Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
This review deals with the functionalization of double bonds carried out in the presence of a chiral catalyst exploiting the intramolecular attack to haliranium ions by nucleophilic nitrogen of amides or carbamates prepared from achiral aminoalkenes, and the C-N bonds formation leads to highly enantioenriched non-aromatic heterocycles. A range of protocols are reported, emphasizing the synthesis of many natural and biologically active products of pharmacological interest prepared according to this methodology.
Keywords:
non-aromatic heterocycles
; haloamination
; haloamidation
; haliranium ion
; stereoselectivity
; chiral catalysts
1. Introduction
In the presence of a halenium ion source [1,2,3] an alkene can give rise to the corresponding intermediate haliranium ion 1 [4,5]. The subsequent nucleophilic attack by a nitrogen atom appropriately tethered on the carbon chain, occurring through an endo- or an exo-mode [6,7,8,9,10,11], leads to a variety of non-aromatic N-heterocycles, whose structure strongly depends on either the substrate geometry and the nucleophilic functionality involved [12,13,14,15,16,17] (Figure 1).
The first intramolecular haloamination reactions of amino alkenes were carried out more than a century ago [18,19,20] and this methodology allowed to increase the molecular complexity of the starting material since a ring is created together with a halide functionality suitable for further derivatizations. In addition, when the nitrogen atom is tethered on a chiral center, two additional chiral centers can be introduced on the framework with definite configuration so that a lot of highly enantioenriched amino alkenes were easily converted into chiral polysubstituted non aromatic heterocycles generally using a source of halenium ions in a basic medium, the stereoselectivity being directed by internal asymmetric induction arising from in-tether chiral centers [21,22,23,24,25,26,27].
According to this methodology, a lot of highly enantioenriched amino alkenes were easily converted into chiral polysubstituted heterocycles exploiting intramolecular haloamination, generally using a source of halenium ions in a basic medium, and the stereoselectivity was directed by internal asymmetric induction due to the chiral centers tethered in the substrate. On the contrary, to the best of our knowledge, starting from achiral amino alkenes, enantioselective intramolecular haloamination reactions were never carried out exploiting external asymmetric induction due to chiral catalysts, mainly derived from Cinchona alkaloids or BINOL, but the amino groups were always protected as sulfonyl amides or carbamates, so haloamidation is the most appropriate definition for this latter process. Within this field recently asymmetric methodologies were devised starting from achiral substrates, directed to prepare enantiomerically enriched non aromatic nitrogen-containing heterocycles, in particular natural products or bioactive molecules of therapeutic interest, and the development of improved ways directed towards the preparation of these compounds continues to be a challenging goal.
2. Asymmetric Synthesis Exploiting Substrate Directed Stereoselectivity
2.1. Polyfunctionalized Pyrrolidines
Many chiral polyhydroxy pyrrolidines isolated from natural sources, otherwise known as iminocyclitols or imino sugars, are able to inhibit glycosidases and other biologically relevant enzymes closely involved with the metabolism of N-linked glycoproteins [28,29]. Among the first examples of chiral amination, the aminoalkene 2, bearing a dioxolanyl group, was used as starting material for the stereoselective synthesis of 1,4-dideoxy-1.4-manno-D-lyxitol, LAB, 5, a potent competitive inhibitor of α-glucosidases [30,31]. The iodine-mediated cyclization proceeded according to a 5-exo mode in moderate yield and with total stereoselectivity leading to the iodomethyl intermediate 3 whose cis-2,3-disubstitution at the pyrrolidine ring, directed by the preexistent oxygenated functionality, can be explained by inspection of the transition states of the process [32,33,34,35,36,37]. Subsequently, this compound without isolation was converted in moderate yield into pyrrolidine 4 that eventually led to the expected iminosugar LAB, 5 (Scheme 1) [38].
Again exploiting the 2,3-cis-directing effect of ta dioxolanyl group [32,33,34,35,36,37], the iodomethyl pyrrolidine 7 was exclusively obtained in good yield with total regio- and stereoselectivity starting from secondary amine 6 and the subsequent metathesis reaction involving both the remaining allyl groups led in good yield to the iodomethyl indolizidine 8 that eventually equilibrated to the regioisomeric iodoquinolizidine 9 via an intermediate aziridine (Scheme 2) [39].
A matching/mismatching effect was observed when polyfunctionalized tertiary amines 10a and 10b underwent stereoselective iodine-mediated cyclization proceeding in a 5-exo mode, together with concurrent cleavage of the phenylethylamino group. In fact, starting from (S)-10a, the product 11, where the iodomethyl group at at C-2 was cis- to the oxygen of dioxolanyl substituent, was isolated in low yield as the major isomer, and the reduced yield might indicate that at the transition state the phenylethyl substituent is displayed in such a manner so as to prevent facile approach of the substrate to the iodenium ions source. On the contrary, starting from (R)-10b, having the opposite configuration at the phenylethylamino group with respect to (S)-10a, the asymmetric induction arising from the configuration of the phenylethylamino group overwhelmed the directing effect of the oxygen atom of the cis-dioxolane moiety and the major isomer was pyrrolidine 12, isolated in good yield, a useful intermediate for the synthesis of the polyhydroxylated pyrrolidine β-amino acid derivative 13 (Scheme 3) [40,41].
The iodoamination of the anti-tertiary homoallylic amine 14, displaying the (E)-configuration at the double bond, was carried out under the same reaction conditions leading to removal of the phenylethylamino group and proceeded as expected in a 5-endo mode to give the corresponding chiral 3-iodopyrrolidines 15 and 16 in moderate yield but with excellent stereoselectivity. In fact, the 2,5-trans isomer 15 was practically the sole product isolated, and eventually converted into the pyrrolidine alkaloid (-)-codonopsinine 17 (Scheme 4), whereas the cyclization of the (Z)-isomer afforded only a complex mixture. The observed stereoselectivity was explained by inspection of the two possible iodiranium ions intermediates taking into account steric interactions at the transition states between substituents at C-4 and C-5 and substituents at nitrogen atom that completely overwhelmed the directing effect of oxygen at C-4 [42,43].
In addition, when the secondary anti-benzylamine 18 underwent iodocyclization according to a 5-endo mode, the reaction proceeded in good yield but with lower stereoselectivity, to give preferentially the isomer 19 with respect to 20. The major isomer displayed 2,5-trans configuration, ascribed to steric interactions occurring at the transition state between the groups lieing at C-2 and C-5 positions, whereas the cis-1,2 directing effect of the acetoxy group was again largely ineffective. Compound 19 was eventually converted into a key intermediate for the synthesis of natural iminosugar (+)-DMDP, 21 [44], an inhibitor of glucosidase I [45] isolated from the leaves of Derris elliptica (Scheme 5) [46].
On the other hand, the cyclization of compound 22, displaying the syn-configuration, proceeded again in a 5-endo mode in good yield but with better stereoselectivity, probably owing to the 3,4-cis-directing effect of the hydroxy functionality matching with the 2,5-trans-disubstitution, leading mainly to the 2,5-trans-disubstituted derivative 23 [47] that was subsequently converted into a key intermediate for the synthesis of the alkaloid (+)-hyacinthacine A1, 25 (Scheme 6) [48].
However, the bis-homoallylic amine 26, on treatment with iodine in a basic medium, underwent cyclization via 5-exo mode to give in very low yield but with nearly total stereoselectivity the polysubstituted pyrrolidine 27 where the 2,3-cis directing effect of the hydroxy group [32,33,34,35,36,37] overwhelmed the strain due to the resulting 2,5-cis-configuration, and this compound was the key intermediate to 1,2,5-trideoxy-1-amino-2,5-imino-D-glucitol, (+)-ADGDP, 28 (Scheme 7) [49].
A different behavior was indeed observed when the bis-homoallylic amine 29, diastereomeric with 26, underwent stereoselective iodoamination to the intermediate 30, followed by in situ conversion into the aziridino derivative 31 that by reaction with TsNCO gave the bicyclic compound 32. Subsequent cleavage of the oxazolidinone ring afforded the cyclic six-membered product 33, eventually converted into (+)-ADANJ, 34, a 2-deoxy-2-amino analogue of (+)-1-deoxyallonojirimycin (Scheme 8) [49].
It is worth mentioning that the haloamination outcome dramatically changed when a primary amine was used in place of a secondary one. In fact, when the aminoalkendiol 35 was treated with iodine in the presence of NaHCO3, the bicyclic compound 37 was isolated in excellent yield and stereoselectivity [32,33,34,35,36,37], arising from insertion of a carbon dioxide molecule at pyrrolidine nitrogen, followed by intramolecular displacement of the iodide functionality of intermediate 36. The eventual cleavage of the oxazolidin-2-one ring in a strong basic medium led in excellent yield and without any racemization to 1,4-dideoxy-1,4-imino-D-xylitol D-DIX, 38 (Scheme 9) [50,51].
Another matching/mismatching effect was observed when polyfunctionalized diastereomeric alkenols displayed different configuration at the carbon atom bearing the amino group. Thus, diastereomeric alkenylamines 39 and 42, displaying the same configuration at C-2 and C-4, underwent cyclization in the presence of NaHCO3 using iodine and NIS, respectively, as halenium sources to give, via the intermediates 40 and 43, the corresponding bicyclic oxazolidin-2-ones 41 and 44 in high to moderate yield but with total stereoselectivity, and compound 44 was eventually converted into the iminosugar 45. The reaction proceeded with total stereoselectivity, owing to the directing effect of the oxygenated functionality at the allylic carbon leading to the 2,3-cis configuration that matched with the formation of the most stable 2,5-trans disubstituted product. (Scheme 10) [52,53]. On the contrary, aminoalkenes 46 and 49, displaying opposite configuration at C-2 with respect to 39 and 42, gave in good yield mixtures of diastereomeric bicyclic oxazolidin-2-ones 47,48 and 50,51, respectively, but with moderate stereoselection due to mismatch between the 2,5-cis unfavourable configuration - with respect to the 2,5-trans-one - and the overwhelming cis-2,3-directing effect exerted by the oxygenated functionality lieing at the chiral allylic carbon (Scheme 11) [32,33,34,35,36,37],[53].
The nitrogen atom of chiral unprotected aziridines was a nucleophile suitable for haloamination reactions leading to polycyclic structures containing the pyrrolidine ring. In fact, starting from compounds 52, treatment with NBS allowed to prepare bicyclic [3.1.0]bromoderivatives 54 in good to moderate yield but and with low to moderate stereoselectivity, and the reaction seemed to proceed through an intermediate bromoaziridine 53 that attacks the double bond to give the cycloamination product [54]. Eventual elimination of HBr, carried out under basic conditions, allowed to obtain the chiral bicyclic compounds 55, whose structure is present in azinocine antibiotics (Scheme 12) [55].
Moreover, chiral N-Boc aziridines 56 bearing an allyl group were treated with NBS to give at first the bicyclic aziridinium [3.1.0]intermediates 57. Although the subsequent attack by NsNH2 proceeded with low regioselectivity, either chiral N-t-Boc protected pyrrolidines 58 and piperidines 59 were isolated in good yield with nearly total stereoselectivity (Scheme 13) [56].
The same reaction was carried out using NBS and nosyl amide (NsNH2) starting from chiral t-Boc aziridines 60 bearing a homoallylic substituent, and the corresponding azepanes 62 displaying three chiral centres were obtained through the bicyclic intermediate 61 with excellent yield and nearly total regio- and stereoselectivity (Scheme 14) [57].
2.2. Pyrrolidines within Polycyclic Structures
Polyhydroxylated indolizidines and quinolizidines containing a pyrrolidine ring are conformationally restricted iminocyclitols and display interesting inhibitory action against glycosidases and found potential therapeutic applications as antidiabetic, antiviral, anticancer, antimetastatic and immunoregulating agents [58]. Thus, the chiral pyrrolidine 63, having a homoallylic substituent at C-2, was treated with NIS to give first the bicyclic intermediate 64 that without isolation, on treatment with an excess silver acetate, afforded the aziridino intermediate 65. Ring enlargement occurring in situ allowed to convert this product in moderate yield but with nearly total stereoselectivity into the bicyclic derivative 66, whose protecting groups were easily removed at once to give the indolizidine 67 (Scheme 15) [59].
Again directed towards preparation of polycyclic structures containing a pyrrolidine ring, the amine 68 was treated with iodine and the tetracyclic intermediate 69 was generated with total stereoselectivity. Then, exploiting a Kornblum oxidation [60], the iodide functionality was converted in moderate yield into a keto group and the cis-fused pyrrolidinocyclopentanone 70, intermediate for the preparation of alkaloid (-)-sinoracutine, 71, was eventually isolated in good yield and total stereoselectivity (Scheme 16) [61].
Another iodoamination reaction carried out with NIS, starting from the chiral amine 72, allowed to build up a pyrrolidine ring in good yield and with total stereoselectivity within the polycyclic compound 73, key intermediate for the synthesis of alkaloid (+)-lyconadine A, 74 (Scheme 17) [62].
Exploiting a tertiary amino group tethered on a chiral center lieing in a seven-membered cycloalkene, chiral bicyclic derivatives were prepared by transannular halocyclization. Thus, within a synthesis of (+)-pseudococaine 77, the bicyclic product 76 was isolated in very high yield and nearly total enantioselectivity starting from the tertiary amine 75 after reaction with iodine (Scheme 18) [63].
In a similar approach, the compound 78, containing a secondary amino group embedded in a eight-membered ring containing a double bond, was treated with iodine in methanol, to afford in good yield and with nearly total stereoselectivity the polyfunctionalyzed bicyclic derivative 79, key intermediate for the synthesis of the alkaloid (-)-hyacinthacine A1, 80 [64]. However, when under the same conditions the structurally similar chiral tertiary amine 81 underwent cyclization, the attack of the nitrogen atom to iodiranium ion occurred on the opposite side of the double bond, with respect to 79, probably due to steric bias arising from the dioxolanyl structure, so that the intermediate 82 displayed the opposite configuration at C-7a, eventually leading to (-)-7a-epi-hyacinthacine A1, 83 (Scheme 19) [65,66].
Within the enantioselective synthesis of the diazatricyclic core of alkaloid TAN1251C, 87, a muscarinic antagonist of potential interest in the treatment of ulcer [67], the spiro derivative 84 underwent cyclization mediated by iodine to provide with low stereoselectivity a mixture of compounds 85 and 86, but the reaction yield was not reported (Scheme 20) [68].
Eventually, within a total synthesis of pyrrolidineindoline alkaloids, the (S)-tryptophane derivative 88 reacted with NBS to afford in good yield a mixture of diastereomers 89 and 90 with low stereoselectivity [69] whereas the reaction of (R)-tryptophane derivative ent-88 with NBS, carried out in the presence of pyridinium p-toluene sulphonate (PPTS), afforded compound ent-89 in good yield and excellent diastereoselectivity (Scheme 21) [70].
2.3. Piperidine, Morpholine and Piperazine Derivatives
In analogy with aminoalkenols 14, 17, 21 and 24 [42,43], the primary amine 91 afforded in good yield but with low regio- and stereoselection the bicyclic oxazolidin-2-ones 92 and 93, generated by nucleophilic substitution of iodine by the intermediate carbamate anion arising from insertion of carbon dioxide at the nitrogen atom. On the other hand, compound 94 arose from attack to the intermediate iodiranium ion by the hydroxy functionality at C-2, followed by nucleophilic substitution by a carbamate anion. However, the major product 92 was eventually converted into 1-deoxygalactonojirimycin (DGJ), 95, (Scheme 22) [40] which is presently undergoing clinical evaluation for the treatment of Fabry’s disease [71].
A variety of natural products and biologically and pharmaceutically active compounds contain a C-substituted morpholine subunit, and in medicinal chemistry trifluoromethyl morpholines deserved particular attention, owing to the substituent that can deeply affect their metabolic properties [72,73]. Thus, enantiopure allylic amino ethers 96, where a trifluoromethyl group lies at a quaternary carbon adjacent to the oxygen atom, underwent cyclization mediated by iodine under basic conditions according to a 6-exo-mode, to give in good yield the corresponding diastereomeric iodomethylmorpholines 97 and 98, but the stereoselectivity of the process was very low or missing (Scheme 23) [74].
Besides morpholine derivatives, many compounds containing disubstituted piperazine ring were reported to display a broad spectrum of pharmacological activities [75]. Thus, chiral unsaturated benzylamines 99, prepared in the enantiomerically pure form starting from (S)-amino acids, were treated with iodine, to afford 2,5-trans-disubstituted piperazine derivatives 100 in good yield and excellent stereoselectivity according to a 6-exo-mode cyclization (Scheme 24). The stereochemical outcome was explained by inspection of the conformational preferences for the chair-like transition states of the reaction, since in the higher energy transition state leading to the cis-isomer a strong interaction between the iodomethyl group and the tosyl group occurs, that is missing in the lower transition state leading to the trans-isomer [76].
2.4. Substituted Guanidines
The marine alkaloids of the batzelladine family, isolated from the Batzella genus, contain a tricyclic guanidine core with substituents of varying complexity, and batzelladines A-F exhibit interesting biological antiviral activity in the inhibition of the binding of HIV gp120 to human CD4 [77]. Within a total synthesis of batzelladine D, 103, the intermediate 101 was treated with iodine in a basic medium and the tricyclic intermediate 102 was isolated in good yield and with total stereoselectivity, the asymmetric induction being due to the chiral centers present in the starting material (Scheme 25) [78,79].
A guanidinium group is present also in saxitoxin (STX) 106 and its analogs, a family of naturally occurring tricyclic guanidium alkaloids produced by some dinoflagellates which share the common chemical feature of high affinity and ion flux blockage capacity for voltage gated sodium channels (Navs), so that these compounds became interesting pharmacological targets [80].
Thus, within a synthesis of saxitoxin (STX), 106, the first representative of this alkaloids family to be isolated, the diprotected homoallyl guanidine 104 underwent cyclization mediated by iodine to give in good yield and with total stereoselectivity the bicyclic compound 105, that was eventually converted into the (+)-saxitoxin, STX, 106 (Scheme 26) [81].
2.5. Penems and Lactams
The bicyclic 1β-methylcarbapenem skeleton was built starting from the β-lactam 107 using a bromoamidation reaction directed by molecular geometry, that was carried out with NBS under mild conditions, to give in excellent yields and with total stereoselectivity the bicyclic compounds 108, [82] eventually converted in good yield into carbapenem 109 (Scheme 27) [83].
However, the intramolecular halolactamization was generally carried out exploiting an imide functionality, since the electron withdrawing tosyl or carboxylate groups favor nucleophilic attack by the nitrogen, whereas simple amides prefer to attack a haliranium ion with the more nucleophilic oxygen atom [84,85,86,87]. Thus, the bromocyclization of the chiral tosylamide 110 was carried out in a basic medium leading to a regioisomeric mixture of tricyclic β-lactams 111 and 112 that were isolated in high yield and with high stereoselectivity although the reaction proceeded preferentially through a SN2’ mechanism at the intermediate bromiranium ion (Scheme 28) [88].
Furthermore, an equimolar inseparable diastereomeric mixture of imides (S,R)- and (S,S)-113 was treated with t-BuOLi, and subsequent addition of NBS [89] allowed to isolate with excellent diastereoselectivity the bromolactam 114, exclusively, whereas N-Boc imide (S,S)-113 remained unchanged and this behavior was attributed to the different conformational flexibility of starting imides 113 (Scheme 29) [90].
Eventually, within a synthesis of indolizidinone dipeptide mimetics, the macrocyclic unsaturated amides 115a,b underwent transannular stereodivergent halocyclization with total regio- and stereoselectivity, depending on the reagents, the solvent employed and the substituent of the nitrogen atom. In fact, treatment of 115a with iodine and (diacetoxyiodo)benzene (DIB) in refluxing MeCN afforded the bicyclic lactam 116, exclusively, whereas the amide 115b by reaction with iodine in refluxing THF led in good yield and total stereoselectivity to the diastereomeric lactam 117 (Scheme 30) [91].
2.6. 1,3-Oxazolidin-2-Ones and 4,5-Dihydrooxazoles
The enantiomerically pure allylic alcohol 118 was treated with methyl thiocyanate followed by iodomethane, to give the intermediate carbonimidothioate 119 that by reaction with N-iodosuccinimide in basic medium afforded in moderate overall yield the corresponding oxazolidinone 120. This latter compound was isolated with total stereoselectivity, directed by the preexisting chiral center, and was further elaborated to give the cis-fused hexahydrofuro [3,2-b]pyran 121, key intermediate [92] of a total synthesis of neuroexcitotoxin (-)-dysiherbaine, 122 (Scheme 31) [93].
The iodocyclization of the enantiomerically pure trichloroacetimidate 123 containing a 1,4-dioxane moiety again occurred with chirality transfer starting from the preexisting allylic chiral center, and the reaction proceeded in good yield and total stereoselectivity to give the trans-4,5-dihydrooxazole 124 that was converted at first into (+)-polyoxamic acid 125 and then to the known lactone 126 (Scheme 32) [94].
2.7. 4,5-Dihydroimidazoles
The chiral imidazolidine 129, prepared by reaction of 2-(cyclohexa-2,5-dien-1-yl)acetaldehyde 127 with chiral diamine 128, underwent bromoamination with desymmetrization through diastereotopic group selection [95,96,97,98,99]. In fact, using an excess NBS this compound was converted at first into the chiral tricyclic imidazolidine 130, whereas a further bromination at the nitrogen atom gave the intermediate 131. The subsequent elimination reaction led in moderate yield but with total stereoselectivity to the tricyclic compound 132, containing a 4,5-dihydroimidazole moiety, that was eventually converted into (-)-γ-licorane, 133 [100], a degradation product of several members of the caranine family of alkaloids (Scheme 33) [101].
3. Asymmetric Synthesis Exploiting Stereoselectivity Directed by an Added Chiral Catalyst
3.1. N-Sulfonyl and Carbamoyl Pyrrolidines, Indolines and Hexahydropyrrolo [2,3,-b]indoles (HPI)
Enantiomerically pure substituted pyrrolidines and their derivatives are components of many pharmaceutically relevant molecules [102,103,104]. Among them, either 2-substituted 3-halopyrrolidine and 2-halomethylpyrrolidine derivatives appeared to be attractive advanced intermediates towards the synthesis of substituted hydroxypyrrolidines that display strong inhibitory activity against a lot of phosphoribosyltransferases [105].
Thus, the homoallylic nosylamides 134 were treated with N-bromopyrrolidin-2-one (NBP) in the presence of the catalyst 136 and the cyclization reaction proceeded in a 5-endo mode, providing 2,3-trans-disubstituted 3-bromopyrrolidine derivatives 135 in excellent yield and good enantioselectivity. After inspection of the possible transition states, where a charge pair formation was hypothised between the quinuclidine nitrogen of the catalyst and bromenium ion, together with binding of the nosyl amide and bromenium ion stabilized by Lewis basic sulphur, the stereoselectivity was ascribed to a strong repulsive interaction between 2,6-diethoxyphenyl group of the catalyst and the aryl or alkyl substituent of the substrate, missing in the most favored TS but occurring in the less favored one (Scheme 34) [106].
Moreover, the compound 134b underwent bromoamidation under the same conditions, but using catalyst 137, pseudoenantiomeric with 136, and the reaction proceeded with high enantioselectivity leading to ent-135b that was eventually converted into the enantiomerically pure pyrrolidine 138, a component of the selective KV1.5 blocker BMS-394136, but the chemical yields of the synthetic steps were not reported (Scheme 35) [107].
Enantioenriched 2-halomethyl pyrrolidine derivatives were useful intermediates for the synthesis of highly bioactive benzazepinones [108,109]. It is worth noting that the cyclization of unsaturated tosylamide 139, carried out with NIS in the presence of catalyst 142, proceeded in a regiodivergent mode on addition of different potassium halides to the reaction mixture. In fact, when a small amount of KI was used, the cyclization according to a 5-exo-trig mode afforded the expected 2-iodomethyl pyrrolidine 140, exclusively, isolated in good yield and high stereoselectivity. Conversely, in the presence of a small amount of KBr, only the corresponding piperidine derivative 141 was obtained, via a 6-endo-trig mode, but the stereoselectivity of the process could not be ascertained owing to the rapid decomposition of the product. In order to obtain a deeper insight about the interaction of the additives with the catalyst, some variable temperature NMR experiments were carried out that evidenced a KBr effect on the binding between the substrate 139 and the catalyst 142. The different regioselectivity of the iodoamidation was ascribed to this interaction, but the real mechanistic changes leading to switch of the regiochemistry remained unclear (Scheme 36) [110].
Tosylamides 143a,b, prepared starting from 2-allylanilines, underwent stereoselective iodoamidation according to the preceeding protocol to give indolines (2,3-dihydro-1H-indoles), whose heterocyclic structure occurs either in the class of natural indole-terpenoid alkaloids [111,112] and in candidates for drugs [113]. The cyclization of tosylamide 143a (R1 = H) proceeded in moderate yield and with high stereoselectivity in the presence of catalyst 142 alone or in the presence of KBr, to give 2-iodomethyl indoline 144 although a better yield was obtained on adding iodine [114]. On the contrary, the tosylamide 143b (R1 = Cl) afforded the indoline 145 in good yield and high stereoselectivity in the absence of KBr, whose addition dramatically decreased the yield of the cyclization, and this result was again ascribed to interactions between the catalyst and the additive. Eventually, it is worth mentioning that the configuration of 144 was opposite to that of 145, but the reason of the different outcome was not ascertained (Scheme 37) [110].
Also NBS was effective for halocyclization of tosylamides 146, carried out in the presence of BINOL-derived catalyst 148 acting as a Lewis base, to give bromomethyl indoline derivatives 147 in good yield and with high enantioselectivity. The stereochemistry of the reaction strongly relied on the electronic density of the aromatic ring, since higher enantioselectivity was observed for tosylamides bearing an ERG at C-4 of the aromatic ring, with respect to tosylamides substituted at C-5, while the opposite effect was observed when an EWG was present (Scheme 38) [115].
Homoallylic tosylamides 149 containing a gem-disubstituted double bond underwent iodoamidation mediated by NIS activated by a small amount iodine [109] in the presence of the chiral thiohydantoin catalyst 142, and N-tosyl 2-iodomethylpyrrolidines 150 were obtained in good yield and high enantioselectivity (Scheme 39) [110].
The bromocyclization of similar (4-nosyl)amino derivatives 151, carried out with NBS in the presence of the catalyst 153, provided in excellent yield and enantioselectivity N-(4-nosyl)pyrrolidines 152 bearing a chiral quaternary center at C-2 only when the substituent of the double bond was an electron-deficient aryl group. On the contrary, when the substituent was hydrogen or an alkyl group, the reaction proceeded with low asymmetric induction (Scheme 40) [116].
Under the same conditions, the nosyl derivative 154 afforded the N-nosyl isoindoline 155 [116] in very high yield, with total regio- and good enantioselectivity, subsequently oxidized to isoindolinone 156 whose framework occurs as a valuable pharmacophore in a wide range of natural compounds displaying different biological activities and therapeutic potential (Scheme 41) [117,118,119].
The chiral Lewis basic amidophosphate catalyst 159, derived from BINOL, was effective for iodocyclization of N-sulfonyl amides 157 bearing a gem-disubstituted double bond, when iodine was used in the presence of Lewis acid N-chlorosuccinimide (NCS) in order to generate a highly reactive iodinating species [120]. In fact, the reaction proceeded in good yield and with excellent enantioselectivity to give N-sulfonyl 2-iodomethyl pyrrolidine derivatives 26 displaying at the quaternary center the configuration opposite to compounds 18 and 20 (Scheme 42) [121].
The (Z)-nosylamides 160 were treated with N-bromopthalimide (NBPhth) as the bromenium ions source, in the presence of the chiral C2-symmetric selenide Lewis base 162. The reaction proceeded in a 5-exo-trig mode exclusively, leading to (3-nosyl) pyrrolidine derivatives 161 in excellent yield and high enantioselectivity. Concerning the reaction mechanism, at first coordination of the Lewis basic selenium of catalyst to NBP was proposed, followed by formation of an electrophilic brominating species whose interaction with the double bond gives a tightly selenium coordinated bromiranium intermediate that, by eventual SN2 attack of the sulfonamide group, leads to the cyclization product (Scheme 43) [122,123].
N-Sulfonyl amides bearing a disubstituted double bond underwent halocyclization mediated by NBS in the presence of the catalyst R-TRIP 165 [(3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl) hydrogenphosphate] using a chiral phase-transfer catalysis (PTC) methodology [124], since exploiting H-bonding interactions it is able to transfer the poorly soluble NBS halogenating reagent into the organic solvent. When the reaction was carried out starting from compounds 163 displaying a (Z)-double bond, the 2-substituted pyrrolidine derivatives 164 were isolated in good yield with high enantioselectivity (Scheme 44) whereas under the same conditions (E)-sulfonamides 166 were converted into pyrrolidine derivatives 167 in moderate yield and stereoselectivity, the configuration of their quaternary center being the same as observed for compounds 161 (Scheme 45) [125].
For this cyclization were proposed transition states where the catalyst 165 activates NBS through a hydrogen bond, whereas the nucleophilic amido group is in turn blocked to the P=O functionality by a hydrogen bond. Since (Z)-alkenes 163 underwent cyclization with higher stereoselectivity with respect to (E)-alkenes 166, the most favored transition states were examined and this outcome was ascribed to unfavourable interactions occurring between the isopropyl groups of the catalyst and the substituent of the (E)-double bond with respect to the (Z)-one [125].
In alternative to BINOL derivatives, the chiral Brønsted acids tethered on Co(III)-complexes Δ-168 and Λ-169 were excellent catalysts able to transfer a slightly soluble brominating reagent to reaction solution generating at the same time a chiral environment with control of the stereochemical outcome. Thus, on treatment of the unsaturated benzenesulfonylamides 170 with NBS in the presence of the chiral Co(III) complex Δ-(S,S)-169, the 2-substituted pyrrolidine derivatives 171 were obtained in excellent yield and enantioselectivity (Scheme 46) [126].
On the contrary, when the reaction was carried out under the same conditions but in the presence of the chiral Co(III) complex Λ-(S,S)-169, diastereomeric with Δ-168, the benzenesulfonamides 172 gave in good yield and with high stereoselectivity the pyrrolidine derivatives 173, that displayed at C-2 the opposite configuration with respect to compounds 171 (Scheme 47) [126].
The asymmetric halocyclization of tryptamine derivatives involved dearomatization of the electron-rich ring [127] leading to derivatives containing the 3-halohexahydropyrrolo [2,3,-b]indole (HPI) framework, a useful and versatile building block for preparation of cyclotryptamine alkaloids that display cytotoxic, neuroprotective and cholinesterase inhibitory activity [128]. Thus, compounds 174 underwent bromoamidation mediated by N-bromoacetamide in the presence of catalyst (DHQD)2PHAL, 176, to give in good yield and moderate enantioselectivity the tricyclic HPI derivatives 175 with the bromine atom suitable for a further substitution (Scheme 48) [129].
Among the available sulfonyl groups, the nosyl substituent was preferred for this cyclization owing to high acidity of the proton on nitrogen [129], and a carbamate was found to be the best protecting group for the indolic nitrogen, with respect to acyl or alkyl substituents (Scheme 48) [129].
The haloamidation of tryptamine derivatives exploited also a chiral anion phase-transfer catalysis (PTC) methodology, where a BINOL-derived phosphate was associated with DABCO-derived poorly soluble cationic halogenating reagents whose solubility in the organic solvent was due to ion-pairing, rather than to H-bonding interactions with the catalyst [130], as it occurred for complexes 168 and 169 and NBS [126]. Thus, compounds 177 were treated with salt 179, that gave the best results among other similar salts, together with Brønsted acid 8H-R-TRIP 180, that with respect to R-TRIP 165 required shorter reaction times coupled with better stereoselectivity, and tricyclic products 178 were isolated in high yield with excellent enantioselectivity (Scheme 49) [131].
Following this methodology, the triptamine derivative 181 afforded on a multigram scale the bromo derivative 182 that through a multistep synthesis gave the C2-symmetric bispyrrolidinoindoline-derived alkaloid (-)-chimonantine 183 [126], component of Chimonanthus praecox, that inhibits tyrosinase and tyrosine-related protein-1 mRNA expression (Scheme 50) [132].
On the other hand, a HPI core displaying the opposite configuration at the chiral center was obtained on treating the compound 184 with the salt 179 and the Brønsted acid 8H-S-TRIP, ent-180. The tetracyclic structure 185 was isolated in excellent yield and stereoselectivity and eventually converted into (-)-conolutinine, 186, an indole terpenoid alkaloid effective to reverse multidrug resistance in vincristine-resistant KB cells (Scheme 51) [133].
It is worth noting that this methodology was changed into an environmentally friendly process that avoided external chemical oxidants and harsh conditions. In fact by oxidation of bromide anion to bromine, that occurred in an undivided electrolytic cell in the presence of the salt 189, allowed to generate in situ the brominating species 178. From its interaction with the Brønsted acid 190 a weak ion pair soluble in the organic solvent arose, which reacted with tryptamine derivatives 187, and the tricyclic compounds 188 were isolated in very good yield and excellent stereoselectivity (Scheme 52) [134]. It is worth mentioning that this metodology was successfully applied also on a multigram scale. In fact, using a reduced amount of 190 (1 mol %) compound 187e was converted into 188e in 99.5% yield and 90% e.e., suitable to be converted into alkaloids (-)-chimonantine 183 [130] and (-)-hodgkinsine [135].
Eventually, exploiting again a chiral phase-transfer catalysis (PTC) methodology, sulfonamides 191 underwent transannular cyclization when the Brønsted acid TRIP 165 was employed together with NBS that was transferred into the organic solvent exploiting H-bonding interactions, to give the tricyclic derivatives 192 in good yield with high stereoselectivity. On the other hand, again exploiting the chiral phase-transfer catalysis methodology, the same compounds 192 were isolated in good yield but with better streoselectivity, when the cationic brominating reagent 193 was used in place of NBS together with TRIP 165 (Scheme 53). The ion-pairing with the catalyst allowed transfer of the poorly soluble salt 193 into the organic solvent and deep insight about the reaction mechanism was obtained by using computational methods [136].
Diprotected tryptamines 194 were easily cyclized with NBS under phase-transfer conditions when the reaction was carried out by using as the catalyst the Brønsted acid chiral Co(III) complex Λ-168 and the corresponding tricyclic derivatives 195a were isolated in good yield and high enantioselectivity [137]. However, when NBS was changed for 1,3-diiodo-5,5-dimethylhydantoin (DIDMH), again in the presence of Λ-168, the conversion of compounds 194 into iododerivatives 195b proceeded with lower yields but with comparable enantioselectivity (Scheme 54) [138].
In addition, for the cyclization of indene (n=1) and 1,2-dihydronaphthalene (n=2) derivatives 196, a chiral anionic phase-transfer methodology exploiting the DABCO-derived cation 198 together with Brønsted acid TRIP 165 was employed, and the corresponding tricyclic products 197, key building blocks for the synthesis of bioactive molecules, were obtained in very good yield and with excellent enantioselectivity (Scheme 55) [139].
Moreover, within the synthesis of the tricyclic compound 201, a potent acetyl cholinesterase (AChE) inhibitor displaying the opposite configuration at the chiral centers with respect to 197 [134], compound 199 was treated under the same conditions but using ent-165 as the catalyst, and the tricyclic derivative 200 was isolated in high yield and enantioselectivity (Scheme 56) [140].
Eventually, a desymmetrization with enantiotopic group discrimination [97,99] [141,142,143,144,145,146,147] was carried out starting from prochiral cyclohexa-1,4-dienes 202 exploiting the bromoamidocyclization mediated by TRIP 165 and the salt 204 under PTC conditions. According this methodology cis-3a-arylhydroindoles 203 were obtained in moderate to good yield but always with excellent stereoselectivity [148], and the usefulness of this methodology was confirmed by the synthesis of (+)-mesembrane, 205, found in plants of the Amaryllidaceae family (Scheme 57) [149,150].
3.2. N-Sulfonyl Piperidines
In the presence of the catalyst 136 the bromoamidation of compounds 134, mediated by N-bromopyrrolidinone (NBP) and proceeding in a 5-endo-trig mode, led to the enantioenriched trans-2-substituted 3-bromopyrrolidine derivatives 135 with total regioselectivity and high stereoselectivity (Scheme 34) [106]. However, under the same conditions, the homolog (E)-substrate 206, biased to cyclize in a 6-exo-mode by electronic factors, afforded in very low yield and neglectable e.e. the trans-2,3-disubstituted N-sulfonyl piperidine 207 that was however isolated with moderate yield and enantioselectivity when the catalyst 136 was changed for 208 and NBS was the bromenium ions source (Scheme 58) [151].
However, a further, significant improvement was obtained using 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) in place of NBS in the presence of catalyst 208, since the cyclization of (E)-sulfonylamino derivatives 209, proceeding in a 6-endo-trig mode, exclusively, allowed to isolate 2,3-trans-disubstituted piperidines 210 in high yield and with good stereoselectivity. It is worth mentioning that these compounds displayed at the chiral centers the configuration opposite to 207, but the reason of this outcome remained unclear (Scheme 59) [151].
Eventually, starting from 206 but using DBDMH in the presence of catalyst 212, pseudoenantiomeric with 208, the piperidine derivative 207 was obtained in the pure enantiomeric form after recrystallization [139], suitable to be converted into bioactive products such as CP 99994, 211, a high affinity NK1 antagonist (Scheme 60) [152].
3.3. [1,2,3] Oxathiazine 2,2-dioxides
The outcome of halofunctionalization of unsaturated sulfamate ester derivatives 213 relied on both the halogen source and the catalyst employed. In fact, when the reaction was carried out with NBS in the presence of ligand 216 and Sc(OTf)3 a diastereomeric mixture of [1,2,3]oxathiazine 2,2-dioxides syn-214 and anti-215 was obtained, the syn-bromoderivatives 83 being isolated as the major products in good yield and high enantioselectivity (Scheme 61). On the contrary, when the compounds 213 were treated with TsNCl2 as the donor of halenium ions, together with the ligand 219 and Lu(OTf)3, diastereomeric mixtures of anti-217 and syn-218 were isolated in good yield, and the major anti-isomers 217 were obtained with excellent enantioselectivity (Scheme 62) [153].
In the event, when the reaction of compounds 213 was carried out with BsNBr2 in the presence of ligand 219 and Lu(OTf)3, followed by treatment with Et3N, the initial bromoamination reaction afforded in good yield diastereomeric mixtures of derivatives anti-220 and syn-221. However, under the basic reaction conditions the minor syn-isomers 221 remained unchanged, whereas the major anti-isomers 220 were easily converted with excellent stereoselectivity into the corresponding (3,3-dioxido-1,8b-dihydroazirino [1,2-c]benzo[e][1,2,3] oxathiazin-1-yl) aryl ketones 222 (Scheme 63) [153].
3.4. N-Sulfonyl 4,5-dihydro-1H-pyrazoles
Recently, 5-halomethyl dihydropyrazoles deserved interest since 1,3-diamine derivatives used as precursors of analogs of anti-influenza agent Peramivir were prepared through cleavage of the N−N bond of polysubstituted pyrazolines bearing a tertiary chiral center [154]. Thus, starting from hydrazones 223, the chiral bromomethyl derivatives 224 were obtained in good yield and excellent stereoselectivity via bromoamidation using the anionic chiral Co(III) complex Λ-(S,S)-225 that, being highly soluble in apolar solvents, was proven to be an efficient phase-transfer catalyst when N-bromoacetamide (NBAc) was the source of bromenium ions (Scheme 64) [155].
In alternative, also Co-complex Λ-(S,S)-169 was highly effective for iodoamidation of unsaturated hydrazones 226 carried out with DIDMH, and the corresponding 5-iodomethyl 4,5-dihydro-1H-pyrazoles 227 displaying at the tertiary center the same configuration as the bromomethyl derivatives 224, were isolated in good yield and high stereoselectivity (Scheme 65) [155].
It is worth noting that some dihydropyrazoles containing a quaternary chiral center were established as potent kinesin spindle protein (KSP) inhibitors, halting the cellular mitosis [156,157]. Thus, many efforts were directed towards asymmetric iodoamidation of unsaturated arenesulfonyl hydrazones directed towards preparation of dihydropyrazoles bearing either a quaternary center and a iodomethyl functionality suitable for further transformations. At first, starting from nosyl hydrazones 228, the source of iodenium ion was N-iodopyrrolidin-2-one (NIPyr) employed together with the chiral amino thiourea 230. This bifunctional catalyst was able to coordinate both the iodiranium ion and the nucleophilic nitrogen, thus generating a chiral environment and the chiral 3,5-disubstituted 5-iodomethyl-1-nosyl-4,5-dihydro-1H-pyrazoles 229 were obtained in high yield and good enantioselectivity (Scheme 66) [158].
In addition, dienyl nosyl hydrazones 231 underwent cyclization mediated by NIS in the presence of difunctional catalyst 233, since under these conditions catalyst 230 was less effective in generating chirality, and the corresponding 1-nosyl-4,5-dihydro-1H-pyrazole derivatives 232 were isolated in moderate yield but with good enantioselectivity (Scheme 67) [159].
3.5. N-Tosyl Lactams
The tosylamides 235 and 238, bearing a sulfonyl functionality at the amidic nitrogen, underwent cyclization mediated by NBS in the presence of the difunctional catalyst 237, to give in excellent yield and enantioselectivity either 3-bromomethyl 236 (Scheme 69) or 3-bromoalkyl 10-bromo-2-tosyl-3,4-dihydropyrazino [1,2-a]indol-1(2H)-ones 239, respectively (Scheme 70), and under the reaction conditions the process was completely regioselective, since products arising from attack of carbonyl oxygen to bromiranium ion were never observed and the tricyclic lactam core formed occurs in bioactive molecules, such as an histamine H3 receptor agonist [161]. Two equivalents of bromenium ion were required for this cyclization, since eventually a bromine atom was transferred to C-3 of the indole ring, and the use of chloroform analytical reagent (AR) grade was compulsory for higher stereoselectivity, due to the presence of a little amount of ethanol, since the stereoselectivity clearly dropped when ethanol was totally removed, although its role in the process was not ascertained. Concerning the reaction mechanism, NMR experiments suggested the initial formation of an intermediate where bromine is directly bonded to the catalyst, unlike catalysts in which a thiocarbamate sulfur interact with the halenium ion as Lewis base, whereas the quinuclidinic nitrogen forces the amide in the enolic form, thus avoiding oxygen attack to the bromiranium ion [162].
3.6. N-Tosyl 1,3-oxazolidin-2-ones and 1,3-oxazin-2-ones
In the presence of the complex generated by chiral phosphine ligand 242 and Sc triflate, N-tosyl carbamates 240 containing a (Z)-double bond were converted in good yield into the corresponding N-tosyl oxazolidin-2-ones 241 through a 5-exo-mode cyclization exploiting NBS as bromenium ions donor. The reaction proceeded with total regioselectivity and excellent enantioselectivity, and 31P NMR spectroscopy evidenced interactions between the ligand 242 and Sc, leading to a chiral reaction environment followed by activation of NBS (Scheme 71) [163].
Both regio- and stereoselectivity of this cyclization strongly relied upon the configuration of the double bond. In fact, under the same reaction conditions, the reaction of (E)-carbamate 243 led to a regioisomeric mixture of 1,3-oxazolidin-2-one 244 and 1,3-oxazin-2-one 245, but only this latter, displaying a six-membered ring, was isolated with good enantioselectivity (Scheme 72) [163].
However, exploiting the same complex arising from phosphine oxide 248 and Sc triflate, but changing dibromodimethylhydantoin (DBDMH) for NBS and using NaCl as an additive, the cyclization the (E)-carbamates 246 proceeded in a 6-endo-mode, exclusively, to afford N-tosyl oxazin-2-ones 247 in good yield, with total regioselectivity and excellent enantioselectivity. It is worth noting that the corresponding (Z)-carbamates under the same reaction conditions gave only oxazolidin-2-ones but in poor yield and low stereoselectivity [164], unlike the results observed with the ligand 242. Furthermore, by addition of KBr in place of NaCl and increasing the amount of the complex, carbamates 249, displaying a trisubstituted double bond, afforded in high yield and excellent stereoselectivity oxazin-2-ones 250 containing a quaternary chiral carbon (Scheme 73) [165].
Accordingly, by reaction of dienyl carbamates 251 under the same conditions, the corresponding oxazin-2-ones 252 were isolated in good yield and high enantioselectivity (Scheme 74) [166].
The cyclization of homoallyl N-tosyl carbamates 253 with (E)-configuration at the double bond required a larger amount of the complex between phosphine oxide 248 and Sc triflate, when N-bromoacetamide was used as bromenium ions source in the absence of halide ions, and the reaction proceeded according to a 6-exo mode, leading to oxazin-2-ones 254 in moderate yield but with nearly total enantioselectivity (Scheme 75) [167].
Eventually, in the presence of an even larger amount of the complex arising from phosphine oxide 248 and Sc triflate, compounds 255 were converted in good yield and with excellent enantioselectivity into the spiro derivatives 256, exploiting dearomatization initiated by attack of a bromenium ion to the electron-rich benzofuran ring (Scheme 76) [168].
3.7. 1,3-Imidazolidin-2-Ones and Tetrahydropyrimidin-2(1H)-Ones
Unsaturated N-tosyl urea intermediates 258 were prepared by reaction of gem-disubstituted allylamines 257 with tosyl isocyanate and the cyclization, carried out in situ using N-iodopyrrolidinone (NIPyr) in the presence of the basic Brønsted catalyst 260, gave the chiral N-tosylimidazolidin-2-ones 259 in good yield with high enantioselectivity (Scheme 77) [157].
Starting from the (Z)-allylamine 261, the cyclization proceeded in a 5-exo-mode leading to imidazolidin-2-one 262 with excellent yield and stereoselectivity, and steric bias due to the double bond configuration overwhelmed electronic factors. On the contrary, proceeding through a 6-endo-mode cyclization directed by electronic factors, the (E)-allylamine 263a led to tetrahydropyrimidin-2(1H)-ones 264a in high yield and stereoselectivity, whereas amine 263b, displaying a trisubstituted double bond, gave the corresponding tetrahydropyrimidin-2(1H)-one 264b with high enantioselectivity but in low yield, probably due to the formation of a quaternary chiral center (Scheme 78) [169].
With the aim of demonstrating the usefulness of this methodology [157], the amine 265 gave in good yield and high enantioselectivity the iodomethyl derivative 266, precursor of the product SCH 388714, 267, a potent and selective NK1 receptor antagonist that is orally active and displays good CNS penetration (Scheme 79) [170].
4. Conclusions
A lot of asymmetric syntheses of non-aromatic nitrogen containing heterocycles were recently developed, exploiting halenium ion initiated cyclofunctionalizations. Thus, starting from chiral intermediates, polyfunctionalized structures were obtained by internal chirality transfer, whereas expensive organocatalysts were very effective in transferring chirality information to achiral starting substrates and the final products were often obtained on multigram scale, within total synthesis of compounds with high medicinal potential, as it occurred for alkaloids or specific inhibitors of biological processes. Moreover, enzymes could provide unique possibilities for chiral induction in highly stereoselective C–N bond formation, but were employed only for C-O bond formation [171,172,173,174,175,176,177]. Thus, introduction of enzymes in a cascade process, that is becoming a very useful and versatile methodology for the synthesis of a broad number of chiral molecules, could lead to new methodologies in this area, and efficient and easy enzymatic approaches protocols for the stereoselective formation of C-N bonds directed towards synthesis of bioactive non aromatic heterocycles can be expected in the next few years [178,179], using green solvents and avoiding the pollution and waste problems arising from halogen containing reagents [134].
Funding
This research received no external funding.
Conflicts of Interest
The author declare no conflict of interest.
References
- Saikia, I.; Borah, A.J.; Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev., 2016, 116, 6837–7042. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, V.S.C.; de Mattos, M.C.S. N-Halo reagents: modern synthetic approaches for heterocyclic synthesis. Synthesis, 2019, 51, 1841–1870. [Google Scholar] [CrossRef]
- Lyubchuk, T.V.; Hordiyenko, O.V. The use of N-halosuccinimides for cyclization with the formation of five-membered heterocyclic compounds. Chem. Heterocycl. Comp., 2020, 56, 1–29. [Google Scholar] [CrossRef]
- Powell, W.H. Revision of the extended Hantzsch-Widman system of nomenclature for heteromonocycles. Pure Appl. Chem., 1983, 55, 409–416. [Google Scholar] [CrossRef]
- Moss, G.P.; Smith, P.A.S.; Tavernier, D. Glossary of class names of organic compounds and reactive intermediates based on structure. Pure Appl. Chem., 1995, 67, 1307–1375. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc., Chem. Commun. 1976, 734. [CrossRef]
- Baldwin, J.E. 5-Endo-trigonal reactions: a disfavoured ring closure. J. Chem. Soc., Chem. Commun., 1976, 736-738. [CrossRef]
- Piccirilli, J.A. Do enzymes obey the Baldwin rules? A mechanistic imperative in enzymatic cyclization reactions. Chem. Biol., 1999, 6, R59–R64. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.; Alabugin, I.V. Cyclizations of alkynes: revisiting Baldwin’s rules for ring closure. Chem. Rev., 2011, 111, 6513–6556. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Gilmore, K. Finding the right path: Baldwin “Rules for Ring Closure” and stereoelectronic control of cyclizations. J. Chem. Soc., Chem. Commun., 2013, 49, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.; Mohamed, R.K.; Alabugin, I.V. The Baldwin rules: revised and extended. WIREs Comput. Mol. Sci., 2016, 6, 487–514. [Google Scholar] [CrossRef]
- Cardillo, G.; Orena, M. Stereocontrolled cyclofunctionalizations of double bonds through heterocyclic intermediates. Tetrahedron, 1990, 46, 3321–3408. [Google Scholar] [CrossRef]
- Orena, M. Amination reactions promoted by electrophiles. In Houben-Weyl Methods in Organic Chemistry: Stereoselective Synthesis, Vol. e-21e, , Helmchen, G., 4th Ed. ed; Hoffmann, R.W.: Mulzer, J.; Schaumann, E., Eds.; Thieme, Stuttgart, 1996; pp. 5291–5355. [Google Scholar]
- Frederickson, M.; Grigg, R. Electrophile mediated heteroatom cyclizations onto C=C π-bonds. Part 1: halogen and chalcogen mediated cyclization Org. Prep. Proc. Int., 1997, 29, 33–62. [Google Scholar] [CrossRef]
- Ranganathan, S.; Muraleedharan, K.M.; Vaisha, N.K.; Jayaraman, N. Halo- and selenolactonisation: the two major strategies for cyclofunctionalisation. Tetrahedron, 2004, 60, 5273–5308. [Google Scholar] [CrossRef]
- Mphahlele, M.J. Molecular iodine-mediated cyclization of tethered heteroatom-containing alkenyl or alkynyl systems. Molecules, 2009, 14, 4814–4837. [Google Scholar] [CrossRef] [PubMed]
- Ashtekar, K.D.; Jaganathan, A.; Borhan, B.; Whitehead, D.C. Enantioselective halofunctionalization of alkenes. In: Organic Reactions, 2021, 105, Evans, P.A., Ed., Wiley, pp. 1–266. [Google Scholar]
- Ladenburg, A. Versuche zur Synthese von Tropin und dessen Derivate. Chem. Ber., 1881, 14, 1342–1349. [Google Scholar]
- https://gallica.bnf.fr/ark:/12148/bpt6k90692z?rk=21459;2.
- Merling, G. Ueber Bromsubstitutionsprodukte des Dimethylpiperidins und einige sich von diesen ableitende Verbingdungen. Chem. Ber., 1884, 17(pt6), 2139–2143. [Google Scholar] [CrossRef]
- Merling, G. Ueber die bei Einwirkung von Brom auf Dimethylpiperidin enstehenden Verbindungen. - Neue Synthese von Piperidinderivaten. Chem. Ber., 1886, 19(pt6), 2628–2632. [Google Scholar] [CrossRef]
- Chemler, S.R.; Bovino, M.T. Catalytic aminohalogenation of alkenes and alkynes. ACS Catal., 2013, 3, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, C.B.; Mukherjee, S. Catalytic enantioselective halocyclizations beyond lactones: emerging routes to enantioenriched nitrogenous heterocycles. Synlett, 2014, 25, 163–169. [Google Scholar] [CrossRef]
- Mizar, P.; Wirth, T. Iodoaminations of alkenes. Synthesis, 2017, 49, 981–986. [Google Scholar] [CrossRef]
- China, H.; Kumar, R.; Kikushima, K.; Dohi, T. Molecules, 2020, 25, 6007–6040. 25. [CrossRef]
- Liu, S.; Zhang, B.-Q.; Xiao, W.-Y.; Li, Y.-L.; Deng, J. Recent advances in catalytic asymmetric syntheses of functionalized heterocycles via halogenation/chalcogenation of carbon-carbon unsaturated bonds. Adv. Synth. Catal., 2022, 364, 3974–4005. [Google Scholar] [CrossRef]
- Yan, J.; Zhou, Z.; He, Q.; Chen, G.; Wei, H.; Xie, W. The applications of catalytic asymmetric halocyclization in natural product synthesis. Org. Chem. Front., 2022, 9, 499–516. [Google Scholar] [CrossRef]
- Yao, C.-Z.; Tu, X.-Q.; Jiang, H.-J.; Li, Q.; Yu, J. Recent advances in catalytic asymmetric haloamination and haloetherification of alkenes. Tetrahedron Lett., 2023, 126, 154639. [Google Scholar] [CrossRef]
- La Ferla, B.; Nicotra, F. Synthetic methods for the preparation of iminosugars. In: Iminosugars as Glycosidase Inibitors - Nojirimycin and Beyond. Stiitz, A.E. Ed.; Wiley-VCH Verlag GmbH, Weinheim, Germany, 1999, pp. 68–92.
- Matassini, C.; Cardona, F. Recent synthetic strategies to access diverse iminosugars. In: Synthetic Strategies in Carbohydrate Chemistry, Tiwari, V.K., Ed.; Elsevier, Amsterdam, 2024, pp. 335–364. [CrossRef]
- Compain, P.; Martin, O.R. Design, synthesis and biological evaluation of iminosugar-based glycosyltransferase inhibitors. Curr. Top. Med. Chem., 2003, 3, 541–560. [Google Scholar] [CrossRef]
- Conforti, I.; Marra, A. Iminosugars as glycosyltransferase inhibitors. Org. Biomol. Chem., 2021, 19, 5439–5475. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Chamberlin, A.R.; Dezube, M.; Dussault, P.; McMills, M.C. Iodocyclization of allylic alcohol derivatives containing internal nucleophiles. Control of stereoselectivity by substituents in the acyclic precursors. J. Am. Chem. Soc., 1983, 105, 5819–5825. [Google Scholar] [CrossRef]
- . [CrossRef]
- Kahn, S.D.; Pau, C.F.; Chamberlin, A.R.; Hehre, W.J. Modeling chemical reactivity. 4. Regiochemistry and stereochemistry of electrophilic additions to allylic double bonds. J. Am. Chem. Soc., 1987, 109, 650–663. [Google Scholar] [CrossRef]
- Chamberlin, A.R.; Mulholland, R.L.; Kahn, S.D.; Hehre, W.J. Modeling chemical reactivity. 7. The effect of a change in rate-limiting step on the stereoselectivity of electrophilic addition to allylic alcohols and related chiral alkenes. J. Am. Chem. Soc., 1987, 109, 672–677. [Google Scholar] [CrossRef]
- Tamaru, Y.; Kawamura, S.-I.; Bando, T.; Tanaka, K.; Hojo, M.; Yoshida, Z.-I. Stereoselective intramolecular haloamidation of N-protected 3-hydroxy-4-pentenylamines and 4-hydroxy-5-hexenylamines. J. Org. Chem., 1988, 53, 5491–5501. [Google Scholar] [CrossRef]
- . [CrossRef]
- Labelle, M.; Guindon, Y. Diastereoselective synthesis of 2,3-disubstituted tetrahydrofuran synthons via the iodoetherification reaction. A transition state model based rationalization of the allylic asymmetric induction. J. Am. Chem. Soc., 1989, 111, 2204–2210. [Google Scholar] [CrossRef]
- Tredwell, M.; Luft, J.A.; Schuler, M.; Tenza, K.; Houk, K.N.; Gouverneur, V. Fluorine-directed diastereoselective iodocyclizations. Angew. Chem. Int. Ed., 2008, 47, 357–360. [Google Scholar] [CrossRef]
- Zhi-cai, S.; Chun-min, Z.; Guo-qiang. L. A synthesis of the polyhydroxylated pyrrolidines: synthesis of 1,4-dideoxy-1,4-imino-D-lyxitol and N-benzyl-4-epi-(-)-anisomycin. Heterocycles, 1995, 41, 277–287. [Google Scholar] [CrossRef]
- Verhelst, S.H.L.; Paez Martinez, B.; Timmer, M.S.M.; Lodder, G.; van der Marel, G.A.; Overkleeft, H.S.; van Boom, J.H. A short route toward chiral, polyhydroxylated indolizidines and quinolizidines. J. Org. Chem., 2003, 68, 9598–9603. [Google Scholar] [CrossRef]
- . [CrossRef]
- Davies, S.G.; Nicholson, R.L.; Price, P.D.; Roberts, P.M.; Smith, A.D. Iodine-mediated ring closing alkene iodoamination with N-debenzylation for the asymmetric synthesis of polyhydroxylated pyrrolidines. Synlett, 1055. [Google Scholar]
- Davies, S.G.; Nicholson, R.L.; Price, P.D.; Roberts, P.M.; Russell, A.J.; Savory, E.D.; Smith, A.D.; Thomson, J.E. Iodine-mediated ring-closing iodoamination with concomitant N-debenzylation for the asymmetric synthesis of polyhydroxylated pyrrolidines. Tetrahedron: Asymmetry, 2009, 20, 758–772. [Google Scholar] [CrossRef]
- Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E.; West, C.J. Asymmetric synthesis of (-)-codonopsinine. Tetrahedron Lett., 2011, 52, 6477–6480. [Google Scholar] [CrossRef]
- Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E.; West, C.J. Ring-closing iodoamination of homoallylic amines for the synthesis of polysubstituted pyrrolidines: application to the asymmetric synthesis of (-)-codonopsinine. Tetrahedron, 2012, 68, 4302–4319. [Google Scholar] [CrossRef]
- Kondo, Y.; Suzuki, N.; Takahashi, M.; Kumamoto, T.; Masu, H.; Ishikawa, T. Enantioselective construction of a polyhydroxylated pyrrolidine skeleton from 3-vinylaziridine-2-carboxylates: synthesis of (+)-DMDP and a potential common intermediate for (+)-hyacinthacine A1 and (+)-1-epi-australine. J. Org. Chem., 2012, 77, 7988–7999. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D.; Mitchell, M.; Sanford, B.A.; Fellows, L.E.; Evans, S.V. The pyrrolidine alkaloid, 2,5-dihydroxymethyl-3,4-dihydroxypyrrolidine, inhibits glycoprotein processing. J. Biol. Chem., 1984, 259, 12409–12413. [Google Scholar] [CrossRef]
- Welter, A.; Jadot, J.; Dardenne, G.; Marlier, M.; Casimir, J. 2,5-Dihydroxymethyl 3,4-dihydroxypyrrolidine dans les feuilles de Derris elliptica. Phytochemistry, 1976, 15, 747–749. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Sintim, H.O.; Hollinshead, J. A noncarbohydrate based approach to polyhydroxylated pyrrolidizines: total syntheses of the natural products hyacinthacine A1 and 1-epiaustraline. J. Org. Chem., 2005, 70, 7297–7304. [Google Scholar] [CrossRef]
- . [CrossRef]
- Davies, S.G.; Figuccia, A.L.A.; Fletcher, A.M.; Roberts, P.M.; Thomson, J.E. Asymmetric syntheses of 2,5-dideoxy-2,5-imino-d-glucitol [(+)-DGDP] and 1,2,5-trideoxy-1-amino-2,5-imino-d-glucitol [(+)-ADGDP]. Tetrahedron, 2014, 70, 3601–3607. [Google Scholar] [CrossRef]
- . [CrossRef]
- Davies, S.G.; Figuccia, A.L.A.; Fletcher, A.M.; Roberts, P.M.; Thomson, J.E. Asymmetric syntheses of (−)-ADMJ and (+)-ADANJ: 2-deoxy-2-amino analogues of (−)-1-deoxymannojirimycin and (+)-1-deoxyallonojirimycin. J. Org. Chem., 2016, 81, 6481–6495. [Google Scholar] [CrossRef] [PubMed]
- Dangerfield, E.M.; Timmer, M.S.M.; Stocker, B.L. Total synthesis without protecting groups: pyrrolidines and cyclic carbamates. Org. Lett., 2009, 11, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Stocker, B.L.; Win-Mason, A.L.; Timmer, M.S.M. I2-mediated carbamate annulation: scope and application in the synthesis of azasugars. Carb. Res., 2012, 356, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Saludes, J.P.; Lievens, S.C.; Molinski, T.F. Occurrence of the α-glucosidase inhibitor 1,4-dideoxy-1,4-imino-d-arabinitol and related iminopentitols in marine sponges. J. Nat. Prod., 2007, 70, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Win-Mason, A.L.; Jongkees, S.A.K.; Withers, S.G.; Tyler, P.C.; Timmer, M.S.M.; Stocker, B.L. Stereoselective total synthesis of aminoiminohexitols via carbamate annulation. J. Org. Chem., 2011, 76, 9611–9621. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Yudin, A.K. Oxidative cycloamination of olefins with aziridines as a versatile route to saturated nitrogen-containing heterocycles. J. Am. Chem. Soc., 2003, 125, 14242–14243. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.S.; Li, J.; Navarro, A. Total synthesis of azinomycin A. Angew. Chem. Int. Ed., 2001, 40, 1736–1739. [Google Scholar] [CrossRef]
- Zhou, J.; Yeung, Y.-Y. Diastereoselective synthesis of functionalized pyrrolidines through N-bromosuccinimide induced aziridine ring expansion cascade of cinnamylaziridine. Org. Biomol. Chem., 2014, 12, 7482–7485. [Google Scholar] [CrossRef]
- Zhou, J.; Yeung, Y.-Y. N-bromosuccinimide-induced aminocyclization-aziridine ring-expansion cascade: an asymmetric and highly stereoselective approach toward the synthesis of azepane. Org. Lett., 2014, 16, 2134–2137. [Google Scholar] [CrossRef]
- . [CrossRef]
- Asano, N. Glycosidase-inhibiting alkaloids: isolation, structure, and application. In: Modern alkaloids: structure, isolation, synthesis and biology, Fattorusso, E.; Taglialatela-Scafati, O., Eds.; Wiley-VCH, Weinheim, 2008, pp. 111–138.
- Salunke, R.V.; Ramesh, N.G. Divergent synthesis of amino-substituted indolizidine alkaloids, decahydropyrazino [2,1,6-cd]pyrrolizine triols, and (-)-pochonicine stereoisomers. Eur. J. Org. Chem, 2020; 2626–2640. [Google Scholar] [CrossRef]
- Itoh, A.; Miura, T.; Tada, N. Oxidation of carbon-halogen bonds. In: Comprehensive organic synthesis, Knochel, P., Ed.; Second Edition, Elsevier Science Ltd., Amsterdam, 2014, Vol. VII, pp. 744–769.
- Volpin, G.; Vepřek, N.A.; Bellan, A.B.; Trauner, D. Enantioselective synthesis and racemization of (-)-sinoracutine. Angew. Chem. Int. Ed., 2017, 56, 897–901. [Google Scholar] [CrossRef]
- Beshore, D.C.; Smith, A.B. III The lyconadins: enantioselective total syntheses of (+)-lyconadin A and (-)-lyconadin B. J. Am. Chem. Soc., 2008, 130, 13778–13789. [Google Scholar] [CrossRef]
- Brock, E.A.; Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E. Asymmetric synthesis of the tropane alkaloid (+)-pseudococaine via ring-closing iodoamination. Org. Lett., 2012, 14, 4278–4271. [Google Scholar] [CrossRef]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids. Nat. Prod. Rep., 2014, 31, 1721–1788. [Google Scholar] [CrossRef] [PubMed]
- Brock, E.A.; Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E. Asymmetric synthesis of polyhydroxylated pyrrolizidines via transannular iodoamination with concomitant N-debenzylation. Org. Lett., 2011, 13, 1594–1597. [Google Scholar] [CrossRef]
- . [CrossRef]
- Brock, E.A.; Davies, S.G.; Lee, J.A.; Roberts, P.M.; Thomson, J.E. Polyhydroxylated pyrrolizidine alkaloids from transannular iodoaminations: application to the asymmetric syntheses of (-)-hyacinthacine A1, (-)-7a-epi-hyacinthacine A1, (-)-hyacinthacine A2, and (-)-1-epi-alexine. Org. Biomol. Chem., 2013, 11, 3187–3202. [Google Scholar] [CrossRef] [PubMed]
- Ciufolini, M.A. Synthetic studies on heterocyclic natural products. Farmaco, 2005, 60, 627–641. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Zhang, H.; Lin, Z.; Huang, H.; Huo, H.; Huang, Y.; Ye, J.; Huang, P. Enantioselective Synthesis of the diazatricyclic core of alkaloid TAN1251C via an iodoaminocyclization reaction. Chin. J. Chem., 2010, 28, 1717–1724. [Google Scholar] [CrossRef]
- . [CrossRef]
- Wada, M.; Murata, T.; Oikawa, H.; Oguri, H. Nickel-catalyzed dimerization of pyrrolidinoindoline scaffolds: systematic access to chimonanthines, folicanthines and (+)-WIN 64821. Org. Biomol. Chem., 2014, 12, 298–306. [Google Scholar] [CrossRef]
- Silva López, C.; Pérez-Balado, C.; Rodríguez-Graña, P.; de Lera, A.R. Mechanistic insights into the stereocontrolled synthesis of hexahydropyrrolo [2,3-b]indoles by electrophilic activation of tryptophan derivatives. Org. Lett., 2008, 10, 77–80. [Google Scholar] [CrossRef]
- . [CrossRef]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med., 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Wijtmans, R.; Vink, M.K.S.; Schoemaker, H.E.; van Delft, F.L.; Blaauw, R.H.; Rutjes, F.P.J.T. Biological relevance and synthesis of C-substituted morpholine derivatives. Synthesis. [CrossRef]
- Shcherbatiuk, A.V.; Shyshlyk, O.S.; Yarmoliuk, D.V.; Shishkin, O.V.; Shishkina, S.V.; Starova, V.S.; Zaporozhets, O.A.; Zozulya, S.; Moriev, R.; Kravchuk, O.; Manoilenko, O.; Tolmachev, A.A.; Mykhailiuk, P.K. Synthesis of 2- and 3-trifluoromethylmorpholines: useful building blocks for drug discovery. Tetrahedron, 2013, 69, 3796–3804. [Google Scholar] [CrossRef]
- Nonnenmacher, J.; Grellepois, F.; Portella, C. Synthesis of enantiopure 2-aryl(alkyl)-2-trifluoromethyl-substituted morpholines and oxazepanes. Eur. J. Org. Chem, 3726. [Google Scholar] [CrossRef]
- Magriotis, P.A. Recent progress toward the asymmetric synthesis of carbon-substituted piperazine pharmacophores and oxidative related heterocycles. RSC Med. Chem., 2020, 11, 745–759. [Google Scholar] [CrossRef]
- Bera, S.; Pandai, G. I2-Mediated diversity oriented diastereoselective synthesis of amino acid derived trans-2,5-disubstituted morpholines, piperazines, and thiomorpholines. ACS Comb. Sci., 2012, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, J.; Ishiwata, T.; Shirai, K.; Koshino, H.; Tanatani, A.; Nakata, T.; Hashimoto, Y.; Nagasawa, K. Total synthesis of (+)-batzelladine A and (-)-batzelladine D, and identification of their target protein. Chem. Eur. J., 2005, 11, 6878–6888. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.A.; Durón, S.G.; Gin, D.Y. Diastereoselective [4+2] annulation of vinyl carbodiimides with N-alkyl imines. Asymmetric synthetic access to the batzelladine alkaloids. J. Am. Chem. Soc., 2005, 127, 6924–6925. [Google Scholar] [CrossRef]
- Arnold, M.A.; Day, K.A.; Durón, S.G.; Gin, D.Y. Total synthesis of (+)-batzelladine A and (-)-batzelladine D via [4+2]-annulation of vinyl carbodiimides with N-alkyl imines. J. Am. Chem. Soc., 2006, 128, 13255–13260. [Google Scholar] [CrossRef]
- Durán-Riveroll, L.M.; Cembella, A.D. Guanidinium toxins and their interactions with voltage-gated sodium ion channels. Mar. Drugs, 2017, 15, 303–331. [Google Scholar] [CrossRef]
- Paladugu, S.R.; James, C.K.; Looper, R.E. A direct C11 alkylation strategy on the saxitoxin core: a synthesis of (+)-11-saxitoxinethanoic acid. Org. Lett., 2019, 21, 7999–8002. [Google Scholar] [CrossRef]
- Sakurai, O.; Takahashi, M.; Ogiku, T.; Hayashi, M.; Horikawa, H.; Iwasaki, T. A new synthesis of 1β-methylcarbapenems using NBS-promoted cyclization as a key step. Tetr. Lett., 1994, 35, 6317–6320. [Google Scholar] [CrossRef]
- Bateson, J.H.; Roberts, P.M.; Snale, T.C.; Southgate, R. Synthesis of 7-oxo-3-sulphinyl-1-azabicyclo [3.2.0]hept-2-ene-2-carboxylates: olivanic acid analogues. J. Chem. Soc., Chem. Commun 1980, 185–186. [Google Scholar] [CrossRef]
- Biloski, A.J.; Wood, R.D.; Ganem, B. A new beta-lactam synthesis. J. Am. Chem. Soc., 1982, 104, 3233–3235. [Google Scholar] [CrossRef]
- . [CrossRef]
- Krook, M.A.; Miller, M.J. The direct cyclization of alpha-acylamino-substituted hydroxamates to beta-lactams. J. Org. Chem., 1985, 50, 1126–1128. [Google Scholar] [CrossRef]
- Rajendra, G.; Miller, M.J. Oxidative cyclization of β,γ-unsaturated O-acyl hydroxamates to β-lactams. Tetr. Lett., 1985, 26, 5385–5388. [Google Scholar] [CrossRef]
- Cheng, Y.A.; Yu, W.Z.; Yeung, Y.Y. An unexpected bromolactamization of olefinic amides using a three-component co-catalyst system. J. Org. Chem., 2016, 81, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.; González, B.; Suescun, L.; Seoane, G.; Carrera, I. Production of enantiopure β-amino-γ-hydroxyesters from benzoic acid by a selective formal aminohydroxylation. Tetr. Lett., 2017, 58, 2182–2185. [Google Scholar] [CrossRef]
- Schulte, A.S.; Janich, S.; Würthwein, E.-U.; Saito, S.; Wünsch, B. Investigation of the Corey bromolactamization with N-functionalized allylamines. J. Heterocycl. Chem., 2016, 53, 1827–1837. [Google Scholar] [CrossRef]
- Schulte, A.; Situ, X.; Saito, S.; Wünsch, B. Bromolactamization: key step in the stereoselective synthesis of enantiomerically pure, cis-configured perhydropyrroloquinoxalines. Chirality, 2014, 26, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Atmuri, P.N.D.; Lubell, W.D. Stereo- and regiochemical transannular cyclization of a common hexahydro-1H-azonine to afford three different indolizidinone dipeptide mimetics. J. Org. Chem., 2020, 85, 1340–1351. [Google Scholar] [CrossRef]
- Snyder, B.B.; Hawryluk, N.A. Synthesis of (-)-dysiherbaine. Org. Lett., 2000, 2, 635–638. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, Y.M. A formal total synthesis of (-)-dysiherbaine. Synlett, 2003, 993-99. [CrossRef]
- Fujioka, H.; Murai, K.; Ohba, Y.; Hirose, H. ; Kita, Y.; Kim, K.K.; Lee, Y.J.; Kim, J.K.; Sung, D.K. Synthesis of (+)-polyoxamic acid and D-sorbitol from simple achiral allylic halides employing (S,S)-hydrobenzoin as a chiral source. Chem. Commun. 2002, 1116–1117. [Google Scholar] [CrossRef] [PubMed]
- Poss, C.S.; Schreiber, S.L. Two-directional chain synthesis and terminus differentiation. Acc. Chem. Res., 1994, 27, 9–17. [Google Scholar] [CrossRef]
- Hoffmann, R.W. Stereoselective synthesis using diastereotopic groups. Synthesis, 2004, 2075-2090.
- . [CrossRef]
- Studer, A.; Schleth, F. Desymmetrization and diastereotopic group selection in 1,4-cyclohexadienes. Synlett 2005, 3033–3041. [Google Scholar] [CrossRef]
- Nakahara, K.; Fujioka, H. Diastereoselective desymmetrization of symmetric dienes and its synthetic application.
- Symmetry2010, 2, 437–454. [CrossRef]
- Horwitz, M.A.; Johnson, J.S. Local desymmetrization through diastereotopic group selection: an enabling strategy for natural product synthesis. Eur. J. Org. Chem 2017, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Murai, K.; Ohba, Y.; Hirose, H.; Kita, Y. Intramolecular bromo-amination of 1,4-cyclohexadieneaminal: one-pot discrimination of two olefins and concise asymmetric synthesis of (-)-γ-lycorane. Chem. Commun., 2006, 832-834.
- . [CrossRef]
- Hall, C.J.J.; Marriott, I.S.; Christensen, K.E.; Day, A.J.; Goundry, W.R.F.; Donohoe, T.J. Extension of hydrogen borrowing alkylation reactions for the total synthesis of (-)-γ-lycorane. Chem. Commun., 2022, 58, 4966–4968. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- O’Hagan, D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep., 2000, 17, 435–446. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Felpin, F.-X.; Lebreton, J. Recent advances in the total synthesis of piperidine and pyrrolidine natural alkaloids with Ring-Closing Metathesis as a key step. Eur. J. Org. Chem 2003, 3693–3712. [Google Scholar] [CrossRef]
- Wen-Fang, X.; Xian-Chao, C.; Qiang, W.; Hao, F. Advances in matrix metalloproteinase inhibitors based on pyrrolidine scaffold. Curr. Med. Chem., 2008, 15, 374–385. [Google Scholar] [CrossRef]
- Harris, L.D.; Harijan, R.K.; Ducati, R.G.; Evans, G.B.; Hirsch, B.M.; Schramm, V.L. Synthesis of bis-phosphate iminoaltritol enantiomers and structural characterization with adenine phosphoribosyltransferase. ACS Chem. Biol., 2018, 13, 152–160. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, L.; Yeung, Y.-Y. A highly enantioselective approach towards 2-substituted 3-bromopyrrolidines. Org. Biomol. Chem., 2012, 10, 3808–3811. [Google Scholar] [CrossRef]
- Lloyd, J.; Finlay, H.J.; Vacarro, W.; Hyunh, T.; Kover, A.; Bhandaru, R.; Yan, L.; Atwal, K.; Conder, M.L.; Jenkins-West, T.; Shi, H.; Huang, C.; Li, W.D.; Sun, H.; Levesque, P. Pyrrolidine amides of pyrazolodihydropyrimidines as potent and selective KV1.5 blockers. Bioorg. Med. Chem. Lett., 2010, 20, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Floyd, D.M.; Kimball, S.D.; Krapcho, J.; Das, J.; Turk, C.F.; Moquin, R.V.; Lago, M.W.; Duff, K.J.; Lee, V.G. Benzazepinone calcium channel blockers. 2. Structure-activity and drug metabolism studies leading to potent antihypertensive agents. Comparison with benzothiazepinones. J. Med. Chem., 1992, 35, 756–772. [Google Scholar] [CrossRef] [PubMed]
- Cushing, T.D.; Baichwal, V.; Berry, K.; Billedeau, R.; Bordunov, V.; Broka, C.; Browner, M.F.; Cardozo, M.; Cheng, P.; Clark, D.; Dalrymple, S.; DeGraffenreid, M.; Gill, A.; Hao, X.; Hawley, R.C.; He, X.; Labadie, S.S.; Labelle, M.; Lehel, C.; Lu, P.-P.; McIntosh, J.; Miao, S.; Parast, C.; Shin, Y.; Sjogren, E.B.; Smith, M.-L.; Talamas, F.X.; Tonn, G.; Walker, K.M.; Walker, N.P.C.; Wesche, H.; Whitehead, C.; Wright, M.; Jaen, J.C.A. A novel series of IKKβ inhibitors part II: Description of a potent and pharmacologically active series of analogs. Bioorg. Med. Chem. Lett., 2011, 21, 423–426. [Google Scholar] [CrossRef]
- Mizar, P.; Burrelli, A.; Günther, E.; Söftje, M.; Farooq, U.; Wirth, T. Organocatalytic stereoselective iodoamination of alkenes. Chem. Eur. J., 2014, 20, 13113–13116. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.S.; Rodrigues, M.T.J.; Santo, H.; Zeoly, L.A.; Almeida, W.P.; Barcelos, R.C.; Gomes, R.C.; Fernandes, F.S.; Coelho, F. Recent advances in indoline synthesis. Tetrahedron, 2019, 75, 2063–2097. [Google Scholar] [CrossRef]
- Hua, T.-B.; Xiao, C.; Yang, Q.-Q.; Chen, J.-R. Recent advances in asymmetric synthesis of 2-substituted indoline derivatives. Chin. Chem. Lett., 2020, 31, 311–323. [Google Scholar] [CrossRef]
- Liu, F.; Su, M. Indole and indoline scaffolds in drug discovery. In: Privileged Scaffolds in Drug Discovery, Yu, B.; Li, N.; Fu, C. Eds.; Academic Press, Cambridge, MA, 2023, Chapter 8, Pages 147-161.
- Breugst, M.; von der Heiden, D. Mechanisms in iodine catalysis. Chem. Eur. J., 2018, 24, 9187–9199. [Google Scholar] [CrossRef]
- . [CrossRef]
- Yu, S.-N.; Li, Y.-L.; Deng, J. Enantioselective synthesis of 2-bromomethyl indolines via BINAP(S)-catalyzed bromoaminocyclization of allyl aniline. Adv. Synth. Catal., 2017, 359, 2499–2508. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Tan, C.K.; Yeung, Y.-Y. Enantioselective bromoaminocyclization using amino-thiocarbamate catalysts. J. Am. Chem. Soc., 2011, 133, 9164–9167. [Google Scholar] [CrossRef]
- Hu, S.; Yuan, L.; Yan, H.; Li, Z. Design, synthesis and biological evaluation of lenalidomide derivatives as tumor angiogenesis inhibitor. Bioorg. Med. Chem. Lett., 2017, 27, 4075–4081. [Google Scholar] [CrossRef]
- Xiao, D.; Wang, Y.-j.; Hu, X.-b.; Kan, W.-j.; Zhang, Q.; Jiang, X.; Zhou, Y.-b.; Li, J.; Lu, W. Design, synthesis and biological evaluation of the thioether-containing lenalidomide analogs with anti-proliferative activities. Eur. J. Med. Chem., 2019, 176, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.P.; Thapa, P.; Sharma, R.; Sharma, M. 1-Isoindolinone scaffold-based natural products with a promising diverse bioactivity. Fitoterapia, 2020, 146, 104722. [Google Scholar] [CrossRef]
- Nakatsuji, H.; Sawamura, Y.; Sakakura, A.; Ishihara, K. Cooperative activation with chiral nucleophilic catalysts and N-haloimides: enantioselective iodolactonization of 4-arylmethyl-4-pentenoic acids. Angew. Chem., Int. Ed., 2014, 53, 6974–6977. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Nakatsuji, H.; Okumura, Y.; Yao, L.; Ishihara, K. Enantioselective halo-oxy- and halo-azacyclizations induced by chiral amidophosphate catalysts and halo-Lewis acids. J. Am. Chem. Soc., 2018, 140, 6039–6043. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tan, C.K.; Yeung, Y.-Y. C2-symmetric cyclic selenium-catalyzed enantioselective bromoaminocyclization. J. Am. Chem. Soc., 2013, 135, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Gieuw, M.H.; Ke, Z.; Yeung, Y.-Y. Lewis base catalyzed stereo- and regioselective bromocyclization. Chem. Rec., 2017, 17, 287–311. [Google Scholar] [CrossRef] [PubMed]
- Sunggi, L.; Chung, W.-j. Enantioselective halogenation via asymmetric phase-transfer catalysis. Bull. Korean Chem. Soc., 2022, 43, 896–911. [Google Scholar] [CrossRef]
- Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Enantioselective bromocyclization of olefins catalyzed by chiral phosphoric acid. Org. Lett., 2011, 13, 6350–6353. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-J.; Liu, K.; Yu, J.; Zhang, L.; Gong, L.-Z. Switchable stereoselectivity in bromoaminocyclization of olefins: using Brønsted acids of anionic chiral Cobalt(III) complexes. Angew. Chem. Int. Ed., 2017, 56, 11931–11935. [Google Scholar] [CrossRef]
- . [CrossRef]
- Liang, X.-W.; Zheng, C.; You, S.-L. Dearomatization through halofunctionalization reactions. Chem. Eur. J., 2016, 22, 11918–11933. [Google Scholar] [CrossRef]
- Crich, D.; Banerjee, A. Chemistry of the hexahydropyrrolo [2,3-b]indoles: configuration, conformation, reactivity, and applications in synthesis. Acc. Chem. Res., 2007, 40, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yin, Q.; You, S.-L. Chiral-amine-catalyzed asymmetric bromocyclization of tryptamine derivatives. Asian J. Org. Chem., 2014, 3, 408–411. [Google Scholar] [CrossRef]
- Qian, D.; Sun, J. Recent progress in asymmetric ion-pairing catalysis with ammonium salts. Chem. Eur. J., 2019, 25, 3740–3751. [Google Scholar] [CrossRef]
- Xie, W.; Jiang, G.; Liu, H.; Hu, J.; Pan, X.; Zhang, H.; Wan, X.; Lai, Y.; Ma, D. Highly enantioselective bromocyclization of tryptamines and its application in the synthesis of (-)-chimonanthine. Angew. Chem. Int. Ed., 2013, 52, 12924–12927. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Morikawa, T.; Nakanishi, Y.; Ninomiya, K.; Matsuda, H.; Nakashima, S.; Miki, H.; Miyashita, Y.; Yoshikawa, M.; Hayakawa, T.; Muraoka, O. Dimeric pyrrolidinoindoline-type alkaloids with melanogenesis inhibitory activity in flower buds of Chimonanthus praecox. J. Nat. Med., 2014, 68, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, G.; Xia, Z.; Hu, J.; Wan, X.; Gao, J.-M.; Lai, Y.; Xie, W. Total synthesis of (-)-conolutinine. Org. Lett., 2015, 17, 4428–4431. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Wang, Q.; Sun, J. Electricity-driven asymmetric bromocyclization enabled by chiral phosphate anion phase-transfer catalysis. Nat. Commun., 2023, 14, 357. [Google Scholar] [CrossRef] [PubMed]
- Lindovska, P.; Movassaghi, M. Concise synthesis of (-)-hodgkinsine,(-)-calycosidine, (-)-hodgkinsine B, (-)-quadrigemine C, and (-)-psycholeine via convergent and directed modular assembly of cyclotryptamines. J. Am. Chem. Soc., 2017, 139, 17590–17596. [Google Scholar] [CrossRef] [PubMed]
- Luis-Barrera, J.; Rodriguez, S.; Uria, U.; Reyes, E.; Prieto, L.; Carrillo, L.; Pedrón, M.; Tejero, T.; Merino, P.; Vicario, J.L. Brønsted acid versus Phase-Transfer Catalysis in the enantioselective transannular aminohalogenation of enesultams. Chem. Eur. J., 2022, 28, e202202267. [Google Scholar] [CrossRef]
- Liu, K.; Jiang, H.-J.; Li, N.; Li, H.; Wang, J.; Zhang, Z.-Z.; Yu, J. Enantioselective bromocyclization of tryptamines induced by chiral Co(III)-complex-templated Brønsted acids under an air atmosphere. J. Org. Chem., 2018, 83, 6815–6823. [Google Scholar] [CrossRef]
- . [CrossRef]
- Sun, T.-T.; Liu, K.; Zhang, S.-X.; Wang, C.-R.; Yao, C.-Z.; Yu, J. Enantioselective halocyclization of indole derivatives: using 1,3-dihalohydantoins with anionic chiral Co(III) complexes. Synlett, 2021, 32, 701–707. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, H.; Xu, X.; Xu, W.; Jiang, X. Catalytic enantioselective bromoaminocyclization and bromocycloetherification. Adv. Synth. Catal., 2020, 362, 5358–5362. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, synthesis, and biological evaluation of conformationally restricted rivastigmine analogues. J. Med. Chem., 2004, 47, 5945–5953. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Ward, R.S. Non-enzymatic asymmetric transformations involving symmetrical bifunctional compounds. Chem. Soc. Rev., 1990, 19, 1–19. [Google Scholar] [CrossRef]
- Magnuson, S.R. Two-directional synthesis and its use in natural product synthesis. Tetrahedron, 1995, 51, 2167–2213. [Google Scholar] [CrossRef]
- Willis, M.C. Enantioselective desymmetrisation. J. Chem. Soc. Perkin Trans. 1, 1999, 1765-1784. [CrossRef]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective enzymatic desymmetrizations in organic synthesis. Chem. Rev., 2005, 105, 313–354. [Google Scholar] [CrossRef]
- Wang, M.; Feng, M.; Tang, B.; Jiang, X. Recent advances of desymmetrization protocol applied in natural product total synthesis. Tetrahedron Lett., 2014, 55, 7147–7155. [Google Scholar] [CrossRef]
- Borissov, A.; Davies, T.Q.; Ellis, S.R.; Fleming, T.A.; Richardson, M.S.W.; Dixon, D.J. Organocatalytic enantioselective desymmetrisation. Chem. Soc. Rev., 2016, 45, 5474–5540. [Google Scholar] [CrossRef]
- Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev., 2016, 116, 7330–7396. [Google Scholar] [CrossRef]
- Long, H.-J.; Li, Y.-L.; Zhang, B.-Q.; Xiao, W.-Y.; Zhang, X.-Y.; He, L.; Deng, J. Asymmetric bromoaminocyclization and desymmetrization of cyclohexa-1,4-dienes through anion phase-transfer catalysis. Org. Lett., 2021, 23, 8153–8157. [Google Scholar] [CrossRef]
- . [CrossRef]
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep., 2009, 26, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Das, M.K.; De, S.; Shubhashish; Bisai, A. Concise total syntheses of (±)mesembrane and (±)-crinane. Org. Biomol. Chem., 2015, 13, 3585–3588. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Tay, D.W.; Chen, J.; Leung, G.Y.C.; Yeung, Y.-Y. Enantioselective synthesis of 2-substituted and 3-substituted piperidines through a bromoaminocyclization process. Chem. Commun., 2013, 49, 4412–4414. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.; von Mentzer, B.; Påhlman, I.; Ahlstedt, I.; Uvebrant, A.; Kristensson, E.; Martinsson, R.; Novén, A.; de Verdier, J.; Vauquelin, G. Neurokinin 1 receptor antagonists: correlation between in vitro receptor interaction and in vivo efficacy. J. Pharmacol. Exp. Ther., 2007, 322, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, P.; Liu, X.; Zhao, J.; Lin, L.; Feng, X. Diastereoselectively switchable asymmetric haloaminocyclization for the synthesis of cyclic sulfamates. Chem. Eur. J., 2015, 21, 6386–6389. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Maji, M.S.; Küçük, H.B.; Atodiresei, I. Asymmetric Brønsted acid catalyzed cycloadditions - Efficient enantioselective synthesis of pyrazolidines, pyrazolines, and 1,3-diamines from N-acyl hydrazones and alkenes. Angew. Chem. Int. Ed., 2012, 51, 12864–12868. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-B.; Gao, Q.; Fan, J.-J.; Zhao, Z.-Y.; Tu, X.-Q.; Cao, H.-Q.; Yu, J. ; Yu, J. Anionic chiral Co(III) complexes mediated asymmetric halocyclization - Synthesis of 5-halomethyl pyrazolines and isoxazolines. Org. Lett., 2021, 23, 9134–9139. [Google Scholar]
- . [CrossRef]
- Sebastian, J. Dihydropyrazole and dihydropyrrole structures based design of Kif15 inhibitors as novel therapeutic agents for cancer. Comput. Biol. Chem., 2017, 68, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Roecker, A.J.; Coleman, P.J.; Mercer, S.P.; Schreier, J.D.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Lobell, R.B.; Tao, W.; Diehl, R.E.; South, V.J.; David, J.P.; Kohl, N.E.; Yan, Y.; Kuo, L.C.; Li, C.; Fernandez-Metzler, C.; Mahan, E.A.; Prueksaritanont, T.; Hartman, G.D. Kinesin spindle protein (KSP) inhibitors. Part 8: Design and synthesis of 1,4-diaryl-4,5-dihydropyrazoles as potent inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett., 2007, 17, 5677–5682. [Google Scholar] [CrossRef]
- Tripathi, C.B.; Mukherjee, S. Catalytic enantioselective iodoaminocyclization of hydrazones. Org. Lett., 2014, 16, 3368–3371. [Google Scholar] [CrossRef]
- Tripathi, C.B.; Mukherjee, S. Catalytic enantioselective 1,4-iodofunctionalizations of conjugated dienes. Org. Lett., 2015, 17, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.J.; Schreier, J.D.; Cox, C.D.; Fraley, M.E.; Garbaccio, R.M.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Lobell, R.B.; Rickert, K.; Tao, W.; Diehl, R.E.; South, V.J.; David, J.P.; Kohl, N.E.; Yan, Y.; Kuo, L.; Prueksaritanont, T.; Li, C.; Mahan, E.A.; Fernandez- Metzler, C.; Salata, J.J.; Hartman, G.D. Kinesin spindle protein (KSP) inhibitors. Part 6: Design and synthesis of 3,5-diaryl-4,5-dihydropyrazole amides as potent inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett., 2007, 17, 5390–5395. [Google Scholar] [CrossRef] [PubMed]
- Richter, H.G.F.; Freichel, C.; Huwyler, J.; Nakagawa, T.; Nettekoven, M.; Plancher, J.-M.; Roche, S.O.; Schuler, F.; Taylor, S.; Ullmer, C.; Wiegand, R. Discovery of potent and selective histamine H3 receptor inverse agonists based on the 3,4-dihydro-2H-pyrazino [1,2-a]indol-1-one scaffold. Bioorg. Med. Chem. Lett., 2010, 20, 5713–5717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.A.; Yu, W.Z.; Yeung, Y.-Y. Carbamate-catalyzed enantioselective bromolactamization. Angew. Chem. Int. Ed., 2015, 54, 12102–12106. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liu, X.; Li, L.; Cai, Y.; Liu, W.; Shi, Y. Enantioselective bromoaminocyclization of allyl N-tosylcarbamates catalyzed by a chiral phosphine–Sc(OTf)3 complex. J. Am. Chem. Soc., 2013, 135, 8101–8104. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Huang, H.; Liu, W.; Tian, H.; Shi, Y. Phosphine oxide–Sc(OTf)3 catalyzed highly regio- and enantioselective bromoaminocyclization of (E)-cinnamyl tosylcarbamates. An approach to a class of synthetically versatile functionalized molecules. Org. Lett., 2016, 18, 896–899. [Google Scholar] [CrossRef]
- Pan, H.; Tian, H.; Shi, Y. Phosphine oxide-Sc(OTf)3 catalyzed enantioselective bromoaminocyclization of tri-substituted allyl N-tosylcarbamates. Sci. China Chem., 2018, 61, 656–659. [Google Scholar] [CrossRef]
- Huang, H.; Pan, H.; Cai, Y.; Liu, M.; Tian, H.; Shi, Y. Enantioselective 6-endo bromoaminocyclization of 2,4-dienyl N-tosylcarbamates catalyzed by a chiral phosphine oxide-Sc(OTf)3 complex. A dramatic additive effect. Org. Biomol. Chem., 2015, 13, 3566–3570. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Pan, H.; Tian, H.; Shi, Y. Enantioselective 6-exo-bromoaminocyclization of homoallylic N-tosylcarbamates catalyzed by a novel monophosphine-Sc(OTf)3 complex. Org. Lett., 2015, 17, 3956–3959. [Google Scholar] [CrossRef]
- Li, Z.; Shi, Y. Chiral phosphine oxide–Sc(OTf)3 complex catalyzed enantioselective bromoaminocyclization of 2-benzofuranylmethyl N-tosylcarbamates. Approach to a novel class of optically active spiro compounds. Org. Lett., 2015, 17, 5752–5755. [Google Scholar] [CrossRef]
- . [CrossRef]
- Struble, T.J.; Lankswert, H.M.; Pink, M.; Johnston, J.N. Enantioselective organocatalytic amine-isocyanate capture-cyclization: regioselective alkene iodoamination for the synthesis of chiral cyclic ureas. ACS Catal., 2018, 8, 11926–11931. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Shue, H.-J.; Chen, X.; Schwerdt, J.H.; Paliwal, S.; Blythin, D.J.; Lin, L.; Gu, D.; Wang, C.; Reichard, G.A.; Wang, H.; Piwinski, J.J.; Duffy, R.A.; Lachowicz, J.E.; Coffin, V.L.; Nomeir, A.A.; Morgan, C.A.; Varty, G.B.; Shih, N.-Y. Cyclic urea derivatives as potent NK1 selective antagonists.Part II: Effects of fluoro and benzylic methyl substitutions. Bioorg. Med. Chem. Lett., 2006, 16, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Naapuri, J.; Rolfes, J.D.; Keil, J.; Manzuna Sapu, C.; Deska, J. Enzymatic halocyclization of allenic alcohols and carboxylates: a biocatalytic entry to functionalized O-heterocycles. Green Chem., 2017, 19, 447–452. [Google Scholar] [CrossRef]
- Younes, S.H.H.; Tieves, F.; Lan, D.; Wang, Y.; Süss, P.; Brundiek, H.; Wever, R.; Hollmann, F. Chemoenzymatic halocyclization of γ,δ-unsaturated carboxylic acids and alcohols. ChemSusChem, 2020, 13, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hoefler, G.T.; But, A.; Younes, S.H.H.; Wever, R.; Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Chemoenzymatic halocyclization of 4-pentenoic acid at preparative scale. ACS Sustainable Chem. Eng., 2020, 8, 2602–2607. [Google Scholar] [CrossRef]
- . [CrossRef]
- Mondal, D.; Fisher, B.F.; Jiang, Y.; Lewis, J.C. Flavin-dependent halogenases catalyze enantioselective olefin halocyclization. Nat. Commun., 2021, 12, 3268. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mondal, D.; Lewis, J.C. Expanding the reactivity of flavin-dependent halogenases toward olefins via enantioselective intramolecular haloetherification and chemoenzymatic oxidative rearrangements. ACS Catal., 2022, 12, 13501–13505. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Jiang, Y.; Lewis, J.C. Asymmetric catalysis by flavin-dependent halogenases. Chirality, 2023, 35, 452–460. [Google Scholar] [CrossRef] [PubMed]
- . [CrossRef]
- Murray, J.; Hodgson, D.R.W.; O’Donoghue, A.C. Going full circle with organocatalysis and biocatalysis: the latent potential of cofactor mimics in asymmetric synthesis. J. Org. Chem., 2023, 88, 7619–7629. [Google Scholar] [CrossRef]
- Qi, C.; Force, G.; Gandon, V.; Leboeuf, D. Hexafluoroisopropanol-promoted haloamidation and halolactonization of unactivated alkenes. Angew. Chem. Int. Ed., 2021, 60, 946–953. [Google Scholar] [CrossRef]
- Stini, N.A.; Gkizis, P.L.; Kokotos, C.G. Cyrene: a bio-based novel and sustainable solvent for organic synthesis. Green Chem., 2022, 24, 6435–6449. [Google Scholar] [CrossRef]
Figure 1.
Formation of heterocyclic compounds via a haliranium intermediate, 1.
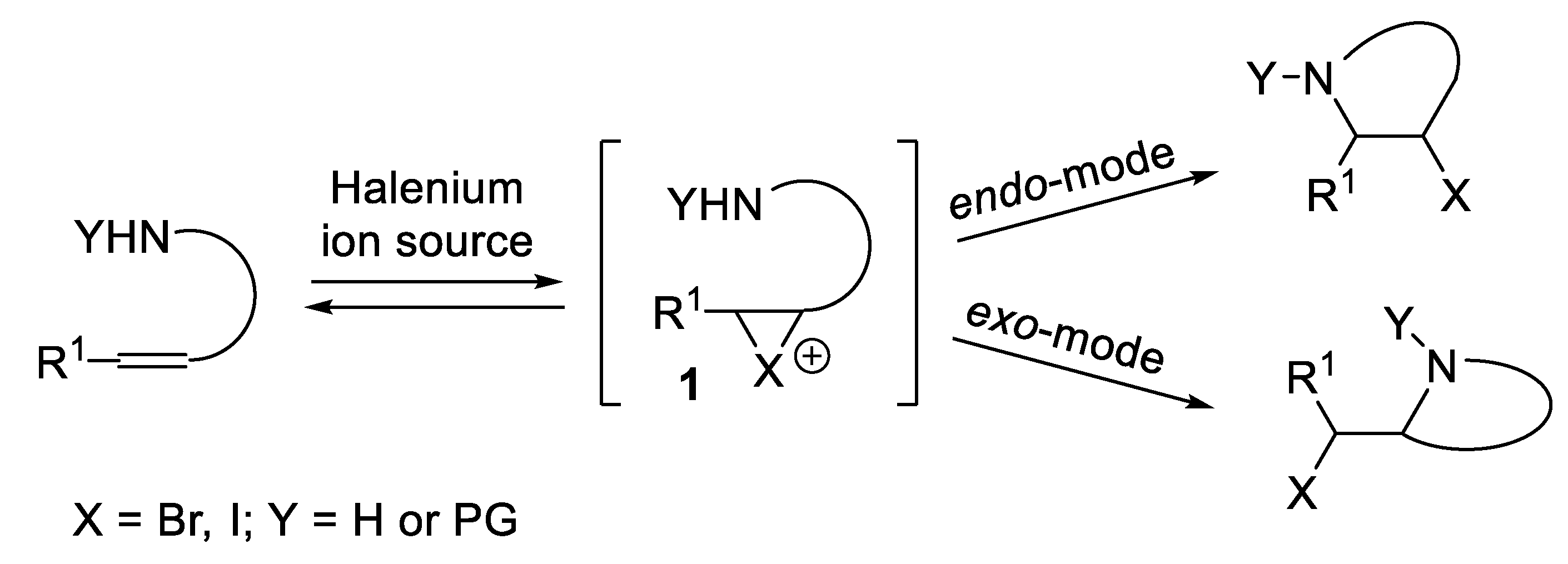
Scheme 1.
Iodocyclization leading to 1.4-dideoxy-1.4-manno-D-lyxitol, LAB, 5.
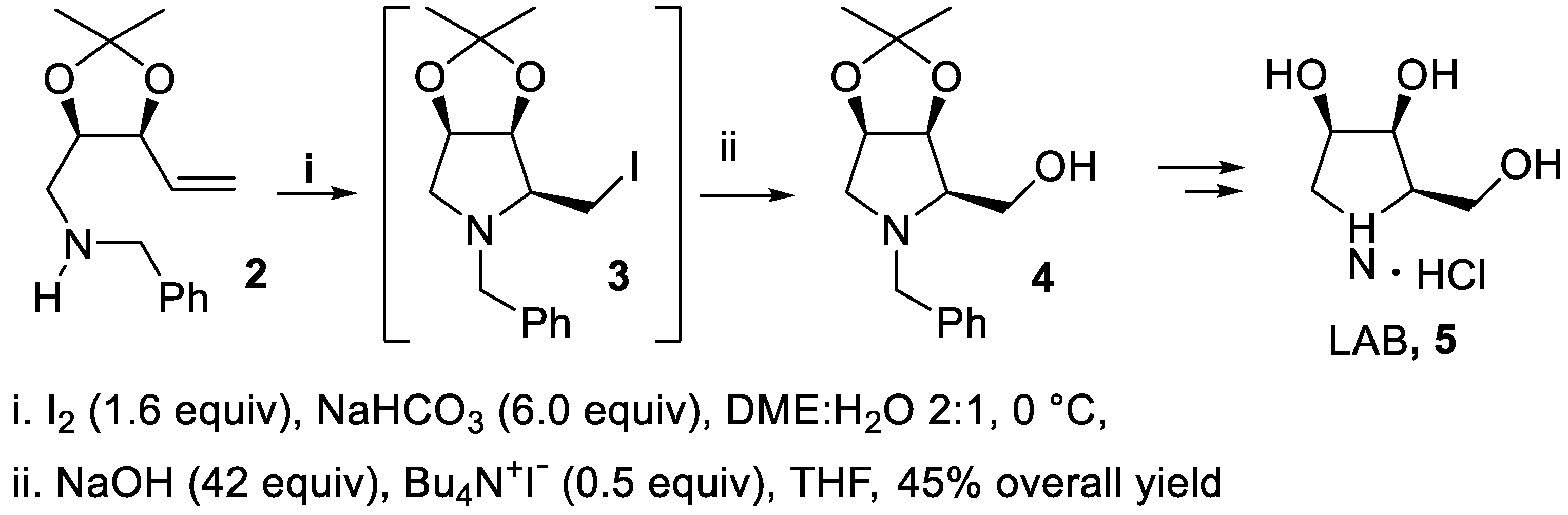
Scheme 2.
Preparation of a regioisomeric mixture of iodomethyl indolizidine 8 and iodoquinolizidine 9.
Scheme 2.
Preparation of a regioisomeric mixture of iodomethyl indolizidine 8 and iodoquinolizidine 9.
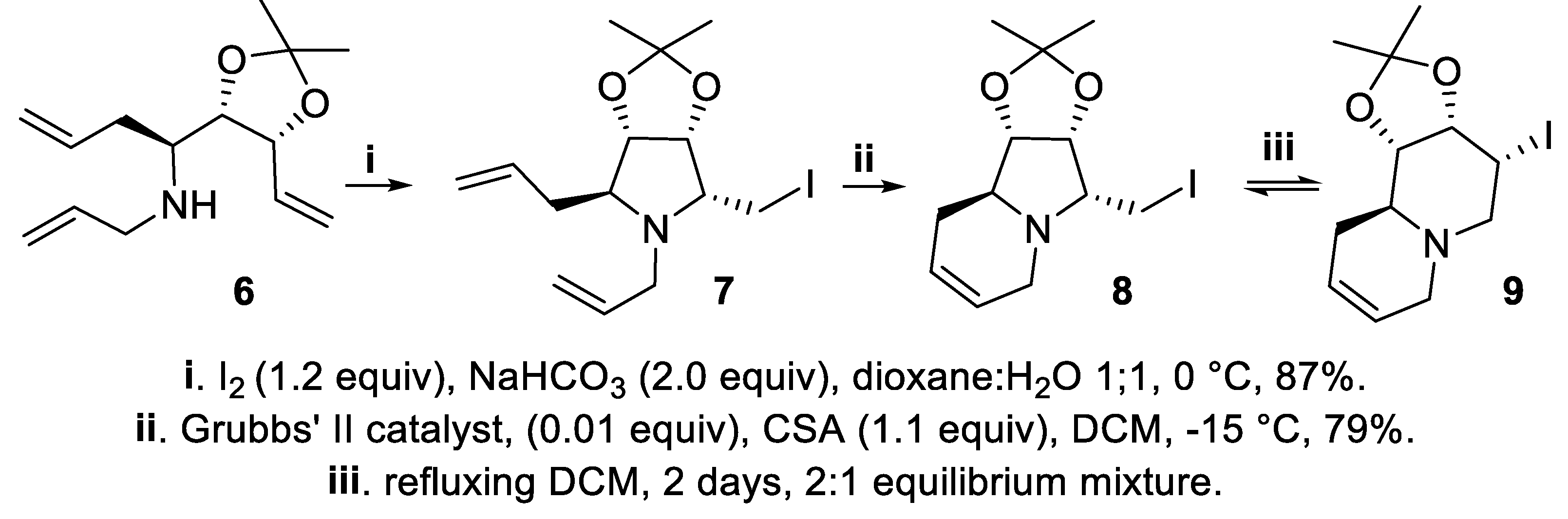
Scheme 3.
Iodoyclization leading to polyhydroxylated pyrrolidine β-amino acid derivative 13.
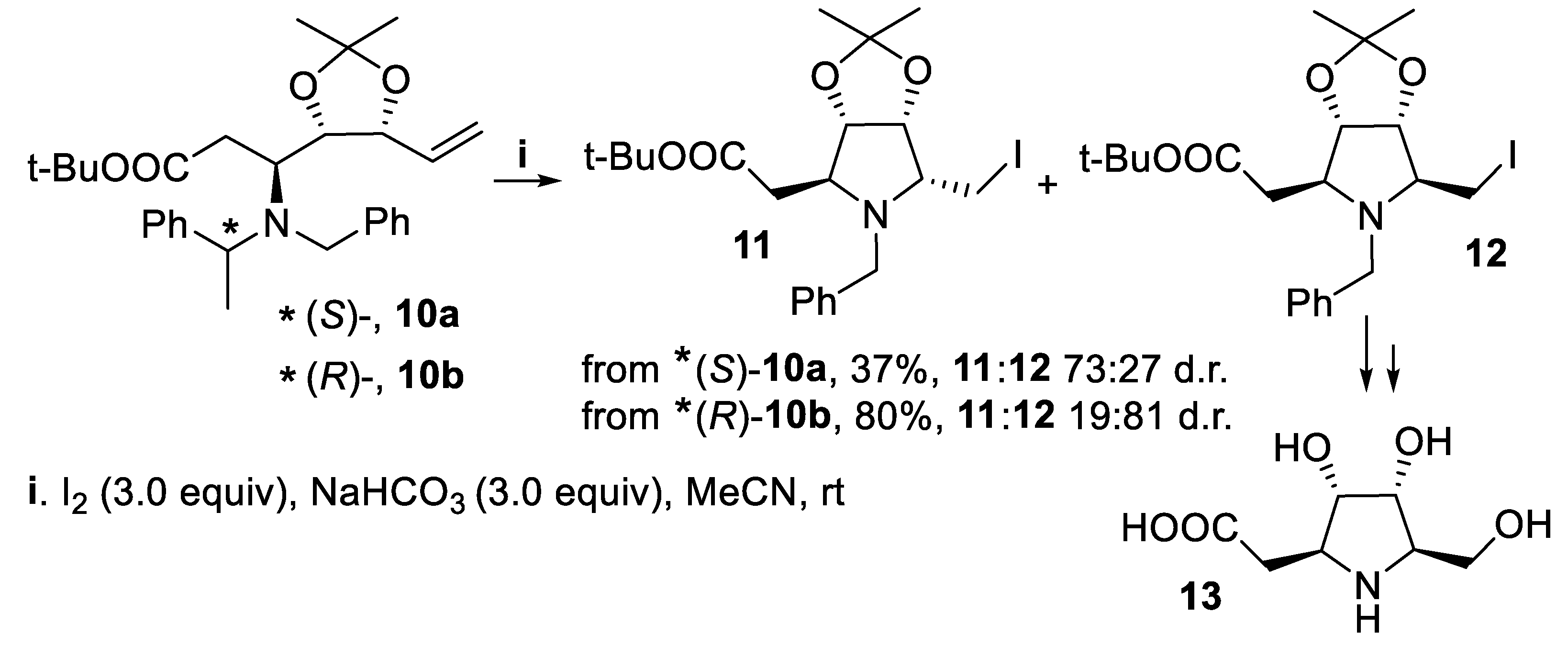
Scheme 4.
Iodoyclization leading to 15, key intermediate to alkaloid (-)-codonopsinine 17.
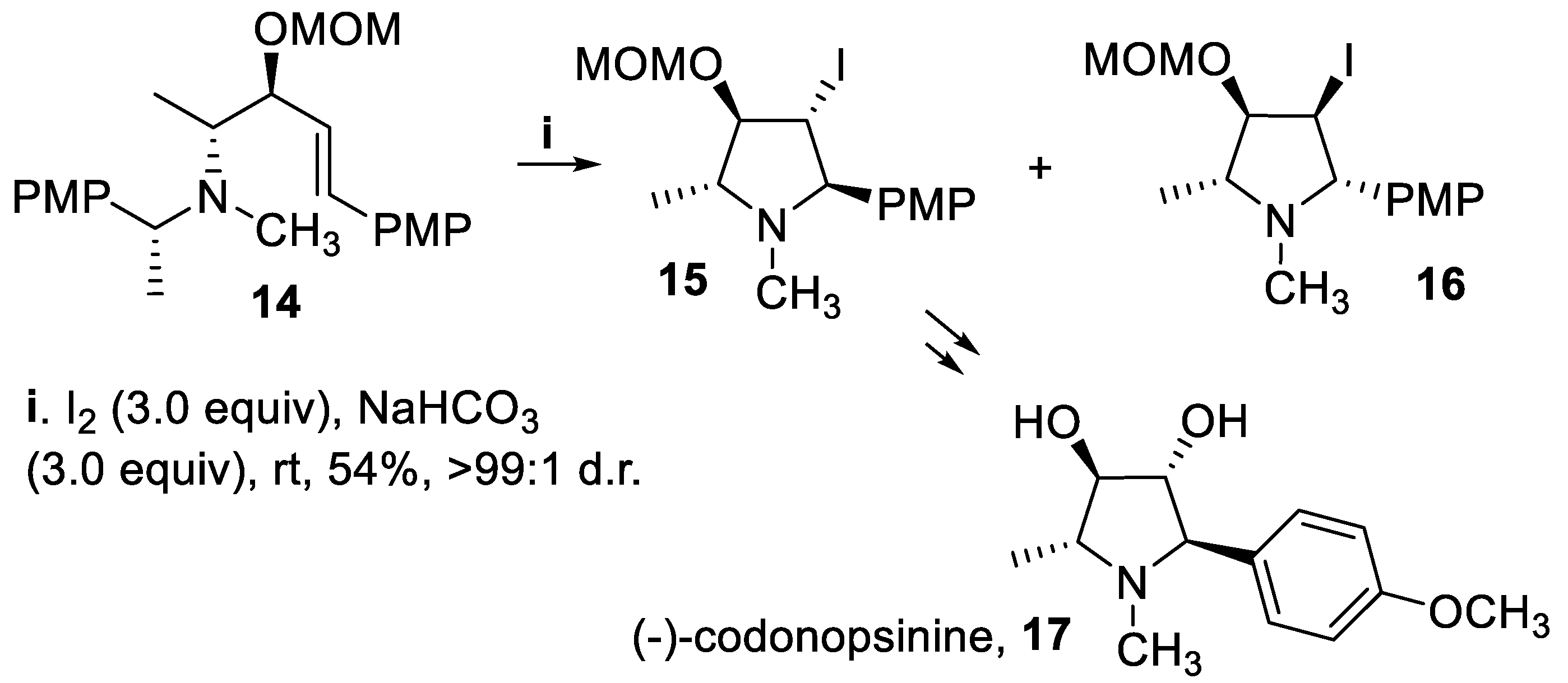
Scheme 5.
Iodoyclization of anti-aminoalkene 18, leading to (+)-DMDP, 21.
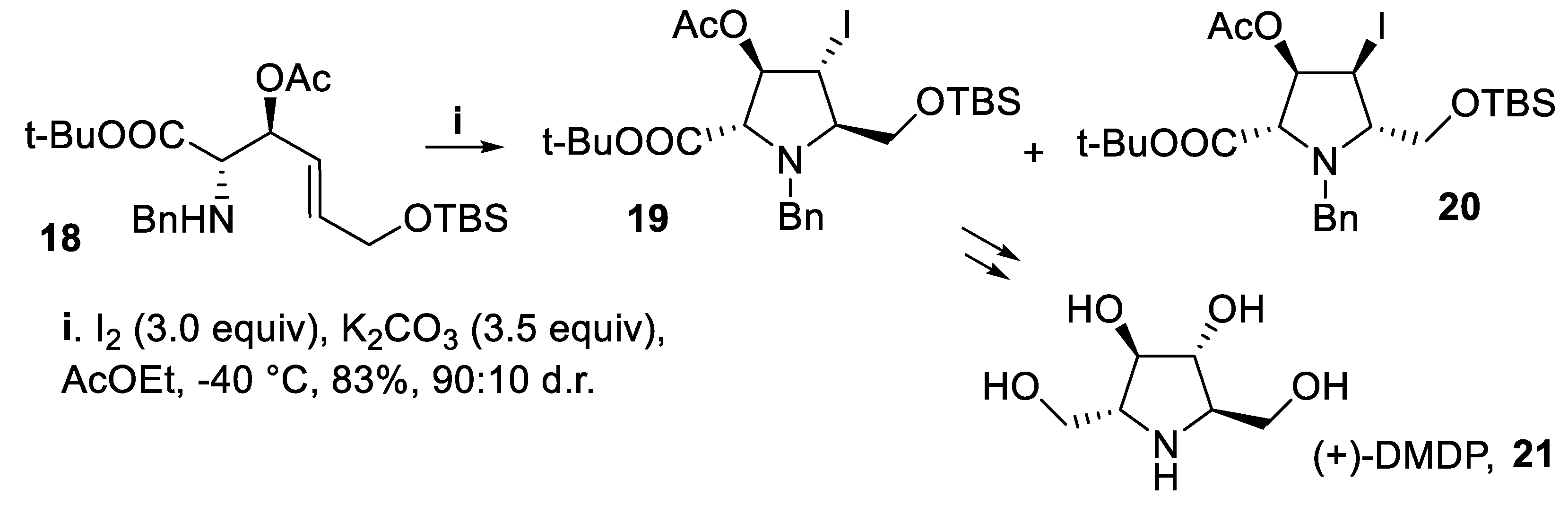
Scheme 6.
Iodoyclization of syn-aminoalkene 22, leading to (+)-hyacinthacine A1, 25.
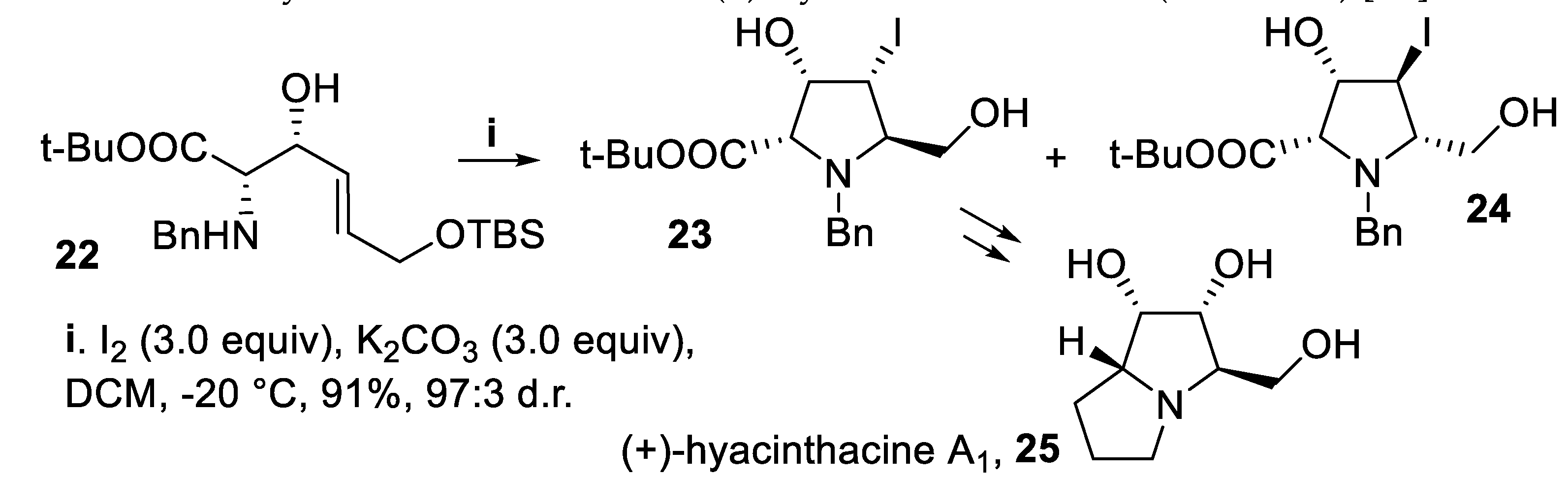
Scheme 7.
Iodoyclization of bis-homoallylic amine 26, leading to (+)-ADGDP, 28.
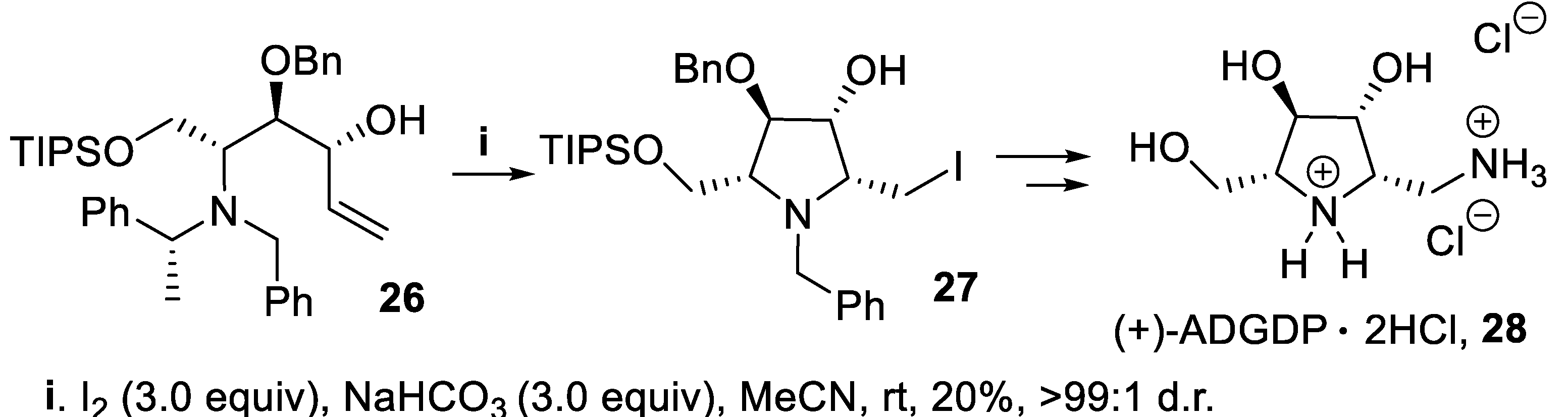
Scheme 8.
Conversion of bis-homoallylic amine 29 to (+)-ADANJ, 34.
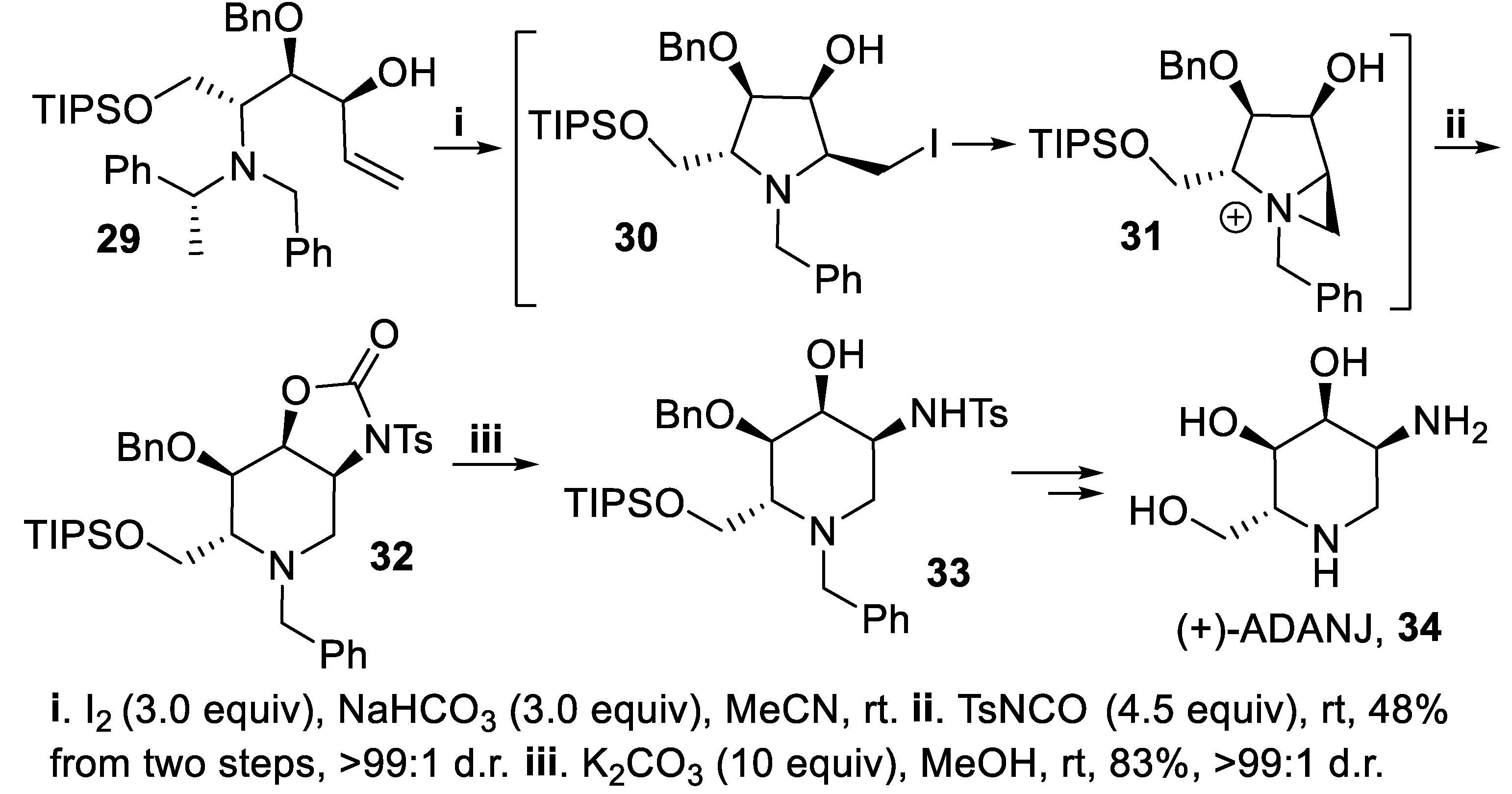
Scheme 9.
Synthesis of 1,4-dideoxy-1,4-imino-D-xylitol D-DIX, 38.
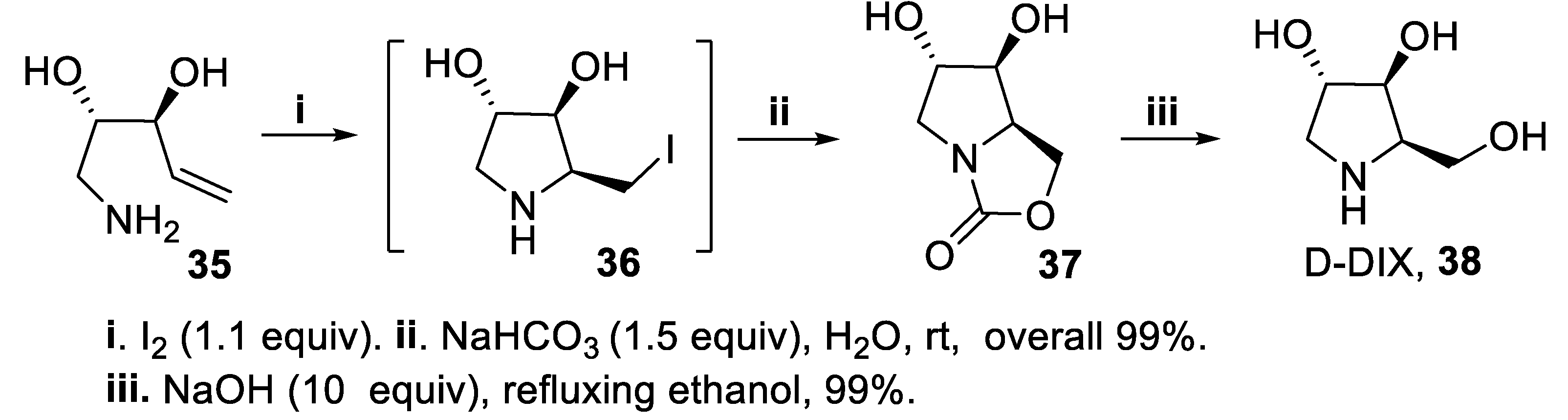
Scheme 10.
Synthesis of bicyclic oxazolidin-2-ones 41 and 44 with matching effects directing stereochemistry.
Scheme 10.
Synthesis of bicyclic oxazolidin-2-ones 41 and 44 with matching effects directing stereochemistry.
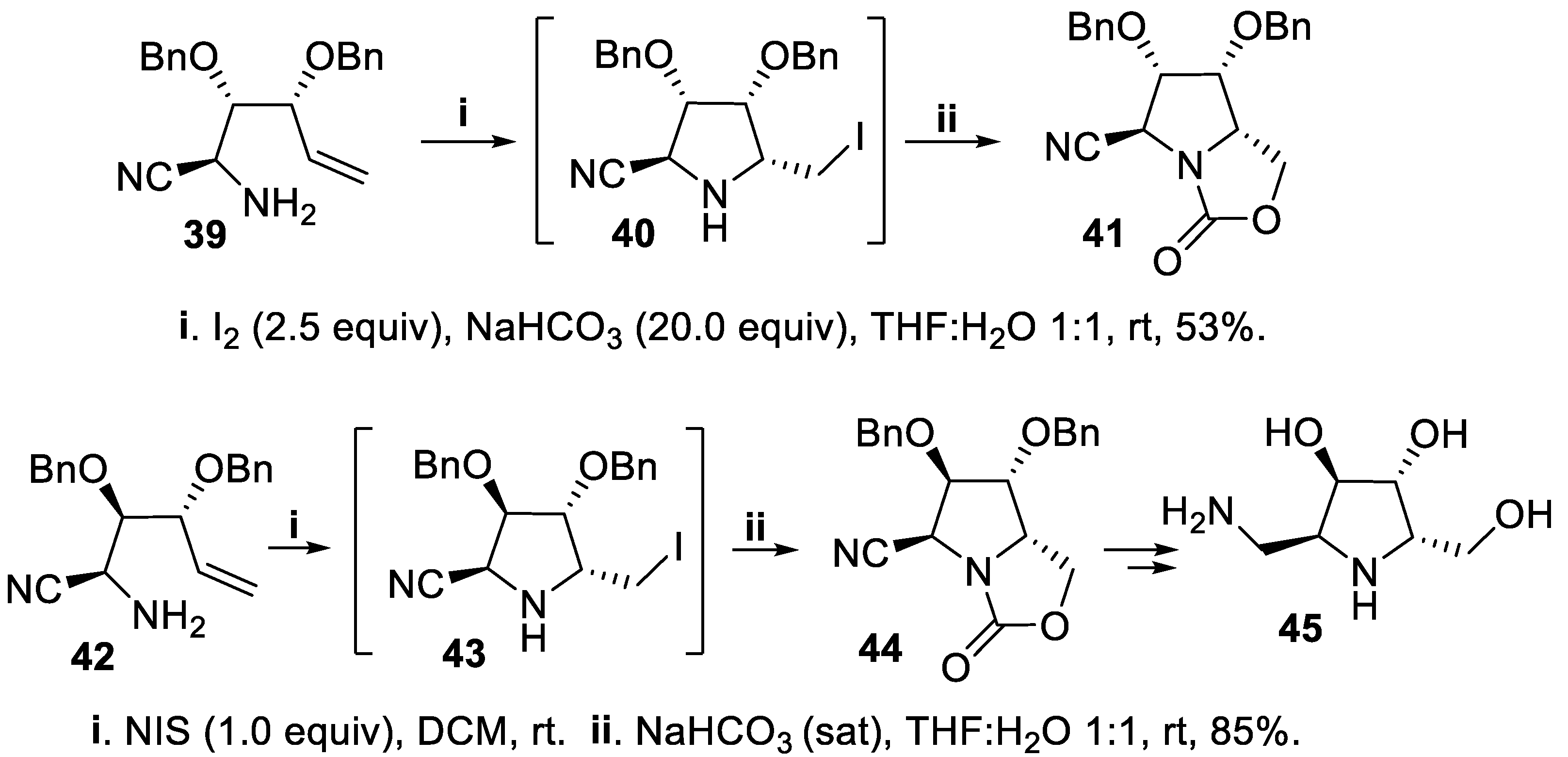
Scheme 11.
Synthesis of mixtures of bicyclic oxazolidin-2-ones 47,48 and 50,51 owing to mismatching effects directing stereochemistry.
Scheme 11.
Synthesis of mixtures of bicyclic oxazolidin-2-ones 47,48 and 50,51 owing to mismatching effects directing stereochemistry.
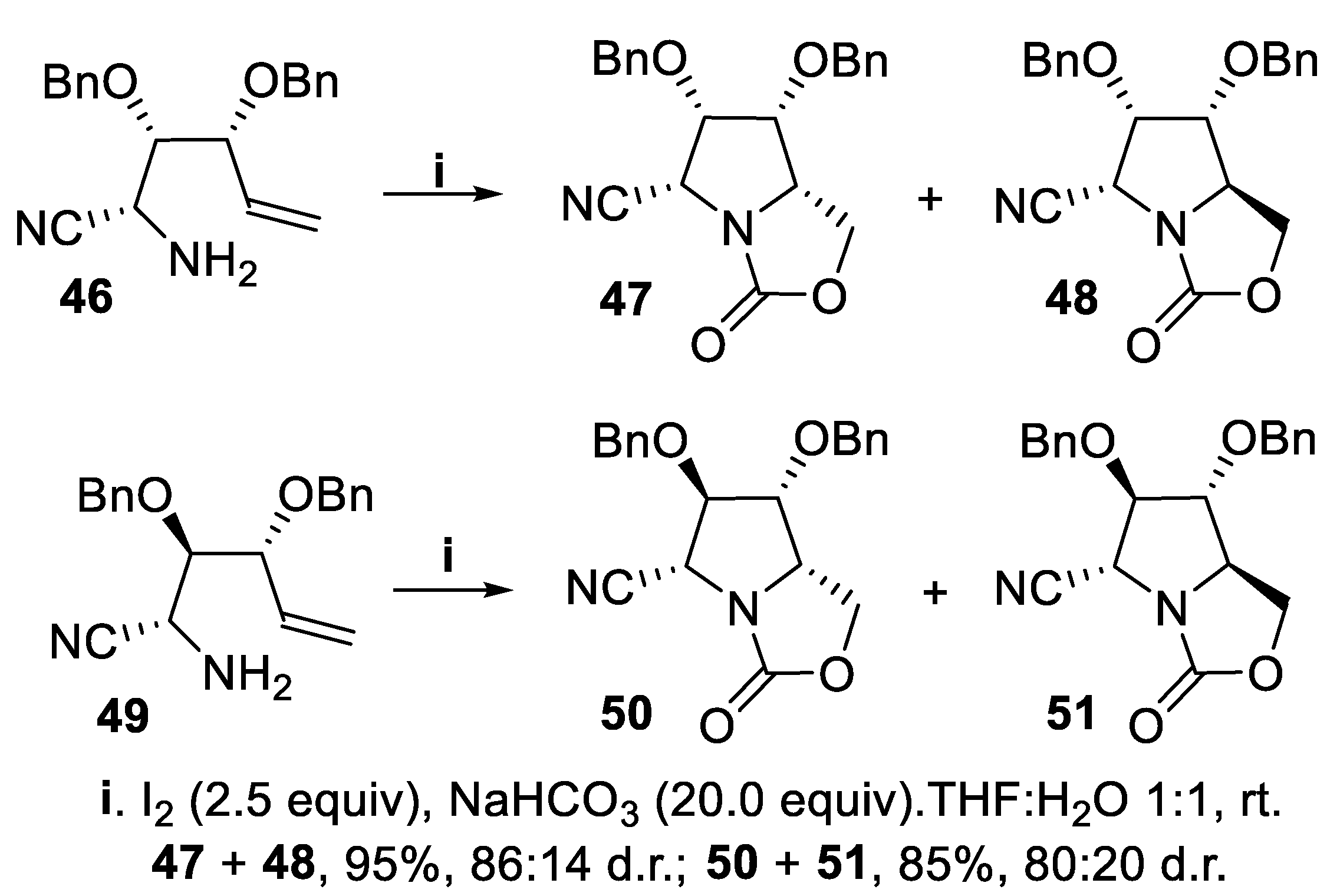
Scheme 12.
Stereoselective cyclization of chiral aziridines 52 leading to bicyclic [3.1.0]bromoderivatives 55.
Scheme 12.
Stereoselective cyclization of chiral aziridines 52 leading to bicyclic [3.1.0]bromoderivatives 55.
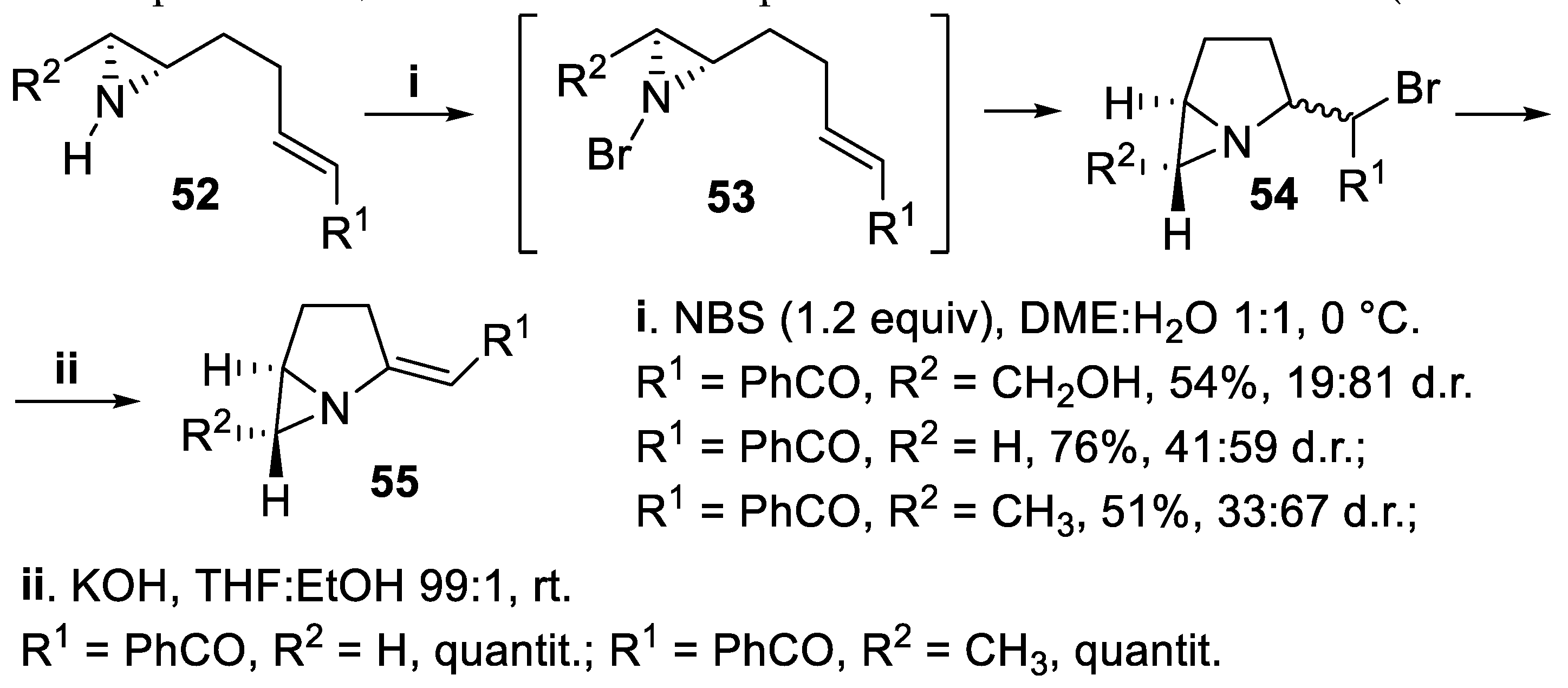
Scheme 13.
t-Boc-aziridines 56 leading to regioisomeric mixtures of chiral pyrrolidines 58 and piperidines 59.
Scheme 13.
t-Boc-aziridines 56 leading to regioisomeric mixtures of chiral pyrrolidines 58 and piperidines 59.
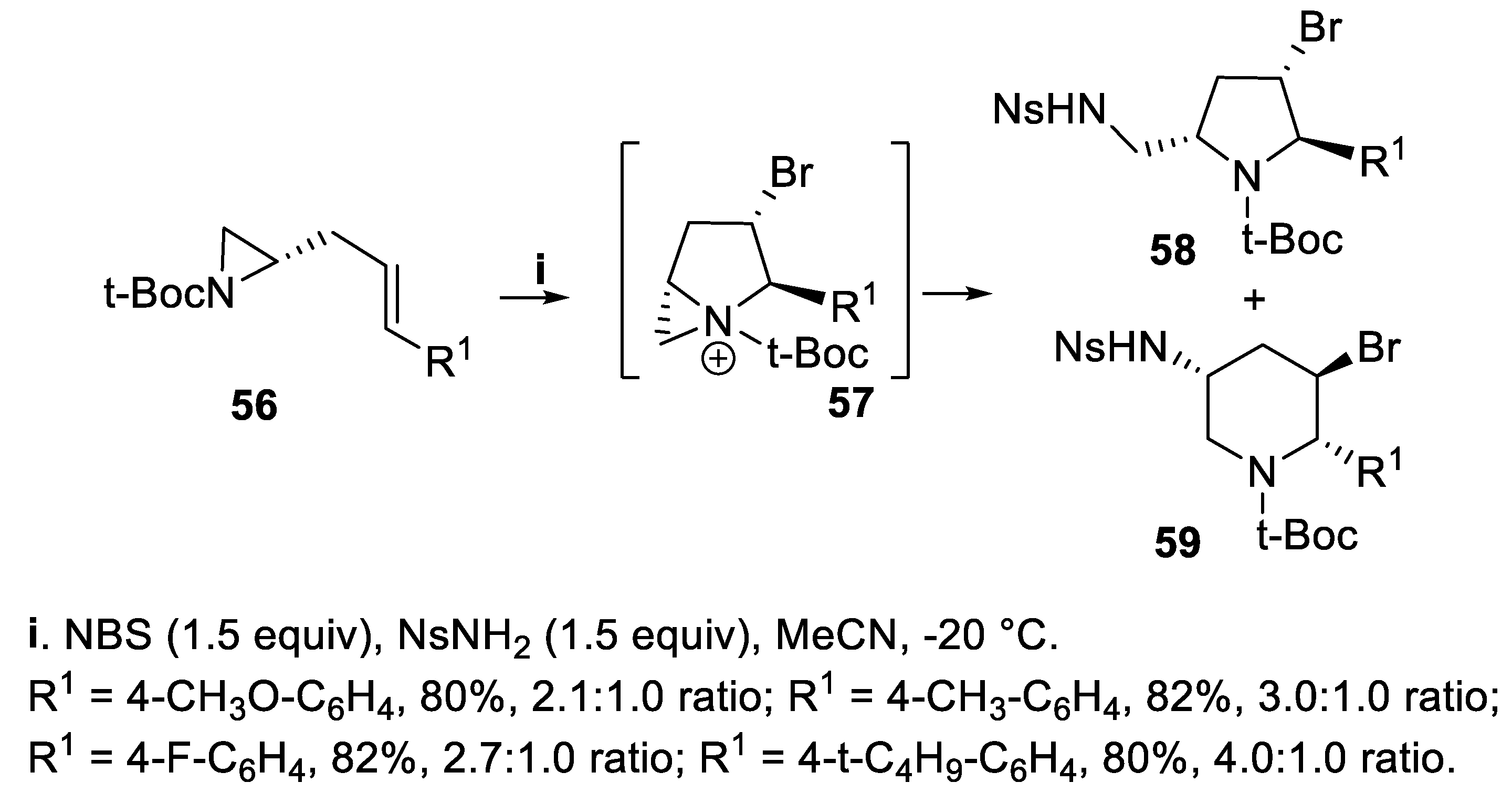
Scheme 14.
Stereoselective synthesis of t-Boc-azepanes 62 starting from chiral aziridines 60.
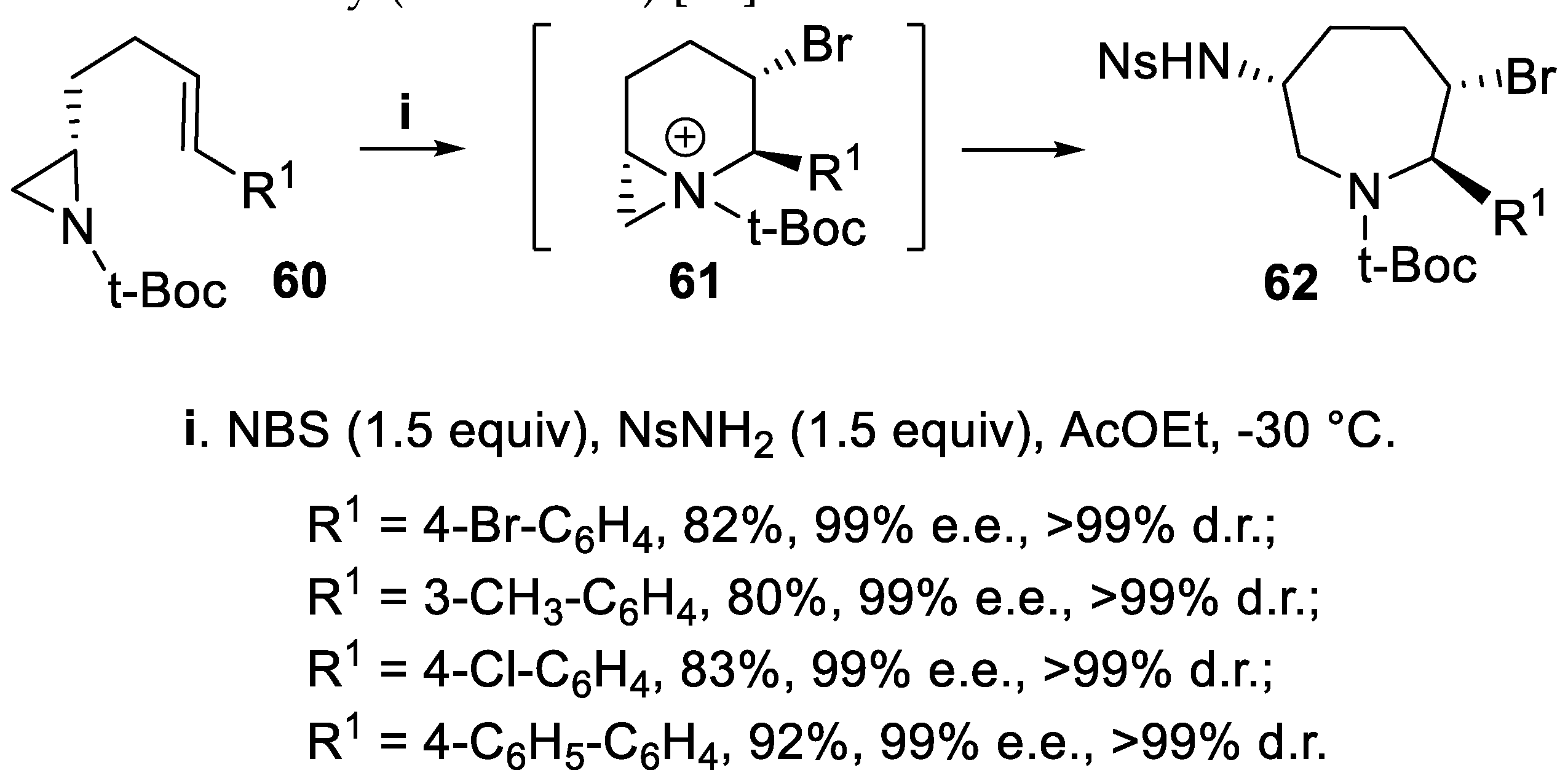
Scheme 15.
Conversion of pyrrolidine 63 to the indolizidine derivative 67.
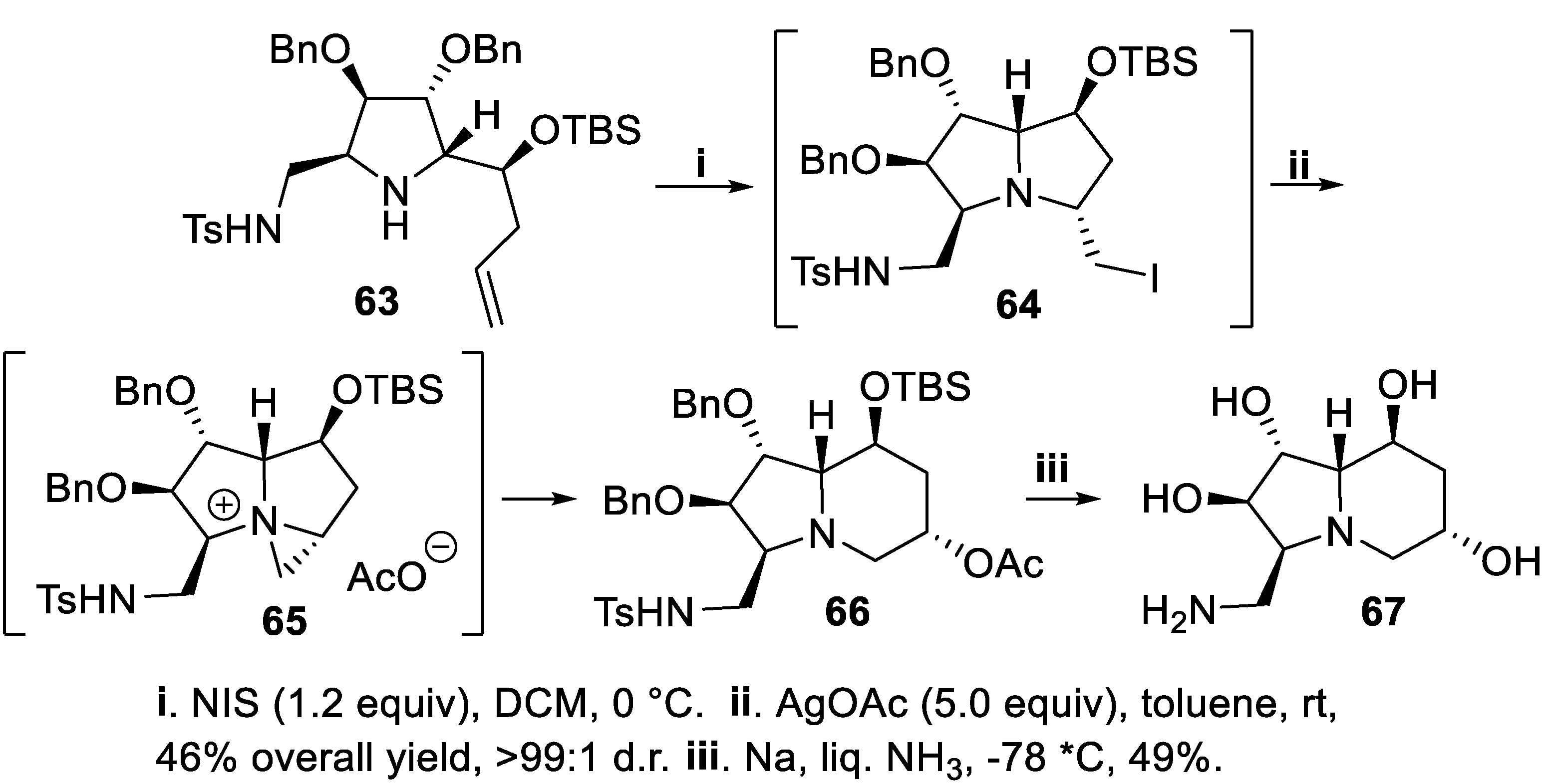
Scheme 16.
Iodoyclization of amine 68 leading to 70, intermediate of the synthesis of alkaloid (-)-sinoracutine, 71.
Scheme 16.
Iodoyclization of amine 68 leading to 70, intermediate of the synthesis of alkaloid (-)-sinoracutine, 71.
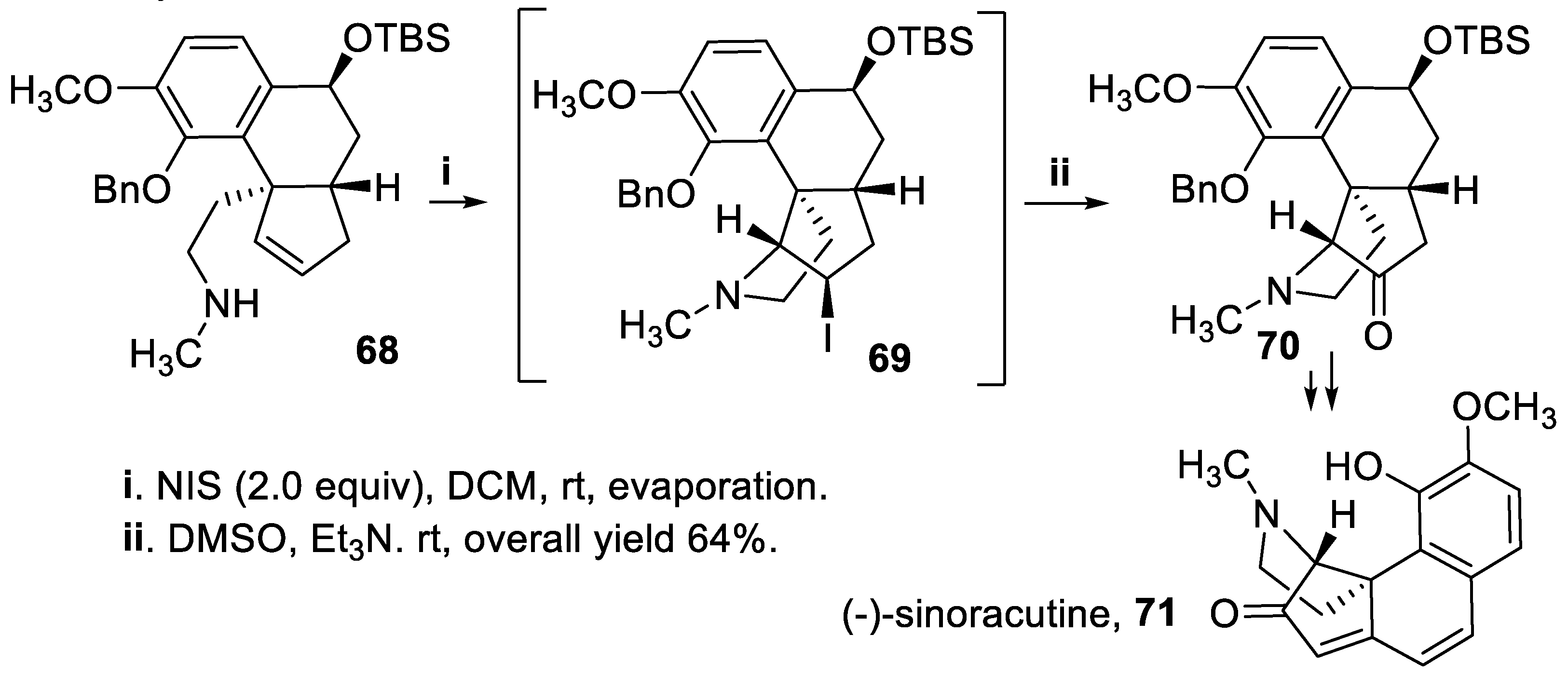
Scheme 17.
Iodoyclization leading to 73, a key intermediate to (+)-lyconadine A, 74.
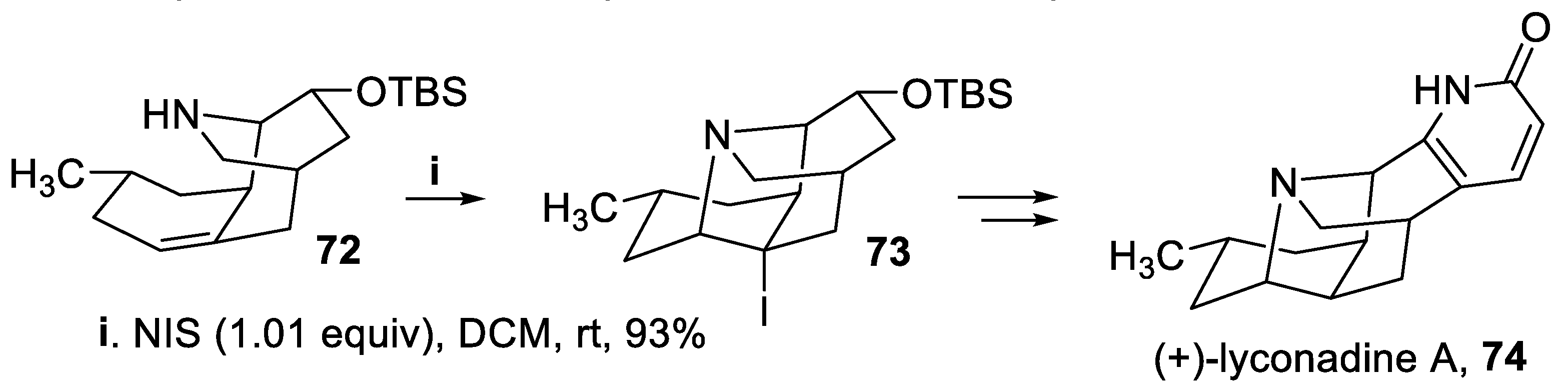
Scheme 18.
Cyclization of amine 75 leading to 76, an intermediate to (+)-pseudococaine, 77.
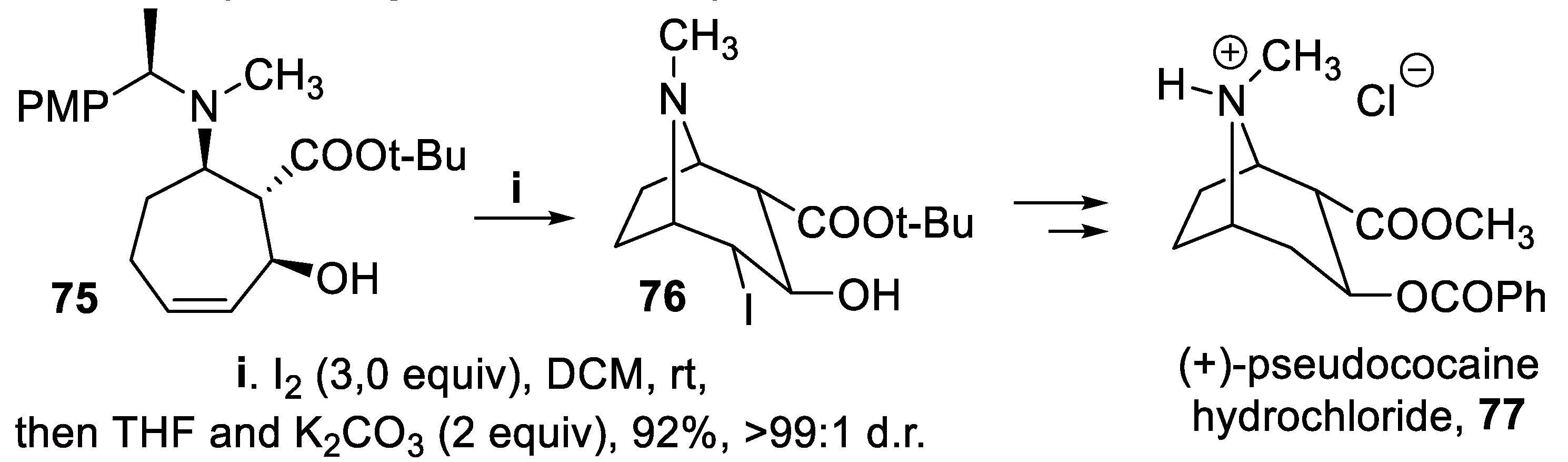
Scheme 19.
Synthesis of (-)-hyacinthacine A1, 80, and (-)-7a-epi-hyacinthacine A1, 83, from cyclic amines 78 and 81.
Scheme 19.
Synthesis of (-)-hyacinthacine A1, 80, and (-)-7a-epi-hyacinthacine A1, 83, from cyclic amines 78 and 81.
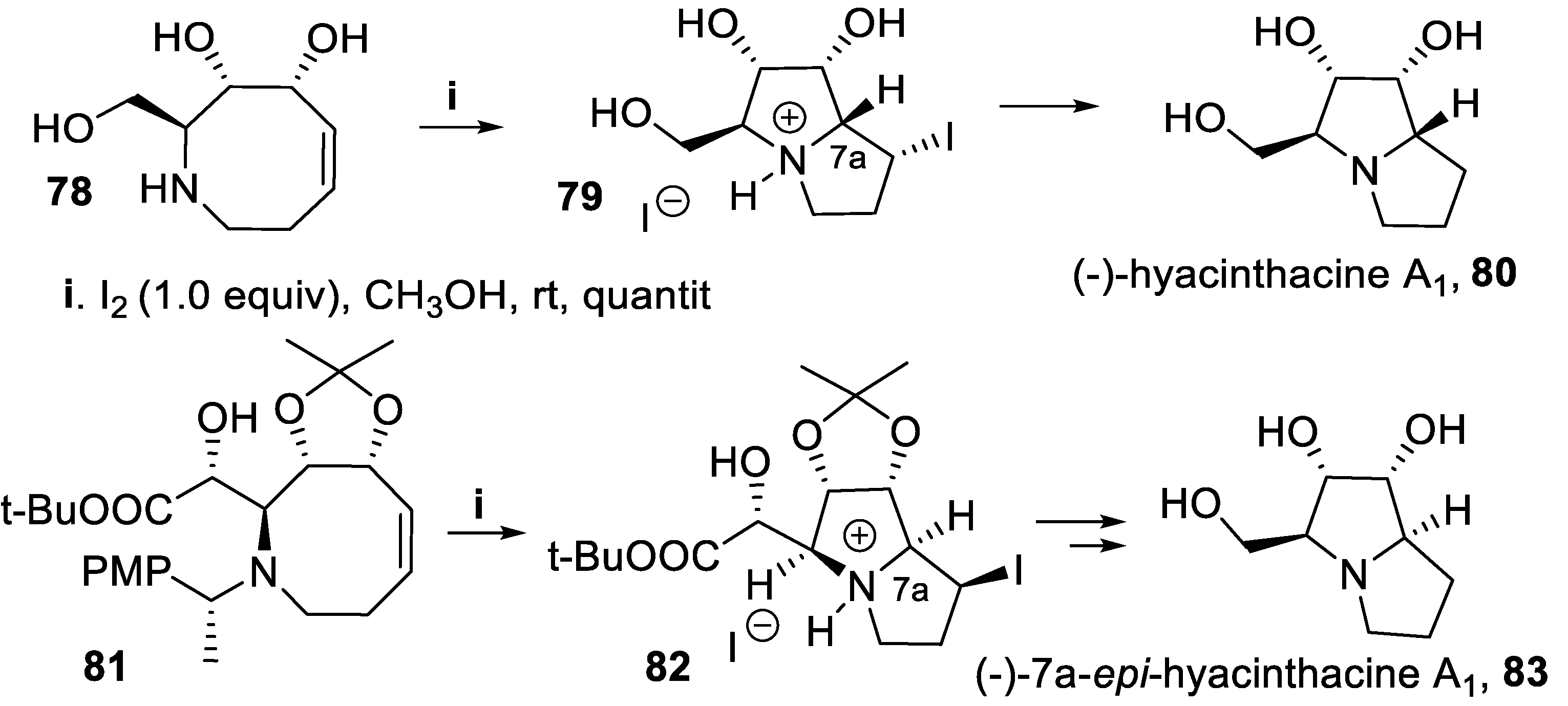
Scheme 20.
Synthesis of spiroderivative 85, intermediate of diazatricyclic core of alkaloid TAN1251C, 87.
Scheme 20.
Synthesis of spiroderivative 85, intermediate of diazatricyclic core of alkaloid TAN1251C, 87.
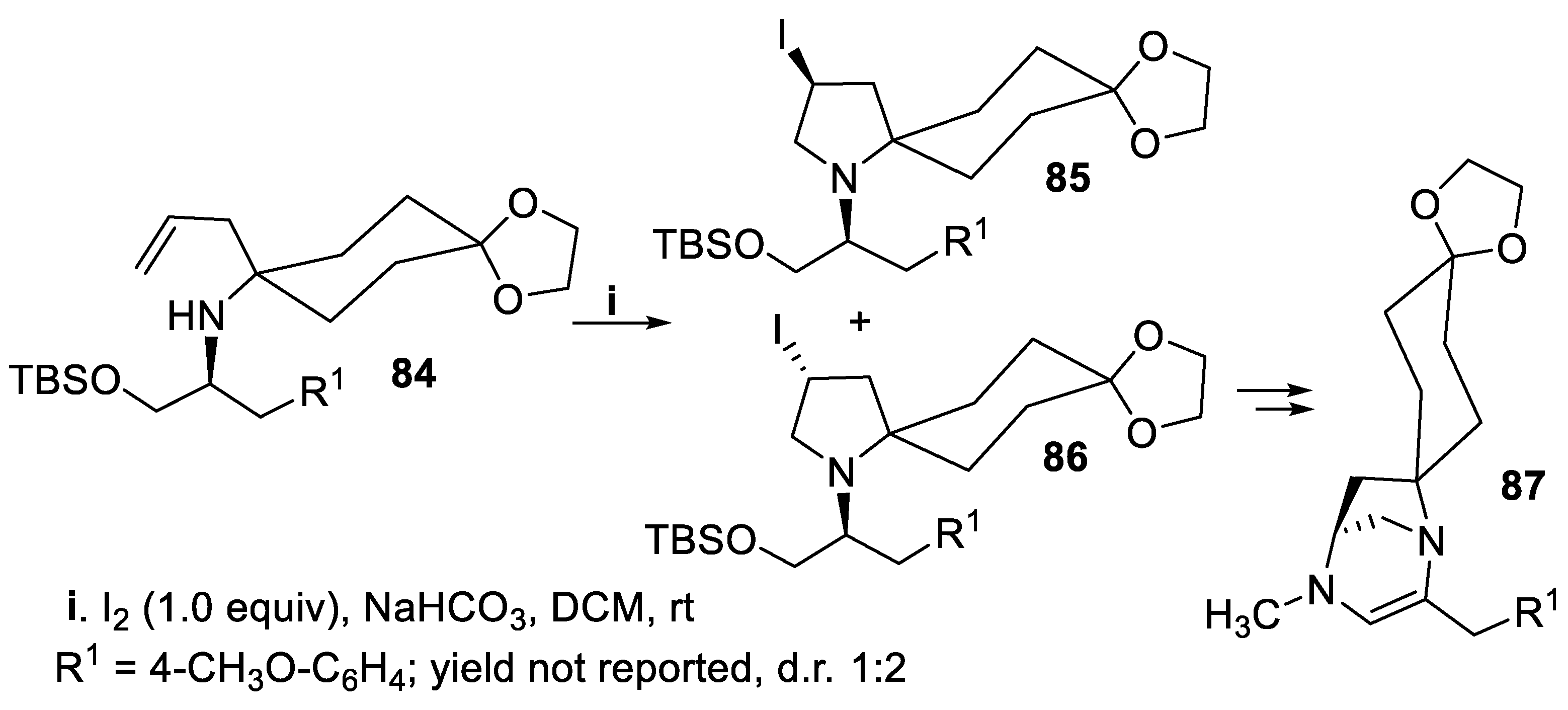
Scheme 21.
Cyclization of tryptophane derivatives 88 and ent-88, intermediates to pyrrolidineindoline alkaloids.
Scheme 21.
Cyclization of tryptophane derivatives 88 and ent-88, intermediates to pyrrolidineindoline alkaloids.
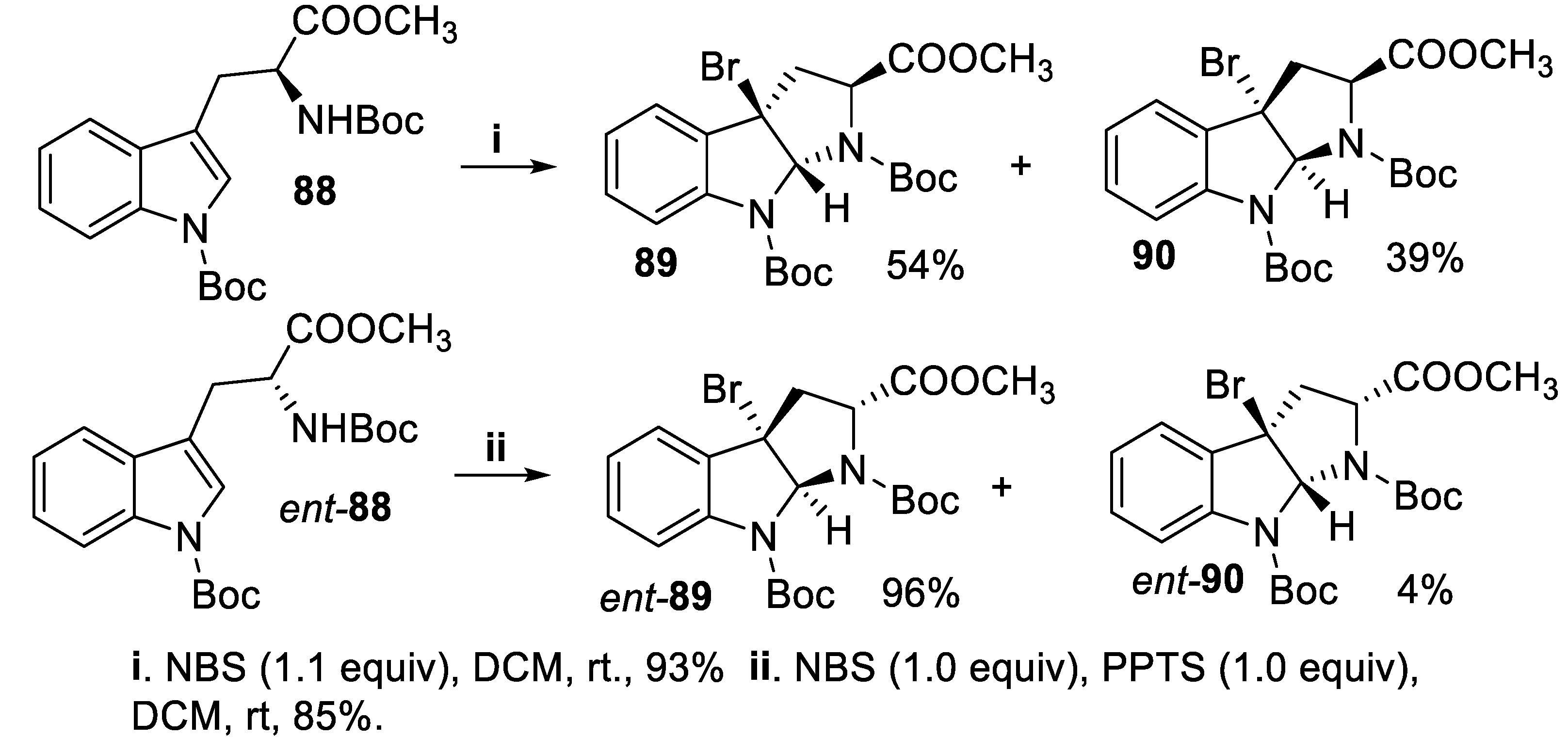
Scheme 22.
Iodoamination leading to 1-deoxygalactonojirimycin (DGJ), 95.
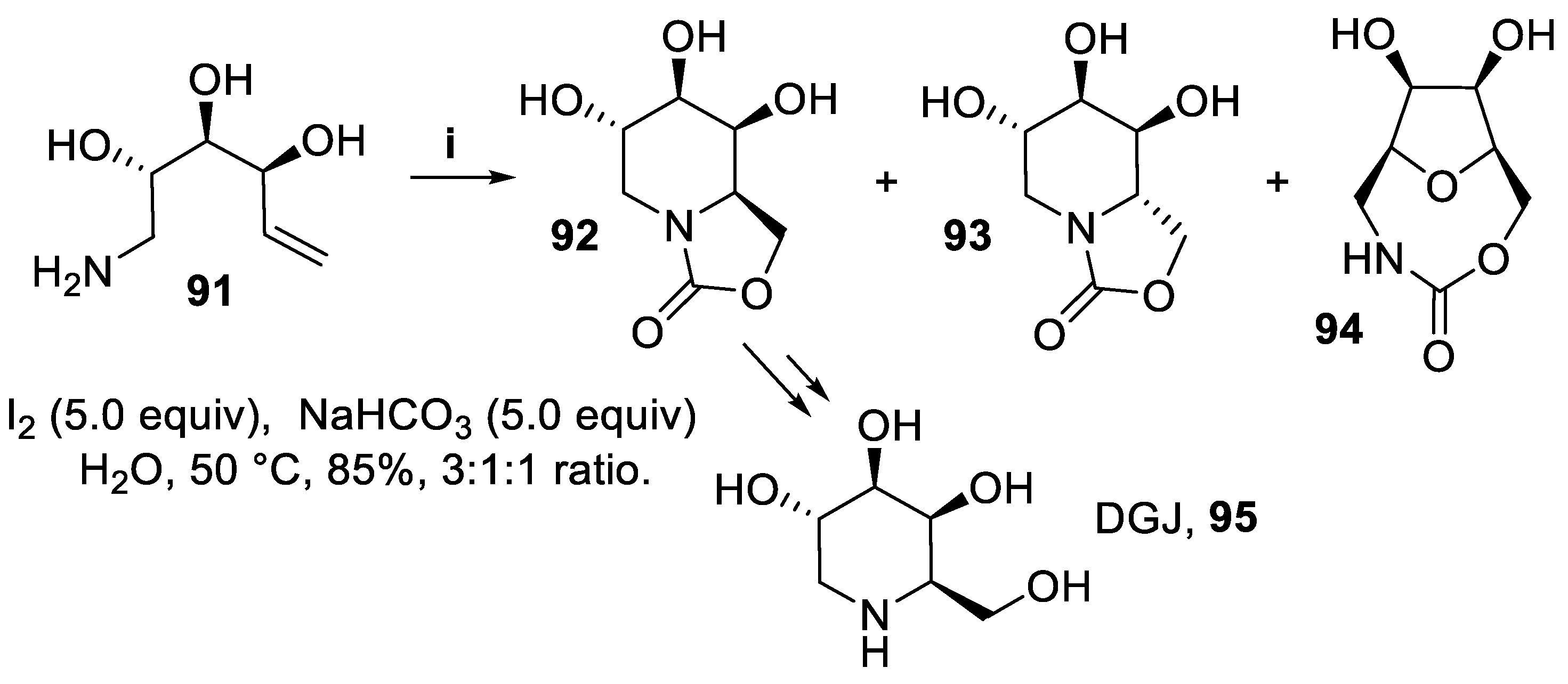
Scheme 23.
Synthesis of chiral trifluoromethyl morpholines by iodocyclization.
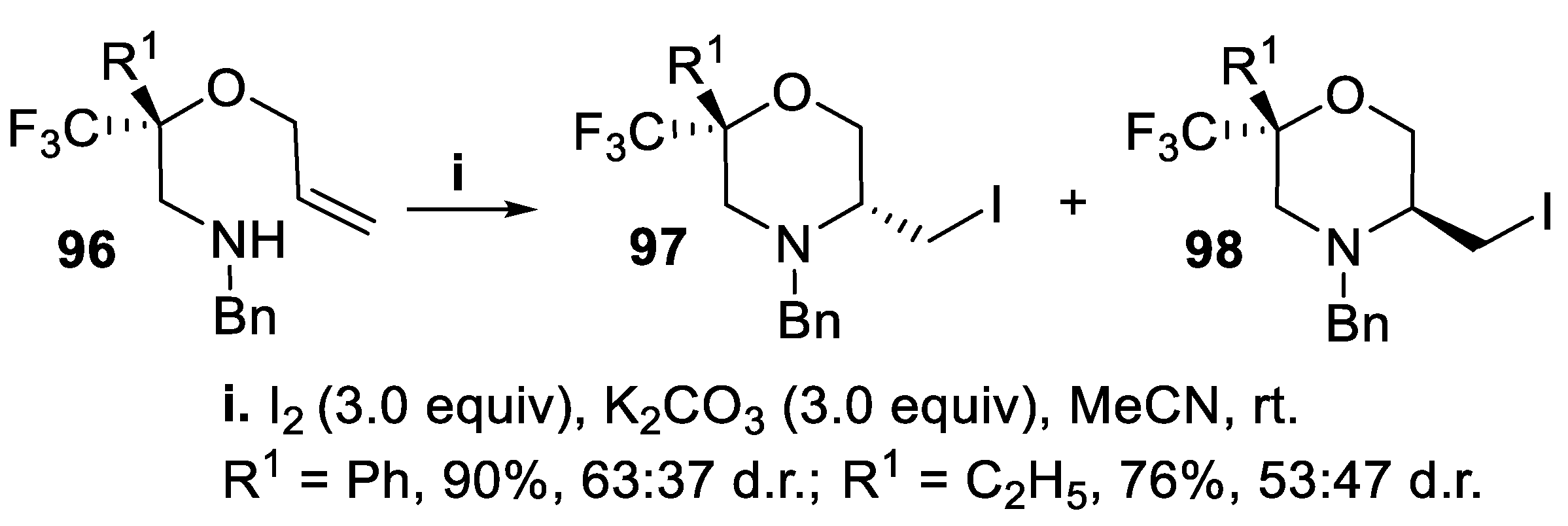
Scheme 24.
Stereoselective iodocyclization of chiral trifluoromethyl morpholines 99.
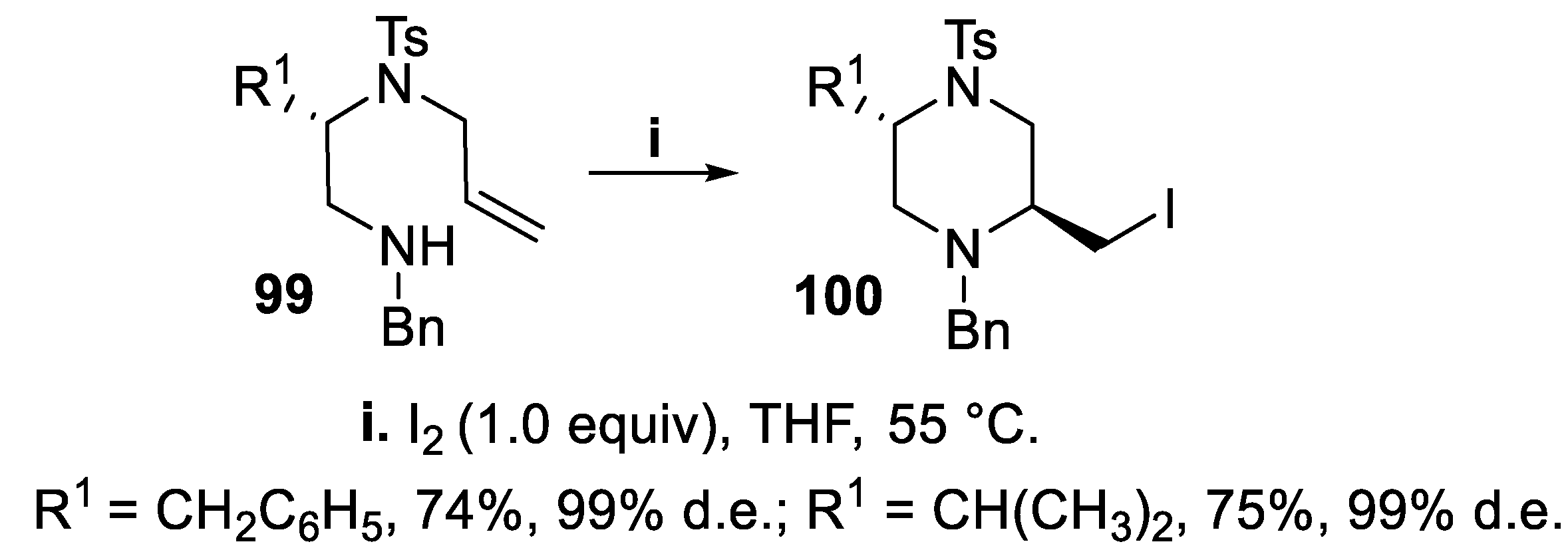
Scheme 25.
Iodoyclization of 101, leading to 102, key intermediate to alkaloid (-)-batzelladine D, 103.
Scheme 25.
Iodoyclization of 101, leading to 102, key intermediate to alkaloid (-)-batzelladine D, 103.
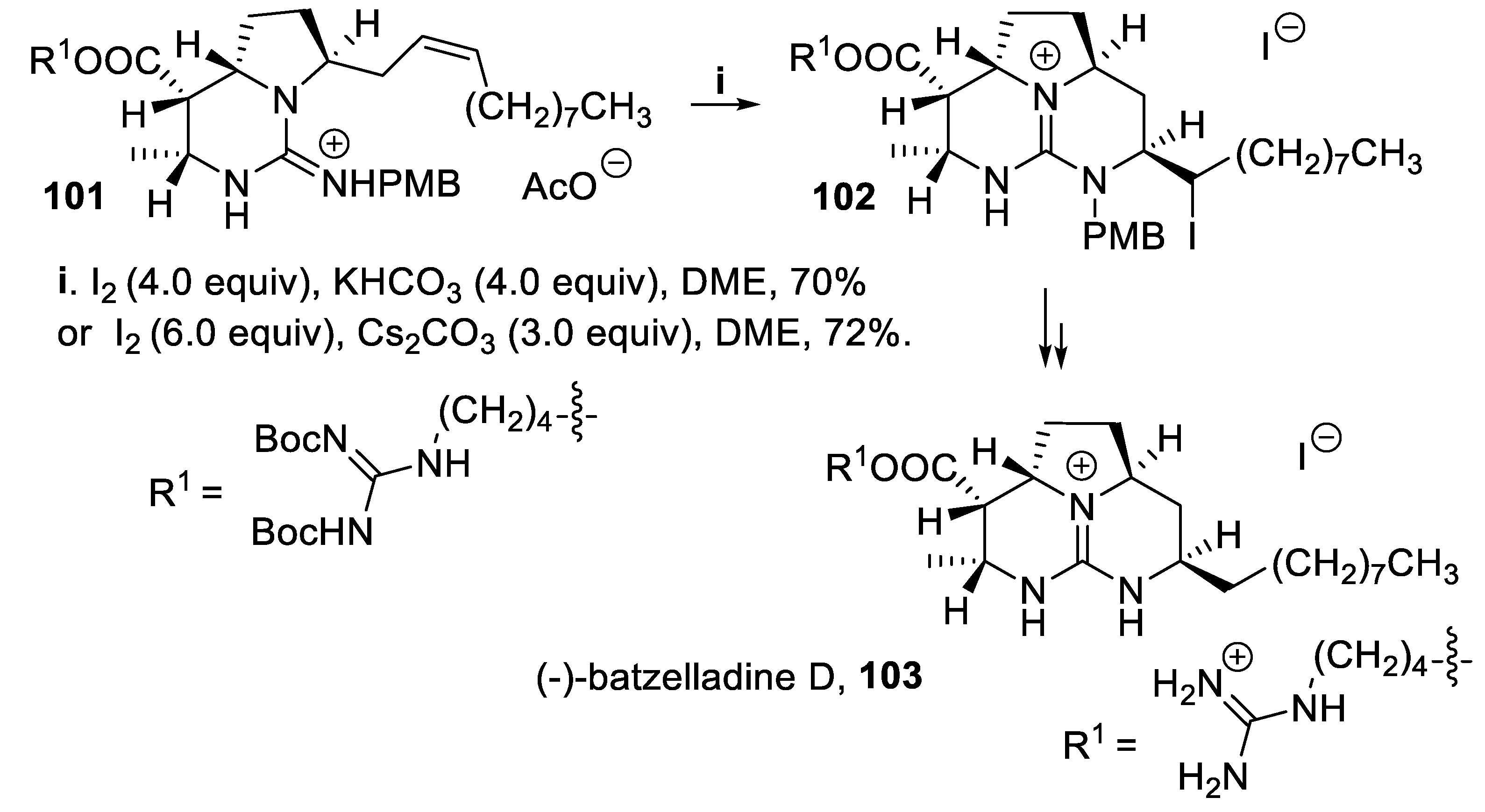
Scheme 26.
Stereoselective synthesis of the bicyclic compound 105, intermediate to (+)-saxitoxin, STX, 106.
Scheme 26.
Stereoselective synthesis of the bicyclic compound 105, intermediate to (+)-saxitoxin, STX, 106.
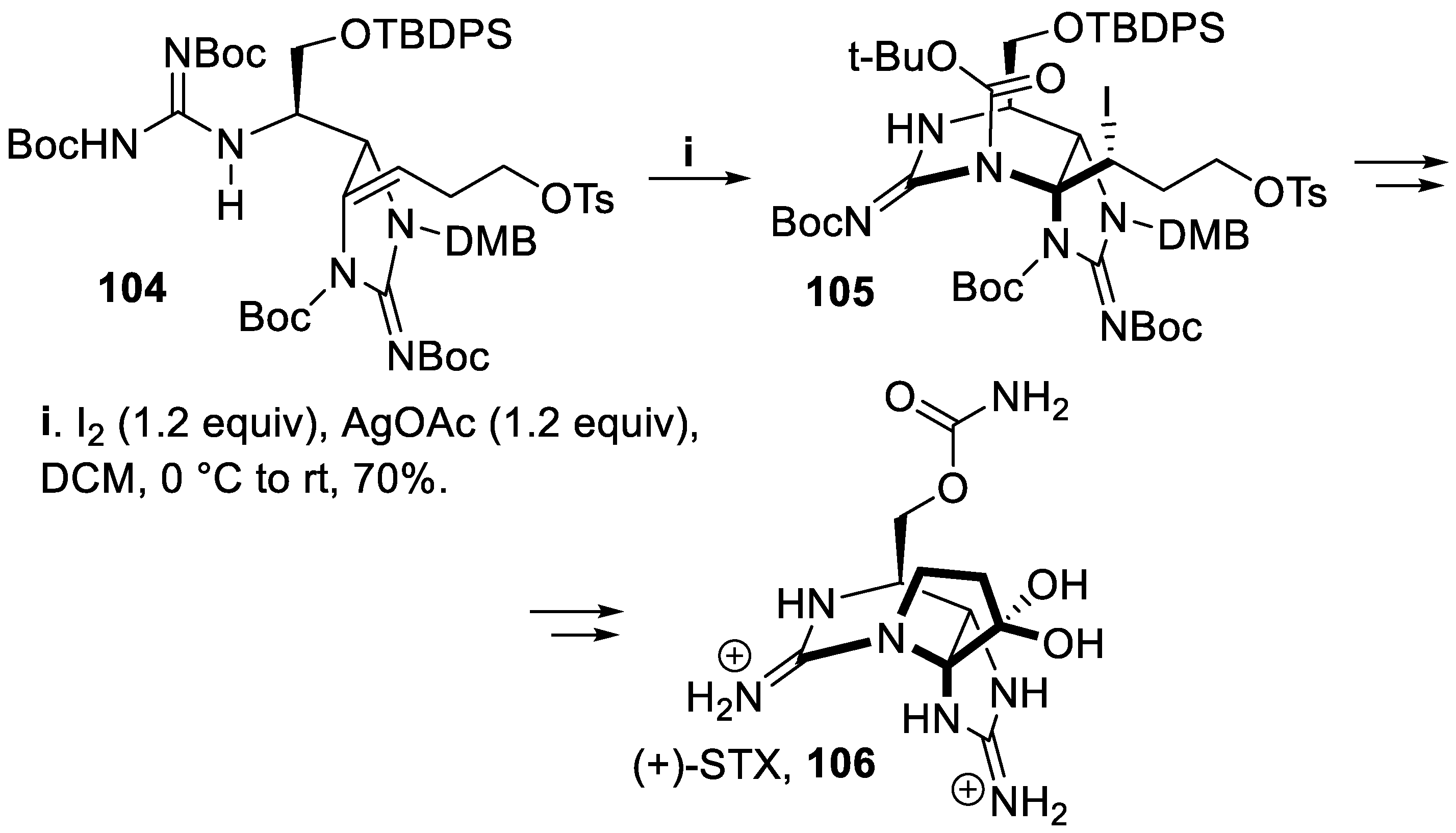
Scheme 27.
Stereoselective synthesis of the bicyclic lactams 108.
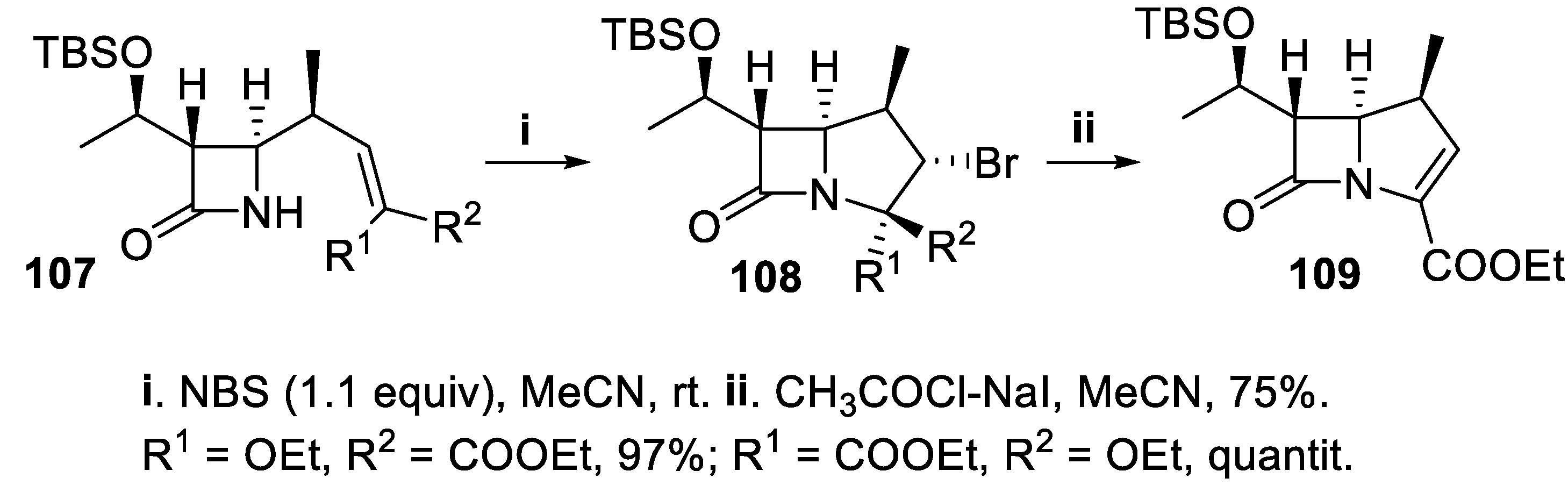
Scheme 28.
Stereoselective synthesis of bicyclic lactams 111 and 112.
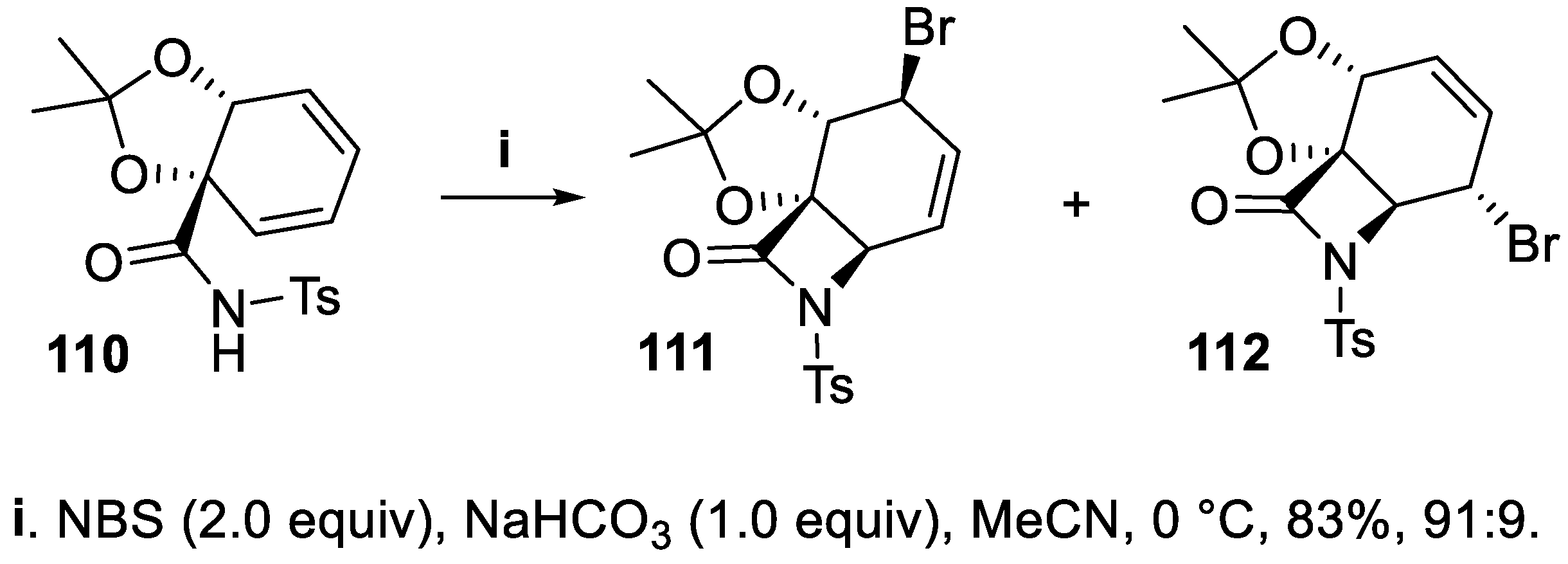
Scheme 29.
Stereodifferentiation of imides (S,R)- and (S,S)-113.
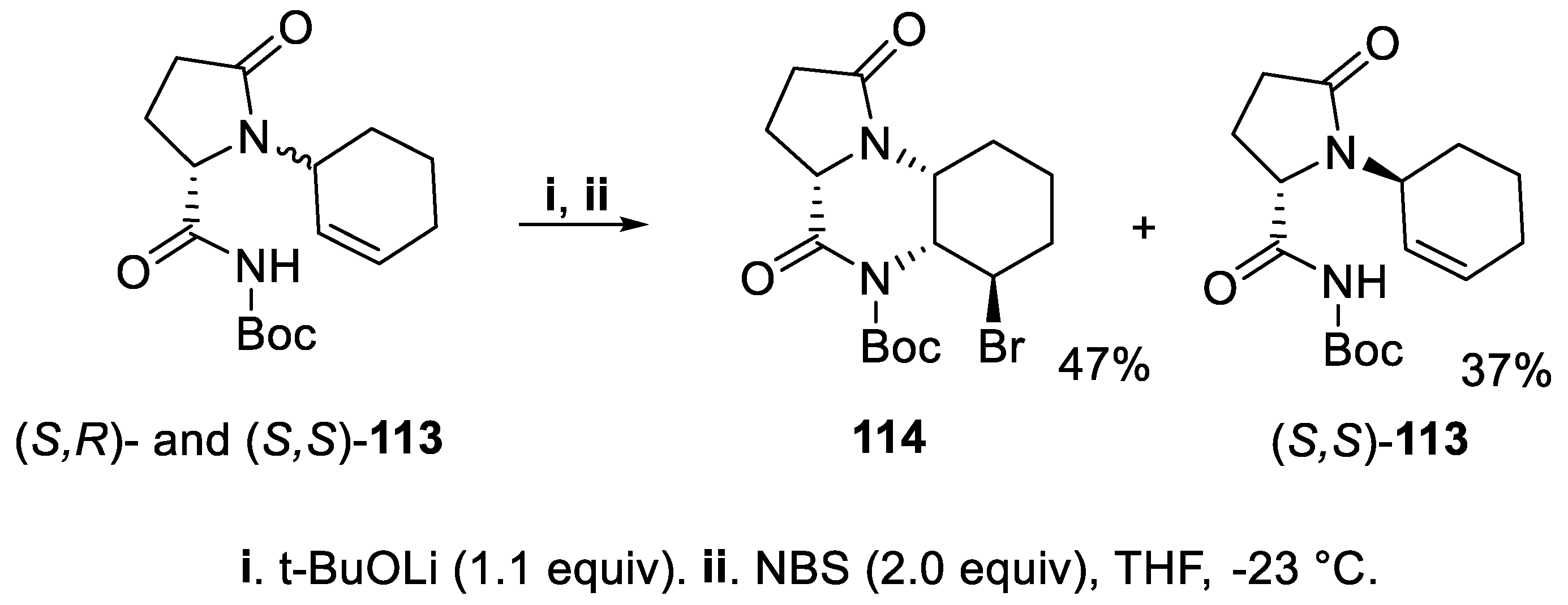
Scheme 30.
Stereodivergent transannular bromoamidation leading to dipeptide mimetics 116 or 117.
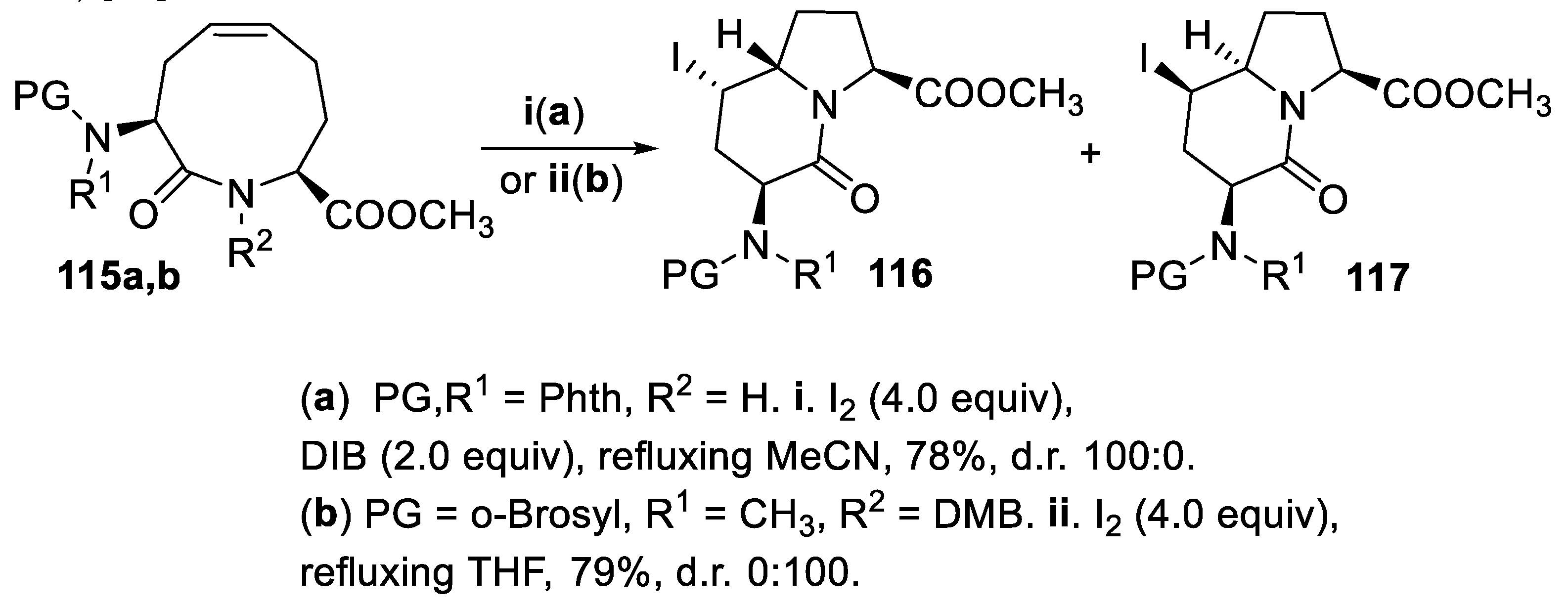
Scheme 31.
Intramolecular cyclization leading to 121, key intermediate to alkaloid (-)-dysiherbaine, 122.
Scheme 31.
Intramolecular cyclization leading to 121, key intermediate to alkaloid (-)-dysiherbaine, 122.
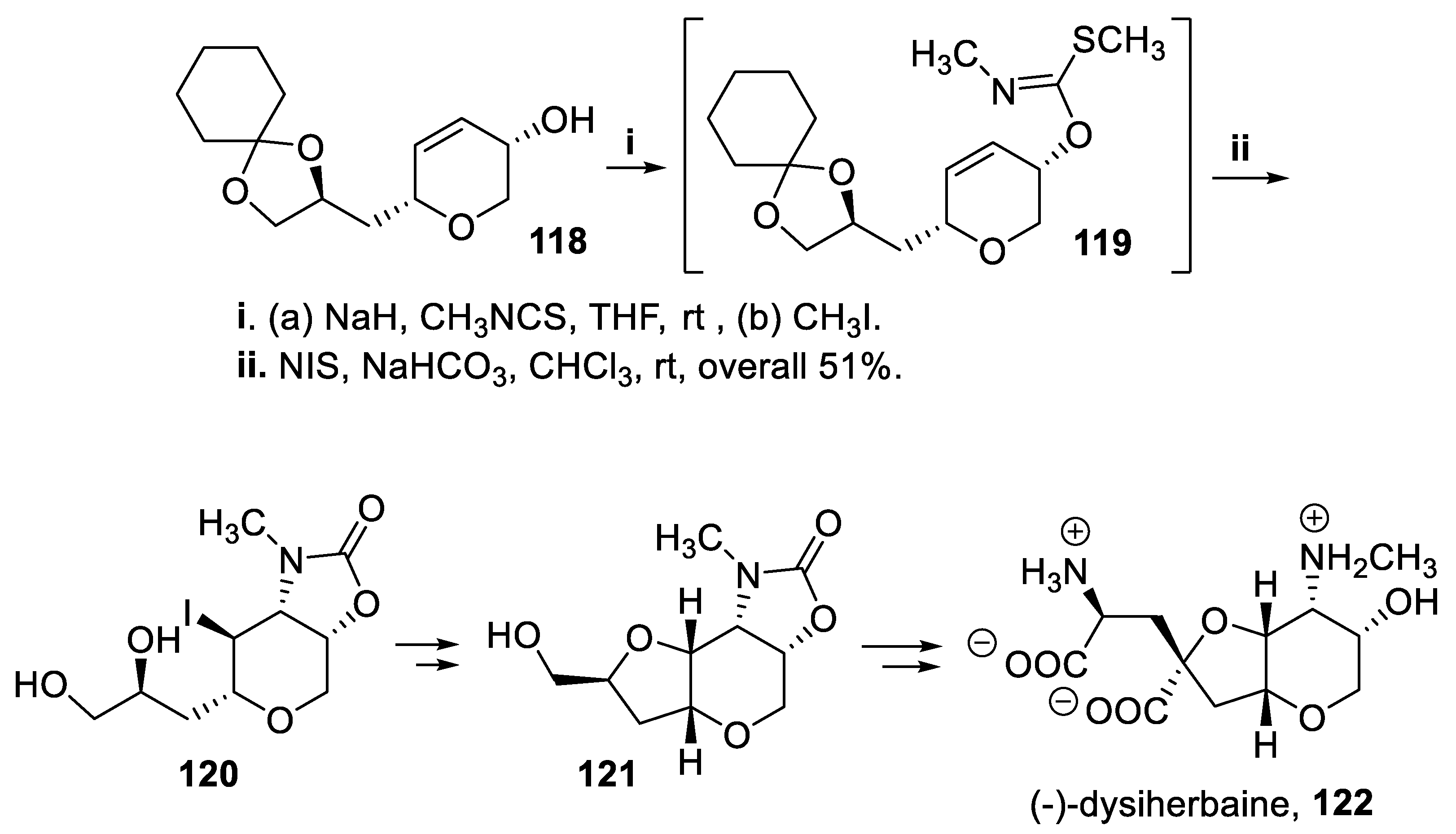
Scheme 32.
Synthesis of (+)-polyoxamic acid 125 and lactone 126 starting from imidate 123.
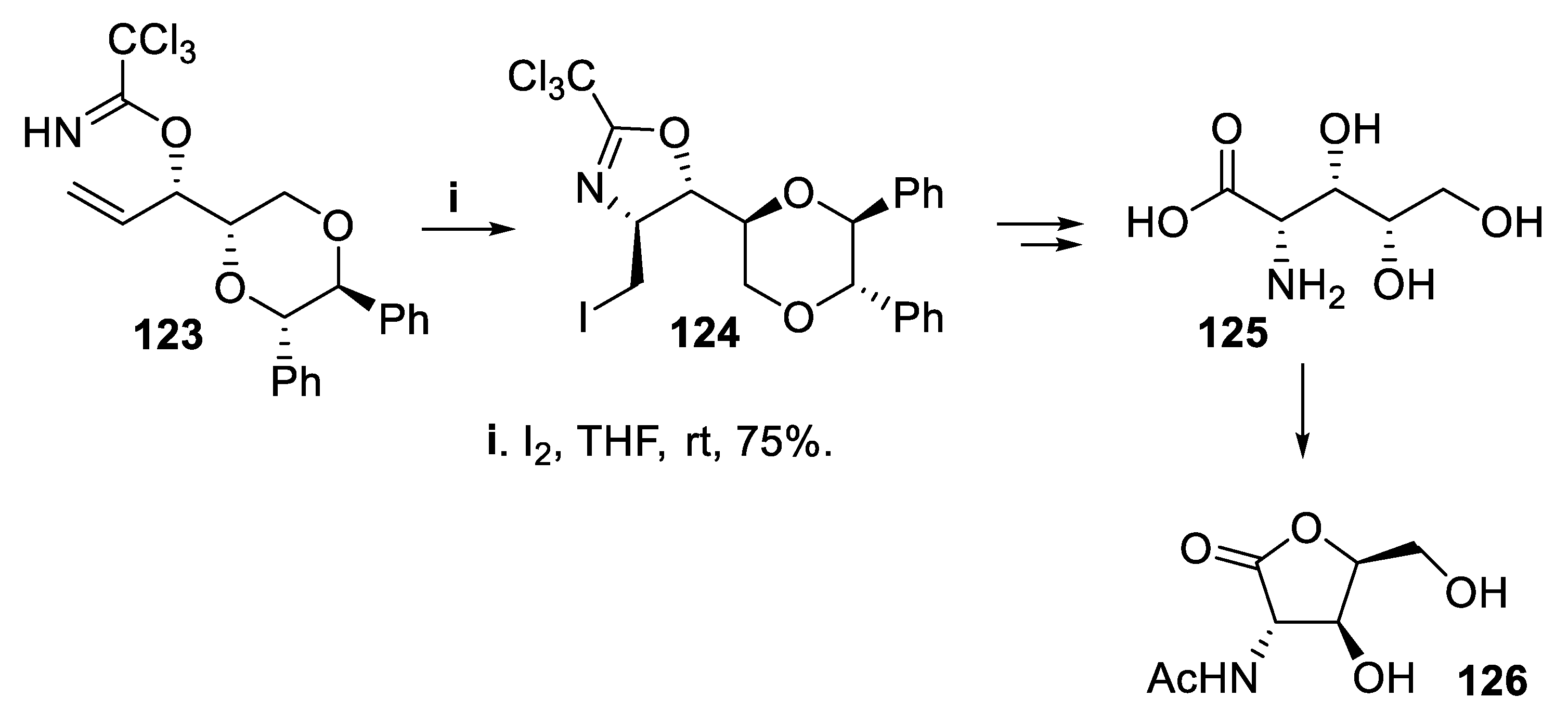
Scheme 33.
Desymmetrization via bromoamination of chiral imidazolidine 129 leading to (-)-γ-licorane 133.
Scheme 33.
Desymmetrization via bromoamination of chiral imidazolidine 129 leading to (-)-γ-licorane 133.
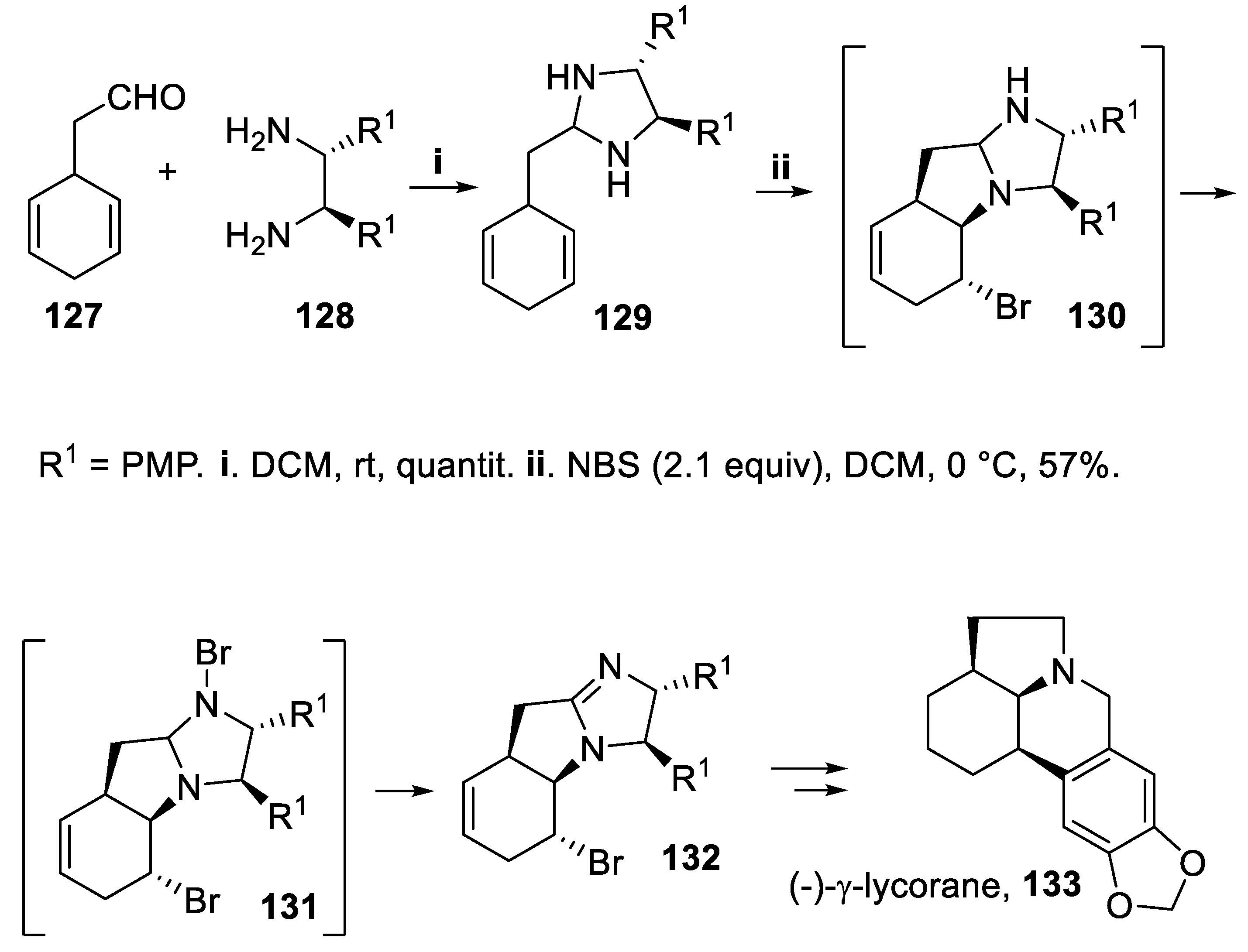
Scheme 34.
Bromocyclization leading to 3-bromopyrrolidine derivatives 134 exploiting catalyst 136.
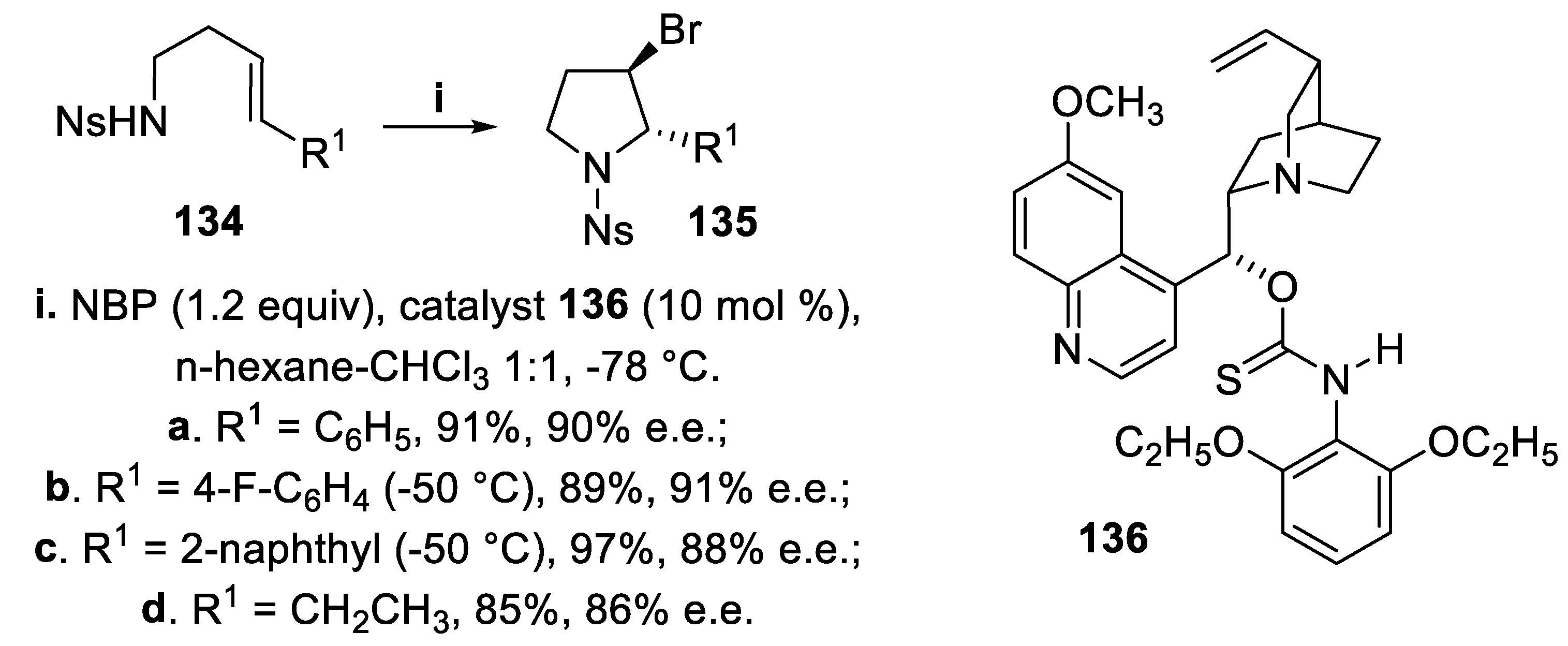
Scheme 35.
Synthesis of pyrrolidine 138, a component of the selective KV1.5 blocker BMS-394136.
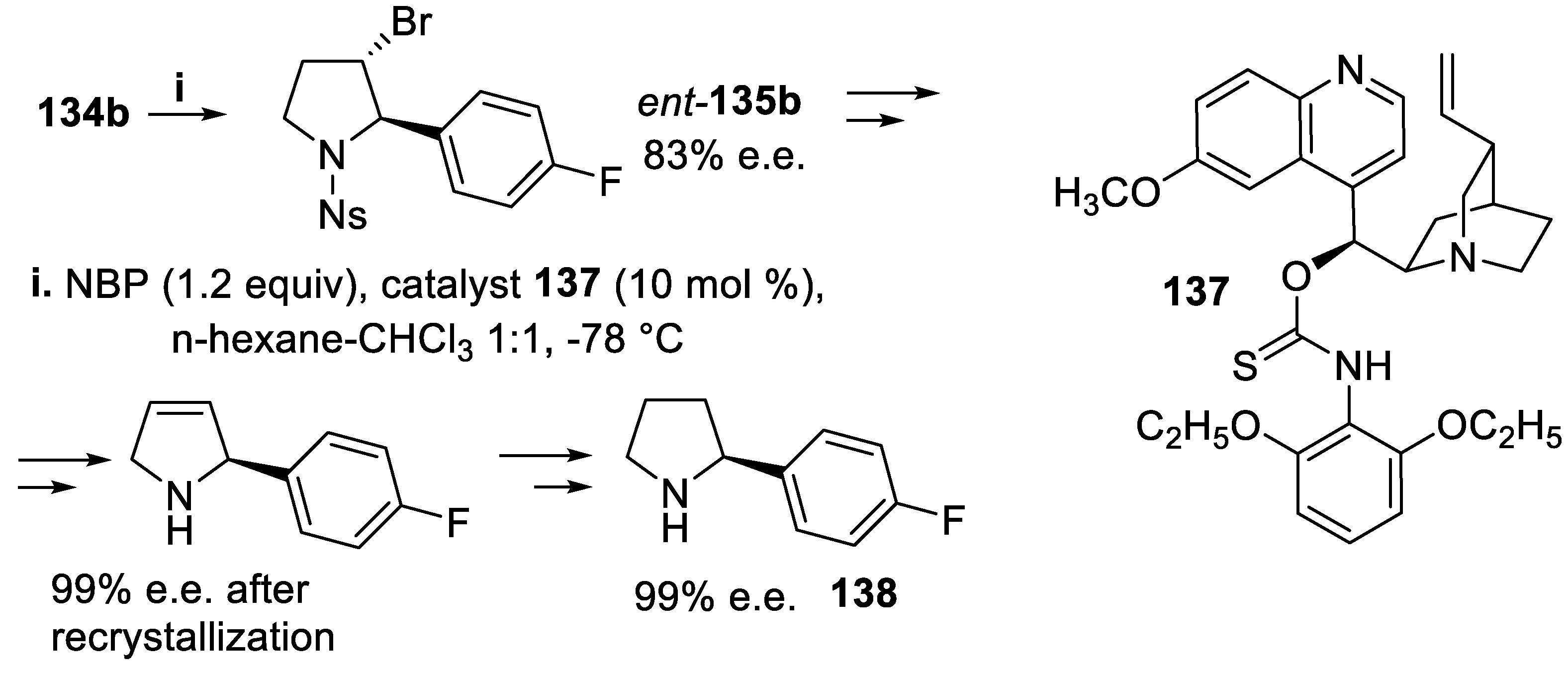
Scheme 36.
Regiodivergent cyclization of tosylamide 139 in the presence of catalyst 142 due to the added halide.
Scheme 36.
Regiodivergent cyclization of tosylamide 139 in the presence of catalyst 142 due to the added halide.
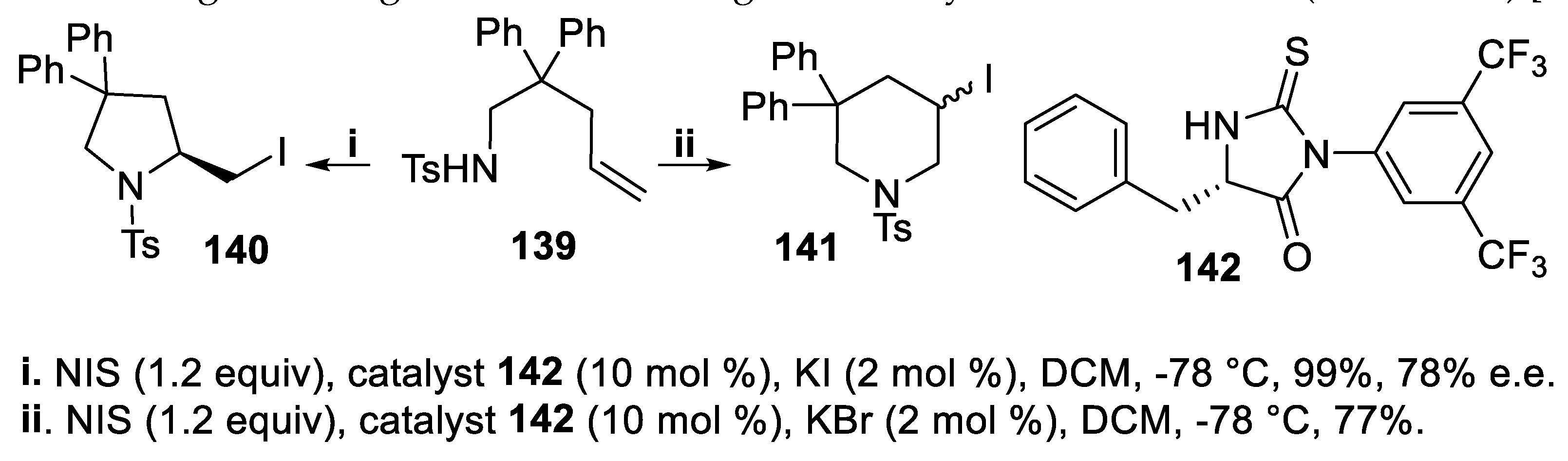
Scheme 37.
Stereodivergent synthesis of 2-iodomethyl indolines 144 and 145.
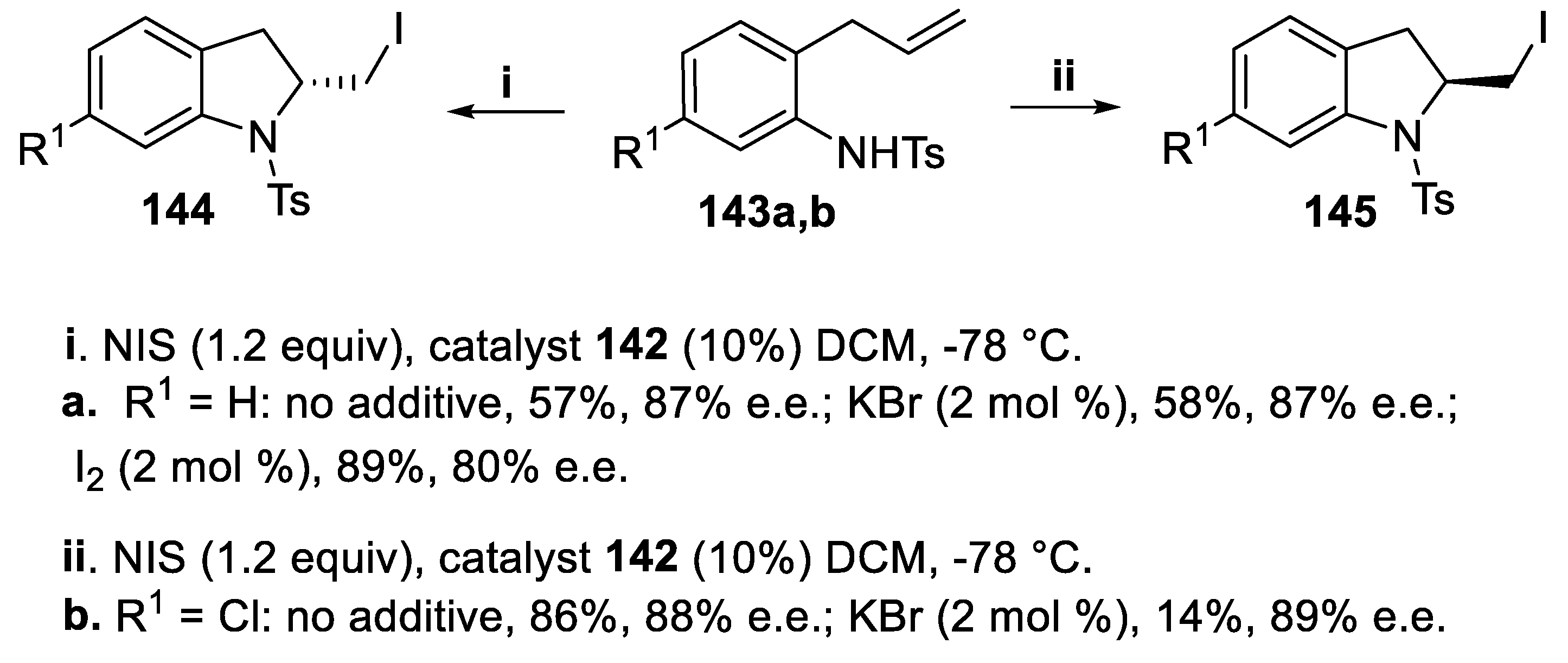
Scheme 38.
Synthesis of 2-bromomethyl indolines 147 mediated by catalyst 148.
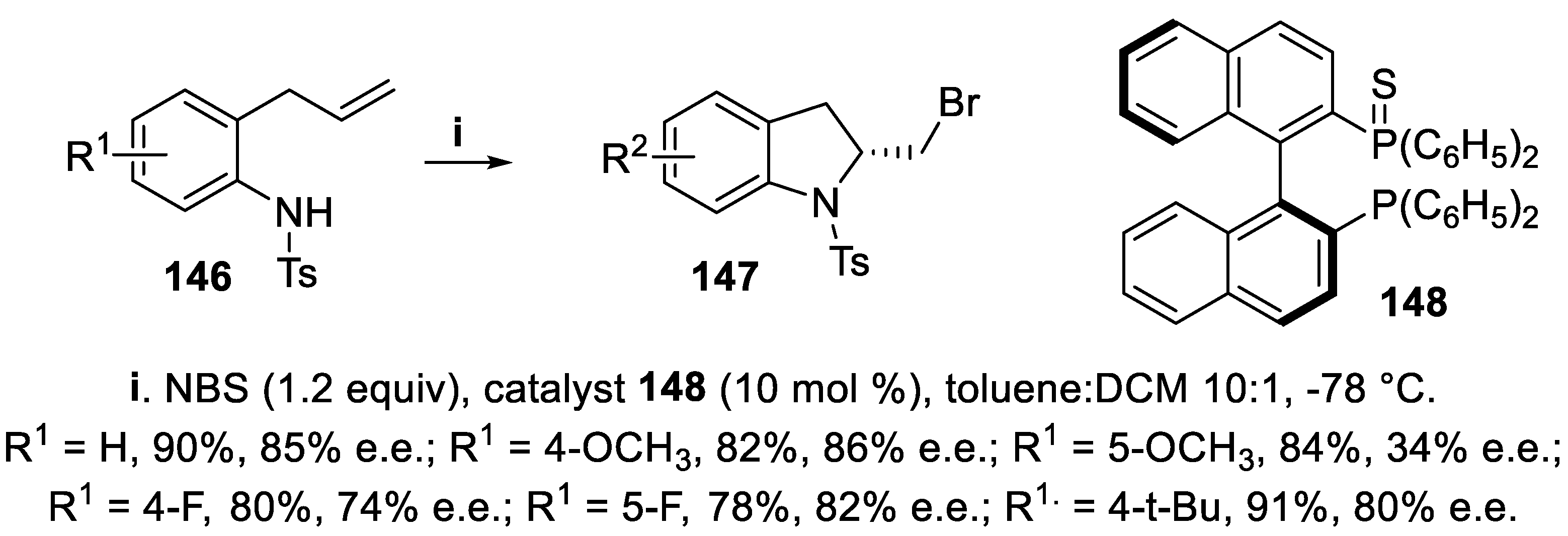
Scheme 39.
Synthesis of 2-iodomethylpyrrolidine derivatives 18 mediated by catalyst 10.
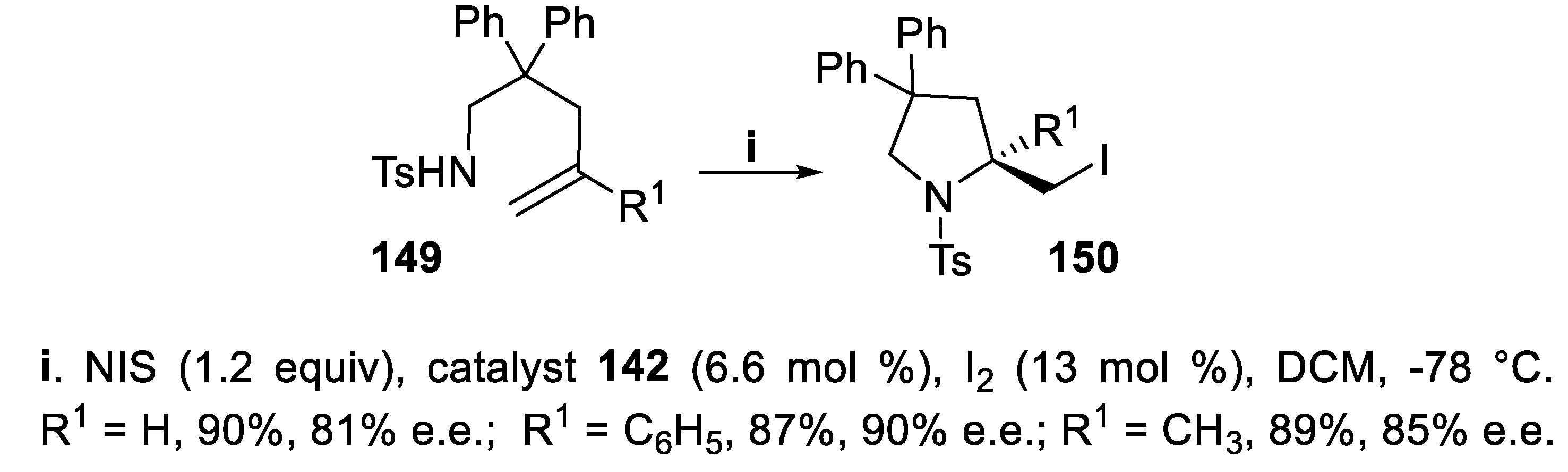
Scheme 40.
Synthesis of 2-bromomethylpyrrolidine derivatives 152 bearing a chiral quaternary center at C-2.
Scheme 40.
Synthesis of 2-bromomethylpyrrolidine derivatives 152 bearing a chiral quaternary center at C-2.
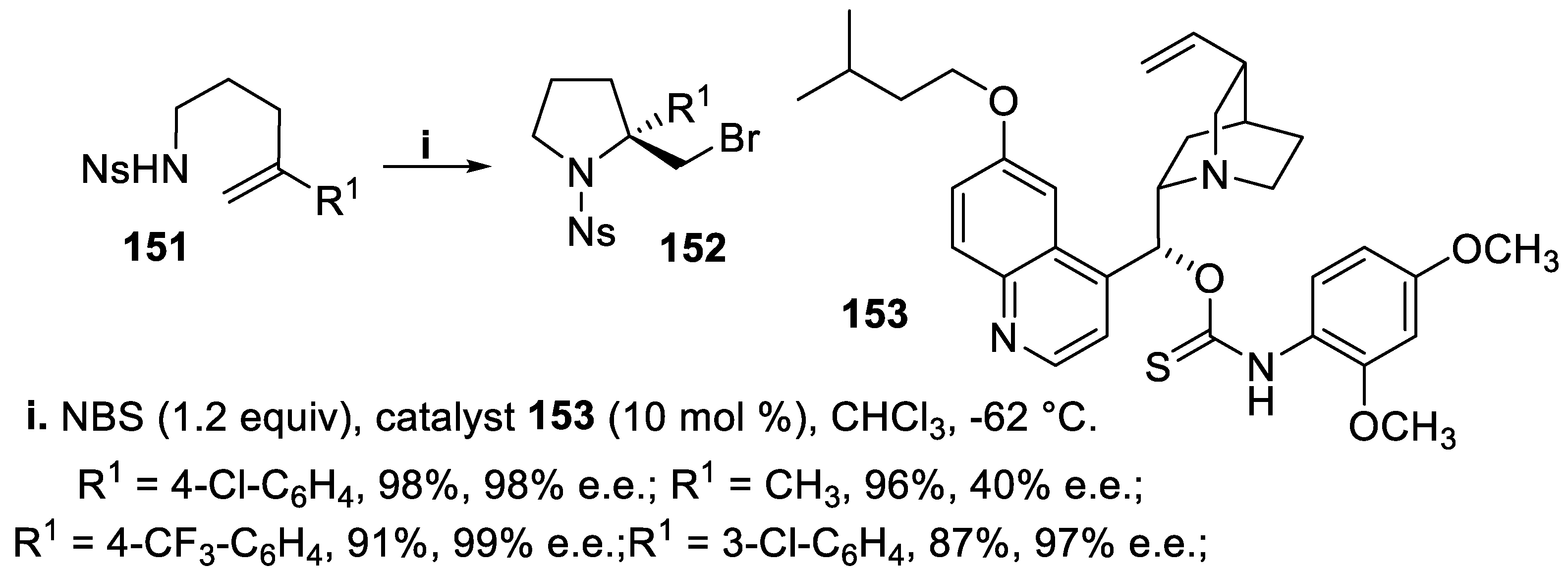
Scheme 41.
Synthesis of chiral isoindolinone 156.
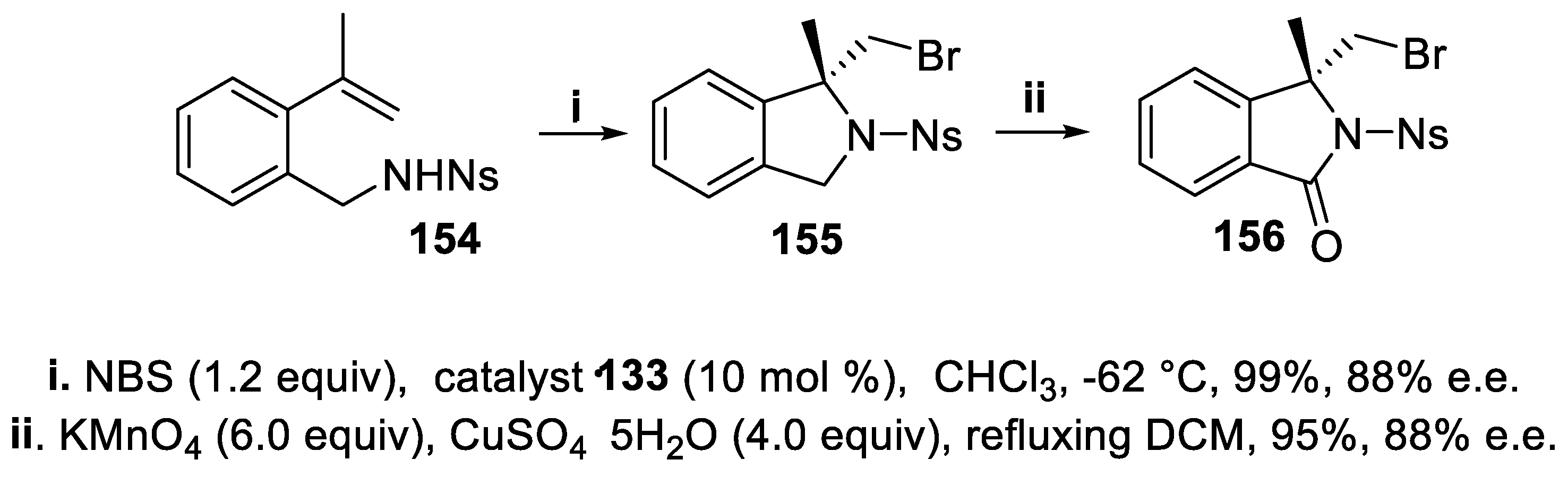
Scheme 42.
Synthesis of 2-iodomethyl pyrrolidine derivatives 158 bearing a chiral quaternary center at C-2.
Scheme 42.
Synthesis of 2-iodomethyl pyrrolidine derivatives 158 bearing a chiral quaternary center at C-2.
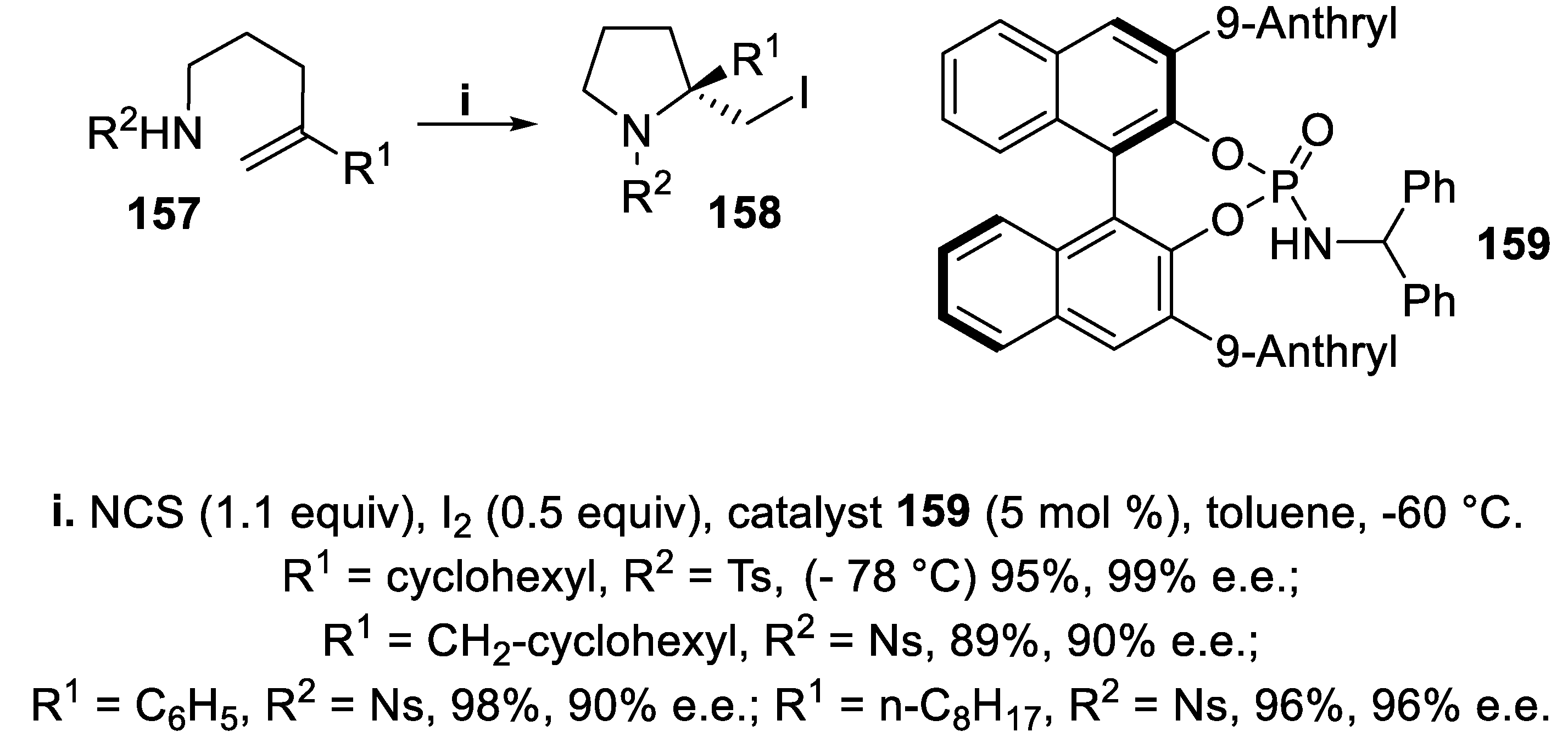
Scheme 43.
Bromocyclization of (Z)-alkenylamides 160 mediated by catalyst 162.
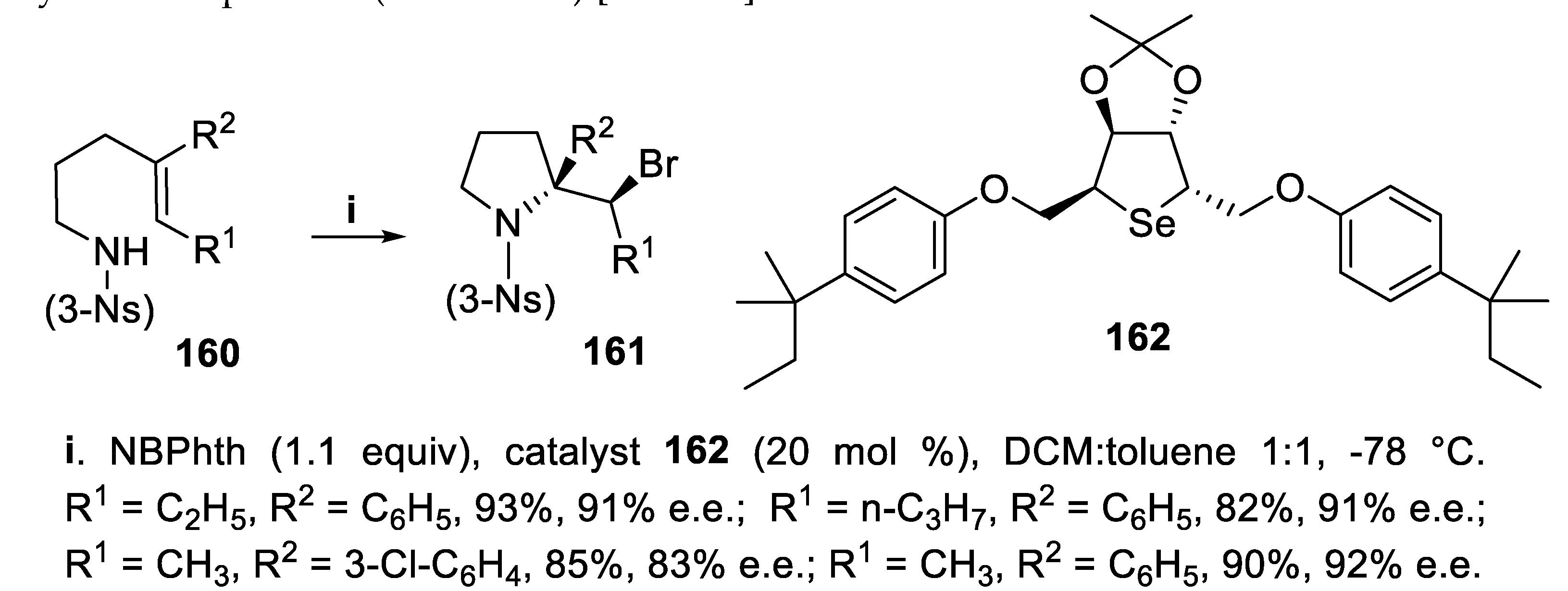
Scheme 44.
Bromocyclization of (Z)-alkenylamides 163 mediated by R-TRIP, 165.
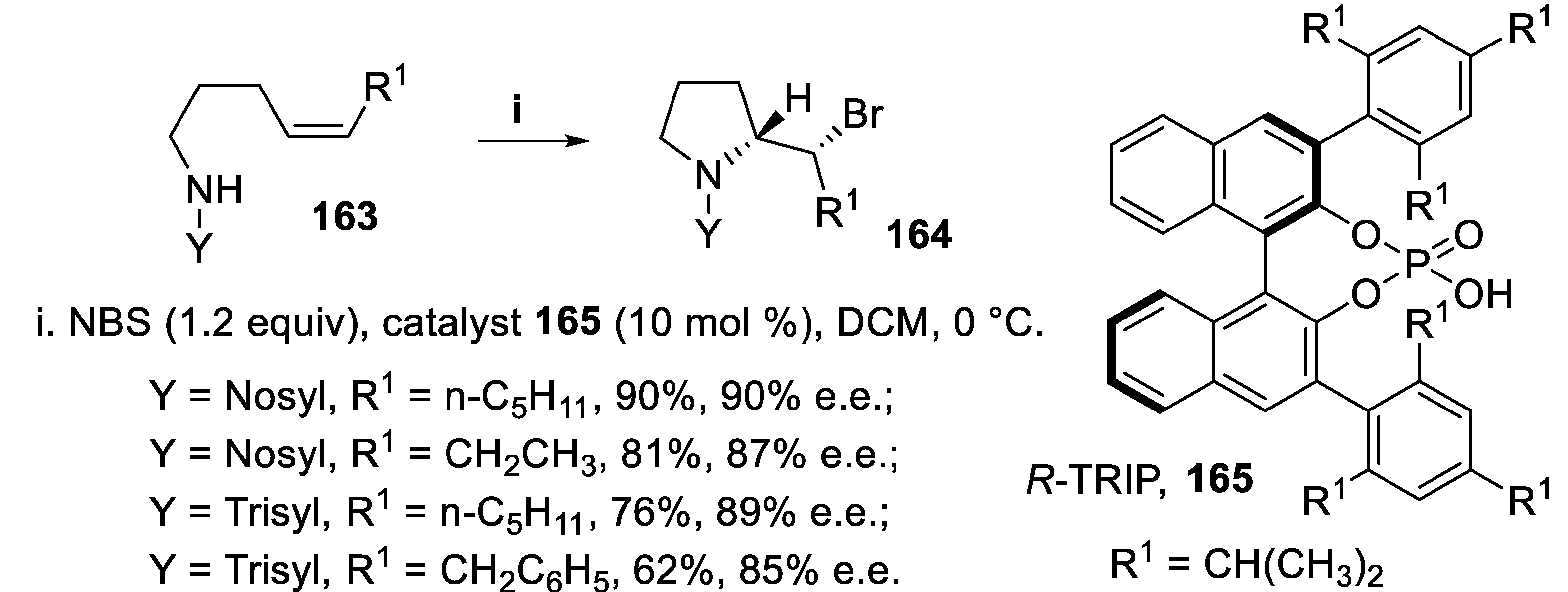
Scheme 45.
Bromocyclization of (E)-alkenylamides 166 mediated by R-TRIP, 165.
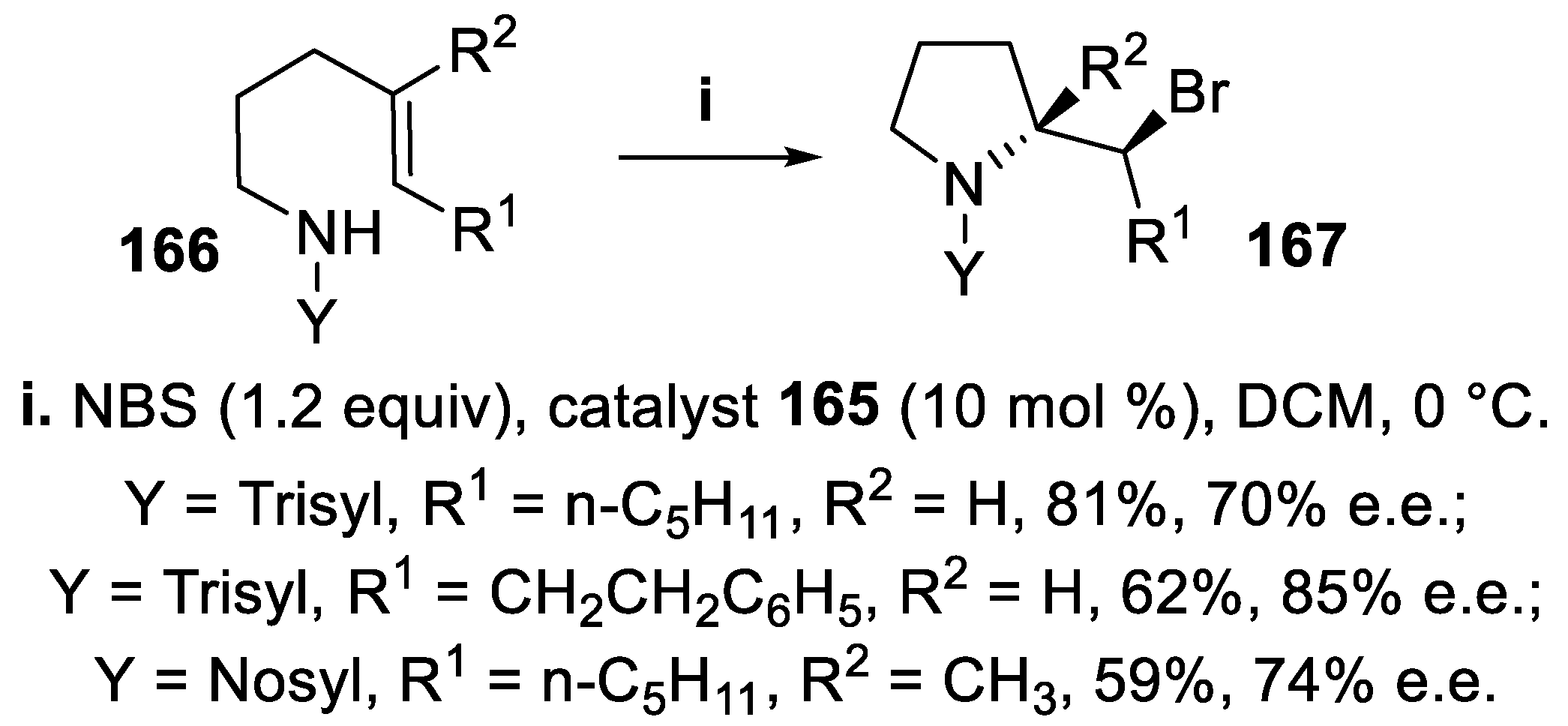
Scheme 46.
Bromocyclization of (E)-alkenylamides 170 mediated by Co(III) complex Δ-(S,S)-168.
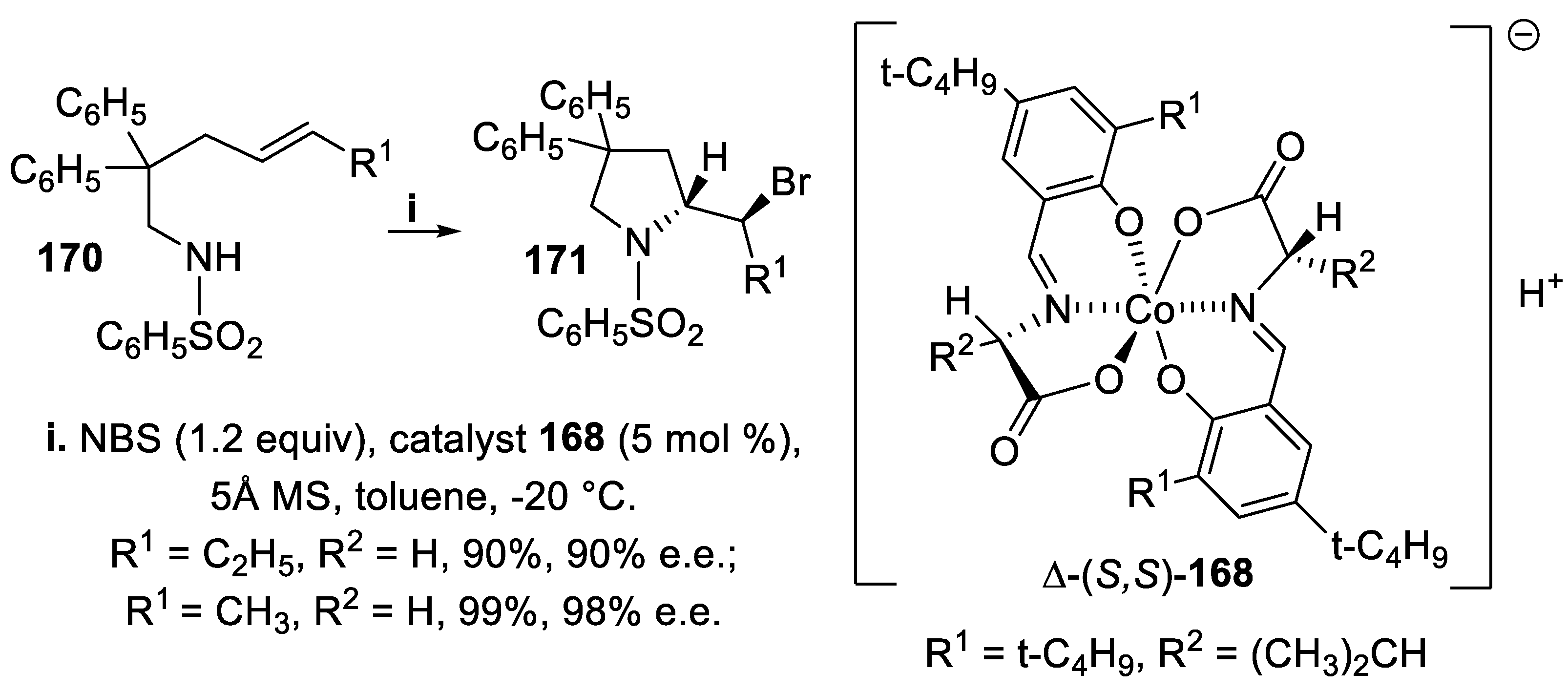
Scheme 47.
Bromocyclization of (Z)-alkenylamides 172 mediated by Co(III) complex Λ-(S,S)-169.
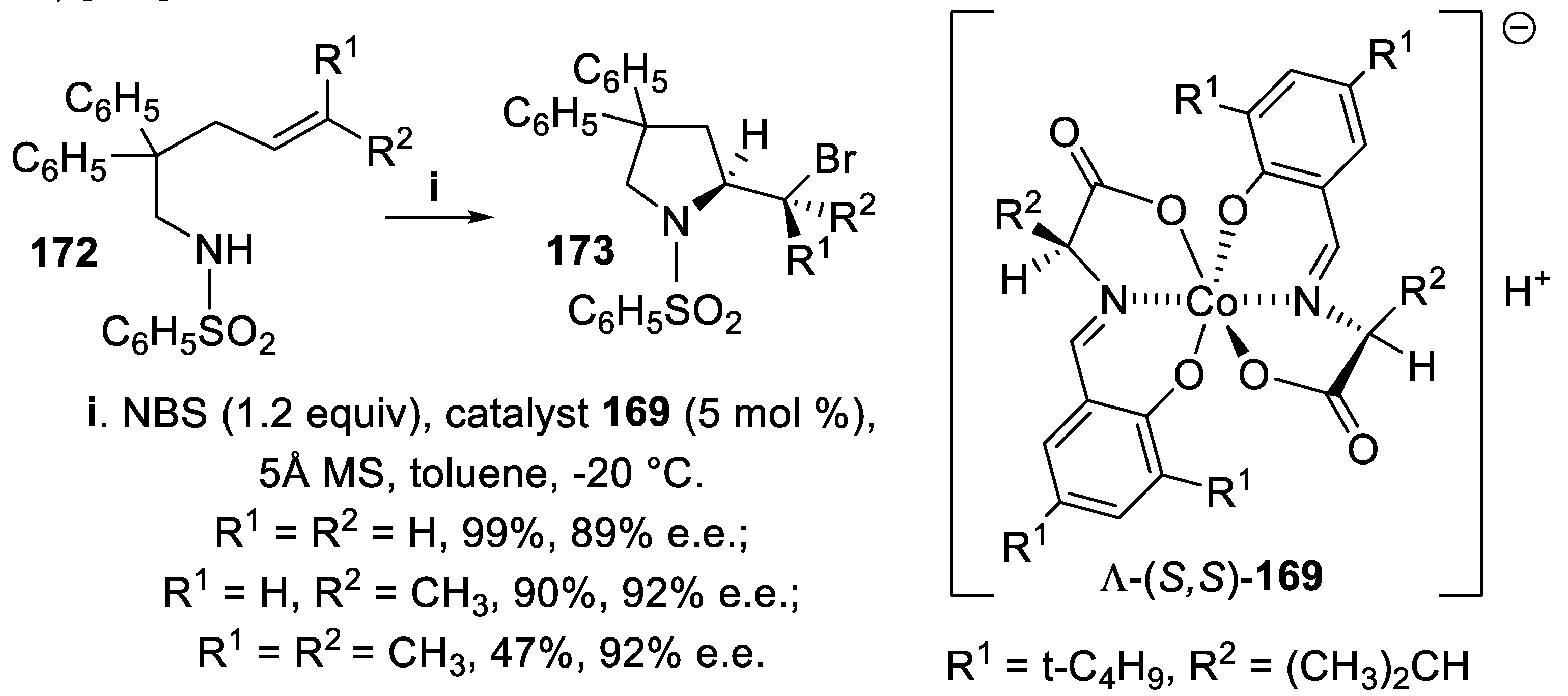
Scheme 48.
Dearomatization by bromoamidation of 174 leading to tricyclic HPI derivatives 175.
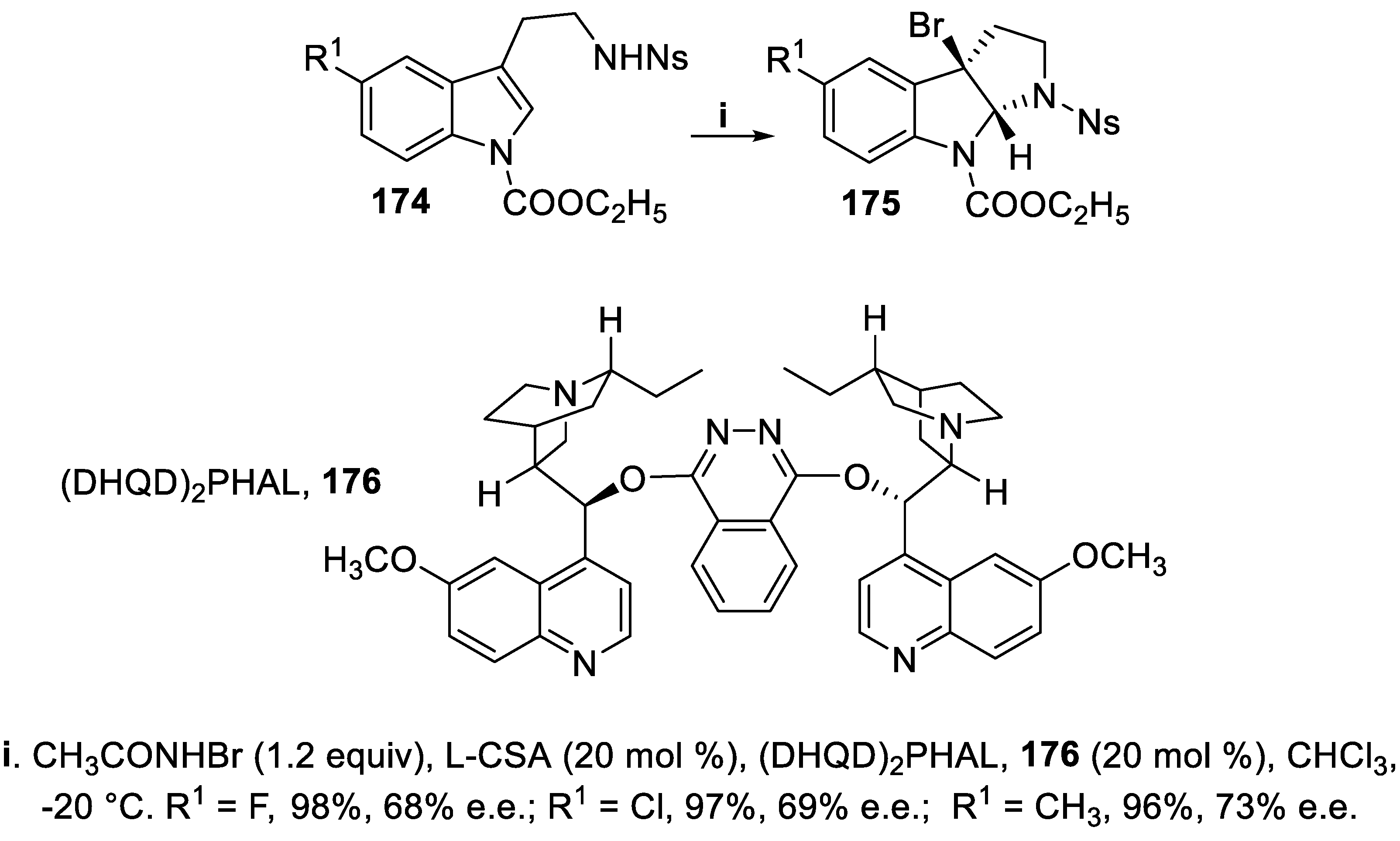
Scheme 49.
Synthesis of tricyclic HPI derivatives 178 mediated by 8H-R-TRIP 180.
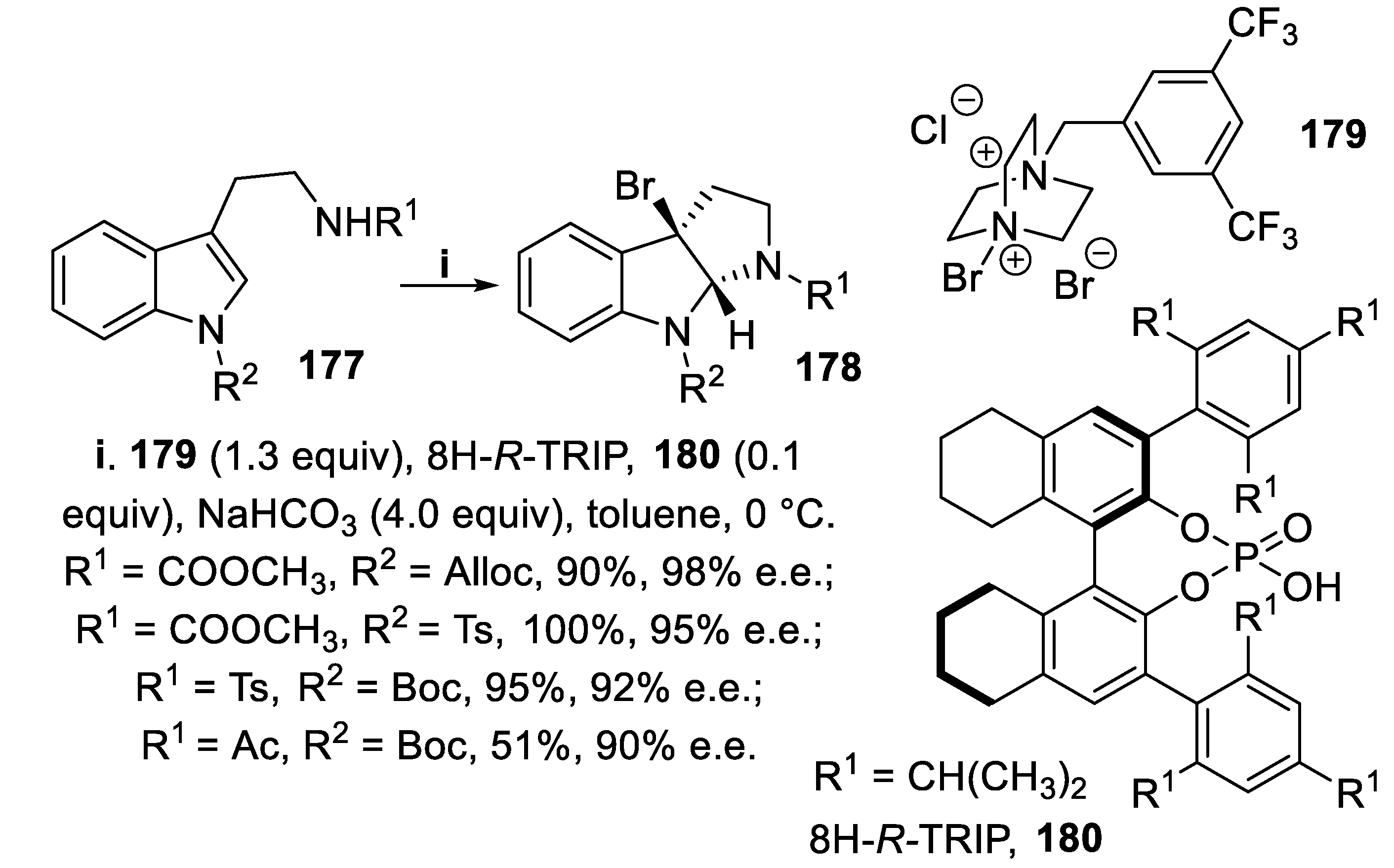
Scheme 50.
Synthesis of tricyclic HPI derivative 182, key intermediate to (-)-chimonantine 183.
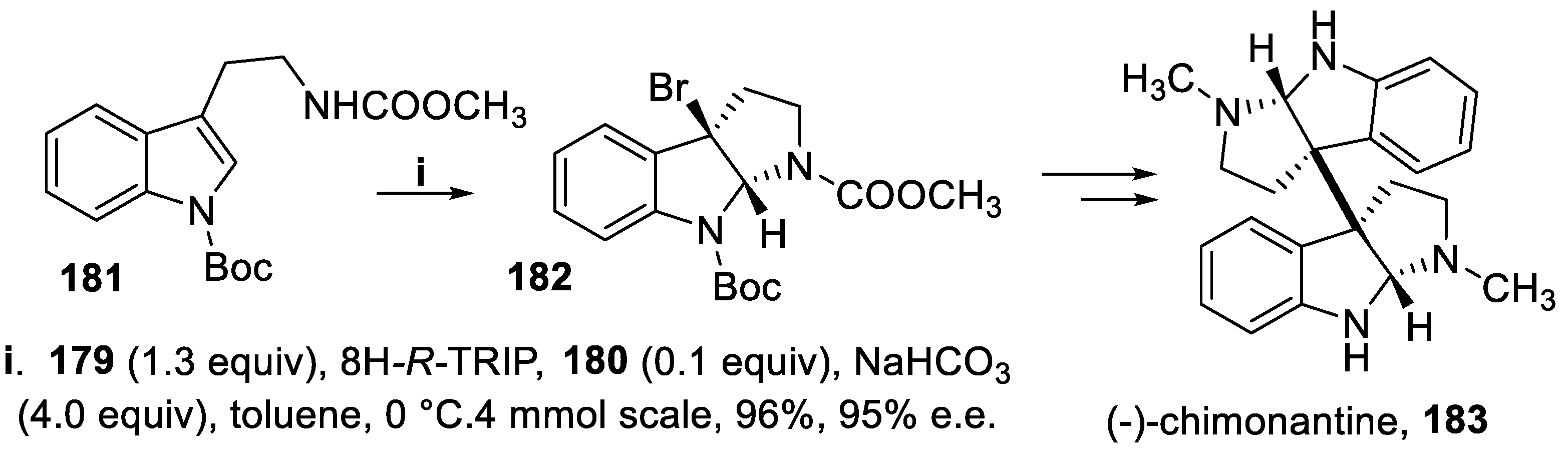
Scheme 51.
Synthesis of tetracyclic HPI derivative 185, intermediate to (-)-conolutinine 186.
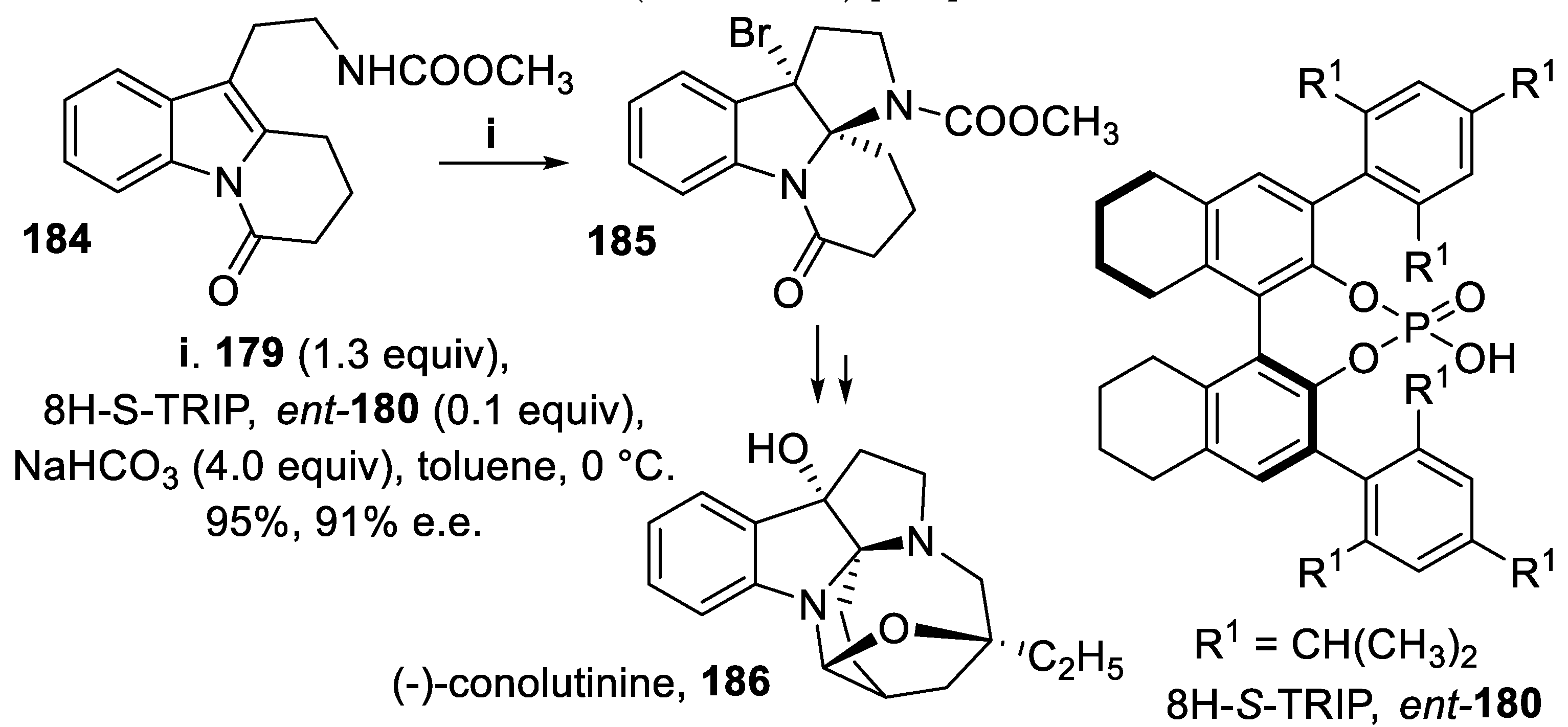
Scheme 52.
Synthesis of tricyclic HPI derivatives 188 in an undivided electrolytic cell.
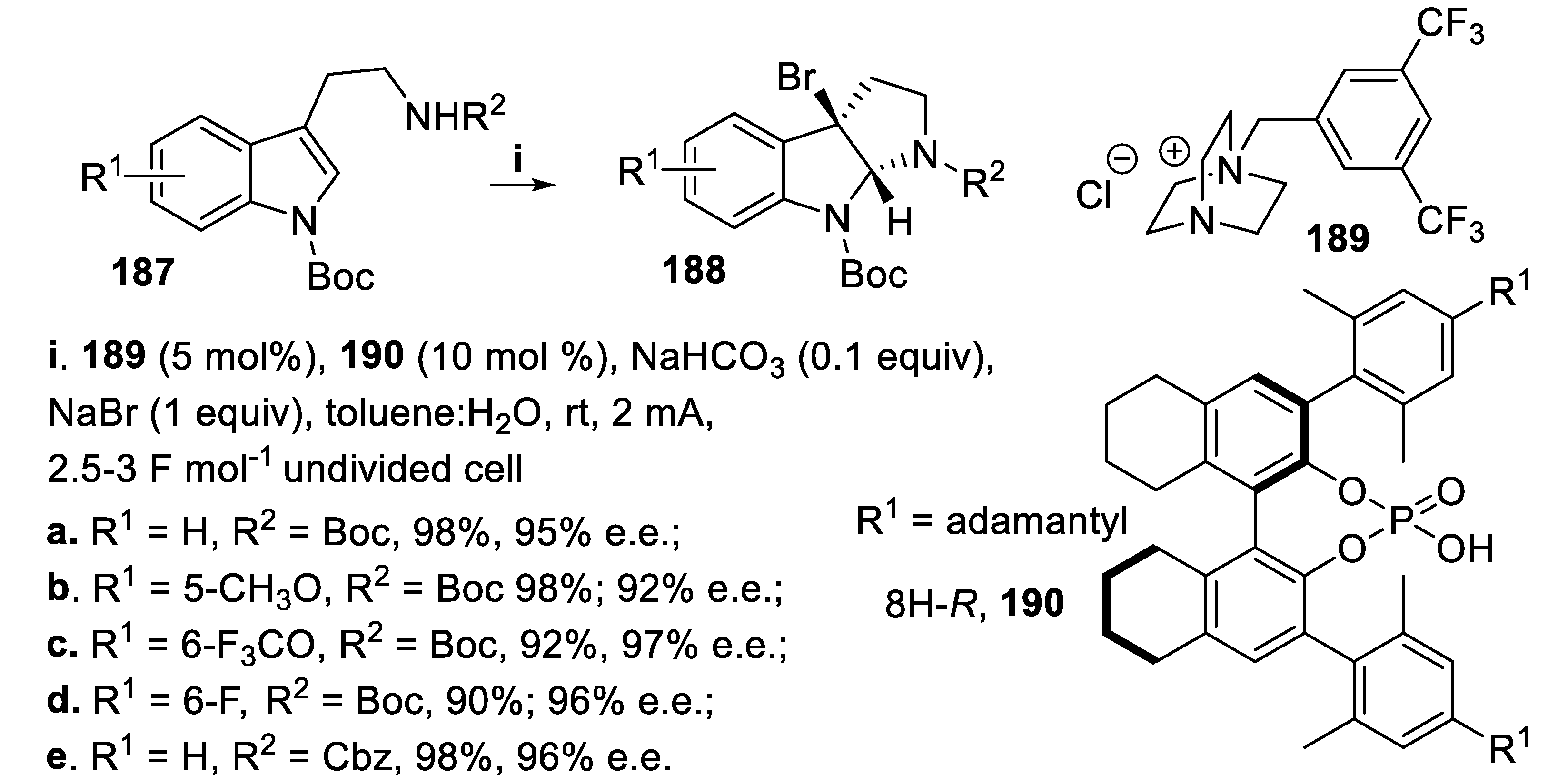
Scheme 53.
Transannular cyclization of sulphonamides 191.
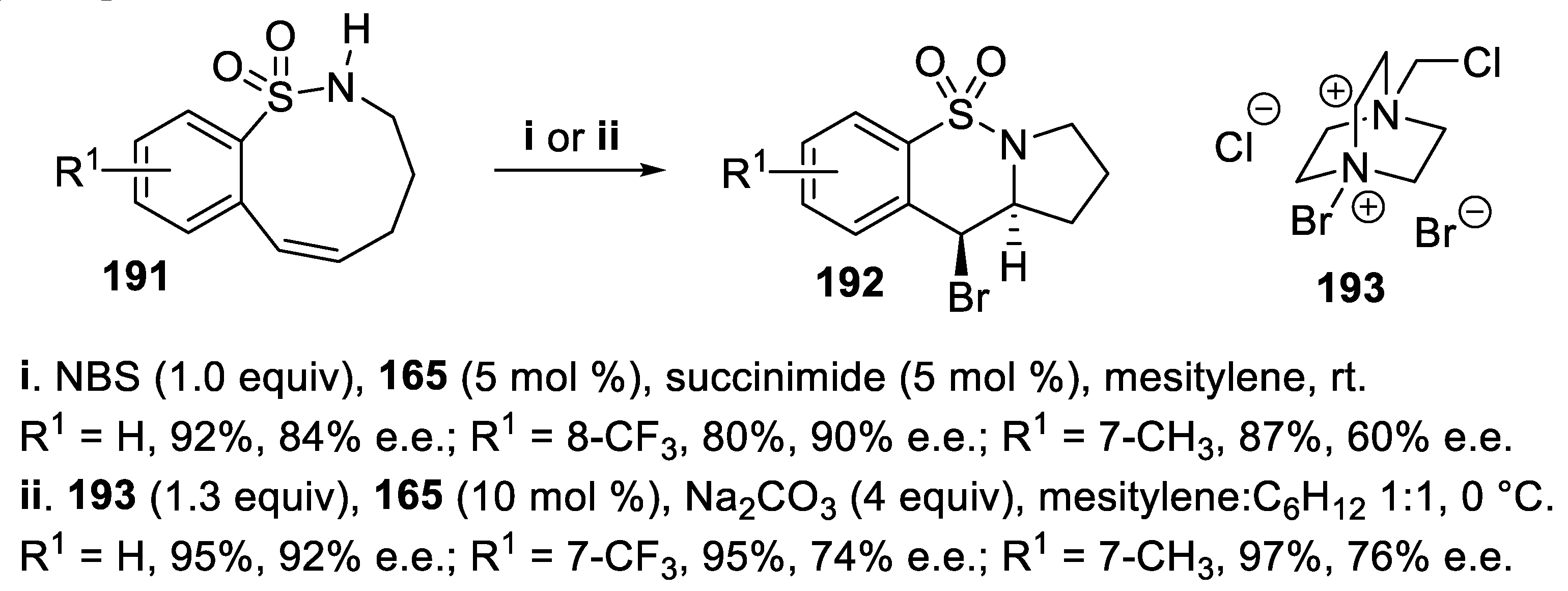
Scheme 54.
Synthesis of HPI derivatives 195a,b mediated by Co(III) complex Λ-(S,S)-168.
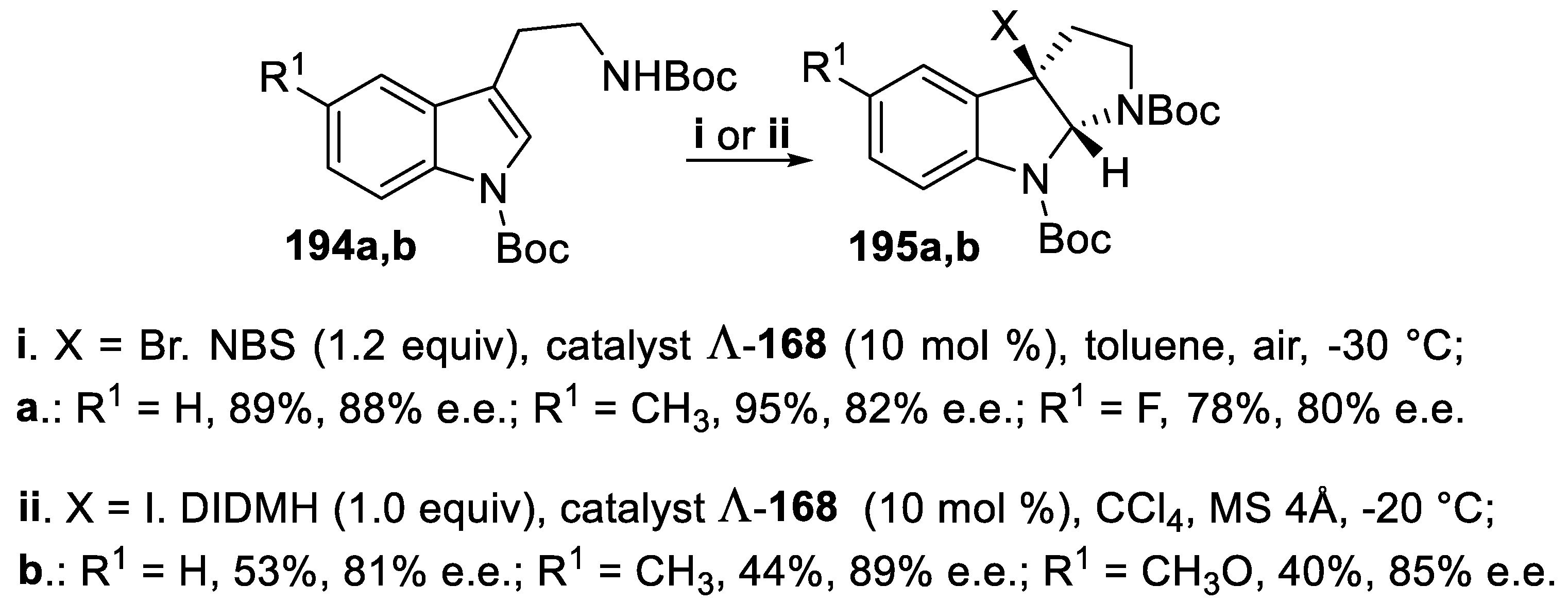
Scheme 55.
Amidocyclization of indene and 1,2-dihydronaphthalene derivatives 196 mediated by TRIP, 165.
Scheme 55.
Amidocyclization of indene and 1,2-dihydronaphthalene derivatives 196 mediated by TRIP, 165.
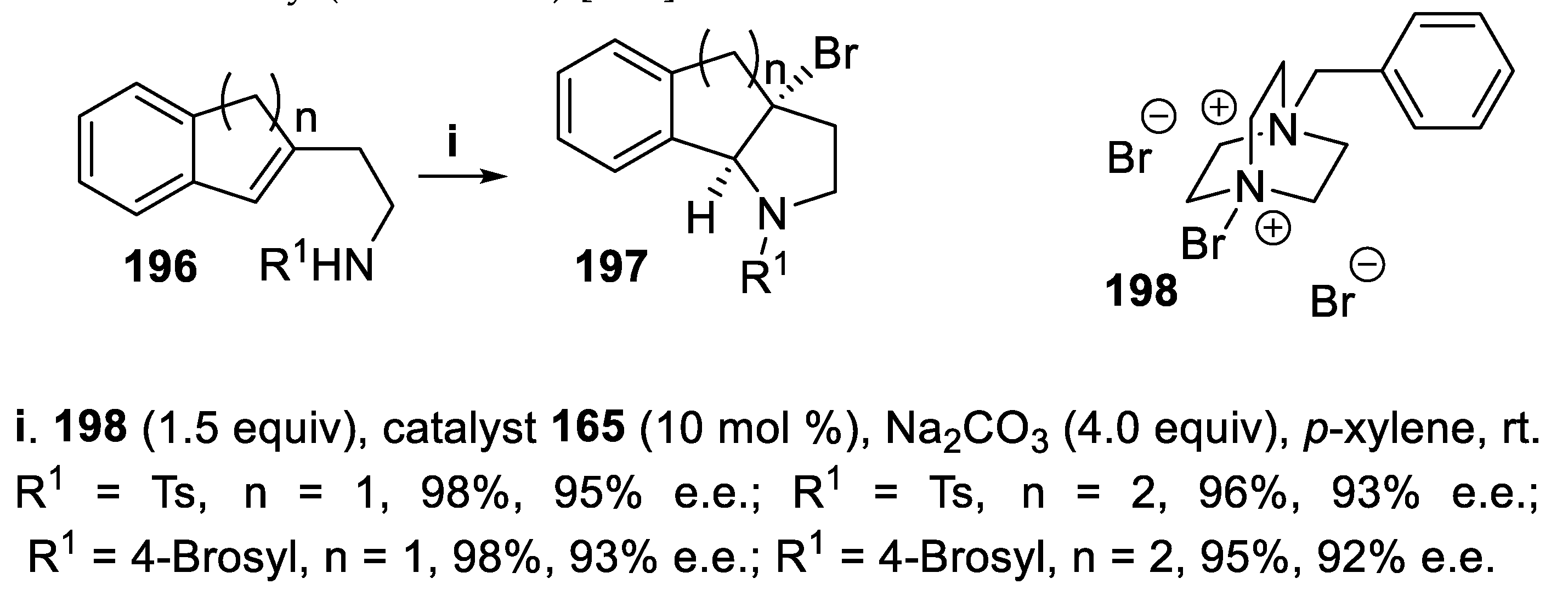
Scheme 56.
Bromocyclization leading to acetyl cholinesterase inhibitor 201 mediated by ent-165.
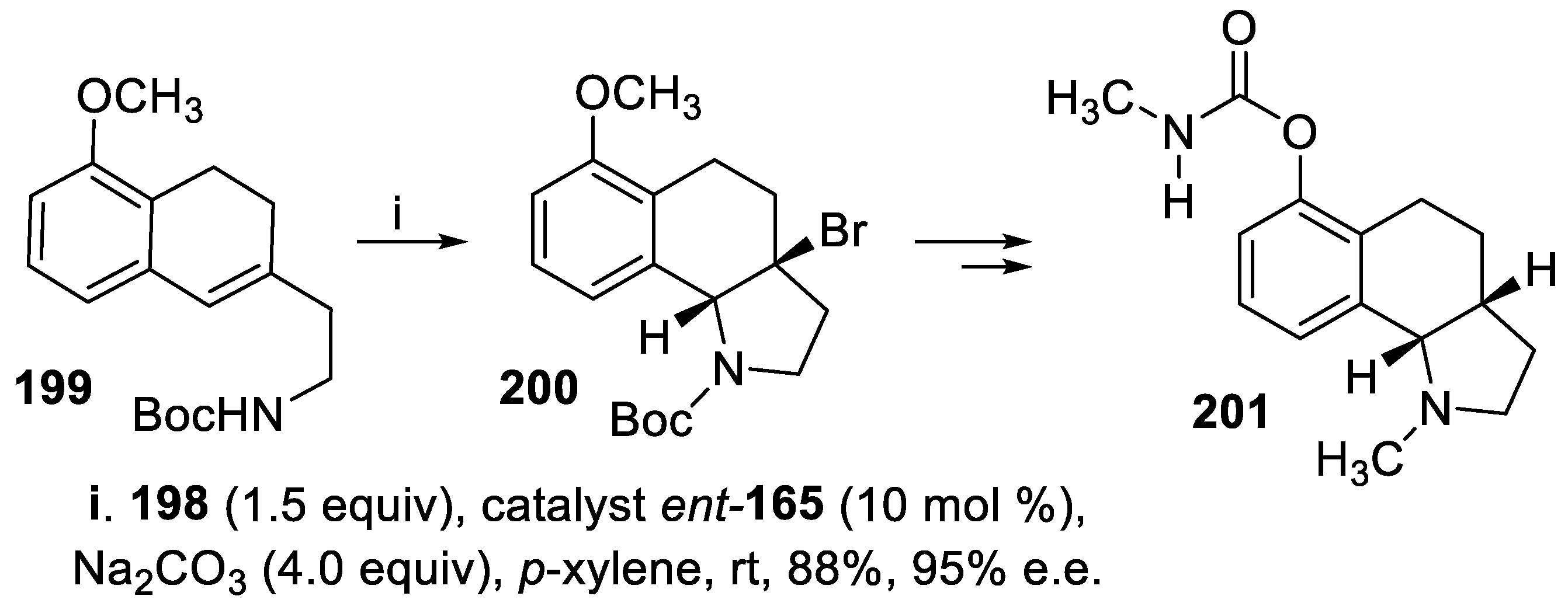
Scheme 57.
Desymmetrization of prochiral cyclohexa-1,4-dienes 202 leading to (+)-mesembrane, 205.
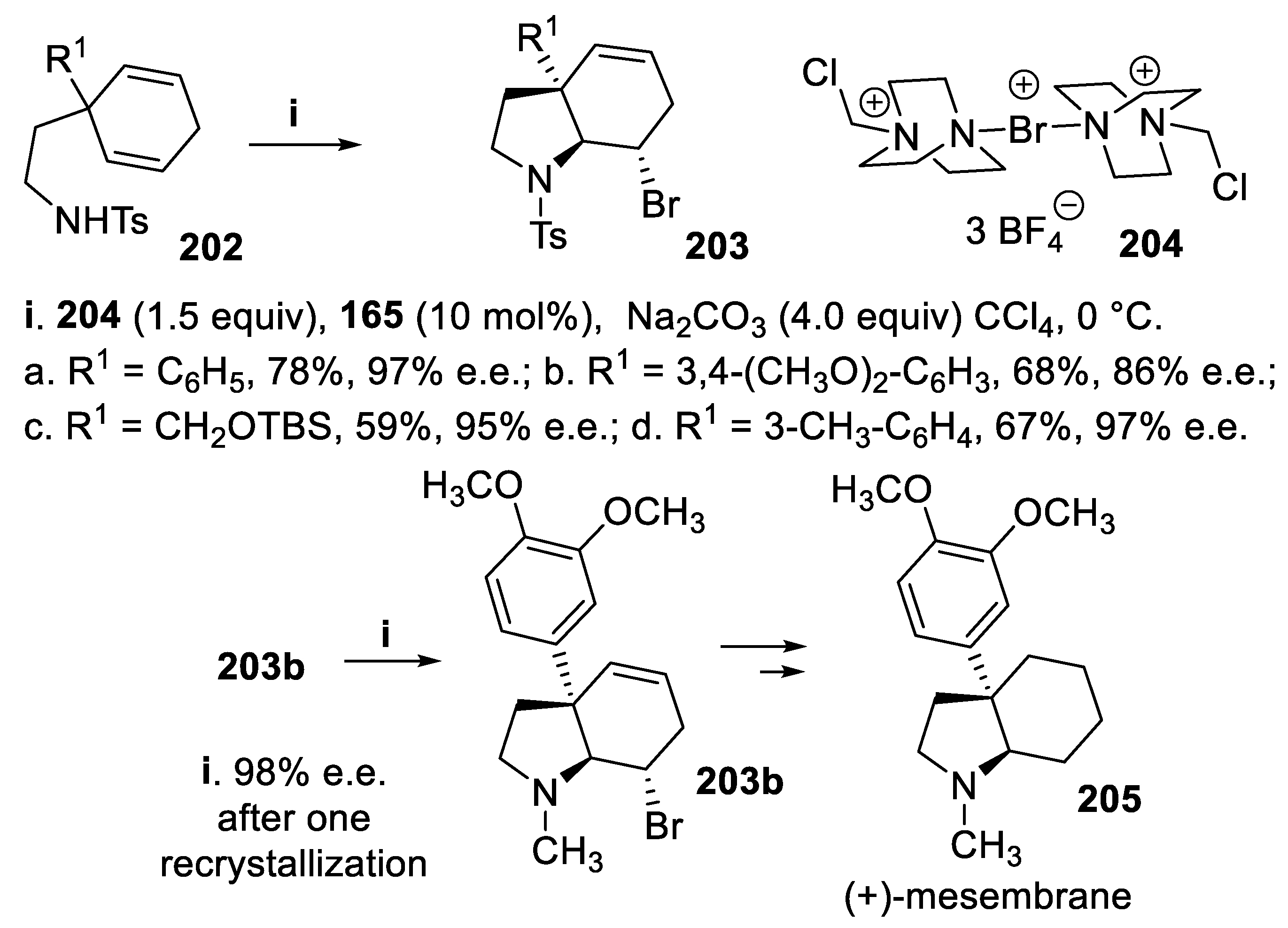
Scheme 58.
Synthesis of 3-bromo piperidine 207 exploiting NBS in the presence of catalyst 208.
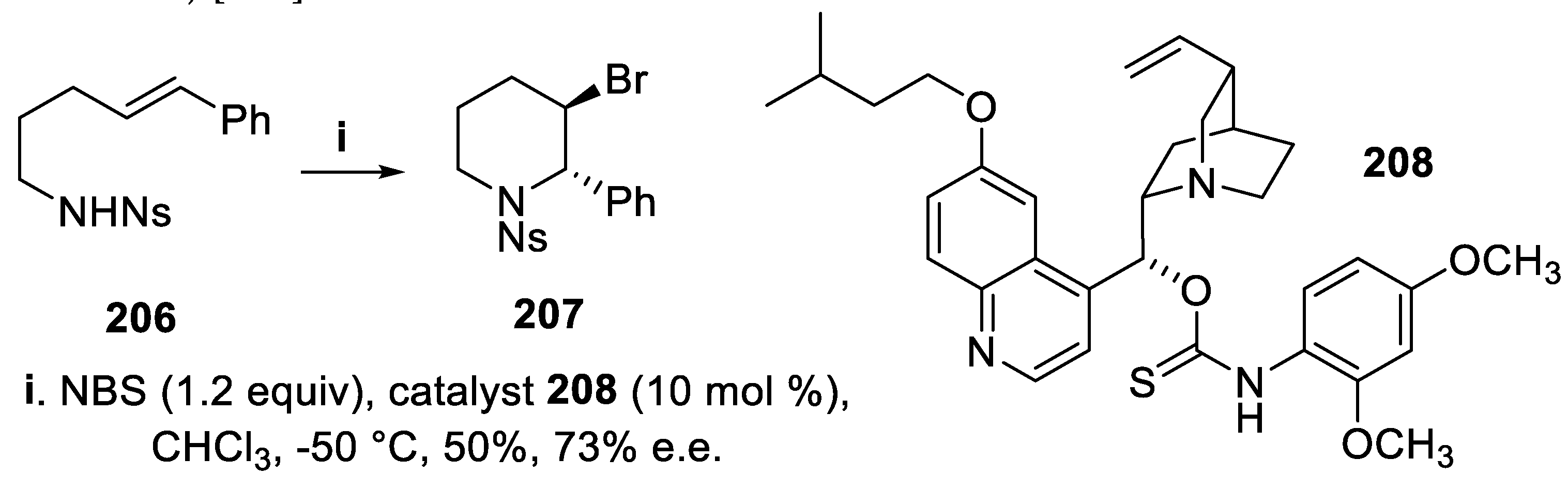
Scheme 59.
Synthesis of 3-bromo piperidines derivatives 210 exploiting DBDMH and the catalyst 208.
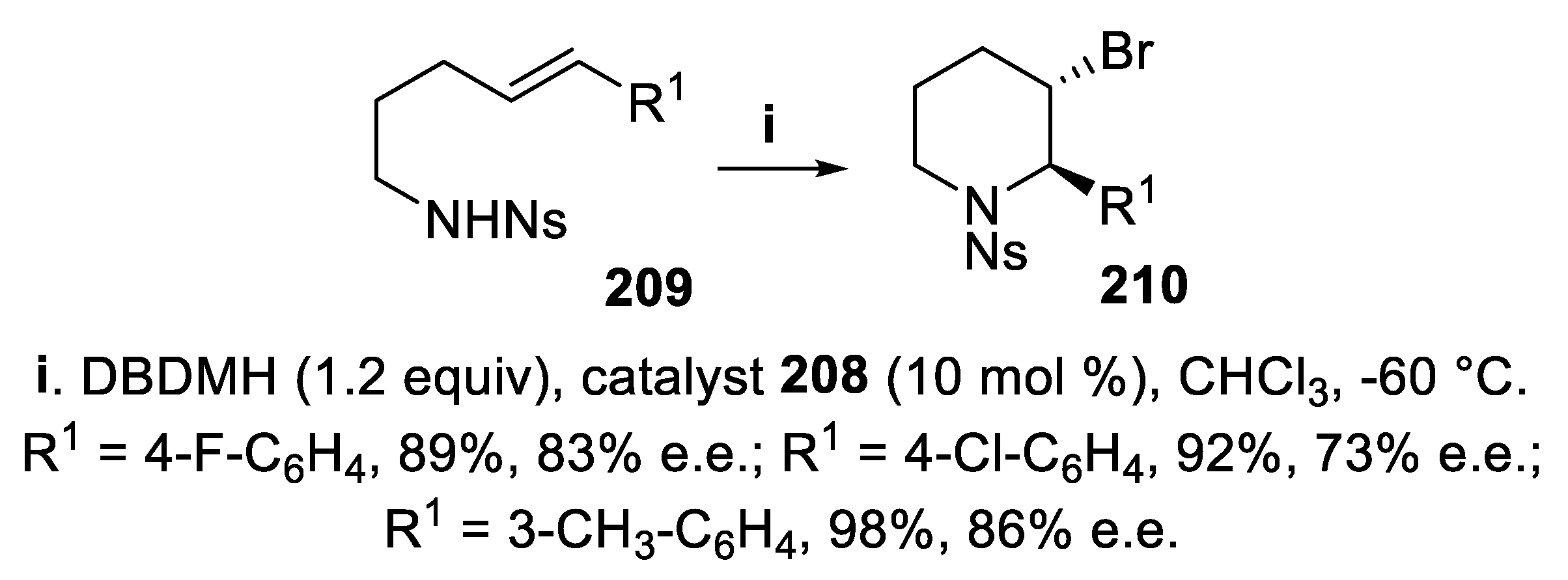
Scheme 60.
Stereoselective synthesis of 3-bromo piperidine 207, key intermediate to NK1 antagonist 211.
Scheme 60.
Stereoselective synthesis of 3-bromo piperidine 207, key intermediate to NK1 antagonist 211.
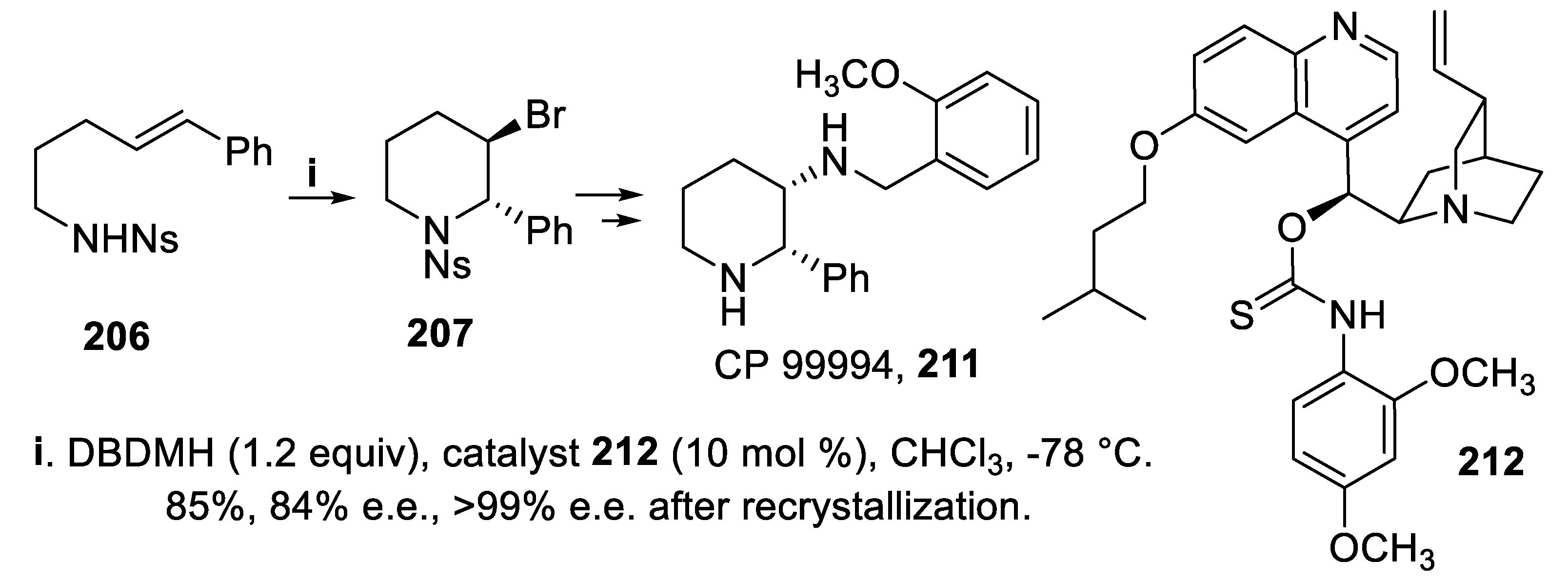
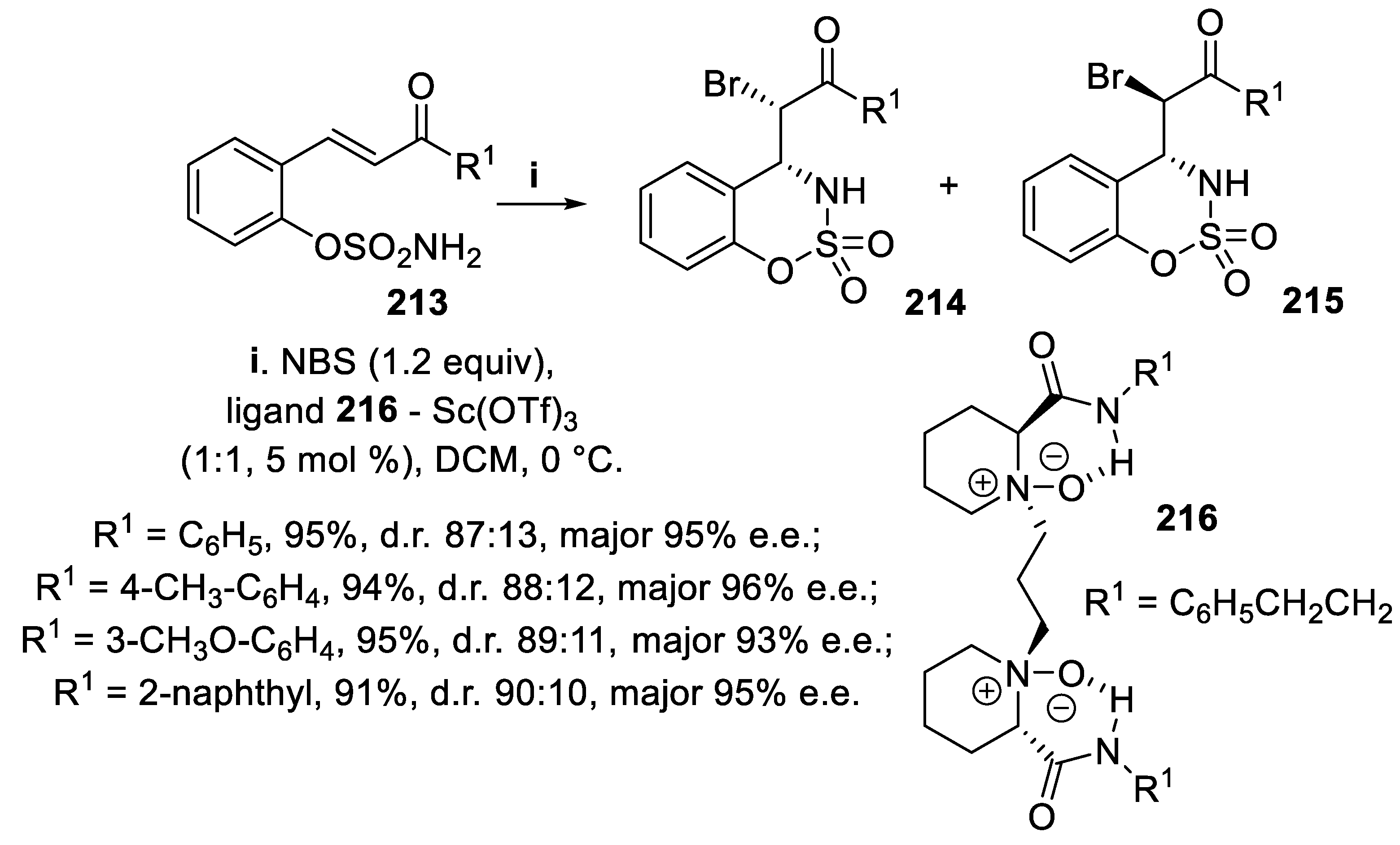
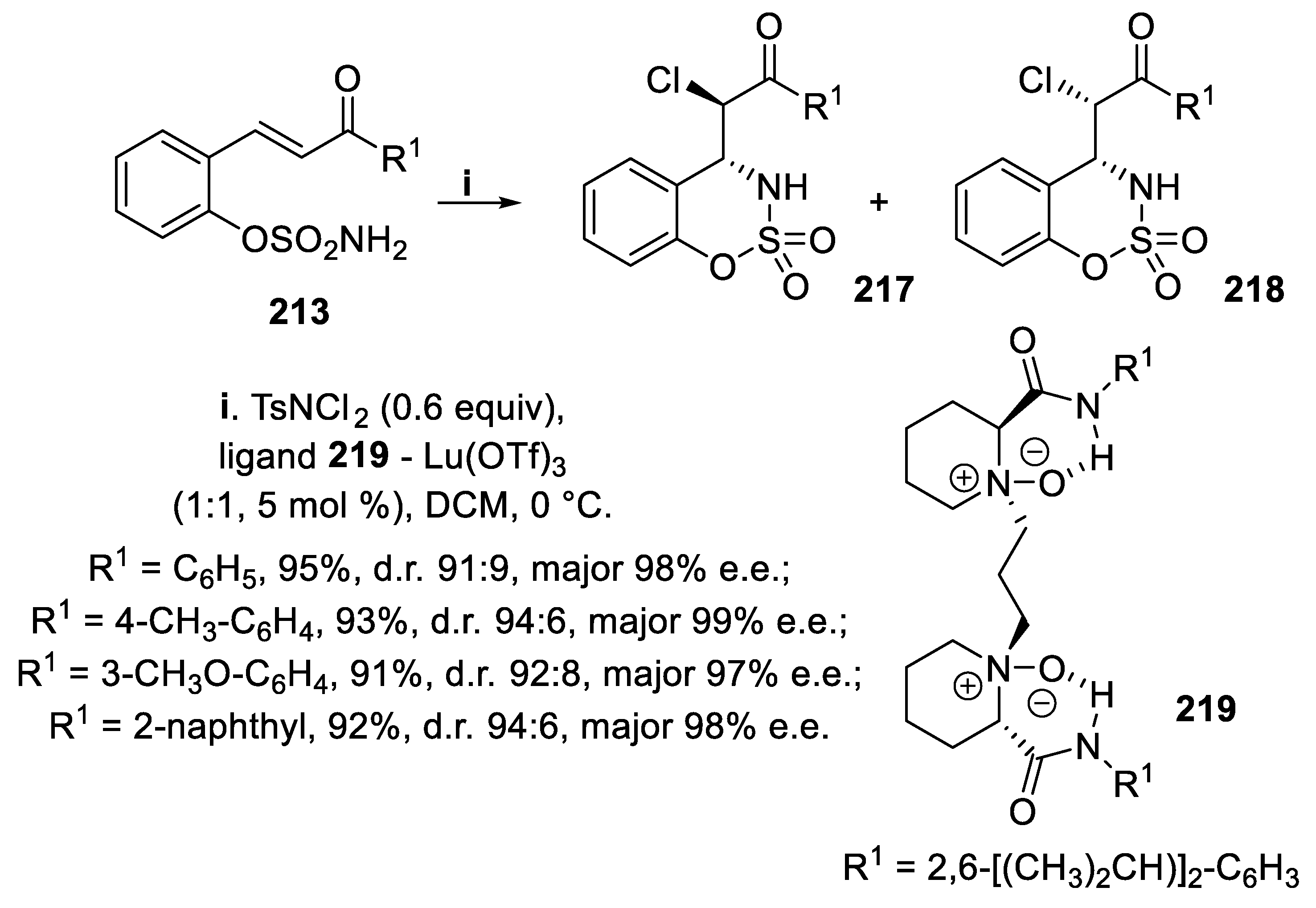
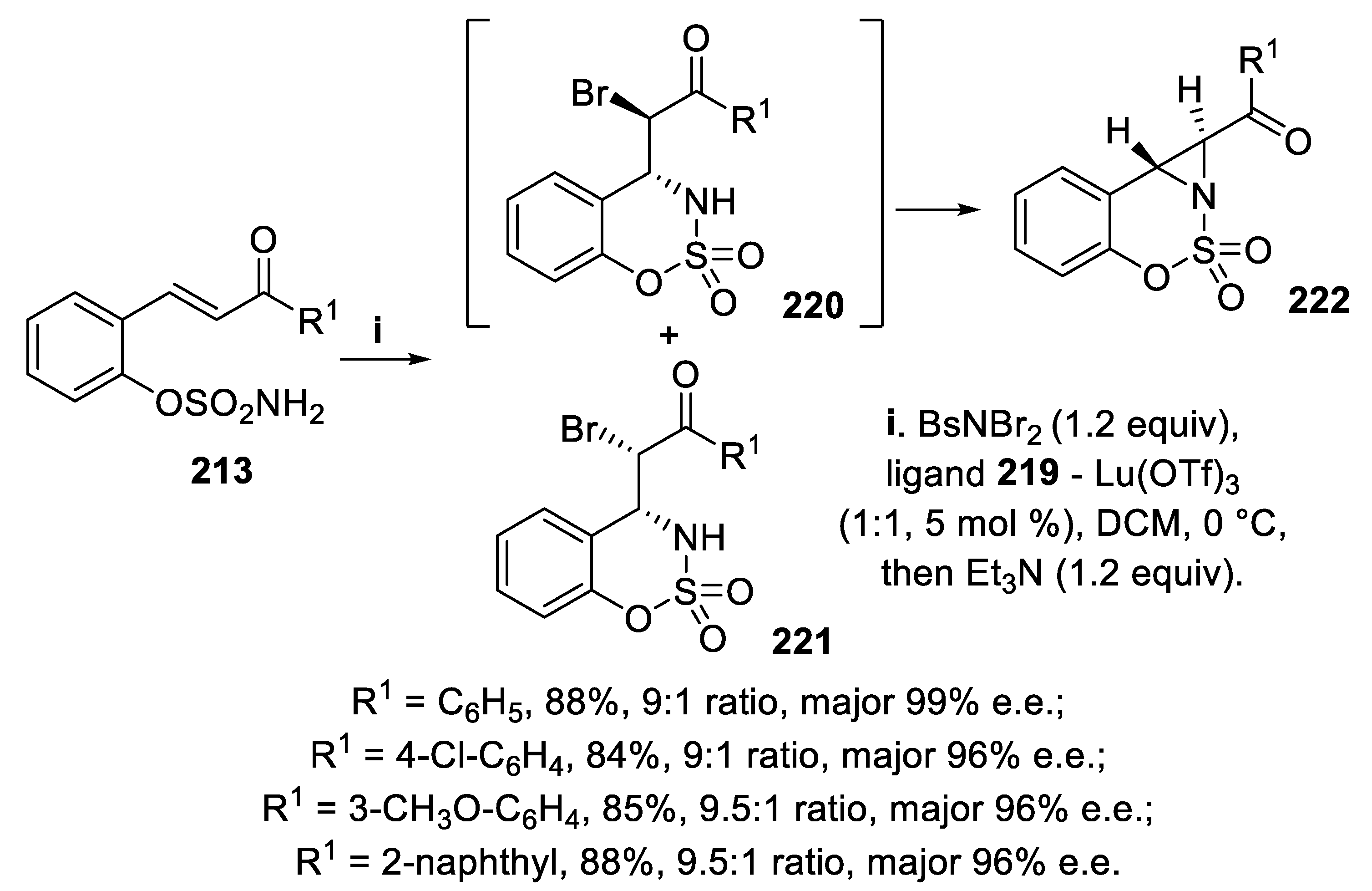
Scheme 64.
Stereoselective synthesis of 5-bromomethyl dihydropyrazoles 224 mediated by Λ-(S,S)-225.
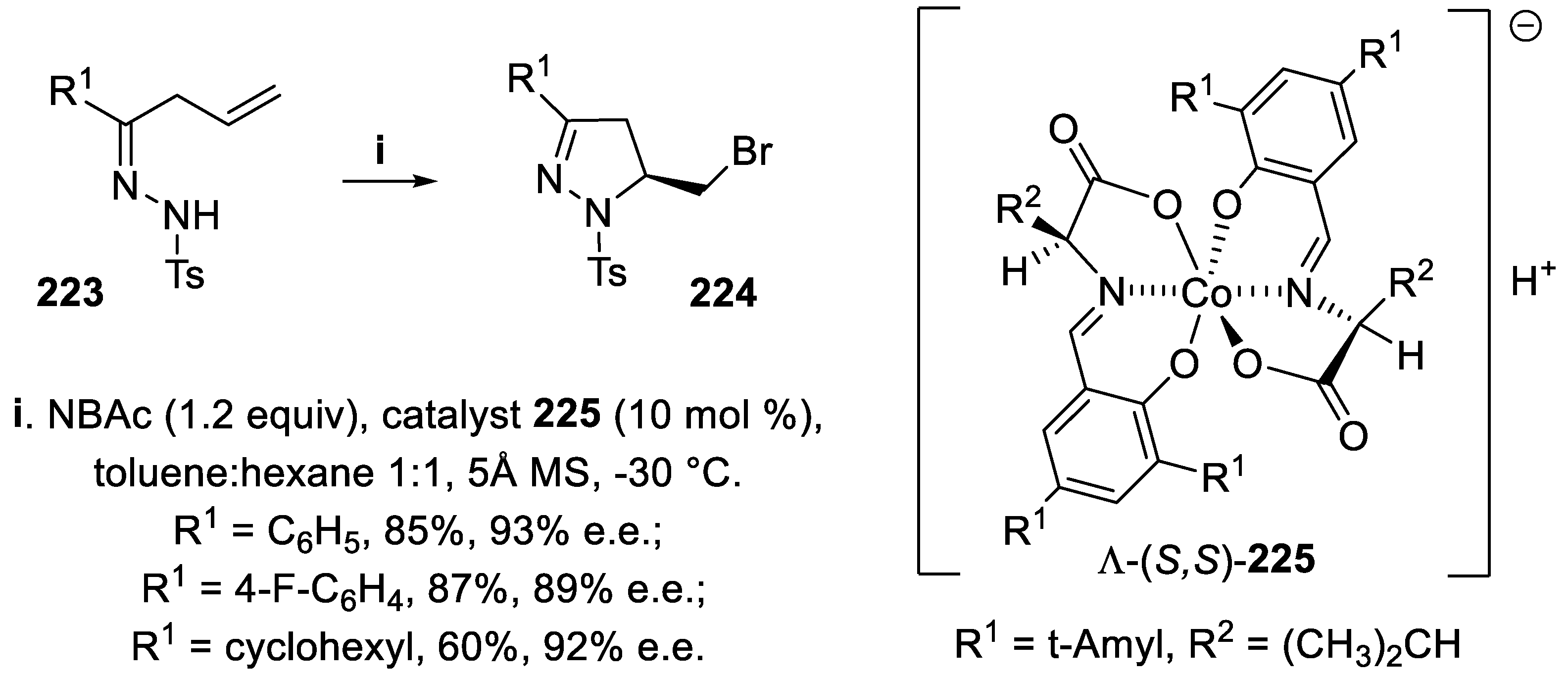
Scheme 65.
Synthesis of 5-iodomethyl dihydropyrazoles 227 mediated by Λ-(S,S)-169.
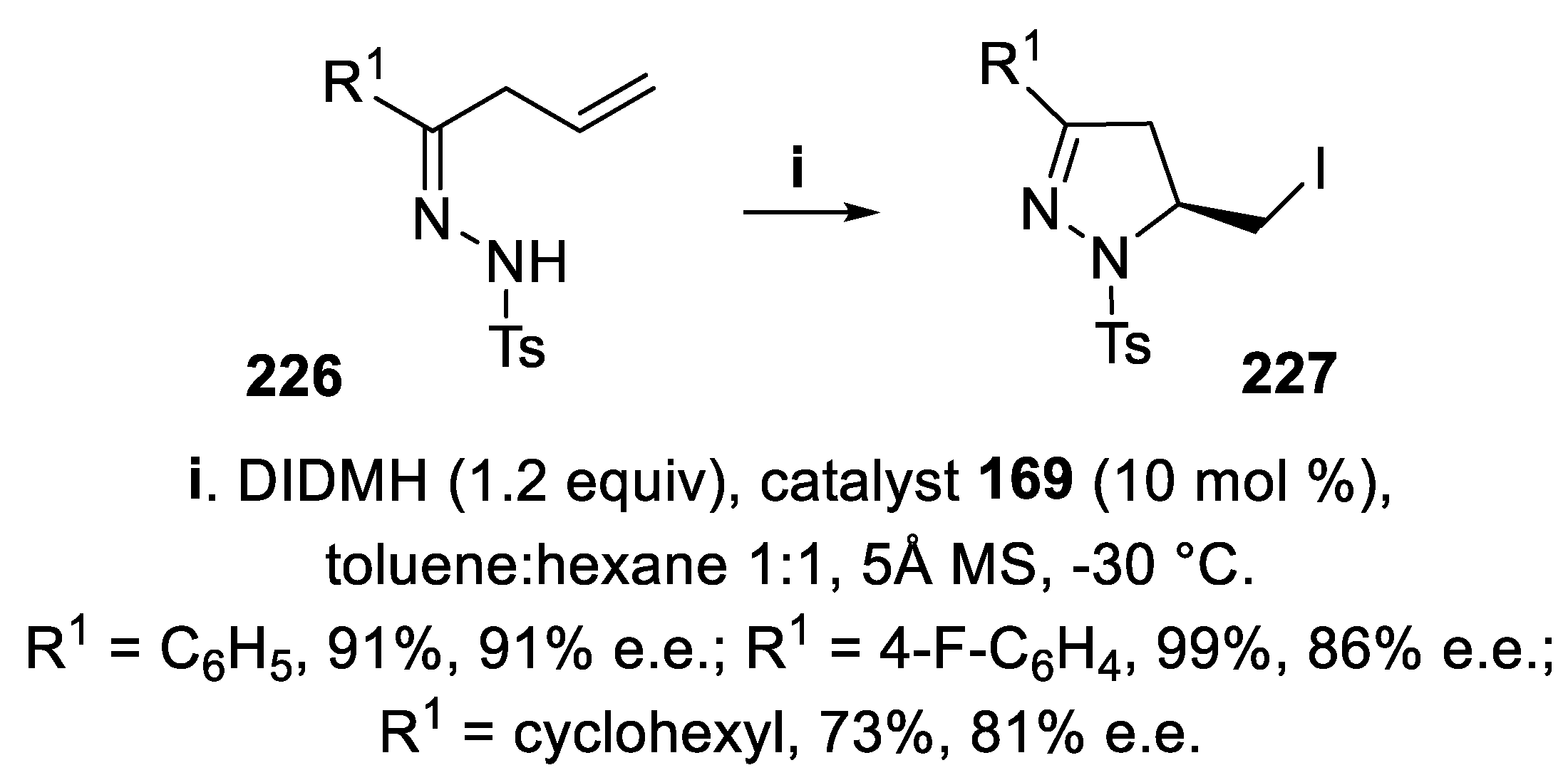
Scheme 66.
Synthesis of 4,5-dihydro-1H-pyrazoles 229 mediated by catalyst 230.
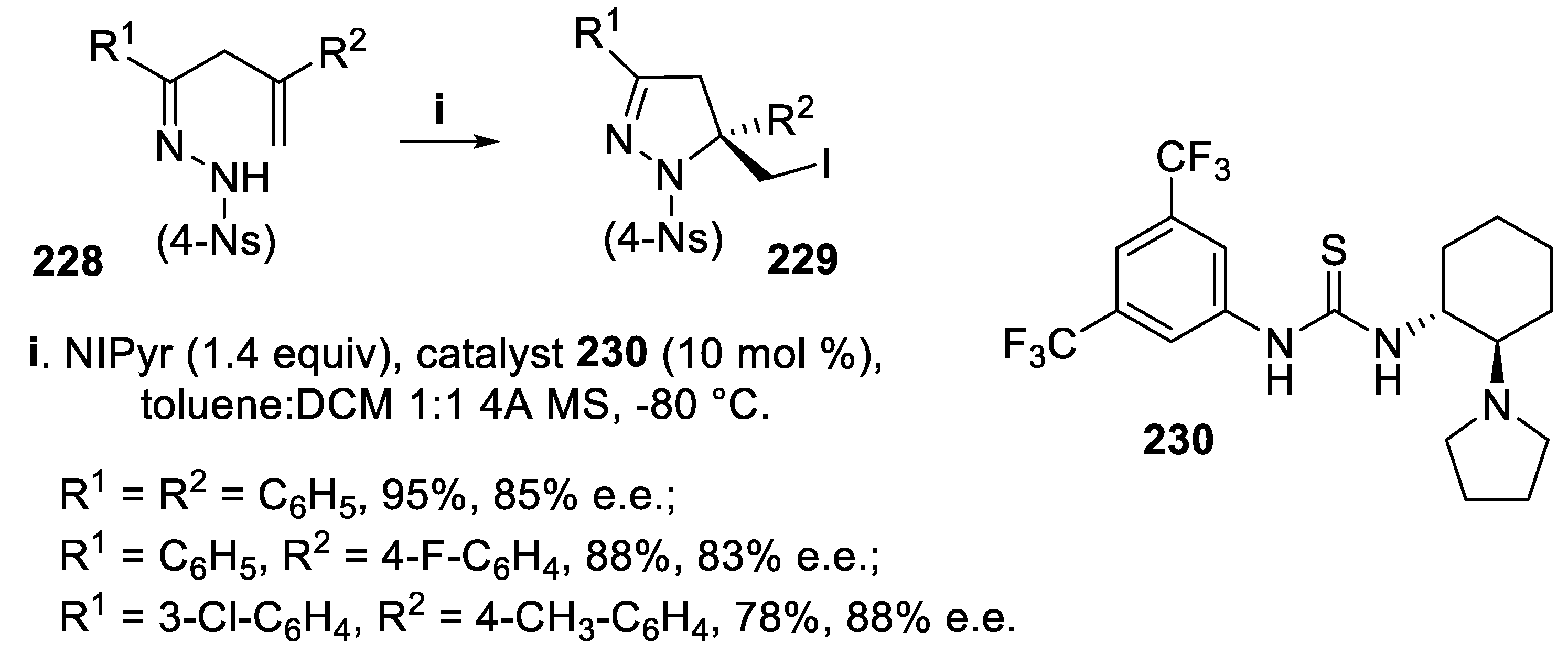
Scheme 67.
Synthesis of 4,5-dihydro-1H-pyrazoles 232 mediated by catalyst 233.
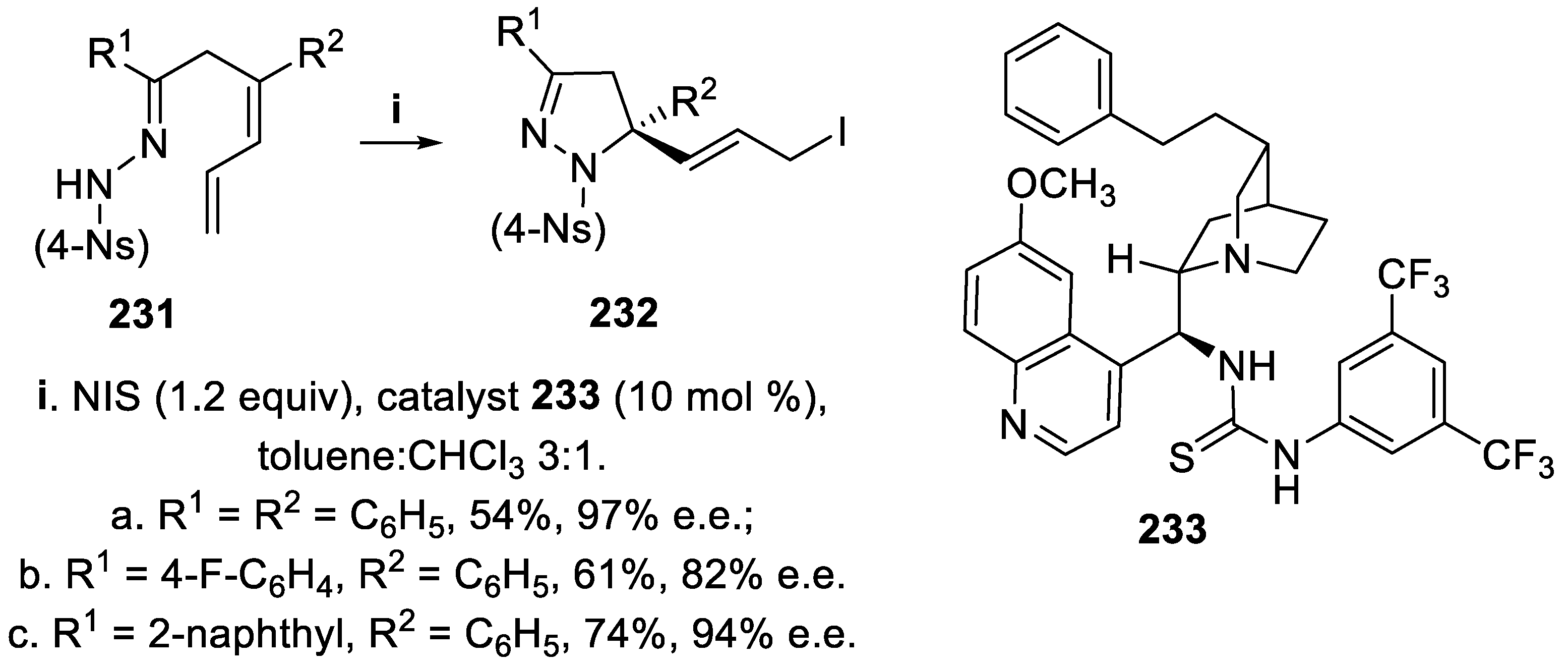
Scheme 68.
Synthesis of compound 234, analog of a kinesin spindle protein (KSP) inhibitor.
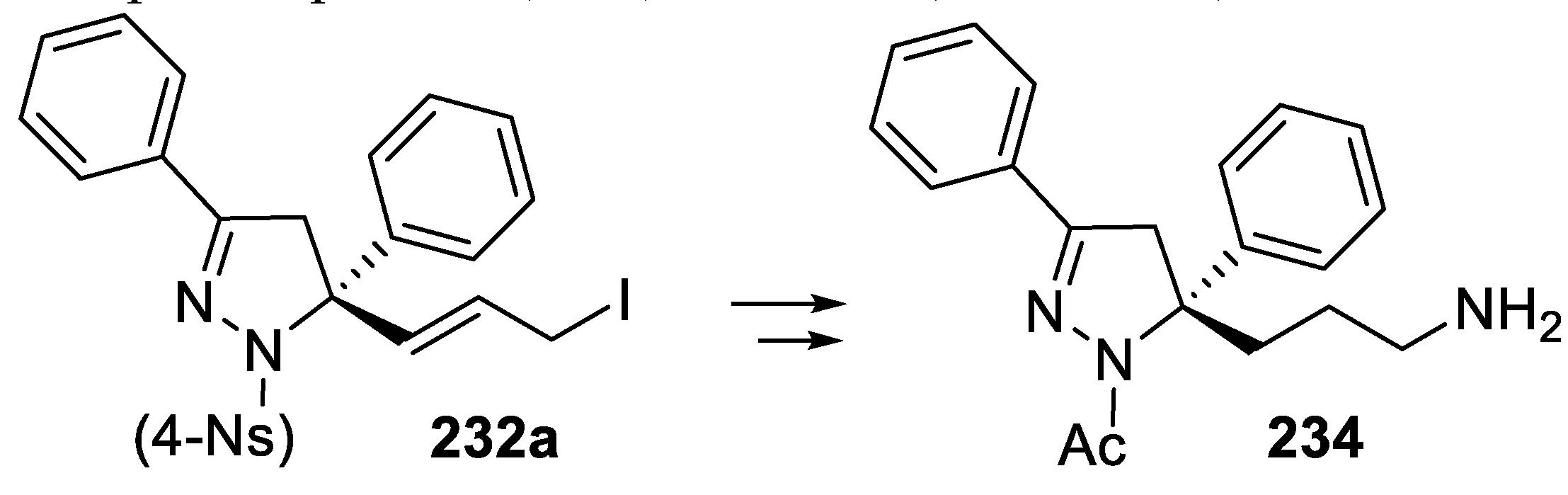
Scheme 69.
Synthesis of N-tosyl lactams 236 bearing a bromomethyl substituent.
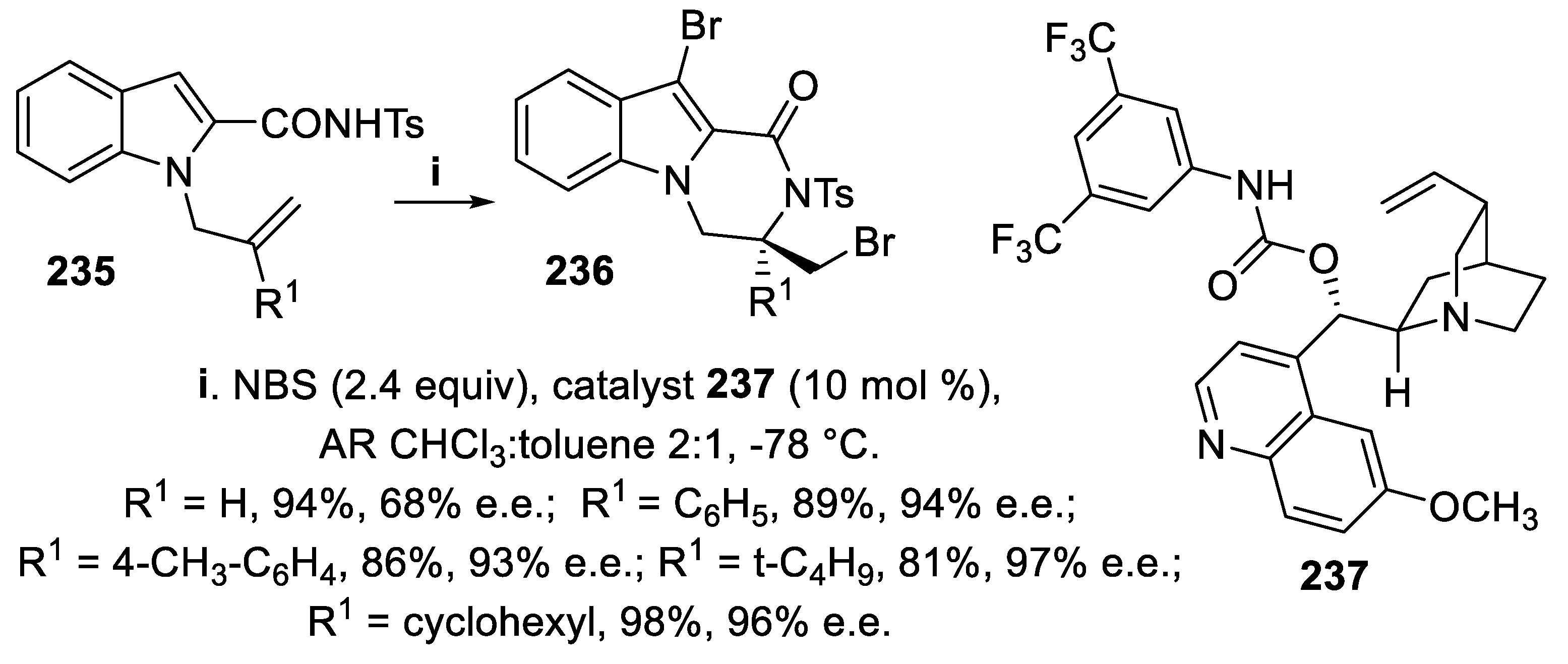
Scheme 70.
Synthesis of N-tosyl lactams 239 bearing a bromoalkyl substituent.
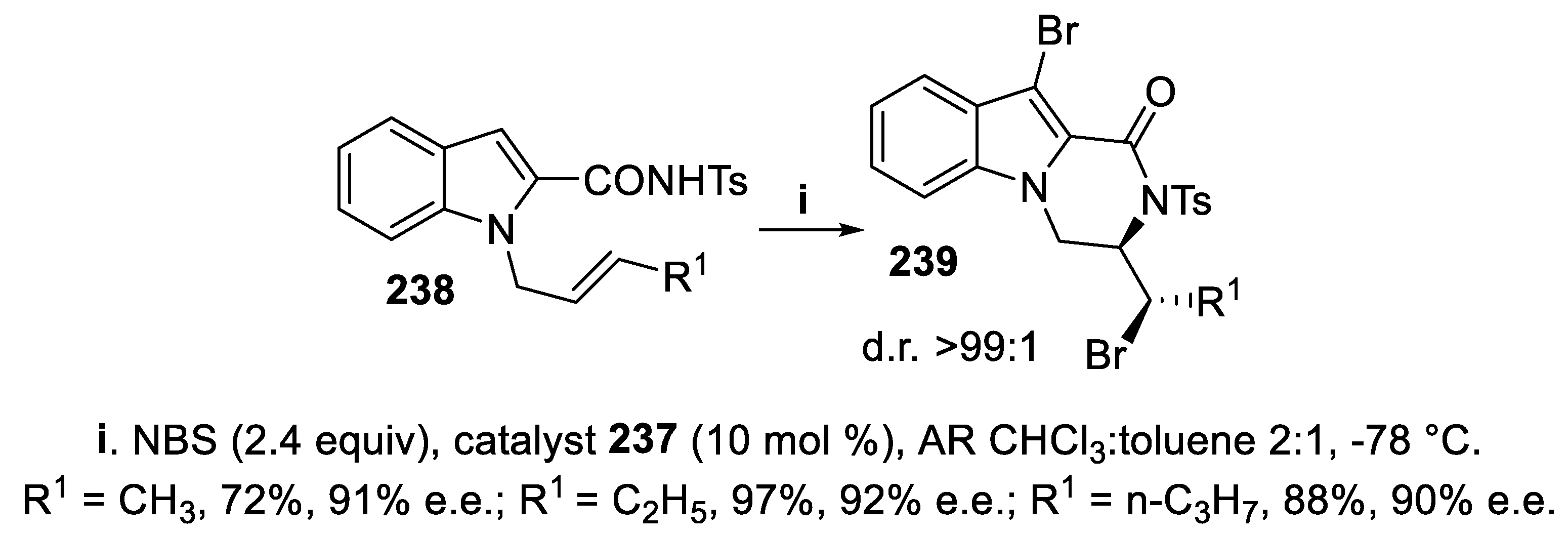
Scheme 71.
Synthesis of chiral oxazolidin-2-ones 241 mediated by ligand 242.
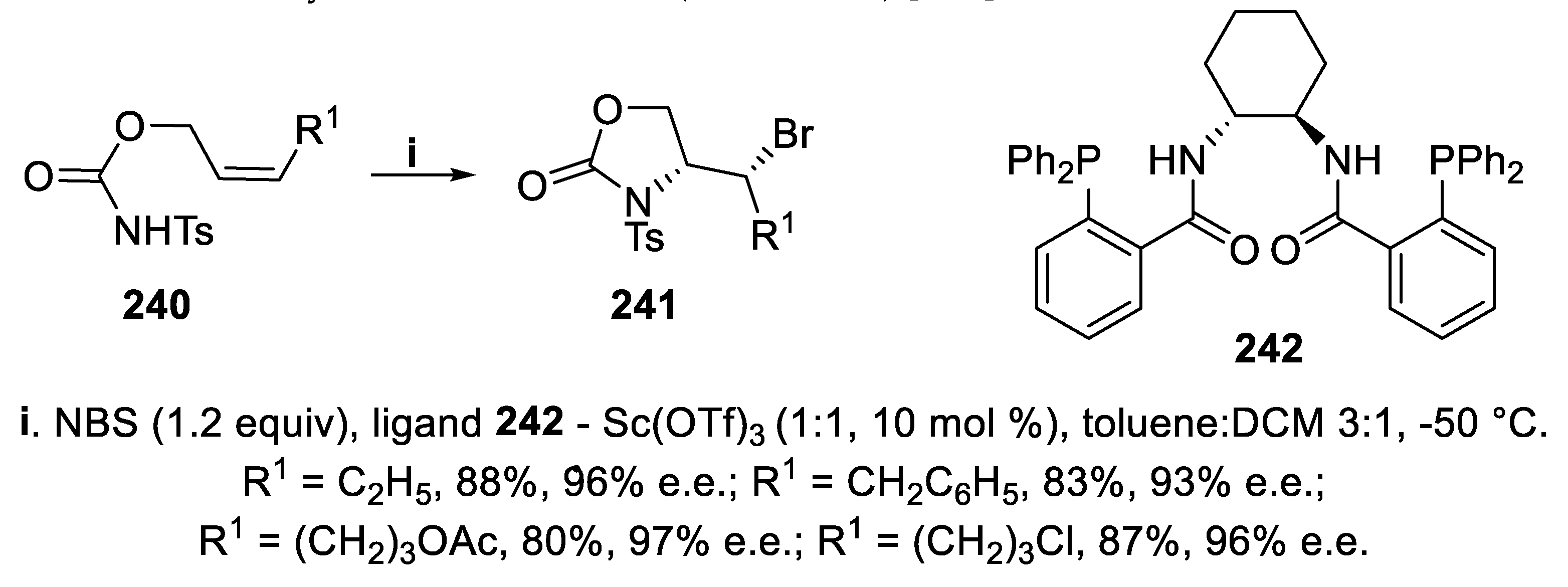
Scheme 72.
Non-regioselective bromocyclization of (E)-tosyl carbamate 243.
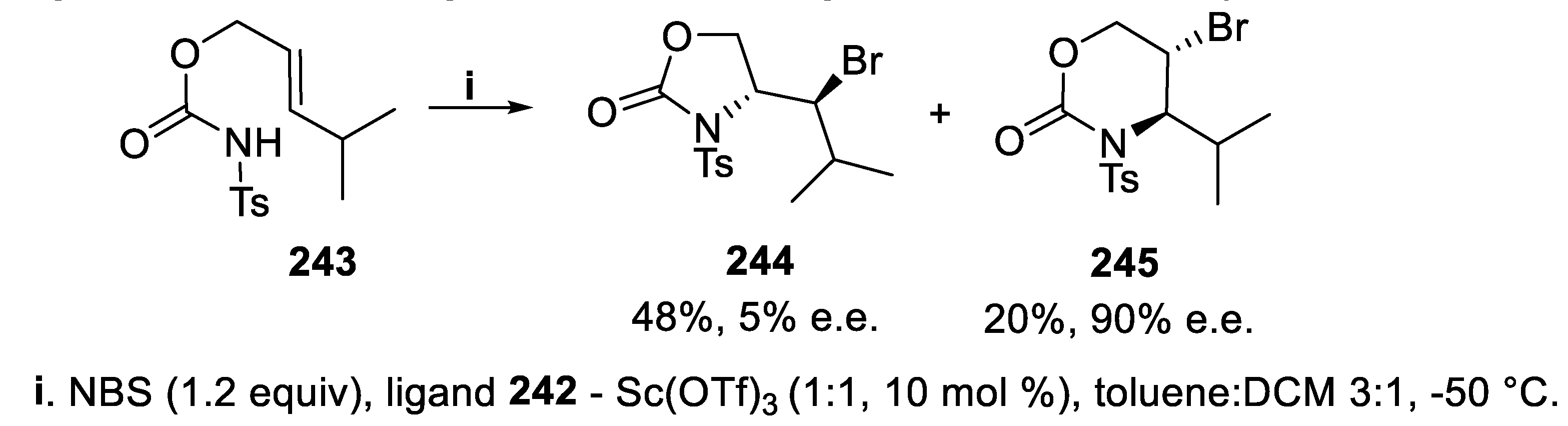
Scheme 73.
Bromoyclization of tosylcarbamates 246 and 249 mediated by ligand 248.
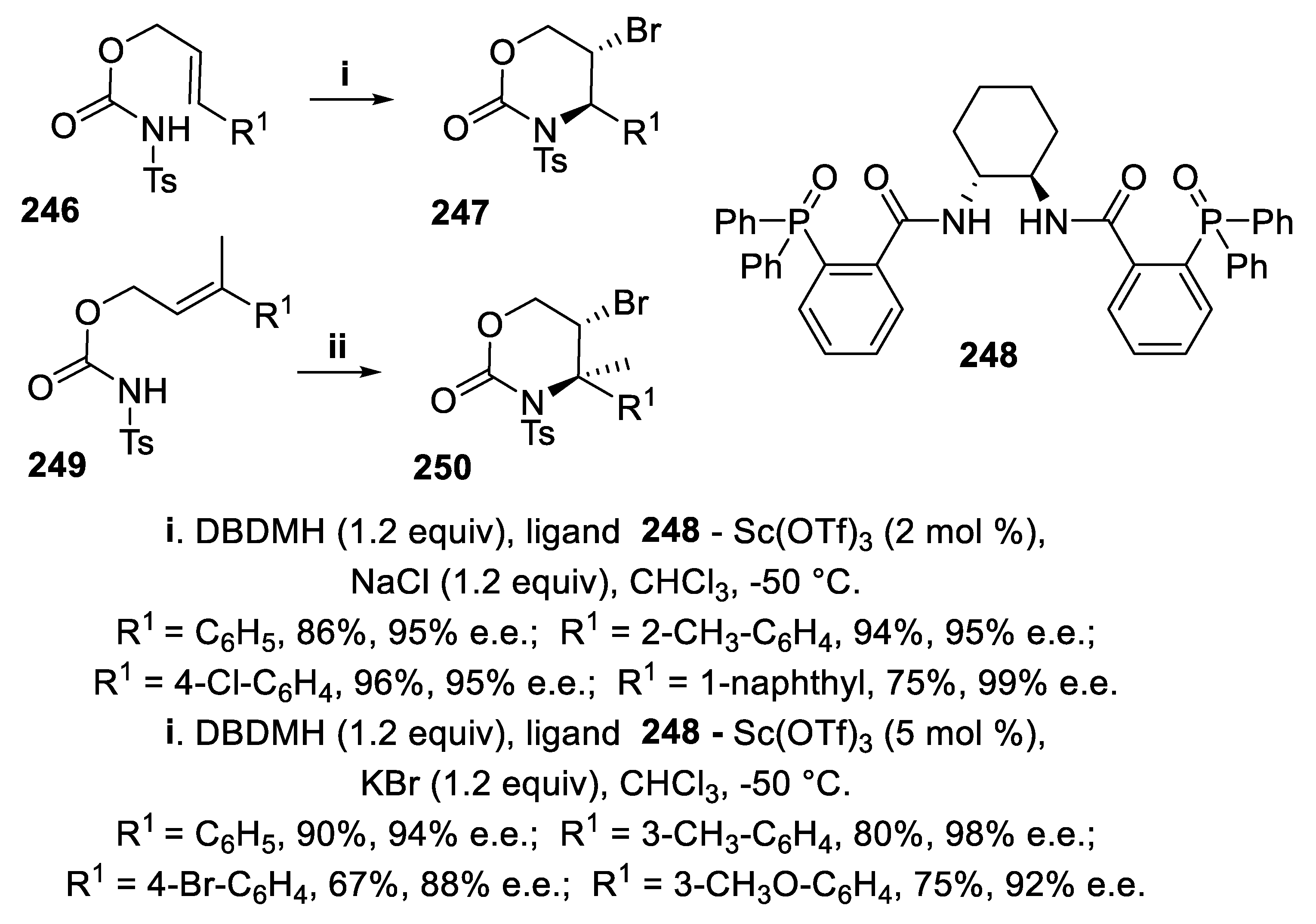
Scheme 74.
Bromoyclization of dienyl carbamates 251 mediated by ligand 248.
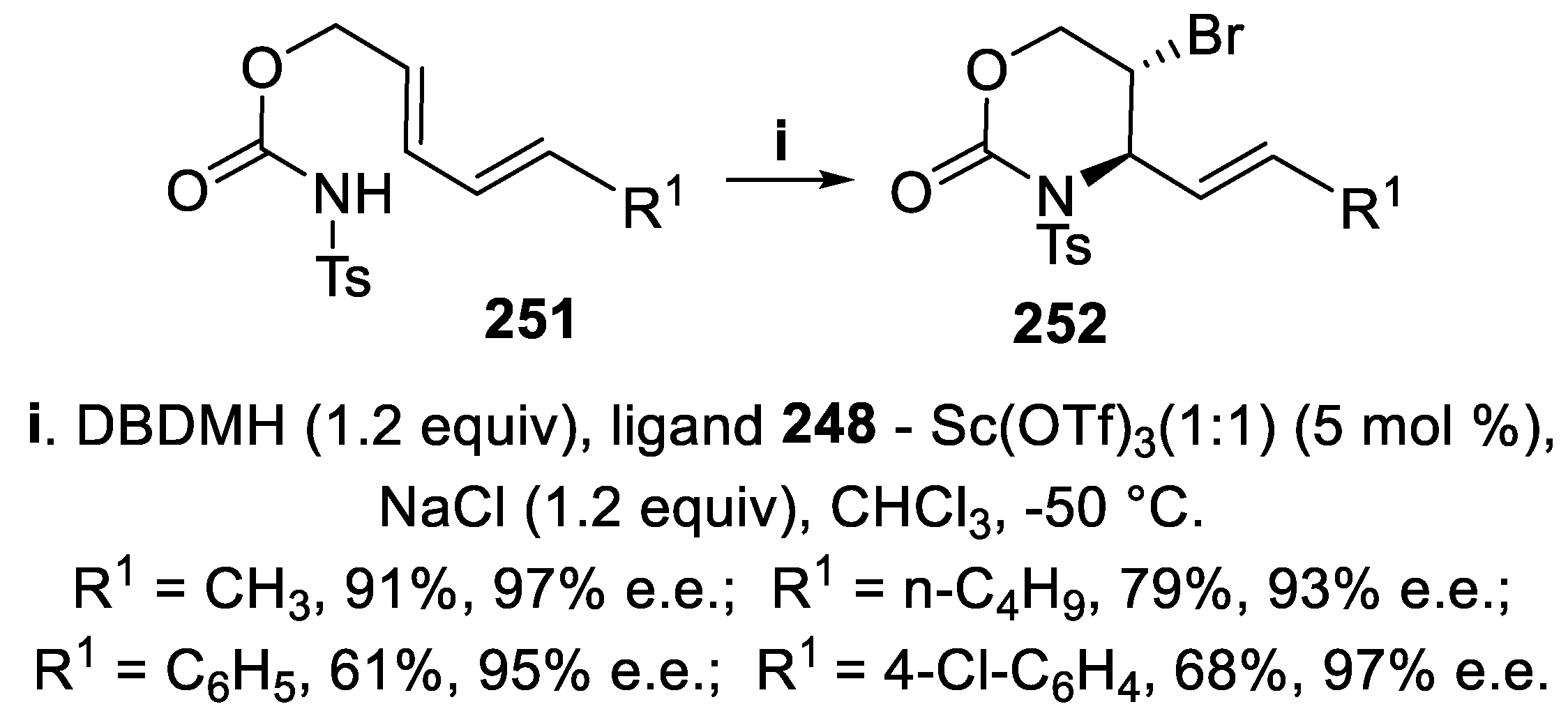
Scheme 75.
Bromoyclization of homoallyl carbamates 253 mediated by ligand 248.
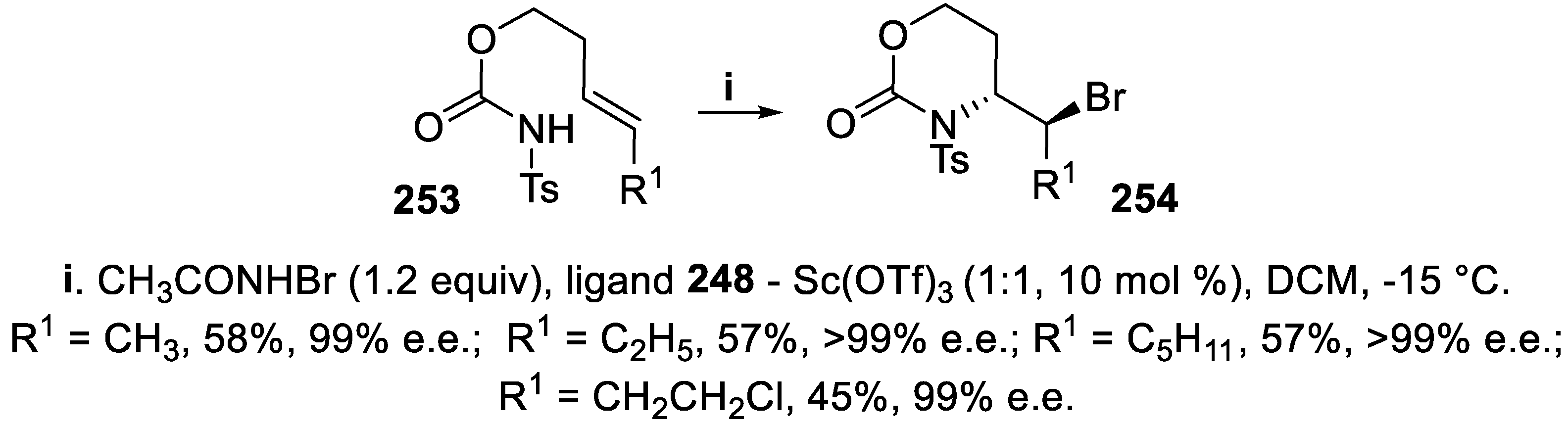
Scheme 76.
Dearomatization of an electron-rich benzofuran ring leading to chiral spiro compounds 256.
Scheme 76.
Dearomatization of an electron-rich benzofuran ring leading to chiral spiro compounds 256.
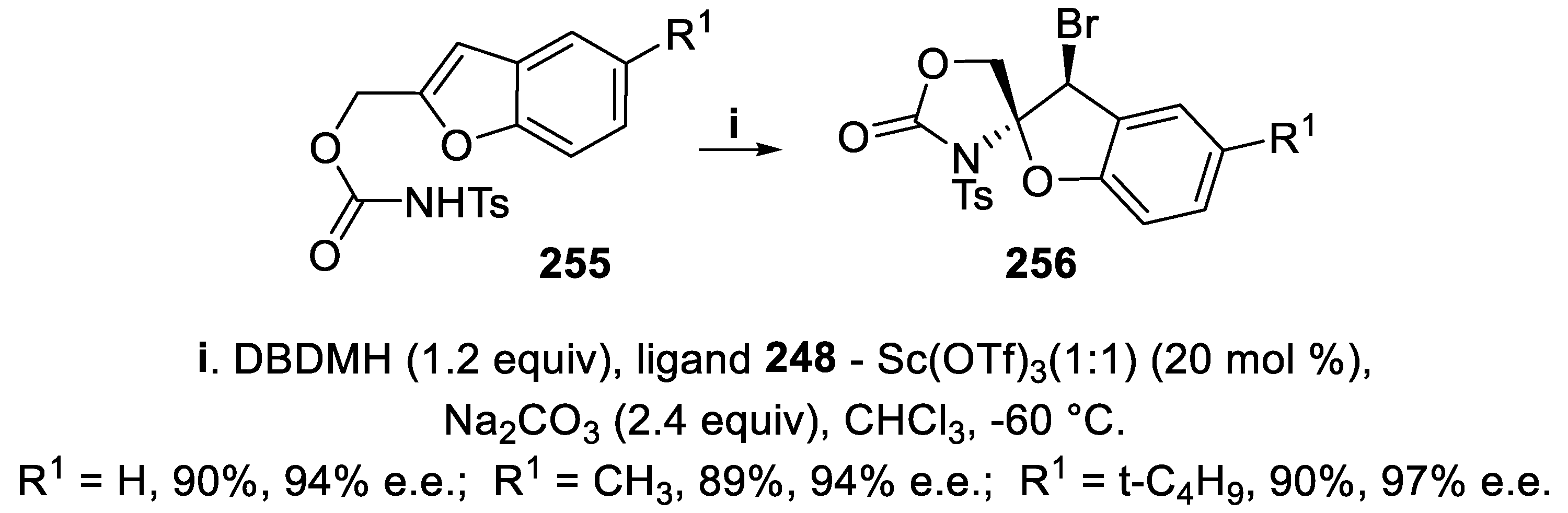
Scheme 77.
Synthesis of N-tosylimidazolidin-2-ones 259 mediated by chiral catalyst 260.
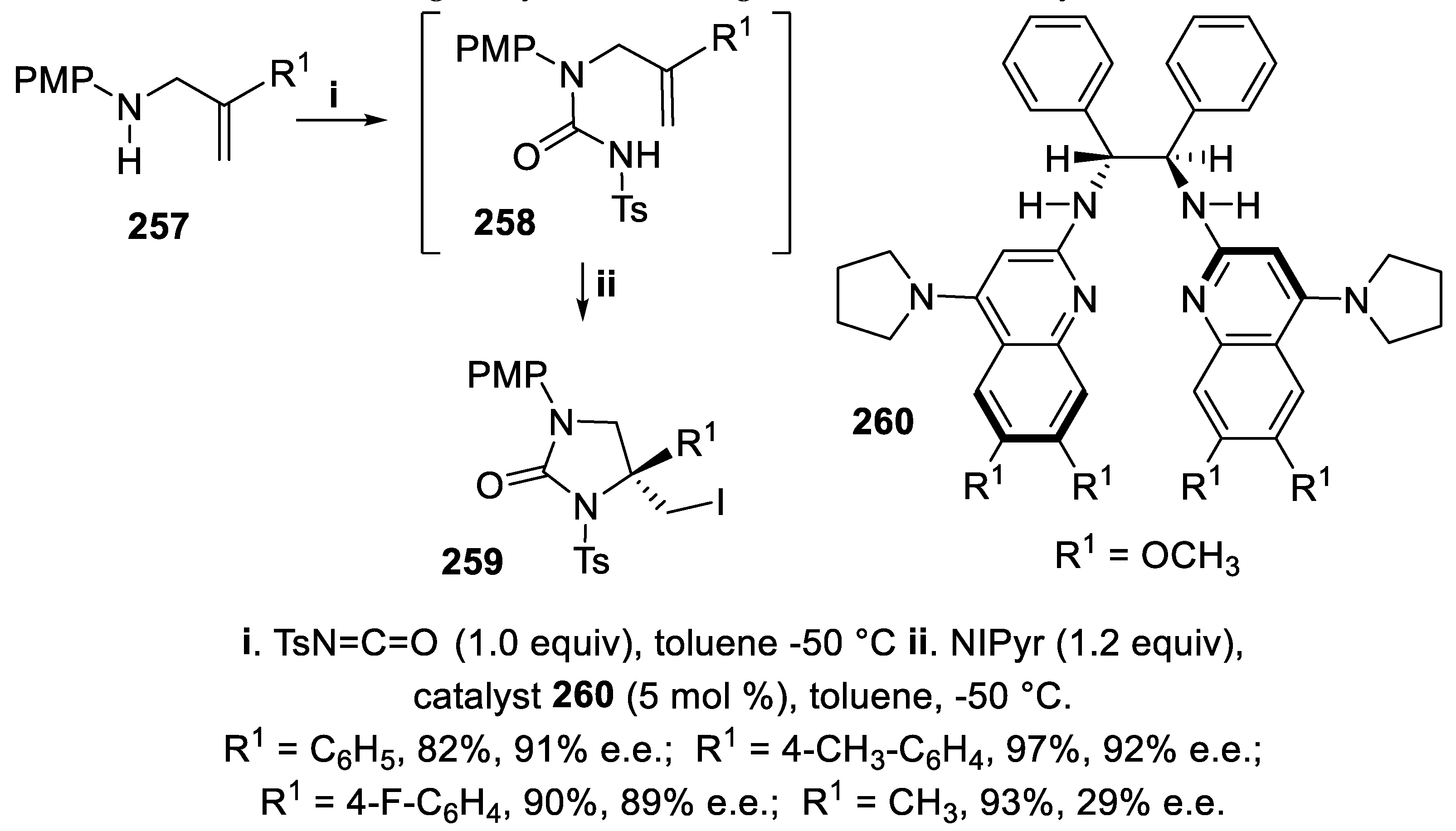
Scheme 78.
Synthesis of imidazolidin-2-one 262 and tetrahydropyrimidin-2(1H)-ones 264a,b.
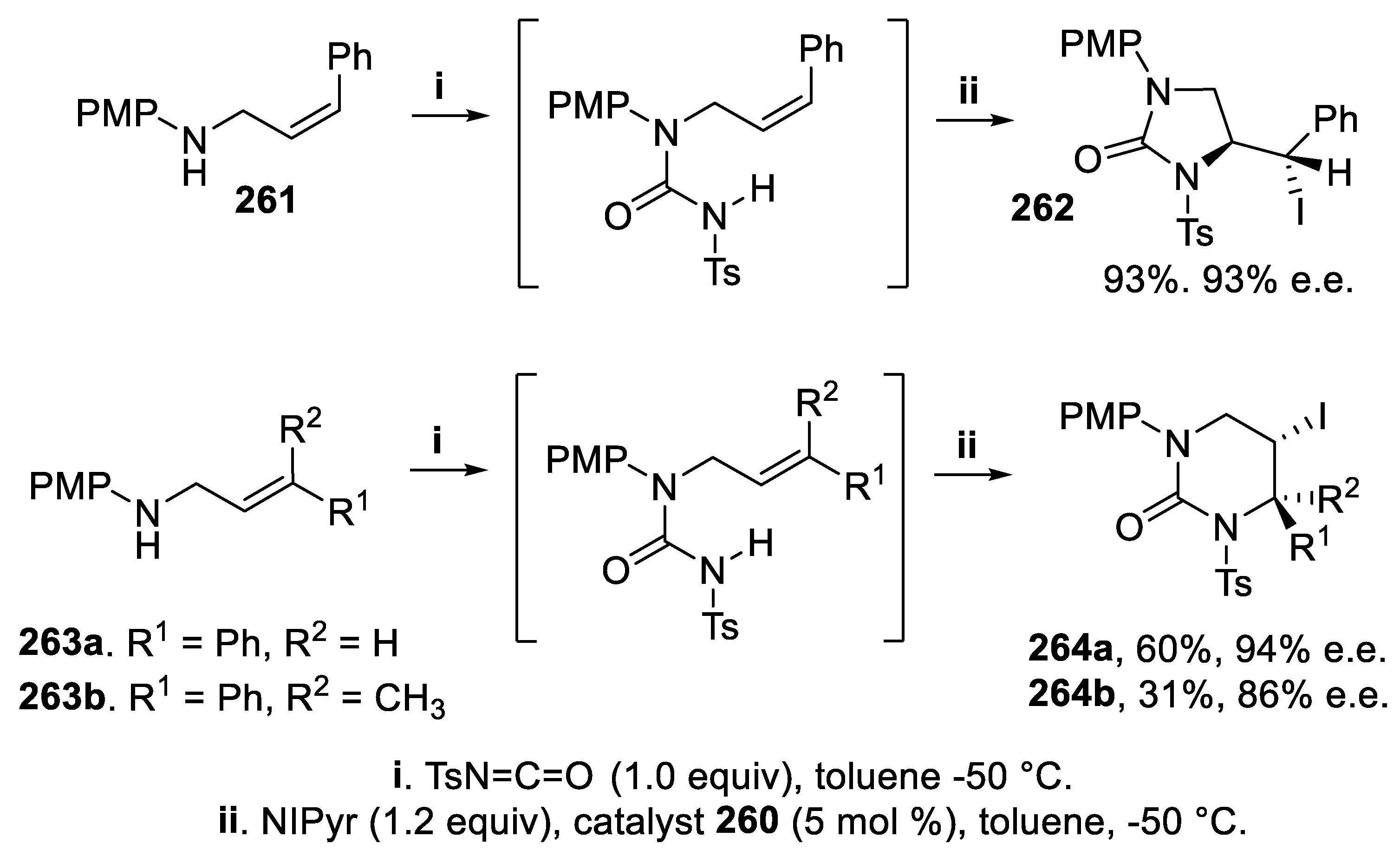
Scheme 79.
Synthesis of SCH 388714, 267, potent and selective NK1 receptor antagonist.
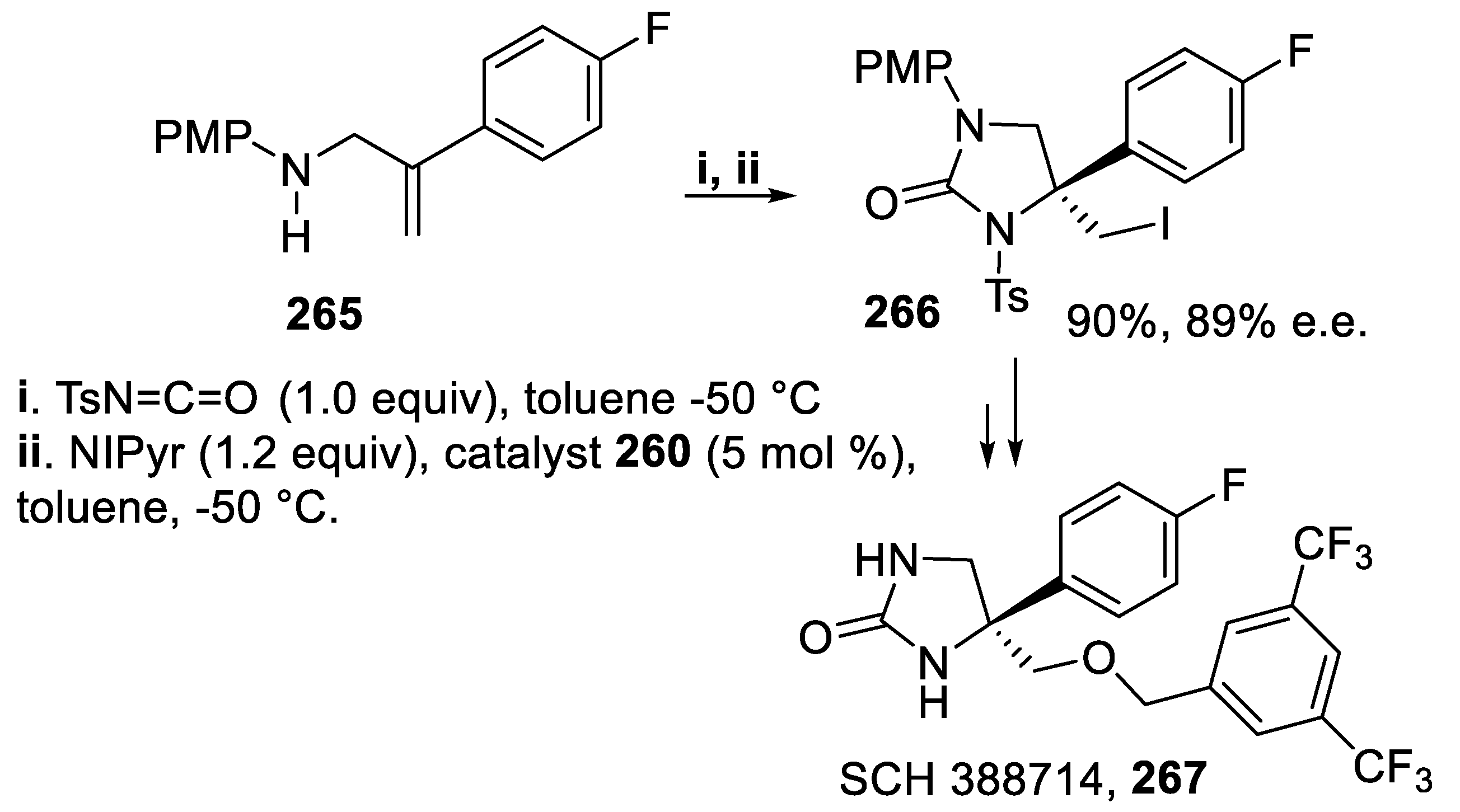
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



