
Submitted:
23 May 2024
Posted:
24 May 2024
You are already at the latest version
Abstract
Personalized cancer vaccines have emerged as a promising avenue for cancer treatment or prevention strategies. This approach targets the specific genetic alterations in individual patient’s tumors, offering a more personalized and effective treatment option. Previous studies have shown that generalized peptide vaccines targeting a limited scope of gene mutations are ineffective, emphasizing the need for personalized approaches. While studies have explored personalized mRNA vaccines, personalized peptide vaccines have not yet been studied in this context. Pancreatic ductal adenocarcinoma (PDAC) remains challenging in oncology, necessitating innovative therapeutic strategies. In this study, we developed a personalized peptide vaccine design methodology, employing RNA sequencing (RNAseq) to identify prevalent gene mutations underlying PDAC development in a specific patient solid tumor tissue. We performed RNAseq analysis for trimming adapters, read alignment, and somatic variant calling. We also developed a Python program, called GeneFinder, which validates the alignment of the RNAseq analysis. The Python program is freely available to download. Using chromosome number and locus data, GeneFinder identifies the target gene along the UCSC hg38 reference set. Based on the gene mutation data, we developed a personalized PDAC cancer vaccine that targets 100 highly prevalent gene mutations in the individual patient. We predicted peptide-MHC binding affinity, immunogenicity, antigenicity, allergenicity, and toxicity for each epitope. Then, we selected the top 50 and 100 epitopes based on our previously published vaccine design methodology. Finally, we generated pMHC-TCR 3D molecular model complex structures which are freely available fo download. The designed personalized cancer vaccine contains epitopes commonly found in PDAC solid tumor tissue. Our personalized vaccine was composed of neoantigens, allowing for a more precise and targeted immune response against cancer cells. Additionally, we identified mutated genes which were also found in the reference study where we obtained the sequencing data, thus validating our vaccine design methodology. This is the first study designing a personalized peptide vaccine using human patient data to identify gene mutations associated with the specific tumor of interest.
Keywords:
Personalized cancer vaccines
; Neoantigens
; Pancreatic Ductal Adenocarcinoma
; peptide based personalized cancer vaccine
; MHC
; HLA
; TCR
1. Introduction
Personalized cancer vaccines are a rising innovation in the field of vaccine design [1]. These vaccines induce an antigen-specific CD8+ and CD4+ T-cell response to enhance anti-tumor activity based on a patient’s individual tumor. Technological innovation has led to the ability to rapidly sequence and analyze patient genome data, which leads into selection of gene targets and on-demand production of a personalized therapy [2]. A phase I clinical trial synthesized personalized mRNA vaccines against PDAC from solid tumors, which led to improved disease-free survival [3]. The trial analyzed a patient population who underwent surgical resection of PDAC tumors. Future development of personalized cancer vaccines direct to demonstrate significant efficacy in patients without major surgical intervention.
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer and projected to be the second-leading cause of cancer mortality by 2030 [4,5]. Current clinical therapies involve neoadjuvant therapy followed by possible surgical resection [6]. However, patients with PDAC suffer from poor prognosis with a median survival rate of 22.1 months with an actual survival rate of 17.0% [7]. PDAC is often diagnosed late, and as a result surgical resection may not be a viable option for many patients [8]. As the cancer progresses and possible treatment options decrease, survival outcomes also significantly worsen. Five-year survival rate for patients diagnosed with late-stage PDAC is less than 10% [8].
PDAC progresses as a complex activation of driver genes and inactivation of tumor suppressor genes [9]. Commonly mutated genes observed in PDAC include KRAS, TP53, CDNK2A, DPC4/SMAD4, and BRCA2. Studies of key mutations in these genes are conducted with the goal of developing targeted gene therapies. One particular mutation, the KRAS G12D mutation, is present in over 40% of PDAC patients [10]. However, this specific mutation has been found to not be significantly associated with overall survival outcomes. The TP53 gene is mutated in about 50% of PDAC patients [11]. These mutations include gain-of-function point mutations and null-mutations as a result of deletions. Mutations of the CDNK2A gene have been found to be significantly associated with poorer survival outcomes for patients with PDAC compared to mutations of KRAS and TP53 [12,13].
Several PDAC vaccines are under development and clinical trials using a variety of immunologic targeting methods [14]. These methods include cell-based, protein-based, microorganism-based, DNA-based, exosome-based, and peptide-based vaccines. Peptide-based vaccines have been growing in popularity due to their ability to be quickly and cheaply developed and for their flexibility in patient populations [15]. For PDAC, the first peptide vaccine to undergo clinical trials was a KRAS-targeting peptide co-administered with GM-CSF to promote a greater immune response [16]. The vaccine successfully induced specific immune response in 58% of patients, contributing to a longer survival time for treated patients. Other peptide vaccines targeting survivin, gastrin, VEGFR-1, VEGFR-2, and WT1 have been ineffective in inducing immune response or contributing to significantly improved survival [14,16,17,18,19,20]. However, the design of personalized based peptide cancer vaccines is completely absent. This study focuses on the development of design protocol to create personalized peptide vaccines with application to PDAC. The protocol identifies genetic variants using RNAseq analysis and designs a personalized peptide vaccine using a vaccine development protocol and omics pipeline previously developed by our group [21,22,23,24,25,26,27,28,29].
2. Materials and Methods
2.1. Patient Genomic Data
We obtained patient genomic data from the Gene Expression Omnibus (GEO) database [26], a publicly accessible repository of comprehensive microarray, next-generation sequencing, and other forms of high-throughput functional genomic data. For this study, we specifically collected raw Illumina sequencing data pertaining to human patient solid tumor samples. These samples were part of a detailed study focused on analyzing long-term heterogeneity in patients with pancreatic ductal adenocarcinoma (PDAC) [27]. This study included genomic data from a cohort of 19 patients, consisting of 10 long-term survivors and 9 short-term survivors, providing a diverse basis for examining genetic variations linked to survival outcomes. For the objectives of this study, we selected one patient classified as a short-term survivor to design personalized vaccines, serving as a proof-of-concept for our approach. This selection was strategic, allowing us to explore the potential of personalized medicine in cases with poorer prognosis and to evaluate the efficacy of targeted therapies based on genomic insights. The design and development of the vaccine were personalized to the unique genetic profile of the chosen patient, focusing on the anomalies most likely to influence tumor behavior and treatment response. To confirm that our personalized vaccine design was rigorous and potentially effective, we compared the targeted genetic components of the vaccine to key genes previously identified as significant in the survival of PDAC patients by Bhardwaj et al. [27]. This comparison enabled us to validate our personalized vaccine design approach and increase the therapeutic relevance of the vaccine design. This proper controlled process of data selection, and comparison with established genetic markers supports our vaccine design methodology detailed further below section.
2.2. RNAseq Analysis of Patient Data
We performed an RNAseq analysis using the Partek Flow genomic analysis suite [28], as shown in Figure 1, which outlines our comprehensive RNAseq workflow to obtain and confirm variant data. Initially, we imported the raw sequence data in fastq format into Partek Flow. This format is widely used for storing the output from high-throughput sequencing instruments and contains both nucleotide sequence data and corresponding quality scores. Following data importation, the first computational step involved trimming the Illumina sequencing adapters. These adapters, which are artificial sequences added during library preparation, can interfere with the analysis if not removed, as they may be misinterpreted as part of the genomic sequence. After trimming, we aligned the reads to a reference genome using the Burrows-Wheeler Aligner (BWA) algorithm. BWA is a software tool that efficiently aligns relatively short sequences (such as those from Illumina sequencers) against a long reference sequence such as a complete genome. This alignment is important for locating the genomic origins of each read and is fundamental to identifying variations from the reference sequence. In the post-alignment, we executed somatic variant calling using the Strelka algorithm, which is specifically designed to detect somatic variants with high sensitivity and accuracy in tumor-normal paired samples. This step was important for identifying potentially significant genetic mutations that could be relevant in the context of disease, herein cancer. To ensure the reliability of our findings, we manually inspected each significant gene variant using the Integrative Genomics Viewer (IGV). IGV is an interactive visualization tool that allows us to visually explore genomic data, thus facilitating the validation of computational predictions through a critical human-oversight step. We excluded gene variants of inadequate quality from further analysis. This quality control step is key to avoid false positives that could skew the results of downstream applications, such as vaccine development. Finally, we focused our efforts on analyzing single nucleotide polymorphisms (SNPs) that hold potential for inclusion in our vaccine development process. SNPs, being the most common type of genetic variation among cancer patients, provide valuable insights into genetic variability which can be exploited to design targeted vaccines.
3. Gene Annotation Confirmation Using GeneFinder Python Program
Development and Application of GeneFinder
After obtaining and processing genomic data through Partek Flow, we advanced to the next step by developing a Python program named ‘GeneFinder’. The code for this innovative tool is comprehensively detailed in Supplementary File S3, and freely available for download. GeneFinder was specifically designed to enhance our analytical capabilities in gene annotation by using both chromosome number and locus information. Using the hg38 reference set accessible via the UCSC Genome Browser [29], GeneFinder systematically identifies corresponding gene names based on their chromosomal location. The tool operates by exploiting web-scraping techniques to extract relevant genomic data directly from the browser. Once the data is retrieved, GeneFinder processes this information to generate a detailed output that includes a table formatted with chromosome numbers, locus details, and the names of associated genes. This functionality not only streamlines the gene identification process but also warrants accuracy by referencing updated genomic data. The application of GeneFinder in our study was twofold. Primarily, it served to externally validate the alignment accuracy and overall reliability of our RNAseq analysis process. By cross-verifying the gene annotations provided by Partek Flow with those extracted by GeneFinder, we could confirm the consistency and validity of our results. Additionally, as shown in Figure 2, we employed a modified version of GeneFinder to specifically extract a list of genes from a given variant file. This adaptation was particularly important for our personalized vaccine as it allowed us to focus on particular genomic variants of interest, facilitating a more targeted approach in our subsequent analyses.
4. Personalized Vaccine Design Protocol
We employed a vaccine design protocol that has been previously outlined in our published studies [21,24,25,26,27,28,29,30]. This protocol integrates cutting-edge bioinformatics tools to predict and select epitopes from mutations identified in genomic data.
4.1. Epitope Prediction and Selection
Initially, we used the IEDB NetMHC 4.1 tool to predict epitopes. NetMHC 4.1 is specifically designed to return potential epitopes along with their predicted binding affinity for the top 27 expressed HLA alleles in the human population. The binding affinity indicated by the IC50 value measured in nanomolar (nM), determines the strength of the interaction between the epitope and the HLA molecules, which is a critical factor in the immune response efficacy.
4.2. Clinical Checkpoint Parameters
Subsequently, we computed several epitope-specific clinical checkpoint parameters. The immunogenicity of each epitope was determined using the IEDB Class I Immunogenicity Tool, which assesses the potential of an epitope to trigger an immune response. The antigenicity, which evaluates the capability of the epitope to be recognized by antibodies, was determined using VaxiJen v2.0.
4.3. Data Filtering and Selection Criteria
With the binding affinity, immunogenicity, and antigenicity data computed for each epitope and its associated HLA allele, we employed stringent filters to select the most promising epitopes. These filters were applied based on the criteria outlined in Table 1, focusing on identifying epitopes that are strong binders, highly immunogenic, and antigenic.
4.4. Physicochemical Property Assessment
In addition to these functional assessments, we analyzed various physicochemical properties of the epitopes using ProtParam. This analysis included determining parameters such as half-life, instability index, isoelectric point, aliphatic index, and GRAVY score. Although these parameters were informative for understanding the physical and chemical characteristics of the epitopes, they were not used in the epitope selection process. Further, we assessed toxicity using ToxinPred and screened for allergenic potential using AllerTOP v2.0, ensuring that only non-toxic and non-allergenic epitopes were considered for further analysis.
4.5. Epitope Selection and Workflow Integration
After applying the filtration restrictions (Table 1), we selected the top 50 and 100 epitopes that met all the specified criteria, warranting a robust selection of candidates for potential vaccine design. We employed binary filters on toxicity and allergenicity to make sure the selection of epitopes that are both non-toxic and non-allergenic.
4.6. Methodological Workflow
Figure 3 shows the comprehensive workflow of our methodology, starting from the collection of Illumina sequencing data, performing RNAseq analysis, and the selection of top epitopes for the development of peptide vaccines. This streamlined workflow integrates multiple stages of data processing and epitope evaluation, indicating the robustness of our approach in vaccine design.
5. Results
We obtained Illumina sequencing data from single patient out of the 19 available in the GEO accession project [27]. The sequencing data represents the genetic landscape of the patient solid tumor sample. We performed RNAseq analysis to determine prevalent mutations. Using these mutations, we determined strong and normal binding MHC class I epitopes that are immunogenic, antigenic, nontoxic, and nonallergenic. We selected the top 50 and top 100 epitopes from this data for a peptide vaccine.
5.1. Determination of Genetic Variants with RNAseq Analysis
We performed RNAseq analysis on Illumina sequencing data to obtain a list of genetic variants identified in a solid PDAC tumor. The RNAseq analysis performed using Partek Flow resulted in 100,819 mutations. These mutations included single nucleotide polymorphisms, multi-nucleotide polymorphisms, deletions, and insertions. Isolating the single nucleotide polymorphisms, we identified 189 unique variants which we could use to develop the peptide vaccine.
5.2. Confirmation of Genetic Variants and Sequencing Alignment Using GeneFinder
We confirmed the alignment of the sequencing data to the hg38 human reference genome using our GeneFinder program. Using GeneFinder, we qualitatively identified the corresponding genes to all 100,819 mutation loci against the hg38 human reference genome. We found 100% similarity between the genes identified through Partek Flow and genes identified using GeneFinder. Therefore, we were confident that the variant genes identified using Partek Flow were correctly aligned to the reference genome.
5.3. Collection of 9-mer and 10-mer Top Epitopes from Genetic Variants
From the pool of identified genetic variants, we curated lists of the top 50 and top 100 epitopes, prioritized based on their binding affinity and immunogenic properties, detailed in Supplementary Files 1-2. All selected epitopes consisted of either 9 or 10 amino acids, representing an epitope capable of binding to an MHC class I molecule. All the top 50 epitopes were classified as having strong binding affinity to their associated HLA allele. The top 100 epitopes included both strong and normal binders. We found no epitopes in the top 100 which were classified as weak binders. Table 2 shows the top 50 epitopes, along with their associated genes, mutations, and binding HLA alleles.
5.4. Population Coverage Analysis of Top 100 Epitopes
We also performed a population coverage analysis to assess the extent of the global population that could potentially benefit from the personalized vaccine. The analysis showed that the vaccine could cover 69.64% of the global population. Table 3 provides this coverage along with average hit rates and PC90 data for various world subregions. While the population coverage may appear relatively low at first glance, it is essential to consider the context of this study. The vaccine was uniquely designed based on the gene expression profile of a specific individual, making it personalized and tailored to the specific mutations and characteristics of their tumor. Consequently, the expectation for widespread coverage across diverse populations is not high. As the patient cohort from whom the vaccine was developed predominantly comprised individuals with European ancestry, the vaccine’s performance in these regional subgroups aligns with the genetic background of the patients involved.
5.5. 3D-Structure Modeling of Epitope-MHC and TCR Interaction Complex
TCR (T-cell receptor) and pMHC (peptide-major histocompatibility complex) interactions play a fundamental role in immunogenicity, which involves the ability of a peptide to initiate an immune response against tumor cells. TCRs on the surface of T cells recognize antigens that are presented by MHC molecules on the surface of antigen-presenting cells. This recognition is specific to the peptide being presented by the MHC. The correct configuration and interaction of a TCR with a pMHC complex is essential for the T cell to become activated and initiate an immune response. Thus, to explore the binding of our designed peptide vaccines, we initiated TCR-pMHC peptide interaction modeling. We found the PDB files for the HLA alleles HLA-B*58:01 on the RCSB protein data bank (https://www.rcsb.org/). Using MDockPeP (https://zougrouptoolkit.missouri.edu/mdockpep/) and CABS-dock [31,32], we attached a top epitope to the binding grooves of the HLA allele. We created two models of the peptide-MHC binding complex (Figure 4). We used TCRModel (https://tcrmodel.ibbr.umd.edu/) to create 3D models of a TCR complex binding to our peptide-MHC complexes. Subsequently, we used PyMOL to edit all of the 3D models. In Figure 4, the color yellow represents HLA alleles and red represents epitopes. The 3D models we obtained were KSFEDIHHY, a mutation of the KRAS gene, binding to the MHC Class I molecule HLA-B*58:01 as well as KTYQGSYGF, a mutation of the TP53 gene, binding to the MHC Class I molecule HLA-B*58:01. All pMHC-TCR 3D molecular model structures generated in this study can be found in Supplementary Files S4–S7.
6. Discussion
We developed a personalized peptide-based vaccine for a patient with pancreatic ductal adenocarcinoma (PDAC). This process began with RNA sequencing (RNAseq) analysis, which enabled the identification of specific genetic mutations driving the development of PDAC in the patient. Based on this analysis, we developed a personalized cancer vaccine using our previously published peptide vaccine development strategy [21,30]. Our approach involved targeting 100 epitopes that were prevalent in the PDAC patient and identified as viable candidates for peptide vaccine design. By focusing on the specific gene targets present in each patient, we intended to improve the specificity of the vaccine, ensuring that it effectively targeted the unique genetic alterations present in the patient’s tumor. This method not only enhances the potential efficacy of the vaccine by adapting it to the individual’s genetic landscape but also minimizes potential off-target effects, thus optimizing the therapeutic outcome.
The final filtered epitopes are predicted to be immunogenic, antigenic, have high or normal binding affinity, and are nontoxic and non-allergenic. The binding affinity restriction used in this study differs from other previous in-silico vaccine design methodologies using the same NetMHC tool [21,30]. Our previous methods of peptide vaccine design used quantitative filters on the percentile rank of the binding affinity value. However, the percentile rank compares the epitopes to a test set of data in IEDB, and therefore is not an accurate nor absolute assessment of binding affinity as necessary for this study. Using the IC50 value instead is a more absolute measure of the binding affinity of the epitopes. We are also able to specify the strength of the binding affinity based on the IC50 value, which provides more qualitative measures for comparison when transitioning to murine studies. By tailoring the vaccine to each patient’s specific genetic makeup, we expect to enhance its effectiveness and improve clinical outcomes. This approach represents a significant step forward in the field of immunotherapy for PDAC, offering a more targeted and personalized treatment option that has the potential to transform the management of this challenging disease.
The top epitopes selected using our novel methodology are all widely recognized in literature as common driver and tumor-suppressor genes in PDAC [9,33,34]. Additionally, these specific epitopes have been identified in trials involving the sequencing of human tumor samples [35,36]. The consistent presence of our top epitopes in both our reference study and other clinical trials of PDAC patients serves as strong validation of our personalized cancer vaccine design methodology. Using RNA sequencing analysis by Partek Flow, along with our peptide cancer vaccine design processes, we created a peptide vaccine derived from the individual’s tumor tissue genetic data. This integrative approach not only emphasizes the relevance of our vaccine targets but also enhances the precision medicine framework by adapting the therapeutic strategy to the genetic individualities of each patient’s tumor. This could potentially lead to improved clinical outcomes by specifically targeting the molecular abnormalities driving the cancer.
Previous studies on the development of peptide vaccines have primarily concentrated on creating generalized vaccines that could be used for a large and broad population [15,37,38,39]. These generalized vaccines target a limited set of gene mutations, to increase sensitivity but often at the expense of specificity. The development of effective global peptide vaccines poses additional challenges. The vast global diversity of HLA alleles complicates the creation of a peptide vaccine that can effectively target a comprehensive population [37]. Each individual’s HLA type influences how well their immune system can recognize and respond to the peptides presented by the vaccine, making it difficult to design a universally effective vaccine. The development of personalized peptide vaccines has historically been limited by the cost and time to produce the peptides [38]. However, implementing a novel design method as described in this study offers a unique and innovative solution to quickly design neoantigen personalized peptide-based vaccines. Recently, with the advent of advanced sequencing technology, neoantigen peptide vaccines are becoming a more viable solution for patients [40]. However, the design process has been complicated with a multitude of software required to design a personalized vaccine. Our methodology using Partek Flow provides a simple and streamlined RNAseq analysis procedure to obtain the list of neoantigens. Our program, GeneFinder, is useful to identify and confirm proper alignment and identification of genes from the RNAseq analysis process. Overall, our methodology employs just two tools throughout the entire design process, significantly simplifying the development of personalized cancer vaccines. This streamlined approach not only reduces the complexity and duration of vaccine design but also enhances the precision with which these vaccines can be personalized to individual genetic profiles.
7. Limitations
While the study presents a promising personalized cancer vaccine strategy targeting neoantigens in pancreatic ductal adenocarcinoma (PDAC) patients, there are several limitations that should be acknowledged. Firstly, the pilot trial size of one patient is relatively small, which could limit the generalizability of the methodology. A larger sample size would provide more robust data and better account for variations and accommodate the heterogeneity inherent in the genetic landscape of PDAC more effectively. While the absence of experimental confirmation may appear as a limitation, the significance of this innovative methodological framework for personalized cancer vaccines, being the first of its kind, corroborates the importance of this work. This framework enables the efficient prioritization of most promising personalized vaccine candidates, thus accelerates the vaccine design process, and enhancing the probability of success in subsequent preclinical and clinical evaluations and also helps to optimize resources by focusing on the candidates for further preclinical studies.
8. Future Directions
We have developed an automation of the peptide vaccine design process using web scraping and API tools [24,25]. Implementation of such a software would further simplify the personalized cancer vaccine process. Furthermore, moving the RNAseq analysis process for a cloud-based solution using Partek Flow to a hardware process using Python or R would allow for complete automation of the personalized vaccine design process. Given such a scaled program and processes, the only limitation to the vaccine design process would be the time to sequence a patient’s tumor tissue.
9. Conclusions
We developed a personalized cancer vaccine targeting specific gene mutations prevalent among PDAC patients by implementing our novel personalized vaccine design workflow. This study addresses the limitations of generalized vaccines and specifically for pancreatic ductal adenocarcinoma (PDAC). By analyzing the genetic alterations driving PDAC in a patient’s tumor tissue, we identified 100 gene mutations as targets for our personalized vaccine strategy. The gene targets were identified and validated using our GeneFinder program, which used the chromosome number and nucleotide position data. By integrating GeneFinder into our workflow, we not only enhanced the precision of our gene annotations but also significantly improved the efficiency of our data analysis process. This development represents a significant step forward in the application of computational tools in personalized vaccine design, providing a robust method for accurate gene identification and the validity of complex genomic analyses.
The top 50 epitopes consisted of only high affinity binding epitopes, indicating the potential efficacy of the vaccine. The use of IC50 values as an absolute measure of binding affinity provided more accurate and quantitative comparisons. To visualize the interactions between epitopes and HLA alleles, 3D models of TCR-peptide-MHC complexes were created. The personalized cancer vaccine developed in this study may hold great promise for PDAC patients. By targeting the unique genetic alterations in each patient’s tumor, this approach offers a more specific and personalized treatment option. Further research is warranted to simplify the variant identification and epitope ranking process.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The author, S.D. (Sivanesan Dakshanamurthy), acknowledges partial support from the Georgetown University Lombardi Comprehensive Cancer Center (LCCC) Cancer Cell Biology Program Pilot Award. Authors participated in the Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Undergraduate Research Program.
References
- Shemesh CS, Hsu JC, Hosseini I, Shen BQ, Rotte A, Twomey P, et al. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol Ther J Am Soc Gene Ther. 2021 Feb 3;29(2):555–70. [CrossRef]
- Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018 Mar 23;359(6382):1355–60. [CrossRef]
- Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023 Jun;618(7963):144–50. [CrossRef]
- Kong X, Cheng D, Xu X, Zhang Y, Li X, Pan W. IFN-γ induces apoptosis in gemcitabine-resistant pancreatic cancer cells. Mol Med Rep. 2024 May;29(5):76.
- Park W, Chawla A, O’Reilly EM. Pancreatic Cancer: A Review. JAMA. 2021 Sep 7;326(9):851–62.
- Anderson EM, Thomassian S, Gong J, Hendifar A, Osipov A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers. 2021 Nov 3;13(21):5510. [CrossRef]
- Strobel O, Lorenz P, Hinz U, Gaida M, König AK, Hank T, et al. Actual Five-year Survival After Upfront Resection for Pancreatic Ductal Adenocarcinoma: Who Beats the Odds? Ann Surg. 2022 May 1;275(5):962–71.
- Tonini V, Zanni M. Pancreatic cancer in 2021: What you need to know to win. World J Gastroenterol. 2021 Sep 21;27(35):5851–89. [CrossRef]
- Hu HF, Ye Z, Qin Y, Xu XW, Yu XJ, Zhuo QF, et al. Mutations in key driver genes of pancreatic cancer: molecularly targeted therapies and other clinical implications. Acta Pharmacol Sin. 2021 Nov;42(11):1725–41. [CrossRef]
- Shen H, Lundy J, Strickland AH, Harris M, Swan M, Desmond C, et al. KRAS G12D Mutation Subtype in Pancreatic Ductal Adenocarcinoma: Does It Influence Prognosis or Stage of Disease at Presentation? Cells. 2022 Oct 10;11(19):3175.
- McCubrey JA, Yang LV, Abrams SL, Steelman LS, Follo MY, Cocco L, et al. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells. 2022 Jul 9;11(14):2155. [CrossRef]
- Sun H, Zhang B, Li H. The Roles of Frequently Mutated Genes of Pancreatic Cancer in Regulation of Tumor Microenvironment. Technol Cancer Res Treat. 2020 May 6;19:1533033820920969. [CrossRef]
- Wartenberg M, Cibin S, Zlobec I, Vassella E, Eppenberger-Castori S, Terracciano L, et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin Cancer Res Off J Am Assoc Cancer Res. 2018 Sep 15;24(18):4444–54.
- Huang X, Zhang G, Tang TY, Gao X, Liang TB. Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine. Mil Med Res. 2022 Oct 13;9:53. [CrossRef]
- Liu W, Tang H, Li L, Wang X, Yu Z, Li J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021 May;54(5):e13025. [CrossRef]
- Gjertsen MK, Bakka A, Breivik J, Saeterdal I, Gedde-Dahl T, Stokke KT, et al. Ex vivo ras peptide vaccination in patients with advanced pancreatic cancer: results of a phase I/II study. Int J Cancer. 1996 Feb 8;65(4):450–3.
- Suzuki N, Hazama S, Ueno T, Matsui H, Shindo Y, Iida M, et al. A Phase I Clinical Trial of Vaccination With KIF20A-derived Peptide in Combination With Gemcitabine For Patients With Advanced Pancreatic Cancer. J Immunother Hagerstown Md 1997. 2014 Jan;37(1):36–42. [CrossRef]
- Suzuki N, Hazama S, Iguchi H, Uesugi K, Tanaka H, Hirakawa K, et al. Phase II clinical trial of peptide cocktail therapy for patients with advanced pancreatic cancer: VENUS-PC study. Cancer Sci. 2017 Jan;108(1):73–80. [CrossRef]
- Nishida S, Ishikawa T, Egawa S, Koido S, Yanagimoto H, Ishii J, et al. Combination Gemcitabine and WT1 Peptide Vaccination Improves Progression-Free Survival in Advanced Pancreatic Ductal Adenocarcinoma: A Phase II Randomized Study. Cancer Immunol Res. 2018 Mar;6(3):320–31.
- Gilliam AD, Broome P, Topuzov EG, Garin AM, Pulay I, Humphreys J, et al. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas. 2012 Apr;41(3):374–9. [CrossRef]
- Kim M, Savsani K, Dakshanamurthy S. A Peptide Vaccine Design Targeting KIT Mutations in Acute Myeloid Leukemia. Pharm Basel Switz. 2023 Jun 27;16(7):932. [CrossRef]
- IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method - PubMed [Internet]. [cited 2024 May 5]. Available from: https://pubmed.ncbi.nlm.nih.gov/36298543/.
- Parn S, Savsani K, Dakshanamurthy S. SARS-CoV-2 Omicron (BA.1 and BA.2) specific novel CD8+ and CD4+ T cell epitopes targeting spike protein. Immunoinformatics Amst Neth. 2022 Dec;8:100020. [CrossRef]
- Samudrala M, Dhaveji S, Savsani K, Dakshanamurthy S. AutoEpiCollect, a Novel Machine Learning-Based GUI Software for Vaccine Design: Application to Pan-Cancer Vaccine Design Targeting PIK3CA Neoantigens. Bioengineering. 2024 Apr;11(4):322. [CrossRef]
- Bautista E, Jung YH, Jaramillo M, Ganesh H, Varma A, Savsani K, et al. AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations. Pharmaceuticals. 2024 Apr;17(4):419. [CrossRef]
- Bass A, Liu Y, Dakshanamurthy S. Single-Cell and Bulk RNASeq Profiling of COVID-19 Patients Reveal Immune and Inflammatory Mechanisms of Infection-Induced Organ Damage. Viruses. 2021 Dec 2;13(12):2418. [CrossRef]
- Policard M, Jain S, Rego S, Dakshanamurthy S. Immune characterization and profiles of SARS-CoV-2 infected patients reveals potential host therapeutic targets and SARS-CoV-2 oncogenesis mechanism. Virus Res. 2021 Aug;301:198464.
- Wathieu H, Issa NT, Fernandez AI, Mohandoss M, Tiek DM, Franke JL, et al. Differential prioritization of therapies to subtypes of triple negative breast cancer using a systems medicine method. Oncotarget. 2017 Nov 3;8(54):92926–42. [CrossRef]
- Issa NT, Kruger J, Wathieu H, Raja R, Byers SW, Dakshanamurthy S. DrugGenEx-Net: a novel computational platform for systems pharmacology and gene expression-based drug repurposing. BMC Bioinformatics. 2016 May 5;17(1):202.
- Savsani K, Jabbour G, Dakshanamurthy S. A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma. Vaccines. 2021 Dec 31;10(1):63. [CrossRef]
- Kurcinski M, Pawel Ciemny M, Oleniecki T, Kuriata A, Badaczewska-Dawid AE, Kolinski A, et al. CABS-dock standalone: a toolbox for flexible protein–peptide docking. Bioinformatics. 2019 Oct 15;35(20):4170–2. [CrossRef]
- Xu X, Yan C, Zou X. MDockPeP: An ab-initio protein-peptide docking server. J Comput Chem. 2018 Oct 30;39(28):2409–13.
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016 Feb 15;30(4):355–85. [CrossRef]
- Saiki Y, Jiang C, Ohmuraya M, Furukawa T. Genetic Mutations of Pancreatic Cancer and Genetically Engineered Mouse Models. Cancers. 2021 Dec 24;14(1):71. [CrossRef]
- Millar DG, Yang SYC, Sayad A, Zhao Q, Nguyen LT, Warner K, et al. Identification of antigenic epitopes recognized by tumor infiltrating lymphocytes in high grade serous ovarian cancer by multi-omics profiling of the auto-antigen repertoire. Cancer Immunol Immunother. 2023;72(7):2375–92. [CrossRef]
- Baleeiro RB, Bouwens CJ, Liu P, Di Gioia C, Dunmall LSC, Nagano A, et al. MHC class II molecules on pancreatic cancer cells indicate a potential for neo-antigen-based immunotherapy. Oncoimmunology. 11(1):2080329.
- Abd-Aziz N, Poh CL. Development of Peptide-Based Vaccines for Cancer. J Oncol. 2022 Mar 15;2022:9749363. [CrossRef]
- Stephens AJ, Burgess-Brown NA, Jiang S. Beyond Just Peptide Antigens: The Complex World of Peptide-Based Cancer Vaccines. Front Immunol [Internet]. 2021 Jun 30 [cited 2024 Mar 24];12. [CrossRef]
- Mizukoshi E, Nakagawa H, Tamai T, Kitahara M, Fushimi K, Nio K, et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat Commun. 2022 Jun 3;13(1):3123. [CrossRef]
- Biswas N, Chakrabarti S, Padul V, Jones LD, Ashili S. Designing neoantigen cancer vaccines, trials, and outcomes. Front Immunol [Internet]. 2023 Feb 9 [cited 2024 Mar 25];14. [CrossRef]
Figure 1.
RNAseq analysis workflow using Partek Flow suite. Created using BioRender.com.
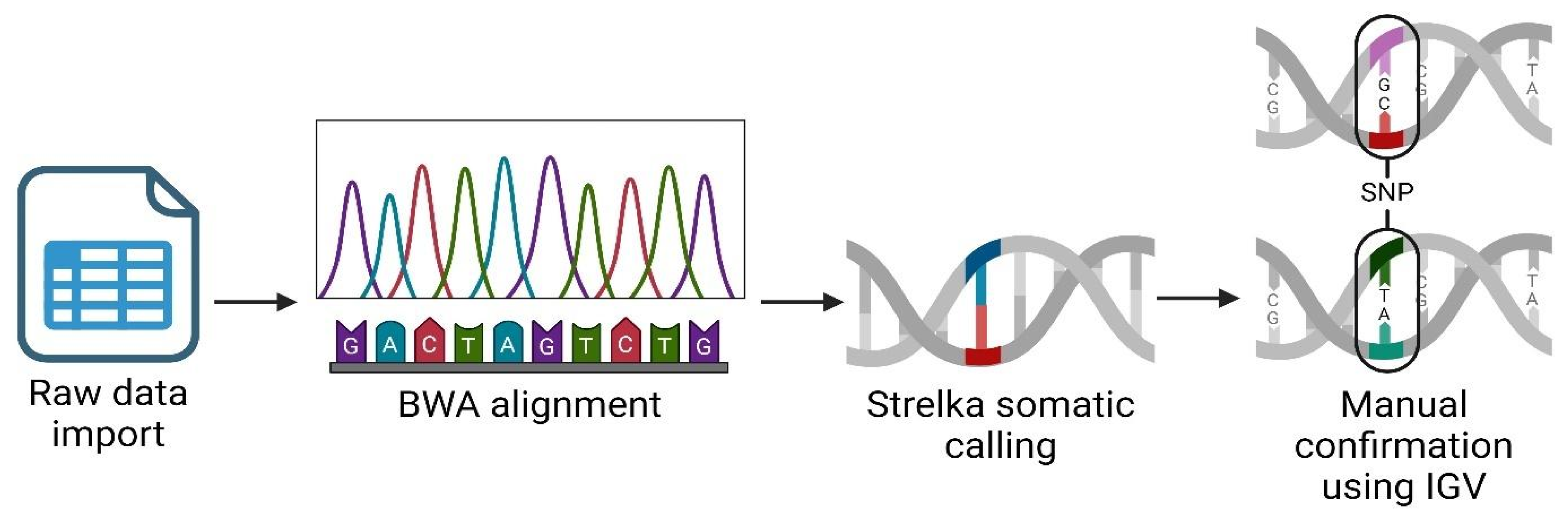
Figure 2.
GeneFinder python program workflow. Created using BioRender.com.
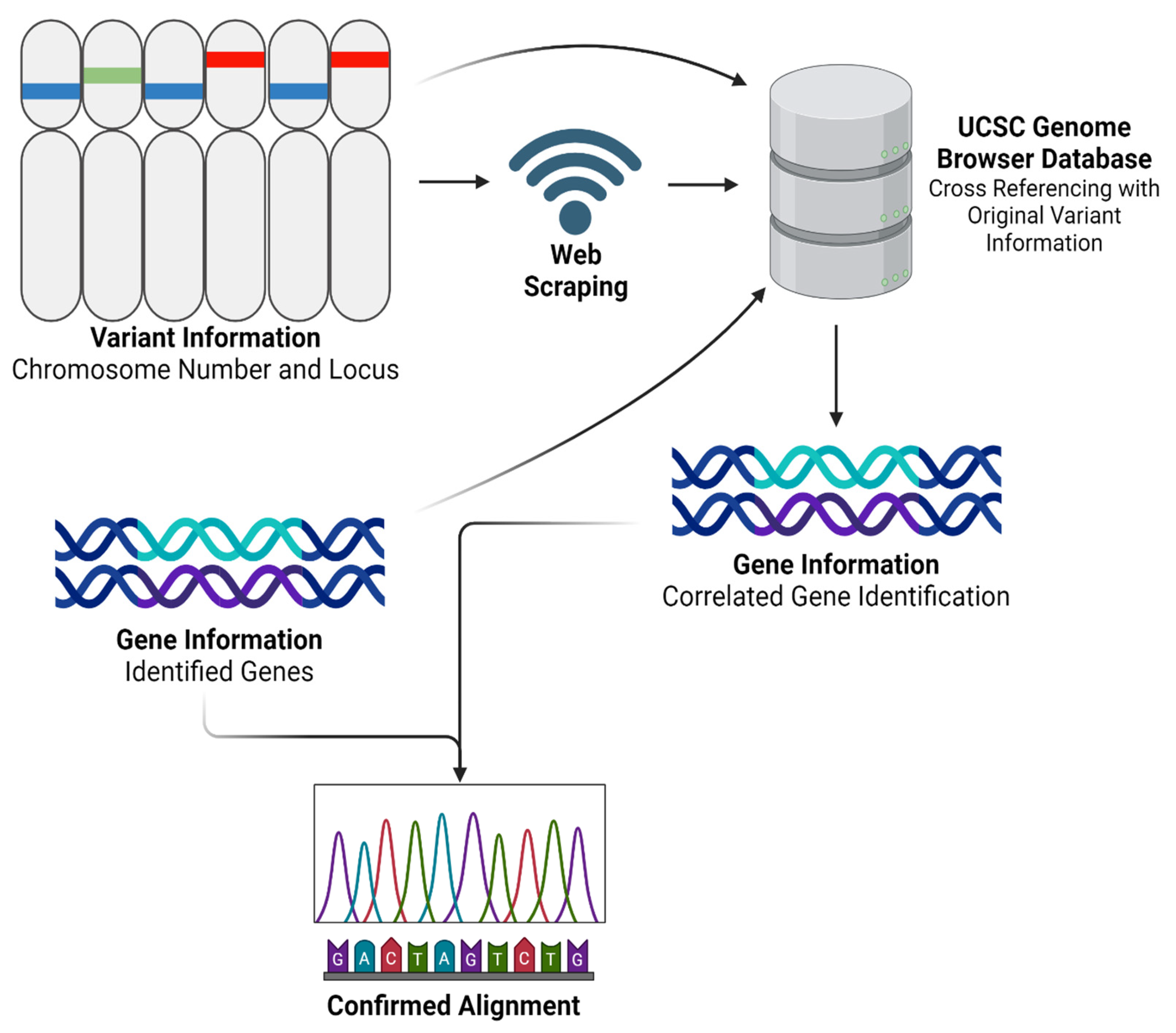
Figure 3.
Overall methodological workflow.

Figure 4.
A. The peptide KSFEDIHHY, a mutation of the KRAS gene, binding to the MHC Class I molecule HLA-B*58:01, B. The peptide, KTYQGSYGF, a mutation of the TP53 gene, binding to the MHC Class I molecule HLA-B*58:01.
Figure 4.
A. The peptide KSFEDIHHY, a mutation of the KRAS gene, binding to the MHC Class I molecule HLA-B*58:01, B. The peptide, KTYQGSYGF, a mutation of the TP53 gene, binding to the MHC Class I molecule HLA-B*58:01.
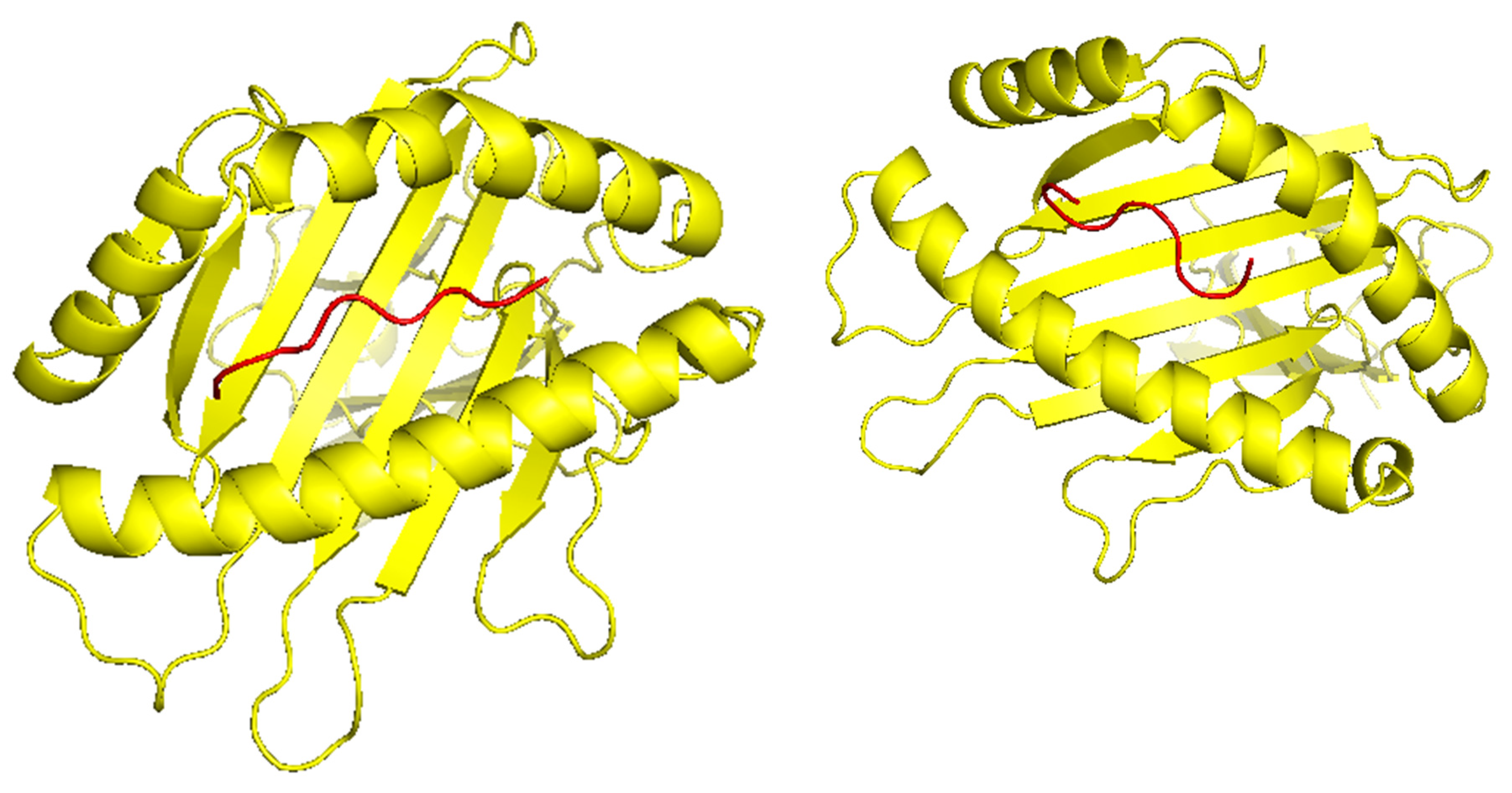
Figure 5.
The peptides KSFEDIHHY and KTYQGSYGF bound to HLA-B*58:01 and their respective TCR complex.
Figure 5.
The peptides KSFEDIHHY and KTYQGSYGF bound to HLA-B*58:01 and their respective TCR complex.
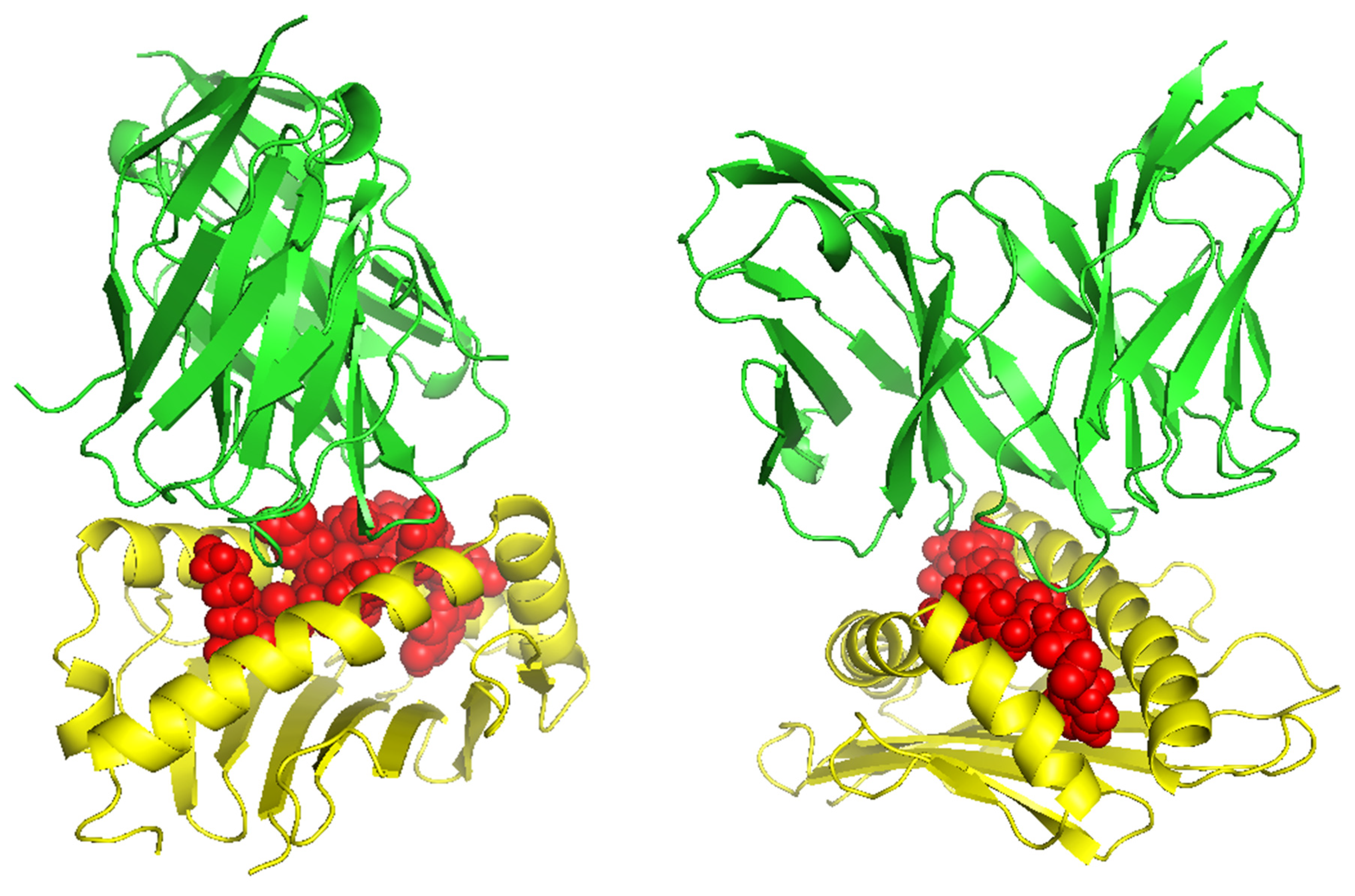

Table 1.
Restriction criteria to quantitatively filter and qualitatively assess each epitope.
| Parameter | Restriction | |
|---|---|---|
| Binding affinity (b) | Strong binder | 0 nM ≤ b ≤ 50 nM |
| Normal binder | 50 nM < b ≤ 500 nM | |
| Weak binder | 500 nM < b ≤ 5000 nM | |
| Immunogenicity (i) | i ≥ 0 | |
| Antigenicity (a) | a ≥ 0.4 | |
| Toxicity | Nontoxic | |
| Allergenicity | Non-allergenic | |
Table 2.
Top 50 epitopes along with their strong-binding associated HLA allele.
| Gene | Mutation | Epitope | HLA Alleles |
|---|---|---|---|
| GNAS | R201C | AMSNLVPPV | HLA-A*02:01 |
| SMAD4 | Y353C | QSIKETPCW | HLA-B*58:01 |
| TP53 | R248Q | CTYSPALNK | HLA-A*03:01 |
| KRAS | G12D | KSFEDIHHY | HLA-B*58:01 |
| SMAD4 | Y353C | MPIADPQPL | HLA-B*39:01 |
| SMAD4 | Y353C | CLSDHAVFV | HLA-A*02:01 |
| SMAD4 | Y353C | KIYPSAYIK | HLA-A*03:01 |
| TP53 | R248Q | LEDSSGNLL | HLA-B*40:01 |
| KRAS | G12D | LARSYGIPF | HLA-B*15:01 |
| TP53 | R248Q | APAAPTPAA | HLA-B*07:02 |
| SMAD4 | Y353C | LLDEVLHTM | HLA-A*02:01 |
| TP53 | R248Q | KTYQGSYGF | HLA-B*58:01 |
| SMAD4 | Y353C | APAISLSAA | HLA-B*07:02 |
| SMAD4 | Y353C | LQSNAPSSM | HLA-B*15:01 |
| TP53 | R248Q | LLGRNSFEV | HLA-A*02:01 |
| KRAS | G12D | KSALTIQLI | HLA-B*58:01 |
| SMAD4 | Y353C | KETPCWIEI | HLA-B*40:01 |
| GNAS | R201C | NQFRVDYIL | HLA-B*39:01 |
| TP53 | R248Q | LQIRGRERF | HLA-B*15:01 |
| SMAD4 | Y353C | LPHHQNGHL | HLA-B*07:02 |
| SMAD4 | Y353C | LQVAGRKGF | HLA-B*15:01 |
| SMAD4 | Y353C | CILRMSFVK | HLA-A*03:01 |
| KRAS | G12D | CLLDILDTA | HLA-A*02:01 |
| SMAD4 | Y353C | LRRLCILRM | HLA-B*27:05 |
| GNAS | R201C | LIDCAQYFL | HLA-A*02:01 |
Table 3.
Population coverage of the personalized PDAC vaccine for regional subgroups.
| Population/Area | Coverage | Average Hit | pc90 |
|---|---|---|---|
| Central Africa | 39.22 | 1.84 | 0.16 |
| Central America | 1.4 | 0.06 | 0.41 |
| East Africa | 41.73 | 2.16 | 0.17 |
| East Asia | 55.26 | 2.8 | 0.22 |
| Europe | 81.05 | 4.98 | 0.53 |
| North Africa | 43.55 | 2.29 | 0.18 |
| North America | 70.36 | 4.09 | 0.34 |
| Northeast Asia | 47.97 | 2.21 | 0.19 |
| Oceania | 38.93 | 1.59 | 0.16 |
| South Africa | 23.99 | 0.93 | 0.13 |
| South America | 36.87 | 1.86 | 0.16 |
| South Asia | 37.28 | 1.66 | 0.16 |
| Southeast Asia | 55.59 | 2.34 | 0.23 |
| Southwest Asia | 43.73 | 2.33 | 0.18 |
| West Africa | 42.65 | 2.14 | 0.17 |
| West Indies | 63.52 | 3.47 | 0.27 |
| Average | 45.19 | 2.3 | 0.23 |
| Standard deviation | 17.8 | 1.13 | 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



