Submitted:
21 May 2024
Posted:
24 May 2024
You are already at the latest version
Abstract
In this study, we utilized the UniProt database to extract breast cancer genes Total 2658 genes in Homo sapiens are obtain. Then 166 genes are taken and after delating duplicates 158 genes were remains. These genes were then analyzed for their biological pathways using Gene Ontology enrichment analysis through the Genecodis platform, resulting in the identification of 12 unique genes involved in disease pathways. Protein-protein interaction (PPI) network analysis was conducted using the STRING database and Cytoscape software, identifying ANLN,TREM1,LZTFL1,OXSM,PLA2G3,TAS2R13,ABCB10,ECT2,TRPC,BCAS3,PLA2G3,ZDHHC7 and ERBB2 as potential gene targets for breast cancer.
Keywords:
breast cancer
; genes
; pathway analysis
; protein protein interaction
; degree and betweenness
Introduction
Breast cancer: breast cancer is a type of cancer that starts in the breast cells and grows out of control. It's more common in women because they are exposed to estrogen throughout their lives. It's a leading cause of cancer death in women, making up about 20-25% of all cancers in women. One in eight women will develop breast cancer during their lives. Breast cancer is the most frequent type of cancer and the second leading cause of mortality for women after lung cancer. Any breast tissue, cell, or gland has the potential to become cancerous. It can start in the ducts that produce milk or in the glandular tissues known as lobules, which produce milk. If cancer cells are not found at an early stage, there is a potential that they will damage other areas of the body and spread. Breast tumours can either be benign or malignant; benign lesions are non-cancerous cell abnormalities that cannot develop into breast cancer, whereas malignant lesions are cancerous lesions.
Network Pharmacology
In 2007, Hopkins et al. first proposed the concept “network pharmacology”. This method analyzes the intervention of drugs and potential treated targets of diseases based on system biology. Network pharmacology highlights a paradigm shift from the current “one target, one drug” strategy to a novel version of the “network target, multi-component” strategy Recently, a new technique known as polypharmacology has emerged which is able to address the limitations with current drug discovery challenges. Poly-pharmacology, also known as network pharmacology, attempts to understand drug action and interactions with multiple targets (Hopkins, 2007). It uses computational power and computer-based virtual high-throughput screening for docking studies to improve the efficiency of discovery process.
Target Identification
Target identification is the process of identifying potential targets for drug development. This process typically involves the use of computational and experimental methods to identify proteins, enzymes, receptors, or other biological molecules that are involved in the pathways of interest, and that could be targeted by small molecule compounds in order to treat a specific disease or condition. By identifying potential targets, researchers can develop and test small molecule compounds that are designed to interact with these targets, in order to modulate their activity and exert a therapeutic effect.
Gene Ontology
Gene Ontology is one of the main resources of biological information since it provides a specific definition of protein functions.Gene Ontology is a powerful tool in bioinformatics that helps in the standardized representation of genes and their functions across different species.
String
Protein networks have become a popular tool for analyzing and visualizing the oftenlong lists of proteins or genes obtained from proteomics and other high-throughput technologies. One of the most popular sources of such networks is the STRING database, which provides protein networks for more than 2000 organisms, including both physical interactions from experimental data and functional associations from curated pathways, automatic text mining, and prediction methods.
Cytoscape
cytoscape utilizes a highly reliable gene functional interaction network combined with human curated pathways derived from Reactome and other pathway databases. Biologists can use this app to uncover network and pathway patterns related to their studies, search for gene signatures from gene expression data sets, reveal pathways significantly enriched by genes in a list, and integrate multiple genomic data types into a pathway context using probabilistic graphical models.
Materials and Methods
Text Mining
The Uniprot database(https://www.uniprot.org/) was utilized to obtain genes involved in breast cancer. In which from a total of 2658 genes in homo-sapiens 158 genes are extracted and dublicate were delated. These genes were analyzed for their biological pathways and involvement in disease pathology.
GO Enrichment Analysis
Gene Ontology is a powerful tool in bioinformatics that helps in the standardized representation of genes and their functions across different species that used in analysis of cellular componants and molecular pathway .The Genecodis(https://genecodis.genyo.es/) platform was utilized in the procedure. Information of 166 genes were acquired. From these pathways 10 unique genes were sorted out as per their involvement in disease pathways and those genes were further utilized for network analysis.
PPI Network Analysis
The STRINGS database (https://string-db.org/)is used to predict protein-protein interaction networks. The Cytoscape software is utilized to visualize and analyze PPI networks in which functional enrichment data for proteins in the network is obtained and also analyzed for PPI network parameters such as degree, betweenness and compartment/tissue score. By network analysis data different potential protein targets are predicted. These protein targets are further utilized for virtual screening by molecular docking.
Lead Identification
Lead identification is a key step in drug discovery where potential compounds with therapeutic effects are identified for further development.We utilized PubChem to explore the chemical constituents relevant to our study. We identified a total of 15 chemical constituents of green Tea, each of which plays a crucial role in our investigation. By leveraging PubChem's extensive database, we were able to comprehensively analyze the properties and characteristics of these constituents, contributing to a more thorough understanding of their potential effects and mechanisms of action within our research context.
Molecular Docking
Molecular docking is a computational technique used in the field of bioinformatics and drug discovery to predict the preferred orientation of one molecule when bound to another molecule to form a stable complex. This method helps in understanding the interactions between small molecules and proteins, which is crucial for designing new drugs or optimizing existing ones. By simulating the docking process, researchers can predict the binding affinity and mode of interaction between a ligand and receptor, aiding in the identification of potential drug candidates.
We use PDB software to download the gene structures necessary for our study.and,we utilized Pyrex software for molecular docking, a crucial step in our computational analysis. By leveraging these tools, we were able to obtain the structural data needed for docking simulations and predict the binding interactions between our ligands and receptors accurately. This approach enabled us to gain valuable insights into the molecular mechanisms underlying our research and facilitated the identification of potential drug candidates.
Results
UNIPROT KB: There are 158 genes are taken out of 2658 genes of breast cancer.
Go Enrichment Analysis
Go enrichment analysis was performed using Genecodis web platform. In the process top 10 pathways are selected which are associated with breast cancer. Significantly enriched genes are selectedTREM1,TRPC4,ERBB2,ANLN,ZDHHC7,LZTFL1,OXSM,PLA2G3,ECT2,TAS2R1 3,ABCB10,BCAS3,ERBB2.
PPI Network Analysis
The STRING database was used to develop the PPI network. The PPI network was visualized and analyzed using the Cytoscape app. Betweenness and Degree of centrality parameters from the Centiscape plugin were used to analyze the network. ANLN,TREM1,LZTFL1,OXSM,PLA2G3,TAS2R13,ABCB10,ECT2,TRPC,BCAS3,PLA2G3,ZDHHC7,ERBB2 this are the potential gene based on string analysis
Degree and betweenness Analysis
Result of target identification with degree and betweenness: Here we have identified the targets and candidate genes (Figure 5). We loaded data into Cytoscape in the “.tsv" format from the STRING EXPORT channel. Then, to evaluate each node's topological characteristics and identify the important nodes, we employed CentiScaPe, a software that computes a greater number of network characteristics. The total number of edges occurring to the node is represented by the node degree.
Molecular Docking
The PubMed database use for obtain chemical constituent of green tea, in that we taken 15 chemical constituent (Table 3). And download their structure from PubChem.
Targeted protein download from PDB data base,in that 5 protein structure download out of 10 protein. Then use Pyrex for molecular docking of protein and lead ,in that 3 protein show binding with 15 leads (Table 4).

Table 1.
genes of breast cancer obtain from text mining.
|
Entry |
Gene name |
|
Q9H2R5 |
KLK15 |
|
Q9H2X6 |
HIPK2 |
|
Q9H2X9 |
SLC12A5 KCC2 KIAA1176 |
|
Q9H3D4 |
TP63 KET P63 P73H P73L TP73L |
|
Q9H3H5 |
DPAGT1 DPAGT2 |
|
Q9H3M9 |
ATXN3L ATX3L MJDL |
|
Q9H3R5 |
CENPH ICEN35 |
|
Q9H3T3 |
SEMA6B SEMAN SEMAZ UNQ1907/PRO4353 |
|
Q9H3V2 |
MS4A5 CD20L2 TETM4 |
|
Q9H3Y6 |
SRMS C20orf148 |
|
Q9H467 |
CUEDC2 C10orf66 HOYS6 |
|
Q9H4A3 |
WNK1 HSN2 KDP KIAA0344 PRKWNK1 |
|
Q9H4B4 |
PLK3 CNK FNK PRK |
|
Q9H4D0 |
CLSTN2 CS2 |
|
Q9H4H8 |
FAM83D C20orf129 |
|
Q9H4L2 |
BRCA2 |
|
Q9H4L3 |
BRCA2 |
|
Q9H4T2 |
ZSCAN16 ZNF392 ZNF435 |
|
Q9H4X1 |
RGCC C13orf15 RGC32 |
|
Q9H582 |
ZNF644 KIAA1221 ZEP2 |
|
Q9H596 |
DUSP21 LMWDSP21 |
|
Q9H5I1 |
SUV39H2 KMT1B |
|
Q9H5J0 |
ZBTB3 |
|
Q9H5V8 |
CDCP1 TRASK UNQ2486/PRO5773 |
|
Q9H6B1 |
ZNF385D ZNF659 |
|
Q9H6R7 |
WDCP C2orf44 PP384 |
|
Q9H6U6 |
BCAS3 |
|
Q9H7D7 |
WDR26 CDW2 MIP2 PRO0852 |
|
Q9H7L9 |
SUDS3 SAP45 SDS3 |
|
Q9H7N4 |
SCAF1 SFRS19 SRA1 |
|
Q9H7R0 |
ZNF442 |
|
Q9H7S9 |
ZNF703 ZEPPO1 ZPO1 |
|
Q9H813 |
PACC1 C1orf75 TMEM206 |
|
Q9H8L6 |
MMRN2 EMILIN3 |
|
Q9H8V3 |
ECT2 |
|
Q9H8Y1 |
VRTN C14orf115 |
|
Q9H910 |
JPT2 C16orf34 HN1L L11 |
|
Q9H9B1 |
EHMT1 EUHMTASE1 GLP KIAA1876 KMT1D |
|
Q9HAU4 |
SMURF2 |
|
Q9HAU8 |
RNPEPL1 |
|
Q9HAW4 |
CLSPN |
|
Q9HAX1 |
DSC3 |
|
Q9HB09 |
BCL2L12 BPR |
|
Q9HB58 |
SP110 |
|
Q9HBL0 |
TNS1 TNS |
|
Q9HBT6 |
CDH20 CDH7L3 |
|
Q9HBW1 |
LRRC4 BAG NAG14 UNQ554/PRO1111 |
|
Q9HBX8 |
LGR6 UNQ6427/PRO21331 VTS20631 |
|
Q9HC10 |
OTOF FER1L2 |
|
Q9HC57 |
WFDC1 PS20 |
|
Q9HC96 |
CAPN10 KIAA1845 |
|
Q9HCE0 |
EPG5 KIAA1632 |
|
Q9HCE3 |
ZNF532 KIAA1629 |
|
Q9HCE6 |
ARHGEF10L GRINCHGEF KIAA1626 |
|
Q9HCH5 |
SYTL2 KIAA1597 SGA72M SLP2 SLP2A |
|
Q9HCI5 |
MAGEE1 HCA1 KIAA1587 |
|
Q9HCS4 |
TCF7L1 TCF3 |
|
Q9HCU0 |
CD248 CD164L1 TEM1 |
|
Q9HCU9 |
BRMS1 |
|
Q9HD15 |
SRA1 PP7684 |
|
Q9HD64 |
XAGE1A GAGED2 XAGE1; XAGE1B XAGE1C XAGE1D XAGE1E |
|
Q9HDB8 |
ERVK-5 ERVK5 |
|
Q9NP09 |
ERBB2 |
|
Q9NP58 |
ABCB6 MTABC3 PRP UMAT |
|
Q9NP60 |
IL1RAPL2 IL1R9 |
|
Q9NP61 |
ARFGAP3 ARFGAP1 |
|
Q9NP99 |
TREM1 |
|
Q9NPC2 |
KCNK9 TASK3 |
|
Q9NPD5 |
SLCO1B3 LST2 OATP1B3 OATP8 SLC21A8 |
|
Q9NPD8 |
UBE2T HSPC150 PIG50 |
|
Q9NPG8 |
ZDHHC4 ZNF374 DC1 UNQ5787/PRO19576 |
|
Q9NPY3 |
CD93 C1QR1 MXRA4 |
|
Q9NQ31 |
AKIP1 BCA3 C11orf17 |
|
Q9NQ48 |
LZTFL1 |
|
Q9NQ66 |
PLCB1 KIAA0581 |
|
Q9NQ88 |
TIGAR C12orf5 |
|
Q9NQB0 |
TCF7L2 TCF4 |
|
Q9NQC3 |
RTN4 KIAA0886 NOGO My043 SP1507 |
|
Q9NQG5 |
RPRD1B C20orf77 CREPT |
|
Q9NQR3 |
BRCA1 |
|
Q9NQU5 |
PAK6 PAK5 |
|
Q9NQW6 |
ANLN |
|
Q9NQX1 |
PRDM5 PFM2 |
|
Q9NR12 |
PDLIM7 ENIGMA |
|
Q9NR30 |
DDX21 |
|
Q9NR80 |
ARHGEF4 KIAA1112 |
|
Q9NR82 |
KCNQ5 |
|
Q9NR96 |
TLR9 UNQ5798/PRO19605 |
|
Q9NRA2 |
SLC17A5 |
|
Q9NRC1 |
ST7 FAM4A1 HELG RAY1 |
|
Q9NRD0 |
FBXO8 FBS FBX8 DC10 UNQ1877/PRO4320 |
|
Q9NRH2 |
SNRK KIAA0096 SNFRK |
|
Q9NRJ1 |
C8orf17 |
|
Q9NRK6 |
ABCB10 |
|
Q9NRM2 |
ZNF277 NRIF4 ZNF277P |
|
Q9NRN9 |
METTL5 DC3 HSPC133 |
|
Q9NRP0 |
OSTC DC2 HDCMD45P HSPC307 |
|
Q9NRP7 |
STK36 KIAA1278 |
|
Q9NRY4 |
ARHGAP35 GRF1 GRLF1 KIAA1722 P190A p190ARHOGAP |
|
Q9NS23 |
RASSF1 RDA32 |
|
Q9NS39 |
ADARB2 ADAR3 RED2 |
|
Q9NS71 |
GKN1 AMP18 CA11 UNQ489/PRO1005 |
|
Q9NS87 |
KIF15 KLP2 KNSL7 |
|
Q9NSC7 |
ST6GALNAC1 SIAT7A UNQ543/PRO848 |
|
Q9NTF0 |
DKFZp727E011 |
|
Q9NTG1 |
PKDREJ |
|
Q9NTK1 |
DEPP1 C10orf10 DEPP FIG |
|
Q9NTK5 |
OLA1 GTPBP9 PRO2455 PTD004 |
|
Q9NTM9 |
CUTC CGI-32 |
|
Q9NTW7 |
ZFP64 ZNF338 |
|
Q9NTX7 |
RNF146 |
|
Q9NTX9 |
FAM217B C20orf177 |
|
Q9NU02 |
ANKEF1 ANKRD5 |
|
Q9NUM3 |
SLC39A9 ZIP9 UNQ714/PRO1377 |
|
Q9NUQ7 |
UFSP2 C4orf20 |
|
Q9NUT2 |
ABCB8 MABC1 MITOSUR |
|
Q9NV12 |
TMEM140 |
|
Q9NV23 |
OLAH THEDC1 |
|
Q9NV64 |
TMEM39A SUSR2 |
|
Q9NVA1 |
UQCC1 BZFB C20orf44 UQCC |
|
Q9NVD3 |
SETD4 C21orf18 C21orf27 |
|
Q9NVE7 |
PANK4 |
|
Q9NVM9 |
INTS13 ASUN C12orf11 GCT1 |
|
Q9NVP1 |
DDX18 cPERP-D |
|
Q9NVW2 |
RLIM RNF12 |
|
Q9NVX2 |
NLE1 HUSSY-07 |
|
Q9NW75 |
GPATCH2 GPATC2 |
|
Q9NWB6 |
ARGLU1 |
|
Q9NWK9 |
ZNHIT6 BCD1 C1orf181 |
|
Q9NWM0 |
SMOX C20orf16 SMO UNQ3039/PRO9854 |
|
Q9NWU1 |
OXSM |
|
Q9NX61 |
TMEM161A UNQ582/PRO1152 |
|
Q9NXF8 |
ZDHHC7 |
|
Q9NXL2 |
ARHGEF38 |
|
Q9NY72 |
SCN3B KIAA1158 |
|
Q9NY74 |
ETAA1 ETAA16 |
|
Q9NYC9 |
DNAH9 DNAH17L DNEL1 KIAA0357 |
|
Q9NYF0 |
DACT1 DPR1 HNG3 |
|
Q9NYV9 |
TAS2R13 |
|
Q9NZ20 |
PLA2G3 |
|
Q9NZ52 |
GGA3 KIAA0154 |
|
Q9NZC7 |
WWOX FOR SDR41C1 WOX1 |
|
Q9NZC9 |
SMARCAL1 HARP |
|
Q9NZJ4 |
SACS KIAA0730 |
|
Q9NZJ5 |
EIF2AK3 PEK PERK |
|
Q9NZL6 |
RGL1 KIAA0959 RGL |
|
Q9NZP6 |
NPAP1 C15orf2 |
|
Q9P032 |
NDUFAF4 C6orf66 HRPAP20 HSPC125 My013 |
|
Q9P0G3 |
KLK14 KLKL6 |
|
Q9P253 |
VPS18 KIAA1475 |
|
Q9P260 |
RELCH KIAA1468 |
|
Q9P283 |
SEMA5B KIAA1445 SEMAG UNQ5867/PRO34001 |
|
Q9P287 |
BCCIP TOK1 |
|
Q9P2J8 |
ZNF624 KIAA1349 |
|
Q9P2U8 |
SLC17A6 DNPI VGLUT2 |
|
Q9T3Q5 |
ND2 NADH2 |
|
Q9UBB6 |
NCDN KIAA0607 |
|
Q9UBF1 |
MAGEC2 HCA587 MAGEE1 |
|
Q9UBF9 |
MYOT TTID |
|
Q9UBK9 |
UXT HSPC024 |
|
Q9UBN4 |
TRPC4 |
|
Q9UBN7 |
HDAC6 KIAA0901 JM21 |
|
Q9UBP0 |
SPAST ADPSP FSP2 KIAA1083 SPG4 |
|
Q9UBS9 |
SUCO C1orf9 CH1 OPT SLP1 |
Figure 1.
GO enrichment analysis chart bar length signifies -log10(pval Ad)and red colour intensity signifies number of input genes involved in pathway.
Figure 1.
GO enrichment analysis chart bar length signifies -log10(pval Ad)and red colour intensity signifies number of input genes involved in pathway.
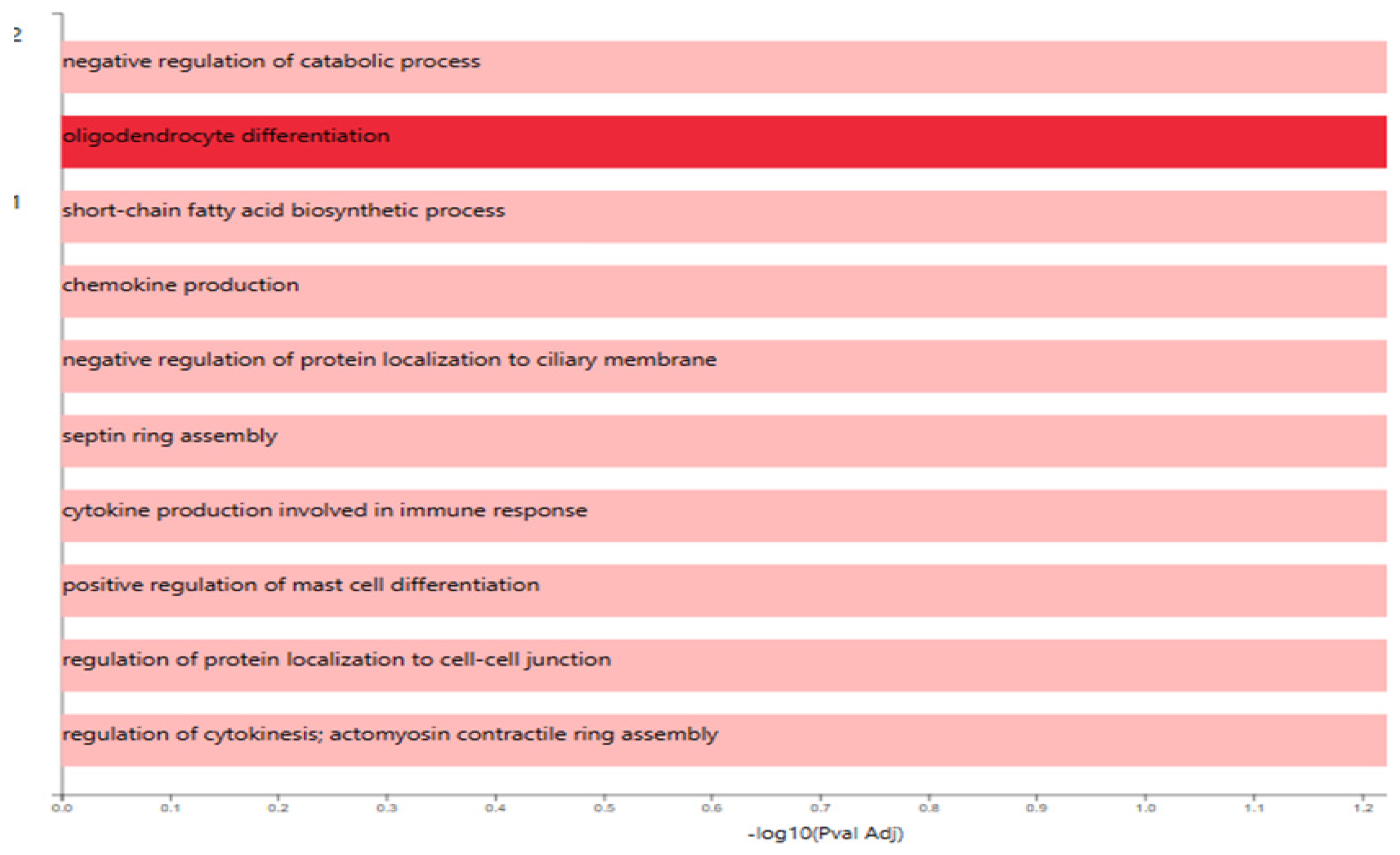
Figure 2.
KEGG enrichment analysis chart bar length signifies -log10(pvalAdj) and red colour intensity signifies number of input genes involved in pathway.
Figure 2.
KEGG enrichment analysis chart bar length signifies -log10(pvalAdj) and red colour intensity signifies number of input genes involved in pathway.
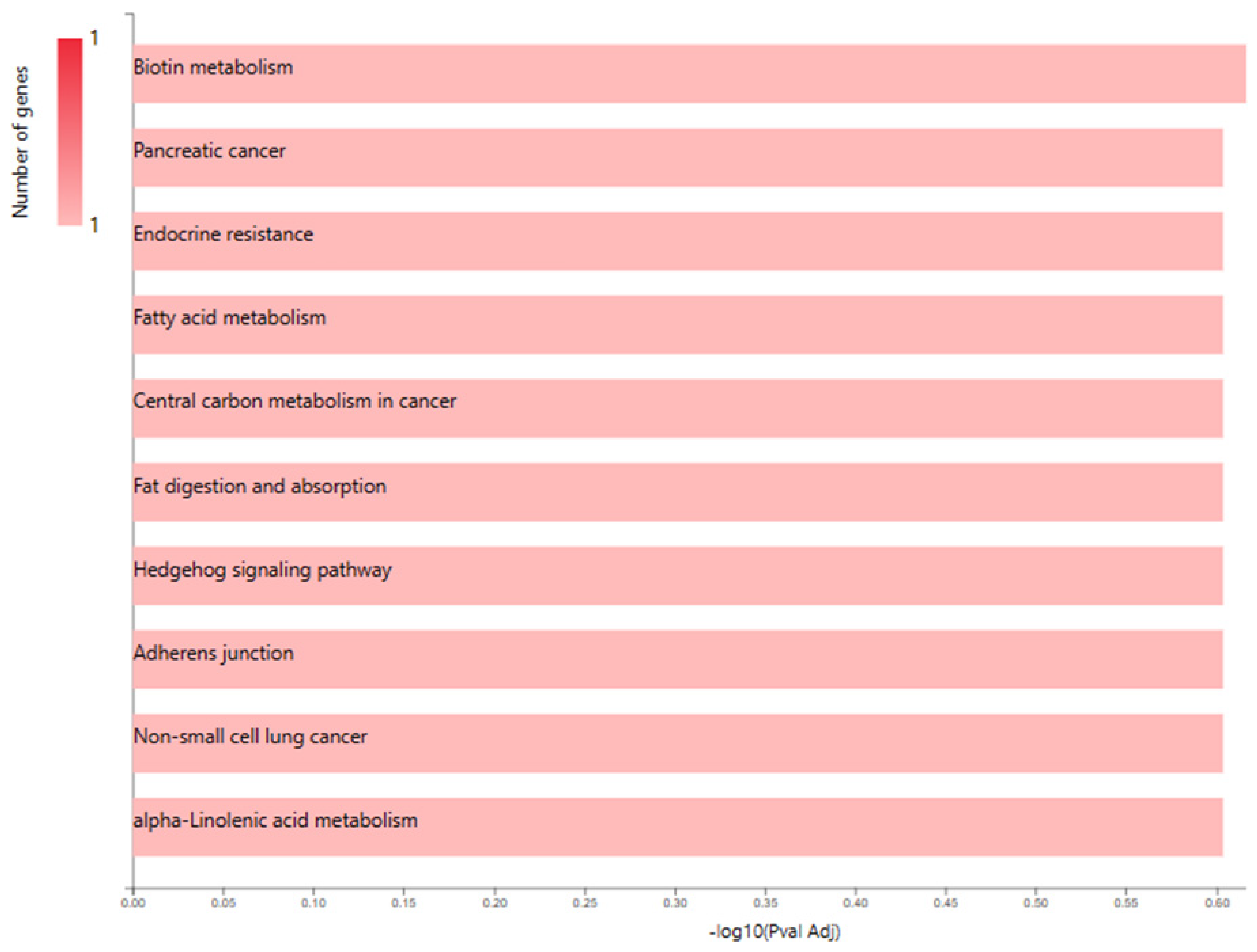
Figure 3.
Protein protein interaction in string database.network nodes represent proteins, different coloured edges represent protein-protein interaction.
Figure 3.
Protein protein interaction in string database.network nodes represent proteins, different coloured edges represent protein-protein interaction.
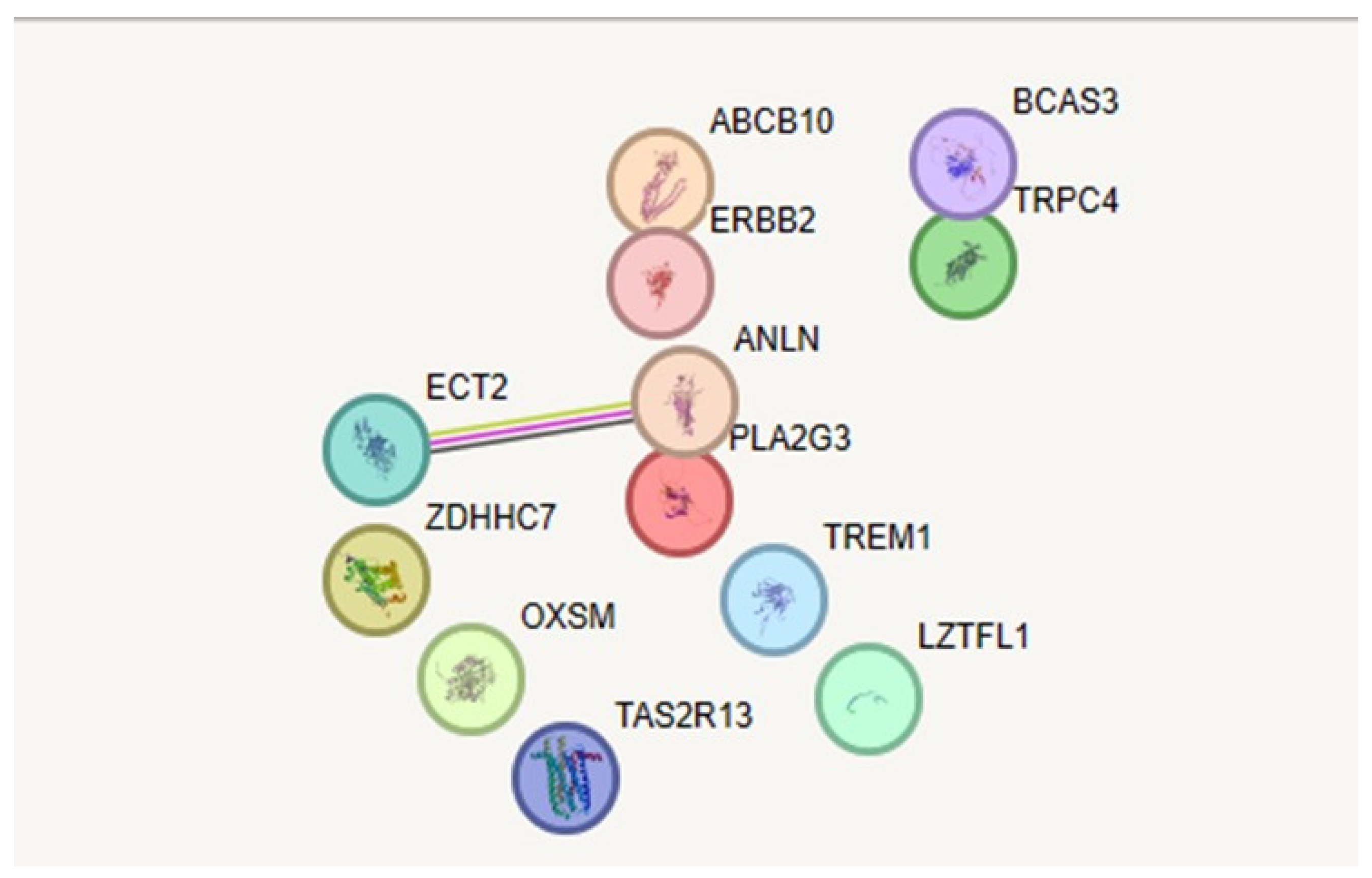
Figure 4.
The protein-protein interaction network of genes and identification of candidate genes, produced using Cytoscape.
Figure 4.
The protein-protein interaction network of genes and identification of candidate genes, produced using Cytoscape.
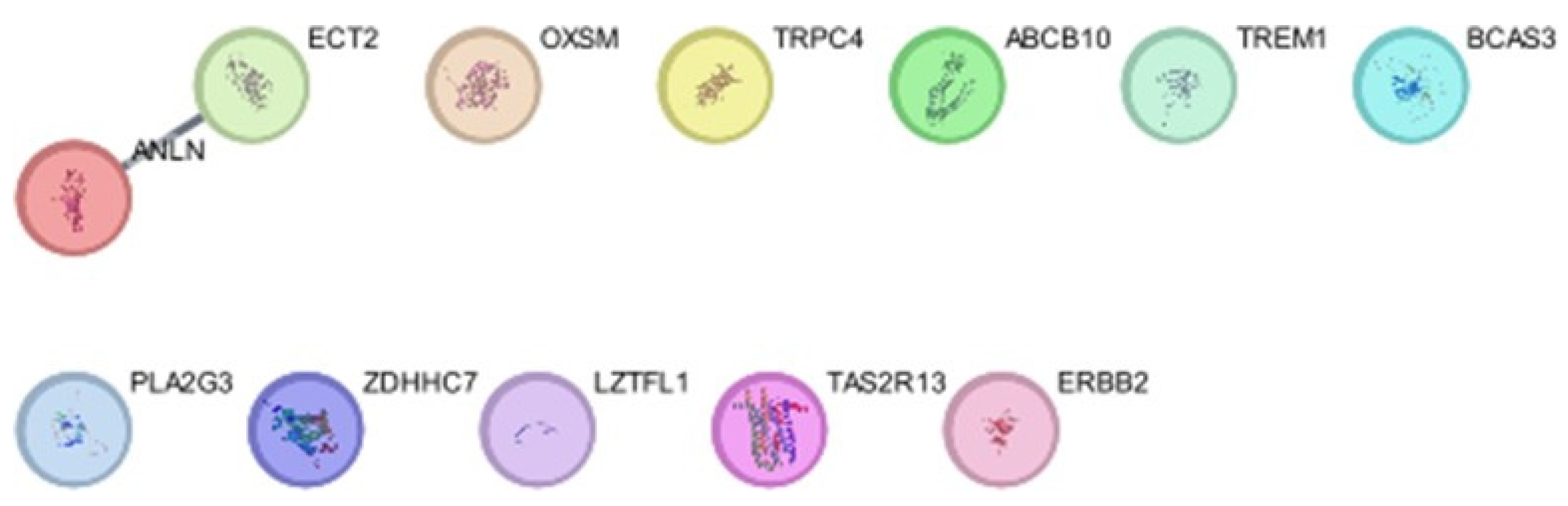
Figure 5.
The degree and betweenness of the protein are produce by using centiscape.
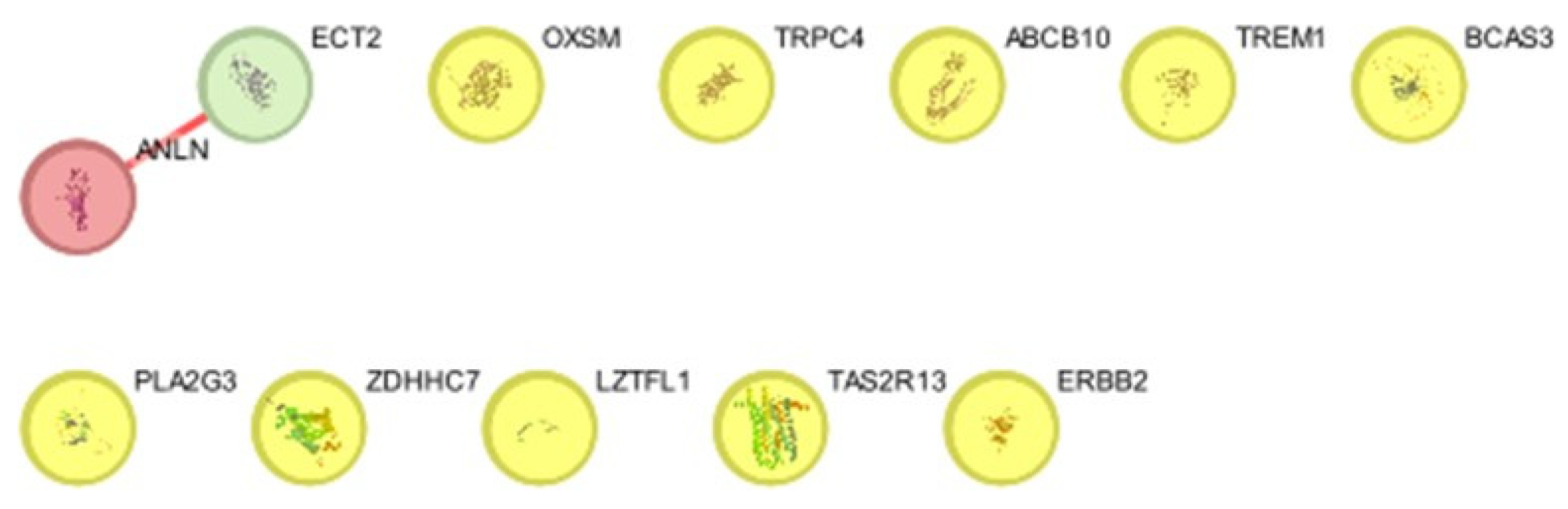
Table 2.
Degree and betweenness obtain using centiscape.
| GENES | DEGREE | BETWEENES |
|
ERBB2 |
0 |
0 |
|
LZTFL1 |
0 |
0 |
|
ZDHHC7 |
0 |
0 |
|
PLA2G3 |
0 |
0 |
|
BCAS3 |
0 |
0 |
|
TREM1 |
0 |
0 |
|
ABCB10 |
0 |
0 |
|
ECT2 |
1 |
0 |
| TRPC4 | 0 | 0 |
| OXSM | 0 | 0 |
|
ANLN |
1 |
0 |
Table 3.
Chemical constituent of green tea obtain from PubChem.
| Epigallocatechin | Gallocatechol |
| Epicatechin gallate | Gallic acid |
| Catechin | Theaflavin |
| Theanine | Chlorogenic acid |
| Theobromine | Epicatechin 3 gallate |
| Catechol | epigallocatechin |
| Gallocatechin | linolool |
| Quinic acid |
Table 4.
Binding Affinity between ligand and protein.
|
Ligand |
Binding Affinity |
Ligand |
Binding Affinity |
|
ABCB10_catechin |
-6.6 |
ERBB2_catechin |
-7.5 |
|
ABCB10_CATECHOL |
-4.4 |
ERBB2_CATECHOL | -5.2 |
|
ABCB10_chlorogenic_acid |
-6.4 |
ERBB2_chlorogenic_acid |
-7 |
|
ABCB10_epicatechin_3_gallate |
-7.3 |
ERBB2_epicatechin_3_galla te |
-8.8 |
|
ABCB10_epicatechin_gallate |
-7.2 |
ERBB2_epicatechin_gallate | -7.6 |
|
ABCB10_epigallocatechin |
-7.2 |
ERBB2_epigallocatechin |
-8.9 |
|
ABCB10_epigallocatechol |
-6.7 |
ERBB2_epigallocatechol |
-7.5 |
|
ABCB10_gallic_acid |
-5 |
ERBB2_gallic_acid |
-6.3 |
|
ABCB10_gallocatechin |
-6.8 |
ERBB2_gallocatechin |
-7.2 |
|
ABCB10_gallocatechol |
-6.8 |
ERBB2_gallocatechol |
-7.2 |
|
ABCB10_linalool |
-5 |
ERBB2_linalool |
-5 |
|
ABCB10_quinic_acid |
-4.9 |
ERBB2_quinic_acid | -5.9 |
|
ABCB10_theaflavin |
-7.9 |
ERBB2_theaflavin |
-9.4 |
|
ABCB10_theanine |
-4.4 |
ERBB2_theanine | -4.6 |
|
ABCB10_theobromine |
-5.1 |
ERBB2_theobromine |
-5.3 |
|
TREM1_catechin |
-6.5 |
TREM1_linalool |
-4.3 |
|
TREM1_CATECHOL |
-5.2 |
TREM1_quinic_acid |
-5.1 |
|
TREM1_chlorogenic_acid |
-7.1 |
TREM1_theaflavin |
-7.7 |
|
TREM1_epicatechin_3_gallate |
-7.3 |
TREM1_theanine |
-4.4 |
|
TREM1_epicatechin_gallate |
-6.9 |
TREM1_theobromine |
-4.6 |
| TREM1_epigallocatechin | -6.9 | TREM1_gallocatechin | -6.6 |
|
TREM1_epigallocatechol |
-6.5 |
TREM1_gallocatechol |
-6.6 |
|
TREM1_gallic_acid |
-5.6 |
||
Discussion
Breast cancer has a high rate of metastasis and a fatality rate of over 70% when it spreads. It is well acknowledged that surgical excision is the primary treatment for most cases of breast cancer.After doing a gene set enrichment analysis, we were able to identify 158 target genes in this study. The carcinogenic process's genetic modifications affect how cells function, enabling self-sufficiency in growth signals, insensitivity to antigrowth signals, escape from apoptosis, infinite replicative potential, invasion, angiogenesis, and metastasis. These changes are consistent with the enriched biological processes—such as "cell differentiation," "cell division," "cell adhesion," and "signal transduction"—that were found using GeneCodis analysis (Table 1). The appropriate genes are then exported to the STRING database to check and analyse the protein-protein interaction along with the study of different types of nodes present. The same genes are as well transferred to the Cytoscape to visualize the interaction of targets more flexibly. With the targeted gene we came across the degree and betweenness of the genes, which helped us to imply the candidate genes for the further analysis with drugs.
Conclusion
The genes that have shown the targeted interaction are: ECT1 and ANLN.
From the above network pharmacology with various analytical tools, we came to the conclusion that, with the incidence of breast cancer on the rise and patient expectations rising in recent years, selecting an appropriate treatment to maximize preservation of function and minimize the risk of metastasis and recurrence remains challenging. In the clinic, surgical excision is frequently considered the best course of action for treating breast cancer. Non-surgical techniques like photodynamic therapy (PDT), radiation therapy, cryotherapy, and chemotherapy are commonly used to treat late-stage breast cancer. However, research on pharmaceutical therapy is still lacking, and more studies are needed to help develop novel therapeutic strategies.
Even though the efficacy of the currently available drug therapies is limited, and the discovery of new drug therapies using traditional methods is likely to take a long time, drug repositioning may speed up the process of discovering additional conditions that existing drugs could treat more effectively and potentially at a lower cost. This study aimed to investigate new drug therapies for breast cancer by means of computational methods including text mining, biological process and pathway analysis, protein-protein interaction (PPI) analysis to mine public databases, and bioinformatics tools to systematically identify interaction networks between drugs and gene targets. We were able to use data analytical techniques to look at the characteristics of possible genes in order to select a medication.
With the help of these targeted candidate genes, we can further take the analysis towards the gene-drug data interpretation and find the genes that can be used as a biomarker in correspondence with the drugs compounded for the breast cancer.
References
- ZJinrui, Huang. (2022). Research on the Growth of Breast Tumor. Advances in social science, education and humanities research. [CrossRef]
- Mr., Jayantkumar, Rathod., Pushvin, Gowda, M, R., Preethi, M., Manila, S, Koddaddi., Bindhu, R. (2023). A Review Paper on Brief Study on Breast Cancer Classification using Deep Learning. International Journal of Advanced Research in Science, Communication and Technology, 18-20. [CrossRef]
- Dong, Y., Tao, B., Xue, X. et al. Molecular mechanism of Epicedium treatment for depression based on network pharmacology and molecular docking technology. BMC Complement Med Ther 21, 222 (2021). [CrossRef]
- Chandran, U., Mehendale, N. E. E. L. A. Y., Tillu, G. I. R. I. S. H., & Patwardhan, B. H. U. S. H. A. N. (2015, June). Network pharmacology: an emerging technique for natural product drug discovery and scientific research on ayurveda. In Proc Indian Natn Sci Acad (Vol. 81, No. 3, pp. 561-8).
- Vijayarani, S., Ilamathi, M. J., & Nithya, M. (2015). Preprocessing techniques for text mining-an overview. International Journal of Computer Science & Communication Networks, 5(1), 7-16.
- Charlie, Mayor., Lyn, Robinson. (2014). Ontological realism and classification: Structures and concepts in the Gene Ontology. 65(4):686-697. [CrossRef]
- Doncheva, N. T., Morris, J. H., Gorodkin, J., & Jensen, L. J. (2018). Cytoscape StringApp: network analysis and visualization of proteomics data. Journal of proteome research, 18(2), 623-632. [CrossRef]
- Wu, G., Dawson, E., Duong, A., Haw, R., & Stein, L. (2014). ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. F1000Research, 3. [CrossRef]
- Yuyan, Pan., Yong, Zhang., Jiaqi, Liu. (2018). Text mining-based drug discovery in cutaneous squamous cell carcinoma. Oncology Reports, 40(6):3830-3842. [CrossRef]
- Noor, F., Tahir ul Qamar, M., Ashfaq, U. A., Albutti, A., Alwashmi, A. S., & Aljasir, M. A. (2022). Network pharmacology approach for medicinal plants: review and assessment. Pharmaceuticals, 15(5), 572. [CrossRef]
- S Azmi, A. (2013). Adopting network pharmacology for cancer drug discovery. Current drug discovery technologies, 10(2), 95-105.
- Pei, C., Yang, K., Chen, Y., Dong, Y., Meng, X., & Song, N. (2024). Mechanisms of action of Qinghao in treating breast cancer and doxorubicin-induced cardiotoxicity based on network pharmacology and molecular docking. [CrossRef]
- Hopkins, A. L. (2008). Network pharmacology: the next paradigm in drug discovery. Nature chemical biology, 4(11), 682-690. [CrossRef]
- Li, Z., Han, P., You, Z. H., Li, X., Zhang, Y., Yu, H., ... & Chen, X. (2017). In silico prediction of drug-target interaction networks based on drug chemical structure and protein sequences. Scientific reports, 7(1), 11174. [CrossRef]
- Robert, C., Goldman. (2020). Target Discovery for New Antitubercular Drugs Using a Large Dataset of Growth Inhibitors from PubChem.. Infectious disorders drug targets. [CrossRef]
- Rupali, Patil, Bhagat., Mitali, B, Ghormade. (2020). Docking of rigid macromolecules using autodok. Journal of emerging technologies and innovative research.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



