
Submitted:
21 May 2024
Posted:
22 May 2024
You are already at the latest version
Abstract
This study investigated the effects of fish oil (FO) treatment, particularly enriched with eicosapentaenoic acid (EPA), on obesity induced by a high-fat diet (HFD) in mice. The investigation focused on elucidating the impact of FO on epigenetic modifications in white adipose tissue (WAT) and the involvement of adipose-derived stem cells (ASCs). C57BL/6j mice were submitted to a control diet or a HFD for 16 weeks, treated (or not) with FO for the last 8 weeks. WAT was removed for RNA and protein extraction, while ASCs were isolated, cultured, and treated with Leptin. All samples were analyzed using functional genomics tools, including PCR-array, RT-PCR and Western Blot assays. Mice receiving HFD displayed increased body mass, fat accumulation, and altered gene expression associated with WAT inflammation and dysfunction. FO supplementation attenuated these effects, a potential protective role against HFD-induced obesity. Analysis of H3K27 revealed HFD-induced changes in histone, which were partially reversed by FO treatment. The study further explored leptin signaling in ASCs, suggesting a potential mechanism for ASC dysfunction in the leptin-rich environment of obese WAT. Overall, FO supplementation demonstrated efficacy in mitigating HFD-induced obesity, influencing epigenetic and molecular pathways, and shedding light on the role of ASCs and leptin signaling in WAT dysfunction associated with obesity.
Keywords:
obesity
; inflammation
; H3K27
; n-3 PUFA
; WAT
; leptin
1. Introduction
The high-fat diet (HFD) obesity induction model triggers inflammation and promotes metabolic dysfunction, contributing to the development of obesity-related complications such as insulin resistance and cardiovascular diseases [1]. One notable characteristic, particularly in the context of white adipose tissue (WAT), is the presence of a chronic state of low-grade inflammation, often termed “metaflammation”, which plays a key role in obesity-related health issues [2]. Natural and synthetic treatments gaining popularity for managing HFD-induced obesity, such as those increasing insulin sensitivity and secretion while reducing glucose levels through actions against alpha-amylase and lipase enzymes, have been explored [3,4]. Along the same line, the beneficial effects of n-3 long-chain polyunsaturated fatty acids (n-3 PUFA), including eicosapentaenoic acid (EPA, 20:5 n-3) and docosahexaenoic acid (DHA, 22:6 n-3), abundantly present in fish oil (FO), have long been demonstrated in addressing dyslipidemia, improving glucose tolerance, enhancing insulin sensitivity, and reducing adipose mass in HFD-induced obese mice, by our group and other researchers [5,6,7,8].
Our previous findings revealed that animals subjected to obesity induced by a HFD and supplemented with FO (for 8 weeks), containing EPA:DHA in a 5:1 ratio, consistently exhibited reductions in both body weight and adipose mass. Additionally, these animals showed improvements in adipocyte function, including reductions in hypertrophy and metabolic and endocrine dysfunctions associated with obesity. Moreover, FO supplementation effectively downregulated the expression of pro-inflammatory cytokines, suggesting its potential in mitigating obesity-related endocrine disorders [9,10,11,12]. Notably, fish oil (FO) treatment appears to modulate the inflammation pathway, suggesting its significance in the epigenetic regulation of WAT cells and providing new insights into obesity management.
It is well known that the proper functioning of WAT requires continuous remodeling to accommodate its rapid and dynamic ability to expand or contract, adjusting lipid stores [13]. This process involves adipocyte differentiation and WAT plasticity, which ensures the supply of nutrients and oxygen to WAT, as well as guaranteeing the transport of fatty acids and appropriate production of adipokines [14]. Adipose tissue-derived mesenchymal stem cells (ASCs) are abundant in WAT, undergoing adipogenesis via epigenetic changes, particularly involving histone 3 lysine 27 (H3K27) marks deposition [15]. Trimethylation of H3K27 (H3K27me3) by EZH2 silences genes, while acetylation (H3K27ac) by CREBBP and EP300 (Cbp/p300) activates transcription[16,17]. H3K27 demethylases, KDM6A and KDM6B, erase the H3H27me3 silencing mark [18]. Therefore, H3K27 modifications play a crucial role in gene expression regulation, controlling which genes are activated or silenced at a given time and in response to environmental and physiological stimuli.
Histone acetylation emerges as a remarkably dynamic chromatin modification, influenced by the availability of acetyl-CoA [19,20,21], where the addition of acetyl groups to lysine residues on histones (such as H3K27ac), results in a more relaxed chromatin structure, accessible for gene transcription [22]. The ATP citrate lyase (ACL) enzyme catalyzes the conversion of citrate and coenzyme A (CoA) into oxaloacetate and acetyl-CoA. Consequently, this enzyme plays a crucial role in generating acetyl-CoA, a pivotal precursor in numerous metabolic pathways, in addition to serving as a substrate for histone acetyltransferases [23], thereby connecting metabolism to protein acetylation processes. Carrer and colleagues demonstrated that a 4-week consumption of a HFD, known to suppress ACL [24,25,26], resulted in decreased acetyl-CoA levels in the whole WAT [24]. Moreover, ACL has been shown to regulate histone acetylation levels and influence gene regulation [21,27,28].
The regulation of ACL expression in ASCs remains to be explored, particularly in the context of obesity. Interestingly, leptin signaling has been shown to reduce ACL expression in hepatocytes and adipocytes [25]. However, whether leptin (which is abundantly expressed in obese WAT) acts paracrinally on ASCs to influence ACL expression, which in turn, could epigenetically mediate differentiation and/or inflammation in these cells to influence dysfunction of WAT, remains to be explored.
On one hand, changes in the deposition of epigenetic marks on H3K27 play a critical role in adipogenesis and depend on the potential action of histone-modifying enzymes. On the other hand, few studies have explored the correlation between the elevated products of chronic low-grade inflammation in obese individuals and the expression of these histone modifiers and ACL expression. Similarly, there is a lack of research on H3K27ac and H3K27me3 expression in WAT and the impact of HFD associated or not with FO supplementation on these epigenetic marks' modulators. In the present study, the inflammation pathway mediated by chemokine and cytokine signaling, emerged as the most prominently affected signaling pathway in our HFD-induced obesity murine model. Significantly, this pathway was modulated by FO treatment. We sought to investigate whether this modulation involves epigenetic modifications of histone 3 lysine 27 (H3K27), as well as the participation of leptin and ACL in the process. To address this question, we employed a combination of functional genomics tools, including RT-PCR, PCR-array, along with Western Blot analyzes in a murine obesity model.
2. Results
2.1. Obesity Model Characterization
Obesity parameters were assessed at the end of the 12-week experimental protocol for obesity induction. As expected, there was a decrease in food and caloric intake but an increase in fat intake (~5x, Figure 1A) in mice receiving HFD. Regarding body mass, a statistical difference between the control group and the groups of animals receiving the HFD became evident from the 3rd week. This difference increased throughout the weeks of the experimental protocol, and by the 12th week of HFD administration, the animals receiving it showed a ~3.5x body mass gain compared to those receiving the CO diet (Figure 1B).
Figure 1 (C-E) also illustrates values related to fasting blood glucose and the glucose tolerance test (GTT). Compared to the CO diet, mice fed with HFD showed an approximately 50% increase in fasting blood glucose (Figure 1C) and glucose intolerance (Figure 1D), with an expressive (82 %) increase in the area under the curve (Figure 1E). Thus, our diet-induced obesity model was characterized, confirming our previous results [10,11,16]. We subsequently supplemented the animals with FO (HFD+FO group) in the last 8 weeks of the 16-week experimental protocol with HFD.
2.2. WAT depots Extracted from Mice Treated with CO, HFD, and HFD+FO Diets: Depot Mass and Gene Expression by PCR Array
As expected [10,11], mice consuming HFD presented a significant increase of 50 % in body mass when compared to the CO group, and the HFD+FO group was still 40 % higher in relation to CO group, but presented a significant reduction of ~30% (P < 0.05) compared to the HFD group (Figure 2A). Moreover, animals in the HFD group exhibited a significant increase in the mass of the visceral epididymal (Epi) fat depot (by ~3x), while treatment with FO completely prevented this increase in fat mass (Figure 2B). However, no significant difference was observed between the HF and HFD+FO groups in the mass of the visceral retroperitoneal (Rp) (Figure 2C) or subcutaneous inguinal (Ing) (Figure 2D) fat depots. The complete metabolic characterization of FO treatment in this model, along with measurements of other tissue weights such as the liver and interscapular brown adipose tissue, has been previously demonstrated and reported in studies conducted by our group [9,10,11,12].
An analysis of Epi WAT depots (whole tissues) was conducted explore their gene expression profiles. To do so, we designed a customized panel of 84 essential genes for adipose tissue using a PCR array gene expression assay. Following the validation of reactions via array control plates, an in-depth comparative analysis was performed on the tissue Ct values to thoroughly examine the gene expression changes induced by the HFD, associated (or not) to FO supplementation. Initially, our emphasis was on scrutinizing genes with differential expression, both up - or down - regulated, attributed to the HFD when compared to the CO diet. These findings, contrasting obese animals with controls, are comprehensively outlined in Table 1.
Among the various genes modulated, we highlight the up-regulation of the genes Lep (+25.48x), Ncor2 (+3.08x), Ccl2 (+5.07x), Tnf (+10.65x), Nfkb1 (+2.52x), and Cd68 (9.16x), triggered by the HFD when compared to CO group (Table 1). With the exception of Ncor2 (anti-adipogenic factor), all these genes encode cytokines (leptin, TNFa) and macrophage chemotactic factors (Ccl2/Mcp-1) or markers of macrophages (Cd68) and inflammation pathways (NFkB). There was also an increase in Dio2 (+3.98x) and Elovl3 (+2.63x), indicating an increase in diet-induced thermogenesis.
We also observed that the HFD suppressed the expression of numerous genes, including those encoding adipokines (adiponectin and resistin), enzymes involved in lipid biosynthesis (Acc1, Scd-1, Lipin1, Pck1, and Fas), factors related to adipogenesis (Cebpa, Cebpd, Fabp4/Ap2, Fgf-2, Fgf-10, AP-1/C-Jun, Sfrp1, Klf15, Adrb2, Dlk1/Pref-1, Foxo-1, Shh, Wnt1, Wnt3a, Gata2), proteins associated with browning, thermogenesis, and fatty acid oxidation (Bmp7, Pgc1-a, Pgc1-b, Sirt3, Tbx1, Ucp1, Nr1h3, Wnt10b), adipocyte receptors (Lepr, Adipor2, Adrb1), pro and anti-inflammatory cytokines (Ifn-γ, Il1-β, Il-6, Il-13), and insulin signaling pathway proteins (Insr, Irs-1, Irs-2, Pik3r1).
The data concerning both up-regulated and down-regulated genes in the samples from the HFD and HFD+FO groups are visualized in a heatmap (clustergram, Figure 3). It was evident that the treatment of obese mice with FO altered the extensive list of genes negatively affected by the HFD, inducing an increase in the expression of most of those genes (and others). These encompass genes encoding adipokines (adiponectin, leptin and resistin), lipolytic and lipogenic enzymes (Scd-1, Lpl, Lipin1, Pck1), pro-adipogenic factors (Cebpa, Pparg, Srebp-1, Fabp4/aP2, sFRP1, Glut4), proteins involved in browning and thermogenesis, mitochondrial biogenesis, and fatty acid oxidation (Dio-2, Elovl3, Foxc2, Ppar-α, Pgc1-α, Tbx1, Tfam, Ucp-1), pro and anti-inflammatory cytokines (Mcp-1, Cxcl10, Il1-Β, Il-4, Il-6, Il-13, Resistin), and signaling transduction factors (Irs-1, Nf-kb).
2.3. Expression of H3K27 Modifiers, H3k27ac, H3k27met3 and Acly/ACL in the Visceral Epi WAT from Mice
Regarding possible alterations in epigenetic marks, we initially assessed the expression of genes encoding enzymes responsible for acetylation (Ep300 and Crebbp), as well as methylation (Ezh2) and demethylation (Kdm6a and Kdm6b) of H3K27 in visceral Epi WAT of control mice, obese mice (HFD), and obese mice treated with FO (HFD+FO). There was no statistically significant difference observed in the transcript levels of Ezh2 (Figure 4B). However, the genes encoding the acetylases Crebbp (Figure 4C) and Ep300 (Figure 4D), as well as the demethylase Kdm6b (Figure 4F), demonstrated a notable increase in expression within the HFD group compared to the control. This effect was completely reversed by FO treatment for Crebbp and Ep300, while partially reversed for Kdm6b. Additionally, no difference was noted between the groups in the expression of the gene encoding the enzyme Kdm6a (Figure 4E).
We next investigated the expression of the Acly (gene) and ACL (protein), as well as histone-modifying proteins. Consistent with the decreased Acly expression (Figure 4A), we observed a reduction in ACL expression in the visceral Epi WAT of obese animals (Figure 5A). Corroborating these findings, we detected a decrease in the expression of H3K27ac in the group of animals with obesity induced by HFD (Figure 5B), and once again, FO was able to completely prevent this effect. Finally, a significant decrease in the expression of the H3K27me3 protein was also observed (Figure 5C), an effect partially prevented by FO.
2.4. Gene Expression of Acly and Leptin Receptors in ASCs from Mice
In Figure 6, we validated the expression of Lepr1 and Lepr2 receptor isoforms in ASCs. We observed that ASCs extracted from the visceral WAT showed no difference in receptor isoform expression between the CO and HFD animal groups (Figure 6A-B). Moreover, Lepr3 isoform was not expressed in these cells. When we treated these mice ASCs with leptin (in vitro for 24 hours) and examined the expression of LEP protein receptor (long isoform, Ob-Rb, responsible for total signal transduction), we found that ASCs fully expressed these receptors, whose expression remained unchanged when the ASCs were exposed to leptin (Figure 6C). Finally, we evaluated the expression of the Acly gene in these ASCs treated in vitro with leptin. Interestingly, we observed a reduction in Acly expression in the HFD mice group. This finding suggests that, in a leptin-rich medium, ASCs exhibit a decrease in Acly expression (Figure 6D).
3. Discussion
We investigated whether FO treatment, rich in EPA, protects against the inflammation pathway triggered by HFD-induced obesity in murine WAT. Activation of this pathway is associated with changes in the expression of several important genes involved in WAT metabolism and cell differentiation. Additionally, we explored whether these effects are modulated by H3K27 modifications and the impact of leptin (whose secretion is high in the WAT of obese individuals) on ASCs. Our results suggest that FO mitigates the negative effects of chronic inflammation associated with obesity, and that its effects involve epigenetic mechanisms by modulating ACL expression and H3K27 acetylation. This study also underscores the role of leptin not only as an important endocrine signal but also as a paracrine one, leading to significant impact on ASCs, which ultimately could affect their adipogenic potential.
Mice fed HFD for 16 weeks displayed a substantial increase in the mass of visceral fat depots and an upregulation of genes encoding cytokines, macrophage chemotactic factors, markers of macrophages, and inflammation pathways, among others. These findings align with existing literature describing numerous pronounced and detrimental effects of the HFD [1]. We also observed a downregulation in the expression of numerous genes, including those encoding adiponectin, enzymes involved in lipid biosynthesis, factors and proteins related to adipogenesis, browning, thermogenesis, and fatty acid oxidation, adipocyte receptors, pro and anti-inflammatory cytokines, and components of the insulin signaling pathway. In total, 41 genes showed decreased expression due to the HFD. Interestingly, the treatment of the animals with omega-3 polyunsaturated fatty acids (FO) prevented the extensive list of genes negatively affected by the HFD. Moreover, a total of 32 genes were up-regulated by the FO treatment, bringing the data from these animals into closer alignment with that of the CO group and distinctly segregating them from the group of obese mice. Taken together, our results clearly indicate a significant impact of FO treatment on the chemokine and cytokine signaling-mediated inflammation pathway in WAT extracted from mice receiving the HFD.
Furthermore, among the genes altered in the array, we were particularly intrigued by the reversal of leptin expression following FO treatment. Leptin, a cytokine crucial for regulating energy expenditure in adipocytes, is significantly elevated in individuals with obesity and chronic inflammation. In this study, leptin expression was upregulated by 25.48-fold in the HFD group compared to the control group. Remarkably, FO treatment not only reversed this increase but also reduced leptin expression in the visceral Epi WAT to values below those of the control group.
Leptin was shown to reduce ACL enzyme expression in hepatocytes and adipocytes [25]. Likewise, animal models of HFD-induced obesity typically exhibit reduced ACL expression in both hepatocytes and adipocytes [24,25,26], supporting our findings. Physiologically, the abundance of dietary lipids (from HFD) leads to decreased demand for de novo lipogenesis and, consequently, reduced ACL expression/ activity, while the esterification of fatty acids to form triglycerides may remain high due to the excess of exogenous fatty acids from the diet.
Acetyl-CoA, the product of ACL, is also a substrate for histone acetyltransferases [23], including CREBBP and EP300, which promote H3K27 acetylation. This connection links its metabolism to protein acetylation processes. Thus, the decreased expression of Acly and ACL may imply a reduction in cellular acetyl-CoA concentration, limiting substrate availability for histone acetylation. This phenomenon is likely to have occurred in our model and is consistent with results observed and published in another study [24]. Furthermore, studies have demonstrated that ACL plays a role in regulating gene expression by influencing histone acetylation [21,28,29].
Herein, simultaneous with the observed exacerbation in leptin expression, we noted a synchronized downregulation of Acly/ACL alongside a decrease in global H3K27ac levels in the visceral WAT of obese animals. Taken together, our findings suggest a plausible correlation between the reduction in ACL (induced by HFD) and the subsequent decrease in substrate availability for acetylase enzymes, which mRNA were upregulated (as observed for Ep300 and Crebbp) probably by a regulatory feedback system, ultimately leading to the observed decrease in H3K27ac. This reduction could potentially impact a wide range of genes negatively affected by the HFD, given the crucial role of H3K27 acetylation in transcriptional activation and chromatin accessibility.
Significantly, the substantial upregulation of leptin expression, as well as the downregulation observed in ACL and H3K27ac, were effectively reversed by FO treatment. Hence, this reversal in H3K27ac may contribute to reinstating the regulation of genes commonly silenced in obesity, as evidenced in previous studies [21] and in our present array analysis. FO treatment not only prevented the extensive list of genes negatively affected by the HFD (a total of 41 genes) but also upregulated 32 genes, aligning the expression profile of these animals more closely with that of the control (CO) group and distinctly segregating them from the group of obese mice. It is worth emphasizing that FO completely reversed the decline in H3K27ac levels, underscoring its potential to mitigate the epigenetic changes associated with obesity and suggesting a protective effect against the detrimental impacts of HFD-induced obesity on gene expression profiles. If the consumption of a HFD affects histone acetylation levels in WAT, and if this effect is mitigated by FO treatment, then gene expression programs related to inflammation and adipogenesis could potentially be influenced, as suggested by our findings.
In line with our findings, diets incorporating n-3 PUFA (EPA and DHA), have demonstrated beneficial effects in cancer cell lines and patients with cancer, attributed to the reduction of inflammation, likely through epigenetic mechanisms (for a recent review, see [30,31]). This lead to global hyperacetylation of histones in N-terminal regions, as well as at specific loci [32]. The n-3 PUFA-rich diet inhibited the enzyme ACC, responsible for converting two acetyl-CoA into malonyl-Co-A, resulting in an increased pool of free acetyl-CoA [24,33], indicating additional mechanisms to elevate the substrate for histone acetylation. Additionally, a side effect of n-3 PUFA-rich diets is the modification of the expression levels of various microRNAs [27], likely mediated by changes in chromatin accessibility through histone hyperacetylation of regulatory elements of target genes.
However, the mechanisms by which n-3 PUFA-rich FO regulates epigenetic marks in WAT, adipocytes or ASCs remain unknown. It's crucial to note that the analyses reported herein were conducted in WAT, making it challenging to dissociate the specific contributions of adipocytes and ASCs in the results. The development of "unhealthy" obesity is associated with significant alterations in WAT. Under "healthy" metabolic conditions, ASCs undergo adipogenesis and differentiate into mature adipocytes to maintain adipocyte renewal [34]. Available evidence suggests that WAT homeostasis is disrupted in an obesogenic context due to dysregulated adipogenesis [35], with ASCs playing a crucial role in WAT remodeling during obesity [36]).
Although little is known about the molecular mechanisms that become dysfunctional in obesity, it has been established that these are epigenetically regulated before disease manifestations. Some studies provide evidence that the epigenetic dysfunction of ASCs is a key and potential regulatory event in obesity, leading to impaired adipocyte maturation and reduced adipogenic potential[37,38]. Moreover, WAT from obese and/or type 2 diabetes individuals contains a dysfunctional pool of ASCs [39,40,41,42]. According to recent work [37], DNA methylation patterns are essentially preserved during cell commitment to adipogenesis, but obesity preconditions ASCs with a dynamic alteration of DNA methylation in selected regions, leading to WAT dysfunction and the development of metabolic syndromes in obesity. They found that most differences in epigenetic marks detected are due to the pre-established obese environment in the ASC niche. While these studies have highlighted the crucial role of ASCs in the process, epigenetic changes in response to potential modulating agents in ASCs, under physiological or pathological conditions, remain largely unexplored.
Our findings in the whole WAT encouraged us to further explore the ASCs niche. Previous studies have indicated the impact of leptin on ASCs proliferation and differentiation into adipocytes [19,43,44]. Herein, ASCs were extracted from CO and HFD groups of animals. In WAT from HFD obese animals, the environment where ASCs reside is chronically enriched with leptin. Leptin is believed to exert its effects also in a paracrine manner on its receptors, which we identified to be readily expressed by these ASCs. We detected the expression of Lepr1 and Lepr2 isoforms in ASCs from visceral WAT. Moreover, we exposed mice ASCs to leptin. In vitro leptin treatment did not alter the protein expression of the long isoform (Ob-Rb) in ASCs from obese mice, suggesting stable receptor expression despite obesity-related conditions.
We next assessed the Acly expression on ASCs. We have previously shown that this enzyme is highly expressed in these cells, and, similar to WAT, it is also negatively regulated by HFD [45]. There is one study demonstrating that ACL links cellular metabolism to histone acetylation during 3T3-L1 adipocyte differentiation, and that Acly silencing impairs histone acetylation and expression of select genes [21].
Interestingly, we observed a significant decrease in Acly expression in ASCs cultivated in the presence of leptin. This finding led us to suggest that chronic exposure of ASCs to a leptin-rich environment, as observed in obese WAT, may contribute to the downregulation of Acly, as detected in our study.
This observation underscores an emerging role for leptin signaling in modulating gene expression patterns in ASCs and unveils a plausible mechanism that influences WAT dysfunction in obesity. Further investigation into the specific mechanisms underlying H3K27 acetylation and H3K27 methylation dissociating ASCs from adipocytes and macrophages, may provide valuable insights into the pathophysiology of obesity-related dysfunction in WAT. We are now advancing further in this context in an ongoing study conducted by our group.
In summary, over a 16-week study designed to induce obesity in mice through a HFD, it was observed significant increases in body mass, fat intake, fasting blood glucose, and glucose intolerance. However, FO supplementation during the last 8 weeks partially counteracted these effects. The HFD led to marked upregulation of genes associated with inflammation, cytokines, and macrophage markers, while suppressing genes related to adipokines, lipid biosynthesis, adipogenesis, and thermogenesis. FO treatment reversed or alleviated many of these changes, suggesting a potential protective effect against HFD-induced obesity. Examination of epigenetic marks revealed alterations in the expression of enzymes responsible for acetylation and methylation of H3K27, with FO treatment partially reversing these effects. Furthermore, FO prevented reductions in ACL expression and H3K27ac levels observed in obese mice, indicating a role in preserving histone modifications. Our investigation into leptin receptor isoforms in ASCs found no differences between the control and HFD groups but revealed a reduction in Acly expression following leptin treatment. Overall, FO supplementation exhibited promise in mitigating HFD-induced obesity and influencing epigenetic and molecular pathways. Nevertheless, the mechanisms by which fish oil attenuates alterations in H3K27 epigenetic marks remain to be investigated.
These collective findings emphasize the multifaceted impact of FO supplementation on metabolic and epigenetic mechanisms within adipose tissue, highlighting its potential therapeutic efficacy in ameliorating obesity-related dysregulation.
4. Materials and Methods
4.1. Animals, Fish Oil Supplementation and Experimental Procedure
Male C57BL/6 mice, aged eight weeks, were sourced from the Center for Development of Experimental Models (CEDEME), Federal University of São Paulo (UNIFESP). They were housed in a controlled environment with a 12-12 hour light-dark cycle and a temperature maintained at 24±1°C. The experimental protocol remained for 16 weeks, where in the first 8 weeks mice were divided into two groups: control (CO, 9% fat, 76% carbohydrates, and 15% proteins) and HFD (26% carbohydrates, 59% fat, and 15% proteins) group. mice were divided into two groups: control (CO, 9% fat, 76% carbohydrates, and 15% proteins) and HFD (26% carbohydrates, 59% fat, and 15% proteins). After the first 8 weeks, the HFD group was further subdivided into the HFD and HFD+FO (supplemented with fish oil) groups for the subsequent 8 weeks. Fish oil supplementation, sourced from HiOmega-3 (5:1 EPA/DHA, Naturalis Nutrição and Farma Ltda, São Paulo, Brazil), was administered three times per week via oral gavage at a dosage of 2 g/kg body weight. The dosage of FO, as well as the 8-week treatment duration, were selected based on our previous studies [9,11], where the CO and HF groups also received water via gavage. Weekly monitoring of body weight and food intake was conducted throughout the 16-week period. Mice were euthanized following a 6-hour fast using isoflurane anesthesia and cervical dislocation. Blood samples were obtained via orbital plexus puncture. Visceral (Epi and Rp) and subcutaneous (Ing) fat depots were excised, weighed, and then processed as detailed below.
4.2. Glucose Tolerance Test
Glucose tolerance tests (GTT) were conducted in the 12th week of the study. Following a 6-hour fast, mice were intraperitoneally injected with glucose (2 g kg-1 BW). Tail-vein glucose was measured at various intervals (15, 30, 45, 60, and 90 min) using a OneTouch® glucometer. The area under the curve was calculated for each animal.
4.3. WAT and SVF Isolation
The Epi depot was immersed in digestion buffer and finely minced before undergoing collagenase digestion, following established protocols [46]. In brief, the minced samples were placed in a digestive buffer solution (consisting of Dulbecco's modified Eagle's medium - D'MEM/HEPES 20 mM/BSA 4%, and collagenase II [Sigma Chemical, St. Louis, MO, USA] at a concentration of 1.0 mg/ml, pH 7.40) and incubated for approximately 45 minutes at 37°C with orbital agitation (150 rpm). Subsequently, the homogenate was filtered through nylon mesh (Corning, NY) and centrifuged at 400g for 1 minute, yielding two distinct fractions: 1. supernatant, containing the isolated mature adipocytes; 2. remaining filtrate containing the stromal vascular fraction (SVF), which was further processed by centrifugation at 1500 g for 10 minutes to form a cellular pellet. This pellet was then washed twice and aspirated. Next, the SVF was incubated on ice for 10 minutes with a red blood cell lysis buffer (Roche Diagnostics GmbH, Mannheim, Germany) before being washed with PBS and subjected to centrifugation once again.
4.4. Isolation of ASCs and Leptin Treatment
We followed the procedure as previously described [45]. In summary, the cellular pellet (SVF) obtained was suspended in culture medium [D'MEM Han's F-12, supplemented with 10% fetal bovine serum (FBS) and 10ml/l penicillin/streptomycin (Gibco BRL, NY, USA)] and plated in culture dishes (100mm), which were then placed in a 5% CO2 incubator at 37°C (with medium change every two days) until reaching 70-80% confluence. Subsequently, the medium was aspirated, and the plates were rinsed with PBS. The cells were detached for the first time (P1), resuspended in the same culture medium, seeded into new culture dishes for expansion, and grown until they reached 70-80% confluence again. The final step for ASC isolation involved selecting the adherent cell population within the SVF. Cell concentration was determined using a Neubauer chamber, and the cells were replated (P2). Between passages P2 and P5, cells were seeded for experiments (1x10⁵ cells/well in 6-well plates (35 mm)), ensuring that confluence did not exceed 80%. When cell density reached 85-90%, cells were harvested for mRNA and protein extraction either before or after treatment with leptin at a concentration of 100 ng/ml (Sigma Chemical, St. Louis, MO, USA), dissolved in culture medium, for 24 hours. Each pooled cell was counted as one sample.
4.5. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from the entire Epi adipose depot or ASCs, utilizing Trizol reagent (Invitrogen Life Technologies). RNA quality was assessed by measuring ratios at 260/280 and 260/230nm using NANODROP (Thermo Scientific). Reverse transcription to cDNA was performed with the Superscript III cDNA kit (Thermo Scientific, USA). Gene expression was quantified using quantitative real-time polymerase chain reaction (PCR) with the Rotor gene (Qiagen) and SYBR Green fluorescent dye, as described in a previously published study [45,47]. Real-time PCR data analysis was conducted using the 2^(-∆∆Ct) method, and the results were expressed as the ratio of target gene expression to housekeeping genes (Gapdh and 36b4). Specific primer sequences are included in Table 2.
4.6. PCR Array Gene Expression Analysis
For PCR Array Gene Expression Analysis, RNA was isolated using an RNA extraction kit following the manufacturer’s instructions, and the assay was conducted as previously described [48]. Briefly, cDNA and RT2 SYBR® Green qPCR Mastermix (Cat. No. 330529) were utilized on a Custom Mouse RT2 Profiler PCR Array (CLAM30774R; Qiagen) consisting of 84 genes. This array allowed us to assess the expression pattern of genes encoding pro/anti-adipogenic, pro/anti-lipogenic and lipolytic, pro/anti-browning, adipokines, receptors, and components of adipocyte transduction pathways (see Table 3). CT values were exported and uploaded to the manufacturer's data analysis web portal at http://www.qiagen.com/geneglobe. Samples were categorized into control and test groups, and CT values were normalized based on a manual selection of reference genes. Fold Change was calculated using the 2^(-∆∆Ct) method via the data analysis web portal (and exported at GeneGlobe®, Qiagen).
4.7. Western Blot
Proteins from Epi WAT depots (whole tissues) or ASCs were resolved by 12% or 15% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes (0.2 μm). The membranes were blocked with 5% albumin for 2 hours at room temperature and incubated with primary antibodies (anti-H3K27ac #ab4729, H3K27me3 #sab5700166, anti-H3 #ab1791, anti-ACL #ab40793, anti-Leptin Receptor #ab5593, Abcam, USA) at 4°C overnight. Membranes were washed and incubated with secondary horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling®, MA, USA - #7074) antibodies at room temperature for 1 hour. Protein blots were visualized by using an enhanced chemiluminescence Western blotting detection kit (ECL Prime Western Blotting System, Amersham Biosciences®, UK) and analyzed by using ImageJ software.
4.8. Statistical Analysis
The data were analyzed using one-way Analysis of Variance (ANOVA), followed by Tukey's post-test for comparisons between-groups or Student's t-test, as indicated in the figures. The results are expressed as mean ± standard error of the mean (SEM). Differences were considered significant for p<0.05. Statistical analysis was performed using GraphPad Prism software version 9.1.2 (GraphPad Software Inc., San Diego, CA, USA). The PCR Array data were analyzed using the RT2 Profiler PCR Array Data Analysis Software version 3.5 (SABiosciences), with tabulation and analysis in databases such as Gene Ontology Analysis and UniProt.
5. Conclusions
Given the limited literature on the link between FO treatment and WAT epigenetics, our study underscores the significance of our findings. It sheds light on the behavior of histone modifiers, pivotal in adipose tissue dysfunctions related to obesity. Furthermore, our research enhances comprehension of ASCs and leptin signaling, while emphasizing FO's potential in modulating epigenetic factors, presenting a promising avenue for addressing obesity-related complications within the scope of this research.
Author Contributions
Conceptualization, methodology, formal analysis and investigation, JJS, AFSB, VTGP and MICAV; writing—original draft preparation, JJS and AFS; writing—review and editing, LMAC and MICAV.; visualization, JJS, AFSB, VTGP and MICAV; supervision, MICAV; project administration, JJS, AFSB and MICAV; funding acquisition, LMAC and MICAV.
Funding
This research was funded by grants received from CNPq and FAPESP (2019/13618-9; 2022/15127-5; 2019/26240-4).
Institutional Review Board Statement
All procedures were approved by the Ethics Committee on Animal Use of the Federal University of São Paulo (CEUA nº 8827141217).
Informed Consent Statement
Not applicable
Data Availability Statement
Data is contained within the article and supplementary material.
Acknowledgments
We would like to thank Dr. Roberta D. C. da Cunha de Sá, who collaborated with the gavages and tissue collections from the animals.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- J. Li, H. Wu, Y. Liu, and L. Yang, “High fat diet induced obesity model using four strainsof mice: Kunming, C57BL/6, BALB/c and ICR,” Exp Anim, vol. 69, no. 3, pp. 326–335, 2020. [CrossRef]
- G. S. Hotamisligil, “Inflammation, metaflammation and immunometabolic disorders,” Nature, vol. 542, no. 7640, pp. 177–185, Feb. 2017. [CrossRef]
- M. Hawash et al., “Characterization and Investigation of Novel Benzodioxol Derivatives as Antidiabetic Agents: An In Vitro and In Vivo Study in an Animal Model,” Biomolecules, vol. 13, no. 10, Oct. 2023. [CrossRef]
- M. Hawash, N. Jaradat, N. A. Salhi, B. Shatreet, A. A. Asbah, and Y. H. Hawash, “Assessing the therapeutic potential and safety of traditional anti-obesity herbal blends in Palestine,” Sci Rep, vol. 14, no. 1, Dec. 2024. [CrossRef]
- Y. Su et al., “Effects of Fish Oil, Lipid Mediators, Derived from Docosahexaenoic Acid, and Their Co-Treatment against Lipid Metabolism Dysfunction and Inflammation in HFD Mice and HepG2 Cells,” Nutrients, vol. 15, no. 2, Jan. 2023. [CrossRef]
- B. Kapoor, D. Kapoor, S. Gautam, R. Singh, and S. Bhardwaj, “Dietary Polyunsaturated Fatty Acids (PUFAs): Uses and Potential Health Benefits,” Curr Nutr Rep, vol. 10, no. 3, pp. 232–242, Sep. 2021. [CrossRef]
- F. Shahidi and P. Ambigaipalan, “Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits,” Annu Rev Food Sci Technol, vol. 9, pp. 345–381, Mar. 2018. [CrossRef]
- S. Yu et al., “Different ratios of DHA/EPA reverses insulin resistance by improving adipocyte dysfunction and lipid disorders in HFD-induced IR mice,” Food Funct, vol. 14, no. 2, pp. 1179–1197, Dec. 2023. [CrossRef]
- R. D. C. da C. de Sá et al., “Fish oil prevents changes induced by a high-fat diet on metabolism and adipokine secretion in mice subcutaneous and visceral adipocytes,” Journal of Physiology, vol. 594, no. 21, 2016. [CrossRef]
- R. D. C. da Cunha de Sá et al., “Fish oil reverses metabolic syndrome, adipocyte dysfunction, and altered adipokines secretion triggered by high-fat diet-induced obesity,” Physiol Rep, vol. 8, no. 4, Feb. 2020. [CrossRef]
- R. D. C. da Cunha de Sá et al., “Fish oil enriched in epa, but not in dha, reverses the metabolic syndrome and adipocyte dysfunction induced by a high-fat diet,” Nutrients, vol. 13, no. 3, pp. 1–18, Mar. 2021. [CrossRef]
- V. J. Antraco et al., “Omega-3 polyunsaturated fatty acids prevent nonalcoholic steatohepatitis (Nash) and stimulate adipogenesis,” Nutrients, vol. 13, no. 2, pp. 1–20, Feb. 2021. [CrossRef]
- K. Sun et al., “Dichotomous effects of VEGF-A on adipose tissue dysfunction,” Proc Natl Acad Sci U S A, vol. 109, no. 15, pp. 5874–5879, Apr. 2012. [CrossRef]
- G. R. Hajer, T. W. Van Haeften, and F. L. J. Visseren, “Adipose tissue dysfunction in obesity, diabetes, and vascular diseases,” European Heart Journal, vol. 29, no. 24. pp. 2959–2971, Dec. 2008. [CrossRef]
- T. S. Mikkelsen et al., “Comparative epigenomic analysis of murine and human adipogenesis,” Cell, vol. 143, no. 1, pp. 156–169, 2010. [CrossRef]
- R. Margueron and D. Reinberg, “The Polycomb complex PRC2 and its mark in life,” Nature, vol. 469, no. 7330, pp. 343–349, Jan. 2011. [CrossRef]
- V. V. Ogryzko, R. L. Schiltz, V. Russanova, B. H. Howard, and Y. Nakatani, “The transcriptional coactivators p300 and CBP are histone acetyltransferases,” Cell, vol. 87, no. 5, pp. 953–959, Nov. 1996. [CrossRef]
- S. H. Hong, Y. W. Cho, L. R. Yu, H. Yu, T. D. Veenstra, and K. Ge, “Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases,” Proc Natl Acad Sci U S A, vol. 104, no. 47, pp. 18439–18444, Nov. 2007. [CrossRef]
- H. Takahashi, J. M. McCaffery, R. A. Irizarry, and J. D. Boeke, “Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription,” Mol Cell, vol. 23, no. 2, pp. 207–217, Jul. 2006. [CrossRef]
- A. A. Cluntun, H. Huang, L. Dai, X. Liu, Y. Zhao, and J. W. Locasale, “The rate of glycolysis quantitatively mediates specific histone acetylation sites,” Cancer Metab, vol. 3, no. 1, Dec. 2015. [CrossRef]
- K. E. Wellen, G. Hatzivassiliou, U. M. Sachdeva, T. V. Bui, J. R. Cross, and C. B. Thompson, “ATP-citrate lyase links cellular metabolism to histone acetylation,” Science, vol. 324, no. 5930, pp. 1076–1080, May 2009. [CrossRef]
- A. G. Evertts, B. M. Zee, P. A. Dimaggio, M. Gonzales-Cope, H. A. Coller, and B. A. Garcia, “Quantitative dynamics of the link between cellular metabolism and histone acetylation,” J Biol Chem, vol. 288, no. 17, pp. 12142–12151, Apr. 2013. [CrossRef]
- L. Galdieri, T. Zhang, D. Rogerson, R. Lleshi, and A. Vancura, “Protein acetylation and acetyl coenzyme a metabolism in budding yeast,” Eukaryot Cell, vol. 13, no. 12, pp. 1472–1483, Dec. 2014. [CrossRef]
- A. Carrer et al., “Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels,” J Biol Chem, vol. 292, no. 8, pp. 3312–3322, Feb. 2017. [CrossRef]
- L. Jiang et al., “Leptin contributes to the adaptive responses of mice to high-fat diet intake through suppressing the lipogenic pathway,” PLoS One, vol. 4, no. 9, Sep. 2009. [CrossRef]
- H. Fukuda, A. Katsurada, and N. Iritani, “Nutritional and hormonal regulation of mRNA levels of lipogenic enzymes in primary cultures of rat hepatocytes,” J Biochem, vol. 111, no. 1, pp. 25–30, 1992. [CrossRef]
- H. M. Jin et al., “MicroRNA-155 as a proinflammatory regulator via SHIP-1 down-regulation in acute gouty arthritis,” Arthritis Res Ther, vol. 16, no. 2, Apr. 2014. [CrossRef]
- M. Peng, N. Yin, S. Chhangawala, K. Xu, C. S. Leslie, and M. O. Li, “Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism,” Science, vol. 354, no. 6311, pp. 481–484, Oct. 2016. [CrossRef]
- J. V. Lee et al., “Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation,” Cell Metab, vol. 20, no. 2, pp. 306–319, Aug. 2014. [CrossRef]
- M. Amatruda, G. Ippolito, S. Vizzuso, G. Vizzari, G. Banderali, and E. Verduci, “Epigenetic Effects of n-3 LCPUFAs: A Role in Pediatric Metabolic Syndrome,” Int J Mol Sci, vol. 20, no. 9, May 2019. [CrossRef]
- P. T. Georgel and P. Georgel, “Where Epigenetics Meets Food Intake: Their Interaction in the Development/Severity of Gout and Therapeutic Perspectives,” Front Immunol, vol. 12, Sep. 2021. [CrossRef]
- A. Abbas et al., “Epigenetic Reprogramming Mediated by Maternal Diet Rich in Omega-3 Fatty Acids Protects From Breast Cancer Development in F1 Offspring,” Front Cell Dev Biol, vol. 9, Jun. 2021. [CrossRef]
- L. Galdieri and A. Vancura, “Acetyl-CoA carboxylase regulates global histone acetylation,” Journal of Biological Chemistry, vol. 287, no. 28, pp. 23865–23876, Jul. 2012. [CrossRef]
- G. Yu et al., “Adipogenic differentiation of adipose-derived stem cells,” Methods Mol Biol, vol. 702, pp. 193–200, 2011. [CrossRef]
- P. Patel and N. Abate, “Role of subcutaneous adipose tissue in the pathogenesis of insulin resistance,” J Obes, vol. 2013, 2013. [CrossRef]
- W. P. Cawthorn, E. L. Scheller, and O. A. MacDougald, “Adipose tissue stem cells meet preadipocyte commitment: going back to the future,” J Lipid Res, vol. 53, no. 2, pp. 227–246, Feb. 2012. [CrossRef]
- M. Ejarque et al., “Adipose tissue mitochondrial dysfunction in human obesity is linked to a specific DNA methylation signature in adipose-derived stem cells,” Int J Obes (Lond), vol. 43, no. 6, pp. 1256–1268, Jun. 2019. [CrossRef]
- L. M. Pérez et al., “Altered metabolic and stemness capacity of adipose tissue-derived stem cells from obese mouse and human,” PLoS One, vol. 10, no. 4, Apr. 2015. [CrossRef]
- G. Pachón-Peña et al., “Obesity Determines the Immunophenotypic Profile and Functional Characteristics of Human Mesenchymal Stem Cells From Adipose Tissue,” Stem Cells Transl Med, vol. 5, no. 4, pp. 464–475, Apr. 2016. [CrossRef]
- C. Serena et al., “Obesity and Type 2 Diabetes Alters the Immune Properties of Human Adipose Derived Stem Cells,” Stem Cells, vol. 34, no. 10, pp. 2559–2573, Oct. 2016. [CrossRef]
- A. B. Crujeiras et al., “Genome-wide DNA methylation pattern in visceral adipose tissue differentiates insulin-resistant from insulin-sensitive obese subjects,” Transl Res, vol. 178, pp. 13-24.e5, Dec. 2016. [CrossRef]
- M. Roldan, M. Macias-Gonzalez, R. Garcia, F. J. Tinahones, and M. Martin, “Obesity short-circuits stemness gene network in human adipose multipotent stem cells,” FASEB J, vol. 25, no. 12, pp. 4111–4126, Dec. 2011. [CrossRef]
- L. Palhinha et al., “Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes,” Front Endocrinol (Lausanne), vol. 10, Dec. 2019. [CrossRef]
- M. J. Crop, C. C. Baan, S. S. Korevaar, J. N. M. Ijzermans, W. Weimar, and M. J. Hoogduijn, “Human adipose tissue-derived mesenchymal stem cells induce explosive T-Cell proliferation,” Stem Cells Dev, vol. 19, no. 12, pp. 1843–1853, Dec. 2010. [CrossRef]
- V. S. da Silva et al., “High-fat diet decreases H3K27ac in mice adipose-derived stromal cells,” Obesity, vol. 30, no. 10, pp. 1995–2004, Oct. 2022. [CrossRef]
- &iartin Rodbell, “Metabolism of Isolated Fat Cells I. EE’li’ECTS OF HORMOXES OX GLUCOSE METABOLISM ASD LII’OLYSIS,” 1961. [Online]. Available: www.jbc.org.
- P. G. Reeves, K. L. Rossow, and J. Lindlauf, “Development and testing of the AIN-93 purified diets for rodents: results on growth, kidney calcification and bone mineralization in rats and mice,” J Nutr, vol. 123, no. 11, pp. 1923–1931, 1993. [CrossRef]
- F. M. Thomaz et al., “Ginkgo biloba Extract Stimulates Adipogenesis in 3T3-L1 Preadipocytes,” Pharmaceuticals (Basel), vol. 15, no. 10, Oct. 2022. [CrossRef]
Figure 1.
Obesity Model Characterization. A) Caloric (kcal/day/animal), Food and Fat (g/day/animal) intake, B) Body mass evolution, C) Fasting glucose, D) Glucose tolerance test or GTT and E) Incremental area under the glycemic curve in control (CO) and obese animals induced by a high fat diet (HFD) for 12 weeks. In A-B, the measurements were performed weekly throughout the experimental protocol. In C-E, the glycemic curve or glucose concentration versus time was calculated after glucose administration (2 g/Kg b.w.). Data were analyzed using Student's t-test, and show mean ± SEM (n=11-12). *p< 0,05 or ****P < 0,0001 versus control.
Figure 1.
Obesity Model Characterization. A) Caloric (kcal/day/animal), Food and Fat (g/day/animal) intake, B) Body mass evolution, C) Fasting glucose, D) Glucose tolerance test or GTT and E) Incremental area under the glycemic curve in control (CO) and obese animals induced by a high fat diet (HFD) for 12 weeks. In A-B, the measurements were performed weekly throughout the experimental protocol. In C-E, the glycemic curve or glucose concentration versus time was calculated after glucose administration (2 g/Kg b.w.). Data were analyzed using Student's t-test, and show mean ± SEM (n=11-12). *p< 0,05 or ****P < 0,0001 versus control.
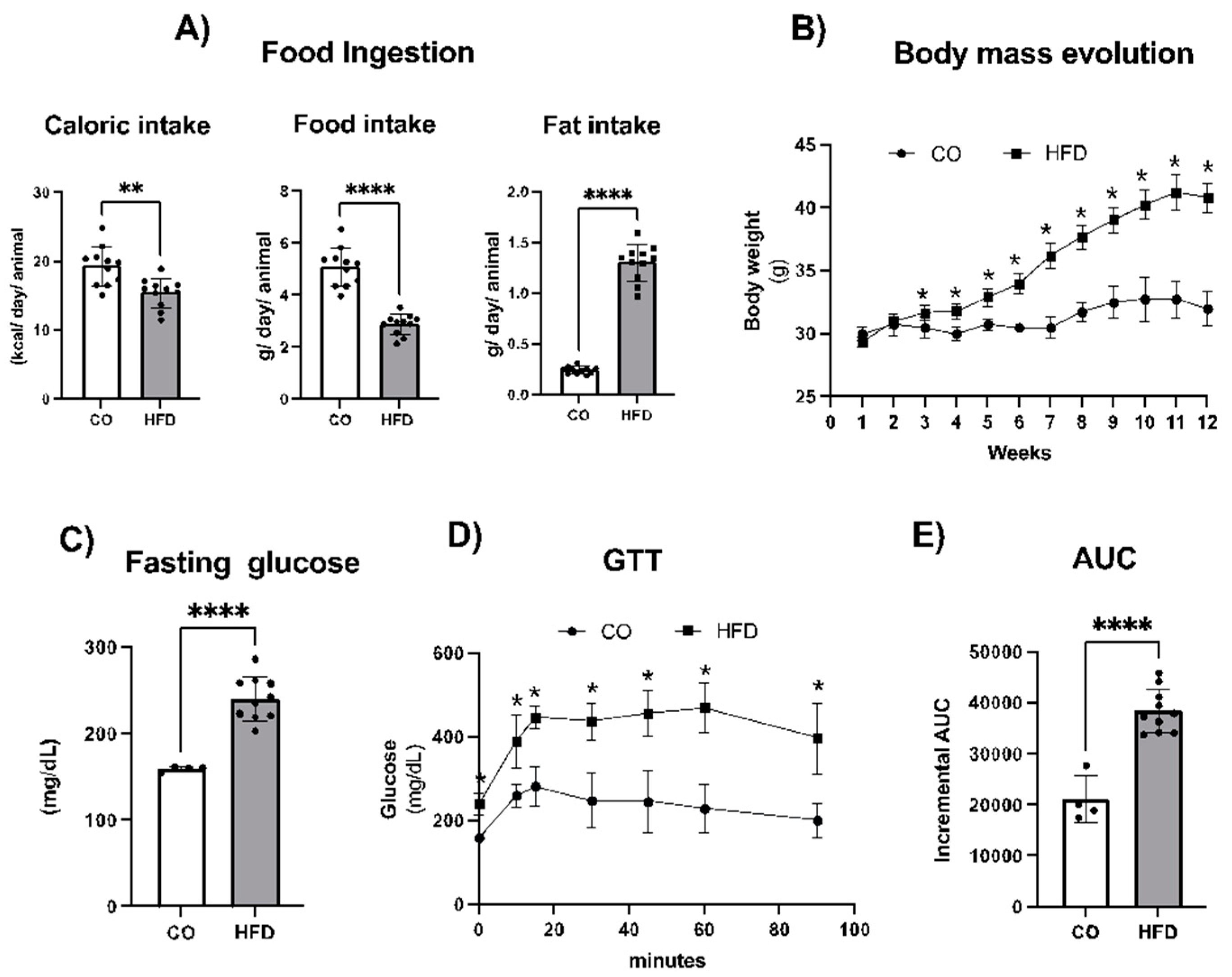
Figure 2.
Body mass evolution (A), depot mass of visceral epididymal (Epi) (B), retroperitoneal (Rp) (C), and subcutaneous inguinal (Ing) (D) adipose tissues in milligrams (mg), after 16 weeks of experimental diets and fish oil (FO) supplementation. In the initial 8 weeks, the animals were submitted to either a control (CO) or high-fat diet (HFD). During the last 8 weeks of the experimental protocol, the diets were continued, and the animals underwent gavage (CO and HFD groups received water, while the HFD+FO group received FO) three times a week. Data were analyzed using one-way Analysis of Variance (ANOVA) followed by Tukey's post-test, and show mean ± SEM (n=5-6). *p< 0,05, **P<0.001 or ****P < 0,0001.
Figure 2.
Body mass evolution (A), depot mass of visceral epididymal (Epi) (B), retroperitoneal (Rp) (C), and subcutaneous inguinal (Ing) (D) adipose tissues in milligrams (mg), after 16 weeks of experimental diets and fish oil (FO) supplementation. In the initial 8 weeks, the animals were submitted to either a control (CO) or high-fat diet (HFD). During the last 8 weeks of the experimental protocol, the diets were continued, and the animals underwent gavage (CO and HFD groups received water, while the HFD+FO group received FO) three times a week. Data were analyzed using one-way Analysis of Variance (ANOVA) followed by Tukey's post-test, and show mean ± SEM (n=5-6). *p< 0,05, **P<0.001 or ****P < 0,0001.
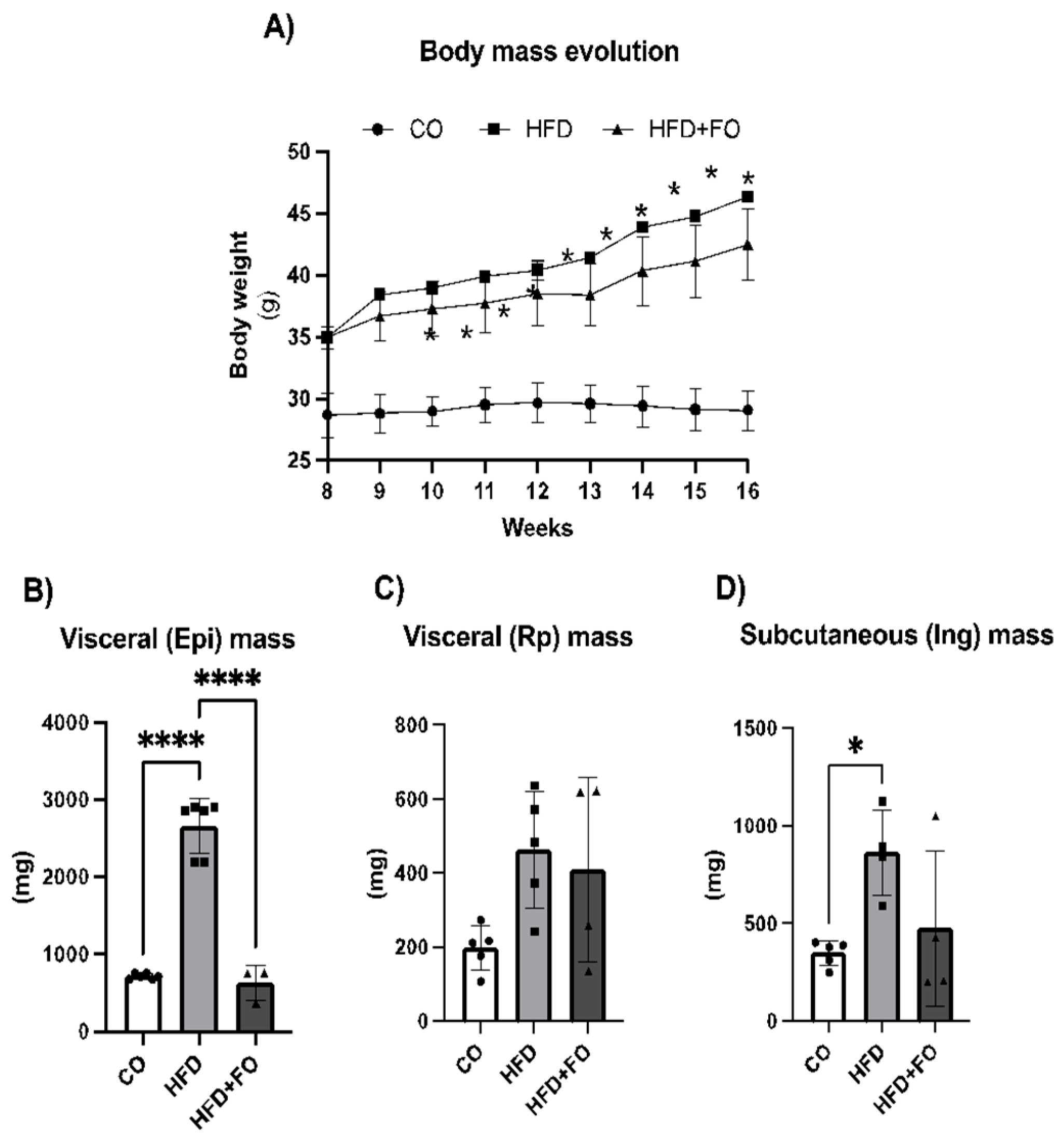
Figure 3.
Heatmap of gene expression in Epi WAT from mice subjected to 16 weeks of experimental diets and fish oil supplementation. Control diet (CO), high-fat diet (HFD), and high-fat diet plus fish oil (HFD+FO). The gene expression level is indicated using a color scale, where red indicates higher expression and green indicates lower expression.
Figure 3.
Heatmap of gene expression in Epi WAT from mice subjected to 16 weeks of experimental diets and fish oil supplementation. Control diet (CO), high-fat diet (HFD), and high-fat diet plus fish oil (HFD+FO). The gene expression level is indicated using a color scale, where red indicates higher expression and green indicates lower expression.
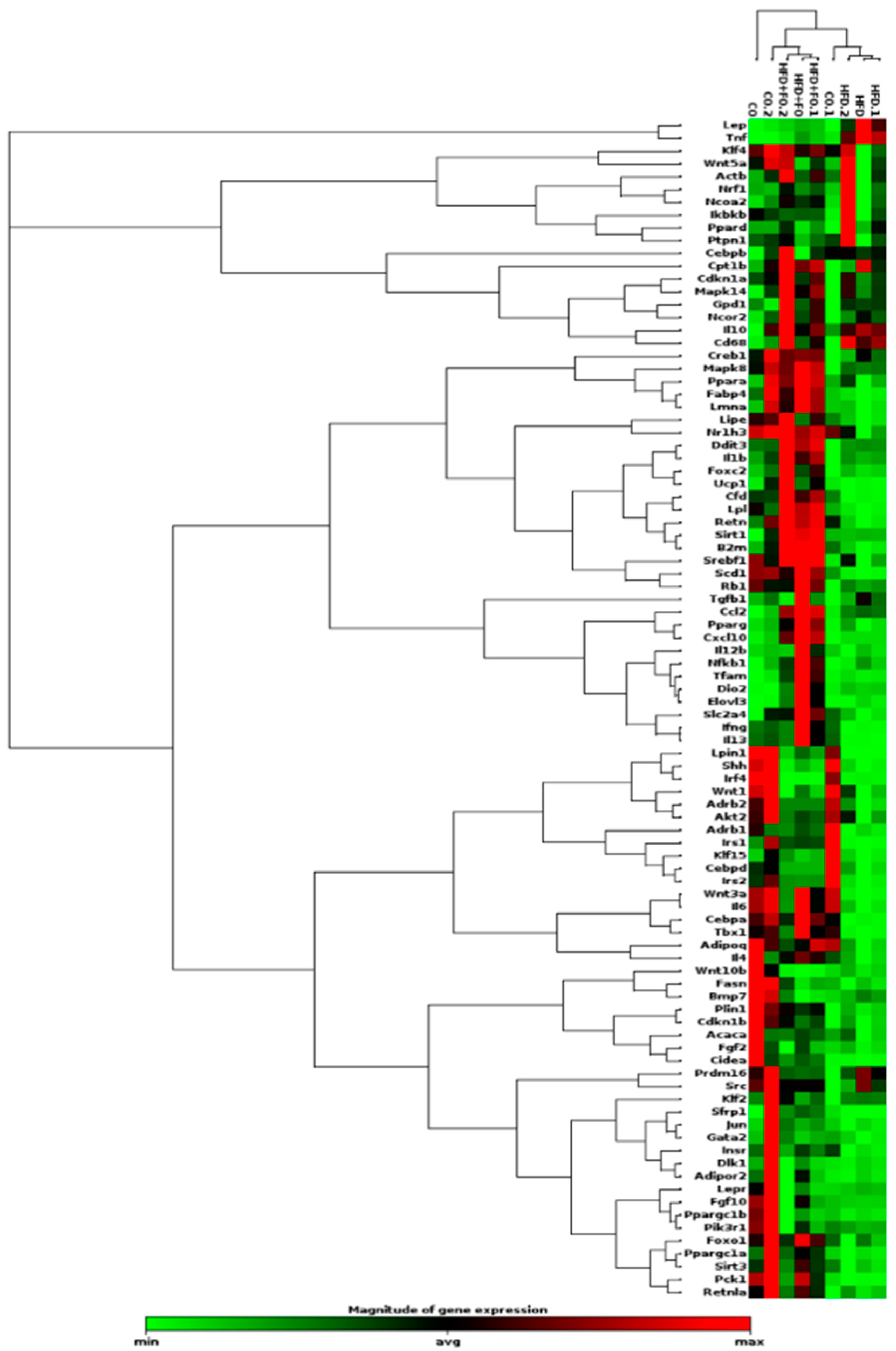
Figure 4.
Gene expression of Acly (A) and genes encoding histone modifiers Ezh2 (B), Crebbp (C), Ep300 (D), Kdm6a (E), and Kdm6b (F), in the visceral Epi WAT from animals that received control diet (CO), high-fat diet (HFD), or HFD and fish oil (HFD+FO). Target genes were normalized by the constitutive Gapdh. Data were analyzed using one-way ANOVA followed by Tukey's post-test, and show mean ± SEM (n=4-6). *p< 0,05 or **P<0.001.
Figure 4.
Gene expression of Acly (A) and genes encoding histone modifiers Ezh2 (B), Crebbp (C), Ep300 (D), Kdm6a (E), and Kdm6b (F), in the visceral Epi WAT from animals that received control diet (CO), high-fat diet (HFD), or HFD and fish oil (HFD+FO). Target genes were normalized by the constitutive Gapdh. Data were analyzed using one-way ANOVA followed by Tukey's post-test, and show mean ± SEM (n=4-6). *p< 0,05 or **P<0.001.
Figure 5.
Graphical representation of the protein content of ACL (A), H3K27ac (B), and H3K27me3 (C), in visceral Epi WAT from animals that received a control diet (CO), a high-fat diet (HFD) or a HFD diet and fish oil (HFD+FO). Data were analyzed using one-way ANOVA followed by Tukey's post-test. Values were expressed as mean ± SEM, in relation to the control and corrected by the expression of total protein by Ponceau (ACL) or constitutive H3 (H3k27ac and H3k27me3). A representative image of protein expression levels from 2 independent experiments is shown above each graph (n= 3 animals) quantified by ImageJ. * P<0,05 or ** P<0,001.
Figure 5.
Graphical representation of the protein content of ACL (A), H3K27ac (B), and H3K27me3 (C), in visceral Epi WAT from animals that received a control diet (CO), a high-fat diet (HFD) or a HFD diet and fish oil (HFD+FO). Data were analyzed using one-way ANOVA followed by Tukey's post-test. Values were expressed as mean ± SEM, in relation to the control and corrected by the expression of total protein by Ponceau (ACL) or constitutive H3 (H3k27ac and H3k27me3). A representative image of protein expression levels from 2 independent experiments is shown above each graph (n= 3 animals) quantified by ImageJ. * P<0,05 or ** P<0,001.
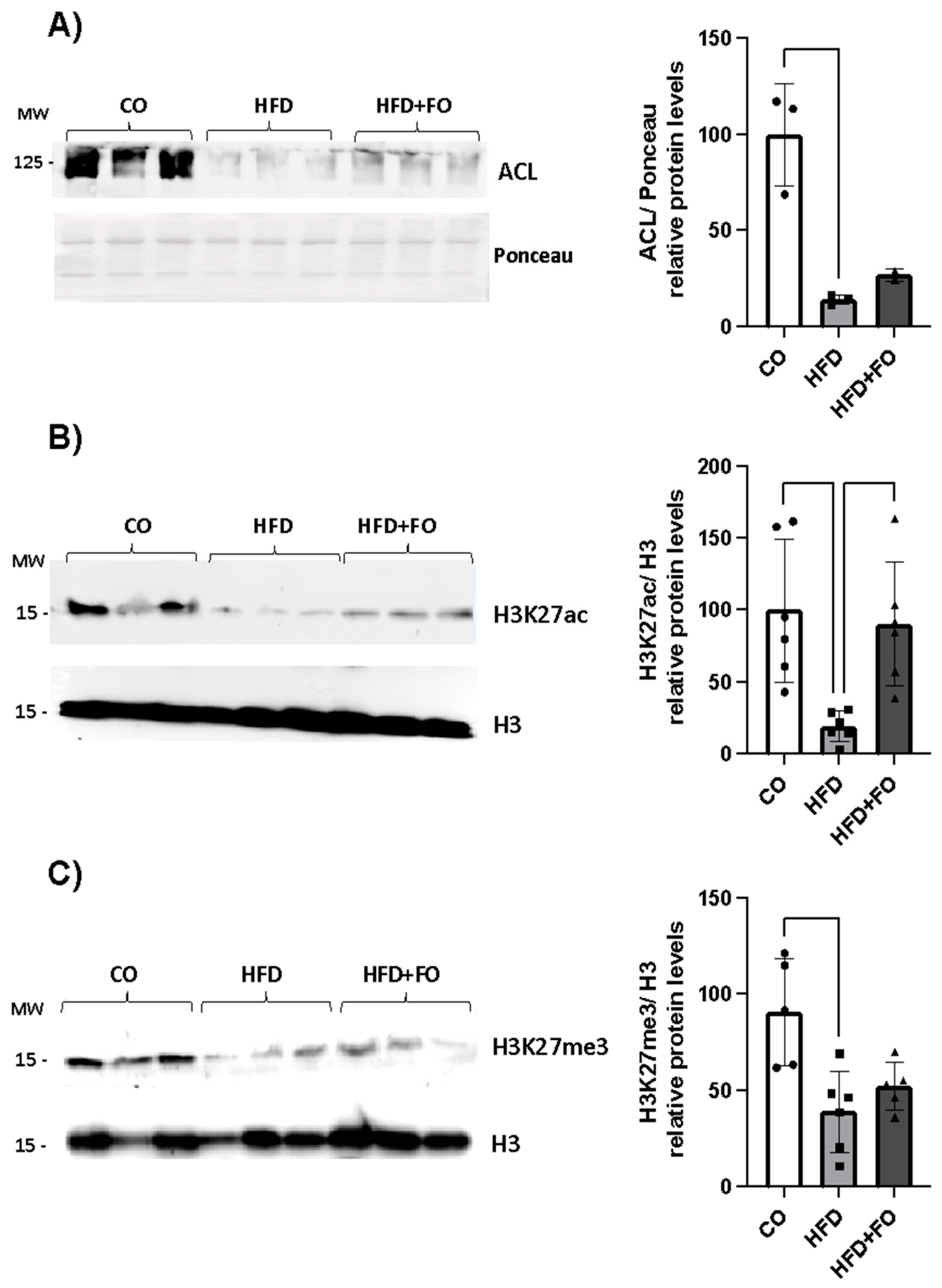
Figure 6.
Gene expression of Lepr1 (A) Lepr2 (B) in ASC isolated from WAT of animals that received a control diet (CO) or a high-fat diet (HFD). Total content of LEP R1 (Ob-Rb) protein (C) and gene expression of Acly (D) in ASC isolated from WAT of HFD-induced obese animals, treated in vitro with 100ng/mL leptin for 24h. Data were analyzed using Student's t-test, and show mean ± SEM (n=4-6). In A, B and C, target genes were normalized by the constitutive 36B4. In C, total content of protein was quantified by ImageJ and expressed in relation to the control and corrected by the expression of the constitutive B-Actin. A representative image of protein expression level is shown above the graphic. * P<0,05 or ** P<0,001.
Figure 6.
Gene expression of Lepr1 (A) Lepr2 (B) in ASC isolated from WAT of animals that received a control diet (CO) or a high-fat diet (HFD). Total content of LEP R1 (Ob-Rb) protein (C) and gene expression of Acly (D) in ASC isolated from WAT of HFD-induced obese animals, treated in vitro with 100ng/mL leptin for 24h. Data were analyzed using Student's t-test, and show mean ± SEM (n=4-6). In A, B and C, target genes were normalized by the constitutive 36B4. In C, total content of protein was quantified by ImageJ and expressed in relation to the control and corrected by the expression of the constitutive B-Actin. A representative image of protein expression level is shown above the graphic. * P<0,05 or ** P<0,001.
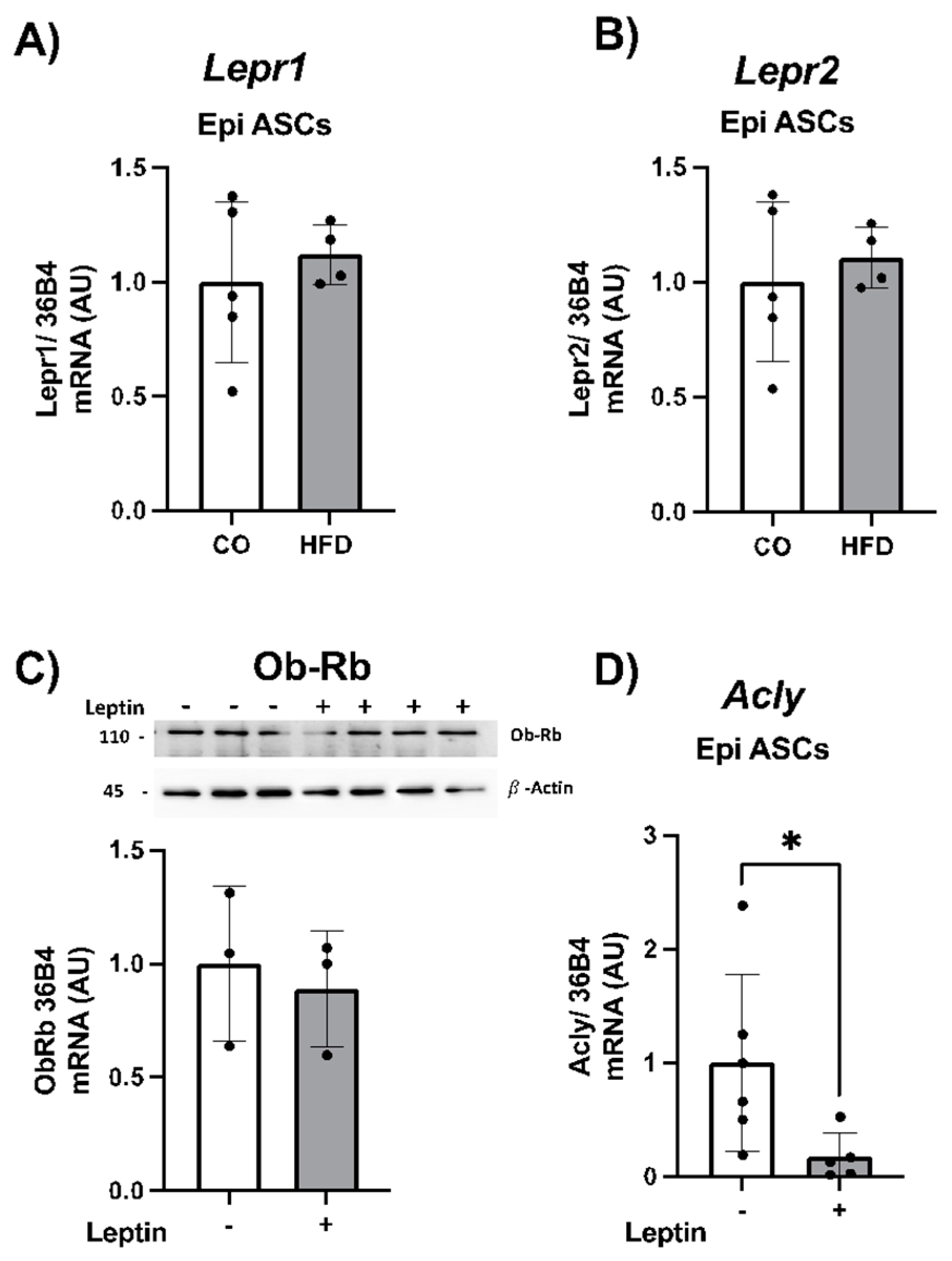

Table 1.
- List of genes that were up-regulated and down-regulated, comparing the obese group to the control group: HFD vs CO.
Table 1.
- List of genes that were up-regulated and down-regulated, comparing the obese group to the control group: HFD vs CO.
| Gene | RefSeq Number | Fold Regulation | p-Value | Pathway related |
|---|---|---|---|---|
| Up- regulated | ||||
| Lep | NM_008493 | 25.48 | 0.046332 | Adipokines |
| Ncor2 | NM_001253904 | 3.08 | ns | Anti-Browning |
| Dio2 | NM_010050 | 3.98 | 0.000554 | Pro-Browning, fatty acid thermogenesis and oxidation |
| Elovl3 | NM_007703 | 2.63 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Ccl2 | NM_011333 | 5.07 | 0.009529 | Cytokines, growth factors and signal transduction |
| Il10 | NM_010548 | 2.08 | ns | Cytokines, growth factors and signal transduction |
| Tgfb1 | NM_011577 | 3.06 | ns | Cytokines, growth factors and signal transduction |
| Tnf | NM_013693 | 10.65 | 0.009005 | Cytokines, growth factors and signal transduction |
| Nfkb1 | NM_008689 | 2.52 | ns | Cytokines, growth factors and signal transduction |
| Cd68 | NM_009853 | 9.16 | 0.000279 | Cytokines, growth factors and signal transduction |
| Down- regulated | ||||
| Adipoq | NM_009605 | -2.82 | 0.010125 | Adipokines |
| Cfd | NM_013459 | -2.54 | 0.004805 | Adipokines |
| Retn | NM_001204959 | -3.30 | 0.016985 | Adipokines |
| Acaca | NM_133360 | -2.55 | ns | Lipases and lipogenic enzymes |
| Scd1 | NM_009127 | -2.90 | 0.042774 | Lipases and lipogenic enzymes |
| Lpin1 | NM_001130412 | -8.55 | 0.001238 | Lipases and lipogenic enzymes |
| Pck1 | NM_011044 | -5.98 | ns | Lipases and lipogenic enzymes |
| Fasn | NM_007988 | -3.87 | ns | Lipases and lipogenic enzymes |
| Cebpa | NM_007678 | -2.45 | 0.002073 | Pro- adipogenesis |
| Cebpd | NM_007679 | -3.64 | 0.041382 | Pro- adipogenesis |
| Fabp4 | NM_024406 | -2.01 | ns | Pro- adipogenesis |
| Fgf2 | NM_008006 | -2.37 | ns | Pro- adipogenesis |
| Fgf10 | NM_008002 | -2.71 | ns | Pro- adipogenesis |
| Jun | NM_010591 | -2.05 | ns | Pro- adipogenesis |
| Sfrp1 | NM_013834 | -2.98 | ns | Pro- adipogenesis |
| Klf15 | NM_023184 | -4.62 | ns | Pro- adipogenesis |
| Adrb2 | NM_007420 | -6.37 | 0.001464 | Anti- adipogenesis |
| Dlk1 | NM_001190703 | -2.36 | ns | Anti- adipogenesis |
| Foxo1 | NM_019739 | -2.26 | ns | Anti- adipogenesis |
| Shh | NM_009170 | -18.41 | 0.000001 | Anti- adipogenesis |
| Wnt1 | NM_021279 | -4.60 | 0.001147 | Anti- adipogenesis |
| Wnt3a | NM_009522 | -11.21 | 0.000001 | Anti- adipogenesis |
| Gata2 | NM_008090 | -2.32 | ns | Anti- adipogenesis |
| Bmp7 | NM_007557 | -2.06 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Ppargc1a | NR_027710 | -2.40 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Ppargc1b | NM_133249 | -2.29 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Sirt3 | NM_001127351 | -2.34 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Tbx1 | NM_011532 | -11.21 | 0.000001 | Pro-Browning, fatty acid thermogenesis and oxidation |
| Ucp1 | NM_009463 | -5.67 | ns | Pro-Browning, fatty acid thermogenesis and oxidation |
| Nr1h3 | NM_001177730 | -2.28 | 0.007073 | Anti-Browning |
| Wnt10b | NM_011718 | -2.50 | ns | Anti-Browning |
| Lepr | NM_001122899 | -2.30 | ns | Adipokines receptors |
| Adipor2 | NM_197985 | -3.03 | ns | Adipokines receptors |
| Adrb1 | NM_007419 | -2.73 | ns | Adipokines receptors |
| Ifng | NM_008337 | -11.06 | 0.000001 | Cytokines, growth factors and signal transduction |
| Il4 | NM_021283 | -2.20 | ns | Cytokines, growth factors and signal transduction |
| Il6 | NM_031168 | -10.93 | 0.000001 | Cytokines, growth factors and signal transduction |
| Il13 | NM_008355 | -11.21 | 0.000001 | Cytokines, growth factors and signal transduction |
| Insr | NM_010568 | -3.52 | ns | Cytokines, growth factors and signal transduction |
| Irs1 | NM_010570 | -4.26 | 0.043055 | Cytokines, growth factors and signal transduction |
| Irs2 | NM_001081212 | -4.61 | 0.017955 | Cytokines, growth factors and signal transduction |
| Pik3r1 | NM_001024955 | -2.33 | ns | Cytokines, growth factors and signal transduction |
| Irf4 | NM_013674 | -11.92 | 0.000978 | Cytokines, growth factors and signal transduction |
*ns = Not significant. This indicates that, although there was a fold-regulation greater than 2 in the array, the 'p' value was higher than 0.5, likely due to a large variation resulting from a low number of biological replicates. Nevertheless, these genes may play an important role in the pathways activated by FO in WAT.
Table 2.
- Sense and antisense primers sequences used for qRT-PCR.
| Gene | 5’ Primer ( 5’-3’) -Sense | 3’ Primer (5’-3’) -Antisense |
|---|---|---|
| Gapdh | AAATGGTGAAGGTCGGTGTG | TGAAGGGGTCGTTGATGG |
| Ep300(p300) | GTTGCTATGGGAAACAGTTATGC | TGTAGTTTGAGGTTGGGAAGG |
| Ezh2 | CAGGATGAAGCAGACAGAAGAGG | TCGGGTTGCATCCACCACAAA |
| Kdm6a | GCTGGAACAGCTGGAAAGTC | GAGTCAACTGTTGGCCCATT |
| Kdm6b | CCTATTATGCTCCTGGGACA | TACGGCTTCCTCACTGTCGT |
| Crebbp (Cbp) | GACCGCTTTGTTTATACCTGC | TCTTATGGGTGTGGCTCTTTG |
| Acly | TCCGTCAAACAGCACTTCC | ATTTGGCTTCTTGGAGGTG |
| 36b4 (Rplp0) | TAAAGACTGGAGACAAGGTG | GTGTACTCAGTCTCCAC AGA |
| Lepr1 | CAGAATGACGCAGGGCTGTA | GCTCAAATGTTTCAGGCTTTTGG |
| Lepr2 | ATTAATGGTTTCACCAAAGATGCT | AAGATCTGTAAGTACTGTGGCAT |
Gapdh, Glyceraldehyde-3-phosphate Dehydrogenase; Ep300(p300), E1A binding protein p300; Ezh2, Enhancer of zest 2 polycomb repressive complex 2 subunit; Kdm6a, Lysine (K)-specific demethylase 6A; Kdm6b, Lysine (K)-specific demethylase 6B; Crebbp, CREB binding protein; Acly, ATP citrate lyase; 36b4 (Rplp0) ribossomal protein lateral stalk subunit P0; Lepr1, leptin receptor 1; Lepr2, Leptin receptor 2.
Table 3.
- List of selected genes in Custom Mouse RT2 Profiler PCR Array.
| Pathways | Genes |
|---|---|
| Adipokines | Adipoq (Acrp30), Cfd (Adipisin), Lep (Leptin), Retn(Resistin) |
| Lipases and lipogenic enzymes | Acaca (Acc1), Gpd1(glycerol-3-phosphate dehydrogenase 1 (soluble), Lipe(HSL),Scd1 (stearoyl CoA desaturase), Lpl, Pnpla2 (Atgl), Lipin 1, Pck1 (phosphoenolpyruvate carboxykinase 1), Fasn |
| Pro- adipogenesis | Cebpa, Cebpb, Cebpd, Pparg (PPAR gamma 2), Srebf1, Fabp4(aP2), Pilin1, Fgf2 (bFGF), Fgf10, Jun (c-jun ou AP1), Lmna (Lamini A), Sfrp1(secreted frizzled-related protein1), Slc2a4 (Glut4), Klf15, Klf4 |
| Anti- adipogenesis | Adrb2, Cdkn1a (p21Cip1, Waf1), Cdkn1b (p27Kip1), Ddit3 (Gadd153, Chop), Dlk1(Pref1), Foxo1, Ncor2, Shh, Sirt1, Wnt1, Wnt3a, Gata2, Klf |
| Pro-Browning, fatty acid thermogenesis and oxidation | Bmp7, Cidea, Cpt1b, Creb1, Dio2, Elovl3, Foxc2, Mapk14 (p38alpha), Nrf1, Ppara, Ppard, Ppargc1a (Pgc1alpha), Ppargc1b (Perc, Pgc1beta), Prdm16, Sirt3, Src, Tbx1, Tfam, Ucp1, Wnt5a |
| Anti-Browning | Ncoa2, Nr1h3, Rb1, Wnt10b |
| Adipokines receptors | Lepr, Adipor2, Adrb1 |
| Cytokines, growth factors and signal transduction | Ccl2 (MCP1), Cxcl10, Ifng, Il1b, Il4, Il6, Il10, Il12b, Il13, Tgfb1, Tnf, Insr, Irs1, Irs2, Akt2, Ptpn1 (PTP1B), Ikbkb (IKKbeta), Mapk8 (JNK1), Nfkb1, Pik3r1 (p85alpha), Irf4, Retnla (Resistin like alpha, Fizz1), Cd68 |
Acaca (Acc1) - Acaca (Acc1) - Acetyl-Coenzyme A carboxylase alpha; Actb or β-actin - Actin, beta; Adipoq, Adiponectin; Adipor2 - Adiponectin receptor 2; Adrb1 - Adrenergic receptor, beta 1; Adrb2 - Adrenergic receptor, beta 2; Akt2 - Thymoma viral proto-oncogene 2; B2m - Beta-2 microglobulin; Bmp7 - Bone morphogenetic protein 7; Ccl2 - Chemokine (C-C motif) ligand 2; Cd68 - CD68 antigen; Cdkn1a - Cyclin-dependent kinase inhibitor 1A (P21); Cdnk1b - Cyclin-dependent kinase inhibitor 1B; Cebpa or C/EBPα - CCAAT/enhancer binding protein (C/EBP), alpha; Cebpb or C/EBPβ - CCAAT/enhancer binding protein (C/EBP), beta; Cebpd - CCAAT/enhancer binding protein (C/EBP), delta; Cfd - Complement factor D (adipsin); Cidea - Cell death-inducing DNA fragmentation factor, alpha subunit-like effector A; Cpt1b - Carnitine palmitoyltransferase 1b, muscle; Creb1 - CAMP responsive element binding protein 1; Cxcl10 - Chemokine (C-X-C motif) ligand 10; Ddit3 - DNA-damage inducible transcript 3; Dio2 - Deiodinase, iodothyronine, type II; Dlk1 (Pref-1) - Delta-like 1 homolog (Drosophila); Elovl3 - Elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 3; Fabp4 (aP2) - Fatty acid binding protein 4, adipocyte; Fasn - Fatty acid synthase; Fgf10 - Fibroblast growth factor 10; Fgf2 - Fibroblast growth factor 2; Foxc2 - Forkhead box C2; Foxo1 - Forkhead box O1; Gapdh - Glyceraldehyde-3-phosphate dehydrogenase; Gata2 - GATA binding protein 2; Gpd1 - Glycerol-3-phosphate dehydrogenase 1 (soluble); Ifng - Interferon gamma; Ikbkb(IKKbeta) - Inhibitor of kappaB kinase beta; Il10 - Interleukin 10; Il12b - Interleukin 12b; Il13 - Interleukin 13; Il1b - Interleukin 1 beta; Il4 - Interleukin 4; Il6 - Interleukin 6; Insr - Insulin receptor; Irf4 - Interferon regulatory factor 4; Irs1 - Insulin receptor substrate 1; Irs2 - Insulin receptor substrate 2; Jun - Jun oncogene; Klf15 - Kruppel-like factor 15; Klf2 - Kruppel-like factor 2 (lung); Klf4 - Kruppel-like factor 4 (gut); Lep – Leptin; Lepr - Leptin receptor; Lipe (HSL) - Lipase, hormone sensitive; Lmna - Lamin A; Lpin1 - Lipin 1; Lpl - Lipoprotein lipase; Mapk14 - Mitogen-activated protein kinase 14; Mapk8(Jnk1) - Mitogen-activated protein kinase 8; Ncoa2 - Nuclear receptor coactivator 2; Ncor2 - Nuclear receptor co-repressor 2; Nfkb1 - Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1, p105; Nr1h3 - Nuclear receptor subfamily 1, group H, member 3; Nrf1 - Nuclear respiratory factor 1; Pck1 -Phosphoenolpyruvate carboxykinase 1, cytosolic; Pik3r1 - Phosphatidylinositol 3-kinase, regulatory subunit, polypeptide 1 (p85 alpha); Plin1 - Perilipin 1; Ppara - Peroxisome proliferator activated receptor alpha; Ppard - Peroxisome proliferator activator receptor delta; Pparg or PPARγ - Peroxisome proliferator activated receptor gamma; Ppargc1a - Peroxisome proliferative activated receptor, gamma, coactivator 1 alpha; Ppargc1b - Peroxisome proliferative activated receptor, gamma, coactivator 1 beta; Prdm16 - PR domain containing 16; Ptpn1 - Protein tyrosine phosphatase, non-receptor type 1; Rb1 - Retinoblastoma 1; Retn – Resistin; Retnla - Resistin like alpha; RTC - Reverse Transcription Control; Scd1 - Stearoyl-Coenzyme A desaturase 1; Scr - Rous sarcoma oncogene; Sfrp1 - Secreted frizzled-related protein 1; Shh - Sonic hedgehog; Sirt1 - Sirtuin 1 (silent mating type information regulation 2, homolog) 1 (S. cerevisiae); Sirt3 - Sirtuin 3 (silent mating type information regulation 2, homolog) 3 (S. cerevisiae); Slc2a4 (Glut4) - Solute carrier family 2 (facilitated glucose transporter), member 4; Srebf1 - Sterol regulatory element binding transcription factor 1; Tbx1 - T-box 1; Tfam - Transcription factor A, mitochondrial; Tgfb1 - Transforming growth factor, beta 1; Tnf - Tumor necrosis factor; Ucp1 - Uncoupling protein 1 (mitochondrial, proton carrier); Wnt1 - Wingless-related MMTV integration site 1; Wnt10b - Wingless related MMTV integration site 10b; Wnt3a - Wingless-related MMTV integration site 3A; Wnt5a - Wingless-related MMTV integration site 5A;.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



