Submitted:
21 May 2024
Posted:
23 May 2024
You are already at the latest version
Abstract
Resistance to the existing drugs and increasing numbers of diseases result in identifying new drug candidates with new forms of activity. Marine sources of alkylglycerols such as the liver oil Shark (SLO) mixture of certain species or rat fish (elasmobranch fishes) contain high levels of these compounds as a mixture of few species varying by length and unsaturation or saturation of the alkyl chain. They have multiple biological activities such as hematopoiesis stimulation, lowering radiotherapy-induced injuries, reducing tumor growth and improving vaccination efficiency. The synthesis of two Alkylglycerols (AKGs) containing at least one alkyne moiety in the alkyl chain has been reported. In this work, we describe the synthesis of others unknown AKGs based alkyne 1-O-(hexadec-11’-ynyl)-sn-glycerol 4 and 1-O-(octadec-9’-ynyl)-sn-glycerol 5 as analogues of bioactive ether lipids found in the SLO mixture.
Keywords:
acetylenic
; alkylglycerol
; lipid0
; solketal
; shark liver oil
1. Introduction
Acetylenic natural or synthetic products include all compounds with a carbon-carbon triple bond or alkynyl functional group. They are widely distributed and occurring in plants, moss and lichens, fungi, marine algae, sponges, insects, frogs, and traces quantities in humans [1]. The earliest isolated alkyne-bearing natural product was dehydromatricaria ester, which was isolated but not fully characterized. No compound was characterized as being acetylenic until 1892 (tariric acid), after which only a handful of compounds were isolated before 1952 [2]. It is only within the 30 years that biologically active polyacetylenes having unusual structural features have been reported from plants, cyanobacteria, algae, invertebrates, and other sources. Naturally occurring aquatic acetylenes are of particular interest since many of them display important biological activities and possess antitumor, antibacterial, antimicrobial, antifouling, antifungical, pesticidal, phototoxic, HIV-inhibitory and immunosuppressive properties [3]. Acetylenic enol ethers of glycerol including bioactive of compounds 1-3, have been isolated from sponge of the genus Petrosia. These compounds have exhibed weak cytotoxicity against the human leukemia cell-line K-562 (LC50 9.2, 57, 29 µg / mL) [4] (Figure 1).
Despite all these beneficials effects of alkyne’s compounds, most of the papers are restricted to the synthesis of 1-O-alkylglycerols (AKGs) with a straight-chain alkane or alkene [5,6,7,8,9,10] with just one article reporting the synthesis of 1-O-alkylglycerols containing at least one alkyne moiety in the alkyl chain [11].
Natural 1-O-alkylglycerols (AKGs) are bioactive ether lipids present in body cells and fluids. They are precursors of ether phospholipids participating in structures and functions of membranes in certain cells such as white blood cells or macrophages. AKGs are also found in bone marrow lipids and in milk [12]. Marine sources of AKGs such as the liver oil of certain shark species or rat fish (elasmobranch fishes) contain high levels of these compounds as a mixture of few species varying by length and unsaturation or saturation of the alkyl chain
They have multiple biological activities such as hematopoiesis stimulation [13], lowering radiotherapy-induced injuries [14], reducing tumor growth [15] and improving vaccination efficiency [16,17]. Currently, resistance to the existing drugs and increasing numbers of diseases result in identifying new drug candidates with new forms of activity. Thus, synthesized non-natural AKGs derivatives could be one new source of drug delivery systems.
Therefore, it was hypothesized that AKGs with one alkyne moiety either at the 9 and 11 position respectively in the alkyl chain could exhibit than their analogues with a straight-chain alkane or alkene more biological activities as it was reported for 8-HETE (hydroxy-(5Z,8Z,11Z,13E)-eicosatetraenoic acids) analogues [18]. In the light of positive attributes of AKGs, acetylenics lipids, herein we describe the synthesis of others acetylenic Alkylglycerols namely 1-O-(hexadec-11’-ynyl)-sn-glycerol 4 from 1,10-decanediol and 1-O-(octadec-9’-ynyl)-sn-glycerol 5 from stearolic acid as analogues of known bioactive glycerol ether lipids 10 and 11 respectively found in the natural oil liver shark mixture (Figure 2).
2. Results and Discussion
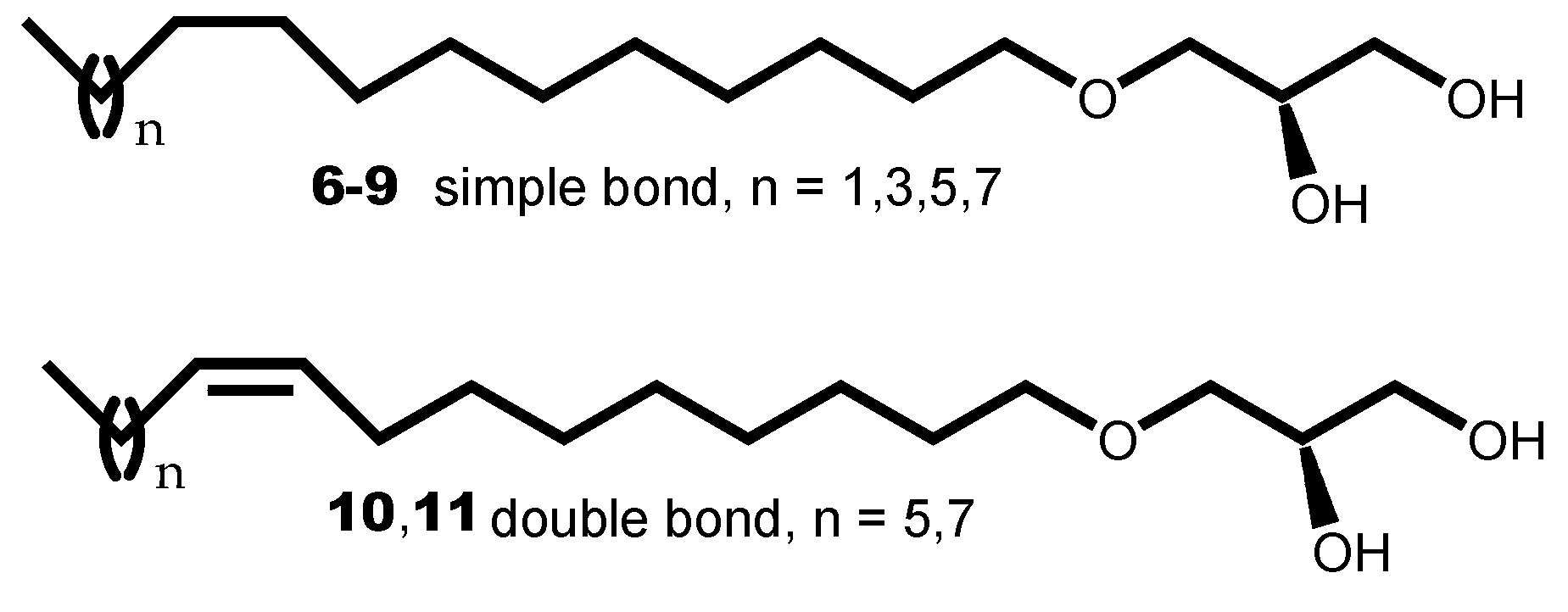
Alkylglycerols 6-11 are the six prominent constituents found in the SLO mixture from Greenland shark (Centrophorus squamosus). The percentage of AKGs in the SLO mixture was determined as follows: 12:0, 1–2% (6); 14:0, 1–3% (7); 16:0, 9–13% (8); 16:1 n-7, 11–13% (9); 18:0, 1–5% (10); 18:1 n-9, 54–68% (11); 18:1 n-7, 4–6%; and minor species (<1%). Beneficial effects of the SLO mixture on health were recognized in traditional medicine of northern countries involved in fishing such as Japan, Norway and Iceland. In these countries, the ancestral use of the SLO mixture was empirically as strengthening or wound healing medication [19]. To assess the biological activity of each prominent AKG from the SLO mixture, derivatives 6-11 were individually obtained in pure form by total synthesis and it was observed that the biological activity was heavily dependent upon the unsaturation of the alkyl chain. When this chain was saturated, the corresponding 1-O-alkylglycerols 6-9 exhibited little or no activity. However, when it was monounsaturated 10,11 a good antitumor activity was observed, thus indicating that the antitumor activity of the SLO mixture was heavily related to its unsaturated components [5]. It was also established that the alkyl chain of a 1-O-alkylglycerol was bound to the glycerol backbone at the sn-1 position; thus leading to an S configuration at the asymmetric carbon [20].
Figure 3.
1-O-alkylglycerols 6-11 from the SLO mixture.
To evaluate the effect position of the triple bound on the biological activity, AKG 4-(16:2) was designed as an analogue of AKG 10-(16:1) with the same alkyl chain length C16 , but with a Δ 11 unsaturation shifted from 3 carbons relative to the usual position Δ 9 unsaturation as found in the SLO. Compound 10 was reported to display antitumor activity [5]. The synthesis of 4 started with the conversion of 1,10-decanediol 12 into 1-bromo-decan-10-ol 13 in 80 % yield by using a two-phase system consisting of 48 % aqueous HBr solution and toluene to assure monobromination [21]. The remaining hydroxyl group was subsequently protected as an acetal 14 (Scheme 1).
Intermediate 14 was employed in the alkyne alkylation using 1-hexyne 15 in THF and n-Buli as a nucleophile resulted in the formation of 16 in 60 % yield. Acetal cleavage under acidic condition using p-toluenesulfonic acid in methanol afforded alcohol 17 in 89 yield. Conversion of the alcohol 17 into mesylate 18 in 94 yield was done through mesyl chloride in DCM with trietylamine. 18 was then alkylated with 2,3-isopropylidene-sn-glycerol 19 in the presence of NaH in NMP afforded 20 in 81% yield. Acetonide 20 cleavage under acid conditions using catalytic amount of p-toluenesulfonic acid monohydrate in MeOH / H2O afforded the targeted 4 in 87% yield (Scheme 2).
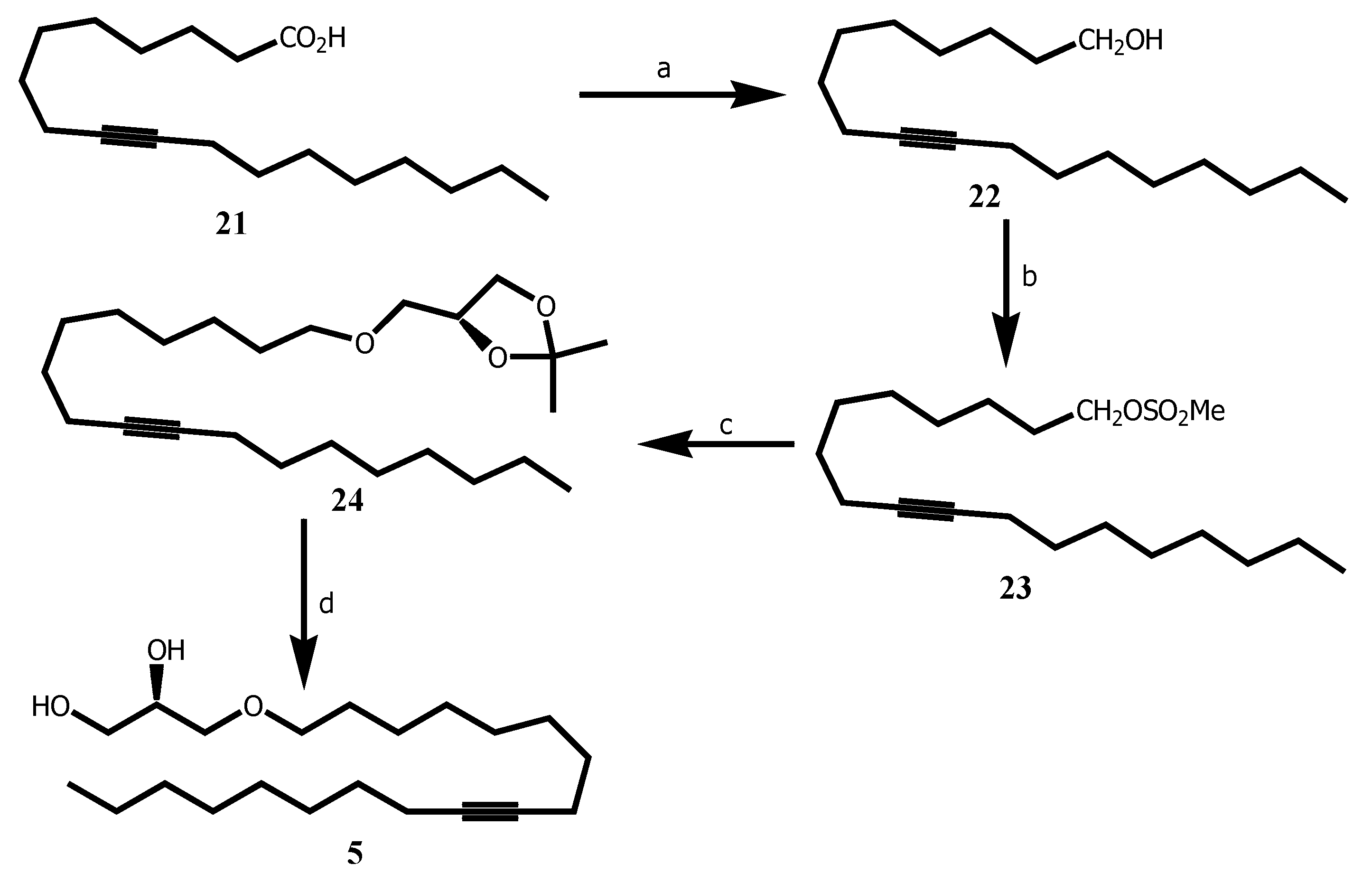
Furthermore, the AKG 5-(18:2) was designed as an analogue of the AKG 11-(18:1) with the same chain length C18, a Δ 9 unsaturation and also as a derivative of stearolic acid 45 which was reported as a novel DNA binding agent [22]. AKG 11 was reported to display antitumor activity [5].
9-octadecynoic acid 21 was reduced into stearolic alcohol 22 in 91 % yield when using Red-Al in THF. 22 was then mesylated using mesyl chloride in DCM with trietylamine afforded 23 in 93 % yield. Alkylation of 23 with 2,3-isopropylidene-sn-glycerol 19 in the presence of potassium hydroxide in DMSO and tetra-n-butylammonium bromide gave 24 in 94 % yield, which under acid conditions using catalytic amount of p-toluenesulfonic acid monohydrate in MeOH/H2O afforded the targeted 5 in 85% yield (Scheme 3).
3. Conclusions
A new series of Alkylglycerols based-acetylenic derivatives analogues of known glycerol ether lipids has been synthesized. Namely 1-O-(hexadec-11’-ynyl)-sn-glycerol 4 from 1,10-decanediol and 1-O-(octadec-9’-ynyl)-sn-glycerol 5 from stearolic acid as analogues of known bioactive glycerol ether lipids 10 and 11 respectively found in the natural oil liver shark mixture. The structures of these compounds were characterized by 1H NMR, 13C NMR and mass spectral studies. We have reported an efficient and flexible strategy for the preparation of these targeted molecules, which will stimulate scientists to further investigate on their biological activities in due course.
4. Experimental
4.1. General Information
Moisture sensitive reactions were performed under nitrogen. Anhydrous THF and diethyl ether were obtained by percolating through a column of a drying resin or by distilling over Na / benzophenone. Anhydrous DMF over molecular sieves was used as commercially supplied (Acros). Room temperature (rt) means a temperature generally in the interval 15-20°C. Basic alumina used for column chromatographies was purchased from Fluka. TLC plates were visualized by UV inspection followed by staining with an acidic ethanolic solution of p-anisaldehyde or with a solution of phosphomolybdic acid (5 g in 100 mL 95% ethanol). IR spectra were measured as films between KBr plates for liquids or as KBr disks for solids on a Thermo Nicolet Avatar 250 FTIR spectrometer. 1H NMR spectra (400.13 and 300.13 MHz) and 13C NMR spectra (100.61 and 75.47 MHz) were recorded on Avance 400 and 300 Bruker spectrometers using TMS as an internal standard. Optical rotations were measured using a Perkin Elmer 341 polarimeter (concentration in g/100 mL). High resolution mass spectra were recorded using a MicrO-Tof-Q II spectrometer under electrospray using methanol as solvent. Microanalyses were performed with a CHNS analyzer. 2,3-Isopropylidene-sn-glycerol 19 (≥ 95% pure) was purchased from Alfa Aesar.
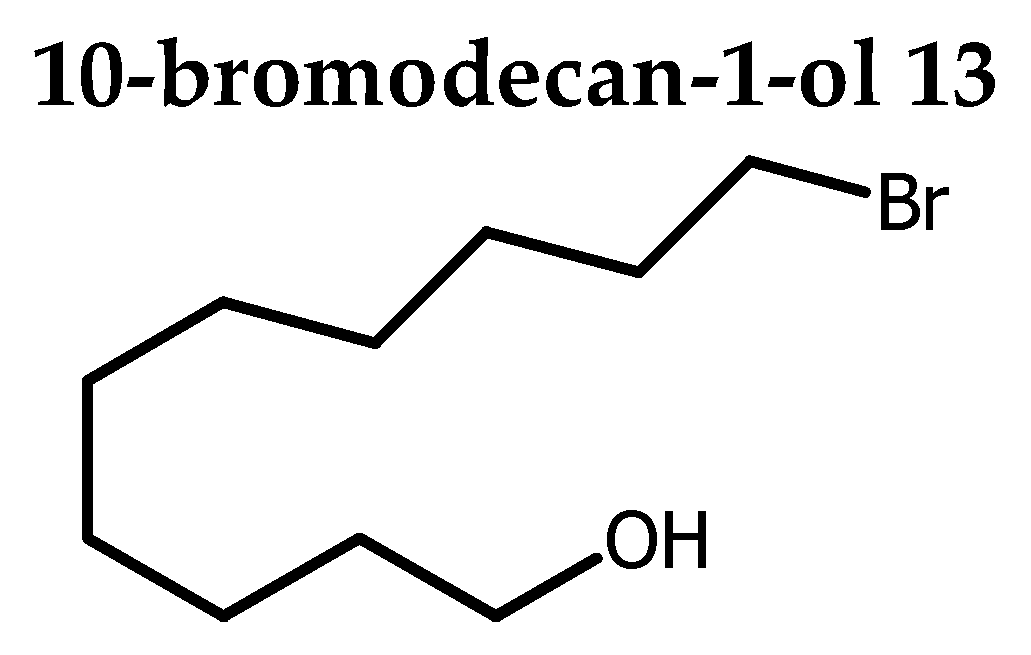
To a solution of 1,10-decandiol 12 (5 g, 28.69 mmol) in toluene (100 mL) under stirring, was added 48% aqueous solution of HBr (4.5 mL, 40.5 mmol, 1.41 eq). The corresponding mixture was refluxed overnight (18 h) and TLC monitoring showed completion of the reaction and cooled at rt. The crude was purified by Kugelrohr distillation (140 °C / 2 Torr) and afforded 13 as colorless oil (5.4 g, 80%). Rf 0.56 (petroleum ether / acetone 70:30).
1H NMR (400 MHz, CDCl3): δ= 3.64 (t, 2H, J = 6.6 Hz), 3.40 (t, 2H, J = 6.9 Hz), 1.85 (tt, 2H, J = 7.4, 6.9, Hz), 1.60 (tt, 2H, J = 7.3, 6.7 Hz), 1.37-1.19 (m, 12H)
13C NMR (100 MHz, CDCl3): δ= 63.15 (CH2), 34.06 (CH2Br), 32.82 (CH2), 32.76 (CH2), 29.47 (CH2), 29.37 (CH2), 29.35 (CH2), 28.74 (CH2), 28.15 (CH2), 25.71 (CH2)
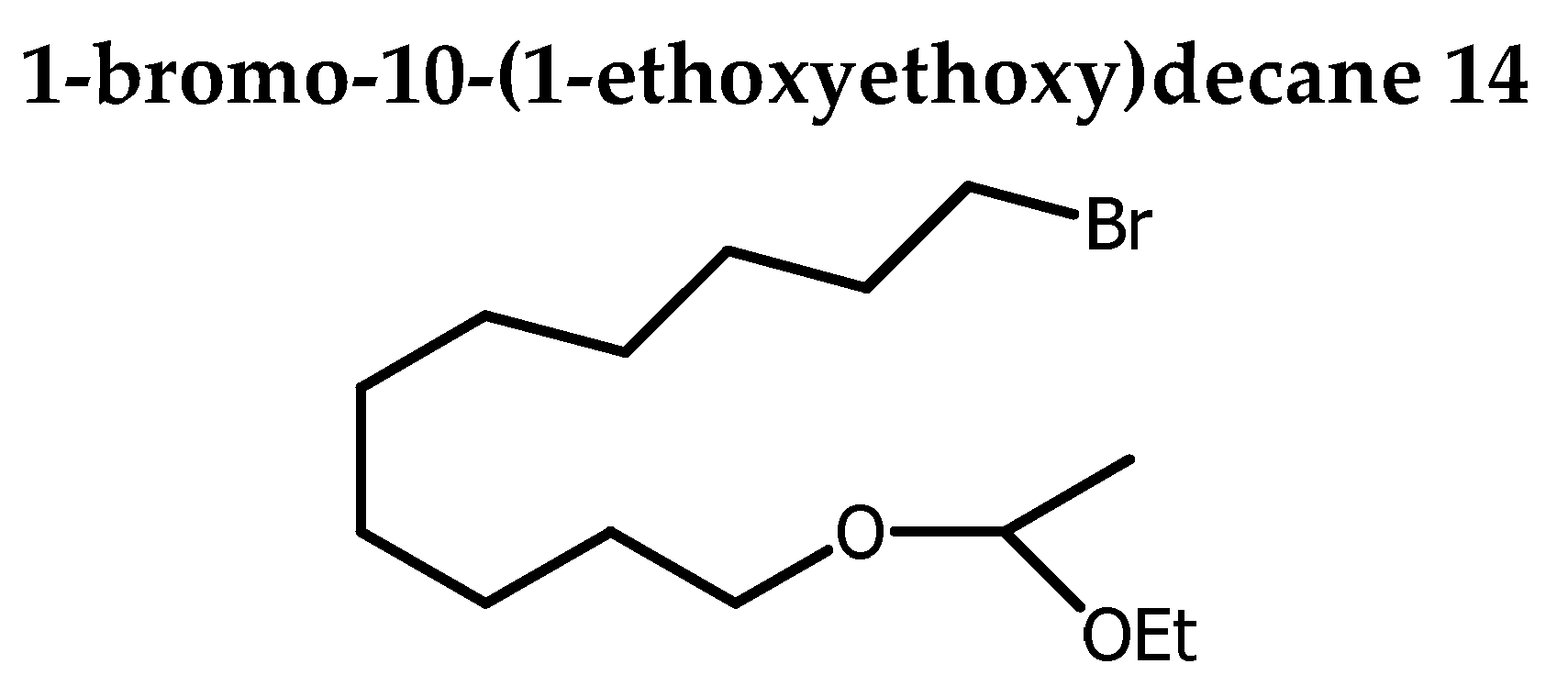
In a flame-dried two necked flask containing a solution of 13 (592 mg, 2.5 mmol) in DCM (5 mL) under stirring and N2 cooled at 0 oC, was added a solution of ethyl vinyl ether (290 µL, 3.0 mmol, 1.2 eq) and p-toluenesulfonic acid (31.2 mg, 0.125 mmol, 0.5 eq). The stirring was maintained 30 min at the same temperature. A saturated solution of NaHCO3 was added to the mixture and the stirring was continued for 10 mn. Extraction was done with DCM (3 x). Organic layer was dried over Na2SO4, concentrated and purified by column chromatography on silica gel (0→1 % acetone in petroleum ether) afforded 14 as a colorless oil (672.1 mg, 87%). Rf 0.59 (petroleum ether / acetone 70:30).
1H NMR (400 MHz, CDCl3): δ= 4.68 (q, 1H, J = 5.3 Hz, CH-CH3), 3.65 (dq, 1H, J = 9.4, 7.1 Hz, OCH2CH3), 3.56 (dt, 1H, J = 9.4, 6.7 Hz, CH2O), 3.48 (dq, 1H, J = 9.4, 7.1 Hz, OCH2CH3), 3.41 (dt, 1H, J = 9.4, 6.6 Hz, CH2O), 3.40 (t, 2H, J = 6.9 Hz, CH2Br), 1.85 (tdd, 2H, J = 7.7, 6.7, 6.6 Hz, CH2CH2Br), 1.56 (tt, 2H, J = 7.0, 6.9 Hz, CH2CH2O), 1.47-1,38 (m, 2H, CH2), 1,38-1,24 (m, 10H, 5 CH2), 1.30 (d, 3H, J = 5.3 Hz, CH-CH3), 1.21 (t, 3H, J = 7.1 Hz, CH2CH3).
13C NMR (100 MHz, CDCl3): δ= 99.52 (CH-CH3), 65.27 (CH2CH2O), 60,65 (OCH2CH3), 34.02 (CH2Br), 32.83 (CH2), 29.89 (CH2), 29.47 (CH2), 29.42 (CH2), 29.38 (CH2), 28.75 (CH2), 28.16 (CH2), 26.24 (CH2), 19.89 (CH-CH3), 15.34 (OCH2CH3).
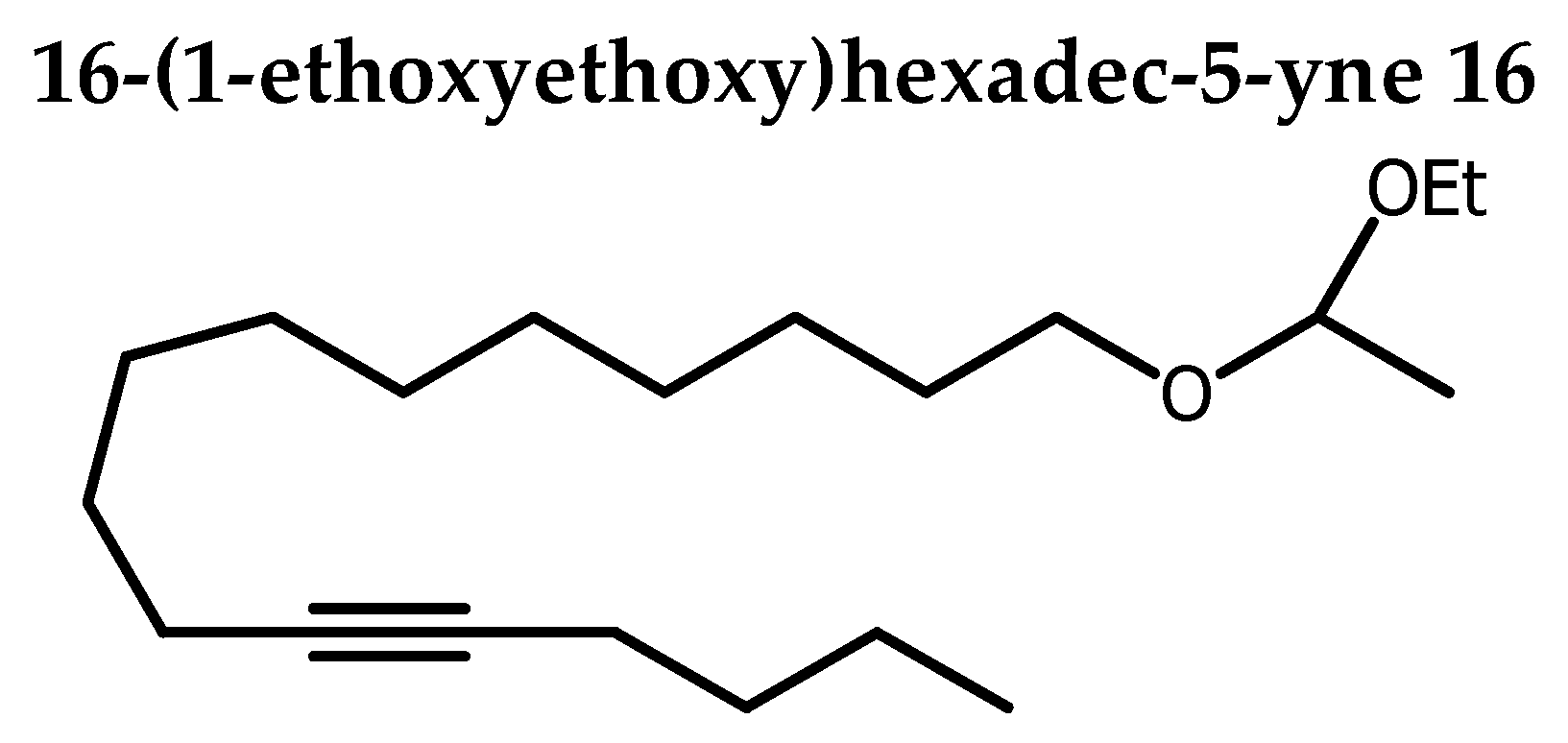
In a flamed-dried flash containing a solution of 1-hexyne 15 (350 µL, 3.0 mmol) in THF (2.5 mL) and HMPA (1 mL) cooled at -80°C , was added a solution of n-Buli (3.26 mL) under N2 and the stirring was continued for 1 h at -78°C. Thereafter, a solution of 14 (427 mg, 1.5 mmol ) in THF (1 mL) was added and the transfer was completed by THF (2 x 0.2 mL). The corresponding mixture was allowed to stir overnight (16 h) at rt. TLC monitoring showed completion of the reaction. Extraction was done with ethyl acetate and organic layer washed with distilled water and dried over Na2SO4. Solvent was removed under reduced pressure to obtain the crude product as yellow light oil. The crude product was purified by silica-gel column chromatography with petroleum ether / Et3N (99/1) and gave 16 as colorless oil (600 mg, 60%). Rf 0.57 (petroleum ether / acetone 90:10).
IR (film) νmax 2930, 2855, 2213, 1466, 1456, 1379, 1338, 1134, 1100, 1086, 1061, 951 cm-1
1H NMR (400 MHz, CDCl3): δ=4.68 (q, 1H, J = 5.3 Hz, CH-CH3), 3.65 (dq, 1H, J = 9.4, 7.1 Hz, OCH2CH3), 3.56 (dt, 1H, J = 9.3, 6.7 Hz, CH2O), 3.48 (dq, 1H, J = 9.4, 7.1 Hz, OCH2CH3), 3.41 (dt, 1H, J = 9.3, 6.7 Hz, CH2O), 2.18-2.10 (m, 4H, CH2CδCCH2), 1,56 (ddt, 2H, J = 8.3, 6.3, 6.7 Hz, CH2CH2O), 1.51-1.24 (m, 18H, 9 CH2), 1.31 (d, 3H, J = 5.3 Hz, CH-CH3), 1.21 (t, 3H, J = 7.1 Hz, OCH2CH3), 0.90 (t, 3H, J = 7.2 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ=99.50 (CH-CH3), 80.21-80.17 (CδC), 65.27 (CH2CH2O), 60.63 (OCH2CH3), 31.27 (CH2), 29.90 (CH2), 29.56 (CH2), 29.50 (CH2), 29.48 (CH2), 29.17 (CH2), 29.16 (CH2), 28.86 (CH2), 26.26 (CH2), 21.94 (CH2, C15), 19.88 (CH-CH3), 18.76 (CH2CδC), 18.45 (CH2CδC), 15.33 (OCH2CH3), 13.65 (CH3).
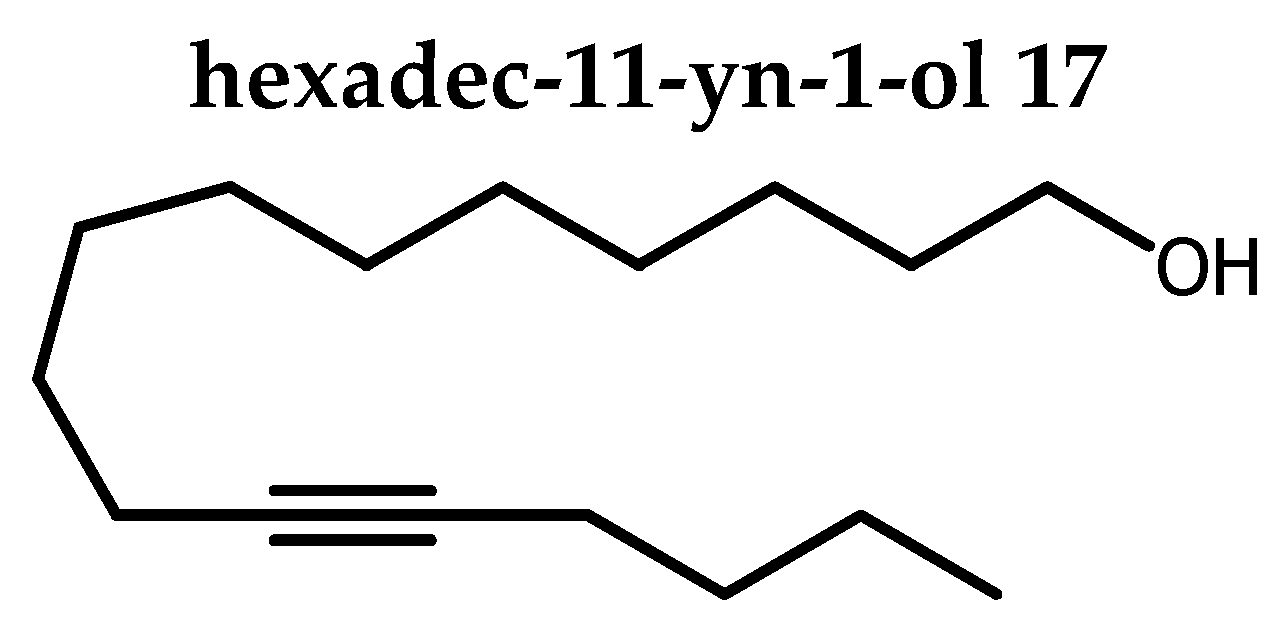
To a solution of 16 (219.1 mg, 0.7 mmol,) in methanol (3.6 mL), was added camphosulfonic acid (9.4 mg, 0.04 mmol, 0.05 eq). The flask was then purged under nitrogen, stoppered and. the stirring was maintained 18 h at rt. TLC monitoring confirmed completion of the reaction and triethylamine (6 drops) was added to neutralise the acid. Methanol was evaporated under reduced pressure and the crude was purified by column chromatography on silica gel (0→2 % acetone in petroleum ether) and afforded 17 as colorless oil (150.5 mg, 89 % yield). Rf 0.16 (petroleum ether / acetone 90:10).
1H NMR (400 MHz, CDCl3): δ=3.64 (t, 2H, J = 6.7 Hz, CH2OH), 2.19-2.10 (m, 4H, CH2CδCCH2), 1.56 (tt, 2H, J = 7.3, 6.7 Hz, CH2CH2OH), 1.52-1.23 (m, 19H, 9 CH2 et OH), 0.90 (t, 3H, J = 7.2 Hz, CH3)
13C NMR (100 MHz, CDCl3): δ=80.21-80.20 (CδC), 63.07 (CH2OH), 32.80 (CH2), 31.27 (CH2), 29.56 (CH2), 29.48 (CH2), 29.43 (CH2), 29.17 (CH2), 29.15 (CH2), 28.85 (CH2), 25.74 (CH2), 21.94 (CH2), 18.76 (CH2CδC), 18.45 (CH2CδC) 13.65 (CH3).
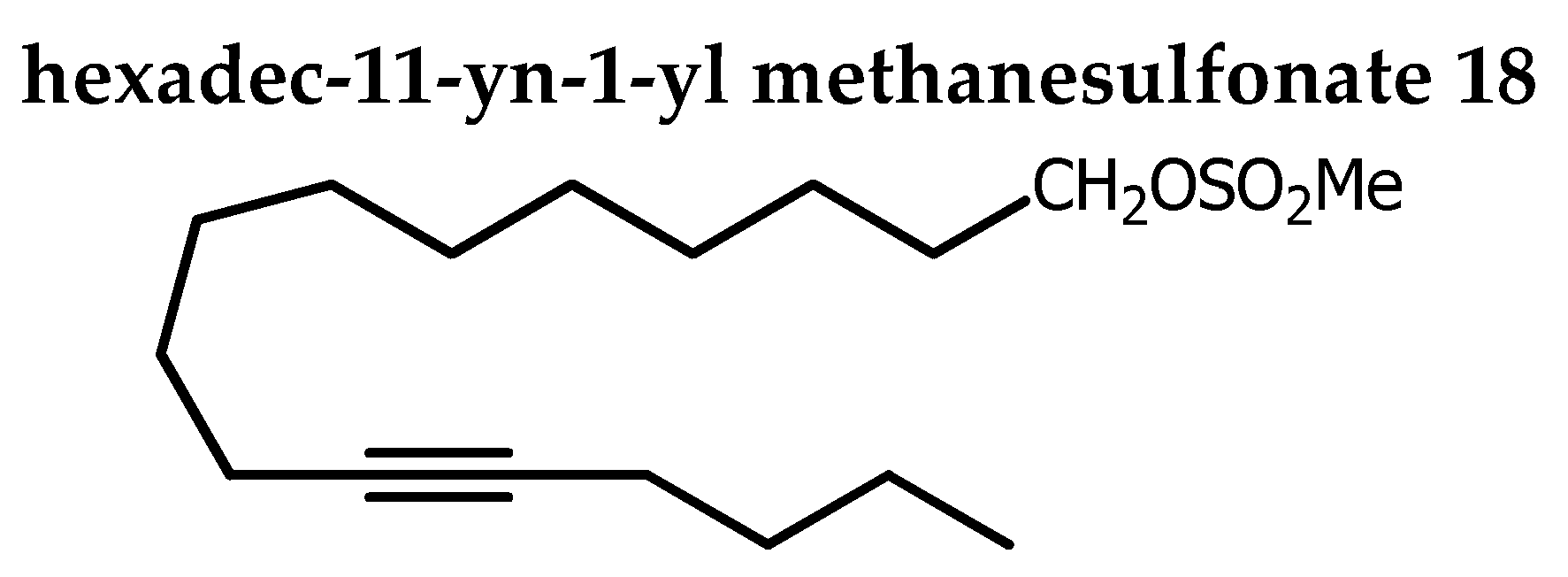
To the stirred solution at -50oC of 17 (121.1 mg, 0.51 mmol), Et3N (0.21 mL, 1.5 mmol, 2.95 eq) in DCM (2.5 mL) under N2, mesyl chloride (94 µL, 1.2 mmol, 1.25 eq) in DCM (9 mL) was added drop wise and the reaction mixture was stirred for an additional 5 h at the same temperature. Distilled water (25 mL) was added to quench the reaction and extraction was done with DCM (3 x). Organic phase was washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure to obtain the crude product as yellow light oil. The crude product was purified by silica-gel column chromatography (0→1 % acetone in petroleum ether) and gave 18 as white solid (114.1 mg, 94%). Rf 0.48 (petroleum ether / acetone 85:15).
1H NMR (400 MHz, CDCl3): δ= 4.22 (t, 2H, J = 6.6 Hz, CH2OMs), 3.01 (s, 3H, OSO2CH3) 2.19-2.10 (m, 4H, CH2CδCCH2), 1.75 (ddt, 2H, J = 8.1, 6.8, 6.6 Hz, CH2CH2OMs), 1.52-1.23 (m, 18H, 9 CH2)
13C NMR (100 MHz, CDCl3): δ=80.20-80.17 (CδC), 70.20 (CH2OMs), 37.35 (OSO2CH3), 31.26 (CH2), 29.40 (CH2), 29.37 (CH2), 29.14 (CH2), 29.12 (CH2),
29.10 (CH2), 29.02 (CH2), 28.81 (CH2), 25.42 (CH2), 21.93 (CH2), 18.74 (CH2CδC), 18.44 (CH2CδC), 13.65 (CH3).
Melting point: 28-30°C.
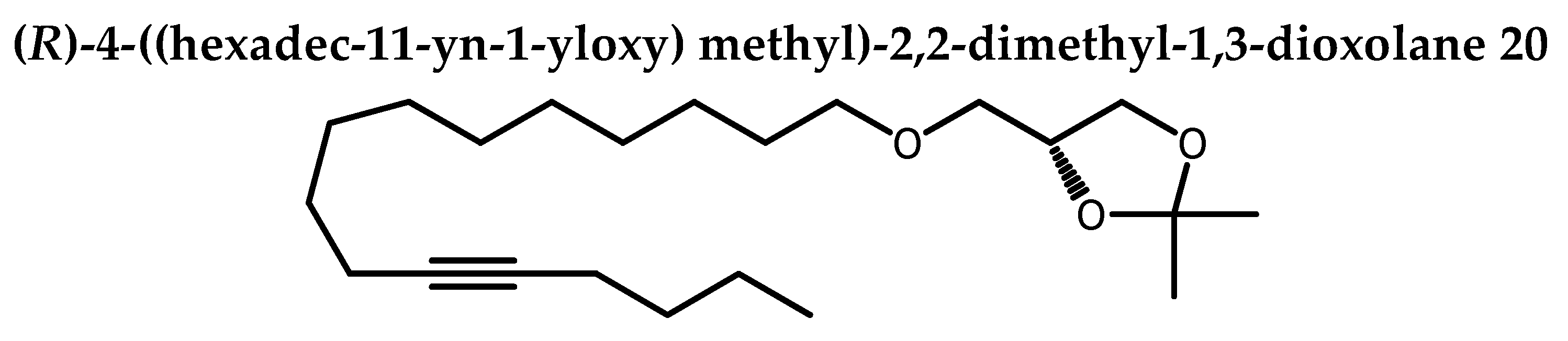
A 60% dispersion of sodium hydride in mineral oil (44 mg, 0.85 mmol, 2.5 eq) was washed three times with petroleum ether under argon. Anhydrous NMP (100 µL) was added and the mixture was cooled at 0°C. A solution of 2,3- isopropylidene-sn-glycerol 19 (56 µL, 0.46 mmol, 1.0 eq) in NMP (50 µL)was added dropwise followed by NMP (2 x 0.2 mL) to complete the transfer of 19. After stirring 10 min at 0°C, a solution of 18 (133.6 mg, 0.42 mmol) in NMP (0.4 mL) was added to the resulting white suspension. Transfer of 18 was completed by rinsing with NMP (2 x 0.2 mL). This mixture was left under good stirring overnight (18 h) at rt. 10 % solution of ammonium acetate (4 mL) and petroleum ether (5 mL) were added to the mixture and the stirring continued for 10 min. Extraction was done with petroleum ether (3 x). Organic phase was washed with brine and dried over Na2SO4. Solvent was removed under reduced and the crude product was purified by silica-gel column chromatography (0→1 % acetone in petroleum ether) afforded 20 as colorless oil (108.1 mg, 81%). Rf 0.56 (petroleum ether / acetone 80:20).
IR (film) νmax 2986, 2928, 2856, 1464, 1456, 1379, 1369, 1255, 1214, 1120, 1056, 847, 514 cm-1.
1H NMR (400 MHz, CDCl3): δ=4.26 (dddd, 1H, J = 6.4, 6.4, 5.7, 5.6 Hz, CHOCMe2), 4.06 (dd, 1H, J = 8.2, 6.4 Hz, CH2OCMe2), 3.72 (dd, 1H, J = 8.2, 6.4 Hz, CH2OCMe2) 3.52 (dd, 1H, J = 9.9, 5.7 Hz, CH2O((CH2)10), 3.50-3.42 (m, 2H, OCH2(CH2)9), 3.42 (dd, 1H, J = 9.9, 5.6 Hz, CH2O(CH2)10), 2.18-2.10 (m, 4H, CH2CδCCH2), 1.57 (tt, 2H, J = 7.3, 6.7 Hz, OCH2CH2), 1.52-1.23 (m, 18H, 9 CH2), 1.42 (s, 3H, C-CH3), 1.36 (s, 3H, C-CH3), 0.90 (t, 3H, J = 7.2 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ=109.36 (C(CH3)2), 80.21-80.18 (CδC), 74.77 (CH α de O), 71.89 (CH2 α de O), 71.83 (CH2 α de O), 66.95 (CH2 α de O), 31.28 (CH2), 29.57 (CH2), 9.54 (CH2), 29.49 (CH2), 29.46 (CH2), 29.18 (CH2), 29.15 (CH2), 28.86 (CH2), 26.78 (C-CH3), 26.06 (CH2), 25.43 (C-CH3), 21.94 (CH2), 18.76 (CH2CδC), 18.45 (CH2CδC), 13.64 (CH3).
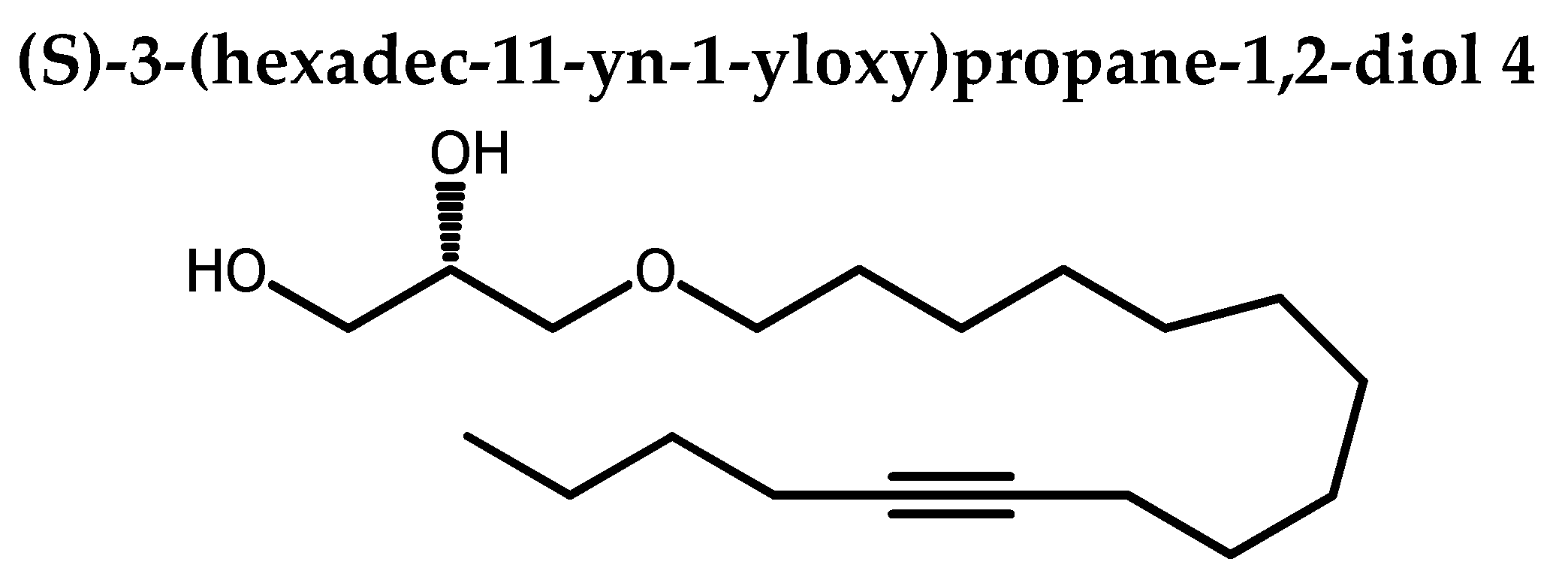
To a solution of 20 (60.4 mg, 0.17 mmol,) in THF (1.2 mL), was added p-toluenesulfonic acid monohydrate (4.8 mg, 0.03 mmol, 0.15 eq) and distilled water (0.51 mL). The flask was then purged under nitrogen, stoppered and dipped in a preheated bath at 80oC. The stirring was maintained 8 h at the same temperature. Sodium carbonate (5.8 mg, 0.07 mmol, 0.04 eq) was added and the stirring was continued for 1 h at 80oC. Extraction was done with EtOAC (3 x). Organic phase was washed with brine and dried over Na2SO4. Solvent was removed under reduced and the crude product was purified by silica-gel column chromatography (0→10 % EtOAC in petroleum ether) afforded 4 as white solid (46.3 mg, 87%). Rf 0.06 (petroleum ether / EtOAC 80:20).
1H NMR (400 MHz, CDCl3): δ=3.85 (ddt, 1H, J = 5.6, 5.5, 4.0 Hz, CHOCMe2), 3.71 (dd, 1H, J = 11.4, 3.8Hz, CH2OCMe2), 3.63 (dd, 1H, J = 11.4, 5.2 Hz, CH2OCMe2), 3.54 (dd, 1H, J = 9.9, 4.0 Hz ), 3.53-3.46 (m, 3H), 2.20-2.08 (m, 4H), 1.56 (ddt, 2H, J = 7.0, 6.8, 6.7 Hz), 1.52-1.18 (m, 21H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ=80.21-80.19 (CδC), 72.52 (CH2), 71.85 (CH2), 70.41 (CH ), 64.30 (CH2), 31.27 (CH2), 29.58 (CH2), 29.52 (CH2), 29.48 (CH2), 29.44 (CH2), 29.17 (CH2), 29.14 (CH2), 28.85 (CH2), 26.08 (CH2), 21.93 (CH2), 18.75 (CH2), 18.44 (CH2), 13.65 (CH3)
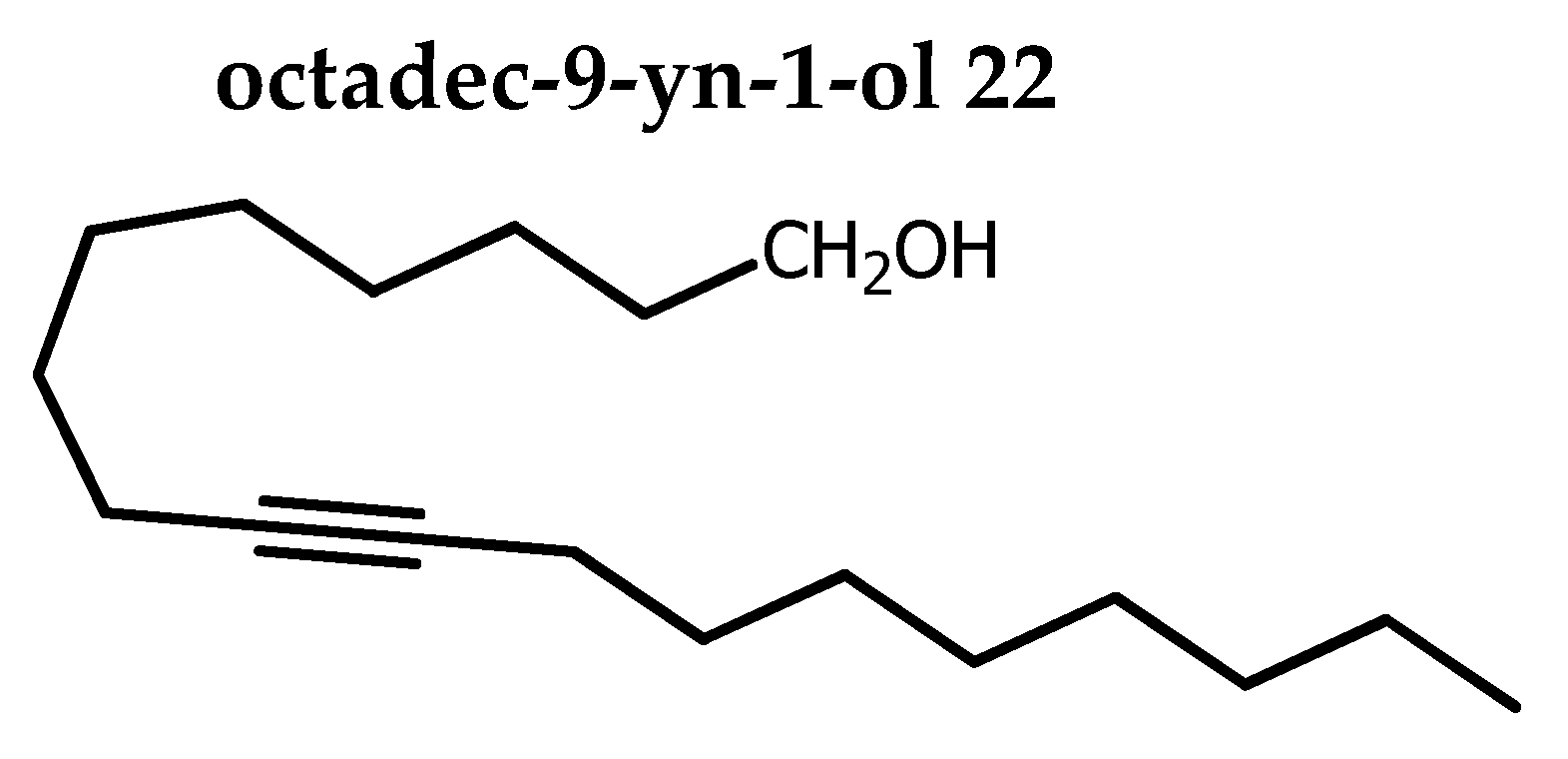
Red-Al (10.2 mL, 33.9 mmol, 1.75 eq) was added dropwise to a solution of 21 (5.43 g, 19.4 mmol) in THF (48 mL) cooled at 0oC under stirring and N2. The stirring was continued overnight at rt. TLC monitoring confirmed completion of the starting material. Citric acid (5.03 g) was added and distilled water (45 mL) and the stirring was continued for 30 mn. Extraction was done with petroleum ether / EtOAC (80:20). Organic layer was dried over Na2SO4, concentrated and purified by column chromatography on alumina gel (0→3 % acetone in petroleum ether) afforded 22 as a colorless oil (4. 69 g, 91 %). Rf 0.48 (petroleum ether / acetone 85:15).
1H NMR (400 MHz, CDCl3): δ=3.64 (t, 2H, J = 6.6 Hz), 2.17-2.10 (m, 4H, CH2CδCCH2), 1.56 (tt, 2H, J = 7.5, 6.8 Hz), 1.54-1.45 (m, 4H), 1.43-1.25 (m, 18H), 0.89 (pseudo t, 3H, J = 6.9 Hz)
13C NMR (100 MHz, CDCl3): δ=80.29 and 80.16 (CδC), 63.06 (CH2), 32.77 (CH2), 31.84 (CH2), 29.31 (CH2), 29.21 (CH2), 29.17 (CH2), 29.14(CH2), 29.13 (2CH2), 28.86 (CH2), 28.77(CH2), 25.69 (CH2), 22.66 (CH2), 18.74 (2CH2CδC), 14.09 (CH3).
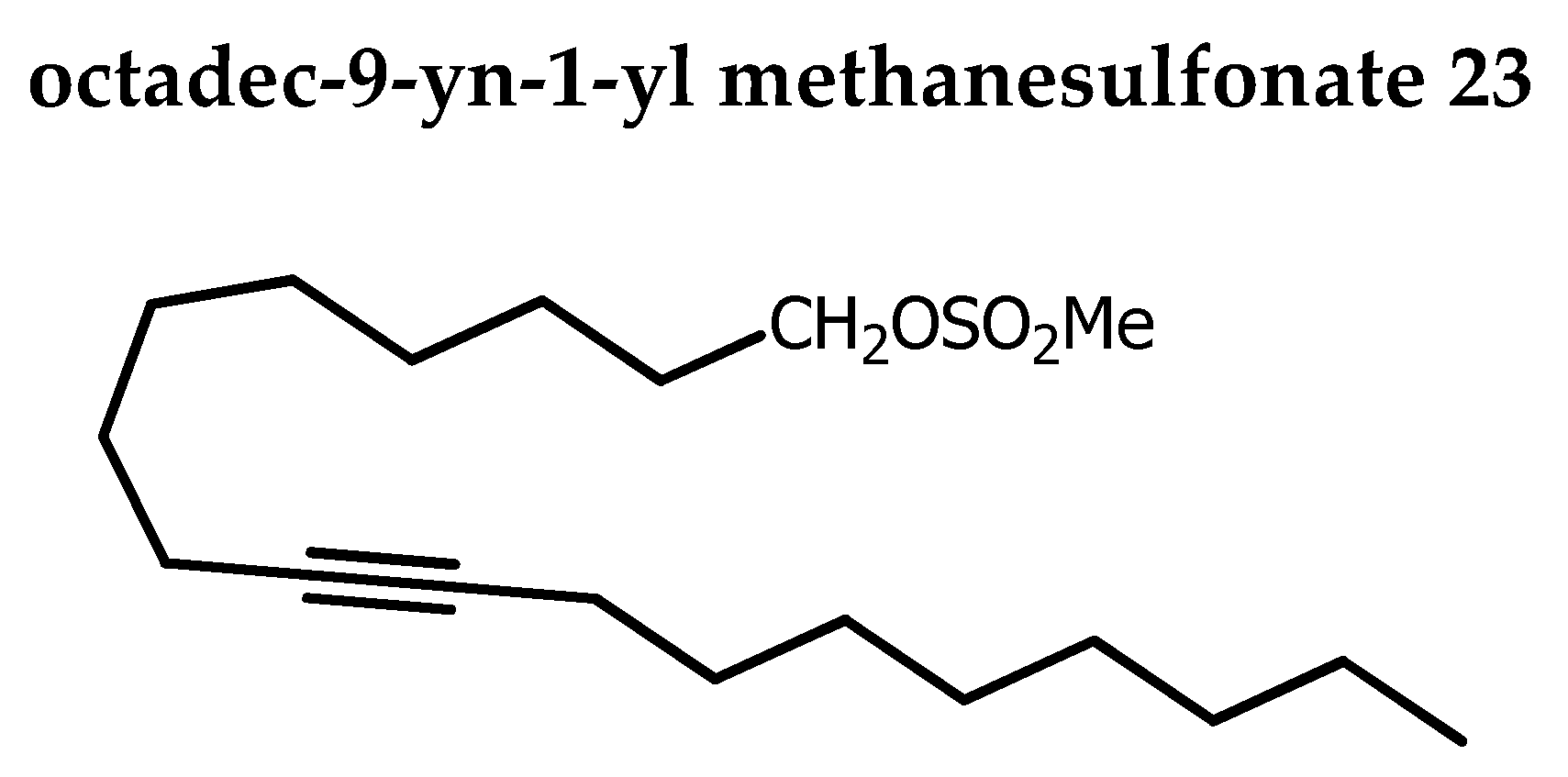
To the stirred solution of 22 (5.14 g, 14.92 mmol), Et3N (4.03 mL, 28.97 mmol, 1.5 eq) in DCM (58 mL) under N2 at -40oC, mesyl chloride (1.94 mL, 25.11 mmol, 1.3 eq) in DCM (8.36 mL) was added drop wise. After addition, the reaction mixture was stirred for an additional 2 h at the same temperature. Distilled water (80 mL) was added to quench the reaction and extraction was done with DCM. Organic phase was washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure and the crude product was purified by silica-gel column chromatography (0→1 % acetone in petroleum ether) gave 23 as colorless oil (6.2 g, 93 %). Rf 0.49 (petroleum ether / acetone 85:15).
1H NMR (400 MHz, CDCl3): δ=4.23 (t, 2H, J = 6.6 Hz, CH2OMs), 3.00 (s, 3H, OSO2CH3) 2.17-2.10 (m, 4H, CH2CδCCH2), 1.75 (tt, 2H, J = 13.2, 6.6 Hz, CH2CH2OMs), 1.52-1.43 (m, 4H), 1.41-1.22 (m, 18H), 0.89 (pseudo t, 3H, J = 6.9 Hz)
13C NMR (100 MHz, CDCl3): δ=80.37 and 80.07 (CδC), 70.14 (CH2OMs), 37.38 (OSO2CH3), 31.86 (CH2), 29.24 (CH2), 29.18 (CH2), 29.14 (CH2), 29.13 (CH2), 29.08 (CH2), 28.95 (2CH2), 28.89 (CH2), 28.70 (CH2), 25.40 (CH2), 22.68 (CH2), 18.76 (CH2CδC), 18.73 (CH2CδC), 14.12 (CH3).
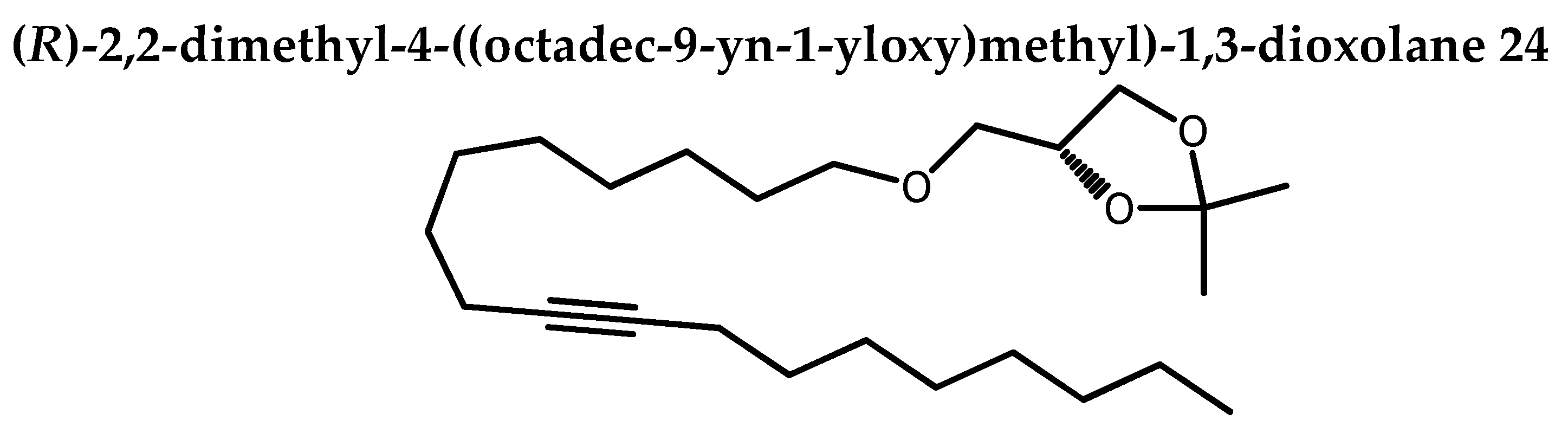
To a stirred solution of 23 (6.19 g, 17.95 mmol), n-Bu4NBr (2.32 g, 7.18 mmol, 0.5 eq) in DMSO (45 mL), and potassium hydroxide (4.74 g, 71.8 mmol, 4 eq), was added a solution of 2,3- isopropylidene-sn-glycerol 19 (2.7 g, 20.64 mmol, 1.15 eq). The corresponding mixture went under stirring overnight (15h) at 45oC. Distilled water was added and extraction was done with petroleum ether / EtOAC (80:20). Organic phase was washed again with brine, dried over Na2SO4. Solvent was removed under reduced pressure. Chromatography of the crude on silica-gel (0→0.2 % acetone in petroleum ether) gave 24 as white solid (6.4 g, 93%). Rf 0.39 (petroleum ether / acetone 80:20).
1H NMR (400 MHz, CDCl3): δ=4.26 (ddt, 1H, J = 6.4, 6.3, 5.7Hz, CHOCMe2), 4.06 (dd, 1H, J = 8.2, 6.4 Hz, CH2OCMe2), 3.73 (dd, 1H, J = 8.2, 6.4 Hz, CH2OCMe2) 3.52 (dd, 1H, J = 10.0, 5.7 Hz), 3.48 (dd, 1H, J = 6.7, 2.7 Hz), 3.45 (dd, 1H, J = 7.0, 6.3 Hz), 3.41(dd, 1H, J = 10.0, 5.7 Hz), 2.16-2.10 (m, 4H, CH2CδCCH2), 1.62-1.53 (m, 2H), 1.52-1.41 (m, 4H), 1.47(q, 3H, J = 0.6 Hz, C-CH3 ), 1.36 (q, 3H, J = 0.6 Hz, C-CH3), 1.40-1.20 (m, 20H), 0.90 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ=109.37 (C(CH3)2), 80.29 and 80.19 (CδC), 74.76 (CH α to O), 71.88 (CH2 α to O), 71.84 (CH2 α to O), 66.95 (CH2 α to O), 31.86 (CH2), 29.55 (CH2), 29.37 (CH2), 29.24 (CH2), 29.18 (CH2), 29.16 (CH2), 29.14 (CH2), 29.12 (CH2), 28.89 (CH2), 28.81 (CH2), 26.78 (C-CH3), 26.03 (CH2), 25.43 (C-CH3), 22.68 (CH2), 18.77 (CH2CδC), 18.76 (CH2CδC), 14.12 (CH3).
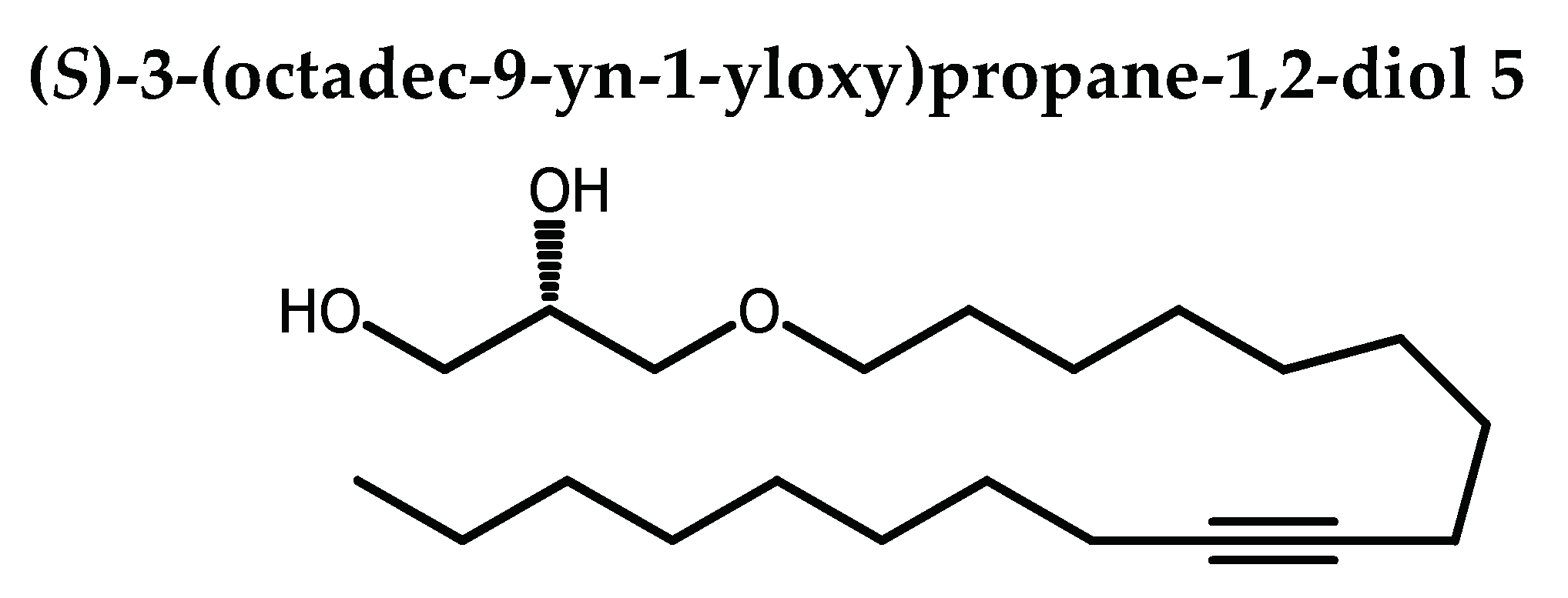
To a solution of 24 (60.4 mg, 0.17 mmol,) in THF (1.2 mL), was added p-toluenesulfonic acid monohydrate (4.8 mg, 0.03 mmol, 0.15 eq) and distilled water (0.51 mL). The flask was then purged under nitrogen, stoppered, and dipped in a preheated bath at 80oC. The stirring was maintained 8 h at the same temperature. Sodium carbonate (5.8 mg, 0.07 mmol, 0.04 eq) was added and the stirring was continued for 1 h at 80oC. Extraction was done with EtOAC (3 x). Organic phase was washed with brine and dried over Na2SO4. Solvent was removed under reduced and the crude product was purified by silica-gel column chromatography (0→10 % EtOAC in petroleum ether) afforded 5 as white solid (46.2 mg, 85%). Rf 0.05 (petroleum ether / EtOAC 80:20).
1H NMR (400 MHz, CDCl3): δ=3.85 (ddt, 1H, J = 5.9, 5.2, 3.9 Hz), 3.71 (dd, 1H, J = 11.4, 3.8 Hz), 3.63 (dd, 1H, J = 11.4, 5.2 Hz), 3.54 (dd, 1H, J = 10.0, 5.7 Hz), 3.48 (dd, 1H, J = 6.7, 2.7 Hz), 3.45 (dd, 1H, J = 7.0, 6.3 Hz), 3.41(dd, 1H, J = 9.7, 4.0 Hz), 3.49 (dd, 9.7, 6.0 Hz), 3.47 (dd, 1H, J = 6.7, 2.5 Hz), 3.44 (dd, 1H, J = 9.5, 6.8 Hz),
2.56-2.52 (enveloppe, 1H, OH), 2.40-2.20 (enveloppe, 1H, OH), 2.17-2.10 (m, 4H, CH2CδCCH2), 1.62-1.54 (m, 2H), 1.52-1.41 (m, 4H), 1.41-1.22 (m, 18H), 0.89 (t, 3H, J = 6.9 Hz, CH3)
13C NMR (100 MHz, CDCl3): δ=80.32 and 80.17 (CδC), 72.52 and 71.84 (CH2OCH2C17H31), 70.44 (CHOH), 64.30 (CH2OH), 31.86 (CH2), 29.57 (CH2), 29.35 (CH2), 29.24 (CH2), 29.18 (CH2), 29.14 (2CH2), 29.10 (CH2), 28.89 (CH2), 28.80 (CH2), 26.05 (CH2), 22.68 (CH2), 18.77 (CH2CδC), 18.76 (CH2CδC), 14.12 (CH3)
Acknowledgments
We also thank CRMPO (Centre Régional de Mesures Physiques de l’Ouest, Rennes, France) for HRMS spectra and microanalyses.
References
- R.E Minto, B. J. Blacklock, Progress in Lipid Resaerch 2008, 47, 233-306.
- A.S. Silva, S. F. Nabavi, M. Saeedi, S. M. Nabavi. ISBN 978-0-12-816455-6, 2020.
- V. M. Dembitsky Lipids, 2006, 41 (10), 883-923.
- Y. Seo, K. W. Cho, H.S. Lee, J.R. Rho, and J. Shin. American Chemical Society and American Society of Pharmacognosy. 1999, 122-126.
- L. Deniau, P. Mosset, F. Pedrono, R. Mitre, D. Le Bot, A. B. Legrand, Mar. Drugs 2010, 8, 2175-2184.
- D. Magnusson, G. G. Haraldsson, Tetrahedron: Asym. 2010, 21, 2841-2847.
- R. Momha, D. Le Bot, P. Mosset, A. B. Legrand. Anticancer Agents in Medicinal Chemistry, 2022, 22 (10), 1913-1920.
- R. Momha, D. E. Pegnyemb; P. Mosset. Synthetic Communications 2020, 50, 1656-1664.
- R. Momha, V. Kuete; J.-M. Pagès; D. E Pegnyemb; P. Mosset, Marine drugs 2020, 18, 113.
- R. Momha, D. E. Pegnyemb; P. Mosset, Tetrahedron 2012, 68, 2973-2983.
- R. Momha, G. B. N. Bayiha; D. E. Penyemb; P. Mosset. ChemistrySelect 2020, 5, 6678-6682.
- B. Hallgren, S. Larsson, J. Lipids Res. 1962, 3, 39–43.
- J.W Linman, M.J Long, D.R. Korst, F.H. Bethell, J. Lab. Clin. Med. 1959, 54, 335–343.
- Brohult, J. Brohult, S. Brohult, I. Joelsson Acta Obstet. Gynecol. Scand. 1977, 56, 441–448.
- Brohult, J. Brohult, S. Brohult. Acta Obstet. Gynecol. Scand. 1978, 57, 79–83.
- B.Z. Ngwenya, D.M. Foster, Proc. Soc. Exp. Biol. Med. 1991, 196, 69–75.
- Brohult, J. Brohult, S. Brohult, S. Experientia 1972, 28, 954–955.
- F. Caijo, P. Mosset, R. Grée, V. Audinot-Bouchez, J. Boutin, P. Renard, D-H. Caignard, C. Dacquet, Eur. J. Org. Chem. 2006, 2006, 2181–2196.
- J. M. Chong, M. A. Heuft, P. Rabbat, J. Org. Chem. 2000, 65, 5837-5838.
- G. Bordier, N. Sellier, A. P. Foucault, F. Le Goffic, Lipids 1996, 31, 521–528.
- E. Baer, H. O. L. Fisher, J. Biol. Chem. 1941, 140, 397-410.
- E. Berry, J. A. Chan, L. MacKenzie, and S. M. Hecht. Chem. Res. Toxicol. 1991, 4, 195-198.
Figure 1.
Acetylenic enol ether of glycerol from the Sponge.
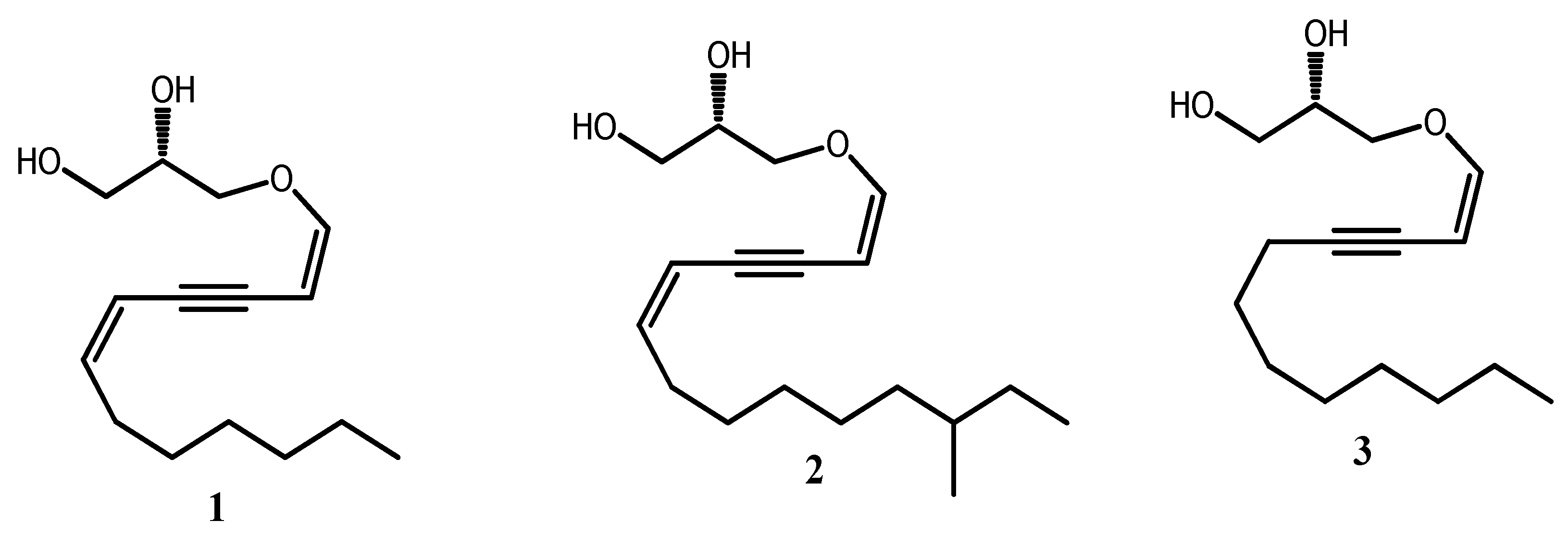
Figure 2.
Unknown synthesized alkylglycerol based-acetylenic derivatives 4-5.
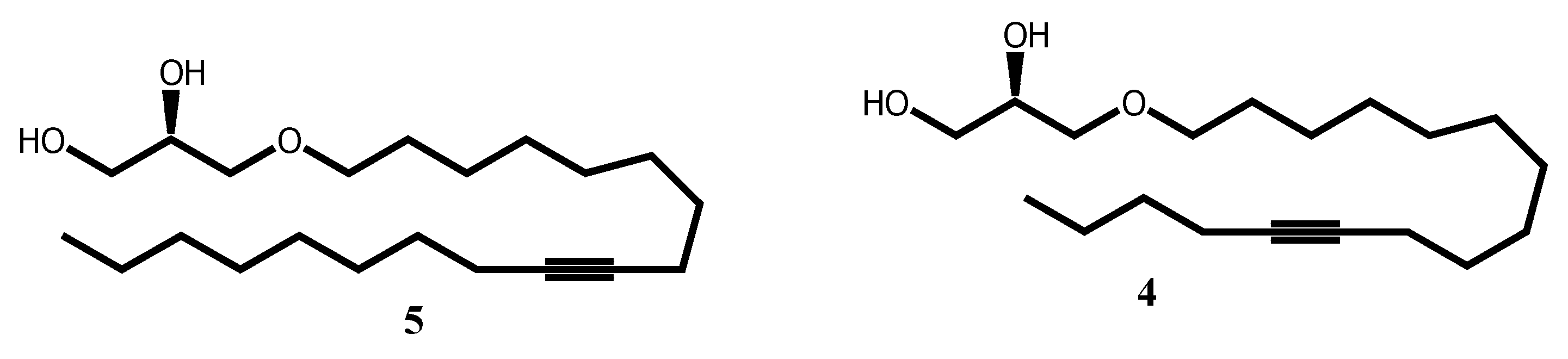
Scheme 1.
Synthesis of bromoacetal 14. Reagents and conditions: (a) 48 % aq. HBr, reflux, 24 h, 80 %; (b) Ethyl vinyl ether, PPTS, CH2Cl2, 0 oC, 30 mn, 87 %.
Scheme 1.
Synthesis of bromoacetal 14. Reagents and conditions: (a) 48 % aq. HBr, reflux, 24 h, 80 %; (b) Ethyl vinyl ether, PPTS, CH2Cl2, 0 oC, 30 mn, 87 %.
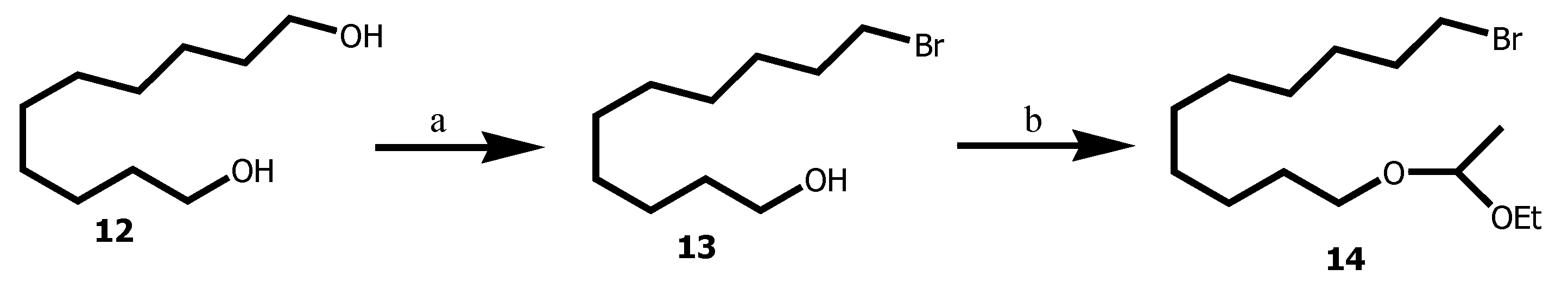
Scheme 2.
Synthesis of the AKG based-acetylenic 4. Reagents and conditions: (c) 14, n-BuLi, THF, 55 min then 15, rt, 16 h, 60 %; (d) PPTS cat., MeOH, rt, 16 h, 89 %; (e) MsCl, Et3N, CH2Cl2, -40°C, 14 h , 94 %; (f) 2,3-isopropylidene-sn-glycerol 19, NaH, NMP, rt, 18 h, 81 %; (g) ) p-TsOH.H2O MeOH / H2O 10:1, 60 °C, 4 h, 85 %.
Scheme 2.
Synthesis of the AKG based-acetylenic 4. Reagents and conditions: (c) 14, n-BuLi, THF, 55 min then 15, rt, 16 h, 60 %; (d) PPTS cat., MeOH, rt, 16 h, 89 %; (e) MsCl, Et3N, CH2Cl2, -40°C, 14 h , 94 %; (f) 2,3-isopropylidene-sn-glycerol 19, NaH, NMP, rt, 18 h, 81 %; (g) ) p-TsOH.H2O MeOH / H2O 10:1, 60 °C, 4 h, 85 %.
Scheme 3.
Synthesis of AKG based-acetylenic 5 an analog of AKG 11. Reagents and conditions: (a) Red-Al, THF, RT, 15 h, 91 % ; (b) MsCl, Et3N, CH2Cl2, -50°C, 2 h, 93 %; (c) 2,3-isopropylidene-sn-glycerol 19, KOH, n-Bu4NBr, DMSO, 45°C, 24 h, 94 %; (d) p-TsOH.H2O MeOH / H2O 10:1, 60 °C, 4 h, 85 %.
Scheme 3.
Synthesis of AKG based-acetylenic 5 an analog of AKG 11. Reagents and conditions: (a) Red-Al, THF, RT, 15 h, 91 % ; (b) MsCl, Et3N, CH2Cl2, -50°C, 2 h, 93 %; (c) 2,3-isopropylidene-sn-glycerol 19, KOH, n-Bu4NBr, DMSO, 45°C, 24 h, 94 %; (d) p-TsOH.H2O MeOH / H2O 10:1, 60 °C, 4 h, 85 %.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



