
Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Detecting volatile organic compounds (VOCs) emitted from different plant species and their or-gans can provide valuable information about plant health and environmental factors that affect them. For example, limonene emission can be a biomarker to monitor plant health and detect stress. Traditional methods for VOC detection encounter challenges, prompting the proposal of novel approaches. In this study, we proposed integrating electrospinning, molecular imprinting, and conductive nanofibers to fabricate limonene sensors. In detail, polyvinylpyrrolidone (PVP) and polyacrylic acid (PAA) served here as fiber and cavity formers, respectively, with multi-walled carbon nanotubes (MWCNT) enhancing conductivity. We developed one-step monolithic molecularly imprinted fibers, where S(-)-limonene was the target molecule using electrospinning technique. The functional cavities were fixed using UV curing method, followed by a target mol-ecule washing. This procedure enabled the creation of recognition sites for limonene within the nanofiber matrix, enhancing sensor performance and streamlining manufacturing. Humidity was crucial for sensor working, with optimal conditions at about 50% RH. The sensors rapidly re-sponded to S(-)-limonene, reaching a plateau within 200 seconds. Enhancing fiber density im-proved sensor performance, resulting in a lower limit of detection (LOD) of 137 ppb. However, excessive fiber density decreased accessibility to active sites, thus reducing sensitivity. Remarka-bly, the thinnest mat on the fibrous sensors created provided the highest selectivity to limonene (Selectivity Index: 72%) compared to other VOCs, such as EtOH (used as a solvent in nanofiber development), aromatic compounds (toluene), and two other monoterpenes (α-pinene and linalo-ol) with similar structure. These findings underscored the potential of the proposed integrated approach for selective VOC detection in applications such as precision agriculture and environ-mental monitoring.
Keywords:
Molecular Imprinting Polymer-MIP
; Molecularly Imprinted Nanofibers-MINF
; Electrospinning
; PVP-PAA-MWCNT composite sensor
; BVOCs
; Terpenes
; Limonene selective detection
; Precision agriculture
; Environmental VOCs monitoring
1. Introduction
Addressing the challenge of developing sustainable agricultural practices has become increasingly imperative. With a growing global population and the widespread impacts of climate change on agriculture, adopting innovative and sustainable approaches is essential to safeguarding food security [1]. Precision resource allocation and stress mitigation strategies are crucial for sustainable productivity. Timely identification of plant stress responses is vital for yield optimization and environmental conservation. However, traditional stress detection methods are often invasive or inefficient, leading to significant shortcomings [2,3]. Advances in understanding plant defense mechanisms have led to sensor-based crop health monitoring, enhancing agroecosystem sustainability. For example, methodologies facilitating prompt and precise detection of biotic plant stressors, such as pathogens and pests, allows for the deployment of remedial measures, to mitigate yield losses [4]. The term biogenic volatile organic compounds (BVOCs) encompasses a wide range of organic substances released by vegetation, soils and the oceans into the atmosphere [5,6]. BVOCs are substances generally emitted by organisms for specific purposes or as a response to external or internal factors and processes [7,8,9]. Among organisms, plants typically release biogenic volatile organic compounds as a response to environmental stresses, diseases and growth status [10,11,12]. Detecting BVOCs, like phenolics, terpenoids, alkaloids, and others, emitted by specific plant organs such as leaves, flowers, stems, and roots is an efficient yet challenging method for monitoring plant health and growth status and offers valuable insights into plant pest attacks, abiotic stressors, and additional diseases [13,14]. Their detection can be achieved through specific sensors or electronic nose-like devices [15,16,17] The latter provides a broader perspective of detecting complex odor profiles and identifying overall patterns in BVOC emissions. However, employing multivariate statistical techniques or machine learning algorithms is necessary to discern characteristic volatile profiles. In contrast, specific sensors offer detailed information about individual BVOCs, enabling targeted analysis of crucial compounds of interest. On the other hand, developing highly selective sensors can be an equally challenging strategy. Terpenes are a class of BVOCs released by plants upon a variety of biotic stresses such as pathogenic microbes, herbivore pests, and weeds, and abotic stresses including water availability, temperature fluctuations, light exposure, and salinity. They are also used by plants as vital signaling molecules for communicating and interacting with other organisms, from bacteria, to fungi, and insects playing critical roles in antagonistic and mutualistic interactions, as well as in combating pest and pathogenic attacks [18,19,20,21]. Hence, a significant opportunity exists to develop selective sensors tailored to specific terpenes. For example, the emission of limonene from plants serves as a potential biomarker for monitoring plant health and detecting early signs of stress or disease. In grapevine limonene is particularly significant, playing a critical role during nematode attacks and in responses to water stress. In olive trees, limonene emissions are related to fruit ripening, and also serve as an efficient repellent against olive fruit fly (Bactrocera oleae (Rossi)) oviposition [13,22,23]. Therefore, the development of selective sensors for monitoring limonene could provide precise and reliable diagnostics directly linked to the agents responsible for plant stress. The proliferation of selective nanosensors for VOCs detection has seen remarkable growth alongside advancements in nanomaterials and nanotechnologies. Molecularly imprinted polymer (MIP) technology has revolutionized the creation of polymeric matrices with tailored binding sites, precisely matching the molecular configurations of target analytes like terpenes. These MIP cavities customized for capturing analytes, can operate according to a lock-and-key sensing mechanism based on size, shape, or functional group ranges. Recognition sites on MIPs accommodate analytes through hydrogen bonding, π-π bonding, and van der Waals interactions, granting sensors distinguished discrimination capabilities for precise detection and quantification of intended compounds. MIP-based sensors boast exceptional chemical and thermal stability, surpassing biomolecules, and ensuring robust performance in diverse environments [24,25]. The development of MIPs to date has involved a variety of disparate approaches, including bulk, precipitation and emulsion polymerization, sol-gel methods, and electro-polymerization [26]. With respect to combinations of MIPs and transducers, either prefabricated MIP nanostructures could be deposited by drop casting, spin-coating, self-coating or growing up the substrate by thermal-, photo-induced polymerization and electro polymerization [27]. Based on transducers, MIP-based VOC sensor have been mainly exploited as chemiresistors, piezoelectric sensors, and optical sensors [28]. According to Hua et al., (2022), chemiresistors using MIPs displayed greater versatility in preparation and superior sensing capabilities compared to other MIP-based sensors for VOC detection [28]. Furthermore, higher-performance chemiresistive sensors for VOCs looked achieved by combining MIPs with many conducting polymers (e.g., polyaniline, polypyrrole, and poly3-hexylthiophene), carbon-based nanomaterials (nanotubes, nanowires, nanoflakes, nanopowder) or metal/metal oxides [29,30,31]. According to Alizadeh et al., (2013), the combination of nanostructured materials (like MWCNTs, carbon black nanoparticles-CBNPs) increased the chance to design more effective VOCs sensors [32,33]. However, despite their excellent properties, MIP-based sensors had rarely transitioned from the academic laboratory to practical application mainly due to several challenges in fabrication, involving costly and complex methods. Optimizing the production process to enable these products to reach consumers is paramount to facilitate efficient mass manufacturing [34]. An effort aimed at this outcome could be achieved by the combination between MIP and electrospinning (ES) nanotechnologies [35]. Indeed, electrospinning technology offers several advantages in the fabrication of chemosensor for VOCs. Firstly, this manufacturing technique is cost-effective, making it an economically viable option for large-scale production of sensors. Additionally, electrospinning is adaptable to various deposition substrates, allowing for versatility in sensor design and integration. Furthermore, electrospinning offers a extensive selection of polymers, including eco-friendly materials, thus promoting sustainability in sensor manufacturing processes [36,37,38,39,40]. The nanoscale features of electrospun fibers enable the resulting fibrous fabrics to mimic natural structures like cilia and hair-like protrusions, pivotal in sensing the environment [41,42] and allow the creation of tunable architectures for sensor design. More specifically, in designing chemiresistors, such continuous structures can result in more efficient conduction of current signals [35]. However, such a combination presents significant challenges due to the distinct processing methodologies involved. Electrospinning embraces forming solid fibers from charged liquid jets directed towards a collector, while molecular imprinting entails selecting a template molecule, functional monomers, and a cross-linker to create specific binding sites within a polymer matrix. Overcoming differences in processing parameters and achieving compatibility between the two techniques is crucial for successful integration. Despite these challenges, ongoing research has developed innovative approaches for an effective combination [43,44,45,46]. Regarding MIP-based electrospun VOC sensors, the most investigated procedure involves fabricating and integrating MIP nanoparticles into nanofibers [47,48,49,50,51,52,53,54,55,56]. Recently, Molinari et al. (2024) reported preliminary results about the feasibility of designing a stereoselective sensor for limonene by incorporating MIPs nanoparticles (MIP NPs) (obtained by photocrosslinking of methacrylic acid and ethylene glycol dimethacrylate monomers) into fibers of PVP containing MWCNTs [57]. The study showed that the sensor exhibited a response ~6 times higher to S(-)-limonene compared to R(+)-limonene. However, embedding MIP-NPs into electrospun nanofibers may present some challenges, including potential aggregation and uneven distribution within the polymer matrix. Moreover, non-specific interactions from the non-imprinted polymer matrix may impact recognition performance, and the incorporation of MIP particles into nanofibers can alter their physicochemical properties and subsequent recognition capacity. Conversely, an alternative approach of molecular imprinting conducted during the electrospinning process and followed by fiber mat washing might be more feasible [58]. Here, the binding sites, in the absence of a cross-linker, could be stabilized by the strong affinity between the polymer and the template, able to form recognition cavities after template removal. To date, this proposed strategy is still in its infancy in sensing applications [59,60]. Macagnano et al. (2024) presented initial results for designing a stereoselective sensor for S(-)-limonene by combining MIP and electrospinning technology in a single step [61]. They employed UV-light to induce in situ cross-linking of PVP and PAA holding S(-)-limonene as template, and the resulting sensitivity to S(-)-limonene was 3.5 times higher than to R(+)-limonene.
The objective of the present study was to investigate and optimize the architecture of the molecularly imprinted nanofibrous sensor using a single-step electrospinning process for the selective detection of limonene. More specifically, we utilized PVP and PAA as the fiber and cavity formers, respectively, while integrating multiwalled carbon nanotubes (MWCNT) to enhance conductivity. Our approach involved the development of one-step monolithic molecularly imprinted fibers via the electrospinning technique, targeting S(-)-limonene as the molecule of interest. The functional cavities were fixed within the fibers using UV curing, followed by a thorough washing process to remove the target molecule residues. This methodology was expected to facilitated the creation of specific recognition sites tailored for limonene within the nanofiber matrix, consequently simplifying the manufacturing process. We delved into the influence of nanofiber density coverage on sensor design, as well as the effect of environmental humidity on sensing characteristics.
2. Materials and Methods
2.1. Materials
Polyvinylpyrrolidone (PVP; Mw 1300000, MW 30000; Merck), Polyacrylic acid (PAA, Mw 450000, Merck), Multi Wall Carbon Nanotubes (MWCNT, Merck), S(-)-limonene (S-lim, 96%, Merck), α-Pinene (α-Pin, 98%, Merck), Linalool (Lin, 97%, Merck), Toluene (Tol, anhydrous, 99.8%), Hyaluronic Acid Sodium Salt from Streptococcus equi (HyA, Mw 750,000-1,000,000, Merck), Absolute Ethanol (EtOH, 99.8%, Merck) and N-N-Dimethylformamide (DMF, 99.8%, Merck) were reagent grade and used without further purification.
2.2. Electrospinning Solutions
The electrospinning-MIP solution was prepared by blending three different mixtures. Initially, PVP was dissolved in ethanol (C=0.12 gmL-1), combined with HyA at a ratio of 1:0.031 (w:w) (Sol A). Separately, PAA was dissolved in 5 ml of EtOH at a concentration of 0,078 gmL-1, under magnetic stirring at 50°C until complete dissolution (Sol B). A dispersion of MWCNT in DMF (0.2% w:w) was prepared by pulsed ultrasonication, vortexing, and magnetic stirring according to a previously described procedure [62]. Solution A, B, and the MWCNT dispersion were then mixed in proportions of 5:0.5:0.2 (v:v:v), respectively, under magnetic stirring until homogenization at room temperature (20°C). Finally, S(-)-limonene was added at the final suspension as the target molecule (0.055:1, v:v). To prevent the zipper-like alignment in EtOH of polymer main chains of PAA and PVP, HyA was added to the PVP solution before complex formation [63]. The NIP solution was prepared following the same procedure as the MIP solution but without the addition of the target molecule.
2.3. Electrospinning Conditions and Device Fabrication
The fibers were deposited using a Fluidnatek® LE-50 electrospinning machine (Bioinicia, Spain). To achieve regular and dry fibers, the distance between the needle and the collector was set at 12 cm, with a solution flow rate of 210 µL/h. In the electrospinning setup, the needle was charged to a voltage of +5.0 kV, while the collector maintained a potential of -2 kV. A rotating drum collector was employed to promote a more ordered arrangement of the fibers during deposition. Once the potential was applied, the polymeric dispersion jet coated the interdigitated electrodes (IDEs) affixed to the collector using conductive tape (500 rpm) and positioned within the deposition cone. The deposition process under the applied electrical field resulted in dry and fine fibers (NFs) collected following the jet bending and stretching processes, solvent evaporation, and subsequent splaying. Deposition times of 5, 10, and 15 minutes were utilized to achieve nanofibrous coverages with increasing density.
2.4. UV Crosslinking Process
Both MIP and NIP fibers were crosslinked using a 500W UV lamp (Polymer Helios Italquartz, emitting a spectrum range from 180 nm to visible) for 6 minutes in air, with a Peltier cell maintaining constant substrate temperature set to 28 °C. Samples were positioned 10 cm away from the light source.
2.5. Scanning Electronic Microscopy
The nanofibers’ size, shape, architecture, and surface characteristics were assessed using Scanning Electron Microscopy (SEM). Specifically, the electrospun nanofibrous fabrics were deposited onto thin SiO2 wafers, sputter-coated with gold using a Balzers MED 010 unit, and observed with a JEOL JSM 6010LA electron microscope (High Equipment Centre, University of Tuscia, Viterbo, Italy). The average fiber diameter was determined using Gwyddion© 2.64 software (GNU General Public License), with measurements conducted on 50 fibers per sample. Microsoft Excel© was employed to calculate the normal probability distribution of these morphological parameters, including means and standard deviations.
2.6. Atomic Force Microscopy
The topographies of the nanostructured layers were examined using Atomic Force Microscopy (AFM, Nanosurf Flex-AFM, version 5 - C3000, Liestal, Switzerland). Measurements were conducted in dynamic force mode (Dyn 190Al) with a resolution of 256 points per line and 256 lines scanning areas of 5 μm×5 μm and 20 μm×20 μm, respectively. Topography images were processed using Gwyddion© 2.64 software, with 3D visualization achieved by representing height variations with different colors, resulting in a visually informative topographical map.
2.7. Transmission Electron Microscope
Transmission Electron Microscopy (TEM) micrographs were captured at 200 keV using a transmission electron microscope equipped with an analytical double-tilt holder. Electrospun nanofibers were placed onto nickel grids (Gilder Grids, 50 mesh, 3.05mm outer diameter, Nickel) in a static mode for a short duration and observed without fixative or staining using a JEOL 1200EXII electron microscope (JEOL, Peabody, MA, USA). Micrographs were then acquired with an SIS VELETA CCD camera (Olympus, Muenster, Germany) equipped with iTEM 2014 software.
2.8. Fourier-Transform Infrared Spectroscopy
Infrared (IR) spectra of nanofibers samples were collected using a Bruker (Billerica, MA, USA) Vertex 70v Michelson Fourier Transform Infrared (FT-IR) spectrometer equipped with a DLaTGS wide range detector. Spectroscopic measurements were performed in transmission mode, at room temperature (26°C) and under vacuum conditions, in order to mitigate the interferences induced by water vapor and CO2 absorptions. For each IR spectroscopic measurement, 128 scans between 30–8000 cm−1 were acquired with a spectral resolution of 4 cm−1. The background spectrum (vacuum) was collected immediately prior to each sample measurement, with the same experimental settings of the samples. IR spectrum of each sample results from the average of five repeated and independent measurements. Raw data were visualized, processed and analyzed (absorbance calculation, baseline correction, background subtraction, cut and average, and 2nd-derivative analysis) using OPUS 8.2. software (Bruker Optics) and MATLAB (ver. 2018, MathWorks Inc., Natick, MA, USA). Background signal was subtracted calculating the IR absorption spectra A(ω) of each sample over the vacuum transmittance spectrum; then a baseline correction was applied. Second derivative analysis was performed in order to identify the positions in frequency of convoluted bands under main absorption peaks [64].
2.9. Electrical Measures
The Interdigitated Electrodes (IDEs), obtained from Micrux Technologies (Spain), were fabricated on borosilicate substrate with IDE dimensions of 10 x 6 x 0.75 mm. These IDEs featured Pt/Ti electrodes comprising 120 pairs, each measuring 10 μm wide x 5 mm long × 150 nm thick, with a 10 μm gap between them. Before electrospinning deposition, the IDEs underwent a cleaning process involving rinsing with soap and a “base piranha” mixture (3:1, v:v, aqueous ammonia and hydrogen peroxide water solution) at 60°C for approximately 30 minutes, followed by rinsing with Milli-Q water (~18 MΩ cm).
The resulting chemiresistors (IDEs + NFs) were sealed in a measuring chamber (~1 mL volume) and connected to an electrometer (Keithley 6517, Solon, Ohio, USA) capable of powering and measuring their electrical outputs, with data transmitted to a PC (LabVIEW 2014 Software, National Instruments, Austin, TX, USA). Clean air (5.0, Nippon gases) was used to record current under controlled humidity percentages and temperature values, applying potential values from −4.0 to +4.0 V. Resistance (R) of the fibrous coating, in the linearity range, was calculated using Ohm’s Law.
Dynamic sensor measurements were conducted at 25 °C by applying a +3 V potential between the interdigitated electrodes and using a 4-channel MKS 247 managing up to four MKS mass flow controllers (MFC), set in the range 0–200 sccm. Pure air humidified through a Nafion tube placed inside a sealed glass jar saturated with water vapors served as the gas carrier. The relative humidity was modulated by adjusting the flow rates through the tubes. Incoming air was blended with increasing concentrations of a set of terpenes (S(-)-limonene, ±linalool, ±α-pinene,) and ethanol and toluene as potential interferents. Both clean and vapor-saturated air flows converged in a mixing chamber (10 mL) before entering the measuring chamber. Each measurement was initiated after the complete recovery of the starting current (the baseline) under clean air flow. IDE responses were calculated as ΔI/I0, where ΔI represents current variation and I0 is the current under clean air flow.
3. Results and Discussion
3.1. Fibers Characterization
Electrospinning technology facilitated the production of nanocomposite nanofibrous layers in a single step utilizing a single needle. A scheme of the entire deposition process and molecularly imprinted sensing fibers fabrication for detecning S(-)-limonene is depicted in Figure 1. Continuous electrospun jet streams guaranteed the formation of a fibrous network within a few minutes. UV light exposure facilitated the formation of new bonds within the single fibers (photocrosslinking), making them insoluble in ethanol (the solvent used for their fabrication), thereby acquiring novel physico-chemical properties and heightened stability [65]. The cross-linking process was verified by observing that after the 5-minute electrospinning of the polymer fibers, when immersed in ethanol to remove the template (S(-)-limonene), appeared to retain their structure without deforming or dissolving in their solvent, and remaining firmly attached to the substrates (Figure 2).
The SEM images in Figure 2 illustrates polymer nanofibers arranged sparsely, displaying smooth surfaces and tubular forms.
These nanofibers exhibited moderate alignment, with interconnected voids which are expected to promote gas diffusion for improved efficacy in gas-VOC sensing. Figure 2B,D depicts the normalized distribution of fiber dimensions. In Figure 2C,D molecularly imprinted nanofibers (MINFs) exhibited a narrow, well-defined diameter distribution, with a mean diameter of 977 nm and a standard deviation(SD) of ±140 nm. In contrast, non-imprinted nanofibers (NINFs) (Figure 2A,B) were approximately 42% thinner on average than MINFs (Ø: 688±122 nm), with a more pronounced and upward-curving Gaussian curve shape indicating the influence of template S(-)-limonene during deposition and photocrosslinking.
The differences between NINFs and MINFs surfaces roughness were emphasized through atomic force microscopy (5x5 μm, achieved in amplitude mode). The AFM micrographs exhibited absence of significant defects and irregularities on the surfaces, especially in the case of MINF (Figure 3,B), where a perfectly tubular and homogeneous fiber diverged from NINFs for their slight wrinkles over the length (Figure 3A).
Carbon nanotubes distribution and orientation inside polymer nanofibers were captured by TEM images. However, TEM micrographs provided information only about the thinnest nanofibers of MINFs and NINFs, because their size enabled the electron beam bombardment under the vacuum to provide images clear enough to display at nanoscale resolution the presence of MWCNT within the polymer matrix. In Figure 4A,B MWCNTs looked successfully embedded in the dispersing polymer matrix and well-oriented along the fiber axis (nevertheless exhibiting some degree of tortuousity) presuming that the original polymer dispersion contained individual nanotubes rather than aggregates or bundles, that is a common big challenge in developing reproducible resistive tools [66]. On the other hand, the imperfect alignment could be due to the electrospinning process, not sufficient for fully stretching the nanotubes (such as the solution viscosity, the applied electric field strength, the flow rate, the collector type, the solvent, and the interactions between the polymer blend (PVP-PAA) and the nanotubes, etc. [67]. Furthermore, in some other regions of the nanofibres, the nanotubes appeared in more irregular conformations, exhibiting bending and even protrusion out of the nanofiber (Figure 4C), mainly in conjunction with the irregularities in the nanofiber.
The SEM images in Figure 5A–E) captured the evolving morphology of electrospun nanofibers (MINFs) as the deposition process proceeded. Initially, after 5 min-growth the field of view showed isolated nanofibers with relatively sparse coverage (Figure 5A,D). These early-stage nanofibers appeared as fine threads extending across the substrate. The dipping of nanofibers in EtOH to remove the template did not appear to have influenced their distribution and shape. As the electrospinning process continued (up to 10 and 15 min, respectively), the SEM images revealed the development of a more complex and interconnected network (Figure 5B,C,E,F).
The nanofibers exhibited a denser packing, leading to the formation of a three-dimensional scaffold (enhanced by AFM in Figure 5H,I). These fibers became entangled and overlaid, creating a mesh-like structure with increased surface area. The individual nanofibers within the network displayed trajectories intersected, resulting in points of contact and potential bonding between adjacent fibers. Smaller bundles, observed in the 10 min-samples (Figure 5B,E,H), consisted of a modest number of merged nanofibers, forming tight aggregations that shared a common alignment, comprising a few closely packed nanofibers. In the 15 min-samples larger bundles occurred, where a higher number of nanofibers merged together, according to a more complex architecture and a thicker and more densely packed network and enhanced in the 3D elaboration of AFM images. Here bundling, branching and surface undulations became more apparent, providing insights into the intricate architecture of the nanofiber network (Figure 5I). Ethanol washing by dipping seemed to affect the arrangement of these fibers, grouping them into bundles where individual nanofibers merged together to form cohesive structures.
3.2. FTIR Spectroscopy
FTIR spectroscopy was used to investigate tThe photocrosslinking effects on the nanofibres (Figure 6). As shown in Figure 6, UV-irradiated MINFs exhibited broader and less intense spectral features between 1100 and 1900 cm-1 compared to the non-irradiated analogous sample. This evidence indicated that UV radiation caused changes in the molecular structure of the nanofibers. In fact, in Figure 6A a decrease in intensity could be observed in the absorption bands associated with the PVP ring (1200-1320 cm-1) [68] and the methylene group (1380-1480 cm-1). This decrease suggests that UV radiation might have induced structural changes in the polymer, through main chain scission [65]. As concerns the variations observed in the shape of the typical C=O stretching band (between 1600 and 1750 cm-1), they could be attributed to either interactions between PVP and PAA or to the effects of UV irradiation. This suggests changes in the chemical environment around the carbonyl groups, indicating interactions between the polymers or modifications induced by UV radiation. Figure 6B compares the C=O stretching band among the samples with and without UV irradiation and following the template elution.
The no UV-treated nanofibres and the UV-treated ones without washes both exhibited a ν(C=O) band at 1680 cm-1.
However, in the no UV-treated sample, this band is narrower and more intense, indicating a higher concentration or more defined structure of carbonyl groups [68,69]. The decrease in intensity of this band, observable in the UV-treated samples, could be linked to photodegradation progress of the polymers, due to side group abstraction or destruction [65]. The UV-treated sample with ethanol washes showed a slightly shifted ν(C=O) band at 1684 cm-1. This shift may suggest changes in the chemical environment around the carbonyl groups induced by the washing process after UV treatment.
After UV treatment, a shoulder is also formed at 1653 cm-1, indicating the presence of additional chemical groups or structural changes induced by UV irradiation. Finally, the different shapes of the C=O bands could be attributed to interactions between PAA and PVP [65], as well as the changes induced by UV radiation [65,68,69].
The second derivative negative minima in Figure 6C shows the positions of convoluted absorption bands. There is an evident difference among sample without and with UV irradiation. There is the formation of a new band around 1655 cm-1, a decrease in intensity of band at 1674 cm-1 and a shift to lower frequency of the band at higher frequencies, from 1688 to 1684 cm-1. These observations confirmed valuable insights into the nanofibers molecular changes induced by both UV treatment and template washing.
3.3. Electrical and Sensing Features
The measuring setup is depicted in Figure 7A. Figure 7B reports the current-voltage (I-V) plot for both the IDEs coated with MINFs and NINFs after 5 min of deposition, exhibiting a semiconductor behavior with a Schottky barrier. In the I-V plot, x-axis represents the applied voltage across the electrode, ranging from +4 V to -4 V, with 0 V at the center. The y-axis corresponds to the current passing through the electrode. According to the following equation:
(where N and L are number and size of the fingers, h and w the electrode thickness and width, respectively, and ρ la resistivity of the overlying material) the whole resistance is ruled by IDEs layout and the resistivity of the overlying material [70].
Depicted in Figure 7B, as the voltage increased positively, both MINFs and NINFs current remained stable until reaching a threshold at around +1 V, signalling the initiation of charge carrier injection into the nanocomposite. Beyond this threshold, a significant increase in current occurred, suggesting Schottky barrier formation at the electrode interface, leading to semiconductor-like behavior with nonlinear characteristics. The trend mirrored, but with a negative sign, was observable when negative voltage was applied. At higher positive or negative voltages as the potential increased, the current increased linearly for both the IDEs but with different slope (RNINF:~1.49·109 Ohm; RMINF:~2,78·109 Ohm). The conductivity mechanism is presumably provided by a combination of charge transport within the polymer matrix generated by the conductive pathways created by MWCNTs [71]. Indeed, the addition of MWCNTs to the PVP matrix enhances electrical conductivity, despite PVP’s natural poor conductivity. The nitrogen heteroatom in PVP may facilitate electron acceptance, forming charge-transfer complexes. However, since MWCNT concentration is below the percolation threshold, charge transport could be likely dominated by tunnelling. The formation of a Schottky barrier could be attributed to heterojunction at MWCNTs-PVP interfaces and imperfections in the nanofiber-electrode boundary. The IV-curves was the same in shape for both MINFs and NINFs, but the resistance appeared still higher in the MINFs, probably affected by various factors, like the density of the nanofiber network over the electrodes, the diameter of the nanofibers, and the overall morphology of the coating [72]. Indeed, NINFs arranged according to a network of thinner fibers on the electrodes, could contribute to a higher surface area per unit volume than MINFs, achieving more contact points between adjacent fibers and a larger overall interfacial area with the IDEs.
Furthermore, such an increased surface area should enhance the opportunities for charge carrier interaction and improve the probability of charge transfer between the nanofibrous layer and the electrodes. On the contrary, MINF layer arranged into in a sparser architecture may have fewer contact points and a less interconnected structure, leading to a higher resistivity.
Figure 8A illustrates the correlation between environmental humidity levels and the conductivity of the MINF sensor. Specifically, as humidity increased, there was an exponential decrease in resistance, transitioning from approximately 1011 Ohms in dry air to 108 Ohms in highly humid conditions. Figure 8A presents the same data on a semilogarithmic scale, facilitating an understanding of how resistance varies with differing humidity levels. The interaction mechanisms occurring at the polymeric surface of fibers with water vapors can be diverse and sometimes contradictory. The observed decrease in resistance is likely attributed to the inherently hydrophilic properties of the polymers under examination. As humidity levels rose, the polar water molecules were readily absorbed by PVP-PAA, facilitated by the layer porosity, the nitrogen atom in PVP and the carboxylic acid groups (-COOH) along the PAA chains. These absorbed water molecules are presumed to participate in conductivity through ions, following the Grotthuss transfer mechanism [73]. Furthermore, PAA, being an acrylic acid-based polymer, could contribute to fiber conductivity through its ionizable groups. According to Pan et al. (2016) [74], ion carriers (H+/H3O+) were responsible of weakening the barrier at heterojunctions between polymers and MWCNTs, thus reducing the resistance in composite nanofibers and improving conductivity. However, it’s worth noting that PVP and PAA are polymers known for their insulating or dielectric properties [75,76]. Therefore, in the absence of moisture and with a concentration of MWCNTs falling below the percolation threshold, the movement of charge carriers is presumably not facilitated [77,78,79]. On the other hand, when the relative humidity exceeded 60%, polymer swelling could occur, counteracting a further decrease in resistance, as depicted in Figure 8A.
The influence of humidity on the sensor features was also investigated. The MINF5min sensor was exposed to a known concentration of S(-)-limonene (55 vpm) while varying the %RH. At approximately 50% relative humidity (RH), the sensor responses demonstrated the highest levels of response, stability, and reproducibility, even amidst fluctuations of %RH up to ±10% (see Figure 8B). Extreme humidity conditions (<20% RH or >60% RH) adversely affected both electrical and sensor functionalities. In lower humidity (<40%), polymer dehydration reduced free ions or charge carriers, resulting in decreased polymer conductivity and subsequent current changes when exposed to the VOC. Conversely, in higher humidity (>60% RH), polymer absorption of water molecules increased conductivity, yet sensor responses to S(-)-limonene were diminished presumably due to factors such as competition between water molecules and limonene for binding sites, and/or alterations in surface properties affecting limonene interaction. Thus, humidity control looks essential for ensuring accurate and reproducible sensor operation. Therefore, in this study, IV curves and sensing measurements were carried out at around 50% RH, representing optimal sensor operating conditions. In order to value the effectiveness of the designed sensor, NINFs and MINFs 5min-chemoresistors were exposed to an air flow containing a concentration of 40 vpm of S(-)-limonene. Both materials exhibited an increase in current, however, the response for MINFs was approximately 55.4 times greater (Figure 9). The substantial disparity in responses appeared to validate the efficacy of the adopted procedure in creating “molecular cavities” within the polymer fibers, showcasing a notable affinity of MINFs for its template.
Figure 10A depicts the IDEs coated with electrospun MINFs, following the three deposition times (5, 10 and 15 min, respectively). After three different electrospinning deposition durations and template washing, the microelectrodes appeared coated with increasing fibers density. The resistance values, calculated outside the Schottky barrier region, exhibited a linear decrease with increasing deposition time (Figure 10B,C). As expected, the increase in density nanofibers over IDEs led to more conductive systems due to the increase in the number of pathways available for charge carriers to travel between the electrodes.
The impact of the 5-10-15 min-nanofibrous layers on S(-)-limonene detection was explored by subjecting the three sensor types to increasing concentrations of the template in air within a range spanning from 15 to 140 vpm. The sensors responses to S(-)-limonene were characterized by a swift increase in current, demonstrating rapid reactivity (Figure 11A–C). All the sensors exhibited a remarkable ability to reach a plateau within a mere 200 seconds according to Langmuir-like kinetics [80], indicating a prompt and effective recognition of the VOC. Additionally, the sensors responses showed a consistent restoration to baseline levels when exposed to clean air, showcasing their repeatability and reliability. As expected, increasing the thickness and the density of the nanofibrous layer (from 5 to 10 min electrospun sensor), the sensor responses looked improved (Figure 11B,E). Such an effect presumably was due to the increasing of the surface area of the sensing material, which allowed for a greater adsorption of S-Limonene and resulted in a more pronounced sensor response.
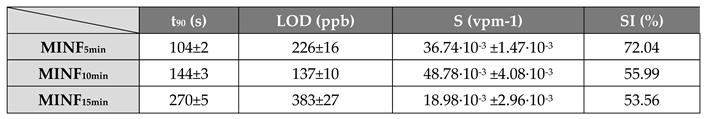
On the other hand, a denser network of polymer nanofibers in a sensor could lead to longer response times due to diffusion limitations and intermolecular interactions. These effects appear to be substantiated by the estimated values presented in the Table 1, detailing the response times (measured as t90, that is the time required by the sensor to reach 90% of the response) and VOC detection limits for each sensor. As the fiber density increased, the response time exhibited an increase of up to 60%. About the sensor responses, they showed a 1.5-fold enhancement when transitioning from the 5-min to the 10-min sensor, followed, conversely, by a significant decrease from the 15-min one (approximately four times smaller than the 10 min- sensor response). Consequently, the Limit of Detection (LOD), calculated as 3 times the standard deviation of the blank, showed a decreasing trend, up to - 39%, from the 5 -min to the 10 min-sensor. This result suggests that a denser fibrous network, although it increased the estimated response time by a few seconds, it allowed the detection of lower S(-)-limonene concentrations down to approximately 137 ppb. However, when an even denser network of fibers occurred (the 15 min one), it reversed this trend by reporting a higher value of LOD, even higher than the sensor with the poorest fiber network (more specifically about +41% and +64% than 5 min- and 10 min-sensors, respectively, as shown in Table 1). The latter trend could be explained by the limited accessibility of S-limonene to the active sites when fibers overlapped and merged together (Figure 5C,F), as observed in SEM and AFM images. As both the transient response shapes and the calibration curves are related to the adsorbing mechanisms resulting in the chemical affinity of the VOCs to the material, the trade-off between sensitivity and response time is a further consideration in optimizing the design of the sensor. The Langmuir-like shaped calibration curves of both 5 min- and 10 min-sensors related to the current changes when the analyte partial pressure increased, undertook that adsorption occurred at specific sites on the sensor’s surface, and these sites could be occupied by only one molecule at a time. As the concentration of the analyte increased, the sensor response initially rised sharply until reaching a saturation point. At this point, the active sites on the sensor’s surface should be predominantly occupied, reaching an equilibrium between adsorption and desorption processes, and leading to a plateau in the response. This shape strongly suggests a high affinity between the sensor and the analyte. The sensor sensitivity, defined as the change in response per unit change in concentration [81], and typically calculated as the slope of the linear portion of the Langmuir-like calibration curve (S=∆Inorm⁄C, where ΔInorm and C represent the change in the current normalized to its baseline value and the analyte concentration, respectively) plays a pivotal role in determining the performance and applicability of a sensor. The calculated sensitivity for S(-)-limonene was notably higher (+25%) in the MINF10min sensor than in MINF5min (Table 1, Figure 12A,B,C,E). Conversely, the flattening of the MINF15min sensor calibration curve (Figure 12F), approaching an almost linear profile with a significantly reduced slope of about 61%, described the decline in sensing performance with a continued increase in the fibrous network. This alteration may result from the slight swelling of the fibers occurring during the template elution by dipping, introducing a more tortuous quality to the fibers (Figure 5C,F). Nevertheless, this effect could also stem from a partial alteration of the molecular imprinted cavities formed along the fiber during the UV-light irradiation. Indeed, during the brief photo-crosslinking procedure, it is plausible that the underlying fibers closer to the substrate may receive less UV-irradiation, potentially diminishing the effectiveness of this treatment. Alternatively, extending the exposure duration was discouraged due to the potential oxidization of limonene, as described in Supplementary Materials paragraph and in Figure S1. To evaluate the sensor’s ability to detect selectively its template, the MINF sensor was exposed once again to increasing concentrations of selected VOCs across the three different architectures (5-, 10-, 15-min). These VOCs included EtOH, used as a solvent in nanofiber development, an aromatic compound (toluene), and two different terpenes with similar molecular structures (α-pinene and linalool).
Calibration curves for these VOCs displayed a linear shape (with the exception of limonene), indicating adsorption behavior resembling a Henry-type isotherm [82], whereas the relationship between the VOC concentration and adsorbed amount was directly proportional and not limited to join the MIP binding sites, then indicating a lower affinity (Figure 12A). The graph in Figure 12B illustrates the estimated sensitivities of all three MINFs to these volatile compounds. While maintaining peak sensitivity and Langmuir-like curve shape, MINF10min exhibited a furtherly reduced selectivity towards compounds with similar molecular structures, such as linalool (Figure 12B), while the other volatile organic compounds (VOCs) remained undetected. Indeed, the sensitivities to toluene and ethanol were minimal, making them imperceptible on the graph when compared to the other values (insets in Figure 12D). The minimal signal responses to EtOH, characterized by a decrease in conductivity (opposite sign in sensitivity value), appeared to result solely from a slight swelling effect. The MINF15min sensor showed the poorest performance (Figure 12C,D). This outcome validates the notion that as the fiber network grows, although the number of available MIP sites was increasing, the washing treatment of the templates jeopardized their sensor functionality.
Here, while confirming in all cases the greater sensitivity of MINFs to their own template (S(-)-limonene), an optimal selectivity seemed to be attained solely in the case of the thinnest fibrous network, where the sensitivity values decreased by −63% for linalool and up to −99% for the other tested VOCs (i.e., α-pinene, EtOH, and toluene).
Based on existing studies, Kikuchi et al. (2006) demonstrated the efficacy of template imprinting technology in designing a thin film sensor based on quartz crystal microbalance (QCM) for limonene detection. This study employed methacrylic acid as a functional monomer, ethylene glycol dimethacrylate (EGDMA) as a cross-linker, and 2,2′-azobisisobutyronitrile (AIBN) as an initiator. The sensor exhibited a limit of detection (LOD) of approximately 10 ppm and a selectivity of around 55%, distinguishing limonene from limonene oxide and α-pinene [Kikuchi, M.; Tsuru, N.; Shiratori, S. Recognition of Terpenes Using Molecular Imprinted Polymer Coated Quartz Crystal Microbalance in Air Phase. Sci. Technol. Adv. Mater. 2006, 7, 156–161]. Ghatak et al., (2019) presented a QCM-MIP sensor prepared from the copolymer of methacrylic acid and ethylene glycol dimethacrylate for detecting R(+)-limonene in varieties of mango with a sensitivity of 0.16 Hz/ppm, repeatability and reproducibility of 98.4% and 98.8%, respectively and with selectivity factor of 58.16% (RH=41%), T=27°C) [B. Ghatak et al., “Selective and Sensitive Detection of Limonene in Mango using Molecularly Imprinted Polymer Based Quartz Crystal Microbalance Sensor,” 2019 IEEE International Symposium on Olfaction and Electronic Nose (ISOEN), Fukuoka, Japan, 2019, pp. 1-3, doi: 10.1109/ISOEN.2019.8823318]. Völke et al. (2022) developed a conductive molecularly imprinted polymer (cMIP) capable of detecting R(+)-limonene down to 50 ppm. Their approach involved blending polystyrene-based MIPs with poly3-hexylthiophene (P3HT) and subsequent deposition on quartz crystal microbalance (QCM) and interdigitated electrodes (IDEs) through spin- and drop-coating, respectively [Julia Völkle, Katarina Kumpf, Adriana Feldner, Peter Lieberzeit, Philipp Fruhmann, Development of conductive molecularly imprinted polymers (cMIPs) for limonene to improve and interconnect QCM and chemiresistor sensing, Sensors and Actuators B: Chemical,356, 2022,131293]. Their investigation revealed a relationship between template concentration and sensor response, with an initial increase in response observed with a limonene to styrene molar ratio of 2.0:2.6. Subsequent declines in response at higher concentrations may be attributed to limonene impeding polymerization and potentially forming covalent bonds with the polymer. Following the determination of optimal proportions between the components of MIPs yielding the most favorable responses to terpenes, Iqbal et al. (2010) proceeded to construct a multiarray device. This device facilitated the monitoring of terpene emanation patterns from both fresh and dry grass, achieving a limit of detection (LOD) below 20 ppm [Iqbal N., et al. Sensors 2010, 10, 6361-6376]. Hawari et al. (2013), designed a membrane MIP based on methacrylic acid and an a gold IDE on PET for designing a capacitive sensor for α-pinene, as a biomarker of the maturity stage of a mango. All MIP coated IDE were then polymerized under UV light at room temperature for 6 hours. The remained molecule on MIP can be removed by immersing it with mixture of methanol and acetic acid for extraction of templates thus allowing the possibility for the sensor to be used repeatedly [Nurul Maisyarah Samsudin, Mohd Noor Ahmad, Ali Yeon Md Shakaff, Supri. A. Ghani, Yufridin Wahab, and Uda Hashim. 2012. Recognition of Limonene Volatile Using Interdigitated Electrode Molecular Imprinted Polymer Sensor. Procedia Engineering 53 (2013) 197 – 202. DOI: 10.1016/j.proeng.2013.02.026].
In the present study, an estimation of sensor selectivity [83] among the tested VOCs was described by the selectivity index (SI):
where Starget is the sensitivity to the defined template and Sinterferents is the sensitivity value to the other chemicals within the measured pattern.
𝑆I=Starget/∑Sinterferent ×100,
This parameter indicated that the MINF5min sensor demonstrated a selectivity index of 72% towards the template, whereas MINF10min and MINF15min exhibited lower values, namely 56% and 53%, respectively.
4. Conclusions
Exploring the intricacies of plant communication, the monitoring of volatile organic compounds (VOCs) released under stress conditions offers a captivating glimpse into ecosystem health and vitality. Terpenes, acting as potent indicators of stressors such as drought, disease, and pollution, provide valuable insights into how plants respond to environmental challenges. By tracking these VOCs, scientists can detect stress early and devise targeted intervention strategies. This not only improves agricultural practices, leading to enhanced crop yields and resilience, but also deepens our understanding of ecosystem dynamics and the impacts of climate change. Based on the findings presented in this study, several conclusions can be drawn regarding the development and performance of molecularly imprinted nanofiber (MINF) sensors for detecting VOCs, particularly focusing on limonene as a biomarker for plant health monitoring. Our study advances the field of VOC sensing through the development and optimization of molecularly imprinted nanofiber (MINF) sensors, with a specific focus on limonene as a key biomarker for plant health monitoring. Overcoming theoretical hurdles, we innovatively address practical challenges associated with molecular imprinting during electrospinning, paving the way for the integration of electrospinning, molecular imprinting, conductive nanofibers, and rapid photopolymerization onto the transducer, successfully exploring an alternative crosslinking methods compatible with electrospinning. This comprehensive approach streamlines sensor fabrication, potentially reducing time and complexity compared to traditional methods, while enhancing sensor performance and expanding application potential in precision agriculture. MIP-based conductimetric sensors for limonene were developed using polyvinyl pyrrolidone (PVP) and polyacrylic acid (PAA) as fiber and functional cavity formers, respectively, with multiwalled carbon nanotubes (MWCNT) for conductivity. The creation of recognition sites for limonene within the nanofiber matrix holds promise for enhancing sensor performance, reducing manufacturing time, and expanding application potential in precision agriculture. Humidity control emerges as a critical factor, ensuring precise and reproducible sensor operation, with optimal conditions identified at approximately 50% relative humidity (%RH). Our sensors exhibit rapid responses to S(-)-limonene, reaching steady-state within 200 seconds, demonstrating their suitability for real-time monitoring applications. The impact of the thickness of the nanofibrous coating on sensor performance was investigated, revealing that increased fiber density improved sensor performance by enhancing surface area for greater adsorption of S(-)-limonene (LOD: 137 ppb). However, excessive fiber density subjected to UV-irradiation and ethanol washing, decreased the accessibility to active sites, leading to reduced sensitivity. Therefore, while all sensors exhibited great sensitivity to their target molecule, optimal selectivity (SI: 73%) is achieved with the thinnest fibrous network, highlighting the intricate interplay between sensor architecture and performance characteristics.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
References
- S. Datta, I. Hamim, D.K. Jaiswal, R. Sungthong, Sustainable agriculture, BMC Plant Biol. 23 (2023) 588. [CrossRef]
- Galieni, N. D’Ascenzo, F. Stagnari, G. Pagnani, Q. Xie, M. Pisante, Past and Future of Plant Stress Detection: An Overview From Remote Sensing to Positron Emission Tomography, Front. Plant Sci. 11 (2021). [CrossRef]
- Cagliero, G. Mastellone, A. Marengo, C. Bicchi, B. Sgorbini, P. Rubiolo, Analytical strategies for in-vivo evaluation of plant volatile emissions - A review, Anal. Chim. Acta. 1147 (2021) 240–258. [CrossRef]
- Kashyap, R. Kumar, Sensing Methodologies in Agriculture for Monitoring Biotic Stress in Plants Due to Pathogens and Pests, Inventions. 6 (2021) 29. [CrossRef]
- M. Cai, C. An, C. Guy, A scientometric analysis and review of biogenic volatile organic compound emissions: Research hotspots, new frontiers, and environmental implications, Renew. Sustain. Energy Rev. 149 (2021) 111317. [CrossRef]
- G.F. Bennett, Volatile organic compounds in the atmosphere, J. Hazard. Mater. 50 (1996) 245–246. [CrossRef]
- J.K. Holopainen, J. Gershenzon, Multiple stress factors and the emission of plant VOCs, Trends Plant Sci. 15 (2010) 176–184. [CrossRef]
- J. Poveda, Beneficial effects of microbial volatile organic compounds (MVOCs) in plants, Appl. Soil Ecol. 168 (2021). [CrossRef]
- L. NERI, F. SILLO, E. ZAMPIERI, E. LUMINI, G. MARTURANO, C.D. ESPOSTI, G. PETRUZZELLI, B. GIOLI, A. ZALDEI, R. BARALDI, R. BALESTRINI, A combined analysis based on microbial communities and volatile organic compounds as a tool to study soil quality in an urban environment, Pedosphere. 33 (2023) 670–675. [CrossRef]
- X. Bao, W. Zhou, L. Xu, Z. Zheng, A meta-analysis on plant volatile organic compound emissions of different plant species and responses to environmental stress, Environ. Pollut. 318 (2023) 120886. [CrossRef]
- C. Faiola, D. Taipale, Impact of insect herbivory on plant stress volatile emissions from trees: A synthesis of quantitative measurements and recommendations for future research, Atmos. Environ. X. 5 (2020) 100060. [CrossRef]
- Mozaffar, N. Schoon, A. Bachy, A. Digrado, B. Heinesch, M. Aubinet, M.L. Fauconnier, P. Delaplace, P. du Jardin, C. Amelynck, Biogenic volatile organic compound emissions from senescent maize leaves and a comparison with other leaf developmental stages, Atmos. Environ. 176 (2018) 71–81. [CrossRef]
- V. Ninkuu, L. Zhang, J. Yan, Z. Fu, T. Yang, H. Zeng, Biochemistry of Terpenes and Recent Advances in Plant Protection, Int. J. Mol. Sci. 22 (2021) 5710. [CrossRef]
- Munawar, Y. Xu, A.S. Abou El-Ela, Y. Zhang, J. Zhong, Z. Mao, X. Chen, H. Guo, C. Zhang, Y. Sun, Z. Zhu, I.T. Baldwin, W. Zhou, Tissue-specific regulation of volatile emissions moves predators from flowers to attacked leaves, Curr. Biol. 33 (2023) 2321-2329.e5. [CrossRef]
- Z. Gan, Q. Zhou, C. Zheng, J. Wang, Challenges and applications of volatile organic compounds monitoring technology in plant disease diagnosis, Biosens. Bioelectron. 237 (2023) 115540. [CrossRef]
- F. De Cesare, E. Di Mattia, S. Pantalei, E. Zampetti, V. Vinciguerra, F. Canganella, A. Macagnano, Use of electronic nose technology to measure soil microbial activity through biogenic volatile organic compounds and gases release, Soil Biol. Biochem. 43 (2011) 2094–2107. [CrossRef]
- R.K. Murali-Baskaran, P. Mooventhan, D. Das, A. Dixit, K.C. Sharma, S. Senthil-Nathan, P. Kaushal, P.K. Ghosh, The future of plant volatile organic compounds (pVOCs) research: Advances and applications for sustainable agriculture, Environ. Exp. Bot. 200 (2022) 104912. [CrossRef]
- N. Dudareva, F. Negre, D.A. Nagegowda, I. Orlova, Plant Volatiles: Recent Advances and Future Perspectives, CRC. Crit. Rev. Plant Sci. 25 (2006) 417–440. [CrossRef]
- M.E. Maffei, Sites of synthesis, biochemistry and functional role of plant volatiles, South African J. Bot. 76 (2010) 612–631. [CrossRef]
- F. Loreto, J.-P. Schnitzler, Abiotic stresses and induced BVOCs, Trends Plant Sci. 15 (2010) 154–166. [CrossRef]
- M. Rosenkranz, Y. Chen, P. Zhu, A.C. Vlot, Volatile terpenes – mediators of plant-to-plant communication, Plant J. 108 (2021) 617–631. [CrossRef]
- Aina, O.O. Bakare, A.O. Fadaka, M. Keyster, A. Klein, Plant biomarkers as early detection tools in stress management in food crops: a review, Planta. 259 (2024) 60. [CrossRef]
- R. Malheiro, S. Casal, S.C. Cunha, P. Baptista, J.A. Pereira, Identification of leaf volatiles from olive (Olea europaea) and their possible role in the ovipositional preferences of olive fly, Bactrocera oleae (Rossi) (Diptera: Tephritidae), Phytochemistry. 121 (2016) 11–19. [CrossRef]
- C. Malitesta, E. Mazzotta, R.A. Picca, A. Poma, I. Chianella, S.A. Piletsky, MIP sensors – the electrochemical approach, Anal. Bioanal. Chem. 402 (2012) 1827–1846. [CrossRef]
- M. Cieplak, W. Kutner, Artificial Biosensors: How Can Molecular Imprinting Mimic Biorecognition?, Trends Biotechnol. 34 (2016) 922–941. [CrossRef]
- L. Chen, X. Wang, W. Lu, X. Wu, J. Li, Molecular imprinting: perspectives and applications, Chem. Soc. Rev. 45 (2016) 2137–2211. [CrossRef]
- M. Ávila, M. Zougagh, Á. Ríos, A. Escarpa, Molecularly imprinted polymers for selective piezoelectric sensing of small molecules, TrAC Trends Anal. Chem. 27 (2008) 54–65. [CrossRef]
- Y. Hua, Y. Ahmadi, K.H. Kim, Molecularly imprinted polymers for sensing gaseous volatile organic compounds: opportunities and challenges, Environ. Pollut. 311 (2022). [CrossRef]
- S. Antwi-Boampong, K.S. Mani, J. Carlan, J.J. BelBruno, A selective molecularly imprinted polymer-carbon nanotube sensor for cotinine sensing, J. Mol. Recognit. 27 (2014) 57–63. [CrossRef]
- X. Tang, J.-P. Raskin, D. Lahem, A. Krumpmann, A. Decroly, M. Debliquy, A Formaldehyde Sensor Based on Molecularly-Imprinted Polymer on a TiO2 Nanotube Array, Sensors. 17 (2017) 675. [CrossRef]
- M.F. Koudehi, S.M. Pourmortazavi, R. Zibaseresht, S. Mirsadeghi, << MEMS-Based PVA/PPy/MIP Polymeric- Nanofiber Sensor Fabricated by LIFT-OFF Process for Detection 2,4-Dinitrotoluene Vapor, IEEE Sens. J. 21 (2021) 9492–9499. [CrossRef]
- T. Alizadeh, F. Rezaloo, A new chemiresistor sensor based on a blend of carbon nanotube, nano-sized molecularly imprinted polymer and poly methyl methacrylate for the selective and sensitive determination of ethanol vapor, Sensors Actuators B Chem. 176 (2013) 28–37. [CrossRef]
- T. Alizadeh, F. Rezaloo, Toluene chemiresistor sensor based on nano-porous toluene-imprinted polymer, Int. J. Environ. Anal. Chem. 93 (2013) 919–934. [CrossRef]
- A.J. Kadhem, G.J. Gentile, M.M. Fidalgo de Cortalezzi, Molecularly Imprinted Polymers (MIPs) in Sensors for Environmental and Biomedical Applications: A Review, Molecules. 26 (2021) 6233. [CrossRef]
- K.D. Patel, H.W. Kim, J.C. Knowles, A. Poma, Molecularly Imprinted Polymers and Electrospinning: Manufacturing Convergence for Next-Level Applications, Adv. Funct. Mater. 30 (2020). [CrossRef]
- M. Keshvardoostchokami, S.S. Majidi, P. Huo, R. Ramachandran, M. Chen, B. Liu, Electrospun Nanofibers of Natural and Synthetic Polymers as Artificial Extracellular Matrix for Tissue Engineering, Nanomaterials. 11 (2020) 21. [CrossRef]
- R.M.D. Soares, N.M. Siqueira, M.P. Prabhakaram, S. Ramakrishna, Electrospinning and electrospray of bio-based and natural polymers for biomaterials development, Mater. Sci. Eng. C. 92 (2018) 969–982. [CrossRef]
- A.R. Borah, P. Hazarika, R. Duarah, R. Goswami, S. Hazarika, Biodegradable Electrospun Membranes for Sustainable Industrial Applications, ACS Omega. 9 (2024) 11129–11147. [CrossRef]
- Macagnano, E. Zampetti, E. Kny, eds., Electrospinning for High Performance Sensors, Springer International Publishing, Cham, 2015. [CrossRef]
- Macagnano, V. Perri, E. Zampetti, A. Bearzotti, F. De Cesare, Humidity effects on a novel eco-friendly chemosensor based on electrospun PANi/PHB nanofibres, Sensors Actuators B Chem. 232 (2016) 16–27. [CrossRef]
- M. Asadnia, A.G.P. Kottapalli, K.D. Karavitaki, M.E. Warkiani, J. Miao, D.P. Corey, M. Triantafyllou, From Biological Cilia to Artificial Flow Sensors: Biomimetic Soft Polymer Nanosensors with High Sensing Performance, Sci. Rep. 6 (2016) 32955. [CrossRef]
- S. Sankar, C.S. Sharma, S.N. Rath, S. Ramakrishna, Electrospun nanofibres to mimic natural hierarchical structure of tissues: application in musculoskeletal regeneration, J. Tissue Eng. Regen. Med. 12 (2018) e604–e619. [CrossRef]
- S. Chigome, G. Darko, N. Torto, Electrospun nanofibers as sorbent material for solid phase extraction, Analyst. 136 (2011) 2879. [CrossRef]
- S. Chigome, N. Torto, A review of opportunities for electrospun nanofibers in analytical chemistry, Anal. Chim. Acta. 706 (2011) 25–36. [CrossRef]
- Ghorani, N. Tucker, M. Yoshikawa, Approaches for the assembly of molecularly imprinted electrospun nanofibre membranes and consequent use in selected target recognition, Food Res. Int. 78 (2015) 448–464. [CrossRef]
- S.A. Zaidi, Recent developments in molecularly imprinted polymer nanofibers and their applications, Anal. Methods. 7 (2015) 7406–7415. [CrossRef]
- K. Yoshimatsu, L. Ye, P. Stenlund, I.S. Chronakis, A simple method for preparation of molecularly imprinted nanofiber materials with signal transduction ability, Chem. Commun. (2008) 2022. [CrossRef]
- K. Yoshimatsu, L. Ye, J. Lindberg, I.S. Chronakis, Selective molecular adsorption using electrospun nanofiber affinity membranes, Biosens. Bioelectron. 23 (2008) 1208–1215. [CrossRef]
- S. Piperno, B. Tse Sum Bui, K. Haupt, L.A. Gheber, Immobilization of Molecularly Imprinted Polymer Nanoparticles in Electrospun Poly(vinyl alcohol) Nanofibers, Langmuir. 27 (2011) 1547–1550. [CrossRef]
- L. Zhang, Y. Guo, W. Chi, H. Shi, H. Ren, T. Guo, Electrospun nanofibers containing p-nitrophenol imprinted nanoparticles for the hydrolysis of paraoxon, Chinese J. Polym. Sci. 32 (2014) 1469–1478. [CrossRef]
- F. Liu, Q. Liu, Y. Zhang, Y. Liu, Y. Wan, K. Gao, Y. Huang, W. Xia, H. Wang, Y. Shi, Z. Huang, B. Lu, Molecularly imprinted nanofiber membranes enhanced biodegradation of trace bisphenol A by Pseudomonas aeruginosa, Chem. Eng. J. 262 (2015) 989–998. [CrossRef]
- Y. Wu, Y. Zhang, M. Zhang, F. Liu, Y. Wan, Z. Huang, L. Ye, Q. Zhou, Y. Shi, B. Lu, Selective and simultaneous determination of trace bisphenol A and tebuconazole in vegetable and juice samples by membrane-based molecularly imprinted solid-phase extraction and HPLC, Food Chem. 164 (2014) 527–535. [CrossRef]
- P. Zahedi, M. Fallah-Darrehchi, S.A. Nadoushan, R. Aeinehvand, L. Bagheri, M. Najafi, Morphological, thermal and drug release studies of poly (methacrylic acid)-based molecularly imprinted polymer nanoparticles immobilized in electrospun poly (ε-caprolactone) nanofibers as dexamethasone delivery system, Korean J. Chem. Eng. 34 (2017) 2110–2118. [CrossRef]
- M. Hassanzadeh, M. Ghaemy, S.M. Amininasab, Z. Shami, An effective approach for fast selective separation of Cr(VI) from water by ion-imprinted polymer grafted on the electro-spun nanofibrous mat of functionalized polyacrylonitrile, React. Funct. Polym. 130 (2018) 70–80. [CrossRef]
- M. Demirkurt, Y.A. Olcer, M.M. Demir, A.E. Eroglu, Electrospun polystyrene fibers knitted around imprinted acrylate microspheres as sorbent for paraben derivatives, Anal. Chim. Acta. 1014 (2018) 1–9. [CrossRef]
- L. Li, H. Liu, X. Lei, Y. Zhai, Electrospun Nanofiber Membranes Containing Molecularly Imprinted Polymer (MIP) for Rhodamine B (RhB), Adv. Chem. Eng. Sci. 02 (2012) 266–274. [CrossRef]
- F.N. Molinari, F. De Cesare, A. Macagnano, Housing Molecularly Imprinted Polymer Nanoparticles in Polyvinylpyrrolidone/Multiwall Carbon Nanotube Nanofibers to Detect Chiral Terpene Vapors, in: Eurosensors 2023, MDPI, Basel Switzerland, 2024: p. 96. [CrossRef]
- I.S. Chronakis, B. Milosevic, A. Frenot, L. Ye, Generation of Molecular Recognition Sites in Electrospun Polymer Nanofibers via Molecular Imprinting, Macromolecules. 39 (2006) 357–361. [CrossRef]
- X. Xue, R. Lu, Y. Li, Q. Wang, J. Li, L. Wang, Molecularly imprinted electrospun nanofibers for adsorption of 2,4-dinitrotoluene in water, Analyst. 143 (2018) 3465–3471. [CrossRef]
- W.J. Kim, J.Y. Chang, Molecularly imprinted polyimide nanofibers prepared by electrospinning, Mater. Lett. 65 (2011) 1388–1391. [CrossRef]
- Macagnano, F.N. Molinari, T. Mancini, S. Lupi, F. De Cesare, UV Light Stereoselective Limonene Sensor Using Electrospun PVP Composite Nanofibers, in: Eurosensors 2023, MDPI, Basel Switzerland, 2024: p. 131. [CrossRef]
- F. Molinari, A. V. Medrano, A. Bacigalupe, M. Escobar, L.N. Monsalve, Different dispersion states of MWCNT in aligned conductive electrospun PCL/MWCNT composites, Fullerenes, Nanotub. Carbon Nanostructures. 26 (2018) 667–674. [CrossRef]
- T. Ito, S. Yamaguchi, D. Soga, T. Yoshimoto, Y. Koyama, Preparation of a Bioadhesive Poly(Acrylic Acid)/Polyvinylpyrrolidone Complex Gel and Its Clinical Effect on Dental Hemostasis, Gels. 8 (2022) 462. [CrossRef]
- D’Arco, M. Di Fabrizio, T. Mancini, R. Mosetti, S. Macis, G. Tranfo, G. Della Ventura, A. Marcelli, M. Petrarca, S. Lupi, Secondary Structures of MERS-CoV, SARS-CoV, and SARS-CoV-2 Spike Proteins Revealed by Infrared Vibrational Spectroscopy, Int. J. Mol. Sci. 24 (2023) 9550. [CrossRef]
- H. Kaczmarek, A. Szalla, A. Kamińska, Study of poly(acrylic acid)–poly(vinylpyrrolidone) complexes and their photostability, Polymer (Guildf). 42 (2001) 6057–6069. [CrossRef]
- W. Salalha, Y. Dror, R.L. Khalfin, Y. Cohen, A.L. Yarin, E. Zussman, Single-Walled Carbon Nanotubes Embedded in Oriented Polymeric Nanofibers by Electrospinning, Langmuir. 20 (2004) 9852–9855. [CrossRef]
- P. Poulin, B. Vigolo, P. Launois, Films and fibers of oriented single wall nanotubes, Carbon N. Y. 40 (2002) 1741–1749. [CrossRef]
- X. Zhu, P. Lu, W. Chen, J. Dong, Studies of UV crosslinked poly(N-vinylpyrrolidone) hydrogels by FTIR, Raman and solid-state NMR spectroscopies, Polymer (Guildf). 51 (2010) 3054–3063. [CrossRef]
- H. Kaczmarek, M. Metzler, F. Ścigalski, Photochemical stability of poly(acrylic acid)/silver nanocomposite, Mater. Lett. 135 (2014) 110–114. [CrossRef]
- J. Janata, M. Josowicz, Conducting polymers in electronic chemical sensors, Nat. Mater. 2 (2003) 19–24. [CrossRef]
- R. Sarabia-Riquelme, J. Craddock, E.A. Morris, D. Eaton, R. Andrews, J. Anthony, M.C. Weisenberger, Simple, low-cost, water-processable n-type thermoelectric composite films from multiwall carbon nanotubes in polyvinylpyrrolidone, Synth. Met. 225 (2017) 86–92. [CrossRef]
- Ding, M. Wang, X. Wang, J. Yu, G. Sun, Electrospun nanomaterials for ultrasensitive sensors, Mater. Today. 13 (2010) 16–27. [CrossRef]
- F.M. Ernsberger, The Nonconformist Ion, 1983.
- X. Pan, Q. Xue, J. Zhang, Q. Guo, Y. Jin, W. Lu, X. Li, C. Ling, Effective Enhancement of Humidity Sensing Characteristics of Novel Thermally Treated MWCNTs/Polyvinylpyrrolidone Film Caused by Interfacial Effect, Adv. Mater. Interfaces. 3 (2016) 1600153. [CrossRef]
- S.K.S. Basha, G.S. Sundari, K.V. Kumar, M.C. Rao, Structural and Dielectric Properties of PVP Based Composite Polymer Electrolyte Thin Films, J. Inorg. Organomet. Polym. Mater. 27 (2017) 455–466. [CrossRef]
- Ismail, N.F.A. Bakar, T.H. Ling, N. Ideris, Z.H.M. Zain, N. Radacsi, Morphology and conductivity evaluation of electrospun polyacrylic acid (PAA) microfiber, Mater. Today Proc. 17 (2019) 574–583. [CrossRef]
- T. Khan, M.S. Irfan, M. Ali, Y. Dong, S. Ramakrishna, R. Umer, Insights to low electrical percolation thresholds of carbon-based polypropylene nanocomposites, Carbon N. Y. 176 (2021) 602–631. [CrossRef]
- P. Rani, M.B. Ahamed, K. Deshmukh, Carbon Nanotubes Embedded in Polymer Nanofibers by Electrospinning, in: Handb. Carbon Nanotub., Springer International Publishing, Cham, 2022: pp. 943–977. [CrossRef]
- J. Abraham, S. Thomas, N. Kalarikkal, Handbook of carbon nanotubes, 2022. [CrossRef]
- T.T. Moore, W.J. Koros, Gas sorption in polymers, molecular sieves, and mixed matrix membranes, J. Appl. Polym. Sci. 104 (2007) 4053–4059. [CrossRef]
- D’Amico, C. Di Natale, A contribution on some basic definitions of sensors properties, IEEE Sens. J. 1 (2001) 183–190. [CrossRef]
- M.A. Al-Ghouti, D.A. Da’ana, Guidelines for the use and interpretation of adsorption isotherm models: A review, J. Hazard. Mater. 393 (2020) 122383. [CrossRef]
- Mujahid, A. Afzal, F.L. Dickert, Transitioning from Supramolecular Chemistry to Molecularly Imprinted Polymers in Chemical Sensing †, Sensors. 23 (2023) 1–13. [CrossRef]
Figure 1.
Layout of the PVP-PAA-MWCNTs sensor fabrication for S(-)-limonene detection based on the combination of electrospinning and molecular imprinting technologies via photocrosslinking and solvent washing.
Figure 1.
Layout of the PVP-PAA-MWCNTs sensor fabrication for S(-)-limonene detection based on the combination of electrospinning and molecular imprinting technologies via photocrosslinking and solvent washing.
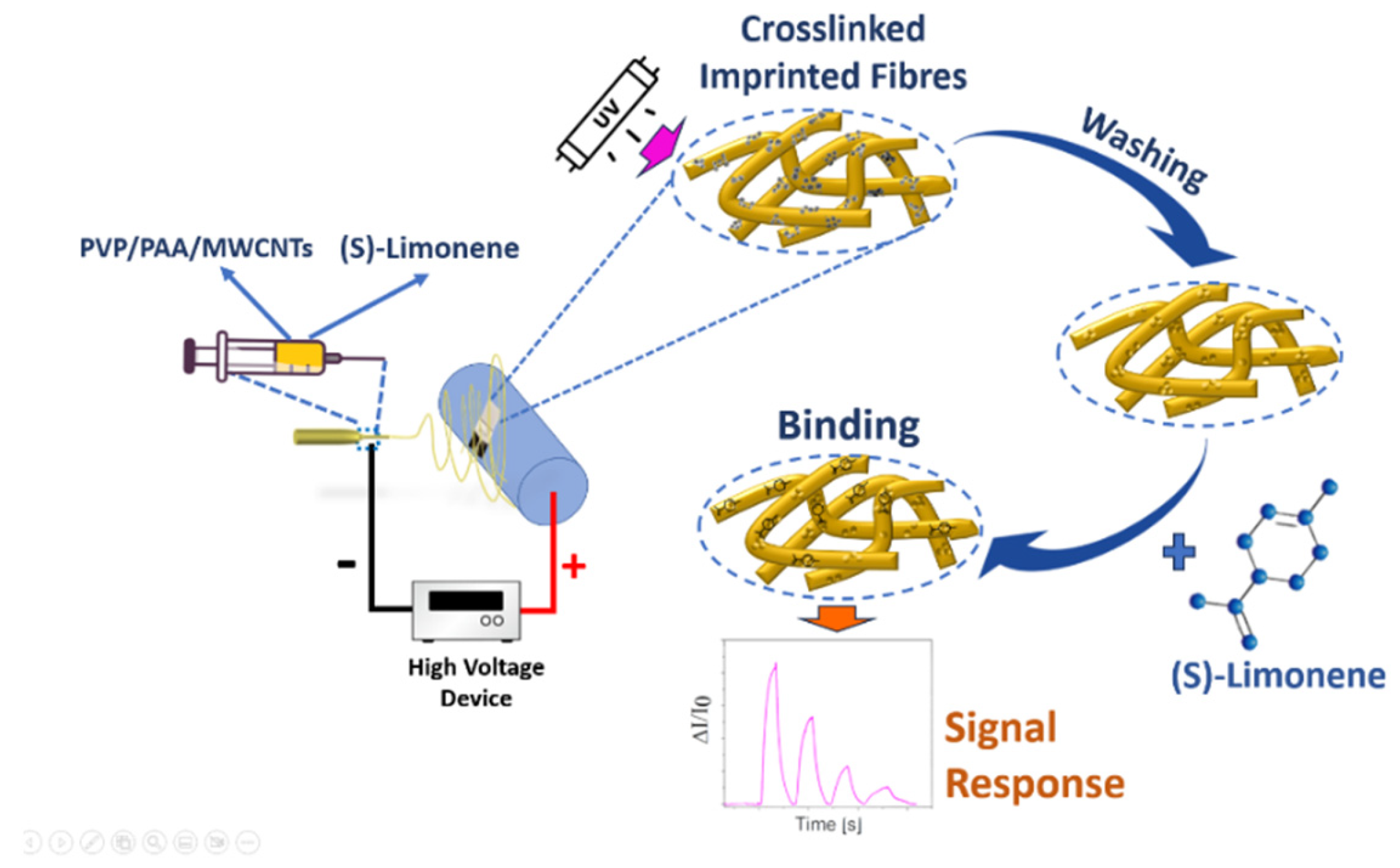
Figure 2.
SEM images of non-imprinted (A) and S(-)-limonene molecularly-imprinted nanofibres (B); Gaussian curves illustrating the size distribution of non-imprinted nanofibers (NINFs) (B) and molecularly imprinted nanofibers (MINFs) (D).
Figure 2.
SEM images of non-imprinted (A) and S(-)-limonene molecularly-imprinted nanofibres (B); Gaussian curves illustrating the size distribution of non-imprinted nanofibers (NINFs) (B) and molecularly imprinted nanofibers (MINFs) (D).
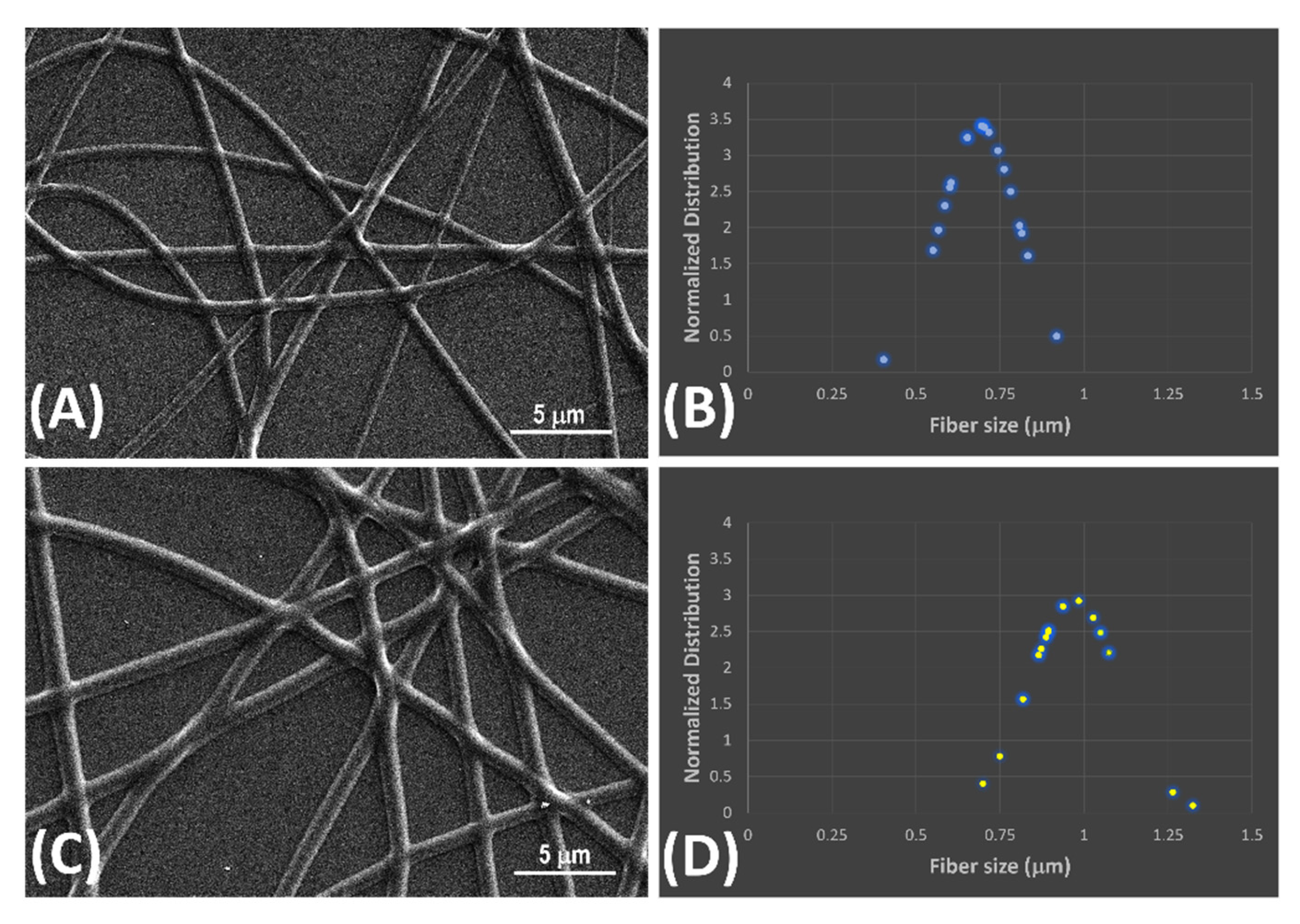
Figure 3.
AFM micrographs of NINFs (A) and MINFs (B) (5x5 mm) achieved in amplitude mode.

Figure 4.
TEM images displaying nanofibers incorporating MWCNT with varying distributions: (A) variable distribution of MWCNTs along the MINFs, from aligned to coiled, until outward protrusions, (B) prevalent alignment of MWCNT inside the NINFs, as (C) a coiled MWCNT within a NINF encircling a cavity and also protruding outwards, while an MWCNT is distributed longitudinally along another nanofiber.
Figure 4.
TEM images displaying nanofibers incorporating MWCNT with varying distributions: (A) variable distribution of MWCNTs along the MINFs, from aligned to coiled, until outward protrusions, (B) prevalent alignment of MWCNT inside the NINFs, as (C) a coiled MWCNT within a NINF encircling a cavity and also protruding outwards, while an MWCNT is distributed longitudinally along another nanofiber.

Figure 5.
SEM micrographs depict the morphology of electrospun MINFs at various deposition durations and magnification: 5 minutes (A,D), 10 minutes (B,E), and 15 minutes (C,F). (G-I) 3D elaborations of AFM images for the same samples (20x20 μm), corresponding to increasing deposition durations, are depicted in sequence.
Figure 5.
SEM micrographs depict the morphology of electrospun MINFs at various deposition durations and magnification: 5 minutes (A,D), 10 minutes (B,E), and 15 minutes (C,F). (G-I) 3D elaborations of AFM images for the same samples (20x20 μm), corresponding to increasing deposition durations, are depicted in sequence.

Figure 6.
(A) IR absorbance spectra between 1100 and 1900 cm-1 of no UV-treated nanofiber (blue) and UV-treated nanofibres without washes (orange) and with washes (yellow). (B) IR absorption band of C=O vibrational mode between 1550 and 1800 cm-1 with the same color code. (C) Second derivative of C=O IR absorption band between 1550 and 1800 cm-1 with the same color code.
Figure 6.
(A) IR absorbance spectra between 1100 and 1900 cm-1 of no UV-treated nanofiber (blue) and UV-treated nanofibres without washes (orange) and with washes (yellow). (B) IR absorption band of C=O vibrational mode between 1550 and 1800 cm-1 with the same color code. (C) Second derivative of C=O IR absorption band between 1550 and 1800 cm-1 with the same color code.

Figure 7.
(A) Measuring setup includes an air cylinder, three mass flow controllers, a bubbler, and a humidifier system using Nafion® tubing, a mixing chamber, the sensor within its measuring chamber, a %RH-T sensor, an electrometer, software, and a PC. (B) Current-voltage (I-V) curves of MINF and NINF sensors, both obtained after 5 minutes of deposition.
Figure 7.
(A) Measuring setup includes an air cylinder, three mass flow controllers, a bubbler, and a humidifier system using Nafion® tubing, a mixing chamber, the sensor within its measuring chamber, a %RH-T sensor, an electrometer, software, and a PC. (B) Current-voltage (I-V) curves of MINF and NINF sensors, both obtained after 5 minutes of deposition.
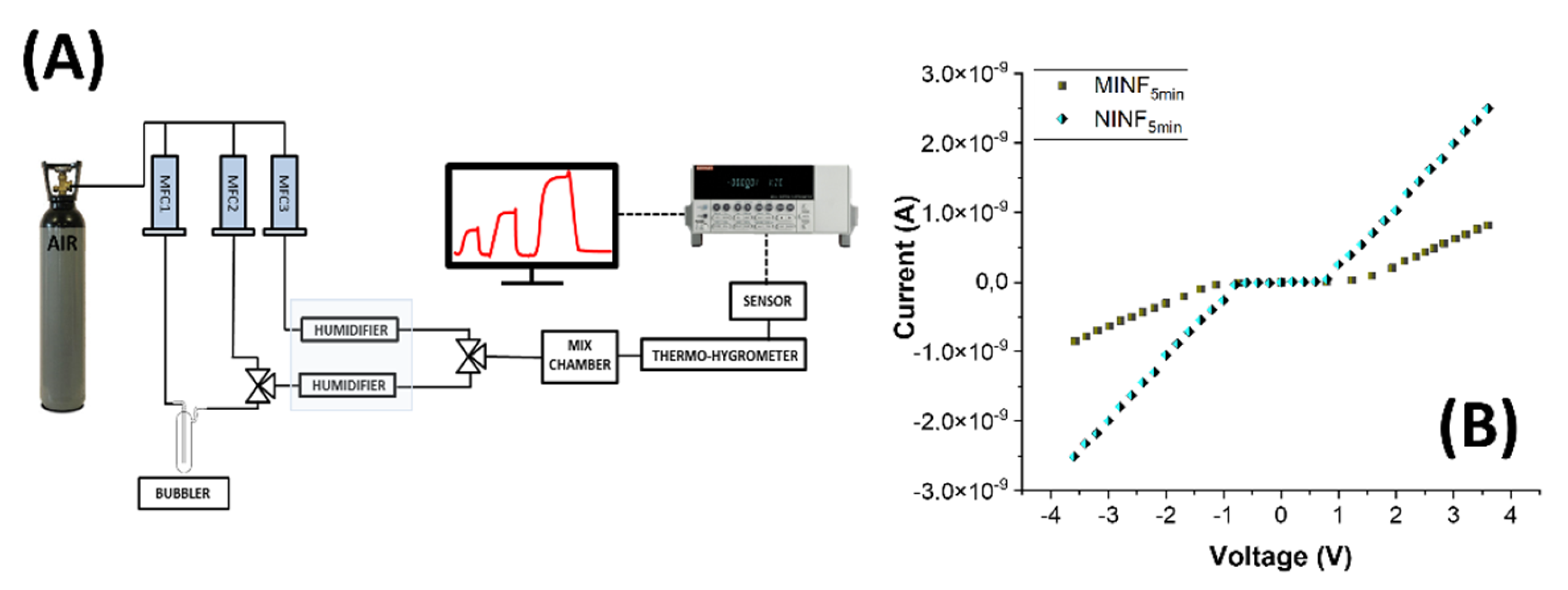
Figure 8.
(A) Electrical resistance values of MINF5min sensor plotted versus increasing relative humidity percentages, and (B) the normalized sensor responses to 55 vpm of S(-)-limonene while changing the %RH.
Figure 8.
(A) Electrical resistance values of MINF5min sensor plotted versus increasing relative humidity percentages, and (B) the normalized sensor responses to 55 vpm of S(-)-limonene while changing the %RH.
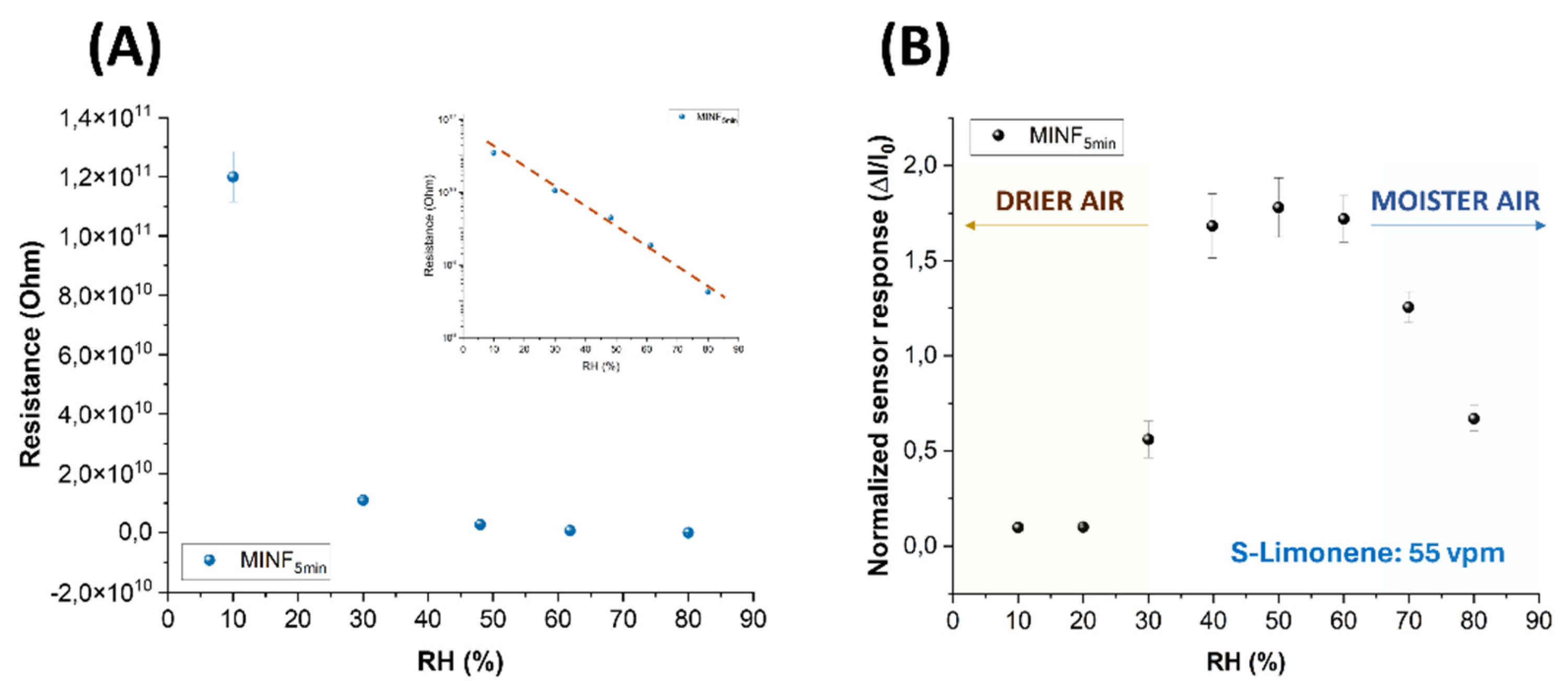
Figure 9.
Bar plot illustrating the normalized response of NINF (Non-Imprinted Nanofiber) and MINF (Molecularly Imprinted Nanofiber) sensors to a known concentration of S-limonene (40 vpm). Measurements were calculated as the ratios between the change in current and the baseline current under clean air conditions.
Figure 9.
Bar plot illustrating the normalized response of NINF (Non-Imprinted Nanofiber) and MINF (Molecularly Imprinted Nanofiber) sensors to a known concentration of S-limonene (40 vpm). Measurements were calculated as the ratios between the change in current and the baseline current under clean air conditions.
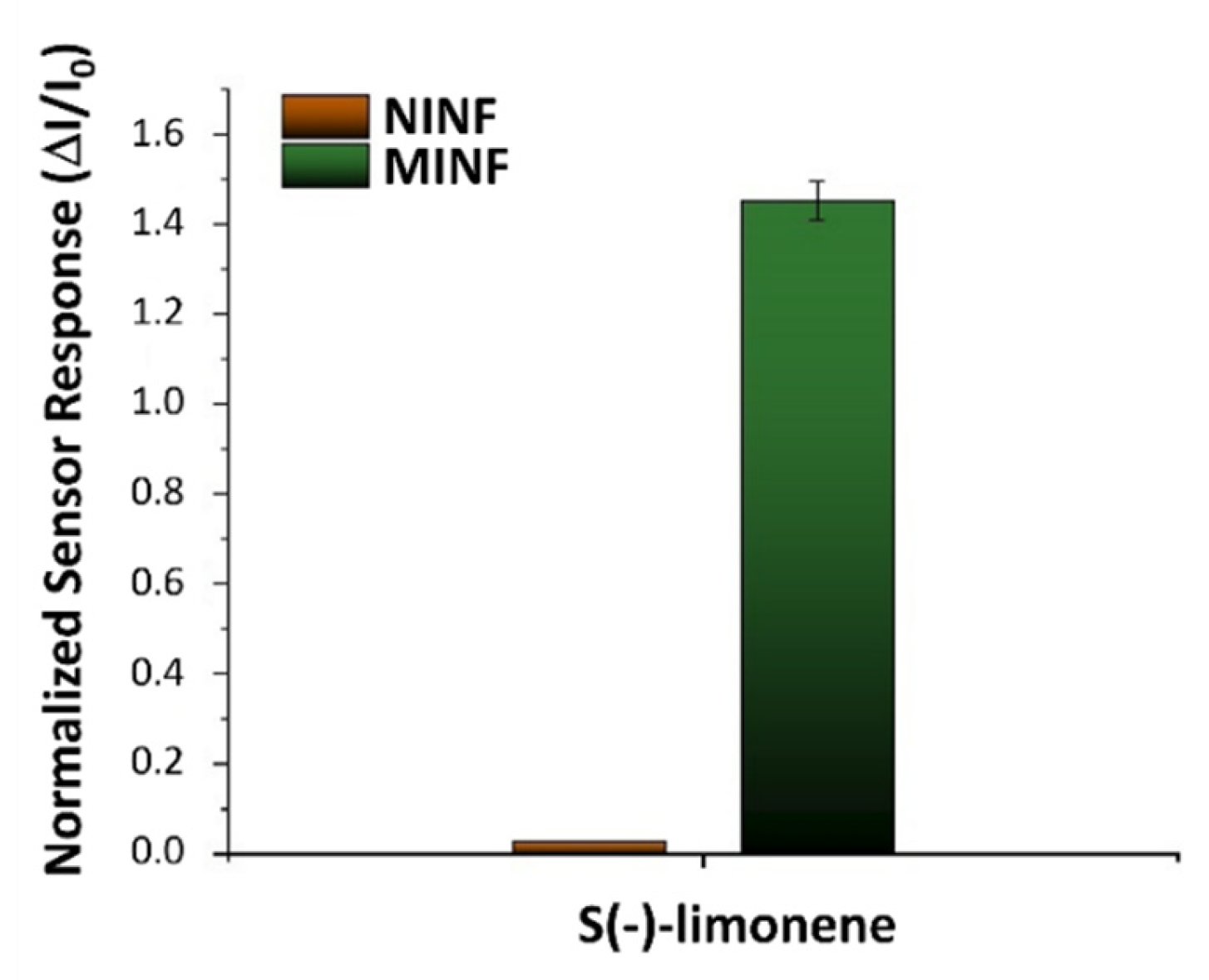
Figure 10.
(A) Photographs of interdigitated electrodes after nanofiber deposition for 5, 10, and 15 minutes, accompanied by optical microscope magnification of a section of each of them, and a bar plot (B) illustrating the electrical resistance values measured for each deposition duration (R5min: ~2,8·109 Ohm, R10min: ~1,76 ·109 Ohm, R15min: ~8,97·109 Ohm) and (C) the corresponding current-voltage curves spanning +4 to -4 Volts.
Figure 10.
(A) Photographs of interdigitated electrodes after nanofiber deposition for 5, 10, and 15 minutes, accompanied by optical microscope magnification of a section of each of them, and a bar plot (B) illustrating the electrical resistance values measured for each deposition duration (R5min: ~2,8·109 Ohm, R10min: ~1,76 ·109 Ohm, R15min: ~8,97·109 Ohm) and (C) the corresponding current-voltage curves spanning +4 to -4 Volts.
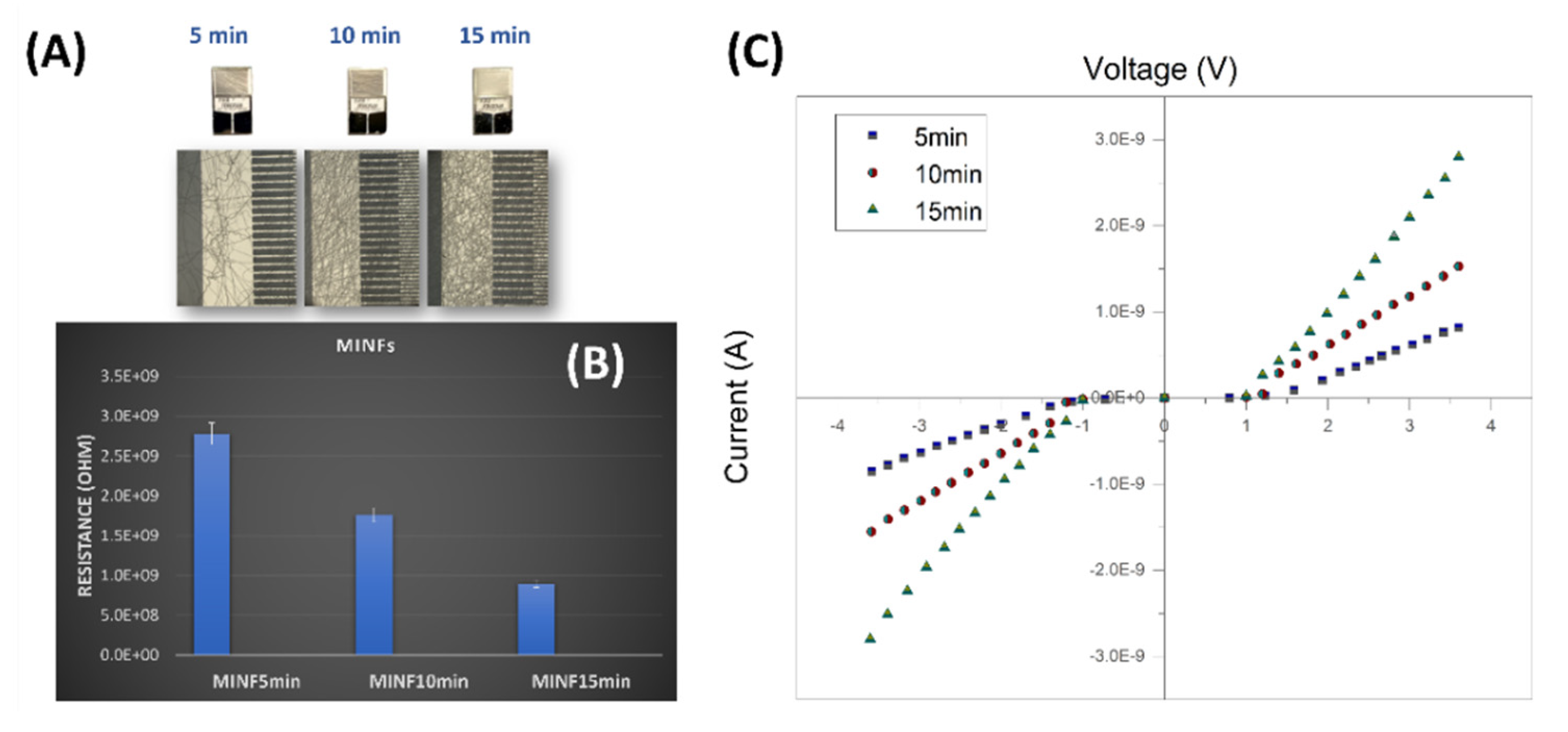
Figure 11.
Transient responses of sensors to known concentrations of S(-)-limonene for MINF5min (A), MINF10min (B), and MINF15min (C), along with the corresponding response curves (D,E,F). The red circles represent the concentration values related to the transient responses plots.
Figure 11.
Transient responses of sensors to known concentrations of S(-)-limonene for MINF5min (A), MINF10min (B), and MINF15min (C), along with the corresponding response curves (D,E,F). The red circles represent the concentration values related to the transient responses plots.
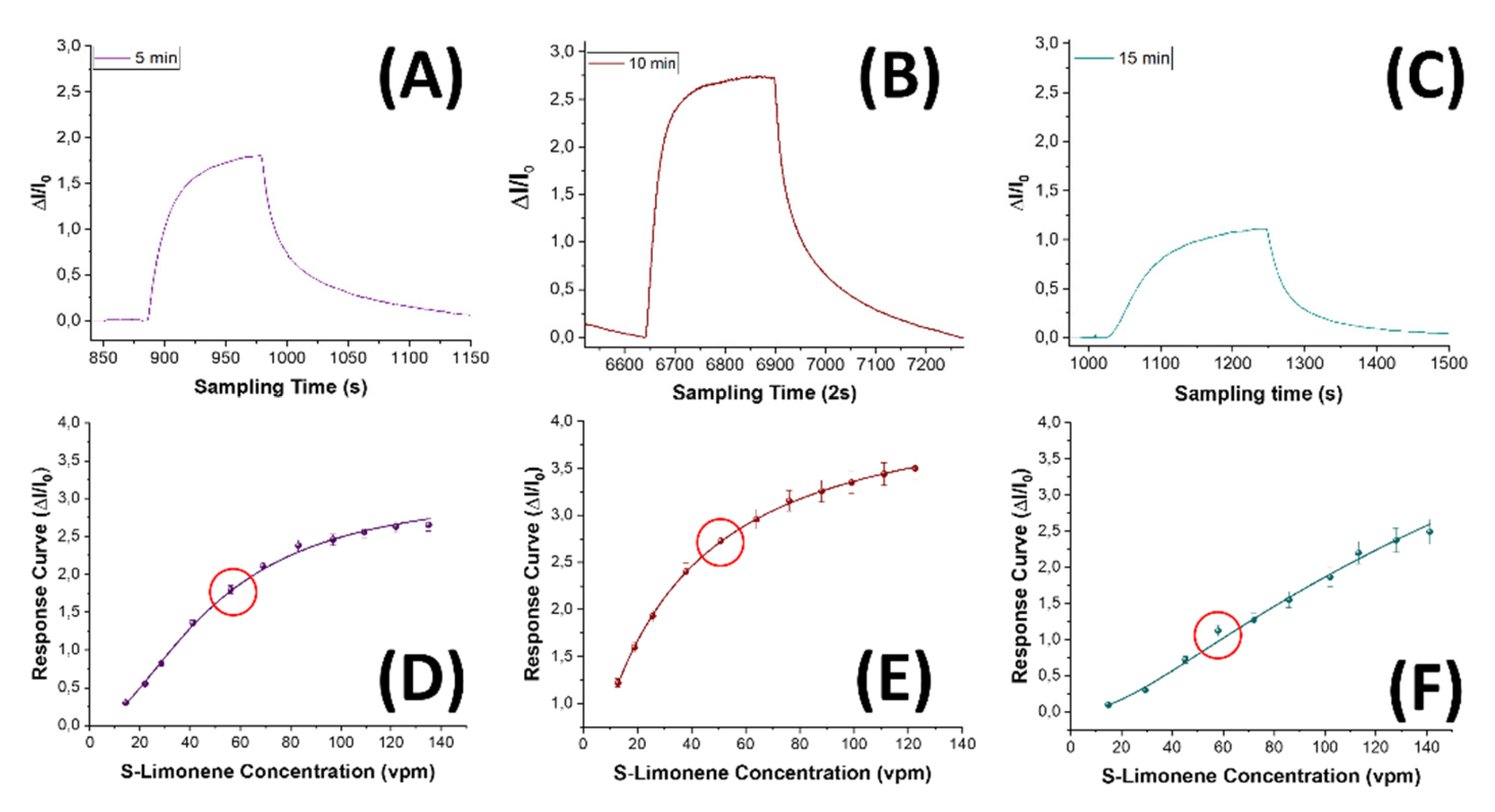
Figure 12.
Sensor response curves of the MINFs (A: 5min; B: 10min; C: 15min) to increasing concentrations of VOCs (S-limonene, linalool, α-pinene, ethanol, toluene) ranging between 15 and 140 parts per million (vpm); (D) bar plot comparing sensitivity values; (E) 3D bar plot illustrating the selectivity index (SI).
Figure 12.
Sensor response curves of the MINFs (A: 5min; B: 10min; C: 15min) to increasing concentrations of VOCs (S-limonene, linalool, α-pinene, ethanol, toluene) ranging between 15 and 140 parts per million (vpm); (D) bar plot comparing sensitivity values; (E) 3D bar plot illustrating the selectivity index (SI).
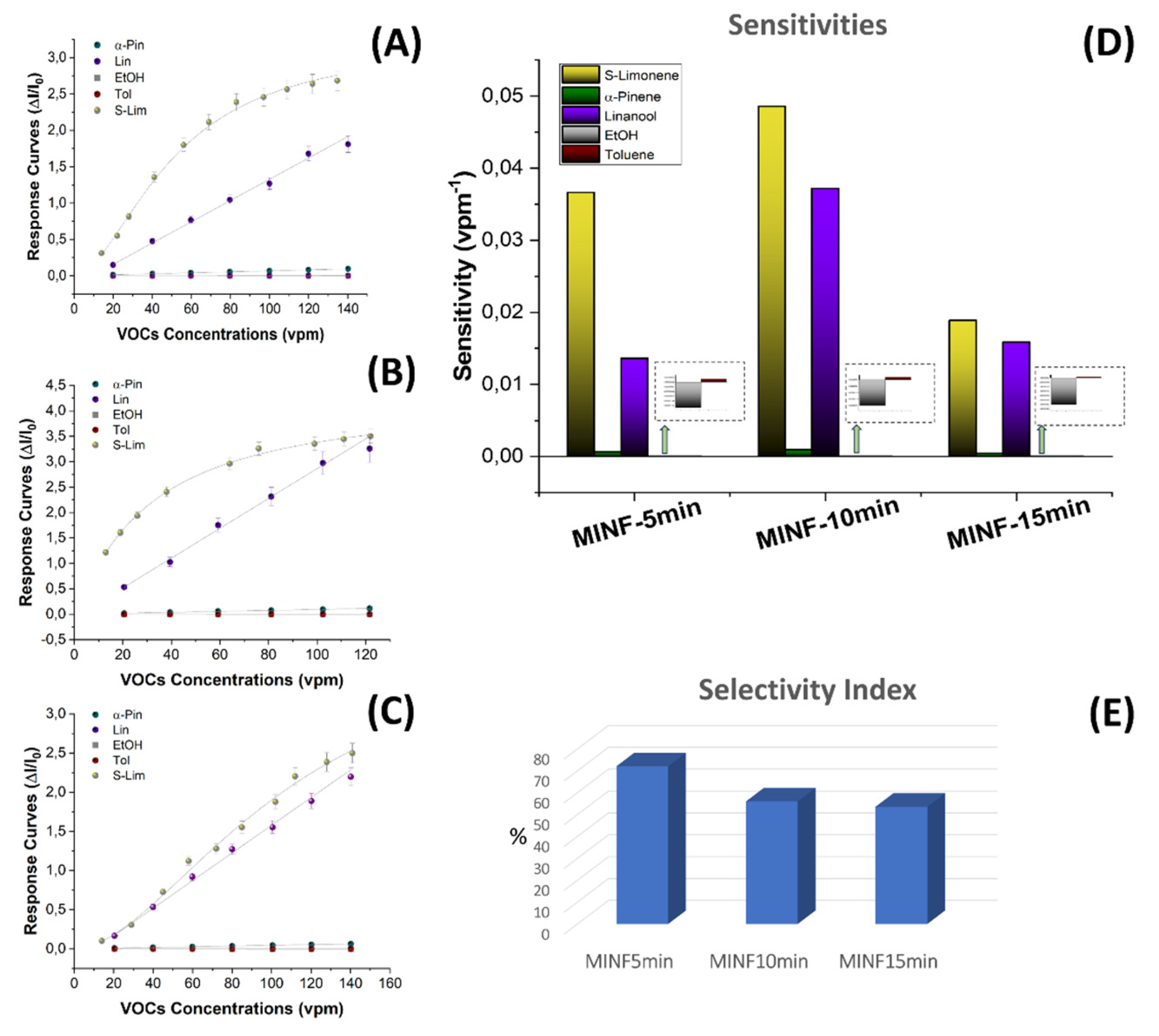

Table 1.
The sensing features of MINFs chemiresistors to S(-)-limonene.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



