
Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Traumatic brain injury (TBI) affects millions of people each year. Previous studies using the central fluid percussion injury (CFPI) model in adult male rats indicated that elevated intracranial pressure (ICP) was associated with long-term effects, including neuronal cell loss and increased sensory sensitivity post-injury and secondary ICP elevation, that were not seen following injury alone. Investigations also indicated that Cathepsin B (Cath B), a lysosomal cysteine protease, may play a role in the pathological progression of neuronal membrane disruption; however, the specific impact of Cath B inhibition following CFPI remains unknown. Thus, the focus of this study was to evaluate the effects of Cath B inhibition via intracerebroventricular infusion of CA-074Me 2w following injury with or without secondary elevation of ICP. This was accomplished using adult male rats continuously infused with CA-074Me or 10% DMSO as a vehicle control for 2w following either sham injury, a CFPI only, or a CFPI with subsequent ICP elevation to 20mmHg. We assessed Cath B activity in the lateral neocortices and liver, evaluated protein levels of Cath B and Cath B binding partners AIF, Bcl-XL, and Bak. We also conducted histological analyses of total cell counts to assess for cell loss, membrane disruption, and Cath B localization. Finally, we investigated somatosensory changes with the whisker nuisance task. Overall, this study demonstrated that Cath B is not a direct driver of membrane disruption, however, administration of CA-074Me alters Cath B localization and reduced hypersensitivity, emphasizing Cath B being an important component in late secondary pathologies.
Keywords:
Cathepsin B
; Intracranial pressure
; Neuronal Membrane disruption
; Rat
; Somatosensory sensitivity
; Traumatic brain injury
1. Introduction
Traumatic brain injury (TBI) affects approximately 60 million people globally [1] and 2.8 million people in the United States each year [2], with a rate of 759 TBIs for every 100,000 individuals reported globally in 2016[3]. The annual number of TBIs is likely even higher as less severe TBIs often go unreported. This life-altering event can produce impairments in motor, cognitive, sensory, and affective function that can persist chronically[4] precipitating significant costs to patients, families, and communities. Moreover, secondary elevations in intracranial pressure (ICP) to levels at or above 20mmHg following TBI can increase TBI-induced morbidity and mortality[5,6]. The current treatments and strategies for managing ICP elevation following a TBI are limited, and the most aggressive treatment involves invasive craniectomies [5]. While we have some indications of the pathological changes induced by elevated ICP following TBI, these changes, particularly the diffuse pathologies that are associated with TBI, are difficult to investigate in human patients and the molecular cascades involved remain nebulous. We, therefore, utilize the central fluid percussion injury (CFPI) model in rodents, which replicates the diffuse pathologies induced by TBI, paired with specific manipulations of ICP [7].
Previous studies from our lab using this CFPI model in adult male rats indicated that elevated ICP to 20mmHg was associated with long-term effects, including neuronal cell loss and increased sensory sensitivity at 4w following CFPI and ICP elevation, that were not seen following injury alone[7]. Additionally, acute neuronal membrane disruption was nearly doubled with secondary intracranial pressure elevation hours following TBI[7,8]. Neuronal membrane disruption refers to the perturbation of the neuronal plasma membrane and is visualized using cell impermeable tracers, including dextran conjugated with fluorescent labels. Membrane disruption has been demonstrated to occur acutely and sub-acutely in vitro and in vivo in mice, rats, and swine following TBI and spinal cord injury[7,8,9,10,11,12,13,14,15,16,17,18,19]. Recently, it was found that neuronal membrane disruption in the lateral neocortex occurs biphasicaly, in which there is significant membrane disruption sub-acutely (6hr-3d post CFPI) that is reduced at 1w post-CFPI and reemerges at more chronic timepoints (2w-4w post CFPI) [20].
Previous investigations indicated that Cathepsin B (Cath B), a lysosomal cysteine protease, may play a role in the pathological progression of neuronal membrane disruption. It was shown that Cath B localizes out of the lysosomal compartment and into the cytosol, specifically in the membrane disrupted neurons at 2w and 4w post-injury [8,21]. Cath B has been known to participate in various cell damage and death processes [22,23,24,25,26,27,28,29,30,31,32,33]. Moreover, Cath B has been implicated in the pathologies seen in other models of TBI, such as protein upregulation, lysosomal permeability, cell death, and behavioral deficits [34,35,36].
Thus, the focus of this study was to evaluate the effects of Cath B inhibition on neuronal membrane disruption 2w following injury and ICP elevation. This was accomplished using adult male rats continuously treated with the Cath B inhibitor, CA-074 methyl ester (CA-074Me), or the vehicle for CA-074Me, 10% dimethylsulfoxide (DMSO) in sterile saline, for 2w following either 1) sham injury, 2) a CFPI, or 3) a CFPI with subsequent ICP elevation to 20mmHg. We found that Cath B does not appear to be a direct driver of membrane disruption at 2w following CFPI; however, Cath B may play a role in TBI-induced and ICP exacerbated sensory hypersensitivity, warranting further investigations. We further found that 10% DMSO reliably reduced neuronal membrane disruption.
2. Materials and Methods
2.1. Animals
Experiments were conducted using protocols in accordance with the Virginia Commonwealth University institutional ethical guidelines concerning the care and use of laboratory animals (Institutional Animal Care and Use Committee, Virginia Commonwealth University), which adhere to regulations including, but not limited to, those set forth in the Guide for the Care and Use of Laboratory Animals, 8th Edition (National Research Council). Animals were housed in individual cages on a 12h light-dark cycle with free access to food and water. A total of 78 adult male Sprague-Dawley rats weighing 320–420g, n=36 for protein analyses and n=36 for histological assessments were used for this study. There were 12 animals in each group (1) Sham injury with 10% DMSO infusion, (2) Sham injury with CA-074Me infusion, (3) TBI with 10% DMSO infusion, (4) TBI with CA-074 infusion, (5) TBI+ ICP elevation with 10% DMSO infusion, and (6) TBI+ ICP elevation with CA-074Me infusion (n = 6/group for histological assessments and n=6/group for molecular assessments). Our a priori exclusion criteria included weight loss of more than 20% or gross brain pathology (contusion, subdural hematoma, or gross tissue loss). Six animals met exclusion criteria in this study and were excluded. All surgeries were conducted by the same surgeon during the same times of day to reduce variability. Animal injury state and drug infusions were randomly determined using a random number generator and prepared by a separate investigator from the surgeon.
2.2. Surgical Preparation and Injury Induction
Animals were anesthetized with 4% isoflurane in 30% O2 and 70% room air and ventilated with 2% isoflurane in 30% O2 and 70% room air throughout the duration of the surgery, injury, and post-injury physiological monitoring. Heart rate, respiratory rate, and blood oxygenation were monitored via a hind-paw pulse oximetry sensor (STARR Life Sciences, Oakmont, PA) for the duration of anesthesia, except during the induction of injury. Body temperature was maintained at 37°C with a rectal thermometer connected to a feedback-controlled heating pad (Harvard Apparatus, Holliston, MA). All animals were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). A midline incision was made, followed by a 4.8mm circular craniectomy made along the sagittal suture midway between bregma and lambda for placement of the injury hub. A 2mm burr hole was also drilled in the left parietal bone overlying the left lateral ventricle (0.8mm posterior, 1.3mm lateral, and 2.5-3mm ventral to bregma) through which a 25-gauge needle, connected to a pressure transducer and micro-infusion pump (11 Elite syringe pump; Harvard Apparatus) via sterile saline-filled PE50 tubing, was placed into the left lateral ventricle. Appropriate placement was verified via a 1.3μl/min infusion of sterile saline within the closed fluid-pressure system during needle placement with the drop in pressure indicating breach of the lateral ventricle [8,10]. This method of validating ventricle cannulation does not increase the intracranial pressure (ICP). The needle was held in the ventricle for at least 5min while recording pre-injury ICP. After this pre-injury reading, the needle was slowly removed, and the burr hole was covered with bone wax before preparation for sham or CFPI [7,37]. Briefly, a Leur-Loc syringe hub was affixed to the craniectomy site, and dental acrylic (methyl-methacrylate; Hygenic Corp., Akron, OH) was applied around the hub and allowed to harden. Anesthetized animals were removed from the stereotaxic frame and injured with a fluid pulse of 2.05±0.16 atmospheres and ~22msec. The pressure pulse was measured by a transducer affixed to the injury device and displayed on an oscilloscope (Tektronix, Beaverton, OR). Immediately after the injury, the animal was reconnected to the ventilator and physiologic monitoring device and the hub and dental acrylic were removed en bloc. Surgifoam was placed over the craniectomy/injury. The animal was then replaced in the stereotaxic device and the ICP probe was reinserted into the lateral ventricle, as described above, for post-injury ICP monitoring. At 15 minutes following injury, animals were given 0.9mg/kg buprenorphine-HCl slow release (Bup-HCl-SR) subcutaneous (SQ) as an analgesic and then monitored until 1 hour following injury. Monitoring of ICP was done for both sham and TBI only animals. For animals in the TBI and 20mmHg ICP elevation group, at 15min post-CFPI ICP was elevated to 20mmHg via infusion of sterile normal saline at 1.3-13µl/minute using the micro-infusion Pump 11 Elite syringe pump controlled by the experimenter. Once 20mmHg was achieved, ICP was maintained at 20mmHg until 1 hour after injury. At 1hr post-CFPI and ICP monitoring or elevation, animals underwent drug or vehicle bolus infusion, followed by subcutaneous implantation of a mini-osmotic pump (Alzet; Model 2002). The animals were then sutured, treated with lidocaine and triple-antibiotic ointment, recovered, and returned to a clean home cage. Identical surgical procedures were followed for sham-injured animals, without release of the pendulum to induce injury [7,20].
2.3. Drug Preparation and Infusion Administration
The solid form of Cath B inhibitor, CA-074 methyl ester (CA-074Me; MedChemExpress, Cat#: HY-100350, Monmouth Junction, NJ), was stored at -20°C until reconstituted in 100% dimethylsulfoxide (DMSO) at a concentration of 100 µg/µL. The 31µL reconstituted aliquots were stored at -80°C. Prior to surgery, an investigator separate from the surgeon used a random number generator to determine the animal’s injury and inhibitor infusion group. For CA-074Me infusion animals, an aliquot was defrosted and diluted in sterile 0.9% saline to a final concentration of 10µg/µl by an investigator separate from the surgeon. The solution (CA-074Me in 10% DMSO-sterile saline or 10% DMSO-sterile saline) was pipetted into tubes labeled with the date of the procedure to maintain surgeon blinding and stored at 37°C. Sterile preparation of the 250 µL mini-osmotic pump (Alzet; Model 2002) with catheter tube and brain cannula was performed by the surgeon the day prior to surgery following infusion blinding. The prepared pump was placed in a sterile tube with saline to initiate osmotic pressure and then incubated overnight at 37°C alongside the tube containing the bolus infusion. One hour following injury, the 12.5µL bolus was infused into the left lateral ventricle. Following bolus infusion, the caudal end of the midline incision was blunt dissected to lift the skin over the shoulder blades allowing for the implantation of the osmotic pump and brain infusion cannula. The cannula was placed into the burr hole for lateral ventricle using a stereotaxic cannula holder (0.8mm posterior, 1.3mm lateral, and 2.5-3mm ventral to bregma). The cannula was held in place with cyanoacrylate. The animals were then sutured, treated with lidocaine and triple-antibiotic ointment, recovered, and returned to a clean home cage. The pump infused at a rate of 0.5µL/hr for the entire 2w post-injury period.
2.4. Tracer Infusion
One hour prior to sacrifice at 2w post-sham or CFPI, tagged dextran (0.6mg/25 µg in sterile 0.9% saline; ~1.6mg/kg) was infused into the lateral ventricle in the animals being prepared for histological assessment (n=6/group) as described previously[8,20]. Briefly, the bur hole overlying the left lateral ventricle was reopened and another bur hole was drilled over the right lateral ventricle (0.8mm posterior, 1.3mm lateral, and 2.5-3mm ventral to bregma). 12.5µl of 10kDa dextran conjugated to 488-Alexa Fluor (Cat#: D22910, Invitrogen, Carlsbad, CA) was infused into the left then right lateral ventricle at 1.3µl/min with continuous ICP monitoring. The tracer was allowed to diffuse throughout the parenchyma for 1hr prior to transcardial perfusion.
2.5. Tissue Processing
At 2w post-injury, the animals were injected with 150mg/kg euthanasia-III solution (Henry Schein, Dublin, OH), then underwent transcardial perfusion with cold 0.9% saline. In half the animals(n=6/group), both left and right lateral neocortices were dissected and frozen for protein expression and activity assessments (n=36). Animals used for histological assessments (n=6/group; n=36) underwent the aforementioned euthanasia process, transcardial perfusion of cold 0.9% saline followed by a switch in transcardial perfusate to 4% paraformaldehyde/ 0.2% glutaraldehyde in Millonig’s buffer (136mM sodium phosphate monobasic/109mM sodium hydroxide) to fix the brain for subsequent immunohistochemical processing and analysis. After transcardial perfusion, the brains were removed, post-fixed for 24-48hr, then sectioned coronally at a thickness of 40µm in 0.1M phosphate buffer with a vibratome (Leica, Banockburn, IL) from the level of bregma to ~4.0mm posterior to bregma. Sections were collected serially in 12 well-plates and stored in Millonig’s buffer at 4°C. A random starting well (wells 1–12) was selected using a random number generator and four serial sections were used for histological analyses. All histological analyses were restricted to layers V and VI of the lateral somatosensory neocortex extending from the area lateral to CA1 to the area lateral to CA3 of the hippocampus.
2.6. Quantification of Cathepsin B Activity
Portions of the dissected left and right lateral neocortices and livers of the sham, TBI, and TBI+ICP animals were mechanically homogenized in 50μM citric acid at a pH 6.0, spun at 12,000xg at 4°C for 10 minutes, and the supernatant of the whole homogenate was collected. Protein concentrations were measured using a NanoDrop Lite (Thermo Fisher Scientific, Wilmington, DE) and Cath B activity was measured in a 96-well plate, each well containing 2x Assay Buffer (100mM sodium acetate pH 5.5, 2mM EDTA, 200mM sodium chloride, 8mM DTT), 2μg of neocortex whole homogenate, and 1mM Z-Phe-Arg-7-amino-4-(trifluoromethyl) coumarin (ZFR-AMC), a substrate for cysteine proteases that fluoresces upon cleavage by Cath B [21,34,35,38]. The plate was read on a PHERAstar microplate reader (BMG Labtech, Cary, NC, USA) at 60-minutes post-substrate addition at 365/450 nm excitation/emission. Each sample was loaded in triplicate per plate and was run on three independent plates to reduce pipetting and run-to-run variability biasing the results. A positive control well with 5ng of purified human liver Cath B and a negative control well with only assay buffer and substrate were included on every plate. The raw arbitrary fluorescent values depicting activity were measured and expressed as fold increase compared to the negative control, which was designated as 0-fold activity.
2.7. Western Blotting
Left lateral neocortices of (1) Sham injury with 10% DMSO infusion, (2) Sham injury with CA-074Me infusion, (3) TBI with 10% DMSO infusion, (4) TBI with CA-074Me infusion, (5) TBI+ ICP elevation with 10% DMSO infusion, and (6) TBI+ ICP elevation with CA-074Me infusion (n = 6/group), were homogenized in western lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% Triton) and protease inhibitor cocktail (AEBSF 10.4mM, Aprotinin 8μM, Bestatin 400μM, E-64 140μM, Leupeptin 8μM, Pepstatin A 150μM, Cat#: P8340, Sigma, Saint Louis, MO). Protein concentrations were measured using a BCA Protein Assay (Pierce Biotechnology, Thermo Fisher Scientific, Rockford, IL, Cat#: 23227) according to manufacturer’s instructions and read on a PHAREstar plate reader (BMG-Lab tech, Cary NC, USA). Protein (20μg for Cath B, 5μg for Bcl-XL, 10μg for Bak and AIF) was boiled for 10min in 50mM dithiothreitol (Cat#: 1610610; Bio-Rad; Hercules, CA), 2x Laemelli loading buffer (Cat#: 1610737; Bio-Rad; Hercules, CA) and run at 200 Volts for 30min on Mini-PROTEAN TGX Stain-Free 4-20% precast polyacrylamide gels (Cat#: 4568096; Bio-Rad, Hercules, CA). Protein was transferred onto 0.45μm PVDF membranes using a Bio-Rad Transblot Turbo transfer system set to the Mixed Molecular Weight manufacturer setting (1.3-2.5 Amps, 25 Volts for 7min). Western blotting was done on an iBind Flex apparatus (Invitrogen, Carlsbad, CA) using primary antibodies rabbit anti-Cath B (1:1000; Cat#: 31718S, RRID:AB_2687580), rabbit anti-Bak (1:1000; Cat#: 12105S, RRID:AB_2716685), rabbit anti-Bcl-xl (1:1000; Cat#: 2764S, RRID:AB_2228008) or rabbit anti-AIF (1:1000; Cat#: 5318S, RRID:AB_10634755). All primary antibodies were purchased from Cell Signaling Technology, Danvers, MA. Secondary antibody anti-rabbit-HRP (1:5000; Cat#: 111-035-003; Jackson ImmunoResearch Laboratories, West Grove, PA, RRID:AB_2313567) was used for all assays. Total protein (Stain Free) and chemiluminescent images were taken on a ChemiDoc imaging system (BioRad) holding the exposure time consistent for each protein assessed. Densitometric analyses of Cath B, AIF, Bak, and Bcl-XL were performed in ImageJ (National Institutes of Health; Bethesda, MD). Cath B, AIF, Bak and Bcl-XL protein bands were normalized to total loaded protein then to a naive control sample that was included in all runs. All western blots were run in triplicates on three separate gels to reduce run-to-run variability biasing the results.
2.8. Membrane Disruption and Total Cell Count Analysis
Consistent with previous studies, we assessed the potential for neuronal membrane disruption via the utilization of 10kDa dextran, which is impermeable to cells with intact membranes [8]. Fluorescently tagged dextran-containing cells, indicative of membrane perturbation, could be visualized via fluorescent microscopy without further processing. To identify individual neurons, four tissue sections from (1) Sham injury with 10% DMSO treatment, (2) Sham injury with CA-074Me treatment, (3) TBI with 10% DMSO treatment, (4) TBI with CA-074 treatment, (5) TBI+ ICP elevation with 10% DMSO, and (6) TBI+ ICP elevation with CA-074Me groups (n= 6/group) were stained with 1:500 dilution of NeuroTrace 435/455 blue fluorescent Nissl Stain (Life Technologies, Eugene, OR, Cat# N21479). The tissue was mounted onto slides using Vectashield Vibrance mounting medium (Cat# H-1700; Vector Laboratories, Burlingame, CA). Sections were imaged with fluorescent optical sectioning microscopy using a Keyence BZ-X800 microscope (Keyence Corporation of America, Itasca, IL, USA). Quantitative analysis was performed as described previously [7]. Briefly, images (20-35/animal) of the left neocortical region of interest were taken at 40X magnification in a systematically random fashion by a blinded investigator using NeuroTrace to verify focus. Image acquisition settings were held constant for comparable regions (layer V or VI) for all groups analyzed. Analyses of neurons exhibiting dextran uptake were performed using the ImageJ cell counting plug-in and verified cellular identity using the NeuroTrace staining. Dextran containing neurons were quantified for each image and averaged for each animal.
To assess for cell loss the total number of NeuroTrace stained neurons were counted using the Hybrid Cell Counter function of the Keyence BZ-X800 Analyzer software. Briefly, images were set to simple thresholding with uniform brightness and no smoothing. Auto thresholding was used to highlight the NeuroTrace positive label followed by watershed cell separation and removal of cell bodies smaller than 15μm in diameter to exclude smaller glial cells and fragments of cell bodies from the analysis.
2.9. Cellular Cathepsin B Localization Analysis
Fluorescently tagged dextran-containing cells, indicative of membrane disruption, could be visualized via fluorescent microscopy without further processing. To visualize Cath B, three sections/ animal and 6 animals/ group were labeled with Cath B. Tissue sections were blocked with 5% normal goat serum (NGS), 2% bovine serum albumin (BSA), and permeabilized with 1.5% triton-X for 2h. This was followed by immunolabeling using primary antibodies rabbit anti-Cath B (1:700; Cat#: 31718; Cell Signaling Technology; Danvers, MA; RRID:AB_2687580) and mouse anti-NeuN (1:500; Cat#: MAB377; MilliporeSigma; Temecula, CA, RRID:AB_2298772). Tissue was incubated in secondary antibodies Alexa-647 conjugated goat anti-mouse(1:700; Cat#: A32728; Life Technologies, Rockford, IL; RRID:AB_10563566) and Alexa-568 conjugated goat anti-rabbit (1:800; Cat#: A11036; Life Technologies, Eugene, OR; RRID:AB_2534102), and the tissue was mounted onto slides using Vectashield hardset mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) (Cat#: H-1500; Vector Laboratories, Burlingame, CA). Quantitative analysis was performed as described previously[8,20]. Briefly, 5-6 images from the 3 sections/animal of the left neocortical region of interest were taken at 40X magnification using a Keyence BZ-X800 microscope (Keyence Corporation of America, Itasca, IL, USA) in a systematically random fashion by an investigator blinded to the animal group using the dextran tag to verify images included neurons with and without membrane disruption. Image acquisition settings were held constant for comparable regions (layer V or VI) and 488-dextran for all groups analyzed. The images including 488-dextran, DAPI, and Cath B were analyzed in FIJI (Image J). DAPI was used to identify nuclei of cells containing and not containing 488 dextran. Ten non-disrupted and ten membrane disrupted cells assessed in each image (n=5-6 images/ animal). If a total of 10 neurons that were membrane disrupted could not be found an equal number of membrane disrupted and non-disrupted neurons were captured in that image; n=6 animals/group (1) Sham injury with 10% DMSO treatment, n=373 cells, (2) Sham injury with CA-074Me treatment, n=424 cells, (3) TBI with 10% DMSO treatment, n=442 cells,(4) TBI with CA-074Me treatment, n=480 cells, (5) TBI+ ICP elevation with 10% DMSO, n=497 cells, and (6) TBI+ ICP elevation with CA-074Me, n=432 cells. Cells were classified by Cath B appearance of punctate (intra-lysosomal) or diffuse (extra-lysosomal) as has been done previously [8,21]. The localization of Cathepsin B was expressed as the percentage of the total membrane disrupted or non-disruption neuronal population assessed in each experimental group that demonstrated punctate (lysosomal) Cath B localization.
2.10. Whisker Nuisance Task
Hypersensitivity in the whisker barrels was assessed using the whisker nuisance task (WNT)[7,39,40]. Briefly, animals were assessed prior to injury and at 2w post sham or CFPI by an investigator blind to animal group. For this assessment the animal was placed inside a clean plastic open field lined with a chux pad and allowed to acclimate for 5 minutes. A wooden applicator stick was brushed along the rat’s whiskers on each side, for a 5-minute trial period followed by 1min rest between each of 3 total trials. During these trials the following behaviors were scored on a scale of 0 to 2 (0 = normal, 1 = exhibits some nuisance behavior, 2 = exhibits profound nuisance behavior): (1) movement, (2) stance and body position, (3) breathing, (4) whisker position, (5) whisker response, (6) evading stimulation, (7) response to stick presentation, (8) grooming, (9) ear position, (10) sniffing, (11) fur ruffling, and (12) urination/defecation (Supplemental Table S1). The individual behavior scores for each animal were summed for each trial with the highest possible score being 24, and the sums were averaged for the three trials. A higher WNT score indicates more pronounced agitation/sensitivity.
2.11. Statistics
Data were tested for normality prior to utilizing parametric or non-parametric assessments, which were conducted using SPSS (IBM Corporation, Armonk, NY) software. Animal numbers for each group were determined by an a priori power analysis using effect size and variability previously observed in the lab when assessing pathology between sham and injured groups using the CFPI model, an alpha =0.05, and a power of 80%. Two-way or one-way analysis of variance (ANOVA) and Bonferroni post hoc tests were performed for all between-group analyses. Statistical significance was set to p<0.05. Data are reported as mean ± standard error of the mean (SEM).
3. Results
3.1. Cathepsin B activity Was Decreased in the Left and Right Cortex after 2w of Continuous CA-074Me infusion
To validate the effectiveness of the intracerebroventricular (ICV) administration of CA-074Me, we measured the activity of Cath B in both the left and right cortices of animals infused with either 10% DMSO vehicle or CA-074Me into the left lateral ventricle (intracerebroventricularly, ICV) over the 2w period following sham, CFPI, or CFPI with secondary ICP elevation to 20mmHg. We also assessed Cath B activity within the liver of each animal to verify that systemic Cath B activity was not significantly impacted with our ICV infusion paradigm. There was no difference in Cath B activity in the liver between the injury groups sham, CFPI, CFPI+ICP elevation (One-way ANOVA injury group F2,93=1.2, p=0.3). As expected, Cath B activity in the brain was significantly decreased in animals following ICV infusion of CA-074Me compared to the 10% DMSO infused animals (One-way ANOVA infusion group F1,93=24.5, p=3.0X10-6; Figure 1). The liver demonstrated much higher Cath B activity compared to either the left or right lateral neocortices (One-way ANOVA region F2,93=41.5, p=1.3X10-13), however, there was no significant difference in Cath B activity within the left cortex compared to the right cortex (p=0.85). This was consistent for each injury group in which Cath B activity in the left and right cortex was significantly lower compared to the liver (liver vs. left cortex p=1.7X10-12; liver vs. right cortex p=3.0X10-10, Figure 1, Supplementary Table S2).
There was no interaction found between the inhibitor infusion and the injury group (Two-way ANOVA F2,93=0.21, p=0.81). However, there was an interaction between infusion of CA-074Me and the region assessed, in which there was a significant decrease in Cath B activity in both the left and right cortices of animals infused with CA-074Me ICV that was not reflected in the liver (Two-way ANOVA F2,93=5.64, p=0.005). There was also a trend toward an interaction between injury group and region (Two-way ANOVA F4,93=2.306, p=0.064) driven by less obvious reductions in Cath B activity in the right cortex of CFPI and CFPI+ICP elevation animals administered CA-074Me compared to vehicle controls. There was no interaction of injury group, inhibitor infusion, and region for Cath B activity (Three-way ANOVA F4,93 = 1.013, p = 0.405).
3.2. Protein Expression of Cathepsin B and Other Signaling Partners; Bcl-XL, Bak, and AIF
The overall expression of Cath B within the left lateral neocortex was evaluated in all injury and infusion groups. Western blot analysis revealed that total Cath B protein levels were consistent across all groups (F5,30=0.64, p=0.67; Figure 2; Supplemental Table S2). There was no effect of injury group (One-way ANOVA injury group F2,30=0.1, p=0.9) or inhibitor infusion (One-way ANOVA infusion group F1,30=2.3, p=0.14) on Cath B protein quantity. There was no interaction between the injury group and infusion of CA-074Me on Cath B protein quantity (Two-way ANOVA F2,30=0.35, p=0.71). As we observed two distinct bands in our Cath B westerns, we also assessed the potential changes in each band. There was no significant difference across groups for the upper Cath B band (One-way ANOVA F5,30=0.41, p=0.83). While, the lower Cath B was not significantly increased in any specific individual group (One-way ANOVA F5,30=1.16, p=0.35), there was a significant effect of infusion group (F1,30=5.25, p=0.029) in which CA-074Me infused. animals had higher expression of the lower Cath B band (Figure 2E). There was no effect of injury group (F2,30=0.03, p=0.97) or interaction between injury and infusion groups (Two-way ANOVA F2,30=0.24, p=0.79) on the lower Cath B band.
Cath B has been shown to participate in various cell damage pathways through downstream proteins such as AIF, Bcl-XL, and Bak [22,23,30,41]. While a previous study indicated that protein expression of AIF, as well as the expression ratio of the antagonizing proteins Bcl-XL and Bak, were consistent from 6h to 4w following CFPI [21], the potential impact of secondary ICP elevation and/or Cath B inhibition following CFPI on such downstream proteins remained unknown. Therefore, we assessed the expression levels of Bcl-XL, Bak, and AIF in the left and right lateral neocortices in all injury and infusion groups.
There were no significant differences in the levels of the pro-survival protein Bcl-XL across all groups (F5,30=0.26, p=0.93; Figure 3, Supplemental Table S2). There was no effect on Bcl-XL protein quantity from the injury group (One-way ANOVA injury group F2,30=0.25, p=0.78) or inhibitor infusion (One-way ANOVA infusion group F1,30=0.69, p=0.41). There was also no interaction between the injury group and infusion group (Two-way ANOVA F2,30=0.06, p=0.94; Figure 3). As Bcl-XL presented as a doublet, we also assessed the potential changes in both the upper and lower bands independently. There were no differences between groups in either the upper band (F5,30=0.32, p=0.89) or lower band (F5,30=0.3, p=0.91) of this doublet. Quantities of the anti-survival Bak protein were also consistent across groups (F5,30=0.05, p=0.99; Figure 4; Supplemental Table S2). Injury group (One-way ANOVA injury group F2,30=0.015, p=0.99) and inhibitor infusion (One-way ANOVA infusion group F1,30=0.05, p=0.83) did not influence Bak protein levels. No interactions between injury group or inhibitor infusion were found (Two-way ANOVA F1,30=0.09, p=0.91).
There were two distinct bands in the AIF western, so both were assessed. Protein levels of both bands of AIF were consistent across all groups (Upper band: F5,30=0.13, p=0.99; Lower band: F5,30=1.24, p=0.33; Figure 5, Supplemental Table S2). AIF protein levels were not directly impacted by the inhibitor infusion (One-way ANOVA Infusion group Upper band F1,30=0.13, p=0.72; Lower band F1,30=0.12, p=0.73) or injury group (One-way ANOVA injury group Upper band F2,30=1.98, p=0.82; Lower band F2,30=0.8, p=0.46). Despite there being a slight decrease in AIF protein levels in the CFPI+ICP elevation group infused with CA-074Me, primarily driven by changes in the lower AIF band for this group, these differences were not significant and there was no interaction between injury group and infusion (Two-way ANOVA Upper band AIF F2,30=0.05, p=0.95; Lower band AIF F2,30=2.2, p=0.13; Figure 5).
3.3. Elevation of ICP and/or Infusion of CA-074Me Did Not Impact the Number of Neurons in the Lateral Neocortex Following CFPI
Fluorescent Neurotrace 435 was used to identify Nissl substance within cells of the left lateral neocortex in sham, CFPI, or CFPI+ICP elevated animals given either 10%DMSO or CA-074Me (Figure 6). The number of NeuroTrace+ cells with cell bodies >15μm2, to enrich for neuronal profiles, in each image (0.098 mm2) was counted to assess the potential for cell loss over the 2w post-injury period. There was no indication of cell loss in layers V and VI of the lateral neocortex in any group (F5,30=0.362, p=0.87; Figure 7; Supplemental Table S2). There was no effect on the total cell count from the injury group (One-way ANOVA injury group F2,30=0.63, p=0.54) or from inhibitor infusion (One-way ANOVA infusion group F1,30=0.47, p=0.49). There was no interaction between the injury group and infusion group (Two-way ANOVA F2,30=0.043, p=0.96; Figure 7).
3.4. Neuronal Membrane Disruption Was Not Significantly Altered Following CFPI in the Presence of 10% DMSO or CA-074Me Regardless of ICP Elevation
To determine the impact of Cath B inhibition via CA-074Me ICV infusion and/or secondary elevations in ICP following CFPI at 2w post-injury, we infused tagged dextran ICV prior to sacrifice at 2w. Fluorecent Nissl NeuroTrace+ neurons (>15um in diameter) containing the normally cell impermeable dextran were considered membrane disrupted and were quantified throughout layers V and VI of the left lateral neocortex (Figure 6). Neuronal membrane disruption was consistent in all groups (F5,30=0.93 p=0.47; Figure 8; Supplemental Table S2). Sham animals demonstrated levels of neuronal membrane disruption consistent with previous studies [7,20,40] in which the sham injury group infused with 10% DMSO had 7.83% ± 2.16 neurons sustaining membrane disruption and the sham injury group infused with CA-074Me had 12.09% ± 3.91 neurons sustaining membrane disruption (Figure 6 A & D, Figure 8). Rats sustaining CFPI demonstrated membrane disruption in a much lower percentage of neurons than anticipated based on previous findings [7,20,40] in which the CFPI group infused with 10% DMSO had 6.56% ± 1.13 neurons sustaining membrane disruption and the CFPI group infused with CA-074Me had 10.86% ± 3.08 neurons sustaining membrane disruption (Figure 6 B & E; Figure 8). At percentages similar to those in sham and CFPI groups, animals sustaining CFPI with secondary ICP elevation to 20mmHg also demonstrated minimal neuronal membrane disruption, in which the TBI+ ICP elevation group infused with 10% DMSO had 10.72% ± 2.60 neurons sustaining membrane disruption and the TBI+ ICP elevation group infused with CA-074Me had 6.34% ± 1.28 neurons sustaining membrane disruption (Figure 6 C & F; Figure 8). There were no significant effects of injury group (One-way ANOVA injury group F2,30=0.19 p=0.83) inhibitor infusion (One-way ANOVA F1,30=0.45 p=0.51), or interactions between injury group and inhibitor infusion (Two-way ANOVA F2,30=1.92 p=0.16).
3.5. Cathepsin B Re-Localizes from Lysosomes to Cytosol in Disrupted Neurons at 2 Weeks Following Injury
Re-localization/redistribution of Cath B out of the lysosome and into the cytosol has previously been seen in neuronal membrane disrupted populations both acutely and weeks following CFPI [8,21]. The longer-term effects of secondary ICP elevation and/or Cath B inhibition on Cath B localization; however, remain nebulous. To investigate potential changes in the localization of Cath B within neuronal membrane disrupted and non-disrupted neurons immunohistochemistry for Cath B paired with quantitative image analysis was done [8,21]. The localization of Cath B within lysosomal puncta was assessed for membrane disrupted and non-disrupted neurons in each group at 2w post injury (Figure 9). As expected, there was an effect of membrane
disruption on Cath B localization (One-way ANOVA membrane disruption state F1,2633= 152.06, p=5.2X10-34) in which Cath B was localized primarily within lysosomal puncta in non-disrupted neurons compared to neurons sustaining membrane disruption in most groups, in which Cath B was diffusely distributed throughout the cytoplasm of the soma (One Way ANOVA F11,2632=20.46 p=2.96 X10-40, Bonferroni post hoc non-disrupted vs. membrane disrupted; Sham CA-074Me p= 2.6X10-17; TBI 10% DMSO p= 4.67X10-4; TBI CA-074Me p=0.025; TBI+ICP elevation 10% DMSO p=0.014; TBI+ICP elevation CA-074Me p=3.0X10-6; Figure 10; Supplemental Table S2). The only exception was the sham injured group infused with 10% DMSO, in which there were no difference in Cath B localization between the non-disrupted and membrane disrupted neuronal populations (p=0.084).
There was also an effect of injury group on Cath B localization (One-way ANOVA injury group F2,2633=9.24, p=1.00X10-4) in which animals sustaining CFPI only had significantly fewer cells displaying punctate Cath-B localization than sham-injured animals (p=0.025) or animals sustaining a CFPI and ICP elevation to 20mmHg (p=2.20X10-5). Specifically, non-disrupted neurons in animals infused with CA-074Me and sustaining a CFPI had significantly lower Cath B localization within puncta compared to sham animals infused with CA-074Me (p=3.59X10-7). In addition, there were differences in the membrane disrupted neuronal population between the CFPI and the CFPI+ICP elevated groups infused with 10% DMSO (p=0.014) and in the non-disrupted neuronal populations between the TBI and TBI+ICP elevated groups infused with CA-074Me (p=0.035).
There was a significant effect of inhibitor infusion (One-way ANOVA infusion group F1,2633= 5.50 p=0.019) on Cath B localization and there was a significant interaction between neuronal membrane disruption state and inhibitor infusion (Two-way ANOVA F1,2633= 5.80, p=0.016). There was also an interaction between inhibitor infusion and injury group (Two-way ANOVA F2,2633=3.53, p=0.03). There was also a significant interaction of membrane disruption state, injury group, and inhibitor infusion (Three-way ANOVA F2,2633=10.11, p=4.20X10-5). Specifically, the non-disrupted neuronal population in the sham group infused with CA-074Me had significantly higher punctate localization of Cath B compared to the sham group infused with 10% DMSO (p=0.021). There was also a significant shift in Cath B localization in the non-disrupted neuronal population in the TBI group infused with CA-074Me compared to the TBI group infused with 10% DMSO (p=0.029).
3.6. Secondary ICP Elevation Exacerbates Somatosensory Sensitivity at 2w Post-CFPI Which Is Ameliorated with CA-074Me Infusion
To investigate the potential changes in somatosensory sensitivity following CA-074Me infusion, the Whisker Nuisance Task (WNT) [7,39,40] was performed in animals prior to injury and at 2w following sham, CFPI, or CFPI and secondary ICP elevation to 20mmHg for all groups in which a higher score indicated higher somatosensory sensitivity to whisker stimulation. Pre-injury scores were comparable across all groups: Sham injury with 10% DMSO (3.33 ± 0.43; n=11), Sham injury with CA-074Me (3.43 ± 0.45; n=13), TBI with 10% DMSO (4.58 ± 0.66; n=12), TBI with CA-074Me (4.36 ± 0.42; n=13), TBI+ ICP elevation with 10% DMSO (3.79 ± 0.37; n=11), TBI+ ICP elevation with CA-074Me (3.68 ± 0.53; n=12). There was an effect between pre and post WNT score from injury group (Repeated measures ANOVA F1,68= 87.24 p=8.2X10-14; Figure 11; Supplemental Table S2). While the Sham injury group infused with CA-074Me maintained a low WNT score at 2w (5.17 ± 0.63), the 2w post-injury WNT scores for animals in all other groups were significantly higher than the pre-injury scores (Paired T-Test; Sham injury with 10% DMSO p=0.009; TBI with 10% DMSO p=0.02; TBI with CA-074Me p=0.004; TBI+ ICP elevation with 10% DMSO p=8.5 X10-8; TBI+ ICP elevation with CA-074Me p=0.004; Figure 11; Supplemental Table S2). There was a significant effect of injury group (Repeated Measure ANOVA time and injury group F2,68= 8.01 p=7.5X10-4) driven primarily by increases in WNT score at 2w in injured groups compared to shams (Sham vs. TBI p=5.1X10-4; Sham vs. TBI+ICP elevation p=0.001). There was an effect of inhibitor infusion on WNT score (Two-way repeated measures ANOVA F1,68= 6.66 p=0.012), in which groups given CA-074Me had lower WNT scores at 2w post-injury compared to those given 10% DMSO. There was no interaction of time, inhibitor infusion and injury group (Three-way repeated measures ANOVA F2,661.96 p=0.148).
4. Discussion
Overall, the findings from this study show that CA-074Me, when given as a bolus followed by continuous osmotic pump infusion into the left ventricle, successfully inhibits Cath B activity in the left and right lateral neocortex (Figure 1). Cath B activity was not significantly different between injury categories. The protein quantifications for Bak, Bcl-XL, and AIF also revealed no changes regarding CA-074Me treatment (Figure 3, Figure 4 and Figure 5). However, there was a small but significant increase in the protein levels of the lower band of Cath B in the groups given CA-074Me. (Figure 2). This could indicate a compensatory mechanism in which cells express increased levels of Cath B in the face of reduced Cath B activity, however, further investigations regarding expression and degradation of this lower Cath B band would be needed to rigorously investigate this possibility. Our histological assessments uncovered no cell loss in the lateral neocortex in layers V and VI in any group (Figure 7). We also did not observe a difference in the percentage of neuronal membrane disruption in any group regardless of injury or inhibitor infusion (Figure 8). Assessments of Cath B localization recapitulated the reduced punctate Cath B localization in membrane disrupted neurons compared to non-disrupted neurons (Figure 10), as has been shown previously [21]. Inhibition of Cath B also significantly altered Cath B localization (Figure 10). Somatosensory hypersensitivity, which was increased with injury and injury with elevated ICP did appear to be reduced by CA-074Me infusion in animals sustaining CFPI with secondary ICP elevation (Figure 11).
The neuronal membrane disruption findings in the current study, particularly in the 10% DMSO vehicle control groups were unanticipated based on previous studies. We previously found that while sham injury precipitated very little neuronal membrane disruption, approximately 20% of neurons were membrane disrupted at 6hr following CFPI in adult male Fisher rats [8]. The percentage of neurons sustaining membrane disruption doubled to 40% in rats with naturally high ICP above 20mmHg following CFPI [8]. A follow-up study using Sprague-Dawley rats found similar percentages of neuronal membrane disruption in which injured rats had ~25% membrane disrupted neurons and injury with ICP elevation to 20mmHg demonstrated ~62% membrane disrupted neurons[7]. A study investigating the time course of membrane disruption from 6h to 4w following a CFPI in male Sprague Dawley rats also found significantly higher percentages of neuronal membrane disruption following injury both acutely and weeks following injury [20]. Therefore, it was surprising to see no increase in neuronal membrane disruption following CFPI and no exacerbation with secondary ICP elevation (Figure. 8). However, while DMSO is mostly noted for its potentially toxic effects if used in high concentrations[42,43,44], DMSO has also been shown to have a neuroprotective effects. In a study conducted by DiGiorgio and colleagues, using the lateral fluid percussion model (LFPI) in rats, they tested drugs alpha-tocopherol and curcumin, with DMSO as the vehicle control as well as including saline controls[45]. In their assessments of cell damage/death they found a significant reduction in Fluoro-Jade staining in all drug treated groups suspended in DMSO vehicle, including the DMSO only group which was not observed in the saline LFPI controls [45]. Earlier preclinical studies saw that intravenous administration of this amphipathic molecule precipitated a reduction in ICP [46,47]. DMSO was also assessed in the clinic as a potential treatment for intractable ICP elevation and was shown to rapidly reduce ICP[42,43,44,48]. However, it was challenging to administer clinically as the DMSO tended to break down IV tubing. In addition, DMSO could lead to hemolysis and hypernatremia/hyperosmolarity, which counteracted any positive impacts of lowering ICP [42,43,44].
Biophysical assessments of DMSO have demonstrated increased permeability/flexibility in membrane dynamics [49,50]; however, the full effects of DMSO on cellular membranes is not fully understood. DMSO’s effects on membranes was initially interrogated using molecular simulations in which Notman et al., found increased membrane flexibility which was postulated to be linked to formation of water pores[50]. Another group arrived at similar conclusions using atomic simulations [49]. These simulations were eventually tested in DC-3F Chinese hamster lung fibroblast cells exposed to different percentages of DMSO (Ménorval et al., 2012). Membrane undulations, but not membrane permeability, was documented with low doses of DMSO[51]. With DMSO concentrations closer to 10-20% small membrane blebs were seen in which Ca2+ diffused into the cells, however, there was no increased diffusion of the larger (630Da) cell impermeable Oxazole Yellow (Yo-Pro-1) molecule for this concentration of DMSO [51]. Increased Yo-Pro-1 diffusion into cells was not significantly higher until ≥25% DMSO was administered to cells[51]. Indeed, addition of 5% DMSO to transected guinea pig spinal cords axons reduced permeability of axonal membranes to a cell impermeable tracer, even under conditions that the group had previously determined to significantly hinder membrane resealing[52]. In the current study it may be possible that DMSO altered membrane flexibility thereby promoting membrane resealing by 2w post-injury, which could explain the lower amounts of membrane disruption we observed compared to previous studies [7,20,40].
We did, however, observe whisker sensory sensitivity increases following CFPI that was exacerbated by ICP elevations to 20mmHg in our 10% DMSO infused group, consistent with previously published studies [7,40]. Notably, CA-074Me administration in animals sustaining TBI and secondary ICP elevation lowered sensory hypersensitivity to levels consistent with the TBI only groups infused with either 10% DMSO or CA-074Me (Figure 11). Previous groups found similar rescues in deficits with Cath B inhibition. Mice administered CA-074 prior to a focal TBI showed recovery of motor function and spatial learning compared to vehicle treated animals[36]. Another study in which Cath B was knocked out of mice sustaining a focal TBI, or inhibited via administration of E64d, a cysteine protease inhibitor, observed better motor behavior in animals with absent or inhibited Cath B compared to controls[35]. Based on these studies and our current finding, it is possible that Cath B inhibition may play a role in the exacerbation of whisker hypersensitivity following secondary ICP elevation. Overall, while the mechanisms behind membrane disruption are not directly coupled with Cath B, Cath B seems to have a role in the exacerbation of behavioral morbidities following TBI and secondary ICP elevation, offering a molecular target in treating the after-effects of elevated ICP following injury. Therefore, further study into this possibility is warranted.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Full Blot Representative Westerns; Table S1: Behavior Criteria rubric; Table S2: Full data set for all data presented in figures 1-9 and Figure 11; Table S3: Full data set for all data presented in Figure 10.
Author Contributions
MH generated all animals, carried out the microscopy, microscopy analysis, WNT, western and protein activity assays, and wrote the manuscript. SR carried out microscopy, microscopy analysis, and western assays. RA carried out microscopy and microscopy analysis. ALW carried out microscopy analysis and edited the manuscript. RL carried out physiology analysis and edited the manuscript. CL carried out microscopy analysis. KG carried out western assays and microscopy analysis. AL carried out microscopy analysis, conceived, designed, and coordinated the study, and wrote the manuscript.
Funding
This work was supported by NINDS grants 1R01NS096143 and F99NS125825. The APC was funded by a Blick fellowship awarded by Virginia Commonwealth University.
Data Availability Statement
The raw data associated with this manuscript is appended as supplemental tables. In addition, this data, along with protocols will be submitted to the Open Science Framework for open data access.
Acknowledgments
The authors would like to recognize Dr. Gladys Shaw for her help refining the WNT rubric. This work was supported by NINDS grants 1R01NS096143 and F99NS125825.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Maas, A.I.R.; Menon, D.K.; Manley, G.T.; Abrams, M.; Åkerlund, C.; Andelic, N.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. Traumatic Brain Injury: Progress and Challenges in Prevention, Clinical Care, and Research. The Lancet Neurology 2022, 21, 1004–1060. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury–Related Emergency Department Visits, Hospitalizations, and Deaths — United States, 2007 and 2013. MMWR. Surveillance Summaries 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- James, S.L.; Theadom, A.; Ellenbogen, R.G.; Bannick, M.S.; Montjoy-Venning, W.; Lucchesi, L.R.; Abbasi, N.; Abdulkader, R.; Abraha, H.N.; Adsuar, J.C.; et al. Global, Regional, and National Burden of Traumatic Brain Injury and Spinal Cord Injury, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 2019, 18, 56–87. [Google Scholar] [CrossRef] [PubMed]
- Dams-O’Connor, K.; Juengst, S.B.; Bogner, J.; Chiaravalloti, N.D.; Corrigan, J.D.; Giacino, J.T.; Harrison-Felix, C.L.; Hoffman, J.M.; Ketchum, J.M.; Lequerica, A.H.; et al. Traumatic Brain Injury as a Chronic Disease: Insights from the United States Traumatic Brain Injury Model Systems Research Program. Lancet Neurol 2023, 22, 517–528. [Google Scholar] [CrossRef]
- Farahvar, A.; Gerber, L.M.; Chiu, Y.-L.; Carney, N.; Härtl, R.; Ghajar, J. Increased Mortality in Patients with Severe Traumatic Brain Injury Treated without Intracranial Pressure Monitoring. Journal of Neurosurgery 2012, 117, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Becker, D.P.; Ward, J.D.; Sullivan, H.G.; Adams, W.E.; Rosner, M.J. Significance of Intracranial Hypertension in Severe Head Injury. Journal of Neurosurgery 1977, 47, 503–516. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; Krahe, T.E.; Povlishock, J.T. Moderately Elevated Intracranial Pressure after Diffuse Traumatic Brain Injury Is Associated with Exacerbated Neuronal Pathology and Behavioral Morbidity in the Rat. Journal of Cerebral Blood Flow & Metabolism 2014, 34, 1628–1636. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; McGinn, M.J.; Povlishock, J.T. Increased Intracranial Pressure after Diffuse Traumatic Brain Injury Exacerbates Neuronal Somatic Membrane Poration but Not Axonal Injury: Evidence for Primary Intracranial Pressure-Induced Neuronal Perturbation. Journal of Cerebral Blood Flow & Metabolism 2012, 32, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Cullen, D.K.; Vernekar, V.N.; LaPlaca, M.C. Trauma-Induced Plasmalemma Disruptions in Three-Dimensional Neural Cultures Are Dependent on Strain Modality and Rate. Journal of Neurotrauma 2011, 28, 2219–2233. [Google Scholar] [CrossRef]
- Farkas, O.; Lifshitz, J.; Povlishock, J.T. Mechanoporation Induced by Diffuse Traumatic Brain Injury: An Irreversible or Reversible Response to Injury? Journal of Neuroscience 2006, 26, 3130–3140. [Google Scholar] [CrossRef]
- Geddes, D.M.; Cargill, R.S.; LaPlaca, M.C. Mechanical Stretch to Neurons Results in a Strain Rate and Magnitude-Dependent Increase in Plasma Membrane Permeability. Journal of Neurotrauma 2003, 20, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Geddes, D.M.; LaPlaca, M.C.; Cargill, R.S. Susceptibility of Hippocampal Neurons to Mechanically Induced Injury. Experimental Neurology 2003, 184, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.P.; Mietus, C.J.; Browne, K.D.; Wofford, K.L.; Keating, C.E.; Brown, D.P.; Johnson, B.N.; Wolf, J.A.; Smith, D.H.; Cohen, A.S.; et al. Neuronal Somatic Plasmalemmal Permeability and Dendritic Beading Caused by Head Rotational Traumatic Brain Injury in Pigs–An Exploratory Study. Frontiers in Cellular Neuroscience 2023, 17. [Google Scholar] [CrossRef] [PubMed]
- Keating, C.E.; Browne, K.D.; Duda, J.E.; Cullen, D.K. Neurons in Subcortical Oculomotor Regions Are Vulnerable to Plasma Membrane Damage after Repetitive Diffuse Traumatic Brain Injury in Swine. Journal of Neurotrauma 2020, 37, 1918–1932. [Google Scholar] [CrossRef] [PubMed]
- LaPlaca, M.C.; Lessing, M.C.; Prado, G.R.; Zhou, R.; Tate, C.C.; Geddes-Klein, D.; Meaney, D.F.; Zhang, L. Mechanoporation Is a Potential Indicator of Tissue Strain and Subsequent Degeneration Following Experimental Traumatic Brain Injury. Clinical Biomechanics 2019, 64, 2–13. [Google Scholar] [CrossRef] [PubMed]
- LaPlaca, M.C.; Thibault, L.E. Dynamic Mechanical Deformation of Neurons Triggers an Acute Calcium Response and Cell Injury Involving the N-Methyl-D-Aspartate Glutamate Receptor. Journal of Neuroscience Research 1998, 52, 220–229. [Google Scholar] [CrossRef]
- Prado, G.R.; LaPlaca, M.C. Neuronal Plasma Membrane Integrity Is Transiently Disturbed by Traumatic Loading. Neuroscience Insights 2020, 15, 263310552094609. [Google Scholar] [CrossRef]
- Singleton, R.H.; Povlishock, J.T. Identification and Characterization of Heterogeneous Neuronal Injury and Death in Regions of Diffuse Brain Injury: Evidence for Multiple Independent Injury Phenotypes. Journal of Neuroscience 2004, 24, 3543–3553. [Google Scholar] [CrossRef] [PubMed]
- Wofford, K.L.; Harris, J.P.; Browne, K.D.; Brown, D.P.; Grovola, M.R.; Mietus, C.J.; Wolf, J.A.; Duda, J.E.; Putt, M.E.; Spiller, K.L.; et al. Rapid Neuroinflammatory Response Localized to Injured Neurons after Diffuse Traumatic Brain Injury in Swine. Experimental Neurology 2017, 290, 85–94. [Google Scholar] [CrossRef]
- Hernandez, M.L.; Chatlos, T.; Gorse, K.M.; Lafrenaye, A.D. Neuronal Membrane Disruption Occurs Late Following Diffuse Brain Trauma in Rats and Involves a Subpopulation of NeuN Negative Cortical Neurons. Frontiers in Neurology 2019, 10. [Google Scholar] [CrossRef]
- Hernandez, M.L.; Marone, M.; Gorse, K.M.; Lafrenaye, A.D. Cathepsin B Relocalization in Late Membrane Disrupted Neurons Following Diffuse Brain Injury in Rats. ASN Neuro 2022, 14, 17590914221099112. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Babu, P.P. Multiple Apoptogenic Proteins Are Involved in the Nuclear Translocation of Apoptosis Inducing Factor during Transient Focal Cerebral Ischemia in Rat. Brain Research 2008, 1246, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.D.; Sarkar, A.; Chatterjee, S.; Patra, D.; Sengupta, D.; Banerjee, S.; Chakraborty, P.; Sadhukhan, G.C. Cathepsin B Mediated Scramblase Activation Triggers Cytotoxicity and Cell Cycle Arrest by Andrographolide to Overcome Cellular Resistance in Cisplatin Resistant Human Hepatocellular Carcinoma HepG2 Cells. Environmental Toxicology and Pharmacology 2019, 68, 120–132. [Google Scholar] [CrossRef] [PubMed]
- de Castro, M.; Bunt, G.; Wouters, F. Cathepsin B Launches an Apoptotic Exit Effort upon Cell Death-Associated Disruption of Lysosomes. Cell Death Discovery 2016, 2, 16012. [Google Scholar] [CrossRef]
- Ellis, R.C.; O’Steen, W.A.; Hayes, R.L.; Nick, H.S.; Wang, K.K.W.; Anderson, D.K. Cellular Localization and Enzymatic Activity of Cathepsin B after Spinal Cord Injury in the Rat. Experimental Neurology 2005, 193, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jäättelä, M. Cathepsin B Acts as a Dominant Execution Protease in Tumor Cell Apoptosis Induced by Tumor Necrosis Factor. Journal of Cell Biology 2001, 153, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B Contributes to TNF-α–Mediated Hepatocyte Apoptosis by Promoting Mitochondrial Release of Cytochrome c. Journal of Clinical Investigation 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Kobayashi, M.; Hoshino, S.; Tagawa, R.; Itagawa, R.; Hoshino, A.; Okita, N.; Sudo, Y.; Nakagawa, Y.; Shimano, H.; et al. Cathepsin B Overexpression Induces Degradation of Perilipin 1 to Cause Lipid Metabolism Dysfunction in Adipocytes. Scientific Reports 2020, 10. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsin B Overexpression Due to Acid Sphingomyelinase Ablation Promotes Liver Fibrosis in Niemann-Pick Disease. Journal of Biological Chemistry 2012, 287, 1178–1188. [Google Scholar] [CrossRef]
- Oberle, C.; Huai, J.; Reinheckel, T.; Tacke, M.; Rassner, M.; Ekert, P.G.; Buellesbach, J.; Borner, C. Lysosomal Membrane Permeabilization and Cathepsin Release Is a Bax/Bak-Dependent, Amplifying Event of Apoptosis in Fibroblasts and Monocytes. Cell Death and Differentiation 2010, 17, 1167–1178. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Zhang, X.; Xu, Y.; Chen, L.; Zhang, W.; Liu, E.; Xiao, C.; Kou, Q. Cathepsin B Aggravates Acute Pancreatitis by Activating the NLRP3 Inflammasome and Promoting the Caspase-1-Induced Pyroptosis. International Immunopharmacology 2021, 94, 107496. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Sheng, R.; Zhang, L.S.; Han, R.; Zhang, X.; Zhang, X.D.; Han, F.; Fukunaga, K.; Qin, Z.H. Neuronal Injury in Rat Model of Permanent Focal Cerebral Ischemia Is Associated with Activation of Autophagic and Lysosomal Pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-Q.; Xu, M.; Yuan, Y.; Li, F.-F.; Yang, Z.; Liu, Y.; Zhou, M.-Q.; Bian, Z.-Y.; Deng, W.; Gao, L.; et al. Cathepsin B Deficiency Attenuates Cardiac Remodeling in Response to Pressure Overload via TNF-α/ASK1/JNK Pathway. American Journal of Physiology-Heart and Circulatory Physiology 2015, 308, H1143–H1154. [Google Scholar] [CrossRef]
- Boutté, A.M.; Hook, V.; Thangavelu, B.; Sarkis, G.A.; Abbatiello, B.N.; Hook, G.; Jacobsen, J.S.; Robertson, C.S.; Gilsdorf, J.; Yang, Z.; et al. Penetrating Traumatic Brain Injury Triggers Dysregulation of Cathepsin B Protein Levels Independent of Cysteine Protease Activity in Brain and Cerebral Spinal Fluid. Journal of neurotrauma 2020, 37, 1574–1586. [Google Scholar] [CrossRef]
- Hook, G.R.; Yu, J.; Sipes, N.; Pierschbacher, M.D.; Hook, V.; Kindy, M.S. The Cysteine Protease Cathepsin B Is a Key Drug Target and Cysteine Protease Inhibitors Are Potential Therapeutics for Traumatic Brain Injury. Journal of Neurotrauma 2013, 31, 515–529. [Google Scholar] [CrossRef]
- Luo, C.-L.; Chen, X.-P.; Yang, R.; Sun, Y.-X.; Li, Q.-Q.; Bao, H.-J.; Cao, Q.-Q.; Ni, H.; Qin, Z.-H.; Tao, L.-Y. Cathepsin B Contributes to Traumatic Brain Injury-Induced Cell Death through a Mitochondria-Mediated Apoptotic Pathway. Journal of Neuroscience Research 2010, 88, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.E.; Lyeth, B.G.; Povlishock, J.T.; Findling, R.L.; Hamm, R.J.; Marmarou, A.; Young, H.F.; Hayes, R.L. A Fluid Percussion Model of Experimental Brain Injury in the Rat. Journal of Neurosurgery 1987, 67, 110–119. [Google Scholar] [CrossRef]
- Yoon, M.C.; Solania, A.; Jiang, Z.; Christy, M.P.; Podvin, S.; Mosier, C.; Lietz, C.B.; Ito, G.; Gerwick, W.H.; Wolan, D.W.; et al. Selective Neutral pH Inhibitor of Cathepsin B Designed Based on Cleavage Preferences at Cytosolic and Lysosomal pH Conditions. ACS Chemical Biology 2021, 16, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.C.S.S.; Lisembee, A.M.; Lifshitz, J. The Whisker Nuisance Task Identifies a Late-Onset, Persistent Sensory Sensitivity in Diffuse Brain-Injured Rats. Journal of Neurotrauma 2010, 27, 695–706. [Google Scholar] [CrossRef]
- Ryu, J.; Jeizan, P.; Ahmed, S.; Ehsan, S.; Jose, J.; Regan, S.; Gorse, K.; Kelliher, C.; Lafrenaye, A. Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats. Pharmaceutics 2022, 14, 2068. [Google Scholar] [CrossRef]
- Boya, P.; Andreau, K.; Poncet, D.; Zamzami, N.; Perfettini, J.L.; Metivier, D.; Ojcius, D.M.; Jäättelä, M.; Kroemer, G. Lysosomal Membrane Permeabilization Induces Cell Death in a Mitochondrion-Dependent Fashion. Journal of Experimental Medicine 2003, 197, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.F.; Camp, P.E.; Bowers, S. Dimethyl Sulfoxide for the Treatment of Intracranial Hypertension: A Preliminary Trial. Neurosurgery 1984, 14, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Waller, F.T.; Tanabe, C.T.; Paxton, H.D. Treatment of Elevated Intracranial Pressure with Dimethyl Sulfoxide. Annals of the New York Academy of Sciences 1983, 411, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Simon, M. Dimethyl Sulphoxide (DMSO) Induced Serum Hyperosmolality. Clinical Biochemistry 1983, 16, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, A.M.; Hou, Y.; Zhao, X.; Zhang, B.; Lyeth, B.G.; Russell, M.J. Dimethyl Sulfoxide Provides Neuroprotection in a Traumatic Brain Injury Model. Restorative Neurology and Neuroscience 2008, 26, 501–507. [Google Scholar] [PubMed]
- de la Torre, J.C.; Kawanaga, H.M.; Rowed, D.W.; Johnson, C.M.; Goode, D.J.; Kajihara, K.; Mullan, S. Dimethyl Sulfoxide in Central Nervous System Trauma. Annals of the New York Academy of Sciences 1975, 243, 362–389. [Google Scholar] [CrossRef] [PubMed]
- Tung, H.; James, H.E.; Laurin, R.; Marshall, L.F. Modification of the Effect of Dimethyl Sulfoxide on Intracranial Pressure, Brain Water, and Electrolyte Content by Indomethacin. Acta Neurochir (Wien) 1983, 68, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Karaca, M.; Bilgin, U.Y.; Akar, M.; de la Torre, J.C. Dimethly Sulphoxide Lowers ICP after Closed Head Trauma. Eur J Clin Pharmacol 1991, 40, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Gurtovenko, A.A.; Anwar, J. Modulating the Structure and Properties of Cell Membranes: The Molecular Mechanism of Action of Dimethyl Sulfoxide. J. Phys. Chem. B 2007, 111, 10453–10460. [Google Scholar] [CrossRef]
- Notman, R.; Noro, M.; O’Malley, B.; Anwar, J. Molecular Basis for Dimethylsulfoxide (DMSO) Action on Lipid Membranes. J. Am. Chem. Soc. 2006, 128, 13982–13983. [Google Scholar] [CrossRef]
- Ménorval, M.-A. de; Mir, L.M.; Fernández, M.L.; Reigada, R. Effects of Dimethyl Sulfoxide in Cholesterol-Containing Lipid Membranes: A Comparative Study of Experiments In Silico and with Cells. PLOS ONE 2012, 7, e41733. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Qiao, X.; Emerson, N.; Malcom, A. Dimethylsulfoxide Enhances CNS Neuronal Plasma Membrane Resealing after Injury in Low Temperature or Low Calcium. Journal of Neurocytology 2001, 30, 829–839. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cathepsin B (Cath B) activity was reduced following 2w of CA-074Me infusion into the left lateral ventricle. Cath B activity was significantly decreased in the left and right lateral neocortex following CA-074Me infusion (filled boxes) compared to 10% DMSO control (vehicle; open boxes). Box and whisker graph depicting the average fluorescent intensity indicating Cath B activity in the right and left side of the lateral neocortex and the liver of sham injured control animals (n=12; grey opened boxes for n=6 saline treated animals and filled boxes for n=6 CA-074Me treated animals), animals sustaining a TBI only (n=13; light blue opened boxes for n=6 saline treated animals and filled boxes for n=7 CA-074Me treated animals), or animals sustaining a TBI followed by secondary ICP elevation (n=12; dark blue opened boxes for n=6 saline treated animals and filled boxes for n=6 CA-074Me treated animals). Note that Cath B activity in the liver was significantly higher than that in the cortex regardless of injury group. Additionally, ICP infusion of CA-074Me did not impact Cath B activity in the liver; however, CA-074Me infusion did significantly lower Cath B activity in the left and right cortex, compared to DMSO controls. * p<0.05 compared to injury type-matched vehicle control, # p<0.05 compared to injury type-matched liver. Mean ± S.E.M.
Figure 1.
Cathepsin B (Cath B) activity was reduced following 2w of CA-074Me infusion into the left lateral ventricle. Cath B activity was significantly decreased in the left and right lateral neocortex following CA-074Me infusion (filled boxes) compared to 10% DMSO control (vehicle; open boxes). Box and whisker graph depicting the average fluorescent intensity indicating Cath B activity in the right and left side of the lateral neocortex and the liver of sham injured control animals (n=12; grey opened boxes for n=6 saline treated animals and filled boxes for n=6 CA-074Me treated animals), animals sustaining a TBI only (n=13; light blue opened boxes for n=6 saline treated animals and filled boxes for n=7 CA-074Me treated animals), or animals sustaining a TBI followed by secondary ICP elevation (n=12; dark blue opened boxes for n=6 saline treated animals and filled boxes for n=6 CA-074Me treated animals). Note that Cath B activity in the liver was significantly higher than that in the cortex regardless of injury group. Additionally, ICP infusion of CA-074Me did not impact Cath B activity in the liver; however, CA-074Me infusion did significantly lower Cath B activity in the left and right cortex, compared to DMSO controls. * p<0.05 compared to injury type-matched vehicle control, # p<0.05 compared to injury type-matched liver. Mean ± S.E.M.
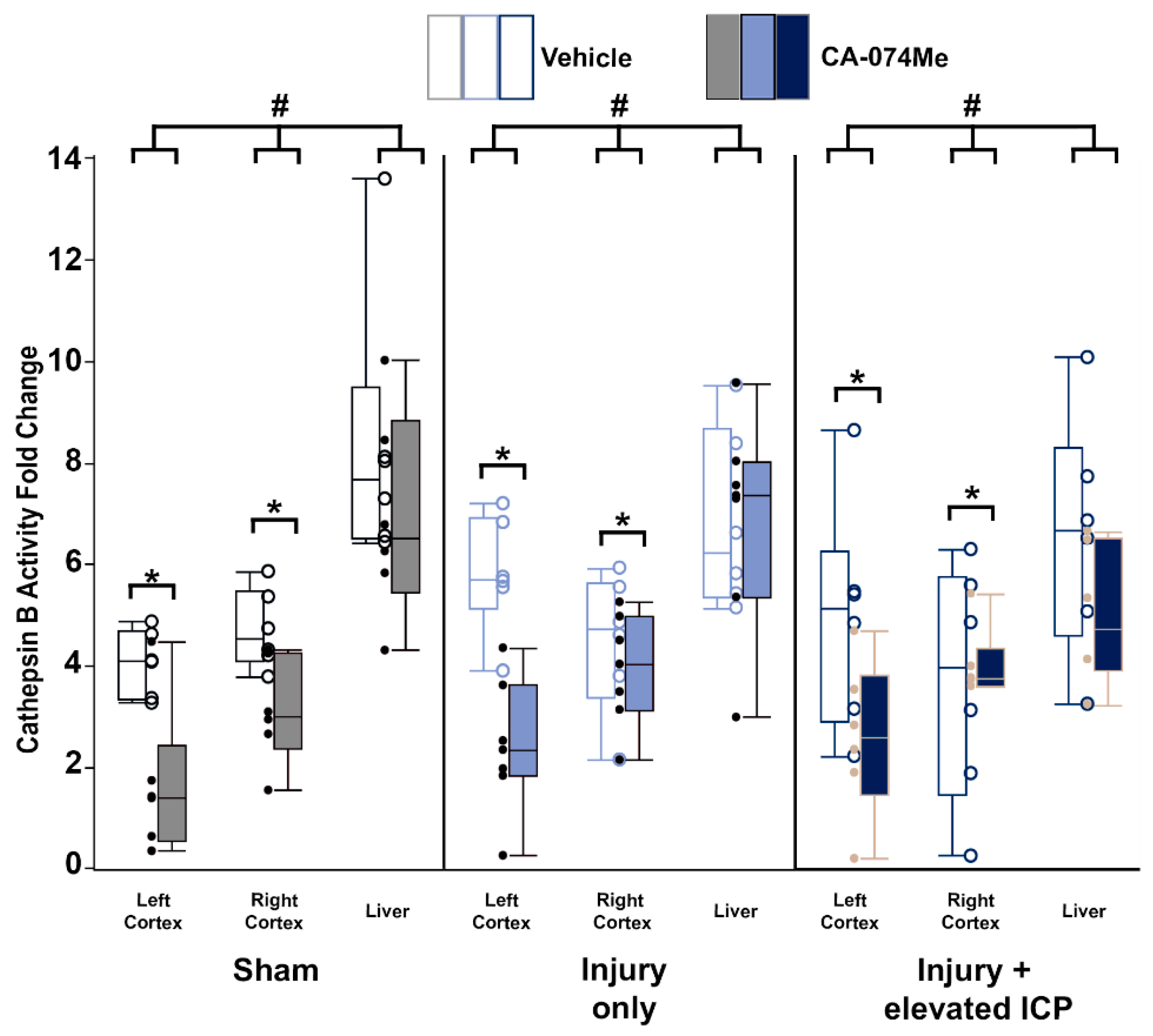
Figure 2.
Cathepsin B (Cath B) protein levels were slightly increased in animals infused with CA-074Me. (A) Representative chemiluminescent blot image of the two mature Cath B bands at 24/27 kDa, which was normalized to (B) total protein. Box and whisker graphs depicting average (n=6/group) quantities of (C) total Cath B protein, (D) the upper band of the Cath B doublet, and (E) the lower band of the Cath B doublet for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Amount of Cath B for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. While there were no signifiant differences found for total Cath B protein or the upper band of the observed doublet, there was a significant increase in Cath B levels of the lower band of the doublet in animals infused with CA-074Me compared to their vehicle counterparts. * p<0.05 compared to vehicle. Mean ± S.E.M.
Figure 2.
Cathepsin B (Cath B) protein levels were slightly increased in animals infused with CA-074Me. (A) Representative chemiluminescent blot image of the two mature Cath B bands at 24/27 kDa, which was normalized to (B) total protein. Box and whisker graphs depicting average (n=6/group) quantities of (C) total Cath B protein, (D) the upper band of the Cath B doublet, and (E) the lower band of the Cath B doublet for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Amount of Cath B for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. While there were no signifiant differences found for total Cath B protein or the upper band of the observed doublet, there was a significant increase in Cath B levels of the lower band of the doublet in animals infused with CA-074Me compared to their vehicle counterparts. * p<0.05 compared to vehicle. Mean ± S.E.M.
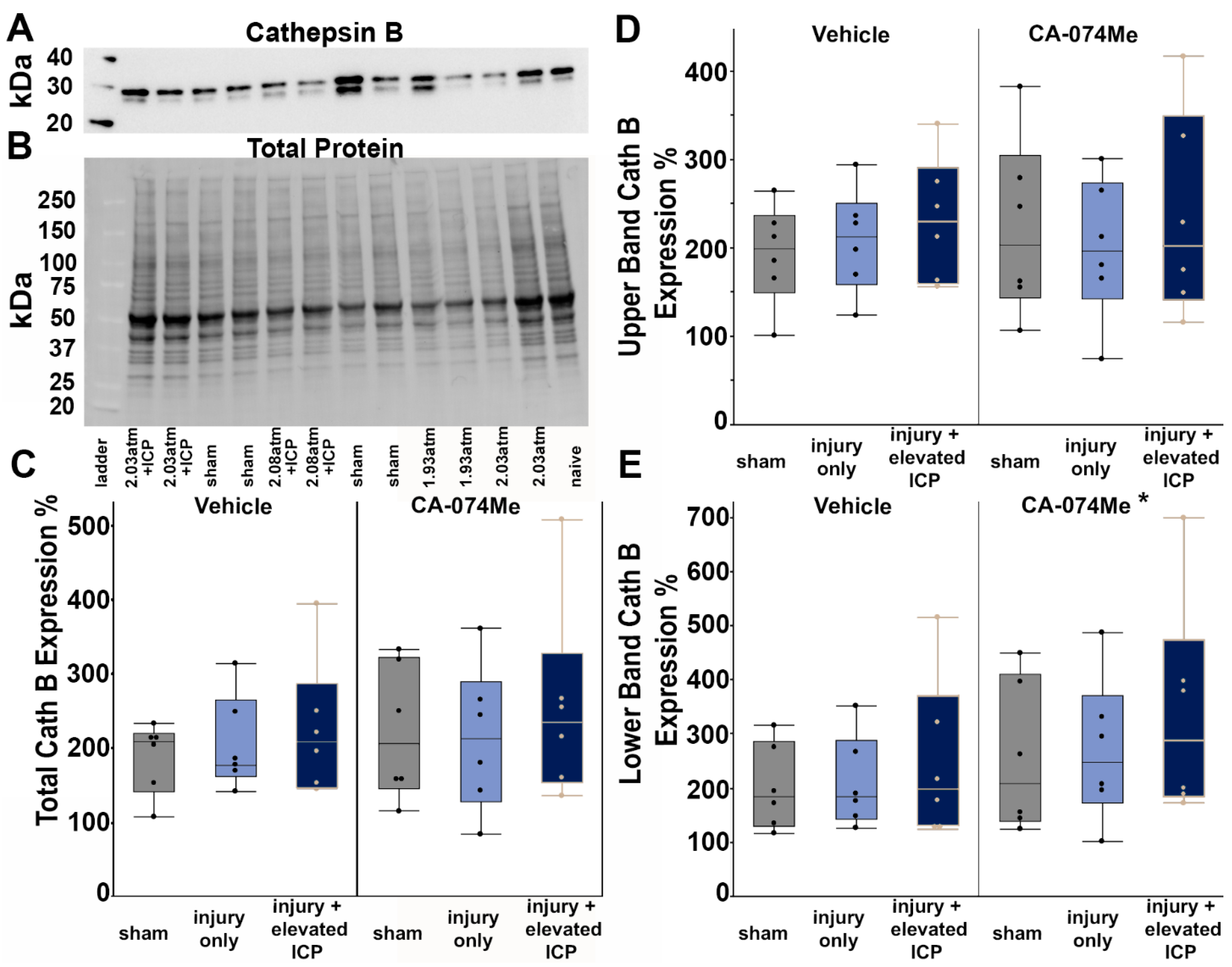
Figure 3.
Protein levels of Bcl-XL were unchanged regardless of injury or infusion group. (A) Representative chemiluminescent blot image of Bcl-XL at 30 kDa, which was normalized to (B) total protein. (C) Box and whisker graph depicting average (n=6/group) quantities of Bcl-XL protein for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graph) or the Cath B inhibitor, CA-074Me, (right half of graph). Amount of Bcl-XL for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
Figure 3.
Protein levels of Bcl-XL were unchanged regardless of injury or infusion group. (A) Representative chemiluminescent blot image of Bcl-XL at 30 kDa, which was normalized to (B) total protein. (C) Box and whisker graph depicting average (n=6/group) quantities of Bcl-XL protein for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graph) or the Cath B inhibitor, CA-074Me, (right half of graph). Amount of Bcl-XL for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
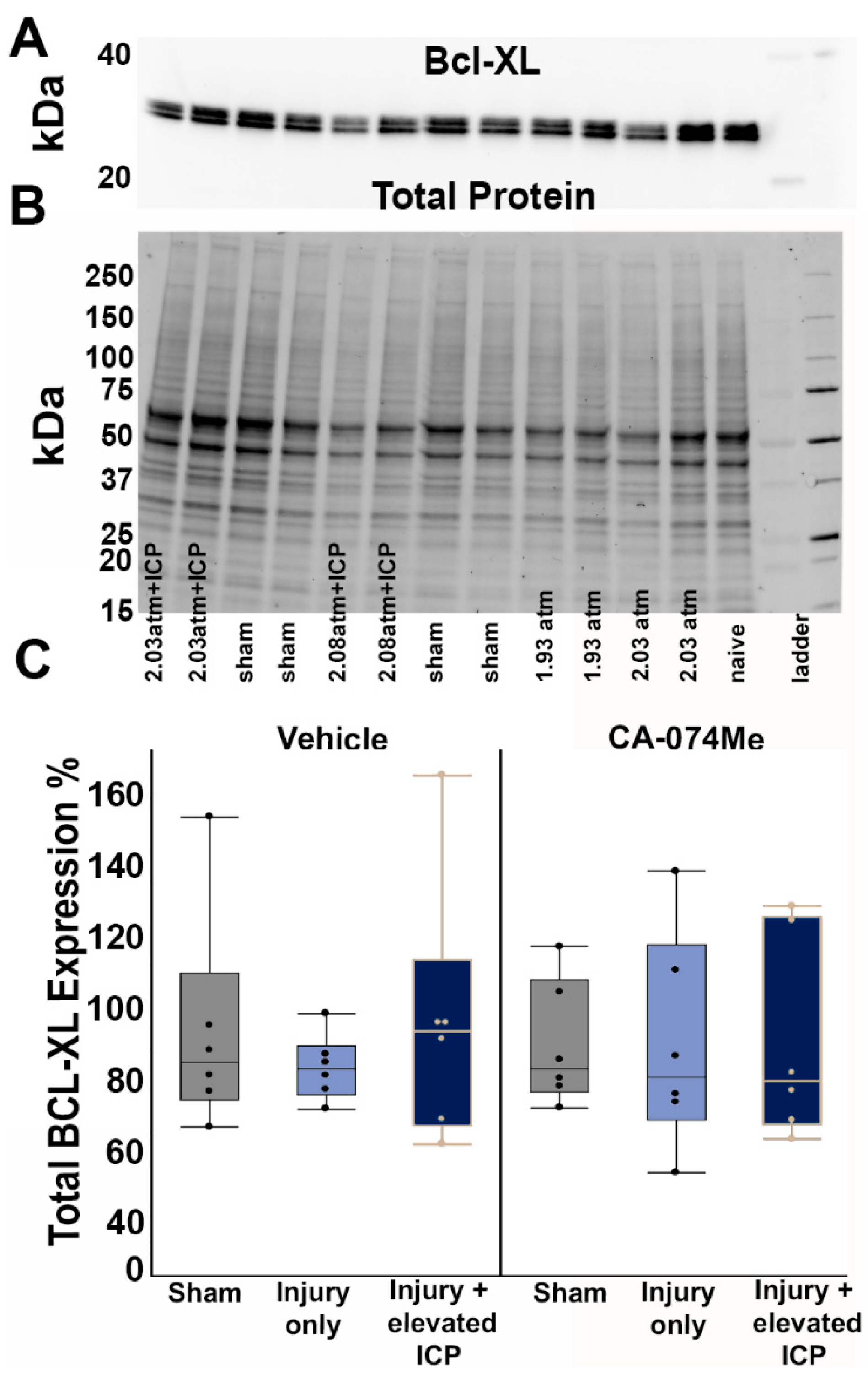
Figure 4.
Protein quantification of BAK revealed no differences in the protein quantity regardless of group. (A) Representative chemiluminescent blot image of BAK at 25kDa, which was normalized to (B) total protein. (C) Box and whisker graph depicting average (n=6/group) quantities of BAK protein for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graph) or the Cath B inhibitor, CA-074Me (right half of graph). Amount of BAK for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
Figure 4.
Protein quantification of BAK revealed no differences in the protein quantity regardless of group. (A) Representative chemiluminescent blot image of BAK at 25kDa, which was normalized to (B) total protein. (C) Box and whisker graph depicting average (n=6/group) quantities of BAK protein for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICF infusion of either 10% DMSO (vehicle, left half of graph) or the Cath B inhibitor, CA-074Me (right half of graph). Amount of BAK for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
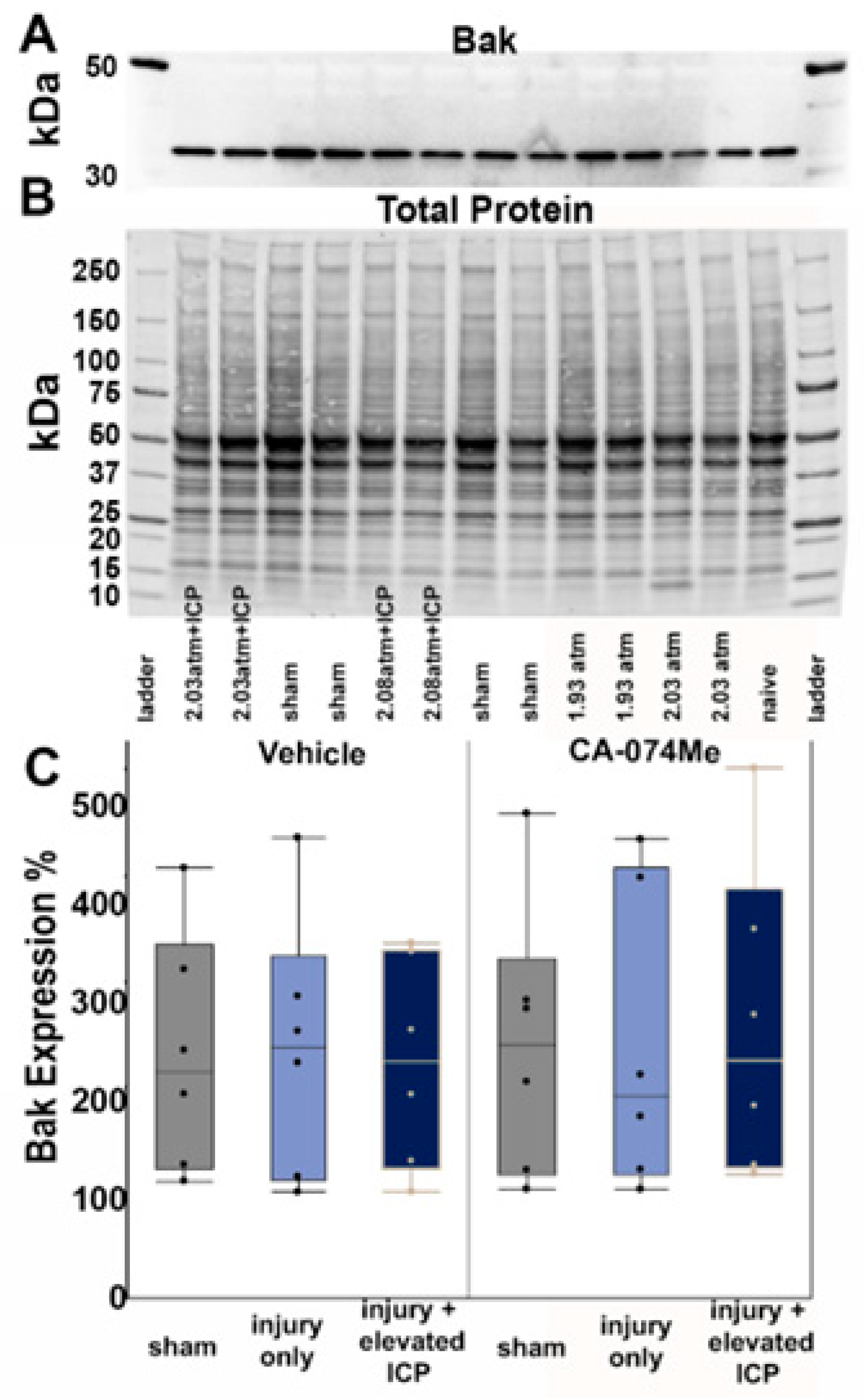
Figure 5.
Protein quantification of AIF revealed no differences in the protein quantity regardless of group. (A) Representative chemiluminescent blot image of the two observed AIF bands at ~67/72kDa, which was normalized to (B) total protein. Box and whisker graphs depicting average (n=6/group) quantities of (C) the upper band of the AIF doublet and (D) the lower band of the AIF doublet for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Amount of AIF for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
Figure 5.
Protein quantification of AIF revealed no differences in the protein quantity regardless of group. (A) Representative chemiluminescent blot image of the two observed AIF bands at ~67/72kDa, which was normalized to (B) total protein. Box and whisker graphs depicting average (n=6/group) quantities of (C) the upper band of the AIF doublet and (D) the lower band of the AIF doublet for sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Amount of AIF for each case was calculated as a percentage compared to a consistent naïve control that was run on all membranes. Mean ± S.E.M.
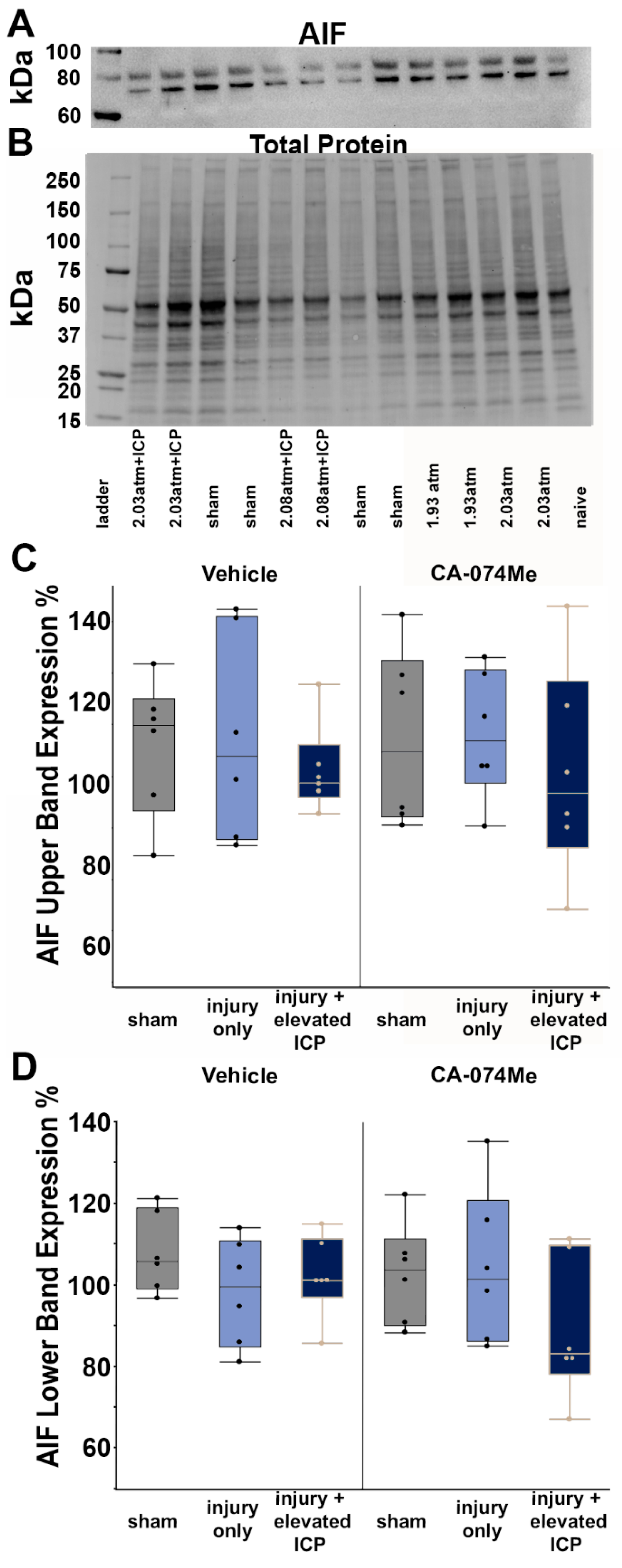
Figure 6.
Representative fluorescent micrographs of membrane disruption in sham injured animals (Sham, A and D), animals sustaining a CFPI (TBI, B and E), and animals sustaining a CFPI followed by secondary ICP elevation (TBI+ 20 mmHg ICP elevation, C and F) paired with 2w of ICV infusion of 10% DMSO (vehicle, A-C) or the Cath B inhibitor, CA-074Me (D-F). The left panel in blue depicts NeuroTrace Nissl stained cells and the middle panel in green depicts cells containing a cell impermeable Alexa fluor-488-tagged dextran (AF-Dextran-488). The right panel is the overlay of the NeuroTrace and membrane disrupted dextran images. The arrow heads indicate representative membrane disrupted neurons. Scale bar 20μm.
Figure 6.
Representative fluorescent micrographs of membrane disruption in sham injured animals (Sham, A and D), animals sustaining a CFPI (TBI, B and E), and animals sustaining a CFPI followed by secondary ICP elevation (TBI+ 20 mmHg ICP elevation, C and F) paired with 2w of ICV infusion of 10% DMSO (vehicle, A-C) or the Cath B inhibitor, CA-074Me (D-F). The left panel in blue depicts NeuroTrace Nissl stained cells and the middle panel in green depicts cells containing a cell impermeable Alexa fluor-488-tagged dextran (AF-Dextran-488). The right panel is the overlay of the NeuroTrace and membrane disrupted dextran images. The arrow heads indicate representative membrane disrupted neurons. Scale bar 20μm.
Figure 7.
The total number of cells in the lateral neocortex layers V and VI is unaffected across injury and infusion groups. Bar graph depicting the average number of fluorescent NeuroTrace Nissl-stained neurons in sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). The mean number of cells was quantified per unit area (0.098 mm2) and averaged for each animal (n=6/group). Mean ± S.E.M.
Figure 7.
The total number of cells in the lateral neocortex layers V and VI is unaffected across injury and infusion groups. Bar graph depicting the average number of fluorescent NeuroTrace Nissl-stained neurons in sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). The mean number of cells was quantified per unit area (0.098 mm2) and averaged for each animal (n=6/group). Mean ± S.E.M.
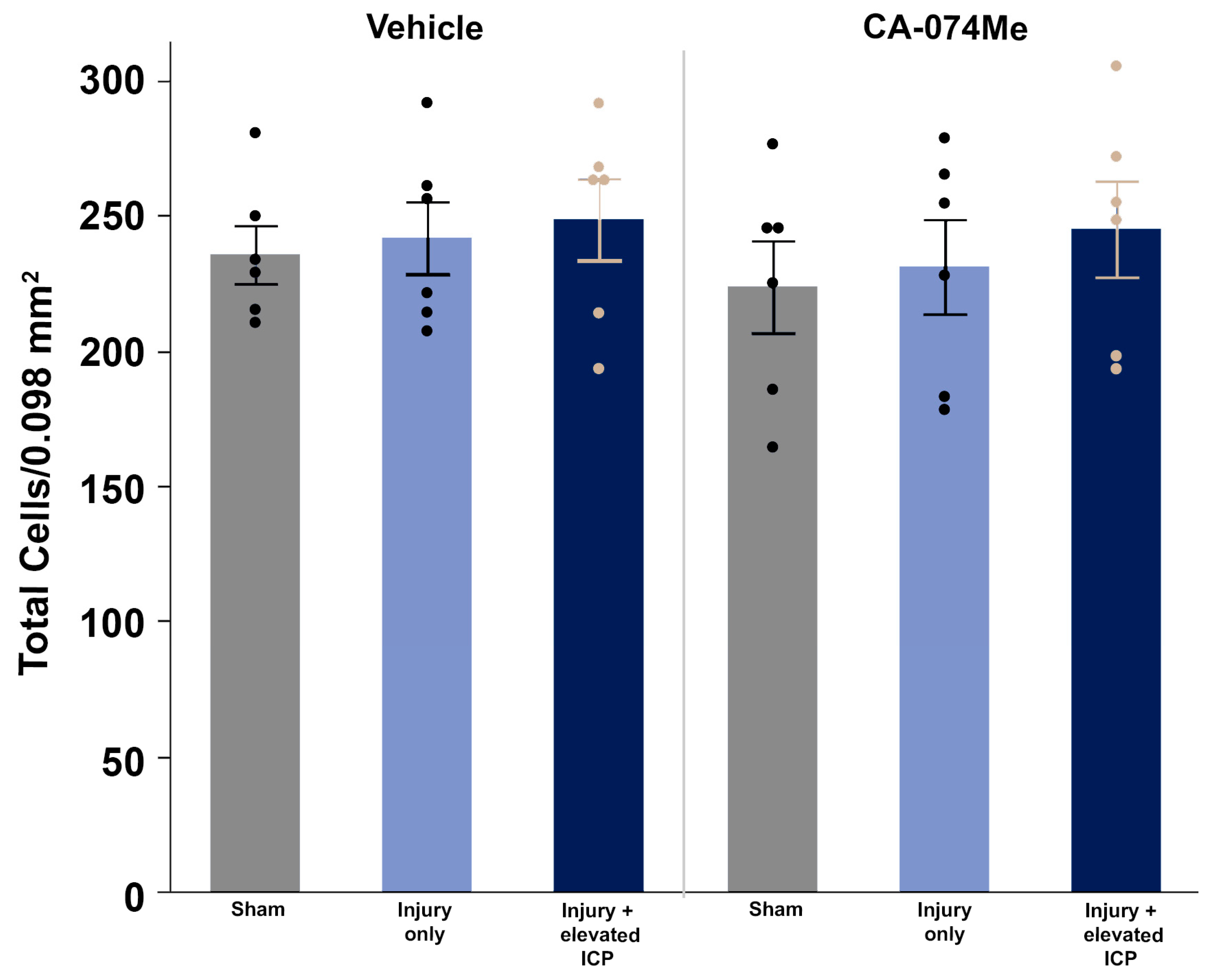
Figure 8.
There were no significant changes in membrane disruption regardless of injury or Cath B inhibitor infusion. Bar graph highlighting a consistently low average (n=6 group) percentage of membrane disrupted neurons in sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Mean ± S.E.M.
Figure 8.
There were no significant changes in membrane disruption regardless of injury or Cath B inhibitor infusion. Bar graph highlighting a consistently low average (n=6 group) percentage of membrane disrupted neurons in sham injured animals (grey boxes), animals sustaining a CFPI (light blue boxes), and animals sustaining both CFPI and secondary ICP elevation (dark blue boxes) followed by a 2w ICV infusion of either 10% DMSO (vehicle, left half of graphs) or the Cath B inhibitor, CA-074Me, (right half of graphs). Mean ± S.E.M.
Figure 9.
Representative fluorescent micrographs of Cathepsin B (Cath B) localization in membrane disrupted and non-disrupted neurons within the lateral neocortex layers V and VI in sham injured animals (Sham, A and D), animals sustaining a CFPI (TBI, B and E), and animals sustaining a CFPI followed by secondary ICP elevation (TBI+ 20 mmHg ICP elevation, C and F) paired with 2w of ICV infusion of 10% DMSO (vehicle, A-C) or CA-074Me (Cath B inhibitor, D-F). The left most panel contains DAPI labeled nuclei (blue). Neurons were identified as non-disrupted or membrane disrupted based upon uptake of cell impermeable Alexa-fluor488 tagged 10kDa dextran (AF-Dextran-488; second panel in green). Immunolabeling for Cath B (third panel in red) allowed investigation of Cath B localization inside lysosomal puncta (punctate) or outside lysosomes. The right most panel is an overlay of the single channel images. Scale bar 20μm.
Figure 9.
Representative fluorescent micrographs of Cathepsin B (Cath B) localization in membrane disrupted and non-disrupted neurons within the lateral neocortex layers V and VI in sham injured animals (Sham, A and D), animals sustaining a CFPI (TBI, B and E), and animals sustaining a CFPI followed by secondary ICP elevation (TBI+ 20 mmHg ICP elevation, C and F) paired with 2w of ICV infusion of 10% DMSO (vehicle, A-C) or CA-074Me (Cath B inhibitor, D-F). The left most panel contains DAPI labeled nuclei (blue). Neurons were identified as non-disrupted or membrane disrupted based upon uptake of cell impermeable Alexa-fluor488 tagged 10kDa dextran (AF-Dextran-488; second panel in green). Immunolabeling for Cath B (third panel in red) allowed investigation of Cath B localization inside lysosomal puncta (punctate) or outside lysosomes. The right most panel is an overlay of the single channel images. Scale bar 20μm.
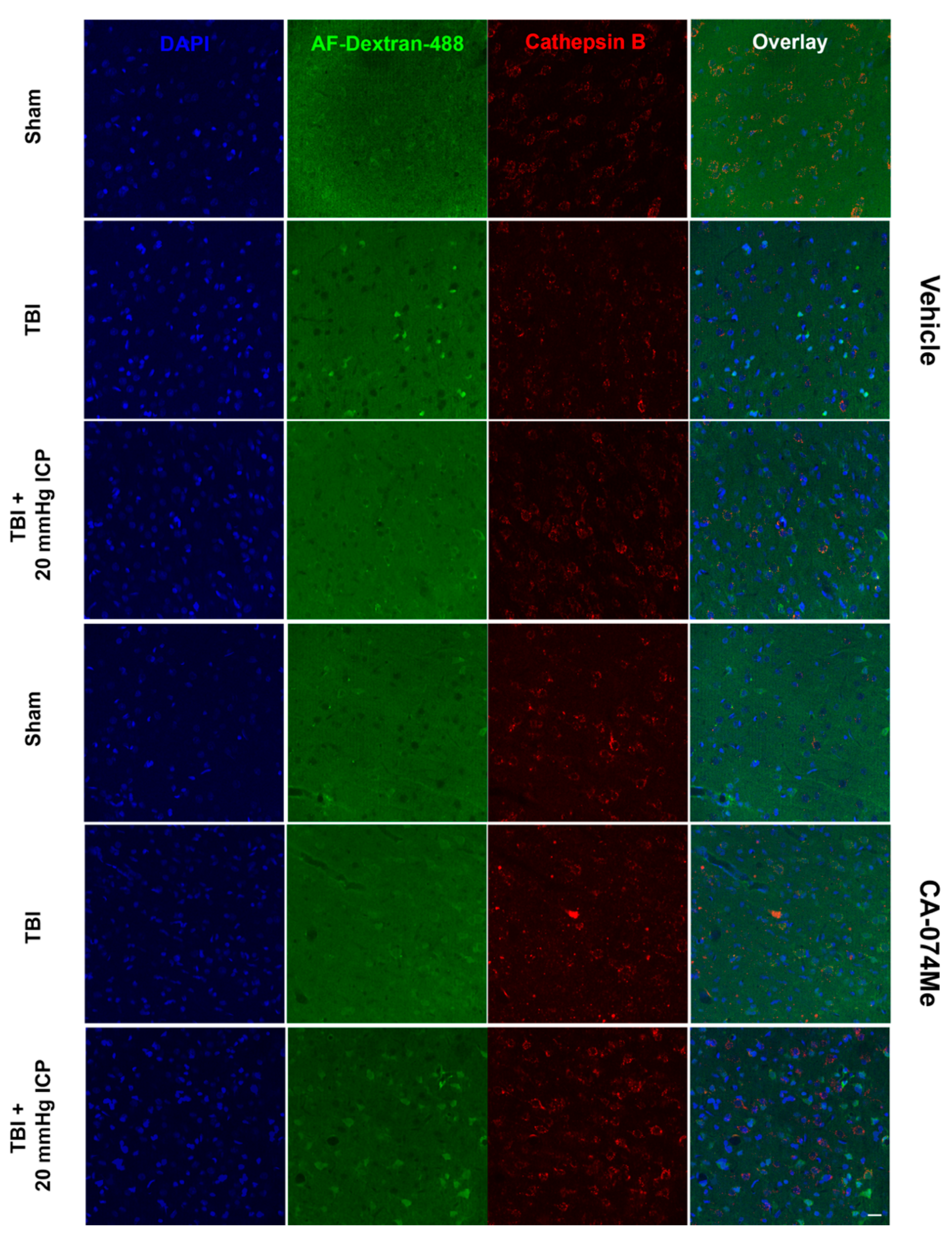
Figure 10.
Cathepsin B (Cath B) inhibition with CA-074Me impacted the distribution of Cath B within lysosomal puncta. Bar graph depicting the percent of neurons within the non-disrupted (left half of graph; Sham DMSO n=214 neurons [dark green bars], Sham CA-074Me n=248 [light green bars], TBI DMSO n=238 neurons [dark blue bars], TBI CA-074Me n=275 neurons [light blue bars], TBI+ICP DMSO n=226 neurons [dark pink bars], and TBI+ICP CA-074Me n=351 neurons [light pink bars]) and membrane disrupted (right half of graph; Sham DMSO n=159 neurons [dark green bars], Sham CA-074Me n=176 neurons [light green bars], TBI DMSO n=204 neurons [dark blue bars], TBI CA-074Me n=205 neurons [light blue bars], TBI+ICP DMSO n=153 neurons [dark pink bars], TBI+ICP CA-074Me n=199 neurons [light pink bars]) populations that exhibited punctate localization of Cath B. Note that Cath B localization within puncta was impacted by inhibitor infusion, injury group, and membrane disruption status of the neurons analyzed. * p<0.05 compared to non-disrupted counterpart, # p<0.05 compared to sham & CA-074Me, $ p <0.05 compared to vehicle-treated counterpart, ^ p<0.05 compared to TBI only infusion group counterpart. Mean ± S.E.M.
Figure 10.
Cathepsin B (Cath B) inhibition with CA-074Me impacted the distribution of Cath B within lysosomal puncta. Bar graph depicting the percent of neurons within the non-disrupted (left half of graph; Sham DMSO n=214 neurons [dark green bars], Sham CA-074Me n=248 [light green bars], TBI DMSO n=238 neurons [dark blue bars], TBI CA-074Me n=275 neurons [light blue bars], TBI+ICP DMSO n=226 neurons [dark pink bars], and TBI+ICP CA-074Me n=351 neurons [light pink bars]) and membrane disrupted (right half of graph; Sham DMSO n=159 neurons [dark green bars], Sham CA-074Me n=176 neurons [light green bars], TBI DMSO n=204 neurons [dark blue bars], TBI CA-074Me n=205 neurons [light blue bars], TBI+ICP DMSO n=153 neurons [dark pink bars], TBI+ICP CA-074Me n=199 neurons [light pink bars]) populations that exhibited punctate localization of Cath B. Note that Cath B localization within puncta was impacted by inhibitor infusion, injury group, and membrane disruption status of the neurons analyzed. * p<0.05 compared to non-disrupted counterpart, # p<0.05 compared to sham & CA-074Me, $ p <0.05 compared to vehicle-treated counterpart, ^ p<0.05 compared to TBI only infusion group counterpart. Mean ± S.E.M.
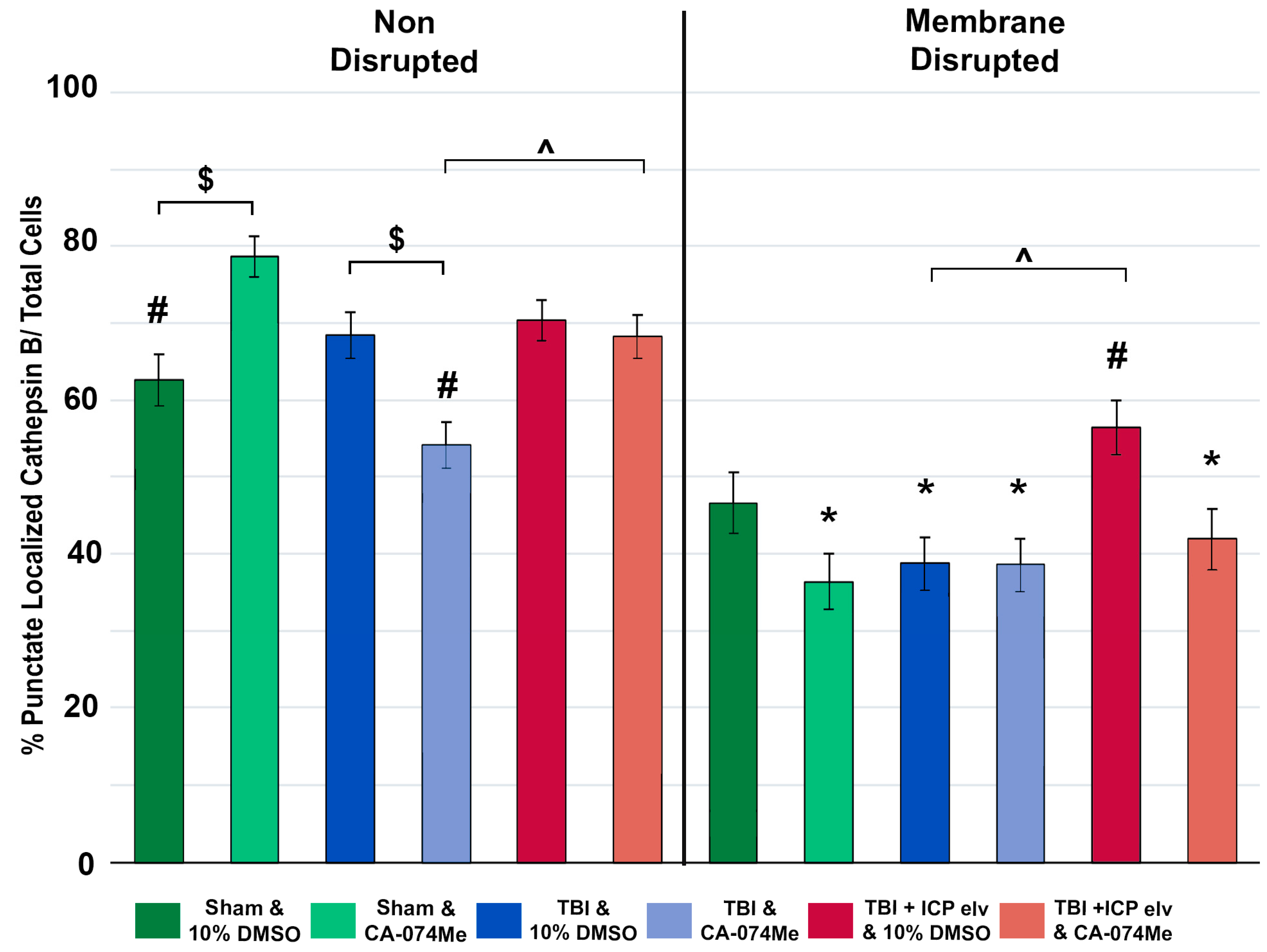
Figure 11.
Somatosensory sensitivity was impacted by injury, particularly with secondary ICP elevation, and further impacted by cathepsin B inhibition with CA-074Me. Bar graph depicting the average whisker nuisance task (WNT) score pre-injury (tan bars) and at 2w post-injury or sham (blue bars). Sham animals infused with 10% DMSO (n=11) had slightly higher WNT scores post-injury compared to sham animals infused with CA-074Me (n=13). Animals sustaining TBI infused with 10% DMSO (n=12) demonstrated similar post-injury WNT scores as TBI animals infused with CA-074Me (n=13). Injured animals with secondary ICP elevations (TBI + 20mmHg ICP) infused with CA-074ME (n=12) had lower WNT scores compared to injured and ICP elevated animals infused with 10% DMSO (n=11) resulting in an overall reduction in post-injury WNT score in animals infused with CA-074Me. * p<0.05 compared pre-injury WNT score for that group, # p<0.05 compared to sham 10% DMSO. Mean ± SEM.
Figure 11.
Somatosensory sensitivity was impacted by injury, particularly with secondary ICP elevation, and further impacted by cathepsin B inhibition with CA-074Me. Bar graph depicting the average whisker nuisance task (WNT) score pre-injury (tan bars) and at 2w post-injury or sham (blue bars). Sham animals infused with 10% DMSO (n=11) had slightly higher WNT scores post-injury compared to sham animals infused with CA-074Me (n=13). Animals sustaining TBI infused with 10% DMSO (n=12) demonstrated similar post-injury WNT scores as TBI animals infused with CA-074Me (n=13). Injured animals with secondary ICP elevations (TBI + 20mmHg ICP) infused with CA-074ME (n=12) had lower WNT scores compared to injured and ICP elevated animals infused with 10% DMSO (n=11) resulting in an overall reduction in post-injury WNT score in animals infused with CA-074Me. * p<0.05 compared pre-injury WNT score for that group, # p<0.05 compared to sham 10% DMSO. Mean ± SEM.
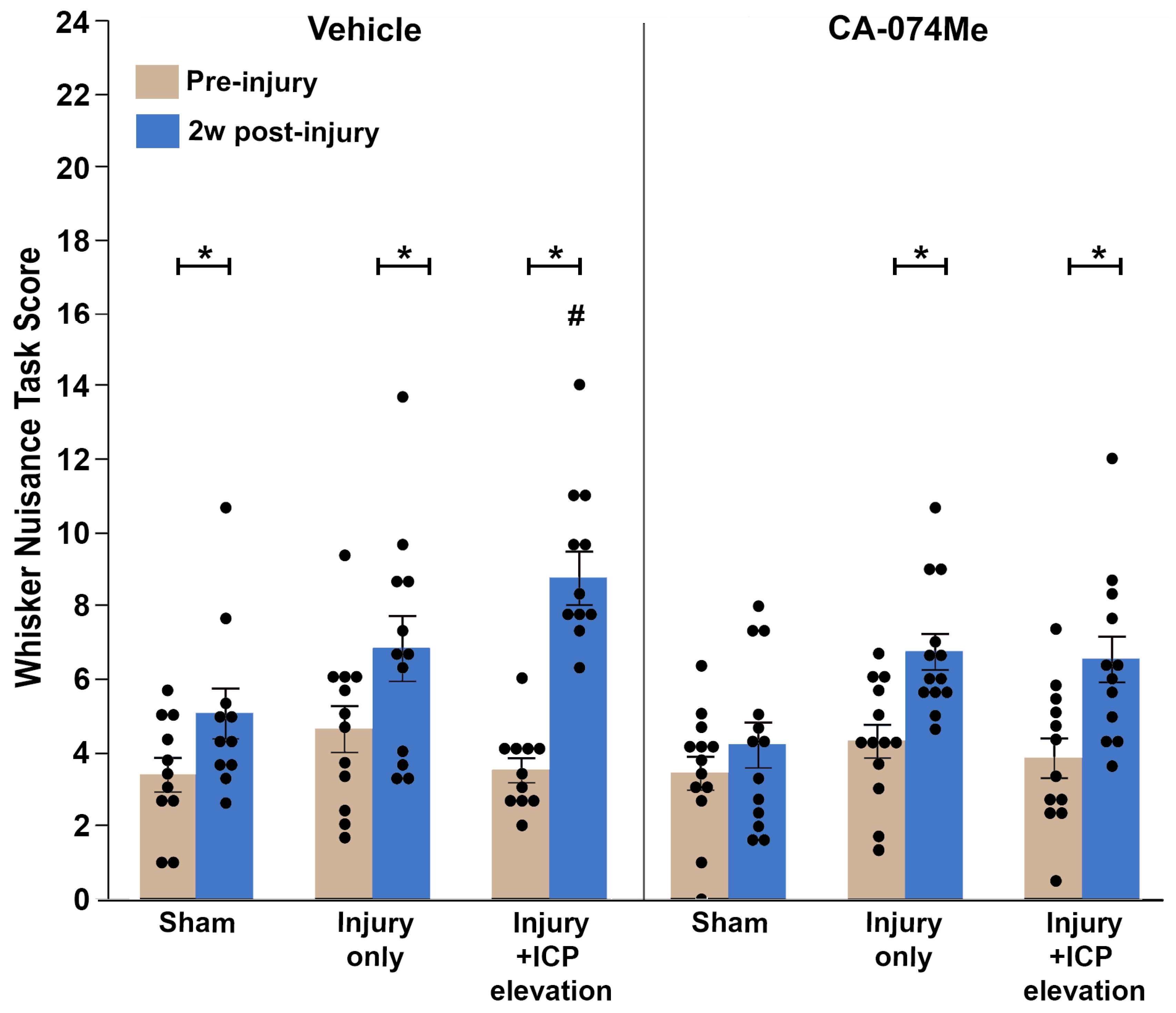
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



