
Submitted:
22 July 2024
Posted:
23 July 2024
You are already at the latest version
Abstract
Traditional photodynamic therapy (PDT) is often limited in its efficacy by insufficient light penetration, inefficient photosensitizer energy transfer, and inadequate target tissue oxygenation. X-ray activated PDT (XPDT), using nanoparticle (NPs) photosensitizers comprised of europium-doped yttrium oxide cores encased within silica shells (Y2O3:Eu@SiO2), offers the potential of overcoming most of these limitations. Aimed at maximizing the generation of reactive oxygen species (ROS), this study reports methodologies for investigation of the effects of synthesis conditions on the characteristics of Y2O3:Eu@SiO2 NP. Transmission electronic microscope (TEM) revealed that the NP core diameter was linearly, positively correlated with condensation time (R2 = 0.7357, p < 0.05) and linearly, negatively correlated with urea concentration (R2 = 0.958, p < 0.05). The addition of cetyltrimethylammonium bromide (CTAB) during core synthesis demonstrated enhanced particle dispersion. Silica coating via APTES-1 and TEOS-3 conditions resulted in Y2O3:Eu@SiO2 NPs that generated the highest ROS, with 8.7-fold and 8.9-fold increase respectively in relative fluorescence intensity at 8 Gy irradiation compared to a 5.2-fold increase in PBS under the same condition. In vivo evaluation using 18F-fluorothymidine positron emission tomography (18F-FLT PET) of mouse xenografts of a human prototypical cancer (ovarian) revealed a 55% reduction in tumor proliferation of radiation-sensitive CAOV3 tumors treated with Y2O3:Eu@SiO2 NPs compared to a 19% reduction in tumor proliferation of radiation-resistant SKOV3 tumors on the fourth day post-treatment. Taken together, these findings suggest Y2O3:Eu@SiO2 NPs can serve as effective photosensitizers for XPDT and their performance characteristics can be optimized by selecting synthesis conditions and parameters to deliver the desirable therapeutic effects, offering novel approaches to circumventing many of the limitations of conventional PDT in the treatment of cancer.
Keywords:
Cancer
; Y2O3:Eu@SiO2 Nanoparticles
; Photodynamic Therapy
; Photosensitizer
1. Introduction
Photodynamic therapy (PDT) is a cancer treatment modality based on the combination of three factors: photosensitizer (PS), light with a specific wavelength, and the presence of molecular oxygen to drive the production of highly cytotoxic reactive oxygen species (ROS) [1]. While conventional PDT shows promise in the effective treatment of cancers, it is hindered by the limited photon penetration in tissues, suppressed efficiency of PS due to multiple energy transfers, and insufficient oxygen in tumor microenvironments. These limitations generally restrict PDT to only superficial tissues and those that can be readily accessed either endoscopically or laparoscopically, which are due to the intrinsic scattering and absorption of photons by intervening tissues [2].
X-ray induced photodynamic therapy (XPDT) is a nascent therapeutic aproach designed to impart precisely targeted, relatively low-dose radiation directly to the tumor microenvironment using x-ray activated photosensitizers to generate ROS for deep-tissue cancer therapy [3]. For a molecule to function as an X-ray activated PS the excitation energy is first absorbed by the PS lattice. The electrons of the PS are then promoted across the band gap into the conduction band which creates holes in the valence band. The observed ROS generation is a result from electrons in the conduction band and holes in the valence band exhibiting high reducing and oxidizing power, respectively [4].
In our published review article, a list of x-ray PDT nanpplatforms developed and evaluated in recent years has been compared and discussed in detail [5]. We choose to study europium-doped yttrium oxide (Y2O3:Eu) nanoparticle (NPs) as nano-PS because our previous research showed a promise for it to be developed for clinical applications [6]. The use of Y2O3:Eu as a scintillator for medical imaging, radiation detection, and dosimetry has been extensively studied [7,8,9,10]. Indeed, Y2O3:Eu NPs have demonstrated to be promising materials for in situ, in vivo x-ray dosimetry [11]. Studies from ours and others have also shown that silica-coated Y2O3:Eu (Y2O3:Eu@SiO2) NPs act as independent nano-PS for XPDT by generating an increased amount of ROS under X-ray irradiation, leading to cancer cell death [6].
While individual studies have been conducted to study the efficacy of Y2O3:Eu@SiO2 NP, the outcomes for cytotoxicity and tumor suppression have been found to vary significantly [12,13,14]. We speculated that these variations in outcome arise from correspondingly different levels of ROS generation as a consequence of subtle inconsistencies in chemistry and methdology during Y2O3:Eu@SiO2 NP synthesis. However, little is known as to how such synthesis variations influence ROS generation. This study focuses on characterizaing and mitigating the impact such variations in synthesis have on the morphology of Y2O3:Eu@SiO2 NP and their ability to produce ROS upon x-ray activation, with in vivo assessment of outcome using a murine xenograft model of a prototypical human malignancy – ovarian cancer. Ovarian cancer was selected for in vivo assessment of Y2O3:Eu@SiO2 NP XPDT due to the cancer radiation sensistive model (CAOV3) and radiation resistant model (SKOV3) of human ovarian cancer.
The ovarian cancer model was chosen because ovarian cancer is the sixth most common cancer worldwide among women in developed countries and the most lethal of all gynecological malignancies. Despite aggressive surgery, chemotherapy, and radiotherapy the prognosis is poor, with a five-year survival rate of less than 30% [15]. The poor prognosis is evident by a recurrence rate in almost 25% of cases with early-stage disease and in more than 80% with more advanced stages [16,17,18] due to the resistance to the standard therapies. Through this study, we establish that Y2O3:Eu@SiO2 NP could improve the radiotherapy efficacy by means of elevated ROS generation, thus demonstrating potential application of Y2O3:Eu@SiO2 NP in XPDT to overcome radiation resistance in treating ovarian cancer.
We hypothesize that the strategy of XPDT combining Y2O3:Eu@SiO2 NP as a nano-PS and low dose X-ray will provide a new modality in combating deep tissue cancer for an improved and efficient treatment outcome. To accomplish this, we have developed methodologies for synthesizing, modifying, refining, and characterizing Y2O3:Eu@SiO2 NP capable of generating extensive levels of ROS under X-ray activation.
2. Materials and Methods
2.1. Chemicals
Y(NO3)3 (99.9%), urea (99%), dihydroethidium (DHE), dimethyl sulfoxide (DMSO, 99.7%), tetraethyl orthosilicate (TEOS, 98%), toluene (99.5%), hexane (98.5%), ethanol (EtOH, 100%), and phosphate buffer solution (PBS, pH 7.4) were purchased from Thermo Scientific (Waltham, MA, USA). Eu(NO3)3 (99.9%), cetyltrimethylammonium bromide (CTAB, 99%), ammonium hydroxide (NH4OH), and (3-aminopropyl)triethoxysilane (APTES, 99%) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Syntheses of Y2O3:Eu Core Nanoparticles
The protocols to synthesize Y2O3:Eu core NP are adapted from Chuang et al. [6]. Two variations of the core synthesis protocols are specified and detailed in Figure 1. For Protocol 1, chemicals were mixed in a 250 mL round-bottomed flask, stirred at 775 rpm for five minutes at room temperature and then heated to 85°C with continuous stirring for a specified condensation time. To understand the factors that affect the core size, Y2O3:Eu core NPs were first produced with the various urea concentrations (1.5, 2, 2.5, and 3 M) in the reaction with the condensation time at two hours. Then the condensation time was varied (1, 2, 3, 4 hours) with urea concentration at 2 M.
In Protocol 2, CTAB was introduced in the reaction mixture. The reaction mixture of Y(NO3)3, Eu(NO3)3, and CTAB was heated at 100°C for one hour under vigorous stirring. Urea (4 M) was simultaneously heated at 40°C for one hour. The two mixtures were then combined and heated at 100°C for one hour while stirring vigorously.
At the end of the reactions, the white precipitate was collected through centrifugation at 15,000 g for 15 minutes (Sorvall ST8, Thermo Fisher, Waltham, MA, USA) and then washed with 15 mL double-distilled water (ddH2O) three times, followed by one wash with 15 mL ethanol. The resulting Y2O3:Eu core NPs were dried overnight at 80°C.
2.3. Silica Encapsulation of Y2O3:Eu Core Nanoparticles
Five variations of the protocols were tested for the silica encapsulation of the Y2O3:Eu core NPs as depicted in Figure 2. All of the Y2O3:Eu core NPs produced without CTAB were encapsulated according to the Variation 5 protocol only, while the Y2O3:Eu core NPs produced with CTAB were encapsulated with all 5 variations. At the end of reaction, the encapsulated Y2O3:Eu@SiO2 NPs were collected through centrifugation and washed three times with 15 mL of a 1:1 solution of ethanol and ddH2O. The obtained particles were then dried at 80°C overnight.
2.4. Calcination of the NP
Dried NPs were calcinated at 800°C for three hours. Calcination was performed either before and/or after the silica encapsulation. For the protocol when CTAB was present in the reaction, the calcination was done at 600°C for 4 hours prior to silica encapsulation. The final Y2O3:Eu@SiO2 NPs were stored at 4°C for further studies.
2.5. Transmission Electron Microscopy Measurement
Particle morphology of Y2O3:Eu@SiO2 NPs was characterized under transmission electron microscopy (TEM) (FEI, Hillsboro, Oregon, USA) at the Advanced Electron Microscopy Facility of the University of Chicago. Y2O3:Eu@SiO2 NPs were first suspended in 100% ethanol (20 mg/mL), sonicated for 15 minutes, diluted 10 times in 100% ethanol, and sonicated for another 15 minutes – to facilitate dispersion. A final additional dilution of 10 times with 100% ethanol and sonication for 30 minutes was done before the Y2O3:Eu@SiO2 NPs were deposited onto grids and imaged. Y2O3:Eu NP core diameters and SiO2 coating thicknesses were determined using the FIJI imaging analysis software.
2.6. Measurement of ROS
ROS measurements were performed as described in Chuang et al. [6] with the following modifications. Briefly, Y2O3:Eu@SiO2 NPs were suspended in phosphate buffered solution (PBS) (10 mg/mL) and sonicated for 15 minutes at 80 Hz and 100% power (FB11205 Ultrasonic Cleaner, Thermo Fisher, Waltham, MA, USA). To measure ROS production, the Y2O3:Eu@SiO2 NP suspension (125 μg/mL) was mixed with DHE (10 μM) and irradiated at 1, 2, 4, and 8 Gy using a Phillips RT250 irradiator (Amsterdam, Netherland) followed by centrifugation for 5 minutes at 15,000 g. Supernatants (150 μL) were transferred to a black 96-well plate. The fluorescence was measured at a plate reader (Biotek, Winooski, VT, USA) with excitation/emission 480/576 nm. The results were expressed as the ratio (I/I0) of the fluorescent intensity between the irradiated (I) and non-irradiated (I0) samples. PBS without NPs was run parallel with Y2O3:Eu@SiO2 NPs as negative control. The reaction mixtures were kept in the dark at all times.
2.7. Cell Culture
Human ovarian cancer cell lines, SKOV3 and CAOV3, were purchased from American Type Culture Collection (ATCC, Manassas, Virginia). SKOV3 and CAOV3 cells were cultured in McCoy’s 5A (Thermo Fisher, Waltham, MA, USA) and DMEM (Thermo Fisher, Waltham, MA, USA) media supplemented with 10% fetal bovine serum respectively. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2.
2.8. Tumor Xenograft
All animal studies were performed in accordance with the guide for the care and use of laboratory animals from the Institutional Animal Care and Use Committee (IACUC) of National Health Research Institutes and approved by the IACUC at the University of Chicago. In vivo experiments were conducted using five to six-week-old Nu/Nu Athymic female mice (Envigo, Indianapolis, IN). Two million SKOV3 or CAOV3 cells were injected subcutaneously in the right hind leg. Tumors were monitored twice a week by caliper measurement and allowed to reach 250-400 mm3 in volume before the treatment of Y2O3:Eu@SiO2 NPs.
2.9. Treatments of Y2O3:Eu@SiO2 NP and Radiation
The Y2O3:Eu@SiO2 NPs were first conjugated with trimethyl-ammonia (TA) to improve solubility of the NPs before inoculation into the tumors as outlined in Figure 3A. The mice with CAOV3 tumors (n=5/group) were assigned to nanoparticle-free sham (CAOV3), and nanoparticle-treatment (CAOV3-Y2O3:Eu@SiO2) groups randomly. The treatment group received intra-tumoral injection of Y2O3:Eu@SiO2-TA NPs (16 mg/kg BW) one hour prior to radiation therapy while the sham tumors received same volume of saline intra-tumoral injection. Corresponding cohorts of animals (n=5), possessing SKOV3 tumors, served as controls. Image guided tumor irradiation was performed on all mice on an XRAD225Cx (Precision X-ray, North Branford, CT). Mice were irradiated with 2 Gy/day for 4 consecutive days.
2.10. 18F-FLT PET Imaging
To determine tumor therapeutic response, 18F-fluorothymidine (tumor cell proliferation radiotracer) positron emission tomography (18F-FLT PET) imaging was conducted at baseline and 1-, 4-, 7-, and 14-days post NP and radiation treatment (Figure 3B). Mice received ~150 μCi (5.55 MBq) of 18F-FLT (Cyclotron Facility, University of Chicago, Chicago, IL, USA) in 100 μL isotonic saline solution by tail vein injection. For each time point, CT images were acquired on the X-Cube preclinical microCT imaging system, (Molecubes, Gent, Belgium). CT images were reconstructed with a 200 μm isotropic voxel size and used for PET attenuation correction and anatomical reference. One hour after 18F-FLT injection, 20 minutes static PET scans were acquired on the β-Cube preclinical microPET imaging system (Molecubes, Gent, Belgium) with 133 mm × 72 mm field of view (FOV) and an average spatial resolution of 1.1 mm at the center of FOV. List-mode data were reconstructed using a three-dimensional OSEM algorithm with a 400 μm isotropic voxel size. Animals were maintained under ~2% isoflurane anesthesia in oxygen during imaging procedures. Respiratory rate and body temperature were constantly monitored and maintained using Molecubes on-board physiological monitoring interface.
2.11. Image analysis
All image analysis was performed with VivoQuant software (Invicro, LLC, Boston, MA, USA). 3D regions of interest (ROIs) were drawn and standardized uptake values (SUVs) and percent injected dose per gram of tissue (%ID/g) were calculated for each ROI and time point. SUV is defined as:
%ID/g was calculated as a ratio of tissue radioactivity concentration (mCi/g) at time of scan to total injected activity (mCi) at time of scan.
2.12. Statistics
Statistical analyses were performed using Prism software (GraphPad, La Jolla, CA, USA). Average data were presented as means ± SD or means ± SEM as specified in the results. Differences between groups were determined by one-way or two-way ANOVA using the Bonferroni post hoc test. The correlation analyses were done using simple linear regression. P < 0.05 was considered statistically significant.
3. Results
To understand the impact of synthesis conditions on Y2O3:Eu@SiO2 NP morphology, TEM imaging was performed on final NPs after the SiO2 encapsulation is completed. Y2O3:Eu@SiO2 NP core diameters and silica coat thickness were measured using FIJI image analysis software on the TEM images. Similarly, the impact that variations in NP synthesis conditions have on ROS generation were quantified using DHE fluorescent probe. Finally, the efficacy of X-ray activated Y2O3:Eu@SiO2 NPs were evaluated in vivo using human ovarian tumor xenograft tumor model in mice. Results are presented in the following sections.
3.1. Effects of Synthesis Condition on Size of Y2O3:Eu@SiO2 Core
As shown in Figure 4A, under the fixed urea concentration of 2 M, a significant direct linear correlation (R2 = 0.7357, p < 0.05) was observed between condensation time of the Y2O3:Eu@SiO2 NP synthesis reaction and the diameter of the resulting NP cores. On average, 98±3 (mean ± SEM) samples of Y2O3:Eu@SiO2 NP cores were measured. NP cores increased in size from 36±6 nm (mean ± SD) for 1 hour of condensation to 205±24 nm (mean ± SD) for four hours of condensation. In contrast, Figure 4B shows a significant linear negative correlation (R2 = 0.958, p < 0.05) between urea concentrations in the synthesis solution and the resulting NP core diameters with a fixed condensation time of four hours. Core diameter decreased substantially in size from 176±13 nm (means ± SD) for 1.5 M urea to 75±6 nm (means ± SD) for 3.5 M urea.
3.2. Effects of Silica Coating Conditions and Calcination on Thickness of Y2O3:Eu@SiO2 Shell
As shown in Figure 5, examination of the silica coating thickness revealed notable differences between Y2O3:Eu@SiO2 NP encapsulation conditions with different solvents during silica deposition. NP made using toluene and hexanes as the solvent consistently had silica shells of approximately 4±1 nm(mean ± SEM) thickness. In comparison, particles made using ethanol as the solvent without hexanes demonstrated thicker silica shells averaging 9±1 nm (mean ± SEM) thickness. In addition, as Figure 5 illustrates, the timing of calcination influenced the silica deposition on to the surface of the Y2O3:Eu core. Both calcination before (CB) the SiO2 encapsulation and calcination before and after (CBCA) the SiO2 encapsulation, produced thicker silica shell than calcination after (CA) SiO2 encapsulation. No differences in shell thickness were observed between the calcination timing of CB and CBCA.
3.3. Effects of Synthesis Conditions on the Dispersibility of Y2O3:Eu@SiO2 NP
A representative TEM image from each synthesis condition is displayed in Figure 6. The cores of Y2O3:Eu@SiO2 NP in CB, CA and CBCA groups were synthesized without CTAB, while the other groups included CTAB in the reaction mixture. In these images, the Y2O3:Eu@SiO2 NP in CB, CA and CBCA group (without CTAB) appear to be clustered while Y2O3:Eu@SiO2 NP in the other groups (with CTAB) appear evenly distributed, suggesting that the presence of CTAB in the NP core sythesis improves the dispersity of the NP. Figure 6 displays an representative TEM image of NPs produced with TOES-3 protocol at higher magnification than those images in Figure 6, where the SiO2 shell and the Y2O3:Eu cord are visualized clearly. Corresponding distributions of Y2O3:Eu core diameter are presented as insets in Figure 6.
3.4. Effects of Synthesis Condition on the ROS Generation
The x-ray induced ROS production was quatified by the DHE (superoxide indicator) fluorescent probe. The higher relative fluorescent ratio indicates the higher levels of ROS production. In Figure 7, all Y2O3:Eu@SiO2 NPs generated progressively higher ROS with increasing radiation dose from 0 to 8 Gy. However, the extent of ROS production varied between NPs. Figure 7A shows the ROS generation by different silica coating protocols on the Y2O3:Eu@SiO2 NP produced with CTAB addition. Under similar coating conditions, using APTES as silica source, enhanced x-ray induced ROS generation (APTES-1 vs TEOS-1 and APTES-2 vs TEOS-2). Higher ROS generation was also observed from NPs with addition of the silica at the beginning of the reaction (APTES-1 vs APTES2 and TEOS-1 vs TEOS-2). Overall, TEOS-3 and APTES-1 demonstrated the highest relative fluorescence intensity that is statistically significant while TEOS-1 and APTES-2 showed modestly elevated ROS over the PBS control that was not statistically significant, and TEOS-2’s ROS generation is similar to that of PBS (Figure 7A.) ROS generation for Y2O3:Eu@SiO2 NPs synthesized with different calcination protocols is shown in Figure 7B. CA showed a small increase in ROS compared to PBS control at 8 Gy but not at other radiation doses with no statiscal difference overall. The ROS generation of CB and CBCA is similar to that of PBS. Figure 7C highlights the significantly greater ROS production of TEOS-3 and APTES-1 than Y2O3:Eu@SiO2 NP produced with other formulations or coating protocols at both 4 Gy and 8 Gy radiation doses. No correlation was found between the ROS production and the size of Y2O3:Eu@SiO2 NP core, nor the silica coat thickness (data not shown).
3.5. In vivo Evaluation of the Y2O3:Eu@SiO2 NP in a Human Ovarian Cancer Xenograft Model
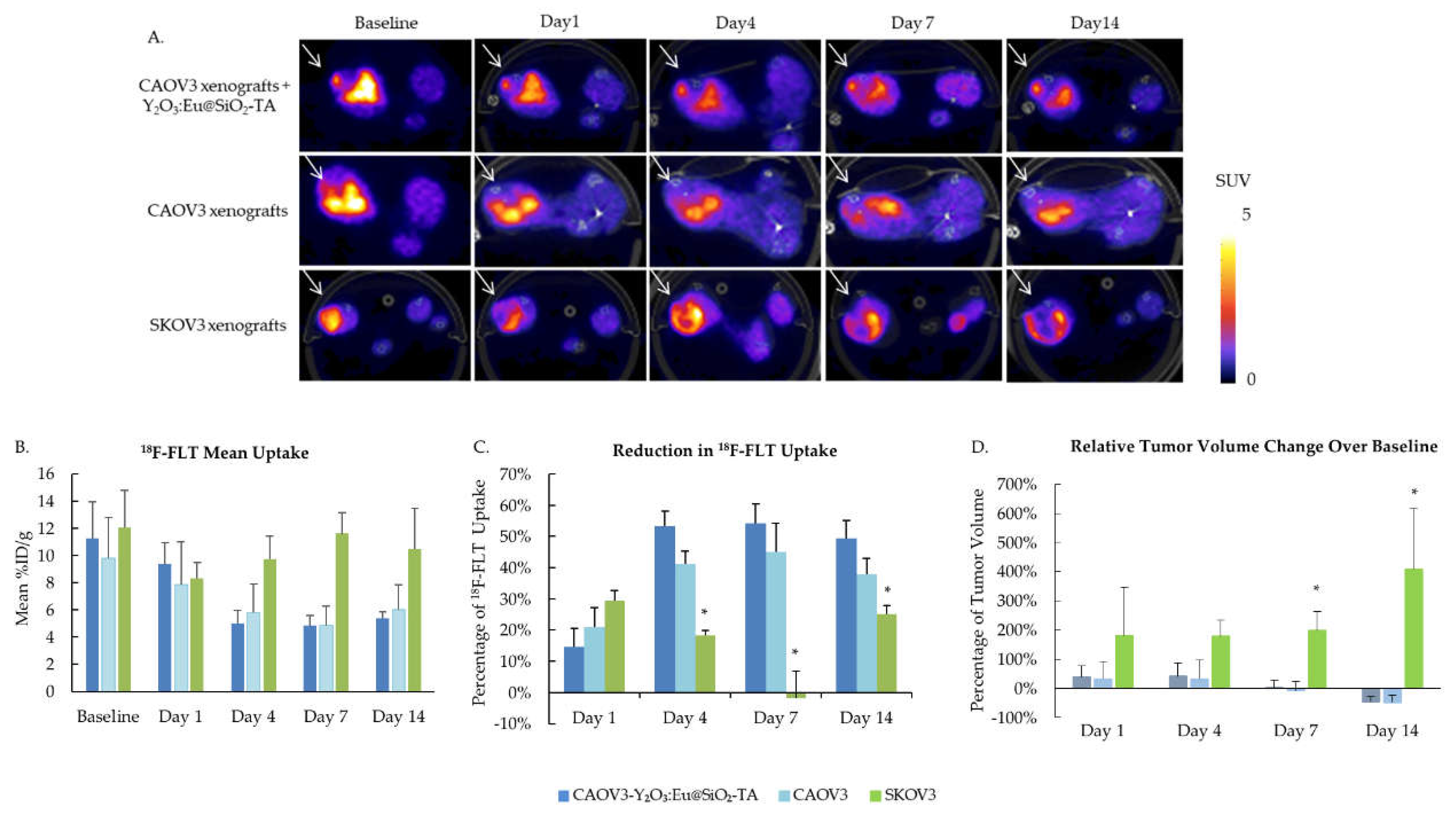
To evaluate the efficacy of Y2O3:Eu@SiO2 NP XPDT, tumor cell proliferation was evaluated before XPDT treatment and up to 14 days after XPDT treatment by PET imaging using 18F-FLT. Figure 8A shows representative axial sections of SUV scaled PET images at each time point for the three animal groups. 18F-FLT uptake decreased for both sham and Y2O3:Eu@SiO2 NP CAOV3 tumors from Day 1 post treatment and remained suppressed. Radiation resistant SKOV3 xenografts showed a similar but temporary response at Day 1 and overall uptake was not significantly different across the 14 days (Figure 8B). However, when comparing both radiation sensitive CAOV3 groups to the SKOV3, the decrease in uptake at Day 4, 7, and 14 was significantly different, measuring about 55% for the CAOV3 xenografts treated with NPs, about 40% for the sham CAOV3, and 19%, 3% and 13% at Day 4, 7, and 14 respectively for SKOV3 (Figure 8C). Tumor volume was monitored by caliper measurement, and changes in volume relative to baseline show significant growth in SKOV3 xenograft, with over 400% increase over 14 days, while CAOV3 tumors grew at a slower rate, reaching about 40% increase at Day 4, before reducing in size, measuring about 45% lower volume at Day 14 (Figure 8D).
4. Discussion
4.1. Assessment of Synthesis Protocols and the Relevant Conditions and Factors Affecting the Characteristics of Y2O3:Eu@SiO2 NPs
Several approaches are available for yttrium oxide syntheses [19]. Wet chemistry method using urea-assistend deposition precipitation followed by calcination provides many advantages and has emerged as a widely applied trend in the generation of metallic NPs [20]. We adapted this techinque to produce Y2O3:Eu@SiO2 NPs previously and showed effectiveness in the generation of ROS, increased cytotoxicity, and enhanced tumor suppression in a mouse model of ovarian cancer xenografts [6]. In the current study, we sough to evaluate a variety of factors in this procedure that may likely influence the therapeutic efficacy of Y2O3:Eu@SiO2 NP based XPDT. We found that the size of NP core is significantly inversely correlated with the urea concentration and positively related to condensation times (Figure 1). The phenomenon observed between urea concentration and particle size may be due to the use of urea as a precipitation agent and its impact in controlling the nucleation and growth of the nanoparticles. Similar to our finding, Sung et al. and Yao et al. found that increased urea concentration is related to decreased sizes of Y2O3:Tb3+ NP and gold NP (AUNP) respectively [21,22]. However, the change of Y2O3 size associated with urea concentration was not observed by Abdalkreem and Kroon [23]. The machanisms behind the effects of urea concentration on the size of NP may be due to its ability to change the pH of reaction solution gradually during the heating period [20]. The decomposition via hydrolysis of urea under heat produces ammonium ions and hydroxide ions leading to an increase in pH of the solution [24]. More study is warranted to clarify the specific condition that urea concentration affects the size of metallic NP and its machenisms.
We also tested a synthesis condition in which CTAB was incorporated for the Y2O3:Eu@SiO2 NP production. CTAB is a cationic surfactant that is often used in the synthesis of nanoparticles. CTAB forms a bilayer on the surface of the nanoparticles, with the hydrophobic tails facing inward and the hydrophilic heads facing outward, which helps to disperse and to stabilize the nanoparticles in aqueous solutions [25]. Our results from TEM imaging analyses show Y2O3:Eu@SiO2 NP syntheses with CTAB in the reaction dispersed better than those without CTAB, which correlate well with the property of CTAB. Future study will investigate whether incorporation of CTAB will lead to cytotoxicity of normal cells.
In addition to characterizing the synthesis conditions of Y2O3:Eu@SiO2 NP core formation, we also compared the effects of five different silica coating protocols for the thickness of encapsulation. The coating condition with ethanol solvent yielded thicker silica coats than four other conditions using toluene and hexane as solvents. This observation implies that solvent polarity impacts the deposition of the silica coating with solvents like toluene and hexanes appearing to restrict silica deposition compared to polar solvents like ethanol. Interestingly, the use of either TEOS or APTES as a silica source did not impact the thickness of silica encapsulation. Calcination timing, however, did affect silica deposition. Calcination prior to silica coating facilitated silica deposition. A second calcination after silica encapsulation, had no further effect on the coating thickness. Calcination after silica coating presented with thinner silica shell.
Silica is believed to be one of the ideal coatings to preserve the quantum property of nanoparticles [26]. However, conflicting results have been reported. In 2007, Liu et al. [27] showed that a silica coating increased Y2O3:Eu luminescent intensity by potentially decreasing the surface defects of NP. In contrast, in 2019 Ansari et al. [28] reported the luminescent efficiency decreased with the increase of the thickness of the silica coating. We found no correlation between the X-ray induced ROS generation and thickness of the silica coating, or the size of the Y2O3:Eu@SiO2 NP core. Instead, under similar reaction conditions, NPs coated via APTES have enhanced the ROS generation then using TEOS as a silica source, especially when it is added at the beginning of the silica deposition reaction. Although this study did not show any significant impact of silica shell thickness on X-ray induced ROS generation, the shell thickness directly affects nanoparticle morphology and could adversely affect the ligation of functional groups such as antibodies specifically targeted to tumor surface protein if too thin or nonuniform. Further studies to understand the relationship of silica coating and functional group conjugation are warranted. Overall, our study concluded that Y2O3:Eu@SiO2 NP synthesized with CTAB addition (Figure 1, Protocol 2) and silica coating using either APTES, toluene and hexane, or TEOS and NH4OH (Figure 2, Variation 1 and 5) generated the greatest levels of ROS. Future studies will include analyses of the in vivo efficacy of tumor therapy using these NPs synthesized through the select protocols indicated above – to investigate and ultimately establish quantitative correlation between therapeutic outcomes and ROS generation levels, and in turn, the optimal choice of Y2O3:Eu@SiO2 NP synthesis protocol and related parametric options. As such, these nanoplatforms could be tailored to the specific therapeutic requirements of individual patients, paving the way to “personalized” XPDT.
4.2. In vivo Evaluation of the Y2O3:Eu@SiO2 NP in a Human Ovarian Cancer Xenograft Model
The in vivo evaluation of Y2O3:Eu@SiO2 NPs using a murine xeonograft model of human ovarian cancer demonstrated significant tumor suppression capabilities of our nanoplatform (Figure 8). These preliminary results suggest the potential of the engineered Y2O3:Eu@SiO2 NPs to suppress both radiation sensitive and radiation resistant tumor progression in vivo via ROS production. It is important to note, however, that this preliminary evaluation study was conducted prior to our systematic investigation of the impact that variations in NPs synthesis protocol can have upon ROS generation and thus were not optimized. Hence, the therapeutic effects demonstrated in this preliminary in vivo study were also not optimized. As discussed above, systematic investigation and correlation of optimized synthesis protocols and treatment methodologies with therapeutic outcomes are planned for the future,and will include in vivo experiments employing humanized animal models to include host immune response and aid in the as translation of these tools to clinical trials.
5. Conclusion
Our studies and results reported here provide important information on the syntheses of Y2O3:Eu@SiO2 NPs for XPDT, a promising approach for the treatment of deep-seated, hypoxic, or radiation-resistant tumors. This nano-photosensitizer has also been demonstrated in preliminary in vivo evaluation to offer significant promise for overcoming limitations of conventional PDT and potentially capable of combating ovarian cancer in clinical applications. The significance of this study lies in its contribution to the understanding of the relationship between nanoparticle synthesis, reactive oxygen species generation, and therapeutic efficacy, which is crucial for the design of personalized treatment strategies. The implications of this research extend well beyond ovarian cancer, as the principles and methodologies established here are not cancer-type-specific. But conjugation of ligands targeted to specific cancer surface molecule, this nano-photosensitizer platform has the potential to be applied to a wide range of malignancies. This work represents an important step towards the clinical translation of X-ray activated photodynamic therapy, offering promising potentials for improved patient care and outcomes in the fight against cancer.
Author Contributions
Conceptualization, L.-W.L.; C.-T.C.; methodology, H.J.Z.; M.S.; L.L.; J.S.S.; S.-H.C.; N.-T.C.; L.-W.L.; C.-T.C.; investigation, H.J.Z.; T.I.; M.S.; L.L.; J.S.S.; S.-H.C.; N.-T.C.; L.-W.L.; C.-T.C.; data curation, H.J.Z.; T.I.; M.S.; L.L.; E.T.; A.P..; formal analysis, H.J.Z.; T.I.; L.L.; D.B.; E.T.; A.P.; writing—original draft preparation, H.J.Z.; T.I.; L.L.; D.B.; writing—review and editing, M.S.; J.S.S.; E.T.; A.P.; S.-H.C.; N.-T.C.; L.-W.L.; C.-T.C.; resources, H.J.Z., M.S., L.L.; L.-W.L.; C.-T.C.; supervision, L.-W.L.; C-T.C.; project administration, H.J.Z.; L.L.; L.-W.L.;C.-T.C.; funding acquisition, J.S.S.; L.-W.L.; C.-T.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the University of Chicago (UChicago) Department of Radiology Pilot Seed Grant; by the UChicago Comprehensive Cancer Center (UCCCC) Program-Specific Pilot Grant through the NIH Cancer Center Support Grant P30 CA014599; by the NIH Shared Instrumentation Grant S10 OD025265 to the UChicago Integrated Small Animal Imaging Research Resource (iSAIRR); by the National Science and Technology Council, Taiwan, Grant 12D2-BNNSTC04; and by the National Health Research Institutes, Taiwan, Grant 13A1-BNPP04-014.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of The University of Chicago (ACUP-72231).
Data Availability Statement
The data presented in this study are available on request from the corresponding authors. The data are not publicly accessible because they are not available in a format that is sufficiently accessible or reusable by other researchers.
Acknowledgments
We are grateful for the support from The Cyclotron Facility and the iSAIRR of the University of Chicago. We are also thankful for the contribution by Dr. Hye-Yeong Kim to the project.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results. Certain commercial equipment, instruments, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation by the authors or their affiliated institutions, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References
- Lee, C.-N.; Hsu, R.; Chen, H.; Wong, T.-W. Daylight Photodynamic Therapy: An Update. Molecules 2020, 25, 5195. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liang, M.; Lei, Q.; Li, G.; Wu, S. The Current Status of Photodynamic Therapy in Cancer Treatment. Cancers 2023, 15, 585. [Google Scholar] [CrossRef] [PubMed]
- Bulin, A.-L.; Truillet, C.; Chouikrat, R.; Lux, F.; Frochot, C.; Amans, D.; Ledoux, G.; Tillement, O.; Perriat, P.; Barberi-Heyob, M.; Dujardin, C. X-Ray-Induced Singlet Oxygen Activation with Nanoscintillator-Coupled Porphyrins. J. Phys. Chem. C 2013, 117, 21583–21589. [Google Scholar] [CrossRef]
- Lecoq, P. Scintillation Detectors for Charged Particles and Photons. In Particle Physics Reference Library: Volume 2: Detectors for Particles and Radiation; Fabjan, C.W., Schopper, H., Eds.; Springer International Publishing: Cham, 2020; pp. 45–89. ISBN 978-3-030-35318-6. [Google Scholar]
- Souris, J.S.; Leoni, L.; Zhang, H.J.; Pan, A.; Tanios, E.; Tsai, H.-M.; Balyasnikova, I.V.; Bissonnette, M.; Chen, C.-T. X-Ray Activated Nanoplatforms for Deep Tissue Photodynamic Therapy. Nanomaterials (Basel) 2023, 13, 673. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Chu, C.-H.; Cheng, S.-H.; Liao, L.-D.; Chu, T.-S.; Chen, N.-T.; Paldino, A.; Hsia, Y.; Chen, C.-T.; Lo, L.-W. Annealing-Modulated Nanoscintillators for Nonconventional X-Ray Activation of Comprehensive Photodynamic Effects in Deep Cancer Theranostics. Theranostics 2020, 10, 6758–6773. [Google Scholar] [CrossRef]
- Cavouras, D.; Kandarakis, I.; Panayiotakis, G.S.; Evangelou, E.K.; Nomicos, C.D. An Evaluation of the Y2O3:Eu3+ Scintillator for Application in Medical x-Ray Detectors and Image Receptors. Medical Physics 1996, 23, 1965–1975. [Google Scholar] [CrossRef]
- Skandani, A.A.; Pham, T.; Luhrs, C.C.; El-Genk, M.S.; Al-Haik, M. Effects of Composition and Transparency on Photo and Radioluminescence of Y2O3:Eu Complexes. Radiation Effects & Defects in Solids 2011. [Google Scholar]
- Cavouras, D.; Kandarakis, I.; Maris, T.; Panayiotakis, G.S.; Nomicos, C.D. Assessment of the Gain Transfer Function of Phosphors for Application in Medical Imaging Radiation Detectors. European Journal of Radiology 2000, 35, 70–77. [Google Scholar] [CrossRef]
- Shivaramu, N.J.; Lakshminarasappa, B.N.; Nagabhushana, K.R.; Singh, F.; Swart, H.C. Synthesis, Thermoluminescence and Defect Centres in Eu3+ Doped Y2O3 Nanophosphor for Gamma Dosimetry Applications. Materials Research Express 2017, 4. [Google Scholar] [CrossRef]
- Souris, J.S.; Cheng, S.-H.; Pelizzari, C.; Chen, N.-T.; La Riviere, P.; Chen, C.-T.; Lo, L.-W. Radioluminescence Characterization of in Situ X-Ray Nanodosimeters: Potential Real-Time Monitors and Modulators of External Beam Radiation Therapy. Appl Phys Lett 2014, 105, 203110. [Google Scholar] [CrossRef]
- Porosnicu, I.; Butnaru, C.M.; Tiseanu, I.; Stancu, E.; Munteanu, C.V.A.; Bita, B.I.; Duliu, O.G.; Sima, F. Y2O3 Nanoparticles and X-Ray Radiation-Induced Effects in Melanoma Cells. Molecules 2021, 26, 3403. [Google Scholar] [CrossRef]
- Cannas, C.; Casu, M.; Mainas, M.; Musinu, A.; Piccaluga, G. Synthesis and Characterization of Y2O3/SiO2 Composites. Zeitschrift für Naturforschung A 2004, 59, 281–287. [Google Scholar] [CrossRef]
- Goswami, B.; Rani, N.; Ahlawat, R. Structural and Optical Investigations of Nd3+doped Y2O3-SiO2 Nanopowder. Journal of Alloys and Compounds 2018, 730, 450–457. [Google Scholar] [CrossRef]
- Modugno, F.; Edwards, R.P. Ovarian Cancer: Prevention, Detection, and Treatment of the Disease and Its Recurrence. Molecular Mechanisms and Personalized Medicine Meeting Report. Int J Gynecol Cancer 2012, 22, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Salani, R.; Backes, F.J.; Fung, M.F.K.; Holschneider, C.H.; Parker, L.P.; Bristow, R.E.; Goff, B.A. Posttreatment Surveillance and Diagnosis of Recurrence in Women with Gynecologic Malignancies: Society of Gynecologic Oncologists Recommendations. Am J Obstet Gynecol 2011, 204, 466–478. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Gynecologic Oncology Group Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer. N Engl J Med 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Trimbos, J.B.; Parmar, M.; Vergote, I.; Guthrie, D.; Bolis, G.; Colombo, N.; Vermorken, J.B.; Torri, V.; Mangioni, C.; Pecorelli, S.; Lissoni, A.; Swart, A.M. International Collaborative Ovarian Neoplasm Trial 1 and Adjuvant ChemoTherapy In Ovarian Neoplasm Trial: Two Parallel Randomized Phase III Trials of Adjuvant Chemotherapy in Patients with Early-Stage Ovarian Carcinoma. J Natl Cancer Inst 2003, 95, 105–112. [Google Scholar] [CrossRef]
- Rajakumar, G.; Mao, L.; Bao, T.; Wen, W.; Wang, S.; Gomathi, T.; Gnanasundaram, N.; Rebezov, M.; Shariati, M.A.; Chung, I.-M.; Thiruvengadam, M.; Zhang, X. Yttrium Oxide Nanoparticle Synthesis: An Overview of Methods of Preparation and Biomedical Applications. Applied Sciences 2021, 11, 11. [Google Scholar] [CrossRef]
- Chrouda, A.; Mahmoud Ali Ahmed, S.; Babiker Elamin, M. Preparation of Nanocatalysts Using Deposition Precipitation with Urea: Mechanism, Advantages and Results. ChemBioEng Reviews 2022, 9, 248–264. [Google Scholar] [CrossRef]
- Sung, J.M.; Lin, S.E.; Wei, W.C.J. Synthesis and Reaction Kinetics for Monodispersive Y2O3:Tb3+ Spherical Phosphor Particles. Journal of the European Ceramic Society 2007, 27, 2605–2611. [Google Scholar] [CrossRef]
- Yao, S.; Xu, L.; Wang, J.; Jing, X.; Odoom-Wubah, T.; Sun, D.; Huang, J.; Li, Q. Activity and Stability of Titanosilicate Supported Au Catalyst for Propylene Epoxidation with H2 and O2. Molecular Catalysis 2018, 448, 144–152. [Google Scholar] [CrossRef]
- Abdalkreem, T.M.; Swart, H.C.; Kroon, R.E. Comparison of Y2O3 Nanoparticles Synthesized by Precipitation, Hydrothermal and Microwave-Assisted Hydrothermal Methods Using Urea. Nano-Structures & Nano-Objects 2023, 35, 101026. [Google Scholar] [CrossRef]
- Sahu, J.N.; Mahalik, K.; Patwardhan, A.V.; Meikap, B.C. Equilibrium and Kinetic Studies on the Hydrolysis of Urea for Ammonia Generation in a Semibatch Reactor. Ind. Eng. Chem. Res. 2008, 47, 4689–4696. [Google Scholar] [CrossRef]
- Morsi, R.E.; El-Salamony, R.A. Effect of Cationic, Anionic and Non-Ionic Polymeric Surfactants on the Stability, Photo-Catalytic and Antimicrobial Activities of Yttrium Oxide Nanofluids. Journal of Molecular Liquids 2020, 297, 111848. [Google Scholar] [CrossRef]
- Mulvaney, P.; Liz-Marzán, L.M.; Giersig, M.; Ung, T. Silica Encapsulation of Quantum Dots and Metal Clusters. J. Mater. Chem. 2000, 10, 1259–1270. [Google Scholar] [CrossRef]
- Liu, G.; Hong, G.; Dong, X.; Wang, J. Silica-Coated Y2O3:Eu Nanoparticles and Their Luminescence Properties. Journal of Luminescence 2007, 126, 702–706. [Google Scholar] [CrossRef]
- Ansari, A.A.; Khan, A.; Labis, J.P.; Alam, M.; Aslam Manthrammel, M.; Ahamed, M.; Akhtar, M.J.; Aldalbahi, A.; Ghaithan, H. Mesoporous Multi-Silica Layer-Coated Y2O3:Eu Core-Shell Nanoparticles: Synthesis, Luminescent Properties and Cytotoxicity Evaluation. Materials Science and Engineering: C 2019, 96, 365–373. [Google Scholar] [CrossRef]
Figure 1.
Protocols for Y2O3:Eu core nanoparticle syntheses.
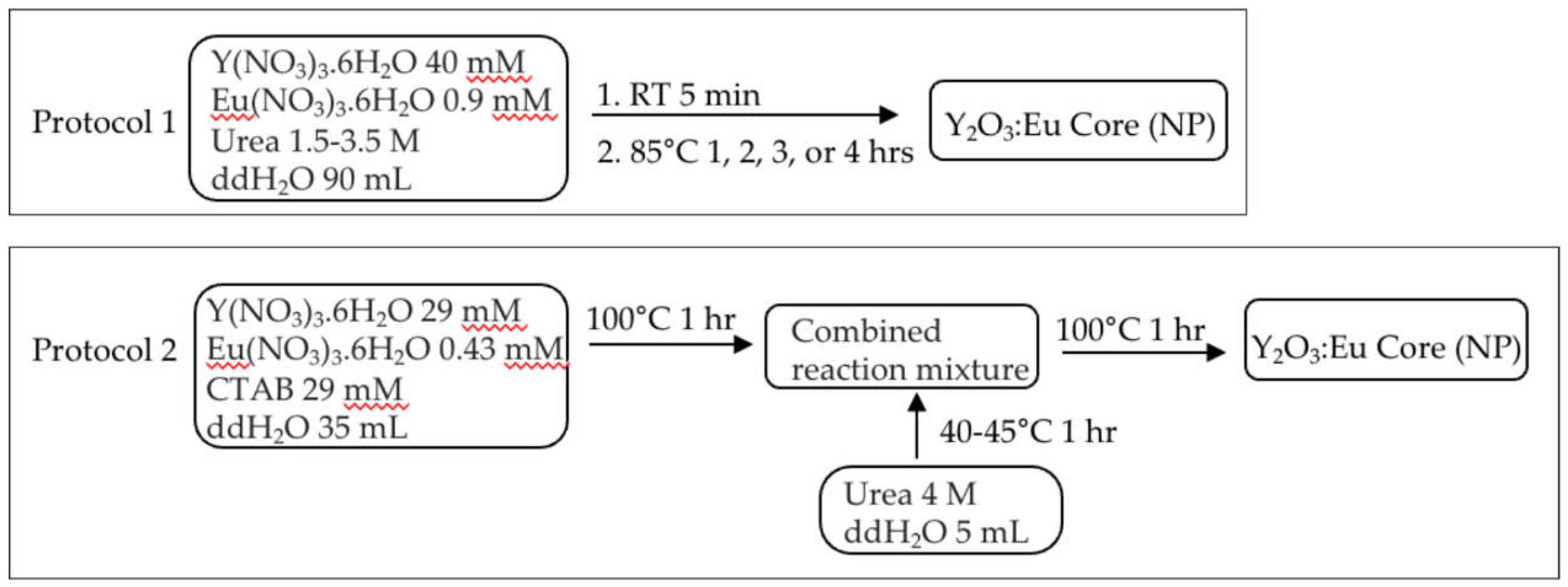
Figure 2.
Protocols for silica encapsulation of Y2O3:Eu core nanoparticles.
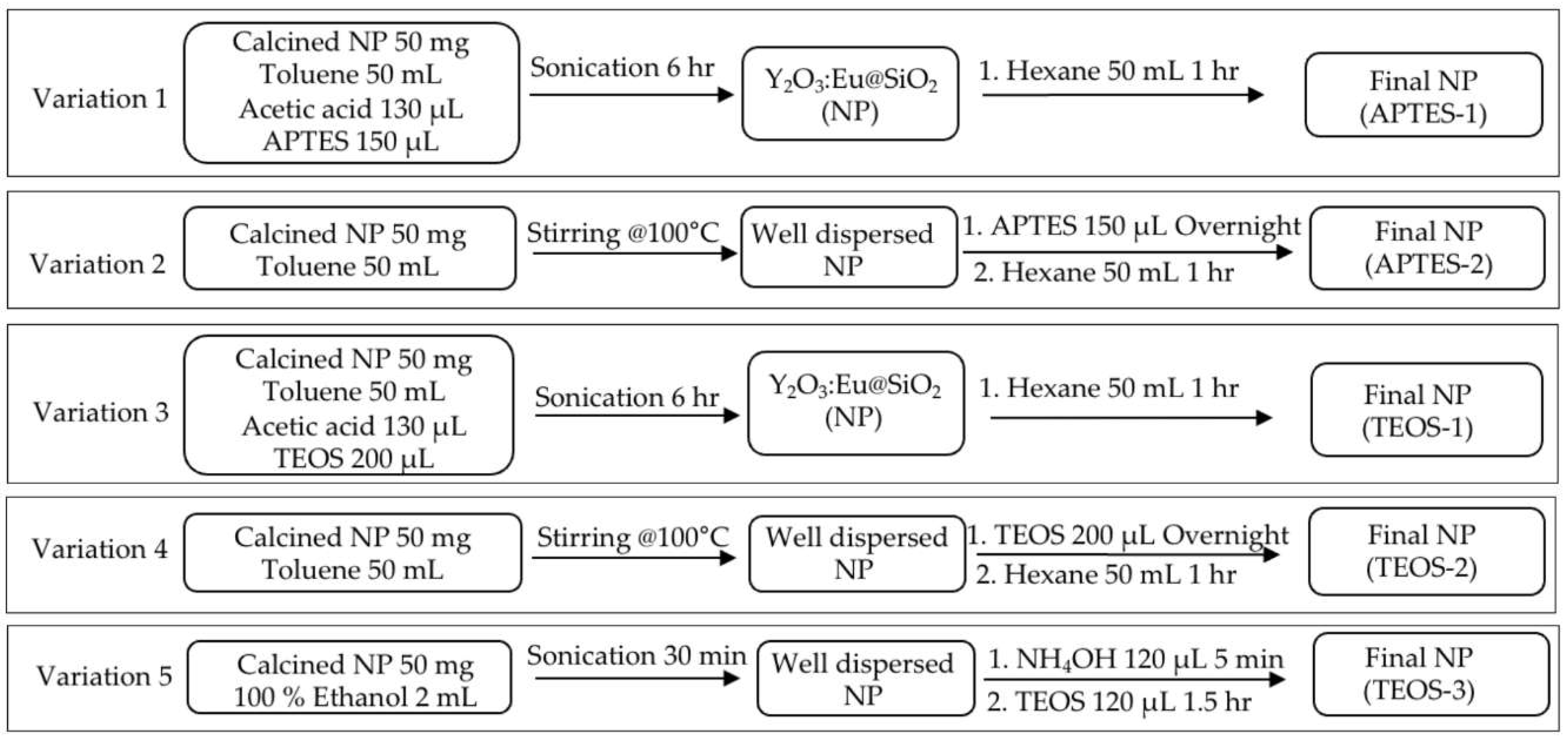
Figure 3.
In vivo study protocol. A. Y2O3:Eu@SiO2 NP functionalization protocol. B. Y2O3:Eu@SiO2 NP treatment of tumor xenograft of human ovarian cancer and 18F-FLT PET imaging.
Figure 3.
In vivo study protocol. A. Y2O3:Eu@SiO2 NP functionalization protocol. B. Y2O3:Eu@SiO2 NP treatment of tumor xenograft of human ovarian cancer and 18F-FLT PET imaging.
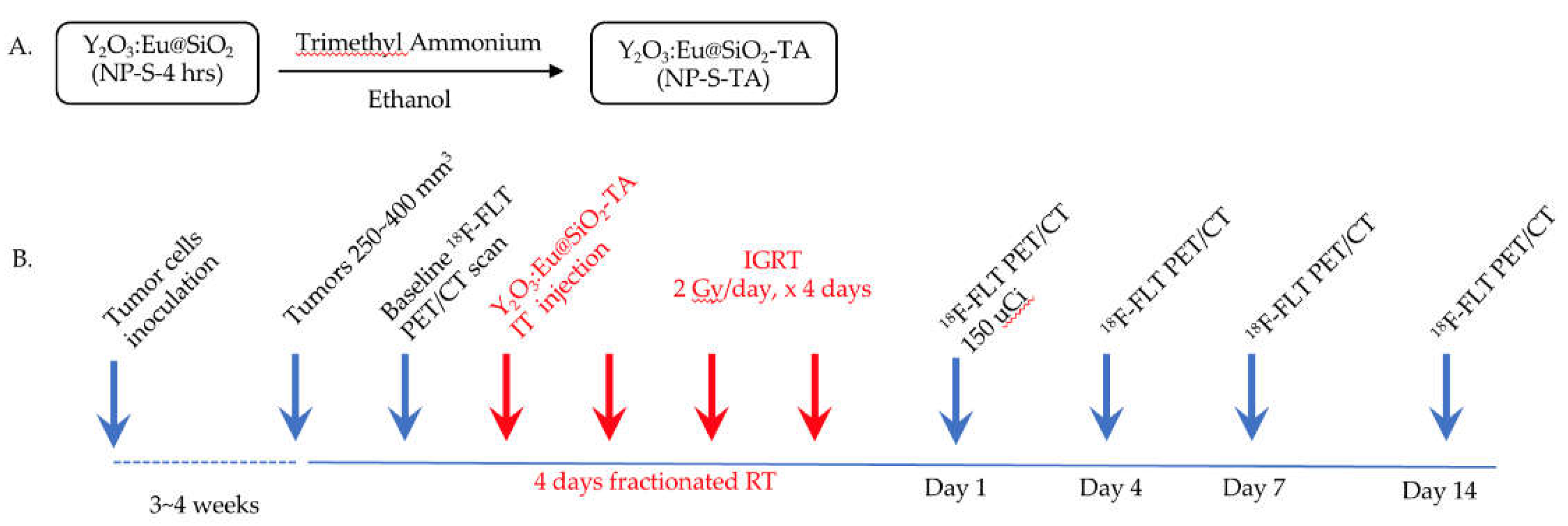
Figure 4.
Effects of condansation time and urea concentration on core size. A. Plot of Y2O3:Eu@SiO2 NP core diameter (nm) versus condensation time (hours) shows that core diameter increases with longer condensation period (R2 = 0.7357, p < 0.05). B. Plot of Y2O3:Eu@SiO2 NP core diameter (nm) versus urea concentration (M) shows an inverse relationship between the urea concentration and core diameter (R2 = 0.958, p < 0.05).
Figure 4.
Effects of condansation time and urea concentration on core size. A. Plot of Y2O3:Eu@SiO2 NP core diameter (nm) versus condensation time (hours) shows that core diameter increases with longer condensation period (R2 = 0.7357, p < 0.05). B. Plot of Y2O3:Eu@SiO2 NP core diameter (nm) versus urea concentration (M) shows an inverse relationship between the urea concentration and core diameter (R2 = 0.958, p < 0.05).
Figure 5.
Effects of various silica coating techniques and calcination placements on thickness of silica coat on Y2O3:Eu@SiO2 NP. A. Silica coating conditions for APTES-1, APTES-2, TEOS-1, and TEOS-2 yeilded similar thickness while the condition for TEOS-3 produced a thicker coating than the others (p < 0.0001). Calcination condition of CA yielded thinner silica coats than CB and CBCA (p < 0.0001 & < 0.0005) (CA: Calcination after silica coating; CB: Calcination before silica coating; CBCA: Calcination before and after silica coating). The particel numbers (n) measured are deplayed on top of the bars. B. A representative TEM image of Y2O3:Eu@SiO2 NP (magnification: 96,000x0) from the APTES-1 group showing the shell thickness and core diameter.
Figure 5.
Effects of various silica coating techniques and calcination placements on thickness of silica coat on Y2O3:Eu@SiO2 NP. A. Silica coating conditions for APTES-1, APTES-2, TEOS-1, and TEOS-2 yeilded similar thickness while the condition for TEOS-3 produced a thicker coating than the others (p < 0.0001). Calcination condition of CA yielded thinner silica coats than CB and CBCA (p < 0.0001 & < 0.0005) (CA: Calcination after silica coating; CB: Calcination before silica coating; CBCA: Calcination before and after silica coating). The particel numbers (n) measured are deplayed on top of the bars. B. A representative TEM image of Y2O3:Eu@SiO2 NP (magnification: 96,000x0) from the APTES-1 group showing the shell thickness and core diameter.
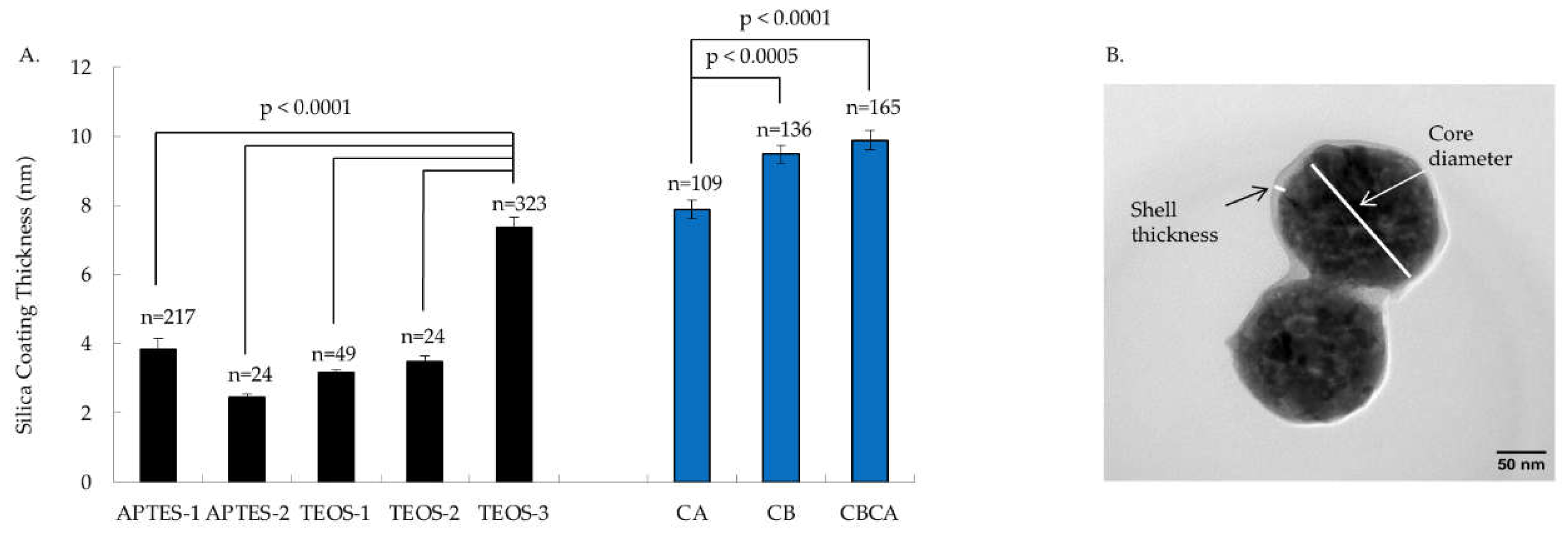
Figure 6.
Characterization of Y2O3:Eu@SiO2 NPs by TEM. Representative TEM images of Y2O3:Eu@SiO2 NPs produced under various conditions shows that the addition of CTAB during core synthesis produced NPs with better dispersity. (TEM magnifications: CA, CB, CBCA, CTAB TEOS-3, CTAB APTES-1: 39,000x; CTAB TEOS-1: 40,000x; CTAB TEOS-2, CTAB APTES-2: 50,000x). Coresponding core diameter distributions are presented as insets.
Figure 6.
Characterization of Y2O3:Eu@SiO2 NPs by TEM. Representative TEM images of Y2O3:Eu@SiO2 NPs produced under various conditions shows that the addition of CTAB during core synthesis produced NPs with better dispersity. (TEM magnifications: CA, CB, CBCA, CTAB TEOS-3, CTAB APTES-1: 39,000x; CTAB TEOS-1: 40,000x; CTAB TEOS-2, CTAB APTES-2: 50,000x). Coresponding core diameter distributions are presented as insets.
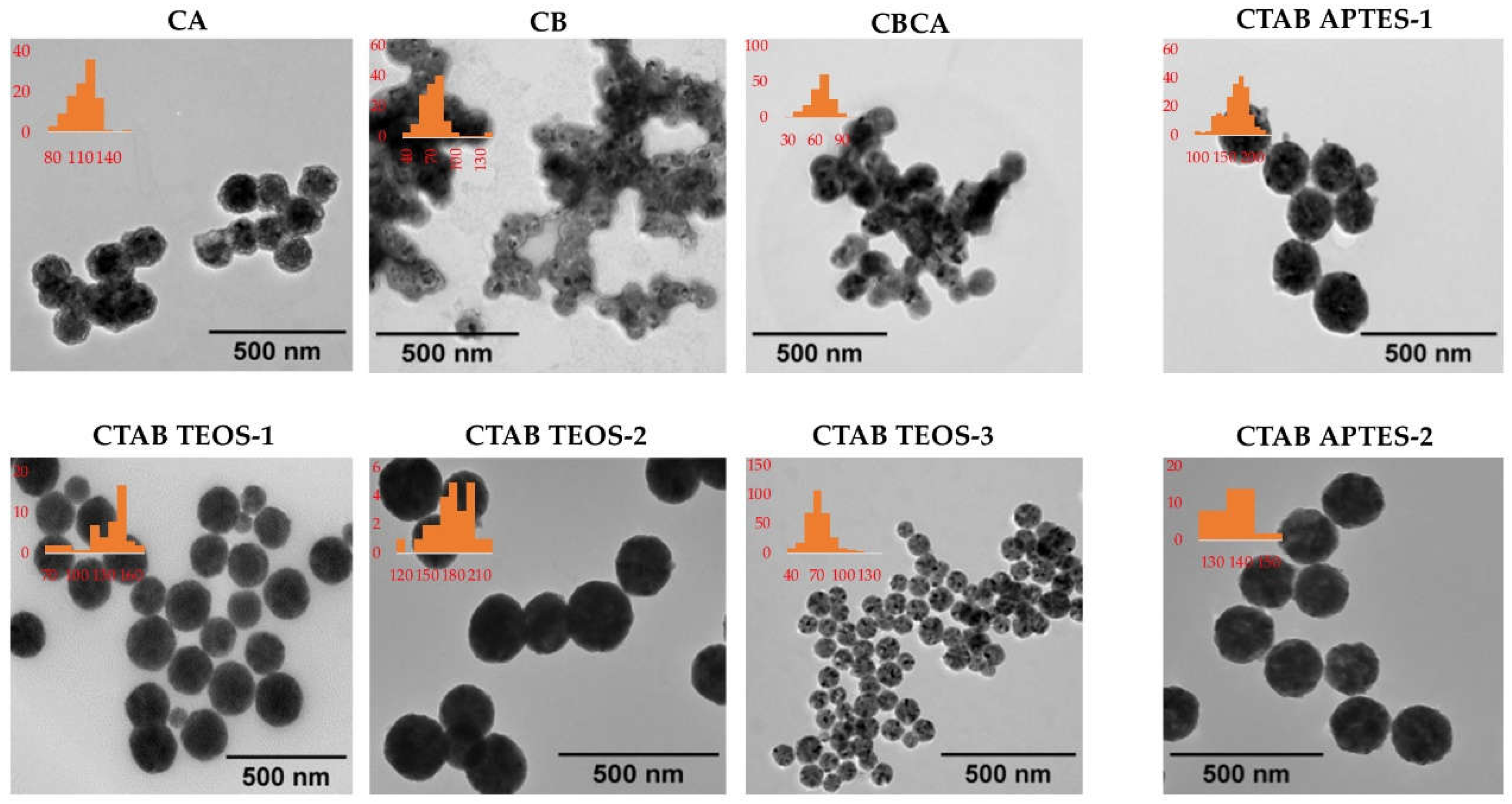
Figure 7.
Effect of NP synthesis on x-ray induced ROS production measured by DHE fluorescent probe. A. Effects of silica coating on the normalized fluorescent intensity under 0-8 Gy irradiation. B. Effects of calcination on the normalized fluorescent intensity under 0-8 Gy irradiation. C. Normalized fluorescent intensity prodeced at 4 & 8 Gy irradiation demonstrates significance of TEOS-3 and APTES-1.
Figure 7.
Effect of NP synthesis on x-ray induced ROS production measured by DHE fluorescent probe. A. Effects of silica coating on the normalized fluorescent intensity under 0-8 Gy irradiation. B. Effects of calcination on the normalized fluorescent intensity under 0-8 Gy irradiation. C. Normalized fluorescent intensity prodeced at 4 & 8 Gy irradiation demonstrates significance of TEOS-3 and APTES-1.
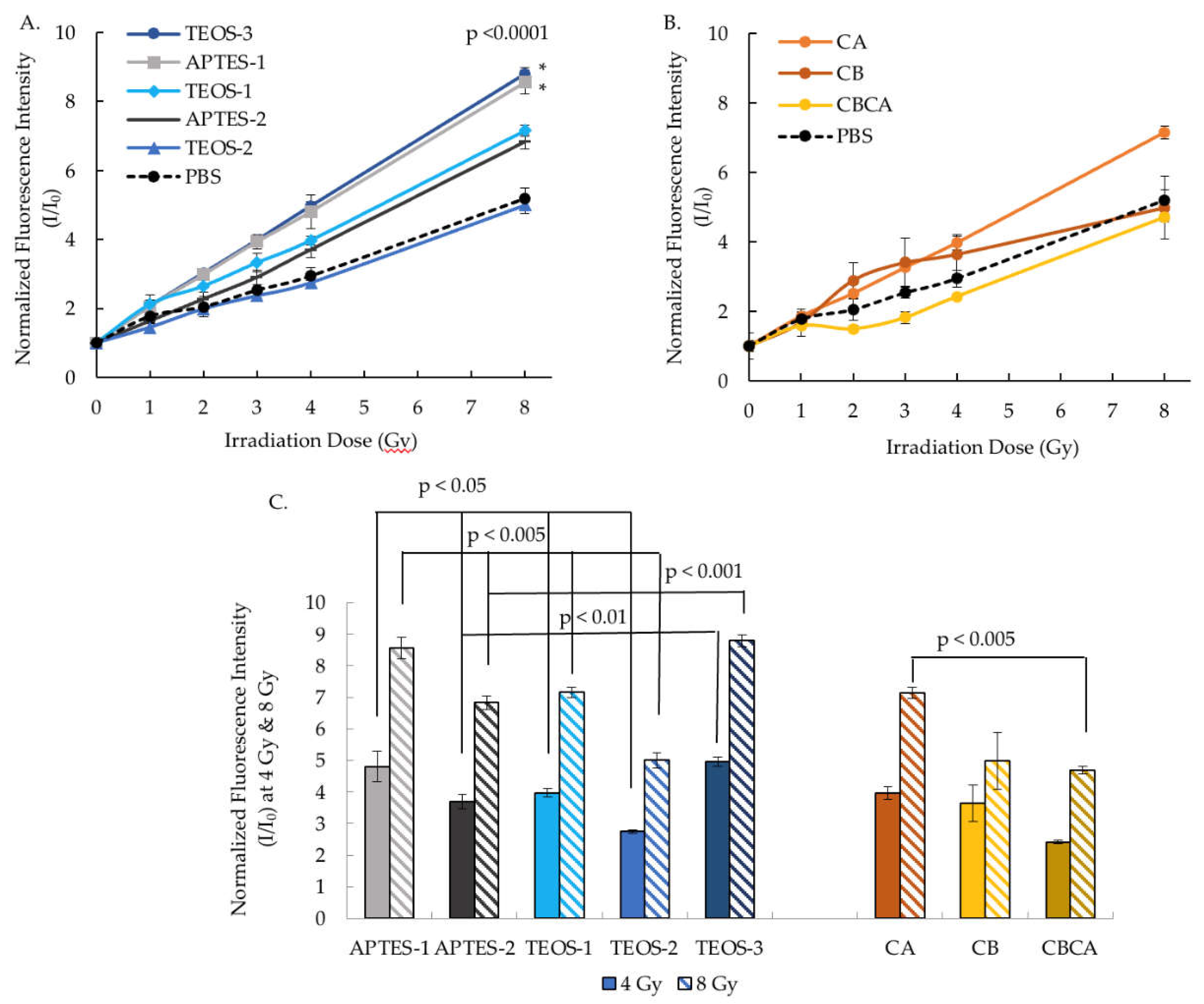
Figure 8.
Intra-tumor Y2O3:Eu@SiO2 treatment enhanced the radiation induced growth suppression of radiation sensitive CAOV3 xenografts. A. Representative images of 18F-FLT PET imaging demonstrate the lowered tumor proliferation in NP treated tumor xenograft. B. Mean 18F-FLT uptake and C. Reduction in 18F-FLT uptake reveals enhanced tumor suppression effects of Y2O3:Eu@SiO2 NP (*p < 0.05). D. Reduction of relative tumor volume in CAOV3 xenograft measured by caliper. (n=3, mean ± SD).
Figure 8.
Intra-tumor Y2O3:Eu@SiO2 treatment enhanced the radiation induced growth suppression of radiation sensitive CAOV3 xenografts. A. Representative images of 18F-FLT PET imaging demonstrate the lowered tumor proliferation in NP treated tumor xenograft. B. Mean 18F-FLT uptake and C. Reduction in 18F-FLT uptake reveals enhanced tumor suppression effects of Y2O3:Eu@SiO2 NP (*p < 0.05). D. Reduction of relative tumor volume in CAOV3 xenograft measured by caliper. (n=3, mean ± SD).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



