
Submitted:
27 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
In the domain of drug discovery and design, the acquisition of precise structural and intermolecular Kd data for biological molecules is paramount. However, conventional methods for obtaining such data are often beset by challenges including high costs, time intensiveness, and inherent limitations in scalability and diversity. This article puts forward a high-throughput approach for in silico generation of structural and intermolecular binding affinity (Kd) data, which keeps exploring the uncharted territories of drug discovery & design by harnessing computational methodologies to synthetically generate diverse datasets. Through a meticulously designed workflow encompassing molecular structural modeling, structural biophysics-based calculations of intermolecular binding affinity (Kd), this innovative methodology transcends the constraints of traditional experimentation, offering a cost-effective, scalable, and efficient alternative. By simulating molecular interactions and binding interfaces and predicting binding affinities with reasonable accuracy, this approach not only expedites the drug development process but also enables the exploration of vast molecular space, thereby facilitating the discovery of novel therapeutics beyond conventional drug modality as a form factor. Moreover, the versatility of synthetic data extends beyond virtual screening and lead optimization, encompassing applications such as dataset augmentation, model validation, and benchmarking against experimental data. This article elucidates the conceptual underpinnings, methodological intricacies, validation strategies, and potential ramifications of in silico generated data, heralding a paradigm shift in drug discovery paradigms. By fostering synergy between computational and experimental research domains, this innovative approach promises to accelerate the pace of drug discovery, enhance the robustness of predictive models, and pave the way for transformative advancements in the entire pharmaceutical industry.
Keywords:
Drug Discovery & Design
; Synthetic Data Generation
; Intermolecular Binding Affinity (Kd)
; GIBAC
1. Challenges and Limitations with Traditional Acquisition of Structural and Binding Affinity Data
Traditional experimental methods of acquiring structural and binding affinity (Kd) data for biological molecules in drug discovery pose significant challenges [1]. First, they entail substantial financial investments due to the need for specialized equipment, reagents, and skilled personnel. This financial burden often impedes smaller research laboratories and organizations with limited resources [2]. Moreover, traditional experimental techniques are time-consuming and labor-intensive. Techniques like X-ray crystallography and NMR spectroscopy require meticulous sample preparation, data collection, and analysis, spanning weeks or months. This prolonged timeline delays drug discovery efforts and hinders bringing new therapeutics to market [3,4,5]. Scalability and throughput are additional challenges. Methods such as X-ray crystallography and NMR spectroscopy are limited in scale, restricting simultaneous sample analysis. This limitation obstructs screening large compound libraries for potential drug candidates and exploring diverse molecular (e.g., chemical) space [6,7,8,9].
Furthermore, traditional techniques may struggle to accurately capture the dynamic nature of biological molecules [10]. Proteins and nucleic acids exhibit conformational flexibility and dynamic behavior, which traditional methods may not fully capture, providing only static snapshots of molecular structures [11]. Characterizing weak or transient interactions is another challenge. Kd, crucial in drug discovery, quantifies the strength of interaction between a ligand and its target. Techniques like SPR and ITC may struggle to measure binding affinities for weak or transient complexes, limiting the assessment of potential drug candidates’ efficacy and specificity [12,13]. In summary, traditional methods of acquiring structural and Kd data face significant challenges, including financial constraints, time intensiveness, scalability limitations, and difficulties in capturing dynamic interactions. These challenges necessitate alternative approaches to accelerate the drug discovery process [14,15].
2. Concept of In Silico Generation of Structural and Intermolecular Kd Data
On August 11, 2022, the concept of a general intermolecular binding affinity calculator (GIBAC) was for the first time proposed in an MDPI preprint [8] and defined as below:
where represents the molecular system described either in strings (e.g., amino acid sequences, strings of SMILES to represent small molecules [16,17]), or in graphs to describe PTMs (e.g., glycosylated proteins) and PEMs (e.g., insulin icodec of Novo Nordisk [18,19,20]).
On October 19, 2023, the concept of GIBAC (Equation (1)) was for the first time updated, including its inception, definition (Equation (1)), construction, practical applications, technical challenges and limitations, and future directions [9,21]. As defined in [9], a real GIBAC (Equation (1)) is able to meet the criteria listed as below:
- a real GIBAC needs to take genetic variations into account; and
- a real GIBAC needs to work even without structural information; and
- for a real GIBAC, a variety of factors need to be taken into account, such as temperature, pH [22,23], site-specific protonation states (e.g., side chain pKa of protein) [24,25], post-translational modifications (PTMs) [26,27,28], post-expression modifications (PEMs) [29,30], buffer conditions [31], et cetera; and
- a real GIBAC requires a general forcefield for all types of molecules [3]; and
- a real GIBAC is able to be used the other way around, i.e., to be used as a search engine for therapeutic candidate(s). With such a GIBAC-based search engine, a list of therapeutic candidates can be retrieved and ranked according to drug-target Kd value(s), with input parameters including drug target(s) and a desired drug-target Kd value or a range of it.
In [8] and [9], the concept of in silico generation of structural and intermolecular Kd data was for the first time proposed, where in silico data generation refers to the computational simulation and modeling of biological molecules and their interactions to generate synthetic structural and Kd data. Unlike traditional experimental methods, which rely on costly and time-consuming laboratory techniques, in silico approaches leverage computer algorithms and simulations to predict molecular structures, simulate molecular interactions, and estimate binding affinities (Kd) between molecules.
3. Rationale Behind In Silico Generation of Structural and Intermolecular Kd Data
The rationale behind using computational approaches for in silico data generation stems from several key advantages over traditional experimental methods. Firstly, computational methods offer a cost-effective and efficient alternative to laboratory experimentation. By harnessing the power of high-performance computing and sophisticated algorithms, researchers can perform complex simulations and modeling studies at a fraction of the cost and time required for experimental techniques [?]. Moreover, computational approaches provide unparalleled insights into the molecular mechanisms underlying biological processes and drug interactions as molecular structural modeling techniques allow researchers to explore the dynamic behavior of biomolecular systems with atomic-level precision, revealing crucial structural insights that may not be accessible through experimental methods alone [11,33].
Furthermore, computational approaches offer the flexibility to simulate and study complex biological systems in silico. Unlike experimental techniques, which may be limited by the availability of reagents or the feasibility of experimental setups, computational methods can simulate virtually any molecular system, from protein-ligand interactions to protein-protein complexes, in a highly controlled and customizable manner [34]. Overall, the use of computational approaches for in silico data generation represents a paradigm shift in drug discovery and design [?], offering unprecedented opportunities to accelerate the pace of research, optimize drug development pipelines, and advance our understanding of molecular biology and disease mechanisms [35,36].
4. Workflow Steps Involved in Generating Synthetic Structural Data and Intermolecular Kd Data
In short, the workflow for generating synthetic structural data and intermolecular Kd data through in silico methods typically involves two main steps: homology structural modeling (Modeller [37]) based on experimental structures deposited inside Protein Data Bank (PDB) [38,39,40,41] and structural biophysics-based calculations of intermolecular binding affinity (Kd) using Prodigy [34,42], as shown in Figure 1.
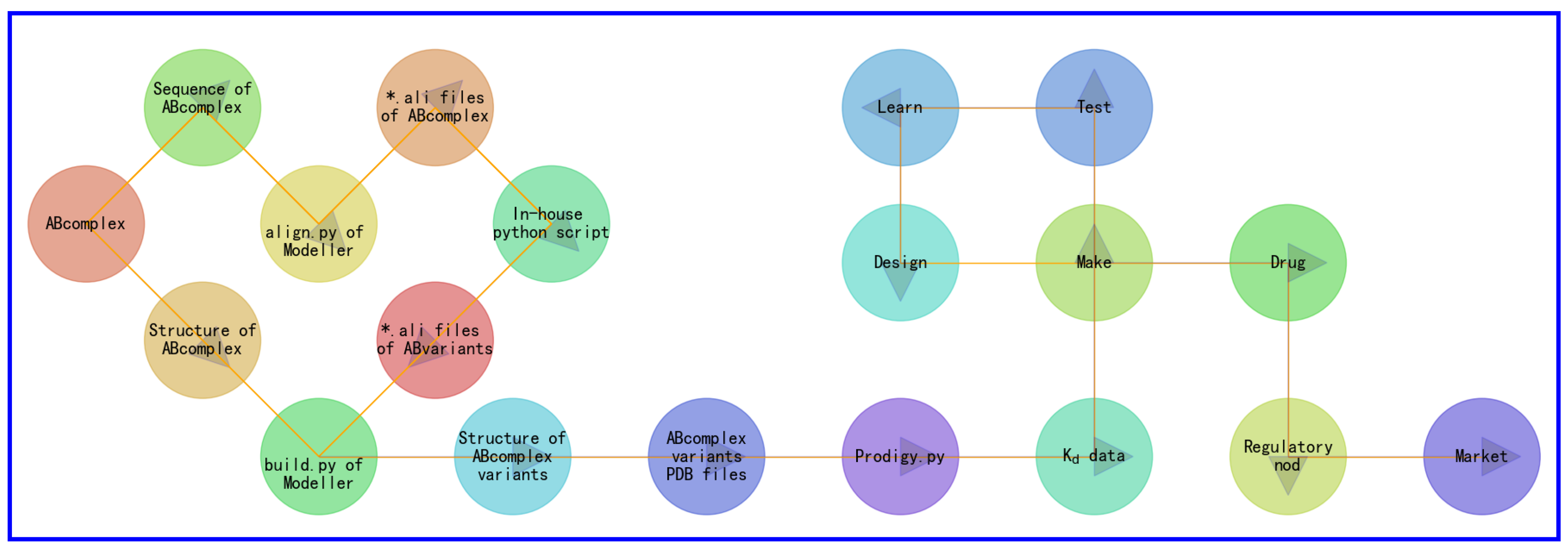
Specifically, the process begins with experimental structures deposited inside Protein Data Bank (PDB) [38], as shown in Figure 2:
- the experimental complex structure of molecule A and molecule B (ABcomplex) is retrived from PDB.
- amino acid sequences of molecules A and B are retrieved from the PDB file of ABcomplex.
- amino acid sequences of molecules A and B are plugged into align.py of Modeller [37] to generate alignment files of ABcomplex (*.ali files) for each set of site-specific mutations for the amino acid sequences of molecules A and B. Here, for each set of site-specific mutations for the amino acid sequences of molecules A and B, the number of missense mutations are restricted to ensure that the overall accuracy of the subsequent structural modeling and Kd calculations.
- alignment files of ABcomplex (*.ali files) are plugged into a set of in-house python scripts to produce a set of *ali files for variants of molecules A and B.
- the structure of ABcomplex and the set of *ali files for variants of molecules A and B are plugged into build.py of Modeller [37] to generate a set of homology complex structural models for variants of molecules A and B for each set of site-specific mutations for the amino acid sequences of molecules A and B.
5. Synthetic Structural Data and Intermolecular Kd Data: Validation and Benchmarking
Validating the accuracy and reliability of in silico generated data is crucial to ensuring its usefulness and relevance in drug discovery and design [43,44]. Specifically, several strategies can be employed to validate the performance of the workflow and assess the quality of the generated data.
- cross-validation is a widely used technique to assess the robustness of predictive models and evaluate their generalization performance. In the context of in silico data generation, cross-validation involves partitioning the dataset into training and validation sets and iteratively training the model on different subsets of the data. This process allows researchers to assess the model’s performance on unseen data and identify any potential biases or overfitting issues [45].
- external validation involves testing the predictive model on an independent dataset that was not used during the model training phase. By evaluating the model’s performance on unseen data from different sources or experimental conditions, researchers can assess its ability to generalize and make accurate predictions in real-world scenarios. External validation provides a more rigorous assessment of the model’s performance and its applicability to diverse biological systems [46].
- various performance metrics can be used to quantify the agreement between in silico predictions and experimental data. For structural data, metrics such as root-mean-square deviation (RMSD) or pairwise atomic distance distributions can assess the similarity between predicted and experimental structures. For Kd data, metrics such as Pearson correlation coefficient or mean absolute error can evaluate the accuracy of predicted binding affinities compared to experimental measurements [47,48].
- benchmarking involves comparing the performance of the in silico workflow with other computational methods or experimental techniques. By benchmarking against established methods or gold standard datasets, researchers can assess the relative strengths and weaknesses of the workflow and identify areas for improvement. Benchmarking provides valuable insights into the performance of the workflow in comparison to existing approaches and helps establish its credibility and reliability in drug discovery applications [49].
6. Synthetic Structural Data and Intermolecular Kd Data: Applications and Implications
6.1. Data Augmentation and Training of Machine Learning Models
Synthetic data can also be used to augment real-world datasets, enhancing the diversity and representativeness of training data for machine learning models [50]. By generating additional samples with diverse structural conformations and binding affinities, synthetic data can help mitigate issues related to dataset imbalance and scarcity, thereby improving the generalization and performance of predictive models. Data augmentation techniques can enrich training datasets and enable more comprehensive exploration of chemical space, facilitating the discovery of novel drug candidates and optimizing lead compounds for specific therapeutic targets.
Thus, another of the key applications of synthetic data in drug discovery and design is training machine learning models for virtual screening and lead optimization. Synthetic structural data and Kd data can serve as valuable training examples for developing predictive models that identify potential drug candidates with high binding affinities and specificities for target proteins. By training machine learning models on synthetic data, researchers can build robust computational tools for accelerating the drug discovery process and prioritizing lead compounds for further experimental validation [51].
6.2. Broader Implications of Synthetic Structural Data and Intermolecular Kd Data
In recent years, the application of AI (including DL and ML) is becoming increasingly popular in drug discovery & design [51,52,53,54], particularly in lead optimization and ADMET studies, including carcinogenicity, hepatotoxicity, et cetera [52]. For instance, VenomPred is a promising solution for deriving structural toxicophores and assessing the safety profile of compounds.
Figure 3.
A variety of the practical application of GIBAC, including SME (small molecule inhibitor), Ab (antibody), Ag (antigen), XDC (antibody-drug conjugate (ADC), peptide-drug conjugate (PDC), aptamer-drug conjugate (ApDC) [55]), rPeptide (recombinant peptide drug), rProtein (recombinant protein drug), intrabodies [56], proteolysis-targeting chimeric molecules (PROTAC) [57,58], drug-drug interaction (DDI) [59], chimeric antigen receptor T (CAR-T) cell therapy [60].
Figure 3.
A variety of the practical application of GIBAC, including SME (small molecule inhibitor), Ab (antibody), Ag (antigen), XDC (antibody-drug conjugate (ADC), peptide-drug conjugate (PDC), aptamer-drug conjugate (ApDC) [55]), rPeptide (recombinant peptide drug), rProtein (recombinant protein drug), intrabodies [56], proteolysis-targeting chimeric molecules (PROTAC) [57,58], drug-drug interaction (DDI) [59], chimeric antigen receptor T (CAR-T) cell therapy [60].
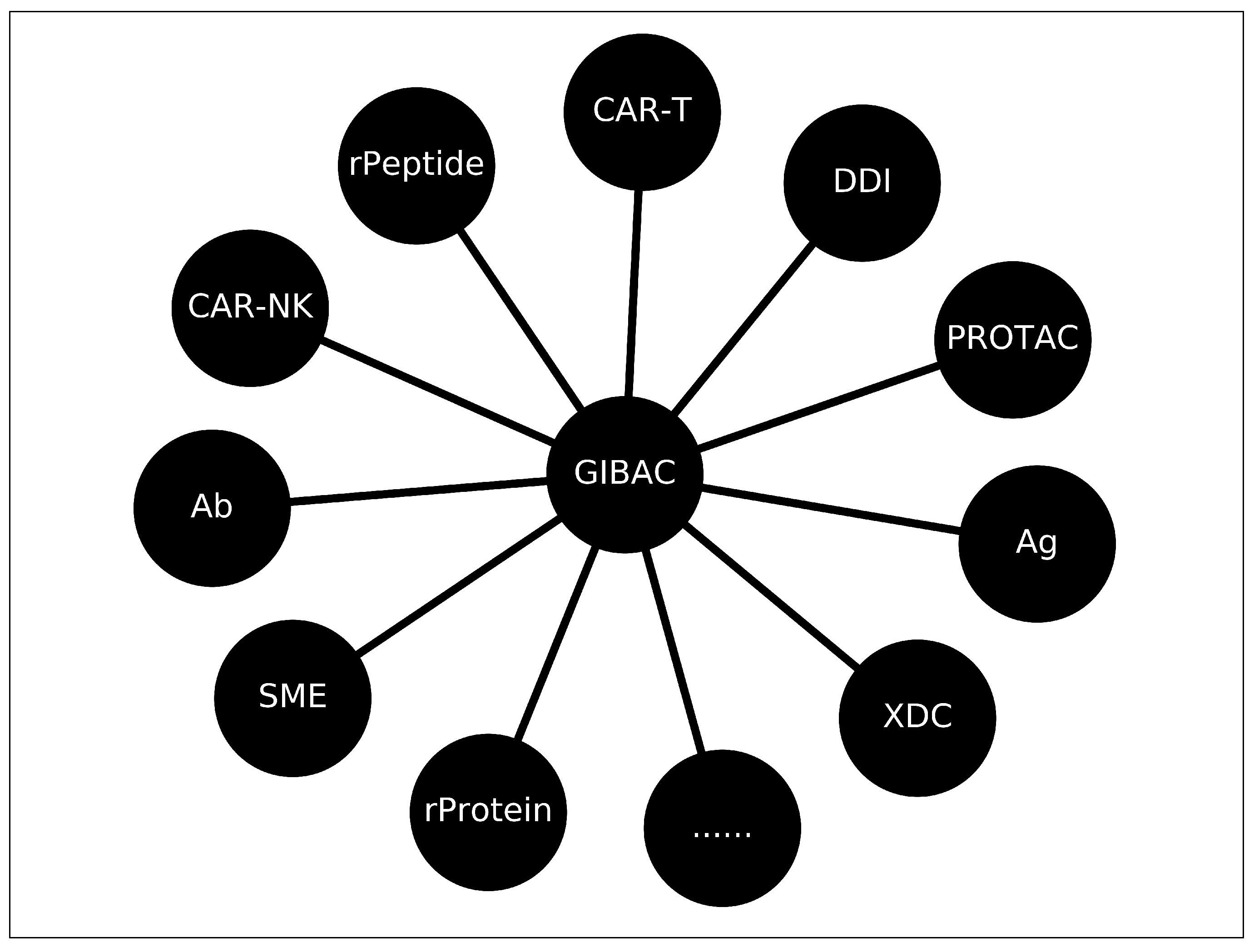
Here, given the definition of GIBAC as in Equation (1), the discussion of the practical application of synthetic structural data and intermolecular Kd data in drug discovery & design focuses on intermolecular binding and interactions. In biological systems, there are a wide range of intermolecular binding pairs, including including enzyme-substrate [61,62], ligand-receptor [63,64], protein-protein [34,65], ion channel-drug [66,67], antibody-antigen [68,69,70], DNA-protein [71], RNA-protein [55,72], RNA-RNA [72], hormone-receptor [73], coenzyme-substrate [74], metal ion-protein [35,75], lipid-protein [76], et cetera. By definition, synthetic structural data and intermolecular Kd data can find its use for any binding pair involved in the molecular pathogenesis of human diseases, infectious or non-communicable, including:
7. In Silico Generation of Structural and Intermolecular Kd Data: Future Directions
While the field of in silico generation of structural and intermolecular Kd data holds immense promise for advancing drug discovery and design efforts, researchers keep pushing the boundaries of computational biology and develop increasingly sophisticated methodologies, several exciting future directions emerge.
7.1. Integration of Multi-Scale Modeling
One promising direction for future research is the integration of multi-scale modeling approaches to capture the complex interplay between different levels of biological organization. By combining techniques such as quantum mechanics/molecular mechanics (QM/MM) simulations, coarse-grained modeling, and cellular simulations, researchers can simulate biological systems across multiple length and time scales, from individual atoms and molecules to entire cells or organisms. This multi-scale modeling framework enables a more comprehensive understanding of biological processes and interactions, facilitating the discovery of novel drug targets and optimization of therapeutic interventions [80,81].
7.2. Incorporation of Structural Dynamics
Incorporating structural dynamics into computational models is essential for capturing the inherent flexibility and conformational changes of biomolecular systems. While traditional static models provide valuable insights into molecular structure and interactions, they often overlook the dynamic nature of biological molecules. Future research directions may involve developing dynamic structural models that explicitly account for protein flexibility, ligand-induced conformational changes, and allosteric regulation. By integrating structural dynamics into computational models, researchers can better predict ligand binding modes, identify allosteric binding sites, and design more effective drugs targeting dynamic protein conformations [82,83,84].
7.3. Integration of Experimental and Computational Approaches
Finally, the integration of experimental and computational approaches represents a promising direction for advancing in silico data generation in drug discovery and design [85]. Combining experimental techniques such as cryo-electron microscopy, X-ray crystallography, and high-throughput screening with computational simulations and modeling enables researchers to leverage the strengths of both approaches and obtain comprehensive insights into biological systems [86,87]. Future research efforts may focus on developing hybrid experimental-computational workflows that seamlessly integrate experimental data with computational predictions, enabling synergistic exploration of drug-target interactions and accelerating the drug discovery process [85].
8. Conclusion
In conclusion, the in silico generation of structural and intermolecular Kd data offers a transformative approach to drug discovery and design. By integrating computational methodologies, this workflow (Figure 1, Figure 2 and Figure 4)enables the efficient generation of diverse datasets, predicting binding affinities accurately and expediting the identification of novel drug candidates beyond drug modality as a form factor. This approach fosters collaboration between computational and experimental researchers, accelerating research efforts and advancing pharmaceutical science. In summary, in silico generated data holds immense promise for revolutionizing drug discovery, enhancing efficiency, and driving innovation for the industry [8,9].
Author Contributions
Conceptualization, W.L.; methodology, W.L.; software, W.L.; validation, W.L.; formal analysis, W.L.; investigation, W.L.; resources, W.L.; data duration, W.L.; writing–original draft preparation, W.L.; writing–review and editing, W.L.; visualization, W.L.; supervision, W.L.; project administration, W.L.; funding acquisition, not applicable.
Funding
This research received no external funding.
Institutional Review Board Statement
No ethical approval is required.
Data Availability Statement
During the preparation of this work, the author used OpenAI’s ChatGPT in order to improve the readability of the manuscript, and to make it as concise and short as possible. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Acknowledgments
The author is grateful to the communities of structural biology, biophysics, medicinal and computational chemistry and algorithm design, for the continued accumulation of knowledge and data for drug discovery & design, and for the continued development of tools (hardware, software and algorithm) for drug discovery & design.
Conflicts of Interest
The author declares no conflict of interest.
References
- Xiong, P.; Zhang, C.; Zheng, W.; Zhang, Y. BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. Journal of Molecular Biology 2017, 429, 426–434. [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Research 2007, 35, D198–D201. [CrossRef] [PubMed]
- Hofmann, D.W.M.; Kuleshova, L.N. A general force field by machine learning on experimental crystal structures. Calculations of intermolecular Gibbs energy with iFlexCryst. Acta Crystallographica Section A Foundations and Advances 2023, 79, 132–144. [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: Struct., Funct., Bioinf. 2005, 59, 687–696. [CrossRef] [PubMed]
- Bezencon, J.; Wittwer, M.B.; Cutting, B.; Smiesko, M.; Wagner, B.; Kansy, M.; Ernst, B. pKa determination by (1)H NMR spectroscopy-an old methodology revisited. J. Pharm. Biomed. Anal. 2014, 1, 147–155. [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 2011, 7, 539–543. [CrossRef] [PubMed]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [CrossRef] [PubMed]
- Li, W. Towards a General Intermolecular Binding Affinity Calculator 2022.
- Li, W.; Vottevor, G. Towards a Truly General Intermolecular Binding Affinity Calculator for Drug Discovery & Design 2023.
- Wang, T.; He, X.; Li, M.; Shao, B.; Liu, T.Y. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics. Scientific Data 2023, 10. [CrossRef]
- Li, W. Characterising the interaction between caenopore-5 and model membranes by NMR spectroscopy and molecular dynamics simulations. PhD thesis, University of Auckland, 2016.
- Liu, W.; Chen, L.; Yin, D.; Yang, Z.; Feng, J.; Sun, Q.; Lai, L.; Guo, X. Visualizing single-molecule conformational transition and binding dynamics of intrinsically disordered proteins. Nature Communications 2023, 14. [CrossRef]
- Kawade, R.; Kuroda, D.; Tsumoto, K. How the protonation state of a phosphorylated amino acid governs molecular recognition: insights from classical molecular dynamics simulations. FEBS Letters 2019, 594, 903–912. [CrossRef]
- Tu, C.L.; Wang, Y.L.; Hu, T.M.; Hsu, L.F. Analysis of Pharmacokinetic and Pharmacodynamic Parameters in EU- Versus US-Licensed Reference Biological Products: Are In Vivo Bridging Studies Justified for Biosimilar Development? BioDrugs 2019, 33, 437–446. [CrossRef] [PubMed]
- Hirata, F.; Sugita, M.; Yoshida, M.; Akasaka, K. Perspective: Structural fluctuation of protein and Anfinsen’s thermodynamic hypothesis. The Journal of Chemical Physics 2018, 148, 020901. [CrossRef] [PubMed]
- Lee, J.; Pilch, P.F. The insulin receptor: structure, function, and signaling. American Journal of Physiology-Cell Physiology 1994, 266, C319–C334. [CrossRef]
- Rahuel-Clermont, S.; French, C.A.; Kaarsholm, N.C.; Dunn, M.F. Mechanisms of Stabilization of the Insulin Hexamer through Allosteric Ligand Interactions. Biochemistry 1997, 36, 5837–5845. [CrossRef] [PubMed]
- Nishimura, E.; Pridal, L.; Glendorf, T.; Hansen, B.F.; Hubálek, F.; Kjeldsen, T.; Kristensen, N.R.; Lützen, A.; Lyby, K.; Madsen, P.; Pedersen, T.Å.; Ribel-Madsen, R.; Stidsen, C.E.; Haahr, H. Molecular and pharmacological characterization of insulin icodec: a new basal insulin analog designed for once-weekly dosing. BMJ Open Diabetes Research & Care 2021, 9, e002301.
- Li, W. Designing Insulin Analogues with Lower Binding Affinity to Insulin Receptor than That of Insulin Icodec 2024. [CrossRef]
- Li, W. How Structural Modifications of Insulin Icodec Contributes to Its Prolonged Duration of Action: A Structural and Biophysical Perspective 2023. [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2018, 20, 273–286. [CrossRef]
- Yang, A.S.; Honig, B. On the pH Dependence of Protein Stability. Journal of Molecular Biology 1993, 231, 459–474. [CrossRef]
- Harris, T.K.; Turner, G.J. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) 2002, 53, 85–98. [CrossRef]
- Li, W. Gravity-driven pH adjustment for site-specific protein pKa measurement by solution-state NMR. Measurement Science and Technology 2017, 28, 127002. [CrossRef]
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proceedings of the National Academy of Sciences 2014, 111, E1705–E1712. [CrossRef]
- Herget, S.; Ranzinger, R.; Maass, K.; Lieth, C.W. GlycoCT—a unifying sequence format for carbohydrates. Carbohydrate Research 2008, 343, 2162–2171. [CrossRef] [PubMed]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.O.; Vizcaíno, J.A. LipidHome: A Database of Theoretical Lipids Optimized for High Throughput Mass Spectrometry Lipidomics. PLoS ONE 2013, 8, e61951. [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Subramaniam, S. Template-based combinatorial enumeration of virtual compound libraries for lipids. Journal of Cheminformatics 2012, 4. [CrossRef] [PubMed]
- Li, W. Strengthening Semaglutide-GLP-1R Binding Affinity via a Val27-Arg28 Exchange in the Peptide Backbone of Semaglutide: A Computational Structural Approach. Journal of Computational Biophysics and Chemistry 2021, 20, 495–499. [CrossRef]
- Weiss, M. Design of ultra-stable insulin analogues for the developing world. Journal of Health Specialties 2013, 1, 59. [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of Chemical Theory and Computation 2011, 7, 525–537. [CrossRef] [PubMed]
- Müller, C.E.; Hansen, F.K.; Gütschow, M.; Lindsley, C.W.; Liotta, D. New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science. ACS Pharmacology & Translational Science 2021, 4, 1712–1713.
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; Zhang, X.; Gao, G.F. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nature Communications 2017, 8. [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4. [CrossRef]
- Li, W.; Shi, G. How CaV1.2-bound verapamil blocks Ca2+ influx into cardiomyocyte: Atomic level views. Pharmacological Research 2019, 139, 153–157. [CrossRef]
- Li, W. How do SMA-linked mutations of SMN1 lead to structural/functional deficiency of the SMA protein? PLOS ONE 2017, 12, e0178519. [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Methods in Molecular Biology; Springer US, 2020; pp. 239–255.
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nature Structural & Molecular Biology 2003, 10, 980–980. [CrossRef]
- Li, W. Half-a-century Burial of ρ, θ and φ in PDB 2021.
- Li, W. Visualising the Experimentally Uncharted Territories of Membrane Protein Structures inside Protein Data Bank 2020.
- Li, W. A Local Spherical Coordinate System Approach to Protein 3D Structure Description 2020.
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, p. btw514.
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance. Journal of Medicinal Chemistry 2007, 50, 726–741. [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. Journal of Chemical Information and Modeling 2009, 49, 169–184. [CrossRef] [PubMed]
- Song, Q.C.; Tang, C.; Wee, S. Making Sense of Model Generalizability: A Tutorial on Cross-Validation in R and Shiny. Advances in Methods and Practices in Psychological Science 2021, 4, 251524592094706. [CrossRef]
- Zeng, Y.; Chen, X.; Luo, Y.; Li, X.; Peng, D. Deep drug-target binding affinity prediction with multiple attention blocks. Briefings in Bioinformatics 2021, 22. [CrossRef] [PubMed]
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based Methods for Binding Mode and Binding Affinity Prediction for Peptide-MHC Complexes. Current Topics in Medicinal Chemistry 2019, 18, 2239–2255. [CrossRef] [PubMed]
- Kastritis, P.L.; Rodrigues, J.P.; Folkers, G.E.; Boelens, R.; Bonvin, A.M. Proteins Feel More Than They See: Fine-Tuning of Binding Affinity by Properties of the Non-Interacting Surface. Journal of Molecular Biology 2014, 426, 2632–2652. [CrossRef] [PubMed]
- Agrawal, P.; Singh, H.; Srivastava, H.K.; Singh, S.; Kishore, G.; Raghava, G.P.S. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinformatics 2019, 19. [CrossRef]
- Zhang, C.; Han, Q.; Chen, R.; Zhao, X.; Tang, P.; Song, H. SSDRec: Self-Augmented Sequence Denoising for Sequential Recommendation, 2024. [CrossRef]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Computational and Structural Biotechnology Journal 2021, 19, 4538–4558. [CrossRef]
- Stefano, M.D.; Galati, S.; Piazza, L.; Granchi, C.; Mancini, S.; Fratini, F.; Macchia, M.; Poli, G.; Tuccinardi, T. VenomPred 2.0: A Novel In Silico Platform for an Extended and Human Interpretable Toxicological Profiling of Small Molecules. Journal of Chemical Information and Modeling 2023. [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; Schneider, R.G.; Senior, A.W.; Jumper, J.; Hassabis, D.; Kohli, P.; Avsec, Ž. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023. [CrossRef] [PubMed]
- Sadybekov, A.A.; Sadybekov, A.V.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; Zvonok, N.; Jain, M.K.; Savych, O.; Radchenko, D.S.; Nikas, S.P.; Petasis, N.A.; Moroz, Y.S.; Roth, B.L.; Makriyannis, A.; Katritch, V. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 2021, 601, 452–459. [CrossRef] [PubMed]
- Kinghorn, A.; Fraser, L.; Liang, S.; Shiu, S.; Tanner, J. Aptamer Bioinformatics. International Journal of Molecular Sciences 2017, 18, 2516. [CrossRef]
- Lobato, M.; Rabbitts, T. Intracellular Antibodies as Specific Reagents for Functional Ablation: Future Therapeutic Molecules. Current Molecular Medicine 2004, 4, 519–528. [CrossRef] [PubMed]
- Weng, G.; Shen, C.; Cao, D.; Gao, J.; Dong, X.; He, Q.; Yang, B.; Li, D.; Wu, J.; Hou, T. PROTAC-DB: an online database of PROTACs. Nucleic Acids Research 2020, 49, D1381–D1387. [CrossRef] [PubMed]
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: an updated database of PROTACs. Nucleic Acids Research 2022, 51, D1367–D1372. [CrossRef]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinformatics 2017, 18. [CrossRef]
- Rahnama, R.; Kizerwetter, M.; Yang, H.; Christodoulou, I.; Fearnow, A.; Vorri, S.; Spangler, J.; Bonifant, C. Poster: AML-496 Study of Affinity Tuning in AML-Targeted CAR-NK Cells. Clinical Lymphoma Myeloma and Leukemia 2022, 22, S136. [CrossRef]
- Fakhrai-Rad, H. Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Human Molecular Genetics 2000, 9, 2149–2158. [CrossRef]
- González-Casimiro, C.M.; Merino, B.; Casanueva-Álvarez, E.; Postigo-Casado, T.; Cámara-Torres, P.; Fernández-Díaz, C.M.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [CrossRef] [PubMed]
- Ferreira, L.G.; Santos, R.N.; Andricopulo, A.D. Computational methods for calculation of protein-ligand binding affinities. Frontiers in chemistry 2019, 7, 559.
- Petukh, M.; Stefl, S.; Alexov, E. The Role of Protonation States in Ligand-Receptor Recognition and Binding. Current Pharmaceutical Design 2013, 19, 4182–4190. [CrossRef]
- Jubb, H.C.; Pandurangan, A.P.; Turner, M.A.; Ochoa-Montaño, B.; Blundell, T.L.; Ascher, D.B. Mutations at protein-protein interfaces: Small changes over big surfaces have large impacts on human health. Progress in Biophysics and Molecular Biology 2017, 128, 3–13. [CrossRef]
- Li, W. Calcium Channel Trafficking Blocker Gabapentin Bound to the α-2-δ-1 Subunit of Voltage-Gated Calcium Channel: A Computational Structural Investigation 2020.
- Elliott, W.J.; Ram, C.V.S. Calcium Channel Blockers. The Journal of Clinical Hypertension 2011, 13, 687–689. [CrossRef]
- Wu, D.; Piszczek, G. Rapid Determination of Antibody-Antigen Affinity by Mass Photometry. Journal of Visualized Experiments 2021. [CrossRef]
- Mason, D.M.; Friedensohn, S.; Weber, C.R.; Jordi, C.; Wagner, B.; Meng, S.M.; Ehling, R.A.; Bonati, L.; Dahinden, J.; Gainza, P.; Correia, B.E.; Reddy, S.T. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nature Biomedical Engineering 2021, 5, 600–612. [CrossRef] [PubMed]
- Li, W. Extracting the Interfacial Electrostatic Features from Experimentally Determined Antigen and/or Antibody-Related Structures inside Protein Data Bank for Machine Learning-Based Antibody Design 2020.
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuñiga, S.; Enjuanes, L. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biology 2011, 8, 237–248. [CrossRef]
- Iida, K.; Itoh, E.; Kim, D.S.; Rincon, J.P.D.; Coschigano, K.T.; Kopchick, J.J.; Thorner, M.O. Muscle mechano growth factor is preferentially induced by growth hormone in growth hormone-deficientlit/litmice. The Journal of Physiology 2004, 560, 341–349. [CrossRef]
- Zhang, S.; Karthikeyan, R.; Fernando, S.D. Evaluating apoenzyme-coenzyme-substrate interactions of methane monooxygenase with an engineered active site for electron harvesting: a computational study. Journal of Molecular Modeling 2018, 24. [CrossRef] [PubMed]
- Ham, S.W.; Jeon, H.Y.; Kim, H. Verapamil augments carmustine- and irradiation-induced senescence in glioma cells by reducing intracellular reactive oxygen species and calcium ion levels. Tumor Biology 2017, 39, 101042831769224. [CrossRef]
- Rhee, S.G. Regulation of Phosphoinositide-Specific Phospholipase C. Annual Review of Biochemistry 2001, 70, 281–312. [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; Larkin, J.; Lorigan, P.; Neyns, B.; Blank, C.U.; Hamid, O.; Mateus, C.; Shapira-Frommer, R.; Kosh, M.; Zhou, H.; Ibrahim, N.; Ebbinghaus, S.; Ribas, A. Pembrolizumab versus Ipilimumab in Advanced Melanoma. New England Journal of Medicine 2015, 372, 2521–2532. [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. Journal of Neural Transmission 2017, 125, 177–191. [CrossRef] [PubMed]
- Pinheiro, F.; Santos, J.; Ventura, S. AlphaFold and the amyloid landscape. Journal of Molecular Biology 2021, p. 167059. [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; Rives, A. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022.
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; Shoichet, B.K.; Urban, L. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [CrossRef] [PubMed]
- Di Russo, N.V.; Estrin, D.A.; Martí, M.A.; Roitberg, A.E. pH-Dependent conformational changes in proteins and their effect on experimental pKas: the case of nitrophorin 4. PLoS Comput. Biol. 2012, 8, e1002761. [CrossRef] [PubMed]
- Shulman, R.; Wüthrich, K.; Yamane, T.; Patel, D.J.; Blumberg, W. Nuclear magnetic resonance determination of ligand-induced conformational changes in myoglobin. Journal of Molecular Biology 1970, 53, 143–157. [CrossRef]
- Masureel, M.; Martens, C.; Stein, R.A.; Mishra, S.; Ruysschaert, J.M.; Mchaourab, H.S.; Govaerts, C. Protonation drives the conformational switch in the multidrug transporter LmrP. Nature Chemical Biology 2013, 10, 149–155. [CrossRef]
- Peirce, S.M.; Gabhann, F.M.; Bautch, V.L. Integration of experimental and computational approaches to sprouting angiogenesis. Current Opinion in Hematology 2012, 19, 184–191. [CrossRef] [PubMed]
- Cramer, P. AlphaFold2 and the future of structural biology. Nature Structural & Molecular Biology 2021, 28, 704–705. [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; Bodenstein, S.W.; Evans, D.A.; Hung, C.C.; O’Neill, M.; Reiman, D.; Tunyasuvunakool, K.; Wu, Z.; Žemgulytė, A.; Arvaniti, E.; Beattie, C.; Bertolli, O.; Bridgland, A.; Cherepanov, A.; Congreve, M.; Cowen-Rivers, A.I.; Cowie, A.; Figurnov, M.; Fuchs, F.B.; Gladman, H.; Jain, R.; Khan, Y.A.; Low, C.M.R.; Perlin, K.; Potapenko, A.; Savy, P.; Singh, S.; Stecula, A.; Thillaisundaram, A.; Tong, C.; Yakneen, S.; Zhong, E.D.; Zielinski, M.; Žídek, A.; Bapst, V.; Kohli, P.; Jaderberg, M.; Hassabis, D.; Jumper, J.M. Accurate structure prediction of biomolecular interactions with AlphaFold3. Nature 2024. [CrossRef] [PubMed]
Figure 1.
Automated in silico generation of synthetic structural and Kd data.
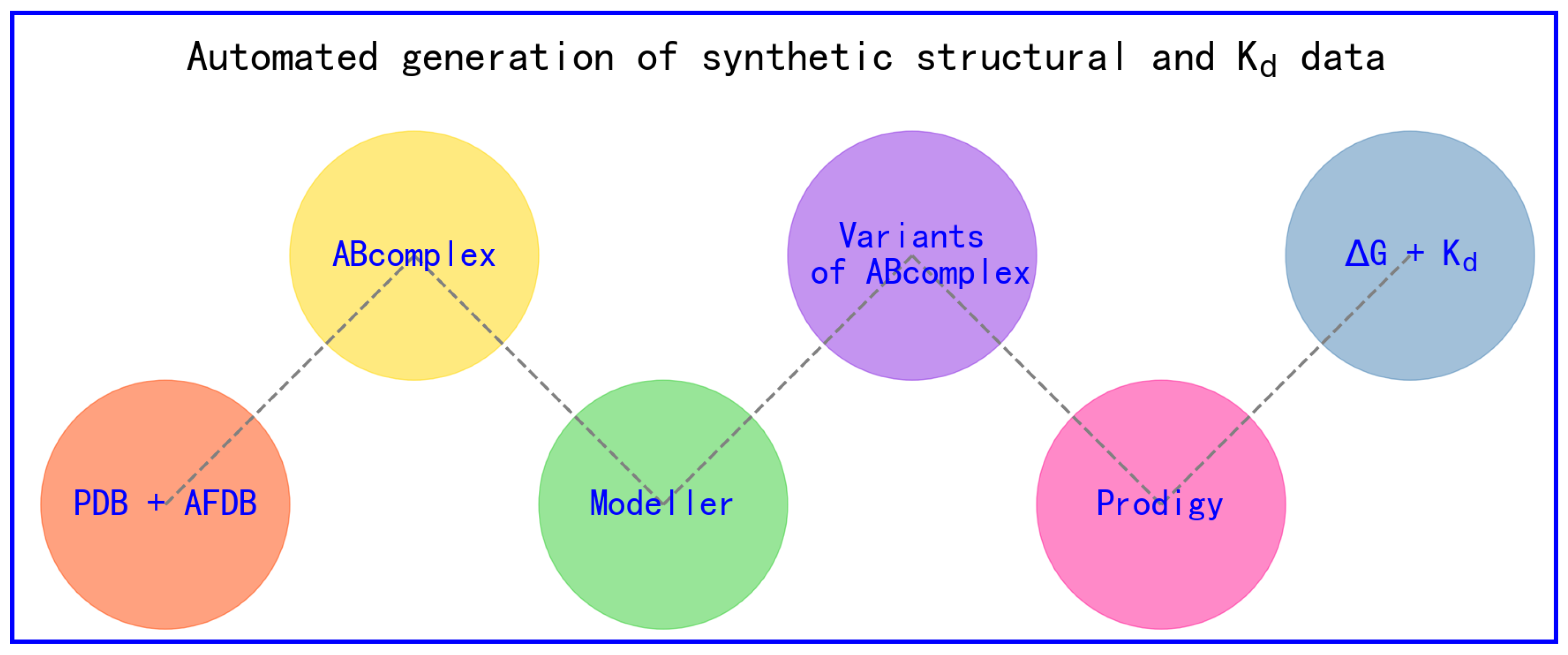
Figure 2.
A detailed flowchart of automated in silico generation of synthetic structural and Kd data. This figure is a detailed version of Figure 1.
Figure 2.
A detailed flowchart of automated in silico generation of synthetic structural and Kd data. This figure is a detailed version of Figure 1.

Figure 4.
A future direction of in silico generation of structural and intermolecular Kd data in drug discovery & design, and the pharmaceutical industry as a whole.
Figure 4.
A future direction of in silico generation of structural and intermolecular Kd data in drug discovery & design, and the pharmaceutical industry as a whole.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



