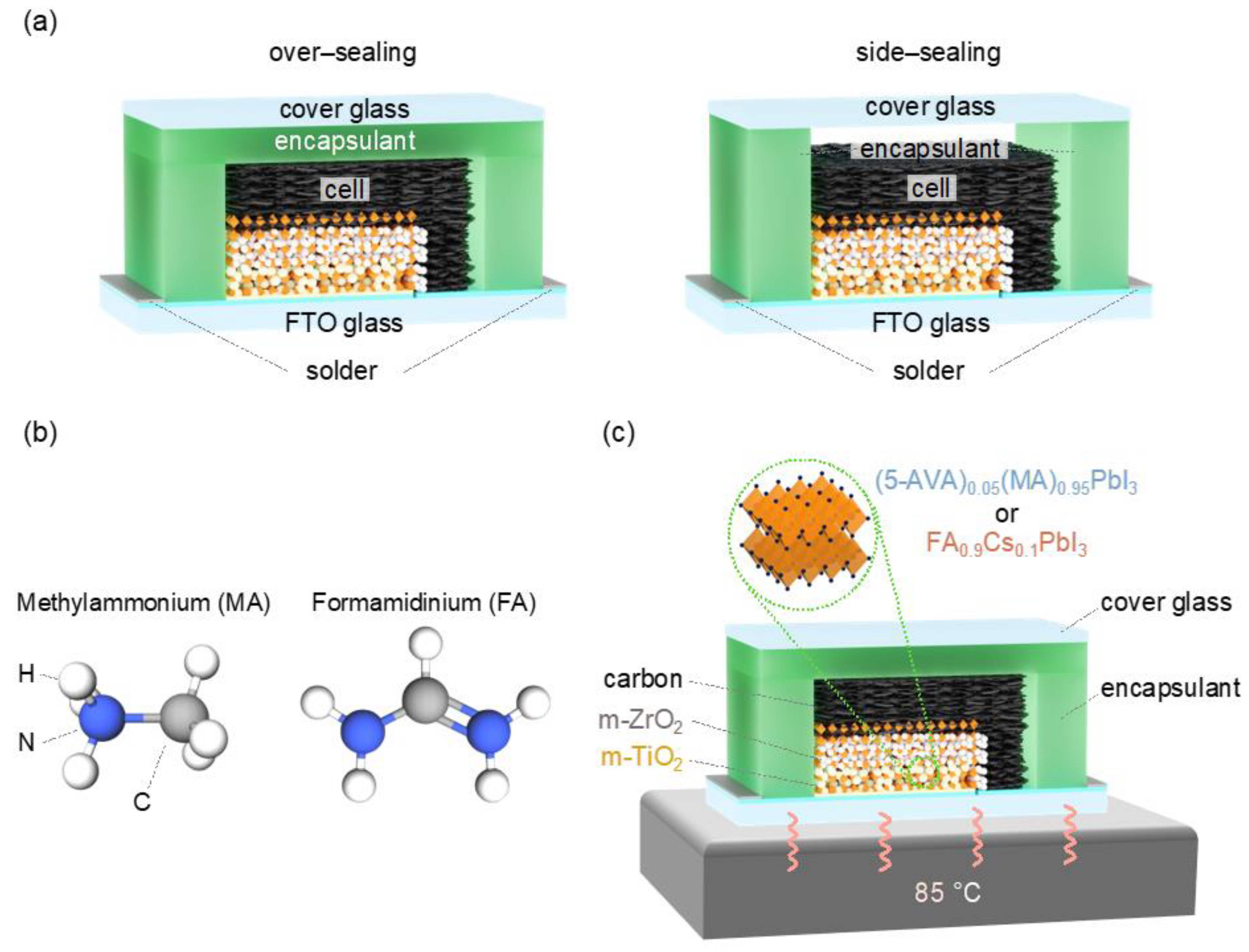
Figure 2.
Device performance of MPLE–PSCs with MA and FACs perovskite. (a) J–V curves of MA perovskite, (b) J–V curves of FACs perovskite, (c) box–and–whisker plots of each J–V parameter, (d) IPCE spectrum and integrated JSC, (e) Nyquist plots (the inset shows the equivalent circuit) of EIS at 0 V bias under the right.
Figure 2.
Device performance of MPLE–PSCs with MA and FACs perovskite. (a) J–V curves of MA perovskite, (b) J–V curves of FACs perovskite, (c) box–and–whisker plots of each J–V parameter, (d) IPCE spectrum and integrated JSC, (e) Nyquist plots (the inset shows the equivalent circuit) of EIS at 0 V bias under the right.
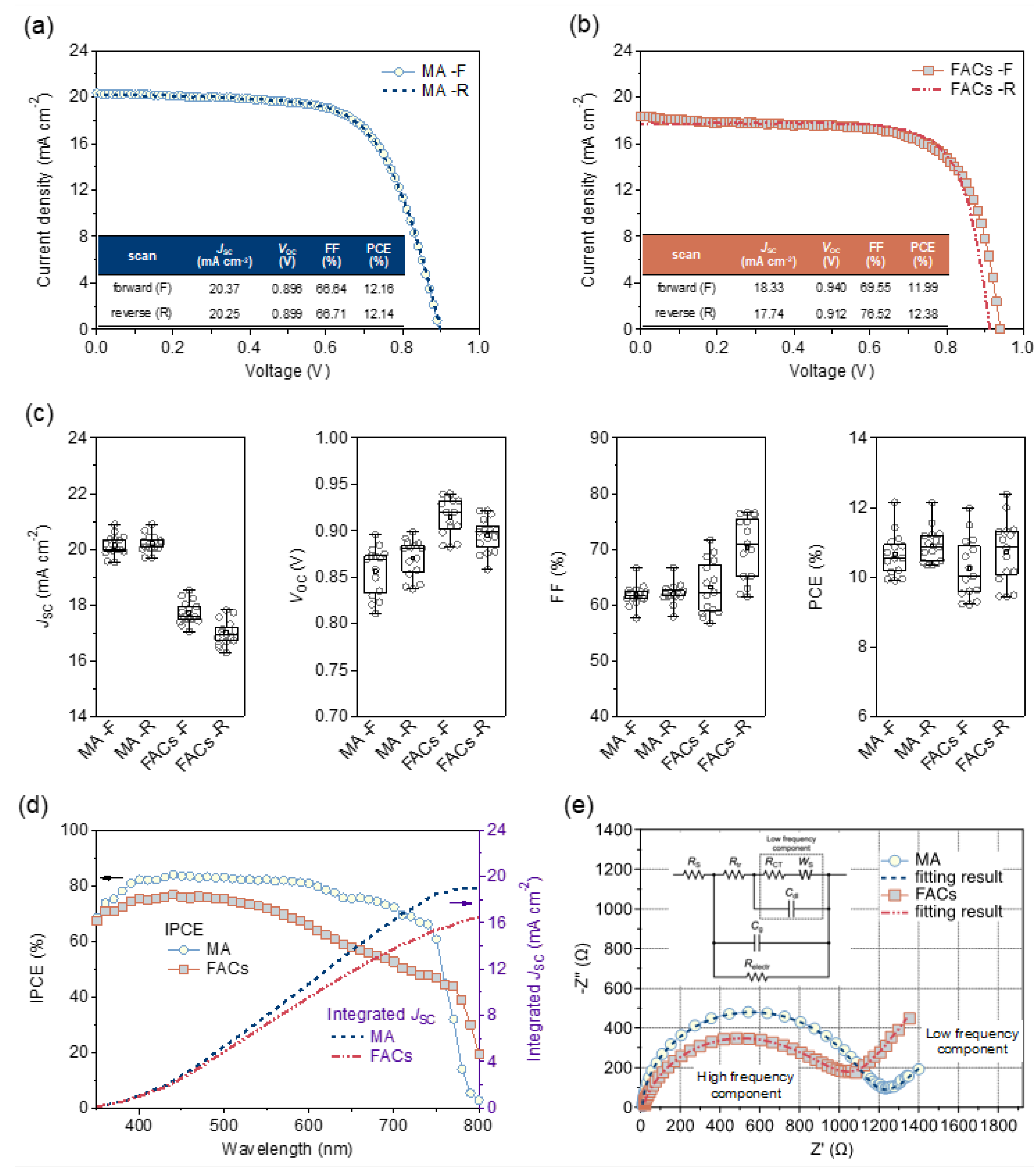
Figure 3.
Thermal stability of MPLE–PSCs without any encapsulation at 85 °C in air. (a) variation of XRD patterns of MAPbI3, (b) XRD–patterns variation of FACsPbI3 (the reflections indicated by * represent Bragg reflections associated FAI.), (c) variation of normalized XRD intensities of MAPbI3 (110) and FACsPbI3 (001) perovskite crystals, (d) variation of normalized PCE of MPLE–PSCs with MA and FACs perovskite.
Figure 4.
(a) Variation for normalized PCE at the initial value of encapsulated MPLE–PSCs at thermal stability test (85 °C (ISOS–D–2) for >5000 h). The number of used devices was five for the statistical data. (b) Photographs of the active area during the thermal test as seen from the FTO glass side (light–receiving surface).
Figure 4.
(a) Variation for normalized PCE at the initial value of encapsulated MPLE–PSCs at thermal stability test (85 °C (ISOS–D–2) for >5000 h). The number of used devices was five for the statistical data. (b) Photographs of the active area during the thermal test as seen from the FTO glass side (light–receiving surface).
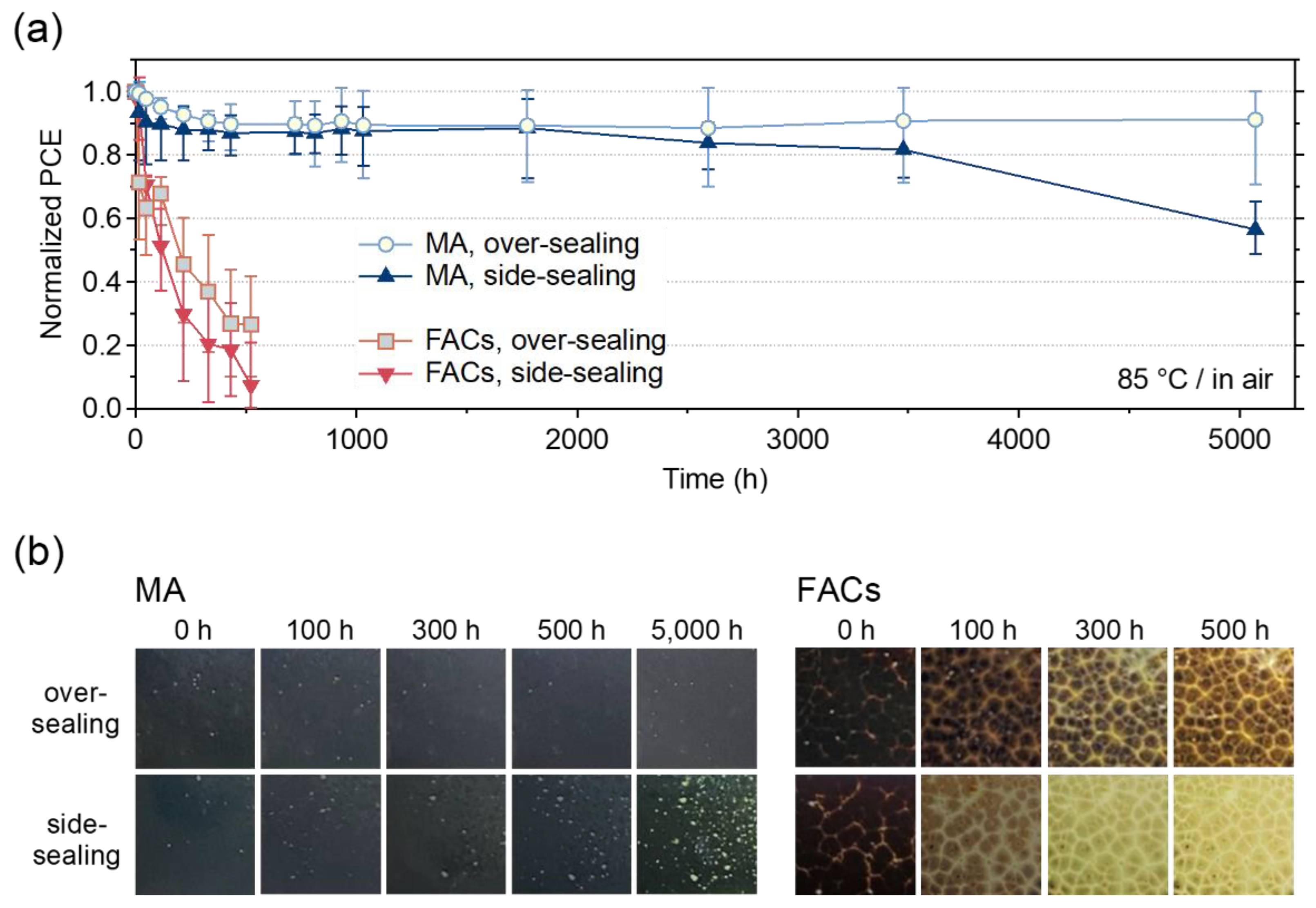
Figure 5.
(a) Calculation of the number of H2O molecules sealed in the 1 cm2 active area device by over– and side–sealing, and (b) the number of perovskite units in the device.
Figure 5.
(a) Calculation of the number of H2O molecules sealed in the 1 cm2 active area device by over– and side–sealing, and (b) the number of perovskite units in the device.
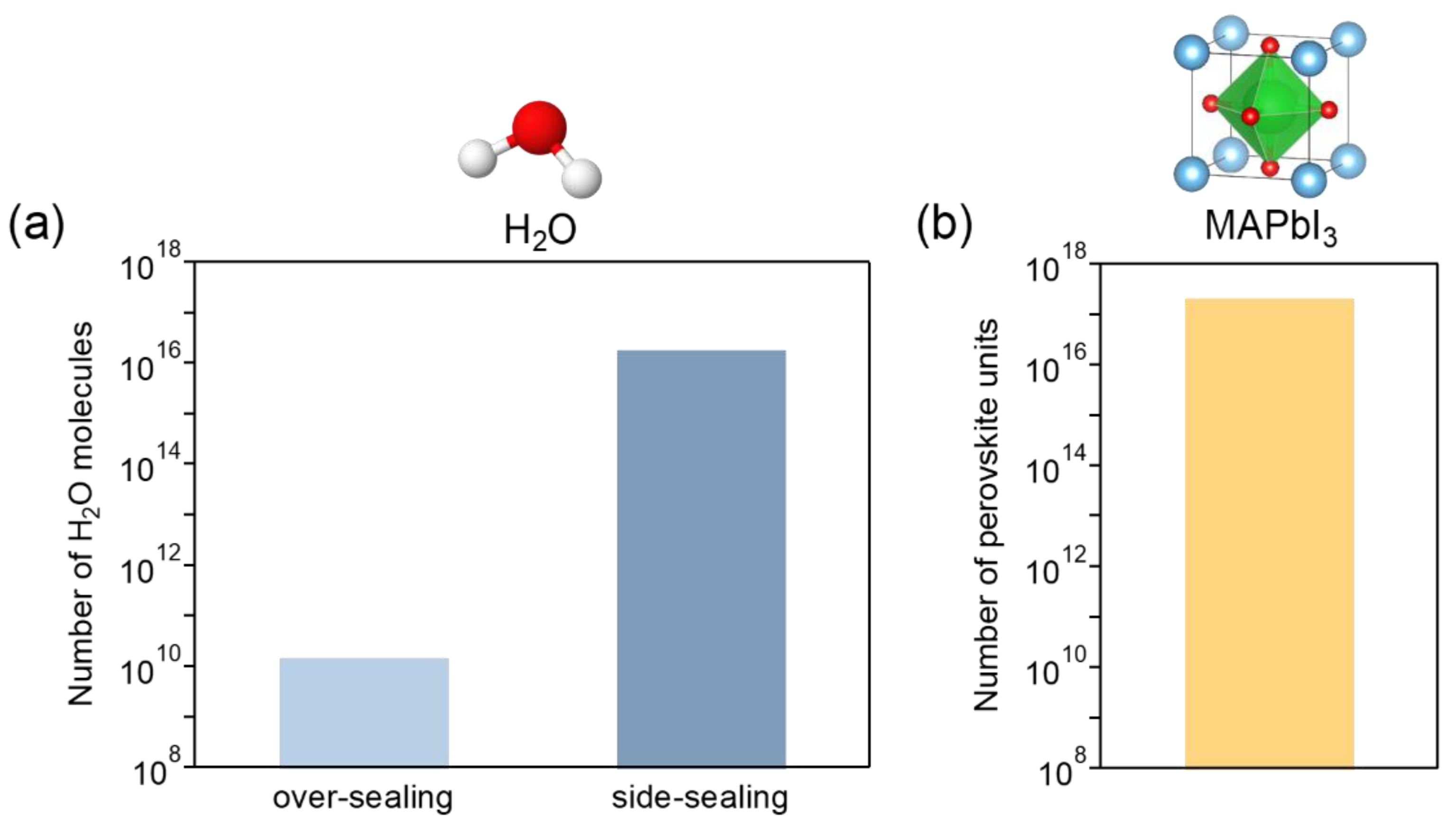
Figure 6.
Thermal degradation model of MPLE–PSC with over– and side–sealing encapsulation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



