
Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Cardiotoxicity is the main side effect of several chemotherapeutic drugs. Doxorubicin (Doxo) is one of the most used anthracyclines in the treatment of many tumors, but the development of acute and chronic cardiotoxicity limits its clinical usefulness. Different studies focused only on the effects of long-term Doxo administration, but recent data show that cardiomyocyte damage is an early event induced by Doxo after a single administration that can be followed by progressive functional decline, that lead to overt heart failure. The knowledge of molecular mechanisms involved in the early stage of Doxo induced cardiotoxicity is of paramount importance to treat and/or prevent it. This review aims to illustrate several mechanisms considered to underlie Doxo-induced cardiotoxicity, such as oxidative and nitrosative stress, inflammation, and mitochondrial dysfunction. Moreover, here we report data from both in vitro and in vivo studies indicating new therapeutic possibilities to prevent Doxo-induced cardiotoxicity.
Keywords:
cardiotoxicity
; doxorubicin
; oxidative stress
; inflammation
1. Introduction
Doxorubicin (Doxo) belongs to the anthracycline family, and it is a highly effective antineoplastic agents derived from the Streptomyces peucetius bacterium. Doxo is a widely used in the treatment of a broad spectrum of cancer but its clinical effectiveness is hampered by cardiotoxicity, the main long-term side effect responsible for increased morbidity and mortality in cancer survivors [1]. A study conducted in the United States on women diagnosed with breast cancer, showed that cardiovascular disease is the leading cause of mortality in patients over 50, exceeding the risk of death from cancer initial itself [2]. So, patients at high risk of developing cardiac complications, including patients with hypertension, diabetes mellitus, liver disease, and previous cardiac disease are not treated with Doxo [3]. Furthermore, Doxorubicin-induced cardiotoxicity (DIC) largely depends on the route of administration, the duration of chemotherapy (cumulative dose), and the dosage regimen [4]. This nowadays limits the recommended maximum lifetime dose of Doxo to <450 mg/m2 to reduce the risk of cardiotoxic side effects [5].
DIC can be classified as acute or chronic. Acute DIC occurs with an incidence of about 11%, it resembles acute myocarditis with myocytes damage, it usually occurs within few days of drug administration, and it is regarded as being reversible [6]. Instead, chronic DIC manifests as cardiomyopathy, it can occur months or years after the initial treatments, and it is clinically characterized by an irreversible reduction of left ejection fraction of >10% and symptomatic heart failure [1]. However, the scientific community agrees that acute and chronic cardiotoxicity are only a spectrum of a single disease and not separate disease entities. Indeed, recent findings suggest that acute and chronic DIC are not separate events, but they are potentially a continuous phenomenon, starting with myocardial cell injury, followed by progressive functional decline, progressively leading to overt heart failure [7]. The time interval between treatment and the onset of chronic cardiotoxicity varies from 30 days to more than 10 years [6]. Therefore, heart failure in patients treated with Doxo may go unnoticed for many years. So, the knowledge of molecular mechanisms involved and the search of therapeutic strategies able to act in the early stage of DIC are of paramount importance. Despite the main molecular mechanism involved in DIC is not yet identified, current understanding includes a central role for oxidative stress and inflammation, which are strictly related to each other and involved in different mechanisms of cell death in Doxo-treated cardiomyocytes. As described in this review, apoptosis, ferroptosis, necrosis, pyroptosis, and mitophagy are all involved in Doxo-induced cell death, and all are responsible for the loss of ventricular and later functional consequence observed in treated patients.
1.1. Involvement of Oxidative Stress in Doxorubicin-Induced Cardiotoxicity
Oxidative stress is a phenomenon caused by an imbalance between production and accumulation of oxygen reactive species (ROS) in cells and tissues and the ability of a biological system to detoxify these reactive products. In Doxo-treated cancer patients, high levels of oxidative stress and a concomitant reduction in the antioxidant status have been observed. The nature of cardiac tissue that exhibits low level of antioxidant enzymes, such as superoxide dismutase (SOD) and catalase (CAT), makes it more susceptible to ROS generation and accumulation of oxidative stress [4].
1.1.1. Interference of Doxorubicin with Redox Cycling and Iron Metabolism
Many studies report that the chemical structure of Doxo itself is involved in free radicals generation. Doxo is well known for its ability to produce ROS through multiple pathways among which the redox cycle of the molecule and the formation of Doxo-iron complexes seem to play a central role.
The redox cycle is related to the presence of the quinone moiety which allows Doxo to act as an electron acceptor. Indeed, a one-electron reduction of the quinone moiety via NADPH and cytochrome P450 reductase, converts Doxo in a semiquinone radical. The latter reacts with oxygen (O2) thus generating the superoxide radical O2•- and the contemporary regeneration of the quinone form. The dismutation of O2•- to hydrogen peroxide (H2O2) is catalysed by SOD or can occur spontaneously. H2O2 is a relatively stable and non-toxic molecule which under physiological conditions is eliminated by catalase or glutathione peroxidase. However, H2O2 and O2•- can generate highly reactive and toxic OH• hydroxyl radicals which react with any oxidizable compound in their vicinity causing damage to all types of macromolecules, including lipids, nucleic acids, and proteins. This occurs during the Haber-Weiss reaction, which is generally very slow unless catalysed by transition metals, particularly iron. The Haber-Weiss reaction catalysed by iron is called the Fenton reaction.
O2•- + H2O2 ⟶ OH− + OH∙ + O2 (Haber-Weiss reaction)
Fe2+ + H2O2 ⟶ Fe3+ + OH− + OH∙ (Fenton reaction) (Figure 1).
The second and most important pathway of free radical production involves formation of the Doxo-Fe3+ complex, which can be reduced to Doxo-Fe2+ by various reducing systems, such as glutathione peroxidase 4 (GPx4), cysteine, etc. The Doxo-Fe2+ complex can react with O2 to generate O2•- which in turn dismutes into H2O2 or undergoes the Haber-Weiss reaction, generating OH• [8,9]. Previous study demonstrated that the Doxo-Fe complex plays a pivotal role in DIC since it could cause lipid peroxidation through interaction with negatively charged membranes [10]. Moreover, Doxo treatment inhibits the fatty acid metabolism enzyme Acyl-CoA thioesterase 1 (Acot1), that reduces lipid peroxidation and ferroptosis by increasing GSH levels [10]. Several evidence demonstrate that the deleterious effects of Doxo on iron metabolism are also mediated by its interference with the proteins that sequester and bind cellular iron, thus leading to an increase in free iron, which could perpetuate the cycle of free radicals’ generation [11,12]. (Figure 2) Indeed, Doxo-treatment induces an increase in transferrin levels, thus allowing for a greater amount of iron into the cell and inactivates the iron regulatory proteins 1 and 2 (IRP1/2) [13]. Furthermore, a downregulation of ABCB8, a protein responsible for iron transport outside the mitochondria, and of ferritin has been demonstrated [14]. Increased free iron accumulation in mitochondria and the consequent production of lipid peroxidation and oxidative stress result in ferroptosis, which has been suggested to be the major form of regulated cell death in Doxo-induced cardiomyocyte death [15].
1.2. Effects of Doxorubicin on Mitochondria
The mitochondria fulfill the huge oxygen demand of cardiomyocytes, but Doxo treatment is responsible for structural changes in the mitochondria which lead to the depletion of energy production [16,17]. The interaction between Doxo and cardiolipin, a membrane phospholipid that resides in the inner lobe of the mitochondrial membrane in cardiomyocytes, seems to be one of the main events responsible for Doxo-associated cardiotoxicity [18]. Indeed, the cationic charge of Doxo and anionic charge of cardiolipin bind irreversibly, resulting in the accumulation of Doxo in mitochondria that lead to mitochondrial dysfunction mediated by oxidative stress [19]. The complex Doxo-cardiolipin affects the role of cardiolipin in electron transport chain whit the consequent inhibition of several enzymes, such as cytochrome c oxidase and cytochrome c oxidoreductase. Furthermore, a lower ATP generation with a consequent acceleration of oxidative phosphorylation has been reported in Doxo-treated cardiomyocytes [20,21,22]. Moreover, as previously described, ROS overproduction is associated with mitochondrial iron overload that leads to cardiomyocytes ferroptosis [6]. Doxo interferes also with mitochondrial dynamics and the maintenance of mitochondrial integrity. It is well known that excessive mitochondrial fission is involved in the initiation of apoptosis, while mitochondrial fusion can inhibit it [23,24]. It has been shown that upon Doxo treatment mitochondrial fission protein 1 (Mtfp1), a GTPase also known as Mtp18 widely involved in mitochondrial fission, is upregulated [25]. Furthermore, it has been proved that Doxo affects mitochondrial fission by enhancing the phosphorylation of the dynamin-related protein 1 (DRP1), which is a GTPase responsible for outer mitochondrial membrane scission [26,27,28]. Mitochondrial fusion, in contrast, is inhibited, since Mitofusin 1 and 2 (MFN 1/2), that are GTPases related to mitochondrial fusion, are downregulated during Doxo treatment [29]. Instead, Optic atrophin 1 (OPA1), another GTPase involved in mitochondrial fusion, is hyperacetylated under Doxo treatment and this effect reduces its GTPase activity and inhibits mitochondrial fusion [30]. Doxo-induced mitochondrial fission, together with oxidative stress, is associated with autophagy, a self-digestion process widely involved in the maintenance of functional mitochondria in DIC but that can exacerbate cardiotoxicity if the autophagic flux exceeds a certain threshold [31]. Indeed, it has been demonstrated that Doxo-induced mitochondrial toxicity can trigger autophagic (including mitophagic) responses, in an attempt to remove damaged mitochondrial and cellular structures [32]. However, chronic Doxo treatment and/or higher doses further increase cellular damage including the extent of oxidative stress and may induce a suppression of autophagy resulting from exhaustion of the autophagic reserve capacity [33]. Doxo also causes a dose-dependent induction of the mitochondrial permeability transition pore (mPTP) opening, a phenomenon that releases mitochondrial Ca2+ and cytochrome c contributing to apoptotic or necrotic cell death [33,34]. Indeed, an increase of the pro-apoptotic protein Bax and a concomitant reduction in antiapoptotic proteins (e.g., Bcl2 and caspase-9 and caspase-3) activation has been well established in Doxo-treated cardiac cells [32].
1.3. Effects of Doxorubicin on NOX Signalling
Another key enzyme involved in Doxo-induced ROS production in cardiomyocytes after Doxo treatment is NADH oxidase (NOX) [35]. All NOX enzymes utilize NADPH as an electron donor and catalyze transfer of electrons to molecular oxygen to generate O2•- and/or H2O2. Under Doxo treatment, NOXs are involved in the semiquinone radical formation in view of the transfer of an electron from NADH to Doxo [11,36,37]. The NOX family oxidases comprise seven members, each characterised by a specific core catalytic subunit, and all need to be associated with specific regulatory subunits for activation, while only NOX4 is constitutively active [38]. Among all NOX isoforms, NOX2 and NOX4, are the most expressed in cardiomyocytes and play a crucial role in DIC since it has been shown that Doxo promotes their expression and ROS generation [39,40]. Indeed, previous study demonstrated that NOX2 and NOX4 genetic disruption attenuates drug-related myocardial dysfunction, reducing myocardial atrophy, cardiac apoptosis, and interstitial fibrosis [41]. Several studies demonstrated that Doxo promotes NOX2 expression and leads to phosphorylation of p47phox, a cytoplasmic subunit of NOX2. The membrane translocation of phosphorylated p47phox activates NOX2 and promotes ROS production [42]. In addition, Doxo induces the interaction between NOX4 and mitochondria, thus leading to further ROS generation. Moreover, NOX4 overexpression promotes the activation of inflammasomes, leading to pyroptosis in Doxo-treated hearts [43].
1.4. Effects of Doxorubicin on Nrf2
Doxo also interferes with Nuclear factor erythroid 2-related factor 2 (Nrf2), a redox-sensitive transcription factor belonging to the Cap ‘n’ collar family transcription factor which plays a pivotal role in the antioxidant defense system [44,45]. In resting conditions, Nrf2 is negatively regulated by Kelch Like ECH associated protein 1 (Keap1) that binds the Neh2 domain of Nrf2 and allow its degradation by CUL3-containing E3 ubiquitin ligase [10,45]. On the contrary, in pro-oxidant conditions, degradation of Keap1 results in the constitutive activation of Nrf2, which enters into nucleus and through the Neh1 domain is able to bind antioxidant response elements (ARE), thus regulating a number of antioxidant enzymes, such as glutathione S-transferase (GST), heme oxygenase-1 (HO-1), and NAD(P)H quinone dehydrogenase 1 (NQO1) [46,47]. Several kinases, such as PKC, PI3K and p38MAPK, are involved in Nrf2 phosphorylation and activation [48]. Other proteins involved in Nrf2 activation are p21 [49], p62 [50], Parkinson’s disease protein 7 (PARK7) [51], and Silent information regulator 1 (Sirt1) [52], that under oxidative stress condition interfere with Keap1-Nrf2 complex thus facilitating Nrf2 activation and translocation into nucleus. The effects of Doxo on Nrf2 are controversial. On one hand, Doxo-induced oxidative stress results in the dissociation of Keap1-Nrf2 complex, which causes an increase of free Nrf2 that enters the nucleus, and bounds ARE thus inducing the expression of downstream antioxidant proteins and biphasic detoxification enzymes [53]. An in vivo study revealed that Nrf2 activation reduces Doxo-induced oxidative impact on cardiomyocytes [41], however, this weak upregulation is not enough to offset the Doxo-induced oxidative stress [18]. On the other hand, Doxo administration can increase Keap1 levels [54] as well as the expression of tripartite motif containing-21 (TRIM21), an E3 ubiquitin ligase that interact with p62 to disturb the dissociation of Nrf2 from Keap1-Nrf2 complex [55]. Overall, another study showed that Doxo can inhibit Nrf2 through p38 MAPK leading to an increase of apoptosis [56]. Furthermore, it is to note that excessive activation of Nrf2, as observed under Doxo treatment, increases heme to ferrous, iron, carbon monoxide, as well as biliverdin, in a HO-1-catalyzed manner, thus triggering ferroptosis [57]. (Figure 3)
1.5. Involvement of Nitrosative Stress in Doxorubicin-Induced Cardiotoxicity
In addition to oxidative stress, there is evidence that also nitrosative stress is involved in DIC, as increase in reactive nitrogen species, most notably peroxynitrite (ONOO-), a potent reactive and cytotoxic free radical, have been reported [4]. Nitric oxide (NO) is required for normal cardiac function, including coronary vasodilatation, inhibition of platelet aggregation and neutrophil adhesion and activation, and modulation of cardiac contractile function. In oxidative stress condition, the inactivation of cytoprotective NO and the formation of the peroxynitrite (ONOO-), produced following the interaction between NO and O2•-, occur [58]. The peroxynitrite-induced increase in myocardial protein nitration may contribute to the Doxo-induced depression of cardiac function [59]. The activity of nitric oxide synthetase (NOS), enzymes responsible for NO formation starting from L-arginine and O2, also contributes to the increase in nitrosative stress. To date, three isoforms of NOS have been identified: neuronal (NOS1), inducible (NOS2), and endothelial (NOS3). NOS1 and NOS3 are constitutive enzymes controlled by Ca2+ concentrations; NOS2, on the other hand, is Ca2+-independent and its expression is induced at the transcriptional level by pro-inflammatory stimuli, including NF-kB, the main nuclear transcription factor involved in inflammation [58]. Treatment with Doxo causes an increase in eNOS transcription: in this regard, Neilan et al. [60] demonstrated that cardiac-specific overexpression of eNOS exacerbates the pathological response to Doxo in the heart, while blockade of eNOS transcription protects from cardiac dysfunction, drug-induced injuries and mortality. Doxo binds to the eNOS enzyme and induces the formation of the semiquinone radical which, in turn, reduces oxygen to the superoxide radical [61]. Specifically, Doxo binds the reductase domain of eNOS, thereby increasing superoxide production and reducing NO formation [62]. Despite data on effects of Doxo treatment on iNOS activity are same controversial, evidence indicate that Doxo increases iNOS protein and mRNA expression in the myocardium which leads to generation of NO [63]. However, in oxidative stress condition, NO reacts with •O2, thus producing the cytotoxic free radical-peroxynitrite anion (ONOO-) [10,64,65,66]. Regarding nNOS, its role in DIC has been minimized because no changes in myocardial nNOS mRNA expression were observed upon Doxo administration [67].
2. Involvement of Inflammation in Doxorubicin-Induced Cardiotoxicity
A growing body of evidence indicate that also inflammation plays a pivotal role in DIC. Previous studies showed that Doxo treatment induces a significant increase in pro-inflammatory cytokines, such as tumor nuclear factor (TNF)-α, interleukin (IL)-1β, and IL-6 via nuclear factor- κB (NF-κB) activation [68,69]. NF-κB is a downstream effector of Toll-like receptor 4 (TLR4) signaling that is part of the innate immune system which can be activated by pathogen-associated molecular patterns (PAMPs) e.g., LPS, or by damage-associated molecular patterns (DAMPs) such as endogenous high-mobility group protein box 1 (HMGB1) and the heat shock protein family (Hsps). Studies showed that Doxo-treatment induces both an up-regulation of TLR4 expression and the release of PAMPs and DAMPs. As a result, an increase in TLR4-mediated cardiac inflammation occurs [70]. It is to note that TLR4 activation is also involved in the apoptotic pathway. Indeed, study reported that Doxo-induced oxidative stress is associated with the increase in TLR4 expression that further promotes inflammation that, in turn, contributes to apoptosis. These data support the notion that both inflammation and oxidative stress contribute to Doxo-induced cardiomyocytes damage creating a vicious circle of impaired pathways [71,72]. Doxo also promotes NLR family pyrin domain containing 3 (NLRP3) inflammasomes formation both in a TLR4-dependent manner and in a TLR4-independent but ROS-dependent manner [16,73,74]. Activation of NLRP3 inflammasomes begins as assembly of NLRP3, apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) and pro-caspase 1, thus leading to caspase-1 activation thereby mediating the event of pyroptosis [73,74,75]. Pyroptosis is a form of pro-inflammatory cell death characterized by cytoplasmic swelling and plasma membrane rupture induced by the cleavage of gasdermin D (GSDMD) or GSDME, which allows the release of IL-1β and IL-18, contributing to cardiac cell damage [76,77,78]. Indeed, pyroptosis can usually result in increased inflammation and can cause the activation of various caspases (e.g., caspase-1, caspase-3, caspase-4, and caspase-11). A study conducted by Ye et al. [78] found that Doxo also directly binds GSDMD, leading to plasma membrane rupture even in absence of inflammatory caspases. Indeed, Doxo-induced cardiomyocytes pyroptosis can occur also through mitochondrial pathway, since Doxo leads to increased expression of Bcl-2/adenovirus E1B interacting protein 3 (Bnip3) protein in mitochondria, which in turn activates caspase-3 [16,79]. Furthermore, Doxo induced GSDMD-N localization in mitochondria, contributes to mtDNA release and cell damage [78].
3. Therapeutic Strategies to Counteract Doxorubicin-Induced Cardiotoxicity
At present, there are no specific clinical practice guidelines for the management of DIC. To reduce Doxo-induced adverse effects, especially at cardiac level, several strategies have been proposed, including the use of substances with antioxidant and/or antiapoptotic activity, and the development of Doxo analogues [13].
Encapsulation of Doxo in nanostructure is one of the strategies applied in recent decades to decrease cardiotoxicity, owing to changes in tissue distribution and rate of drug release. The first Doxo liposomal injection, named Doxil®, has been approved by FDA in 1995. Doxil® consists in a PEGylated liposomal system in which Doxo is incorporated. This formulation shows clinical advantages, such as an increase in the half-life of Doxo, reduction of adverse effects, and accumulation of the drug in tumor tissue [80]. Doxil® has been shown to have reduced cardiotoxic effects than Doxo, however its cumulative dose remains a clinical concern [81]. Indeed, despite a randomized trial reported that less than 2% of patients developed cardiotoxicity when the cumulative dose reached 1061 mg/m2, it is recommended that the Doxil® dose should not exceed 550mg/m2 [82,83]. However, a 10-years follow up showed that the cumulative doses over 550 mg/m2 of Doxil® alone or Doxil® combined with previous Doxo had no drug-related heart failure [84]. Doxil®, was firstly approved for the intravenous treatment of Kaposi’s sarcoma, subsequently, the comforting results obtained led to an extension of the approved treatment (e.g., breast and ovarian cancer) and to the development of similar product (Lipodox®) [80]. In order to reduce DIC, liposomal Doxo- based chemotherapy has also been evaluated. In 2015 a Meta analysis performed by Xing and co-workers analyzed data from ten clinical trials evaluating the clinical efficacy and cardiotoxicity of liposomal Doxo-based chemotherapy compared to conventional Doxo in breast cancer patients. Despite some controversial data between studies, the meta-analysis comes to conclusion that liposomal Doxo-based chemotherapy showed significant advantages in progression-free survival and reduced cardiotoxicity relative to conventional Doxo [85]. Other types of Doxo nano-formulation , such as Doxo-conjugated poly-aspartic acid/polyethylene glycol micelles (NK-911) and Doxo-loaded polyalkylcyanoacrylate nano particles (Livatag), are currently in clinical trials [86]. In addition, it has been demonstrated that peptide-based Hydrogels (HGs) and nanogels (NGs) formulations are convenient approach for drug delivery; since, modulating the ratios of the peptide components resulted in different Doxo release. Indeed, HGs could be used for transepithelial drug delivery or in situ gelation process implants formation, while nanogels could be used for systemic, oral, and pulmonary drug delivery [87,88]. Magnetic iron oxide nanoparticles (IONP) stabilized with trimethoxysilylpropyl-ethylenediamine triacetic acid (EDT) has been evaluated as a Doxo carrier for Glioblastoma multiforme (GBM) [89]. It is to note that Doxo-IONP showed increased Doxo release under acidic conditions, such as those in the tumor microenvironment [90,91]. Based on these encouraging results, several IONPs have been produced as effective nanocarrier for anti-cancer drugs like Doxo [92], paclitaxel [93] and 5-fluorouracil [94], although none of these are yet in clinical trial. The iron chelator Dexrazoxane has been approved by FDA to prevent anthracycline-mediated cardiotoxicity in cancer patients [95]. Dexrazoxane is able to bind iron before it enters cardiomyocytes, thus preventing the formation of Doxo-Fe3+ complex, free radical formation and cardiac damage; however, its use is hampered by severe side effects which are very similar to the side-effect profile of anthracyclines [96,97].
Regardless of the source, an excess of ROS in the myocardium is clearly harmful as it could lead to cardiac hypertrophy, gene expression alterations, cell death activation, extracellular matrix transformation, ventricular remodelling and transient perturbation of calcium, all of which underlie the cardiomyopathy and heart failure [4]. To prevent Doxo-induced oxidative stress the use of some antioxidant drugs has been proposed. Many studies showed that Carvedilol exerts anti-oxidative stress effects in the context of DIC. Indeed, Carvedilol, unlike other β1-receptor antagonists, inhibits the formation of ROS and lipid peroxidation by scavenging oxygen free radicals and preventing the consumption of endogenous antioxidants, such as Vitamin E and glutathione [53,98,99]. Encouraging data from clinical trials support the use of Carvedilol in DIC prevention [100,101], as well as of Vitamin E [102]. Also, Statins have been proposed as cardioprotective agents in view of their pleiotropic effects ranging from upregulation of SOD2 [103] to attenuation of ROS production [104]. An in vivo study showed that Rosuvastatin administration in Doxo-treated rats inhibited the expression of HMGB1, in addition to the decrease of TNF-α and IFN-γ and increase of IL-4 and IL-10 [105]. However, clinical trials showed controversial results. Indeed, while the STOP-CA randomised clinical trial showed that Atorvastatin reduced the incidence of cardiac dysfunction in patients with lymphoma receiving anthracycline, another clinical trial (PREVENT) showed that Atorvastatin demonstrated very modest effects on oxidative/nitrosative stress biomarkers [106,107].
Many other natural compounds with antioxidant activity have been evaluated as cardioprotective agents, although no clinical data are currently available. Both in vitro and in vivo models of DIC showed that Resveratrol reduces ROS levels and enhances the levels of antioxidant enzymes such as SOD and CAT [108]; furthermore, a suppression of Doxo-induced ferroptosis, possibly through modulation of the MAPK signaling pathway, has been shown for Resveratrol [109]. Allicin [110,111] and Ginsenoside Rh2 restore the Doxo-induced reduction of antioxidants enzymes like SOD, CAT and glutathione [112].
In vitro and in vivo studies showed that Neferin [113], Astragaloside IV [114], Acacia hyspadica [115], and Resolvin D1 [116] all reduce DIC by inhibiting NOXs activity. However, even though numerous evidence has indicated the role of NOXs in Doxo-induced oxidative stress, no clinically effective NOXs inhibitors have been developed. It has been proposed that Valsartan, an Angiotensin-converting enzyme inhibitor, could be used as cardioprotective agent in DIC since it has been shown that AngII activates and regulates NOXs expression [117]. Further studies will be needed to determine whether NOXs inhibitors are valuable in clinical settings [10].
Dapaglifozin downregulates oxidative stress accumulation and mitochondrial dysfunction through the restoration of antioxidant enzymes (e.g., HO-1, NQO1 and SOD) via the PI3K/Akt/Nrf2 axis [118]
Reduction of Doxo-induced ROS formation, and enhancement of antioxidants enzymes (SOD, CAT, GSH) have been reported for Ursolic acid [119]. An upregulation of Nrf2/ ARE axis, and so reduced ROS production, has also been reported for Panax Ginseng and its ginsenosides [120]. Furthermore, antiapoptotic effects have been reported for Panax Ginseng in Doxo-treated cardiomyocytes [121]. Quercetin has been demonstrated to counteract ROS production in DIC and to prevent the opening of mPTP [122]. Several in vivo and in vitro studies showed that Rutin administration significantly reduces Doxo-induced ROS production [123], and a decrease in the expression of pro-apoptotic proteins caspase 3 and 9 has been reported [124]. Moreover, it has been reported that Selenium and Pinocembrin reduce Doxo-induced inflammation by enhanced Nrf2 expression which weak the NLRP2 activation [125].
5. Conclusions
Cardiotoxicity remains the main long side effect of many chemotherapeutic drugs, especially Doxo. Indeed, Doxo has detrimental effects, classified as acute and chronic abnormalities, including arrhythmias, heart failure, and ventricular dysfunction. In the last decade, much attention has been paid to the early events involved in cardiomyocytes toxicity, since it has been well established that these are the first step of a continuous escalation that over the years degenerates in heart failure. Growing body of evidence, indicate that oxidative stress and inflammation are the main characters involved in DIC. However, they are closely related to each other, and the activated pathways converge at many time points thus making it complicated to establish a cause-effect relationship and identify the target on which to act. The knowledge of molecular mechanisms involved in the early stage of DIC could help to identify drugs or natural products capable of counteracting these effects so reducing the onset of chronic cardiotoxicity and cardiac dysfunction in Doxo-treated patients. As described in this review, the only FDA approved cardioprotective agent, Dexrazoxane, is not free from side effects. Recently, several Doxo nanoformulations demonstrated better anticancer activity compared to the free drug, however, there are no definitive data on their long-term cardiac safety. A better understanding of the molecular mechanisms involved in cardiotoxicity would allow to use also drugs currently in use for other therapeutic indications, such as Carvedilol or Statins.
Furthermore, it is now clear that cardiotoxicity is also associated with other chemotherapeutic drugs, so, research in this area has a high impact in clinical practice.
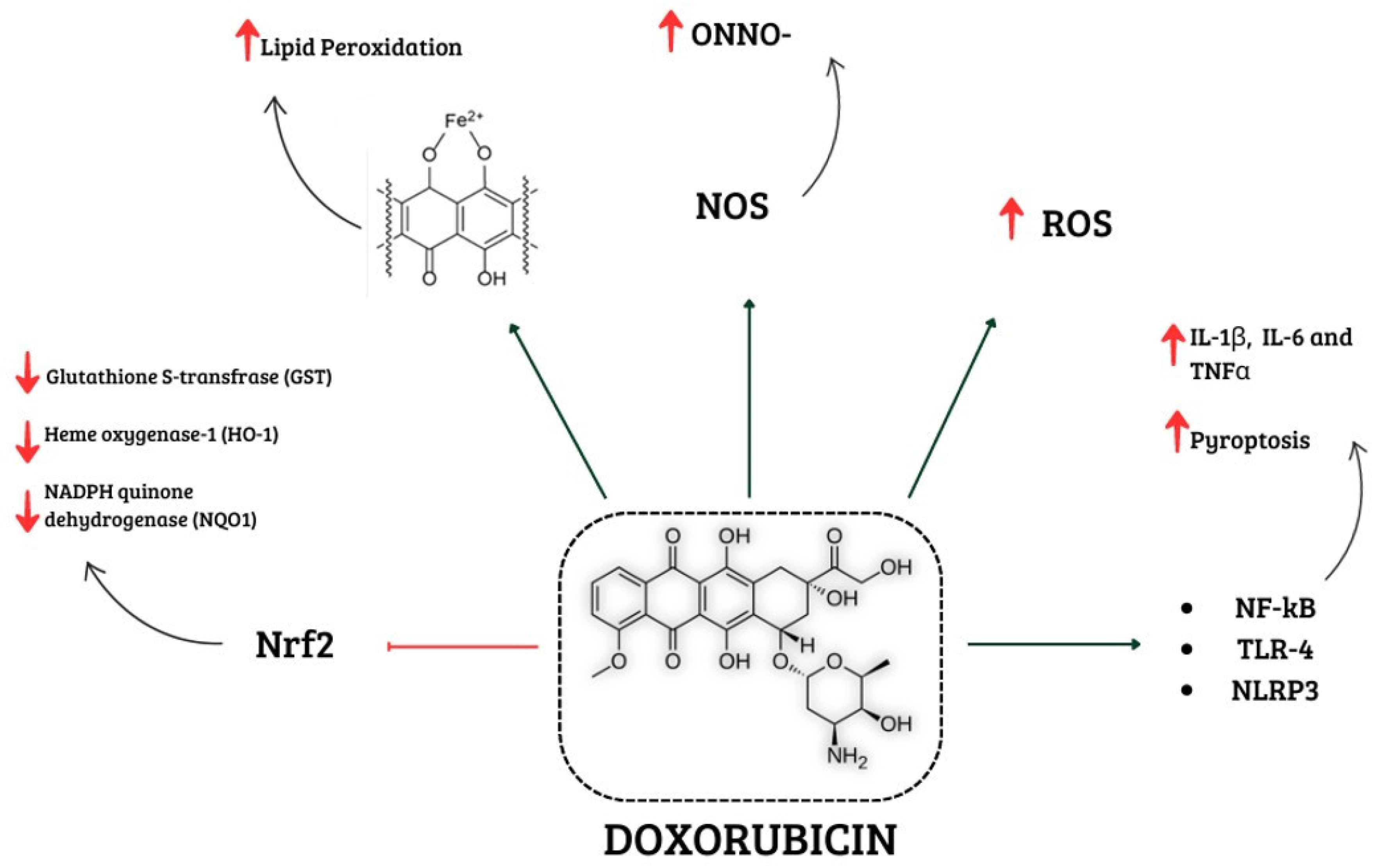
Figure 4.
Schematic representation of different mechanisms involved in Doxo-induced cardiotoxicity Doxorubicin is involved in increased ROS and NOS levels, which promote oxidative stress. In addition, it activates NF-kB, TLR4 and NLRP3, which are involved in enhancing levels of pro-inflammatory cytokines. Lipid peroxidation is caused by Doxo-iron complexes, and the reduction in antioxidant enzymes is related to the inactivation of Nrf2. Abbrevations: ROS, Reactive oxygen species; NOS, Reactive nitrogen species; NF-kB, Nuclear factor kappaB; TLR4, Tool like receptor 4; NLRP3, NLR family pyrin domain containing 3; Nrf2, Nuclear factor erythroid 2 like.
Figure 4.
Schematic representation of different mechanisms involved in Doxo-induced cardiotoxicity Doxorubicin is involved in increased ROS and NOS levels, which promote oxidative stress. In addition, it activates NF-kB, TLR4 and NLRP3, which are involved in enhancing levels of pro-inflammatory cytokines. Lipid peroxidation is caused by Doxo-iron complexes, and the reduction in antioxidant enzymes is related to the inactivation of Nrf2. Abbrevations: ROS, Reactive oxygen species; NOS, Reactive nitrogen species; NF-kB, Nuclear factor kappaB; TLR4, Tool like receptor 4; NLRP3, NLR family pyrin domain containing 3; Nrf2, Nuclear factor erythroid 2 like.
References
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J Clin 2016 66 (4): 309-25. [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics. CA Cancer J Clin 2023 73 (1): 17-48. [CrossRef]
- Hershman, D.L.; Neugut, A.I. Anthracycline cardiotoxicity: one size does not fit all! J Natl Cancer Inst 2008 100 (15): 1046-7.
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid Med Cell Longev 2015 2015:795602.
- Timm, K.N. and Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc Drugs Ther 2020 34 (2): 255-269. [CrossRef]
- Schirone, L.; D’Ambrosio, L.; Forte, M.; Genovese, R.; Schiavon, S.; Spinosa, G.; Iacovone, G.; Valenti, V.; Frati, G.; Sciarretta, S. Mitochondria and Doxorubicin-Induced Cardiomyopathy: A Complex Interplay. Cells 2022 11 (13): 2000. [CrossRef]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.F.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail Rev 2021 26 (4): 881-890. [CrossRef]
- Stěrba, M.; Popelová, O.; Vávrová, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Simůnek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 2013 18 (8): 899-929. [CrossRef]
- Hrdina, R.; Gersl, V.; Klimtová, I.; Simůnek, T.; Machácková, J.; Adamcová, M. Anthracycline-induced cardiotoxicity. Acta Medica (Hradec Kralove) 2000 43(3):75-82. PMID: 11089274.
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng. T.; Yan, L.; Tang, Q.Z.; Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. International Journal of Biological Sciences 2022 18(2) :760-770. [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 2012 52 (6): 1213-25. [CrossRef]
- Milczarek, A.; Starzyński, R.R.; Styś, A.; Jończy, A.; Staroń, R.; Grzelak, A.; Lipiński, P. A drastic superoxide-dependent oxidative stress is prerequisite for the down-regulation of IRP1: Insights from studies on SOD1-deficient mice and macrophages treated with paraquat. PLoS One 2017 12 (5): e0176800. [CrossRef]
- Kciuk, M.; Gieleci’nska, A.; Mujwar, S.; Kołat, D.; Kałuzi´nska-Kołat, Z.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023 12, 659. [CrossRef]
- Bhagat, A.; Shrestha, P.; Kleinerman, E.S. The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity. Int J Mol Sci 2022 23 (23): 14649. [CrossRef]
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells. 2022 11(17):2726. [CrossRef]
- Zheng, X.; Zhong, T.; Ma, Y.; Wan, X.; Qin, A.; Yao, B.; Zou, H.; Song, Y.; Yin, D. Bnip3 mediates doxorubicin induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020 242:117186.
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018 23 (5): 1184. [CrossRef]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal 2023 21 (1): 61. [CrossRef]
- Aryal, B.; Rao, V.A. Deficiency in Cardiolipin Reduces Doxorubicin-Induced Oxidative Stress and Mitochondrial Damage in Human B-Lymphocytes. PLoS One 2016 11 (7): e0158376. [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis 2021 12 (4): 339. [CrossRef]
- Ruggeri, C.; Gioffré, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: an overview of preclinical models and cancer patients. Heart Fail Rev 2018 23 (1): 109-122. [CrossRef]
- Panpan, T.; Yuchen, D.; Xianyong, S.; Meng, L.; Ruijuan, H.; Ranran, D.; Pengyan, Z.; Mingxi, L.; Rongrong, X. Cardiac Remodelling Following Cancer Therapy: A Review. Cardiovasc Toxicol 2022 22 (9): 771-786. [CrossRef]
- Perfettini, J.L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005 Apr;15(4):179-83. [CrossRef]
- Corrado, M.; Scorrano, L.; Campello, S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int J Cell Biol. 2012 2012:729290. [CrossRef]
- Aung, L.H.H.; Li, R.; Prabhakar, B.S.; Li, P. Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J Cell Mol Med. 2017 Dec; 21(12):3394-3404. [CrossRef]
- Antonny, B., Burd, C., De Camilli, P., Chen, E., Daumke, O., Faelber, K., Ford, M.; Frolov, V.A.; Frost, A.; Hinshaw, J.E.; Kirchhausen, T.; Kozlov, M.M.; Lenz, M.; Low, H.H.; McMahon, H.; Merrifield, C.; Pollard, T.D.; Robinson, P.J.; Roux, A.; Schmid, S. Membrane fission by dynamin: What we know and what we need to know. EMBO J. 2016 35 (21): 2270–2284. [CrossRef]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; Ge, J. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2017 108: 138-148. [CrossRef]
- Miyoshi, T., Nakamura, K., Amioka, N., Hatipoglu, O. F., Yonezawa, T., Saito, Y., Yoshida, M.; Akagi, S.; Ito, H. LCZ696 ameliorates doxorubicin-induced cardiomyocyte toxicity in rats. Sci. Rep. 2022 12 (1), 4930. [CrossRef]
- Chen, R.; Niu, M.; Hu, X.; He, Y. Targeting mitochondrial dynamics proteins for the treatment of doxorubicin-induced cardiotoxicity. Front. Mol. Biosci. 2023 10: 1241225. [CrossRef]
- Bell, E. L., and Guarente, L. The SirT3 divining rod points to oxidative stress. Mol. Cell 2011 42 (5): 561–568. [CrossRef]
- He, L.; Liu, F.; Li, J. Mitochondrial Sirtuins and Doxorubicin-induced Cardiotoxicity. Cardiovasc Toxicol 2021 21 (3): 179-191. [CrossRef]
- Wallace R. Fractured Symmetries: Information and Control Theory Perspectives on Mitochondrial Dysfunction. Acta Biotheor. 2021 69(3):277-301. [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res. 2020 126 (7): 926-941. [CrossRef]
- Marechal, X.; Montaigne, D.; Marciniak, C.; Marchetti, P.; Hassoun, S.M.; Beauvillain, J.C.; Lancel. S.; Neviere, R. Doxorubicin-induced cardiac dysfunction is attenuated by ciclosporin treatment in mice through improvements in mitochondrial bioenergetics. Clin Sci (Lond) 2011 121 (9): 405-13. [CrossRef]
- Gilleron, M.; Marechal, X.; Montaigne, D.; Franczak, J.; Neviere, R.; Lancel, S. NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochemical and Biophysical Research Communications 2009 388 (4): 727-731. [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases in mechano-transduction: mechanisms and consequences. Antioxid Redox Signal 2014 20 (6): 887-98. [CrossRef]
- Efentakis, P.; Doerschmann, H.; Witzler, C.; Siemer, S.; Nikolaou, P.E.; Kastritis, E.; Stauber, R.; Dimopoulos, M.A.; Wenzel, P.; Andreadou, I.; Terpos, E. Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib. Int J Mol Sci 2020 21 (15): 5185. [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol 2011 106 (5): 735-747. [CrossRef]
- Tayeh, Z.; Ofir, R. Asteriscus graveolens Extract in Combination with Cisplatin/Etoposide/Doxorubicin Suppresses Lymphoma Cell Growth through Induction of Caspase-3 Dependent Apoptosis. Int J Mol Sci 2018 19 (8): 2219. [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother 2021 139: 111708. [CrossRef]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res 2010 70 (22): 9287-97. [CrossRef]
- Ma, Z.G.; Kong, C.Y.; Wu, H.M.; Song, P.; Zhang, X.; Yuan, Y.P.; Deng, W.; Tang, Q.Z. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics 2020 10 (24): 11013-11025. [CrossRef]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther 2010 10 (3): 223-31. [CrossRef]
- Wang, Y.; Liao, J.; Luo, Y.; Li, M.; Su, X.; Yu, B.; Teng, J.; Wang, H.; Lv, X. Berberine Alleviates Doxorubicin-Induced Myocardial Injury and Fibrosis by Eliminating Oxidative Stress and Mitochondrial Damage via Promoting Nrf-2 Pathway Activation. Int J Mol Sci 2023 24 (4): 3257. [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology 2013 53 :401-26. [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; Ma, J.; Wang, A.; Xu, X.; Shahane, S.A.; Xia, M.; Woo, J.; Mensah, G.A.; Wang, Z.; Ferrer, M.; Gabrielson, E.; Li, Z.; Rastinejad, F.; Shen, M.; Boxer, M.B.; Biswal, S. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem Biol 2016 11 (11): 3214-3225. [CrossRef]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: crosstalk between the Nrf2 pathway and autophagy. Ageing Res Rev. 2021 65:101207.
- Pi, J., Bai, Y., Reece, J. M., Williams, J., Liu, D., Freeman, M. L., Waalkes, M. P. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radical Biology and Medicine 2007 42(12), 1797–1806.
- Chen, W., Sun, Z., Wang, X.-J., Jiang, T., Huang, Z., Fang, D., & Zhang, D. D. Direct interaction between Nrf2 and p21Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Molecular Cell 2009 34(6), 663–673.
- Ichimura, Y., Waguri, S., Sou, Y.S., Kageyama, S., Hasegawa, J., Ishimura, R., Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; Hoshii, T.; Hirao, A.; Takagi, K.; Mizushima, T.; Motohashi, H.; Lee, M.S.; Yoshimori, T.; Tanaka, K.; Yamamoto, M.; Komatsu, M. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Molecular Cell 2013 51 (5): 618–631.
- Milani, P., Ambrosi, G., Gammoh, O., Blandini, F., & Cereda, C. (2013). SOD1 and DJ-1 converge at Nrf2 pathway: A clue for antioxidant therapeutic potential in Neurodegeneration. Oxidative Medicine and Cellular Longevity, 2013 836760. [CrossRef]
- Su, S., Li, Q., Xiong, C., Li, J., Zhang, R., Niu, Y., Zhao, L.; Wang, Y.; Guo, H. Sesamin ameliorates doxorubicin-induced cardiotoxicity: Involvement of Sirt1 and Mn-SOD pathway. Toxicology Letters 2014 224 (2): 257–263.
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol Lett 2019 307: 41-48. [CrossRef]
- Deng, J.; Huang, M.; Wu, H. Protective effect of limonin against doxorubicin-induced cardiotoxicity via activating nuclear factor - like 2 and Sirtuin 2 signaling pathways. Bioengineered 2021 12 (1): 7975-7984. [CrossRef]
- Shi, K.N.; Li, P.B.; Su, H.X.; Gao, J.; Li, H.H. MK-886 protects against cardiac ischaemia/reperfusion injury by activating proteasome-Keap1-NRF2 signalling. Redox Biol 2023 62: 102706. [CrossRef]
- Zhang, Y.; Ahmad, K.A.; Khan, F.U.; Yan, S.; Ihsan, A.U.; Ding, Q. Chitosan oligo saccharides prevent doxorubicin-induced oxidative stress and cardiac apoptosis through activating p38 and JNK MAPK mediated Nrf2/ARE pathway. Chem Biol Interact. 2019 305: 54–65.
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharm Res. 2021 166: 105466.
- Pecoraro, M.; Pala, B.; Di Marcantonio, M.C.; Muraro, R.; Marzocco, S.; Pinto, A.; Mincione, G.; Popolo, A. Doxorubicin induced oxidative and nitrosative stress: Mitochondrial connexin 43 is at the crossroads. Int J Mol Med 2020 46 (3): 1197-1209. [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol 2009 296 (5): H1466-83. [CrossRef]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; Picard, M.H.; Scherrer-Crosbie, M.; Bloch, K.D. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007 116 (5): 506-14. [CrossRef]
- Rawat, D.K.; Hecker, P.; Watanabe, M.; Chettimada, S.; Levy, R.J.; Okada, T.; Edwards, J.G.; Gupte, S.A. Glucose-6-phosphate dehydrogenase and NADPH redox regulates cardiac myocyte L-type calcium channel activity and myocardial contractile function. PLoS One 2012 7 (10): e45365. [CrossRef]
- Vásquez-Vivar, J.; Martasek, P.; Hogg, N.; Masters, B.S., Pritchard, K.A.Jr; Kalyanaraman, B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 1997 36 (38): 11293-7. [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid Med Cell Longev 2017 2017: 1521020. [CrossRef]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; Li, M.; Sorriento, D.; Ciccarelli, M.; Ghigo, A.; Hirsch, E.; Bianco, R.; Iaccarino, G.; Abete, P.; Bonaduce, D.; Calignano, A.; Berni Canani, R.; Tocchetti, C.G. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur J Heart Fail 2019 21 (4): 519-528. [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol 2018 14: 618-625. [CrossRef]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; Vassalli, G. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc Res 2020 116 (2): 383-392. [CrossRef]
- Liu, B.; Li, H.; Qu, H.; Sun, B. Nitric oxide synthase expressions in ADR-induced cardiomyopathy in rats. J Biochem Mol Biol. 2006 39(6): 759-65. [CrossRef]
- Xu, Z.; Lin, S.; Wu, W.; Tan, h.; Wang, Z., Cheng, C.; Lu, L.; Zhang, X. Ghrelin prevents doxorubicin-induced cardiotoxicity through TNF-alpha/NF-kappaB pathways and mitochondrial protective mechanisms. Toxicology 2008 247 (2-3):133-8. [CrossRef]
- Pecoraro, M.; Del Pizzo, M.; Marzocco, S.; Sorrentino, R.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicol Appl Pharmacol 2016 293: 44-52. [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. American Journal of Physiology-Heart and Circulatory Physiology 2011 (6): H2181-90. [CrossRef]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr Atheroscler Rep. 2017 19 (6):27. [CrossRef]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Failure Rev 2018 23 (5): 733-758. [CrossRef]
- Tavakoli, D. Z.; Singla, D.K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am J Physiol Heart Circ Physiol 2019 317(2): H460-H471. [CrossRef]
- Singla, D.K.; Johnson, T.A.; Tavakoli, D. Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019 8(10): 1224. [CrossRef]
- Ren, G.; Zhang, X.; Xiamo, Y.; Zhang, W.; Wang, Y.; Ma, W., Wang, X.; Song, P.; Lai, L.; Chen, H.; Zhan, Y.; Zhang, J.; Yu, M.; Ge, C.; Li, C.; Yin, R.; Yang, X. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deuibiquitination. The EMBO Journal 2019 Mar 15;38(6): e100376. [CrossRef]
- Li, L.L.; Wei, L.; Zhang, N.; Wei, W.Y.; Hu, C.; Deng, W.; Tang, Q.Z. Levosimendan Protects against Doxorubicin-Induced Cardiotoxicity by Regulating the PTEN/Akt Pathway. Biomed Res Int. 2020 2020 :8593617. [CrossRef]
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNγ-mediated metabolic reprogramming in cardiomyocytes. J Pathol 2019 247(3): 320-332. [CrossRef]
- Ye, B.; Shi, X.; Xu, J.; Dai, S.; Xu, J.; Fan, X.; Han, B.; Han, J. Gasdermin D mediates doxorubicin-induced cardiomyocyte pyroptosis and cardiotoxicity via directly binding to doxorubicin and changes in mitochondrial damage. Translational Research. 2022 248: 36-50. [CrossRef]
- Lan, Y.; Wang, Y.; Huang, K.; Zeng, Q. Heat shock protein 22 attenuates doxorubicin induced cardiotoxicity via regulating inflammation and apoptosis. Front Pharm. 2020 11:257.
- D’Angelo, N.A.; Noronha, M.A.; Câmara, M.C.C.; Kurnik, I.S.; Feng, C.; Araujo, V.H.S.; Santos, J.H.P.M.; Feitosa, V.; Molino, J.V.D.; Rangel-Yagui, C.O.; Chorilli, M.; Ho, E.A.; Lopes, A.M. Doxorubicin nanoformulations on therapy against cancer: An overview from the last 10 years. Biomaterials Advances. 2022 133:112623. [CrossRef]
- Li, X.R.; Cheng, X.H.; Zhang, G.N.; Wang, X.X.; Huang, J.M. Cardiac safety analysis of first-line chemotherapy drug pegylated liposomal doxorubicin in ovarian cancer. J Ovarian Res 2022 15 (1):96. [CrossRef]
- Dempke, W.C.M.; Zielinski, R.; Winkler, C.; Silberman, S.; Reuther, S.; Priebe, W. Anthracycline-induced cardiotoxicity — are we about to clear this hurdle? European Journal of Cancer 2023 185: 94-104. [CrossRef]
- Pendlebury, A.; De Bernardo, R.; Rose, P.G. Long-term use of pegylated liposomal doxorubicin to a cumulative dose of 4600 mg/m2 in recurrent ovarian cancer: Anti-Cancer Drugs 2017 28 (7): 815-817. [CrossRef]
- Misra, R.; Das, M.; Sahoo, B.S.; Sahoo, S.K. Reversal of multidrug resistance in vitro by co-delivery of MDR1 targeting siRNA and doxorubicin using a novel cationic poly(lactide-co-glycolide) nanoformulation. International Journal of Pharmaceutics 2014 475 (1-2): 372-384. [CrossRef]
- Xing, M.: Yan, F.; Yu, S.; Shen, P. Efficacy and Cardiotoxicity of Liposomal Doxorubicin Based Chemotherapy in Advanced Breast Cancer: A Meta-Analysis of Ten Randomized Controlled Trials. PLoS ONE 2015 10(7): e0133569. [CrossRef]
- Cagel, M.; Grotz, E.; Bernabeu, E.; Moretton, M. A.; Chiappetta, D. A. Doxorubicin: nanotechnological overviews from bench to bedside. Drug Discov. Today 2017 22, 270–281.
- Gallo, E.; Diaferia C.; Rosa, E.; Smaldone, G.; Morelli, G.; Accardo, A. Peptide-Based Hydrogels and Nanogels for Delivery of Doxorubicin. International Journal of Nanomedicine 2021:16 1617–1630.
- Li, J.; Mooney, D.J.Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016;1:16071.
- Norouzi, M.;Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: a combinational approach for enhanced delivery of nanoparticles Scientific RepoRtS 2020 10:11292 | . [CrossRef]
- Mu, Q.; Kievit, F.M.; Kant, R.J.; Lin, G.; Jeon, M.; Zhang, M. Anti-HER2/neu peptide-conjugated iron oxide nanoparticles for targeted delivery of paclitaxel to breast cancer cells. Nanoscale. 2015;7(43):18010-4.
- Norouzi, M., Nazari, B. & Miller, D. W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016 1(11), 1835–1849.
- Zhu, L.; Wang, D.; Wei, X.; Zhu, X.; Li, J.; Tu, C.; Su, Y.; Wu, J.; Zhu, B.; Yan, D. Multifunctional pH-sensitive superparamagnetic iron-oxide nanocomposites for targeted drug delivery and MR imaging. J Control Release. 2013;169(3):228-38.
- Chang, Y.; Li, Y.; Meng, X.; Liu, N.; Sun. D.; Wang, J. Dendrimer functionalized water soluble magnetic iron oxide conjugates as dual imaging probe for tumor targeting and drug delivery. Polym Chem. 2013, 4, 789-794.
- Shen, B.; Ma, Y.; Yu, S.; & Ji C. Smart multifunctional magnetic nanoparticle-based drug delivery system for cancer thermo chemotherapy and intracellular imaging. ACS Appl. Mater. Interfaces. 2016 , 24502–24508.
- Weiss, G.; Loyevsky, M.; Gordeuk, V.R. Dexrazoxane (ICRF-187). Gen Pharmacol 1999 32 (1): 155-8. [CrossRef]
- Hutchins, K.K.; Siddeek, H.; Franco, V.I.; Lipshultz, S.E. Prevention of cardiotoxicity among survivors of childhood cancer. Br J Clin Pharmacol 2017 83(3): 455-465. [CrossRef]
- Kourek, C.; Touloupaki, M.; Rempakos, A.; Loritis, K.; Tsougkos, E.; Paraskevaidis, I.; Briasoulis, A. Cardioprotective Strategies from Cardiotoxicity in Cancer Patients: A Comprehensive Review. J Cardiovasc Dev Dis 2022 9(8):259. [CrossRef]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther. 1992 263(1):92-8. PMID: 1357162.
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; Barsotti, A.; Brunelli, C. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol 2004 37 (4): 837-46. [CrossRef]
- Tashakori Beheshti, A.; Mostafavi Toroghi, H.; Hosseini, G.; Zarifian, A.; Homaei Shandiz, F.; Fazlinezhad, A. Carvedilol Administration Can Prevent Doxorubicin-Induced Cardiotoxicity: A Double-Blind Randomized Trial. Cardiology 2016 134(1):47-53. [CrossRef]
- Abuosa, A.M.; Elshiekh, A.H.; Qureshi, K.; Abrar, M.B.; Kholeif, M.A.; Kinsara, A.J.; Andejani, A.; Ahmed, A.H.; Cleland, J.G.F. Prophylactic use of carvedilol to prevent ventricular dysfunction in patients with cancer treated with doxorubicin. Indian Heart J 2018 Dec; 70 Suppl 3: S96-S100. [CrossRef]
- Moustafa, I.; Connolly, C.; Anis, M.; Mustafa, H.; Oosthuizen, F.; Viljoen, M. A prospective study to evaluate the efficacy and safety of vitamin E and levocarnitine prophylaxis against doxorubicin-induced cardiotoxicity in adult breast cancer patients. J Oncol Pharm Pract 2024 Mar; 30(2): 354-366. [CrossRef]
- Riad, A.; Bien, S.; Westermann, D.; Becher, P.M.; Loya, K.; Landmesser, U.; Kroemer, H.K.; Schultheiss, H.P.; Tschöpe, C. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer Res 2009 69(2):695-9. [CrossRef]
- Dadson, K.; Thavendiranathan, P.; Hauck, L.; Grothe, D.; Azam, M.A.; Stanley-Hasnain, S.; Mahiny-Shahmohammady, D.; Si, D.; Bokhari, M.; Lai, P.F.H.; Massé, S. Nanthakumar, K.; Billia, F. Statins Protect Against Early Stages of Doxorubicin-induced Cardiotoxicity Through the Regulation of Akt Signaling and SERCA2. CJC Open 2022 4 (12): 1043-1052. [CrossRef]
- Zhang, H.; Lu, X.; Liu, Z.; Du, K. Rosuvastatin reduces the pro-inflammatory effects of adriamycin on the expression of HMGB1 and RAGE in rats. Int J Mol Med 2018 42 (6): 3415-3423. [CrossRef]
- Neilan, T.G.; Quinaglia, T.; Onoue, T.; Mahmood, S.S.; Drobni, Z.D.; Gilman, H.K.; Smith, A.; Heemelaar, J.C.; Brahmbhatt, P.; Ho, J.S.; Sama, S.; Svoboda, J.; Neuberg, D.S.; Abramson, J.S.; Hochberg, E.P.; Barnes, J.A.; Armand, P.; Jacobsen, E.D.; Jacobson, C.A.; Kim, A.I.; Soumerai, J.D.; Han, Y.; Friedman, R.S.; Lacasce, A.S.; Ky, B.; Landsburg, D.; Nasta, S.; Kwong, R.Y.; Jerosch-Herold, M.; Redd, R.A.; Hua, L.; Januzzi, J.L.; Asnani, A.; Mousavi, N.; Scherrer-Crosbie, M. Atorvastatin for Anthracycline-Associated Cardiac Dysfunction: The STOP-CA Randomized Clinical Trial. JAMA. 2023 Aug 8; 330(6): 528-536. [CrossRef]
- Makhlin, I.; Demissei, B.G.; D’Agostino, R.; Hundley, W.G.; Baleanu-Gogonea, C.; Wilcox, N.S.; Chen, A.; Smith, A.M.; O’Connell, N.S.; Januzzi, J.; Lesser, G.J.; Scherrer-Crosbie, M.; Ibáñez, B.; Tang, W.H.W.; Ky, B. Statins Do Not Significantly Affect Oxidative Nitrosative Stress Biomarkers in the PREVENT Randomized Clinical Trial. Clin Cancer Res. 2024 Apr 4. [CrossRef]
- Tatlidede, E.; Sehirli, O.; Velioğlu-Oğünc, A.; Cetinel, S.; Yeğen, B.C.; Yarat, A.; Süleymanoğlu, S.; Sener, G. Resveratrol treatment protects against doxorubicin-induced cardiotoxicity by alleviating oxidative damage Free Radic Res 2009 43(3):195-205. [CrossRef]
- Chen, L.; Sun, X.; Wang, Z.; Chen, M.; He, Y.; Zhang, H.; Han, D.; Zheng, L. Resveratrol protects against doxorubicin-induced cardiotoxicity by attenuating ferroptosis through modulating the MAPK signaling pathway. Toxicol Appl Pharmacol. 2024 Jan; 482:116794. [CrossRef]
- Abdel-Daim, M.M.; Kilany, O.E.; Khalifa, H.A.; Ahmed, A.A.M. Allicin ameliorates doxorubicin-induced cardiotoxicity in rats via suppression of oxidative stress, inflammation and apoptosis. Cancer Chemother Pharmacol 2017 80(4):745-753. [CrossRef]
- Ky, B.; Vejpongsa, P.; Yeh, E.T.; Force, T.; Moslehi, J.J. Emerging paradigms in cardiomyopathies associated with cancer therapies. Circ Res 2013 113 (6): 754-64. [CrossRef]
- Wang, H.; Yu, P.; Gou, H.; Zhang, J.; Zhu, M.; Wang, Z.H.; Tian, J.W.; Jiang, Y.T.; Fu, F.H. Cardioprotective Effects of 20(S)-Ginsenoside Rh2 against Doxorubicin-Induced Cardiotoxicity In Vitro and In Vivo. Evid Based Complement Alternat Med 2012 2012: 506214. [CrossRef]
- Priya, L.B.; Baskaran, R.; Huang, C.Y.; Padma, V.V. Neferine ameliorates cardiomyoblast apoptosis induced by doxorubicin: possible role in modulating NADPH oxidase/ROS-mediated NFκB redox signaling cascade. Scientific Reports 2017 7(1) :12283. [CrossRef]
- Lin, X.; Wang, Q.; Sun, S.; Xu. G.; Wu, Q.; Qi, M.; Bai, F.; Yu, J. Astragaloside IV promotes the eNOS/NO/cGMP pathway and improves left ventricular diastolic function in rats with metabolic syndrome. Journal of International Medical Research 2019 48 (1): 1-15. [CrossRef]
- Afsar, T.; Razak, S.; Batoo, K.M.; Khan, M.R. Acacia hydaspica R. Parker prevents doxorubicin-induced cardiac injury by attenuation of oxidative stress and structural Cardiomyocyte alterations in rats. BMC Complement Altern Med 2017 17(1):554. [CrossRef]
- Wang, M.; Zhang, J.; Zhao, M.; Liu, J.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; Li, D.; Wan, J. Resolvin D1 Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting Inflammation, Oxidative and Endoplasmic Reticulum Stress. Front Pharmacol 2022 12: 749899. [CrossRef]
- Cheng, D.; Chen, L.; Tu, W.; Wang, H.; Wang, Q.; Meng, L.; Li, Z.; Yu, Q. Protective effects of valsartan administration on doxorubicin induced myocardial injury in rats and the role of oxidative stress and NOX2/NOX4 signaling. Mol Med Rep. 2020 22(5): 4151-4162. [CrossRef]
- Hsieh, P.L.; Chu, P.M.; Cheng, H.C.; Huang, Y.T.; Chou, W.C.; Tsai, K.L.; Chan, S.H. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. Int J Mol Sci. 2022 23(17):10146. [CrossRef]
- Draginic, N.; Jakovljevic, V.; Andjic, M.; Jeremic, J.; Srejovic. I.; Rankovic, M.; Tomovic, M.; Nikolic, Turnic T.; Svistunov, A.; Bolevich, S.; Milosavljevic, I. Melissa officinalis L. as a Nutritional Strategy for Cardioprotection. Front Physiol. 2021 12: 661778. [CrossRef]
- Wan, Y.; Wang, J.; Xu, J.F.; Tang, F.; Chen, L.; Tan, Y.Z.; Rao, C.L.; Ao, H.; Peng, C. Panax ginseng and its ginsenosides: potential candidates for the prevention and treatment of chemotherapy-induced side effects. J Ginseng Res. 2021 Nov;45(6):617-630. [CrossRef]
- You, J.S.; Huang, H.F.; Chang, Y.L. Panax ginseng reduces adriamycin-induced heart failure in rats. Phytother Res 2005 19 (12): 1018-22. [CrossRef]
- Syahputra, R.A.; Harahap, U.; Dalimunthe, A.; Nasution, M.P.; Satria, D. The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules 2022 27 (4): 1320. [CrossRef]
- Yu,W.; Sun, H.; Zha, W.; Cui, W.; Xu, L.; Min, Q.; Wu, J. Apigenin Attenuates Adriamycin-Induced Cardiomyocyte Apoptosis via the PI3K/AKT/mTOR Pathway. Evid Based Complement Alternat Med 2017 2017: 2590676. [CrossRef]
- Ma, Y.; Yang, L.; Ma, J.; Lu, L.; Wang, X.; Ren, J.; Yang, J. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim Biophys Acta Mol Basis Dis 2017 Aug; 1863 (8): 1904-1911. [CrossRef]
- Yang, H.B.; Lu, Z.Y.; Yuan, W.; Li, W.D.; Mao, S. Selenium Attenuates Doxorubicin-Induced Cardiotoxicity Through Nrf2-NLRP3 Pathway. Biol Trace Elem Res 2022 200 (6): 2848-2856. [CrossRef]
Figure 1.
Interference of Semiquinone in redox cycle.NADPH oxidase is involved in the generation of the Semiquinone form of Doxo. Quinone form is regenerated by the presence of oxygen, that forms superoxide, a highly reactive specie. Indeed, superoxide interacts with nitric oxide to form the peroxynitrite anion, a potent free radical; in addition, superoxide is converted into peroxide by SOD. Hydrogen peroxide is responsible for the formation of hydroxyl radicals that lead to DNA, proteins, and lipids damage. Abbrevations. SOD, Superoxide dismutase; NOs, nitric oxide synthetase.
Figure 1.
Interference of Semiquinone in redox cycle.NADPH oxidase is involved in the generation of the Semiquinone form of Doxo. Quinone form is regenerated by the presence of oxygen, that forms superoxide, a highly reactive specie. Indeed, superoxide interacts with nitric oxide to form the peroxynitrite anion, a potent free radical; in addition, superoxide is converted into peroxide by SOD. Hydrogen peroxide is responsible for the formation of hydroxyl radicals that lead to DNA, proteins, and lipids damage. Abbrevations. SOD, Superoxide dismutase; NOs, nitric oxide synthetase.
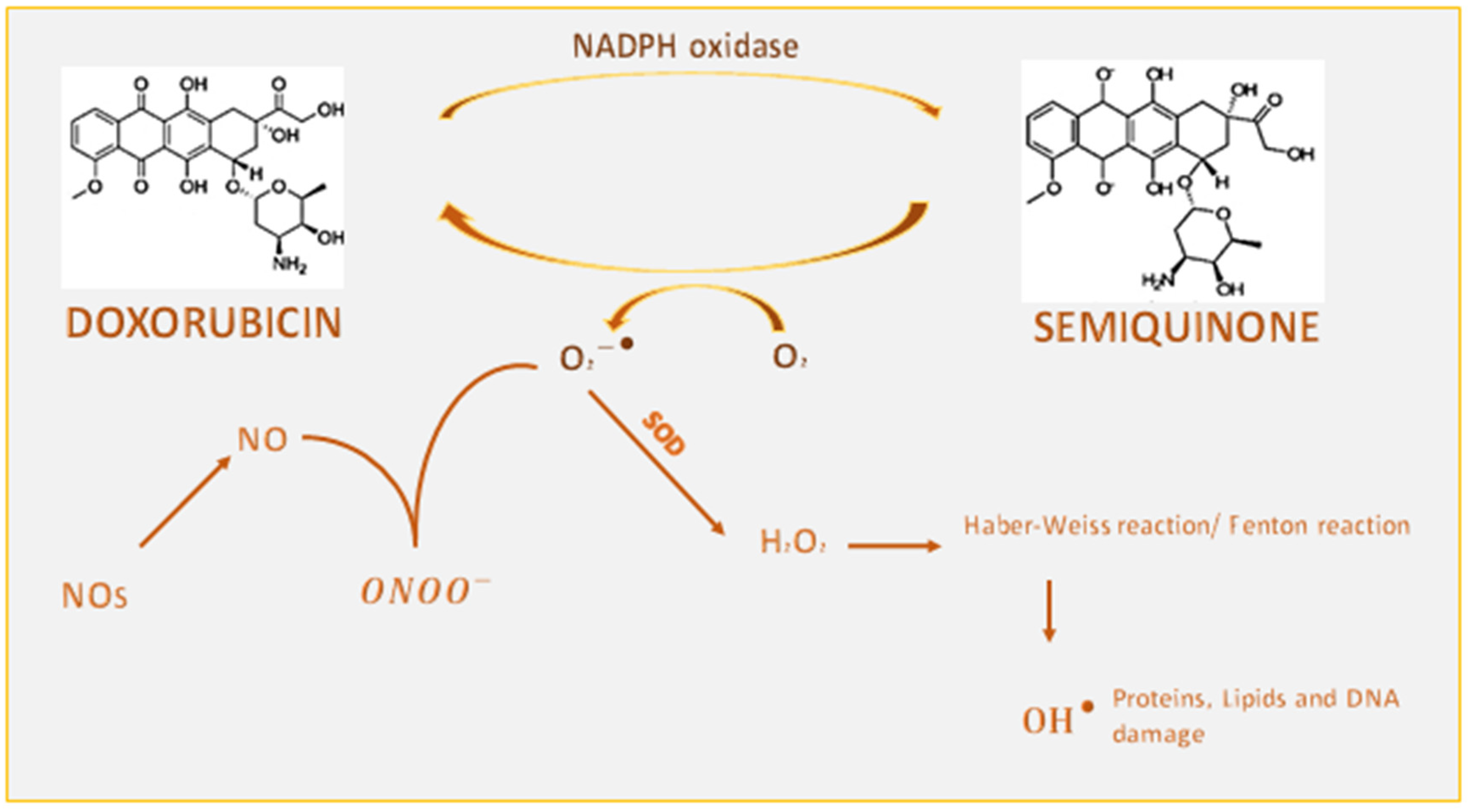
Figure 2.
Involvement of Doxo-Fe complexes in oxidative stress and lipid peroxidation. Doxo has a high affinity for iron this leads to the formation of Doxo-Fe complexes. Doxo-Fe3+ complex can be reduced by several reducing systems such as Gpx4 to form the Doxo-Fe2+ complex. The latter is involved both in increasing oxidative stress through interaction with O2 and inducing lipid peroxidation through the interaction with the negative charge of the membrane.
Figure 2.
Involvement of Doxo-Fe complexes in oxidative stress and lipid peroxidation. Doxo has a high affinity for iron this leads to the formation of Doxo-Fe complexes. Doxo-Fe3+ complex can be reduced by several reducing systems such as Gpx4 to form the Doxo-Fe2+ complex. The latter is involved both in increasing oxidative stress through interaction with O2 and inducing lipid peroxidation through the interaction with the negative charge of the membrane.
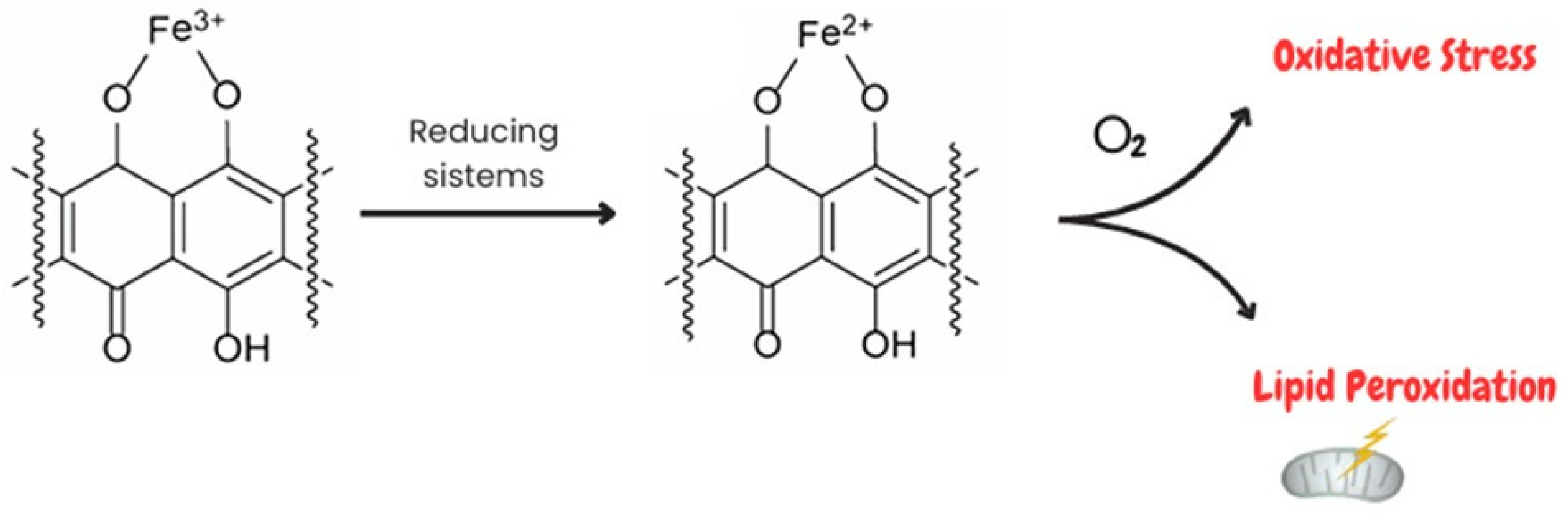
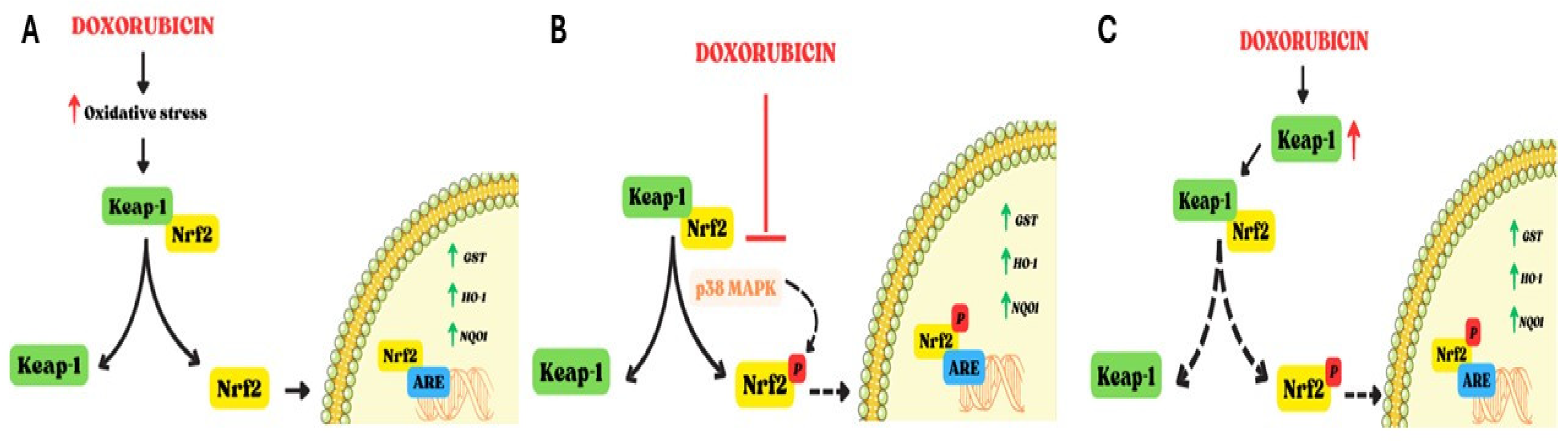
Figure 3.
Involvement of Nrf2 in Doxo-induced cardiotoxicity Nrf2 is negatively regulated by Keap-1; Keap-1 degradation induces activation and nuclear translocation of Nrf2, which thereby upregulates several antioxidant enzymes (GST, OH-1, NQO1). The effects of Doxo on Nrf2 are controversial as reported in panel A, B and C. A. Doxo-induced oxidative stress leads to activation of Nrf2 and its nuclear translocation to increase the levels of antioxidants enzyme. B. Doxo can inhibit Nrf2 by interfering with p38MAPK. The phosphorylation of Nrf2 is reduced and interaction with ARE in the nucleus is hindered. C. Doxo can induce increase in Keap-1 levels, thereby disrupting dissociation of the Keap-1/Nrf2 complex. In this way, nuclear translocation of Nrf2 is inhibited. Dashed lines indicate blocking of the mechanism. Abbrevations. Nrf2, Nuclear factor erythroid 2 like; Keap-1, Kelch Like ECH associated protein 1; GST, Glutathione S-transferase; HO-1, heme oxygenase-1; NQO1, NAD(P)H quinone dehydrogenase 1; ARE, Antioxidant response elements; p38MAPK, p38 mitogen-activated protein kinase.
Figure 3.
Involvement of Nrf2 in Doxo-induced cardiotoxicity Nrf2 is negatively regulated by Keap-1; Keap-1 degradation induces activation and nuclear translocation of Nrf2, which thereby upregulates several antioxidant enzymes (GST, OH-1, NQO1). The effects of Doxo on Nrf2 are controversial as reported in panel A, B and C. A. Doxo-induced oxidative stress leads to activation of Nrf2 and its nuclear translocation to increase the levels of antioxidants enzyme. B. Doxo can inhibit Nrf2 by interfering with p38MAPK. The phosphorylation of Nrf2 is reduced and interaction with ARE in the nucleus is hindered. C. Doxo can induce increase in Keap-1 levels, thereby disrupting dissociation of the Keap-1/Nrf2 complex. In this way, nuclear translocation of Nrf2 is inhibited. Dashed lines indicate blocking of the mechanism. Abbrevations. Nrf2, Nuclear factor erythroid 2 like; Keap-1, Kelch Like ECH associated protein 1; GST, Glutathione S-transferase; HO-1, heme oxygenase-1; NQO1, NAD(P)H quinone dehydrogenase 1; ARE, Antioxidant response elements; p38MAPK, p38 mitogen-activated protein kinase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



