
Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Hereditary cancer syndromes caused by germline mutations account for 5-10% of all cancers. The finding of a genetic mutation could have far-reaching consequences for pharmaceutical therapy, personalized prevention strategies, and cascade testing. According to the National Comprehensive Cancer Network’s (NCCN) and the Italian Association of Medical Oncology (AIOM) guidelines, unaffected family members should be tested only if the affected one is unavailable. This article explores whether germline genetic testing may be offered to high-risk families for hereditary cancer even if a living affected relative is missing. A retrospective study was carried out on 103 healthy subjects tested from 2017 to 2023. We enrolled all subjects with at least two first or second-degree relatives affected by breast, ovarian, pancreatic, gastric, prostate, or colorectal cancer. All subjects were tested by Next Generation Sequencing (NGS) multi-gene panel of 27 cancer-associated genes. In the study population, 5 (about 5%) pathogenic/likely pathogenic variants (PVs/LPVs) were found while 40 (42%) had a Variant of Uncertain Significance (VUS). This study highlights the importance of genetic testing for individuals with a strong family history of hereditary malignancies. This approach would allow women who tested positive to receive tailored treatment and prevention strategies based on their personal mutation status.
Keywords:
hereditary cancer
; unaffected family member
; NGS multigene panel
1. Introduction
Genetic testing for hereditary cancer risk is a strategy increasingly used in risk management and treatment planning. Indeed, it is well established that the identification of individuals with deleterious mutations in cancer susceptibility genes has clinical implications for affected people and their families [1]. Family investigations reveal an increased risk for multiple cancer types among first-degree relatives (parents, siblings, and children) and second-degree relatives (grandparents, aunts or uncles, grandchildren, nieces, or nephews) of affected individuals [2]. This may be due to pathogenic variants in parental germline cells. In the mid-1990s, BRCA1 and BRCA2 gene variants, exposing to a higher risk of breast and ovarian cancer, were discovered [3]. Furthermore, by genetic linkage analysis, DNA sequencing, and positional cloning techniques, additional genes whose mutations are associated with moderate and low risk were identified [4]. Genetic testing is generally indicated when there is a personal or family history consistent with an inherited predisposition to cancer [5]. According to the American Cancer Society’s guidelines, genetic testing should be recommended for people: i) with a strong family history of certain types of cancer; ii) diagnosed with cancer when other factors suggest a likely inherited predisposition to cancer (remarkable familiarity, early-onset cancer, or uncommon cancer, i.e. male breast cancer); iii) relatives of a person known to carry an inherited gene mutation increasing their cancer risk [6]. When a patient has a causative mutation, it's advisable to include first-degree relatives in the analysis, as each family member has a 50% probability of carrying the same mutation [7]. In particular, genetic testing may be recommended for cancer-unaffected individuals with collaterals ovarian tumors or early onset breast cancer, bilateral disease, male breast cancer, numerous primary tumors, or additional malignancies linked to a probable hereditary condition, which are typically autosomal dominant [8]. Healthy carriers can benefit from risk management strategies, such as screening, chemoprevention, and risk-reducing prophylactic surgery for breast and ovarian cancer [7]. National and international guidelines recommend that unaffected family members should be tested only when the affected one is unavailable, emphasizing that testing affected relatives is more informative than testing healthy members [9]. Examining unaffected individuals without examining affected family members can pose significant challenges. Assessing multiple family members might be necessary, as the absence of a pathogenic variant in one unaffected relative doesn't preclude its presence in other family members. It is essential to analyze both the maternal and paternal sides of the family to identify familial cancer patterns accurately [10]. It is critical to address serious limitations in interpreting test results as most are negative or non-informative due to the presence of unknown significance variations. Thus, it is important to emphasize that if a pathogenetic mutation is not inherited, the risk of developing is similar to the general population [7]. In this scenario, genetic counseling is crucial in explaining the limited significance of "uninformative" results, and management should focus on other risk factors, rather than on test result [11]. Few studies have explored genetic testing in unaffected subjects, with the percentage of positive carriers being less than 5%. For example, Trottier et al found that 2.8% of unaffected women with a family history of breast or ovarian cancer had a pathogenic variant in BRCA1/2 [12]. However, in recent years, multigene panel testing has been emerged as a crucial approach for detecting clinically significant variants in individuals at high risk for cancer predisposition genes [13]. The present study involved around 100 unaffected individuals, selected only based on their familiar cancer history in the absence of a positive familiar member. Furthermore, we focused on the potential impact of a Next Generation Sequencing (NGS)-based multi-gene panel of 27 genes, including BRCA1/2 genes, with the aim to understand whether expanding the analysis to a larger panel of genes may result into a percentage of healthy subjects with cancer-predisposing gene variants higher than that reported in previous studies [13,14,15].
2. Materials and Methods
2.1. Study Population
One-hundred and three healthy subjects (95 women and 8 men), who during pre-counseling test reported a significant familiar history for hereditary cancers, were retrospectively retrieved among those consecutively referred to the Medical Genetic Service of the University “G.d’Annunzio” of Chieti-Pescara–Center of Advanced Studies and Technologies (CAST) from 2017 to 2023. In these families, anyone has undergone genetic testing. All subjects with at least two first or second-degree relatives with breast, ovarian, pancreatic, gastric, prostate, or colorectal cancer were enrolled in the study. During genetic counseling, which was provided by a multidisciplinary team of geneticists, psychologists, and physicians, personal and familiar histories were acquired. All subjects were informed about the significance of the genetic test, the possible implications of detecting the gene variant related to increased cancer risk, and available prevention strategies. All subjects signed an informed consent form. The results of the analysis and their implications were explained during the post-test counseling.
2.2. Genomic DNA Extraction
Buccal swabs or blood samples were collected from all subjects during pre-counseling test. Genomic DNA was extracted using the MagPurix instrument and the Forensic DNA Extraction Kit (Zinexts Life Science Corp, Taipei, Taiwan-CatZP01001) or Blood DNA Extraction Kit 200 (Zinexts Life Science Corp, Taipei, Taiwan-CatZP02001), according to the manufacturer’s protocol.
2.3. Next-Generation Sequencing (NGS)
NGS analysis was carried out with a Thermo-fisher Oncomine custom panel developed in our laboratory, including 27 genes (Table 1). NGS was performed through the Ion Torrent S5 system (Thermo Fisher Scientific, Waltham, MA, USA) after automatic library preparation using Ion Chef (Thermo Fisher Scientific, Waltham, MA, USA). The Ion Chef system allows fragmentation and adapter ligation onto the PCR products, called clonal amplification. After quantification of DNA libraries with the Real-Time Step One PCR System (Thermo Fisher Scientific, Waltham, MA, USA), the prepared samples of ion sphere particles (ISP) were loaded onto an Ion 530TM chip with the Ion Chef (Thermo Fisher Scientific, Waltham, MA, USA). Sequencing was performed using the Ion S5TM sequencing reagents (Thermo Fisher Scientific, Waltham, MA, USA). The Torrent Suite 5.14.0 platform and specific plugins were used for NGS data analysis. The uniformity of base coverage was over 99% in all batches, and base coverage was over 20× in all target regions.
2.4. Sanger Sequencing
Specific pathogenic variants identified in each subject via NGS were confirmed via Sanger sequencing. All DNA samples were amplified via polymerase chain reaction (PCR) performed in 30 μL reaction volume, containing 22.25 μL of H2O, 3 μL of 10X PCR buffer, 2.1 μL of MgCl2 solution 25 mM, 0.5 μL of dNTPs 10 mM, 0.15 μL of AmpliTaq Gold polymerase, 1 μL of DNA, and 0.5 μL of Forward and 0.5 μL of Reverse primers. All primers were designed using NCBI designing tools (https://www.ncbi.nlm.nih.gov/tools/primer-blast/ accessed on 12 October 2023). The list of primers used to confirm the analysis is reported in Table 2. Amplification was performed via SimpliAmpTM thermal cycler (ThermoFisher, Applied Biosystem, CA, USA). FastGene Gel/PCR Extraction kit (Nippon Genetics Europe, Düren, Germany) was utilized for the purification of PCR products, according to the manufacturer’s protocol. The amplification products were submitted to direct sequencing procedure using BigDye Term v3.1 CycleSeq Kit (Life Technologies, Monza, Italy) followed by automatic sequencing analysis. All sequences were purified via “NucleoSEQColumns” purification kit (Macherey-Nagel Colonia, Dueren, Germany) and analyzed in forward and reverse directions on a SeqstudioGenetic Analyzer (ThermoFisher, Applied Biosystem, Foster City, CA, USA).
2.5. Genetic Variant Classification
According to the guidelines of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) (https://enigmaconsortium.org/), genetic variants were classified into five classes: benign (C1), likely benign (C2), variant of uncertain significance (VUS, C3), likely pathogenic (C4), and pathogenic (C5). In the present study, we focused on the pathogenic variants that can be used for cancer prevention. The variants were referred to according to the nomenclature recommendations of the Human Genome Variation Society (https://www.hgvs.org). The clinical significance of the genetic variants found in this study was evaluated according to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), Varsome (https://varsome.com), Franklin Genoox (https://franklin.genoox.com) and, for some other susceptibility genes, according to LOVD-InSIGHT (https://www.insight-group.org/variants/databases/).
3. Results
Mean age of the 103 healthy subjects was 49 years (range 28-65). A panel of 27 cancer susceptibility genes was examined. The prevalence of pathogenic variants was 5%. In particular, among five discovered pathogenic variants, two were detected in BRCA1 and BRCA2, one each in CHEK2, POLE, and MUTYH (Table 3). Of these, 4 females resulted positive and only one man with a PV in POLE gene. Out of 103 unaffected individuals tested, 36 (35%) had a VUS in 18 different genes, including ATM, BARD1, BRCA1, BRCA2, PALB2, and CHEK2, for a total of 40 variants classified as C3. The remaining 62 subjects (60%) showed neither deleterious variant nor VUS.
3.1. Genes Variants linked to the Homologous Recombination (HR) and related family history
The study revealed two crucial aspects in the familial history of the tested healthy subjects. In many cases several members of the family were affected by the same or by the different type of cancer while in others at least one family member developed a cancer before the age of 50 years. Starting from the genetic analysis of subject 1, we found the c.4914dupA pathogenic mutation located in exon 10 of the BRCA2 gene, that causes a translational frameshift with a predicted alternate stop codon (V1639fs). The family history of this subject revealed three relatives affected: the mother with gastric cancer at age of 65, the maternal grandfather with colorectal cancer at 77 and the maternal aunt with breast cancer at 37 (Figure 1), all deceased at the time of the pre-test counseling.
Subject 2 showed the c.1427C>T LPV in the CHEK2 gene, resulting in a damaging effect with reduced or absence of kinase activity and DNA damage response [16]. Familial history of this case revealed a grandmother with ovarian cancer at 69 and maternal aunt with ovarian cancer at 51. In other cases, we found the same type of cancer in two family members.
During the pre-counseling test, subject 3 reported a sister and a paternal aunt deceased from ovarian cancer at 54 and 58 years, respectively. In this subject, genetic testing evidenced a pathogenic mutation in BRCA1 gene, the c.1953dup located in coding exon 9, resulting from a duplication of Guanine at nucleotide position 1953, that leads to a translational frameshift with a predicted alternate stop codon (K652fs) [17].
Case number 4 presented a pathogenic mutation in POLE gene, the c.778C>T, which creates a premature translational stop signal and is expected to result in an absent or disrupted protein product with loss of function (R260*) [18]. In this family, there was a significant and heterogeneous presence of tumors. From maternal side, the mother had ovarian cancer at 50 and a first-degree uncle had throat cancer at 55. On the paternal side, the father had throat cancer at 70 and, subsequently, colorectal cancer. Other tumors registered in additional family members were: colorectal cancer in a paternal aunt and in a cousin both at 55, bladder cancer in a first-degree uncle at the age of 55, throat cancer in a paternal cousin at 45, lung cancer in a paternal uncle at 85 and ultimately, another cousin deceased at 45 from brain cancer (Figure 2)
3.2. Genes Variants linked to the base excision repair (BER) and related family history
The father of subject 5 was diagnosed with colorectal cancer at the age of 41, a first-degree aunt had breast cancer at 47 and a first-degree uncle had colorectal cancer at 45. The PV found, in this case, was the c.1187G>A in the MUTYH gene, located in coding exon 13 and causing the substitution of a Glycine with Aspartate in codon position 396. This alteration is frequently reported as founder mutation in multiple populations. M. Nielsen et al. have shown that this missense variant changes the function of MUTYH protein [19].
4. Discussion
In recent years, several genes associated with hereditary cancer syndromes have been identified, and at least 2% of presumably healthy individuals carries highly penetrating pathogenic gene variants predisposing them to cancer [20]. Individuals with hereditary cancer syndromes have a higher risk of developing multiple primary cancers during their lifetime or may develop cancer at a younger age.
Early mutation detection and prevention are key aspects of managing hereditary cancer risks [21], thus supporting the implementation of targeted screening and prevention strategies in this population. In this regard, genetic testing may play a crucial role. However, according to international and national guidelines, when a familial predisposition is suspected, genetic testing should be preliminarily performed on a family member who has already developed the disease (index case). Alternatively, when the index case is unwilling to perform the test, it is possible to offer the genetic test to a healthy relative who has a high probability of mutation (>10%) during the entire lifetime [22]. The present study evaluated, for the first time, the presence of pathogenic mutations in healthy subjects with a strong family history of hereditary cancers, in the absence of affected living relatives. As an inclusion criterion, the presence of at least two first- or second-degree relatives with breast, ovarian, pancreatic, gastric, prostate, or colorectal cancer was used. In this population of healthy subjects, a 5% prevalence of pathogenic mutations in genes correlated with high or moderate risk of developing cancer was found. This finding highlights the need to extend genetic testing to healthy individuals with suggestive familiarity, even in the absence of an index case. In particular, the family trees of two probands (Fig.1 and Fig.2) were reported, in which a pathogenic mutation in BRCA2 c.4914dupA and POLE c.778C>T respectively was detected. In both cases, distinct forms of cancers were found in the generations portrayed. In particular, Family 2 with a POLE mutation exhibits a significant cancer history from both maternal and paternal lines, thus emphasizing the importance of studying the entire family history to select subjects eligible for testing. These results suggest that a change in current eligibility criteria for genetic testing in healthy subjects could be evaluated. Specifically, we propose that testing should be conducted in healthy subjects with the following family history features: (I) at least 2 first-degree relatives with one of the following cancers: breast, ovarian, pancreatic, gastric, prostate and colorectal; (II) at least 1 first-second or third-degree relative with an early-onset tumor (<45 years old); (III) at least two relatives affected by the same type of cancer. Furthermore, we propose to perform the test before the age of 55 rather than 25, since in clinical practice instrumental preventive screening starts at age of 25 or 10 years before the age of cancer beginning in the youngest affected family member [22]. The NCCN guidelines for clinical practice in oncology offer specific recommendations and surveillance programs tailored to the type of mutated gene detected in healthy individuals, such as imaging modalities, frequency of evaluation, and risk-reducing surgery. This proactive approach seeks to diagnose cancer in its earliest, most treatable stages or to entirely prevent its development. Furthermore, genetic testing and the discovery of a mutation associated with an elevated risk of cancers is critical not only for enrolling the proband in tailored surveillance programs, but also for the healthy collaterals. Once the mutation in the family has been identified, testing should be extended to all members. This could allow the assessment of their likely carrier status and their enrollment in surveillance and therapy programs. A recent study by Di Rado et al. underscores the importance of cascade testing in at-risk relatives of probands with PV/LPV in one of 27 cancer susceptibility genes [7]. Segregation among relatives strengthens the association between identified variants and cancer predisposition. Carriers of pathogenic mutations can benefit from appropriate risk management and preventative strategies due to an inherited increased risk of breast, ovarian, prostate, melanoma, and pancreatic cancers. Cascade testing could significantly increase the identification of pathogenic variant carriers, as 70% of probands may inform their family members and 20% may receive genetic testing, potentially increasing the 5% of pathogenetic variants found in the present work. It is important to emphasize how the use of the multigenic panel, including 27 genes associated with hereditary cancers, allowed us to increase the detection rate of unaffected individuals with mutations in genes beyond BRCA1 and BRCA2. In absence of multi-gene panel analysis, a considerable percentage of mutations would have been lost.
The use of a multi-gene panel significantly reduces both the time and cost of analysis from a cost-benefit perspective. Avoiding stringent criteria to select healthy patients with familiarity to submit NGS testing allows a larger cohort and reduced NGS testing costs. In addition, the early detection of a pathogenic mutation and the inclusion of healthy individuals in surveillance programs may significantly reduce cancer-related healthcare costs. So far, our experience with hereditary cancer panel testing in unaffected individuals has been very encouraging. All five families with identified mutations allowed women to make decisions about surgical risk reduction based on their personal mutation state. Interestingly, our research found that women are more likely to request genetic testing, possibly due to their long-standing knowledge of the benefits of being identified as BRCA carriers, while men are more likely to participate for their daughters. Additionally, unaffected individuals under 40 were found to be more likely to request pre-test counseling appointments.
In this view, genetic counseling importantly contributes to the analysis process, by providing people with information on their risk, available preventive measures, chemotherapy protocols and other treatment options, such as preventive surgery. Moreover, genetic counseling represents a strategic tool for identifying the criteria that suggest the presence of a mutation in the proband's family tree and the decision to consider him or her eligible for testing. On these bases, the role of pre-test counseling in explaining the limited significance of "uninformative" results appears crucial. Genetists must clarify how mutations in moderate penetrance genes vary from BRCA1/2 mutations in terms of risks. Certainly, the involvement of multiple family members in the decision regarding a genetic test can potentially lead to tensions and disagreements. In the future, it will be crucial to overcome social and cultural barriers hindering effective communication between families. Additionally, the psychological impact of predictive genetic testing should not be overlooked. Understanding mutation status can cause psychological distress due to the high lifetime risks of cancer development, so psychological support is recommended during pre and post-genetic counselling [23,24,25]. Challenges, and future directions should focus on supporting intrafamilial communication and improving communication processes between professionals and at-risk relatives. In addition, radiological screening, surgeons, gynecologists and “omics” approaches should play a crucial role in identifying high-risk individuals for hereditary cancer predisposition syndromes prevention. [26] It is possible to outline a hypothetical scenario based on the extension of BRCA genetic testing to healthy women in the general population. Population screenings would also reduce the overall costs associated with managing these hereditary syndromes, offsetting the additional costs resulting from increased genetic testing. From a practical clinical point of view, there is the need to develop strategies to improve test uptake by unaffected individuals. In this context, it has been recently proposed that the use of a web-based tool may result in higher quality collection of cancer family history compared to clinician collection, thus improving the percentage of participants completing genetic counseling and testing [27].
In the era of personalized cancer prevention and early detection, the present study is the first to implement hereditary cancer gene panel testing to families at high risk, providing women with mutations with a better foundation for individual decisions about surveillance or risk-reducing surgery. Despite its limitations, our findings offer preliminary insights that can serve as a baseline for future research.
Author Contributions
Conceptualization, L.P., F.A. and I.A.; methodology, L.P., F.A., A.D.E.; software, L.P., F.A.; validation, M.M., R.G., M.C and I.A.; formal analysis, L.P., F.A.; investigation, L.P., F.A., M.M., R.G., M.C and I.A.; resources, S.G., G.C., L.S. and I.A.; data curation, L.P. and F.A.; writing—original draft preparation, I.A.; writing—review and editing, L.P., F.A., N.T., A.B. and I.A.; visualization, A.G., A.P., S.G., N.C., G.C and I.A.; supervision, N.T., A.B., P.B., L.S. and I.A.; project administration, P.B., L.S. and I.A.; X.X.; funding acquisition, L.S. and I.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Our University has an agreement with Italian public health system for genetic testing. Patients underwent genetic testing for diagnostic purposes. They filled out an informed consent where they declared their availability to provide genetic data for research purposes and/or scientific publications.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data available on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Antonucci I., Provenzano M., Sorino L., Rodrigues M., Palka G., Stuppia L. A new case of “de novo” BRCA1 mutation in a patient with early-onset breast cancer. Clin. Case Rep. 2017;5:238–240. Author 1, A.; Author 2, B. Title of the chapter. In Book Title, 2nd ed.; Editor 1, A., Editor 2, B., Eds.; Publisher: Publisher Location, Country, 2007; Volume 3, pp. 154–196. [CrossRef]
- NCCN. Detection, Prevention, and Risk Reduction. Available online: https://www.nccn.org/guidelines/category_2 (accessed on 27 February 2024).
- Knerr S, Guo B, Mittendorf KF, Feigelson HS, Gilmore MJ, Jarvik GP, Kauffman TL, Keast E, Lynch FL, Muessig KR, Okuyama S, Veenstra DL, Zepp JM, Goddard KAB, Devine B. Risk-reducing surgery in unaffected individuals receiving cancer genetic testing in an integrated health care system. Cancer. 2022 Aug 15;128(16):3090-3098. Epub 2022 Jun 9. [CrossRef] [PubMed] [PubMed Central]
- Kamps, R.; Brandão, R.D.; van den Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. Author 1, A.B.; Author 2, C.D.; Author 3, E.F. Title of Presentation. In Proceedings of the Name of the Conference, Location of Conference, Country, Date of Conference (Day Month Year). [CrossRef] [PubMed]
- DQ Cancer Genetics Editorial Board. Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version. 2024 Mar 6. In: PDQ Cancer Information Summaries.
- https://www.cancer.org/cancer/risk-prevention/genetics/genetic-testing-for-cancer-risk/understanding-genetic-testing-for-cancer.html.
- Di Rado S, Giansante R, Cicirelli M, Pilenzi L, Dell'Elice A, Anaclerio F, Rimoldi M, Grassadonia A, Grossi S, Canale N, Ballerini P, Stuppia L, Antonucci I. Detection of Germline Mutations in a Cohort of 250 Relatives of Mutation Carriers in Multigene Panel: Impact of Pathogenic Variants in Other Genes beyond BRCA1/2. Cancers (Basel). 2023 Dec 6;15(24):5730. [CrossRef] [PubMed] [PubMed Central]
- Petrucelli N, Daly MB, Pal T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. 1998 Sep 4 [Updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1247/.
- Forbes C, Fayter D, de Kock S, Quek RG. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manag Res. 2019 Mar 22;11:2321-2337. [CrossRef] [PubMed] [PubMed Central]
- Weitzel JN, Lagos VI, Cullinane CA, Gambol PJ, Culver JO, Blazer KR, Palomares MR, Lowstuter KJ, MacDonald DJ. Limited family structure and BRCA gene mutation status in single cases of breast cancer. JAMA. 2007 Jun 20;297(23):2587-95. [CrossRef] [PubMed]
- E:Bramanti SM, Trumello C, Lombardi L, Cavallo A, Stuppia L, Antonucci I, Babore A. Uncertainty following an inconclusive result from the BRCA1/2 genetic test: A review about psychological outcomes. World J Psychiatry. 2021 May 19;11(5):189-200. [CrossRef] [PubMed] [PubMed Central]
- Trottier M, Lunn J, Butler R, Curling D, Turnquest T, Francis W, Halliday D, Royer R, Zhang S, Li S, Thompson I, Donenberg T, Hurley J, Akbari MR, Narod SA. Prevalence of founder mutations in the BRCA1 and BRCA2 genes among unaffected women from the Bahamas. Clin Genet. 2016 Mar;89(3):328-31. Epub 2015 May 31. [CrossRef] [PubMed]
- Bernstein-Molho R, Singer A, Laitman Y, Netzer I, Zalmanoviz S, Friedman E. Multigene panel testing in unselected Israeli breast cancer cases: mutational spectrum and use of BRCA1/2 mutation prediction algorithms. Breast Cancer Res Treat. 2019 Jul;176(1):165-170. Epub 2019 Apr 12. [CrossRef] [PubMed]
- Ece Solmaz A, Yeniay L, Gökmen E, Zekioğlu O, Haydaroğlu A, Bilgen I, Özkınay F, Onay H. Clinical Contribution of Next-Generation Sequencing Multigene Panel Testing for BRCA Negative High-Risk Patients With Breast Cancer. Clin Breast Cancer. 2021 Dec;21(6):e647-e653. Epub 2021 Apr 12. [CrossRef] [PubMed]
- Anaclerio F, Pilenzi L, Dell'Elice A, Ferrante R, Grossi S, Ferlito LM, Marinelli C, Gildetti S, Calabrese G, Stuppia L, Antonucci I. Clinical usefulness of NGS multi-gene panel testing in hereditary cancer analysis. Front Genet. 2023 Feb 1;14:1060504. [CrossRef] [PubMed] [PubMed Central]
- Desrichard A, Bidet Y, Uhrhammer N, Bignon YJ. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011;13(6):R119. Epub 2011 Nov 24. [CrossRef] [PubMed] [PubMed Central]
- Oros KK, Ghadirian P, Greenwood CM, Perret C, Shen Z, Paredes Y, Arcand SL, Mes-Masson AM, Narod SA, Foulkes WD, Provencher D, Tonin PN. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5 BRCA1 and BRCA2 mutations. Int J Cancer. 2004 Nov 10;112(3):411-9. [CrossRef] [PubMed]
- Thiffault I, Saunders C, Jenkins J, Raje N, Canty K, Sharma M, Grote L, Welsh HI, Farrow E, Twist G, Miller N, Zwick D, Zellmer L, Kingsmore SF, Safina NP. A patient with polymerase E1 deficiency (POLE1): clinical features and overlap with DNA breakage/instability syndromes. BMC Med Genet. 2015 May 7;16:31. [CrossRef] [PubMed] [PubMed Central]
- Nielsen, M., Jones, N., Vogt, S., Carli, M., Vasen, H. F. A., Sampson, J. R., et al. (2009). Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology 136 (2), 471–476. [CrossRef]
- Imyanitov EN, Kuligina ES, Sokolenko AP, Suspitsin EN, Yanus GA, Iyevleva AG, Ivantsov AO, Aleksakhina SN. Hereditary cancer syndromes. World J Clin Oncol. 2023 Feb 24;14(2):40-68. [CrossRef] [PubMed] [PubMed Central]
- Antonucci I., Provenzano M., Sorino L., Balsamo M., Aceto G.M., Battista P., Euhus D., Cianchetti E., Ballerini P., Natoli C., et al. Comparison between CaGene 5.1 and 6.0 for BRCA1/2 mutation prediction: A retrospective study of 150 BRCA1/2 genetic tests in 517 families with breast/ovarian cancer. J. Hum. Genet. 2017;62:379–387. [CrossRef]
- AIOM (Associazione Italiana Oncologia Medica) Linee guida Carcinoma Mammario in stadio precoce -Edizione 2023 (Aggiornata al 20/11/2023)23.
- Bramanti SM, Trumello C, Lombardi L, Cavallo A, Stuppia L, Antonucci I, Babore A. Uncertainty following an inconclusive result from the BRCA1/2 genetic test: A review about psychological outcomes. World J Psychiatry. 2021 May 19;11(5):189-200.
- Lombardi L, Bramanti SM, Babore A, Stuppia L, Trumello C, Antonucci I, Cavallo A. Psychological aspects, risk and protective factors related to BRCA genetic testing: a review of the literature. Support Care Cancer. 2019 Jun 15.
- Babore A, Bramanti SM, Lombardi L, Stuppia L, Trumello C, Antonucci I, Cavallo A. The role of depression and emotion regulation on parenting stress in a sample of mothers with cancer. Support Care Cancer. 2019 Dec 19.
- Rossi C, Cicalini I, Cufaro MC, et al. Breast cancer in the era of integrating "Omics" approaches. Oncogenesis. 2022;11(1):17. Published 2022 Apr 14. [CrossRef]
- Frey MK, Ahsan MD, Webster E, Levi SR, Brewer JT, Lin J, Blank SV, Krinsky H, Nchako C, Wolfe I, Thomas C, Christos P, Cantillo E, Chapman-Davis E, Holcomb K, Sharaf RN. Web-based tool for cancer family history collection: A prospective randomized controlled trial. Gynecol Oncol. 2023 Jun;173:22-30.
Figure 1.
Family tree of subject 1 with BRCA2 c.4914dupA pathogenic mutation

Figure 2.
Familiar history of subject 4 with a PV c.778C>T in POLE gene
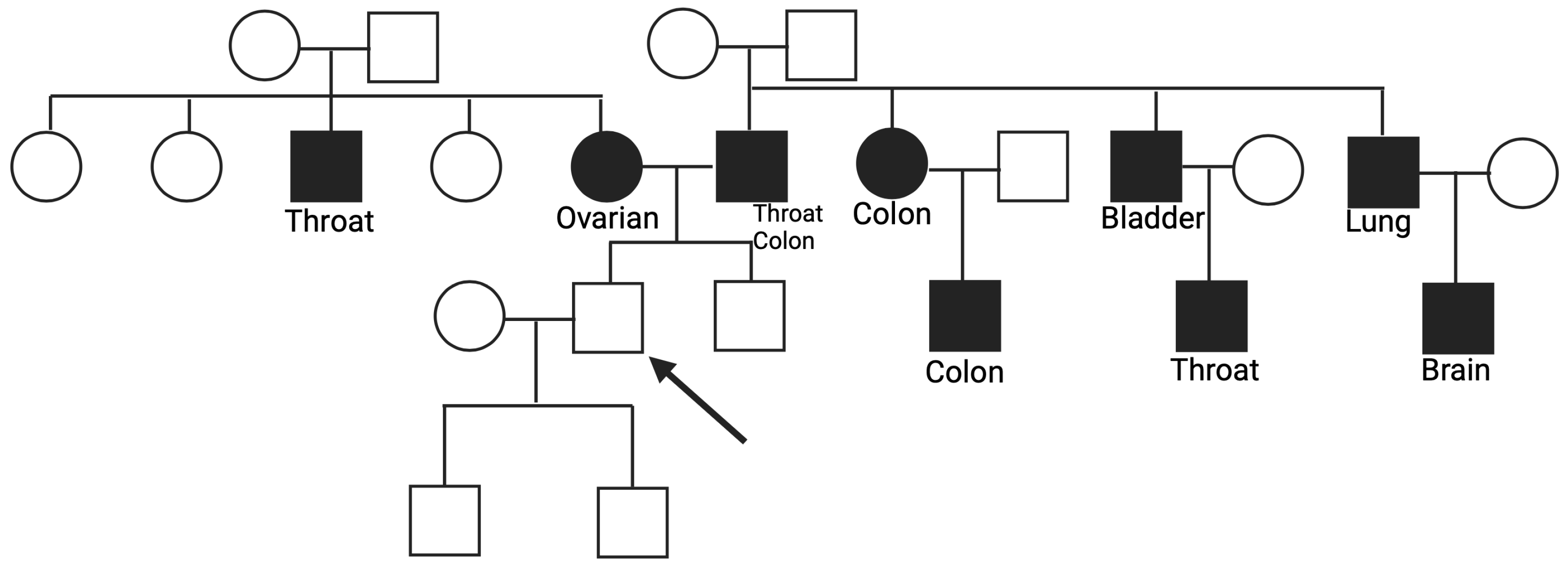

Table 1.
The 27 genes included in the NGS multi-gene panel
| Multi-gene panel | ||
|---|---|---|
| APC | ATM | BARD1 |
| BRCA1 | BRCA2 | BRIP1 |
| CDK4 | CDK12 | CDKN2A |
| CDH1 | CHEK2 | EPCAM |
| MLH1 | MSH2 | MSH6 |
| MUTYH | NBN | NF1 |
| PALB2 | POLE | POLE |
| POLD1 | PTEN | RAD51C |
| RAD51D | SMAD4 | TP53 |
Table 2.
List of primers designed to confirm the PV/LPV found in the 5 healthy subjects
| NAME | EXON | SEQUENCE |
|---|---|---|
| BRCA2_EX11F | Exon 11 | attgagatcacagctgcccc |
| BRCA2_EX11R | Exon 11 | tgaagtctgactcacagaagttt |
| CHEK2_EX13F | Exon 13 | atgtggatgtgagtcagccag |
| CHEK2_EX13R | Exon 13 | atcagctccttaagcccagacta |
| BRCA1_EX10F | Exon 10 | ttggtcagctttctgtaatcg |
| BRCA1_EX10R | Exon 10 | ccataccacgacatttgaca |
| POLE_EX8F | Exon 8 | gtcgctgctcacatgaattt |
| POLE_EX8R | Exon 8 | atttgggggaaaagcagcaa |
| MUTYH_13F | Exon 13 | agggcagtggcatgagtaac |
| MUTYH_13R | Exon 13 | gggtcaaggggttcaaatag |
Table 3.
All PVs/LPVs found in healthy carriers.
| CASE ID GENE | OMIM REFSEQ | CODING PROTEIN |
|---|---|---|
| Subject 1 BRCA2 | 164757 NM_000059.3 | c.4914dupA V1639fs |
| Subject 2 CHEK2 | 604373 NM_007194.4 | c.1427C>T T476M |
| Subject 3 BRCA1 | 113705 NM_007294.4 | c.1953dup K652fs |
| Subject 4 POLE | 174762 NM_006231.3 | c.778C>T R260* |
| Subject 5 MUTYH | 608456 NM_001128425.2 | c.1187G>A G396D |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



