
Submitted:
28 May 2024
Posted:
29 May 2024
You are already at the latest version
Abstract
Multiple myeloma (MM) is a common type of cancer that unfortunately leads to a significant number of deaths each year. The majority of the reported MM cases are detected in the advanced stages, posing significant challenges for treatment. Additionally, all MM patients eventually develop resistance or experience relapse, therefore advances in treatment are needed. However, developing new anti-cancer drugs, especially for MM, requires significant financial investment and a lengthy development process. The study of drug repurposing involves exploring the potential of existing drugs for new therapeutic uses. This can significantly reduce both time and costs, which is typically a major concern for MM patients. The utilization of pre-existing non-cancer drugs for various myeloma treatments presents a highly efficient and cost-effective strategy, considering their prior preclinical and clinical development. The drugs have shown promising potential in targeting key pathways associated with MM progression and resistance. Thalidomide exemplifies the success that can be achieved through this strategy. This review delves into the current trends, the challenges faced by conventional therapies for MM, and the importance of repurposing drugs for MM. This review highlights a noncomprehensive list of conventional therapies that have potentially significant anti-myeloma properties and anti-neoplastic effects. Additionally, we offer valuable insights into the resources that can help streamline and accelerate drug repurposing efforts in the field of MM.
Keywords:
hematological malignancies
; multiple myeloma
; drug repurposing
; drug development
; drug resistance
1. The Significance of Repurposing
Every year, millions of people around the world are diagnosed with cancer, and sadly, many lose their lives to this devastating disease. Globally in 2020, hematological malignancy incidence was almost 1.3 million and mortality was over 700,000, making hematological cancers the fourth highest in terms of cancer mortality [1]. Multiple myeloma (MM) accounts for more than 12% of all hematologic cancers. MM is a complex form of cancer that can be challenging to diagnose and treat. It is a malignancy of plasma B cells and originates in the bone marrow (BM). Despite advancements in treatments, MM remains an incurable disease as itwill inevitably progress or develop resistance to treatments in all patients. Optomistically speaking, however, therapeutic advancements could potentially increase life expectancy for MM patients, addressing both current and future challenges.
Drug research and development, especially in the realm of cancer drugs, have experienced remarkable changes in recent decades. Year after year, the collection of medications to combat cancer continues to grow. Nevertheless, there are a number of challenges that need to be addressed before a drug can be brought to market. These include the extensive drug development process, the significant expenses associated with drug research, the possibility of adverse events, and the limited efficacy of new treatments. In the field of cancer treatment, the search for effective lead compounds has long followed a well-established process. This involves conducting extensive pre-clinical and clinical research to carefully assess and document the compounds' pharmacological properties, anti-cancer effects, and potential toxicity (Figure 1).
Despite the advancements in technology and our improved understanding of human disease, the translation of these advantages into therapeutic breakthroughs has been disappointingly slow [3,4]. The global pharmaceutical industry faces a multitude of challenges, such as high attrition rates [5,6], evolving regulations, and prolonged time to market for new drugs in certain therapeutic fields, all of which have led to increased costs. A prediction has been made that the return on investment for new drug development is expected to be less than one dollar for every dollar spent due to the increasing cost and time required for these endeavors [7]. This could potentially reduce the attractiveness of the pharmaceutical industry as an investment option. As shown in Figure 1, the process of developing a new medicine can span a decade or more and require a significant investment of billions of dollars [8,9]. Thus, it is crucial to explore alternative methods for drug development.
Drug repurposing, also referred to as drug repositioning, re-tasking, or reprofiling, is a strategy employed to uncover new therapeutic uses for medications that have already been approved, tested, or are currently available on the market. These medications were initially developed and trialed for different therapeutic purposes than the ones they are being explored for now. Compared to new drug development, the repurposing approach offers numerous advantages to the research and clinical fields. It can be described as finding alternative applications for familiar medications. Previous clinical trials have confirmed the safety, effectiveness, and efficiency of a drug, considering its pharmacokinetic, pharmacodynamic, and toxicological properties. As a result, it has gained popularity and become more accessible. Looking at a drug molecule from a fresh angle could help us uncover new possibilities for its therapeutic applications beyond the conventional ones. Repurposing a medicine that has already received approval for a different therapeutic indication requires less funding. The process of approving drugs through the repurposing approach is estimated to take anywhere from 3 to 12 years and cost between $40 to $80 million, which is significantly lower than the cost of the traditional drug development method [10,11]. In addition, it is anticipated that the repurposing pathway will have a higher approval rate for medications, estimated at around 30%, compared to the typical drug development method which only yields 10% [11]. Once failures are taken into consideration, these advantages can lead to a faster and safer return on investment for the development of repurposed pharmaceuticals, along with lower average development costs. Only a small fraction, around 5%, of oncology treatments that qualify for phase 1 clinical trials end up getting the green light from the FDA. The chances of potential anticancer therapies being approved are even more dismal, with only 1 in 5,000-10,000 making the cut [12]. Through the implementation of a drug repurposing strategy, it becomes feasible to bypass the initial phase and make swift progress to subsequent phases of clinical trials, resulting in a reduction of concerns related to pharmacodynamics [12]. Therefore, there is a strong attraction towards methods that optimize the utilization of information from drugs that have already been approved and brought to market [13]. In the end, repurposed pharmaceuticals have the potential to uncover new targets and pathways for further exploration. Therefore, researchers are currently focusing on exploring new pharmacological action mechanisms that have emerged from unexpected clinical trial findings. These findings have sparked interest in bridging the gap between clinical practice and laboratory research. Various preclinical investigations are conducted to validate the claim of a new pharmacological indication. This approach focuses on addressing chronic illnesses such as diabetes, cancer, and other rare disorders [13]. There are numerous benefits to repurposing pharmaceuticals in general. This approach is both compelling and practical, especially in today's era in which deep data mining technologies are readily accessible. The frequent approval of repurposed pharmaceuticals suggests that the technique of repurposing has a minimal risk of drug failure, as most of the drugs being repurposed undergo thorough safety testing. The drug repurposing sectors have experienced consistent growth from a business perspective, with approximately 14-16 new companies emerging every five years [14,15].
MM is an incurable cancer in which relapse inevitably occurs even for patients in remission. Typically, the primary treatment for newly diagnosed MM includes bortezomib (a proteasome inhibitor), lenalidomide (an immunomodulatory agent), dexamethasone (a corticosteroid)[16]. This combination of drugs is termed the Vrd regimen and has shown to be effective in treatment of MM upfront [16]. Patients may also undergo autologous stem cell transplantation, if deemed eligible, either initially or later within the treatment process [16]. However, this regime is not permanently curative, and patients must switch drugs when they develop resistance. Malignant plasma cells display high levels of aberrant cell signaling pathways that prevent apoptosis and promote cell survival, ultimately leading to relapse. The NF-κB pathway is activated by cytokines, such as IL-6, and the binding of MM cells to bone marrow stromal cells [17]. This pathway is inhibited by proteasome inhibitors and strongly linked with disease progression as it increases MM cell proliferation and resists apoptosis [17]. Similarly, the antiapoptotic subgroup of Bcl-2 includes Mcl-1, Bcl-2, and Bcl-xL which are commonly overexpressed in MM and are shown to prevent intrinsic apoptosis by suppressing BH-3 activators and competing for binding Bax and Bak proteins [18]. Interaction of myeloma cells with bone marrow microenvironment is integral to malignant myeloma cell proliferation and the development of resistance [17]. Cytokines such as IL-6, IL-3, and IL-5 and growth factors such as VEGF and EGF trigger upregulation of intracellular pathways such as NF-κB, JAK/STAT, PI3-K/Akt, and Bcl-2 antiapoptotic proteins– all of which promote MM cell proliferation and therefore the development of resistance [18].
MM therapy has stood out as an anomaly in terms of the typical balance between risks and benefits. Some medications used in anti-cancer chemotherapy and radiation have severe adverse effects that can be life-threatening. The primary objective of therapy in MM is to ensure the patient's survival. Overcoming resistance to treatment is a significant hurdle when it comes to developing a successful dosage plan. This review explores the potential of various medications to treat MM, highlighting the anticancer properties of conventional drugs such as thalidomide, statins, celecoxib, aspirin, artesunate, leflunomide, rapamycin, nelfinavir, valproic acid, metformin, bisphosphonates, and clarithromycin. We have compiled a comprehensive list of resources that may prove valuable for drug repurposing.
2. Pharmacological Repurposing Strategies and Tools
The drug repurposing strategy involves three stages that occur before a candidate drug progresses through the development pipeline. These stages include generating hypotheses to identify a potential molecule for a specific indication, assessing the drug's effects using preclinical models, and conducting phase II clinical trials to evaluate its efficacy (assuming sufficient safety data has been gathered from phase I trials conducted for the original indication). The first stage is crucial. We must select appropriate molecules whose mechanisms of action would negatively affect the malignant properties of MM cells. This is where contemporary methodologies for generating hypotheses can be most advantageous. There are different types of systematic approaches, such as computational approaches and experimental approaches, that are being increasingly used together (Figure 2). These two core domains encompass clinical data-driven drug repurposing. The majority of computational techniques rely heavily on data. These techniques generate hypotheses for repurposing by thoroughly analyzing various types of data, such as electronic health records (EHRs), genotyping, chemical structure, gene expression, or proteomic information [19]. The process of signature matching involves comparing the unique characteristics or "signature" of a specific pharmaceutical compound with those of another compound, disorder, or clinical phenotype [20,21]. The creation of a drug's signature can be derived from three different sources of data: transcriptome (RNA), proteomic, or metabolomic data; chemical structures; or adverse event patterns.
After the initial sketch of the human genome was produced through the human genome project, research and development expenses in OECD nations have increased by more than 50 percent [22]. With the help of genomics, microbiomics, and metabolomics, it is possible to easily search for information on signal transduction pathways, systems biology, the safety and adverse effects of approved drug libraries, and the additional targets of approved pharmaceuticals. Methods rooted in systems biology, like the Genome-wide Positioning Systems network (GPSnet), are aiding in the comprehension of disease-gene-drug interactions, potentially facilitating drug repurposing for MM [14,23,24,25,26,27]. In addition, there are multinational collaborative programs, like Repurposing Drugs in Oncology (ReDO), that are working to accelerate the repurposing of non-cancer drugs for cancer treatment. Table 1 provides supplementary information and tools that can assist in the drug-repurposing process. This text discusses the role of various technologies in studying signaling networks in cancer cells, particularly MM, and how this knowledge can be applied to understand the interactions between medicines, proteins, and genes. However, there are other techniques that can help in understanding adverse medication effects, disease-disease connections, and drug sensitivity of multiple cancer cells. These approaches can be used to repurpose any authorized or discontinued medicine for use in MM. The role of driver pathways in promoting the survival and proliferation of cancer cells is crucial, while bystander pathways offer support. Through the utilization of biological databases and systems biology technologies, it becomes feasible to identify driver pathways in MM. Subsequently, pre-existing drugs from drug libraries can be discovered, which possess the ability to target these driver pathways [28].
3. Medicines That Could Be Repurposed to Treat MM
MM cells display numerous abberant signaling pathways and protein expression contributing to disease progression, there are numerous agents that have potential to aid in treatment of MM due to the pathways that they directly or indirectly influence. Many of the proposed drugs for repurposing have mechanisms of actions that could treat other cancers as well; however, these agents are particularly of interest in terms of MM. This is because MM cells consistently overexpress the pathways and proteins targeted by these agents, as detailed below, and many of which are crucially involved in disease relapse, meaning the abberant activity heightens despite treatment. Because MM patients inevitably relapse, alternative or additional medications need to be explored and drug repurposing is the most efficient and potentially effective way of doing so. Table 2 displays the medications that have potential to demonstrate effectiveness against MM. Drug repurposing has often occurred by chance and circumstance throughout history. The identification of an off-target impact or a newly identified on-target effect led to the advancement of a pharmaceutical drug towards commercialization. The logic behind estimating the anti-cancer properties of non-cancer drugs was established based on their effectiveness, mechanisms of action, and minimal side effects (Figure 3). Interestingly, the most successful cases of drug repurposing so far have not followed a systematic approach. As an example, the discovery of thalidomide's effectiveness in treating erythema nodosum leprosum (ENL) and MM was purely accidental [3].
3.1. Thalidomide
Thalidomide, a derivative of glutamic acid [52], was initially introduced as a sedative in 1957 [53] and also employed as an antiemetic during pregnancy. However, due to its severe teratogenic effects, it was withdrawn from the market in 1961 [54]. Moving forward, however, Thalidomide's efficacy against erythema nodosum leprosum (ENL), a cutaneous form of leprosy, led to its FDA approval for this condition in 1998 [55]. Subsequently, its anti-angiogenic properties expanded its usage to treat various skin disorders, infectious diseases, immunologic and rheumatologic disorders, hematologic diseases, and several cancer types [56,57,58]. In 2006, the FDA approved thalidomide for newly diagnosed MM in combination with dexamethasone. Thalidomide influences numerous signaling pathways commonly dysregulated in cancer cells [59]. It inhibits tumor necrosis factor (TNF), frequently deregulated in cancers [60], and impedes NF-κB activation, a protein complex crucial for DNA transcription, cytokine production, and cell survival, by inhibiting IκB kinase (IKK) [61]. Additionally, thalidomide inhibits interleukin (IL)-1, IL-6, and IL-12, granulocyte-macrophage colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and interferon (IFN) [62,63], making it potentially effective aga inst various cancers like AIDS-related Kaposi’s sarcoma, renal cell carcinoma, and gliomas [64,65,66]. Thalidomide shows promise in treating hormone-dependent prostate cancer, as evidenced by a phase III clinical trial where it reduced time to prostate-specific antigen (PSA)-based progression in patients receiving androgen deprivation therapy [67]. However, toxicity issues have impeded many clinical trials of thalidomide as a cancer treatment [68,69,70], leading to the development of less toxic thalidomide analogues with similar anti-cancer effects. Thalidomide's repurposing involved off-label usage, pharmacological analysis, and derivative development, resulting in significant clinical and commercial success in MM.
3.2. Statins
The discovery of statins as HMG-CoA reductase inhibitors led to their approval for treating individuals at high risk of heart failure. The transformation of HMG-CoA to mevalonic acid is catalyzed by HMG-CoA reductase, which is the first and rate-limiting step in cholesterol production. Statins inhibit HMG-CoA reductase, which in turn suppresses cholesterol biosynthesis and the mevalonate cascade. Byproducts of this cascade play a vital role in the survival of cancer cells; therefore, the effectiveness of statins in combating various types of cancer cells has been assessed.Studies have demonstrated that Simvastatin can induce apoptosis in CML cells [71]. In addition, it has been found through meta-analyses that statins are associated with a lower risk of hematological malignancies [72,73]. The use of statins has been linked to an enhanced survival rate and a decrease in mortality in MM [74,75,76,77]. Clinically in 4315 patients, statin use is associated with improved survival in MM [75]. A PRISMA-compliant meta-analysis indicated that statin use might be a protective factor for MM incidence [78].
MM cells containing chromosomal translocation t(4;14) is typically a more aggressive subtype of MM. It has been shown that t(4;14)-positive cells have increased reliance on the mevalonate pathway due to its production of on geranylgeranyl pyrophosphate [79]. When treated with statins, t(4;14)-positive cells underwent an integrated stress response due to lack of geranylgeranyl pyrophosphate and, used in conjunction with bortezomib, results showed even greater cytotoxic effects in vivo [79]. MM cells frequently rely heavily on Mcl-1 for their survival, making them resistant to venetoclax, a Bcl-2 inhibitor [18]. Statins have the potential to overcome resistance to venetoclax in MM cell lines and primary cells by blocking the mevalonate pathway. Furthermore, it has been observed that statins can enhance the susceptibility to apoptosis induced by S63845, a potent Mcl-1 inhibitor [80]. In retrospective analyses of venetoclax clinical trials in patients with MM, the use of statins prior to treatment was found to be significantly associated with a greater likelihood of achieving a rigorous complete response prolonging progression free survival[80]. The sensitivity of MM cells to venetoclax is increased by statins through the upregulation of two pro-apoptotic proteins, PUMA (in a mechanism not dependent on p53) and NOXA (via the integrated stress response) [80]. Additional research involving animals and humans suggests that statins may offer some level of protection against the development of MM [77]. This study presents a new avenue for utilizing statins in the treatment of MM and other blood malignancies. It also indicates their potential in preventing cancer in susceptible populations.
3.3. Celecoxib
Several studies have demonstrated a clear link between the overexpression of cyclooxygenase-2 (COX-2) and lower survival rates in patients with MM [81,82]. Hematological malignancies such as Hodgkin's lymphoma, NHL, CLL, CML, and MM show an upregulation of COX-2. The pro-inflammatory cytokine, COX-2, plays a crucial role in angiogenesis, metastasis, cell proliferation, and survival [83,84]. Celecoxib, a non-steroidal anti-inflammatory drug (NSAID), selectively inhibits COX-2. Celecoxib has been used in the clinic since 1998 to treat osteoarthritis, rheumatoid arthritis (RA), and generalized pain. In addition, extensive research has indicated celecoxib as a promising chemopreventive drug for various forms of cancer [85]. Furthermore, studies have indicated that celecoxib has the ability to inhibit crucial survival proteins in various types of cancers, such as Mcl-1, Bcl-2, survivin, and Akt [86]. In MM, these proteins have all been established as overexpressed; additionally, Mcl-1 and Bcl-2 are drivers for the mechanisms of aquired resistance and relapse [18]. The FDA has previously approved celecoxib for use in a number of cancer therapies, including familial adenomatous polyposis (FAP) (800 mg/day), albeit at higher doses than prescribed for RA or osteoarthritis [87]. Higher dosages of celecoxib can lead to various side effects, including cardiotoxicity, gastrointestinal issues, and renal problems. Therefore, It is important to exercise caution when using NSAIDs in cancer treatment. Limited data exist regarding the role of COXs in the onset and progression of MM. To address this, researchers tested seven human MM cell lines representing different disease stages to assess COXs expression levels and cell viability upon treatment with COX inhibitors alone or in combination with conventional anti-MM drugs. The results showed that all tested drugs had moderate antiproliferative effects on MM cell lines [88]. Celecoxib shows great potential as a potential anti-myeloma drug, considering its correlation with decreased survival rates in MM due to COX-2 overexpression.
3.4. Aspirin
Aspirin, also known as acetylsalicylic acid (ASA), is another, more common, NSAID that is utilized for pain relief and reducing fever. In addition, it effectively helps prevent myocardial infarctions and cerebrovascular accidents caused by thromboembolism. As a target, aspirin is not as selective as celecoxib. Aspirin inhibits both COX-1 and COX-2, preventing the synthesis of prostaglandin H2 (PGH2) and prostaglandin E2 (PGE2). These compounds play a role in regulating inflammatory and thrombus formation processes. Aspirin not only inhibits COX-1/2, but also has a suppressive effect on other inflammatory cytokines like NF-κB. Additionally, it impacts pro-survival ERK signaling in cancer cells. It is worth noting that MM cells exhibited an overexpression of both NF- κB and ERK signaling [89,90]. Furthermore, Aspirin demonstrates its anti-tumor effect in MM by inhibiting Blimp1 and activating the ATF4/CHOP pathway [91]. Based on two significant prospective studies, the "Health Professional Follow-up Study" (1986–2008) and the "Nurses' Health Study" (1976–2008), it has been found that taking aspirin after being diagnosed with MM is associated with a lower risk of death, both overall and specifically related to MM [92]. According to the trials, regularly taking 325 mg aspirin five or more times per week was linked to a significant 40% decrease in the rate of MM [93]. However, it is important to consider potential secondary issues like bleeding stomach ulcers and heartburn when using aspirin for an extended period of time.
3.5. Clarithromycin
Clarithromycin (CAM) is a widely used antibacterial treatment belonging to the class of semisynthetic macrolide antibiotics. The first clinical study revealed a significant increase in median survival time for patients with advanced non-small cell lung cancer (NSCLC) who underwent long-term CAM therapy [94]. It has been discovered that CAM can be a helpful addition to the treatment of MM [95]. Although, treating MM with single agent CAM therapy is not effective, there is confirmed efficacy of CAM in combination chemotherapies for the treatment of MM. There have been several documented clinical trials that have explored the use of CAM in combination with other drugs for MM patients. These trials include NCT01745588, NCT01559935, and NCT02248428 [96,97,98,99,100,101,102,103]. However, most recently, a randomized phase III clinical trial found that in combination with steroid dexamethasone and lanalidomide, CAM did not improve progression free survival due to the tosic effects of steroid overexposure (NCT02575144) [104].
It is widely recognized that various myeloma growth factors (MGFs), including IL-6, have a significant impact on the advancement of MM [18,105]. CAM has been shown to inhibit several MGFs, including IL-6 [106]. The treatment of MM with complementary and alternative medicine CAM involves various potential mechanisms of action. These include autophagy inhibition, immunomodulatory activity, reversibility of drug resistance, steroid sparing/enhancing impact, and suppression of MGFs. MM is characterized by the excessive growth of cancerous plasma cells that produce a single type of immunoglobulin (Ig). The presence of excessive misfolded or unfolded Ig can cause considerable stress on the endoplasmic reticulum [107]. Thus, MM arises as a fragile tumor that is particularly susceptible to autophagy inhibitors, proteasome inhibitors, and histone deacetylase 6 inhibitors. The combined effects of CAM play a crucial role in the treatment of MM [108].
3.6. Rapamycin
The antifungal agent rapamycin, also known as sirolimus and commercialized as Rapamune®, was initially discovered by Surendra Nath Sehgal and colleagues. It was isolated from the bacteria Streptomyces hygroscopicus on the island of Rapa Nui [109]. One of the initial signs of its approval was its use in managing graft rejection in kidney transplant recipients, thanks to its immunosuppressant properties [110]. Rapamycin was later identified as an antagonist of the mTOR (mammalian target of rapamycin) signaling pathway. mTOR plays a crucial role in various signaling pathways, such as cytoskeleton maintenance, protein synthesis, autophagy, lipid synthesis, cell growth, and angiogenesis [111]. In addition to its role in preventing graft rejection, rapamycin has been extensively researched as a potential anti-cancer treatment for various types of cancer. Rapamycin sensitized MM cells to apoptosis induced by dexamethasone [112]. In addition, the effectiveness of rapalogs and newer TORC1/TORC2 inhibitors in myeloma models has been supported by preclinical studies. Clinical trials in the initial phases have already begun [113]. Rapamycin, when combined with CC-50 (Revlimid), an immunomodulatory analog (IMiD) of thalidomide, has demonstrated anti-MM activity [114]. These trials serve as the basis for evaluating the effectiveness of mTOR inhibitors in combination with IMiDs in improving patient outcomes in MM. The individuals under investigation experienced negligible adverse effects from rapamycin, which highlights its decreased risk and treatment advantages. In order to further improve the efficiency of this molecule, efforts have been made to enhance its structure.
3.7. Valproic Acid
Valproic acid (VPA, Depakene) is a medication that is commonly prescribed for the treatment of migraine, seizures, epilepsy, and bipolar disorders. Valproic acid is believed to block voltage-gated sodium channels and histone deacetylases (HDAC). HDAC plays a crucial role in the survival of MM cells. It is believed that the impact of bortezomib on apoptosis in MM is partially due to the inhibition of Class-I HDACs [115]. In addition, VPA has been found to inhibit the activation of NF- κB and the production of inflammatory cytokines TNF and IL-6 [116]. It has been found that VPA enhances autophagic flux in human MM cells [117]. In vitro studies demonstrated autophagy activation in MM cell lines RPMI8226 and U266 following VPA treatment [118]. There are indications that VPA could potentially be utilized as a treatment for MM [119].
3.8. Nelfinavir
Nelfinavir, an efficient protease inhibitor, is effective against both HIV-1 and HIV-2. As for ritonavir, principally attributed to the drug's anti-cancer properties include its ability to disrupt Akt signaling and induce endoplasmic reticulum stress. Nelfinavir therapy has been seen to induce regression of prostate xenograft tumors and decrease phosphorylation of both STAT3 and Akt [120]. In MM, treatment induces a reduction in signaling via Akt, STAT3, and Erk1/2 by inhibiting the 26S proteasome [121]. Additionally, Nelfinavir and bortezomib have a synergistic effect. In a trial involving MM and non-small cell lung cancer, the combination of bortezomib and nelfinavir increased endothelial resistance (ER) stress and inhibited growth in vitro and in vivo. The buildup of ubiquitin (Ub)-proteins is the mechanistic consequence of nelfinavir therapy. Bortezomib, on the other hand, inhibits degradation by the proteasome; this interferes with proteotoxic stress and ultimately induces cell death [122]. MM cells may also become sensitive to bortezomib in the presence of nelfinavir. Nelfinavir demonstrates inhibitory effects on the proteasome, an activity that bortezomib fails to target. This leads to the hypothesis that nelfinavir's anti-neoplastic actions in MM cells are mostly mediated by this mechanism in conjunction with suppression of Akt-phosphorylation [123].
3.9. Metformin
Metformin is a commonly prescribed medication for the treatment of insulin-independent DM type 2 (diabetes mellitus). AMPK is crucial for cellular metabolism, specifically in controlling glucose metabolism. Metformin enhances the utilization of blood glucose absorption by muscles and liver by activating AMPK. AMPK suppresses mTORC1, a pathway associated with cell proliferation, while activating p53, a tumor suppressor protein involved in promoting cell death. Metformin has been found to inhibit mTORC1 in a way that is different from AMPK [124]. Metformin has been found to have various effects on cancer cells, including inhibiting epithelial-to-mesenchymal transition (EMT), inducing senescence, and reducing the survival of cancer stem cells [125]. In MM, the development of EMT phenotypes are pertinent and shown to be hypoxic drive and contribute to malignant plasma cell migration and cells aquiring drug resistance [126]. MM cells will also overexpress EMT transcription factors in response to signals from the BMM [126]. Therefore, metformin has the potential to be developed as a therapeutic anti-cancer treatment [127,128]. Based on a comprehensive analysis of observational studies, it was found that individuals with diabetes who were prescribed preventative metformin medication experienced a lower likelihood of developing cancer and succumbing to cancer-related causes [127]. Studies suggest that metformin may have inhibitory effects on various cell cycle regulatory proteins, such as cyclin D1, Rb, ERK1/2, JAK2/STAT signaling, and mitochondrial function, which could potentially explain its observed effects [129]. Metformin suppresses IL-6 signaling by reducing IL-6R expression on MM cells [130]. In addition, metformin is known to stimulate autophagy. Researchers are currently focusing on the AMPK/mTORC1 and mTORC2 pathways to trigger autophagy and cell cycle arrest in myeloma. Importantly, In a comprehensive studies, metformin use among diabetic individuals with MGUS was linked to a decreased risk of developing active MM [131,132]. Several of preclinical studies have also shown anti-myeloma effects of metformin [133,134]. Several studies have demonstrated the synergistic effects of combining metformin with drugs that affect glycolysis, such as ritonavir in MM [134]. The interaction between metformin and a Glut4 glucose transporter inhibitor, which would modulate MM, was predicted in silico [135]. Metformin and FTY720 synergistically triger apoptosis in MM cells [136]. Metformin has been found to potentially speed up cell death in MM cells when combined with the proteasome inhibitor bortezomib, by inhibiting a protective autophagic response and disrupting protein homeostasis [137]. It appears that when metformin is used alongside chemotherapy, MM patients may experience a longer survival period.
3.10. Bisphosphonates
Bisphosphonates are a class of chemical compounds that contain two phosphonate groups and two R-groups, distinguishing them from other bisphosphonates. There are two main classes of bisphosphonates: nitrogenous and non-nitrogenous. Examples of nitrogenous bisphosphonates are clodronate, tiludronate, etidronate, and nitrousous bisphosphonates. On the other hand, non-nitrogenous bisphosphonates include zoledronate, neridronate, alendronate, pamidronate, ibandronate, olpadronate, and risedronate. Due to its early presence in bone metabolism research, bisphosphonates were later recommended for postmenopausal women as a preventive measure against bone loss (osteoporosis). In addition, its application was broadened to encompass patients suffering from bone loss caused by metastatic lung cancer, breast cancer, and MM. The mechanisms governing nitrogenous and non-nitrogenous bisphosphonate compounds have distinct differences. The integration of first-generation non-nitrogenous bisphosphonates with the ATP of osteoclasts results in the production of ATP analogs that exhibit resistance to hydrolysis. The activity of farnesyl pyrophosphate synthase (FPPS), a crucial enzyme in the HMG-CoA pathway responsible for cholesterol production, is suppressed by fourth and third generation nitrogenous bisphosphonates. Based on extensive clinical and laboratory research, it has been found that nitrogenous bisphosphonates are effective in treating both solid and hematological cancers [138]. Osteoclast cells undergo apoptosis when exposed to bisphosphonates [139]. The growth of MM is facilitated by the favorable conditions found in bone microenvironments. The interaction between osteoclasts and MM cells is reciprocal, with each one influencing the other's activity and survival [140,141]. The collaboration between MM cells and osteoclasts is disrupted by bisphosphonates, which creates an unfavorable microenvironment for MM cell growth. In MM, bisphosphonate has potential to serve as a valuable supplemental treatment.
3.11. CuET
Diethyldithiocarbamate-copper complex (CuET) is a product of metabolism combining Disulfiram (DSF) and cofactor copper[142]. DSF is an FDA approved drug for alcohol abuse, and it causes adverse, immediate, and unpleasant reactions to alcohol consumption [143]. This is because DSF inhibits enzyme aldehyde dehydrogenase (ALDH) which is crucial in the liver’s metabolism of alcohol [143]. CuET is a biologically active complex, due to the copper cofactor, and is shown to produce intracellular and extracellular reactive oxygen species (ROS) which promotes apoptosis [144]. Additionally, in cancer cells CuET caused levels of proteasome inhibition, which MM cells are extremely sensitive to due to their function in antibody production, and aggregates proteins such as IκB (which inhibits NF-κB) , p27, Kip1 and c-Myc [144]. Multiple Myeloma stem cells (MMSCs), cells integral in the progression of disease, display increased ALDH activity and have higher tumorigenic rates [145]. Additionally, the dominant isoform of ALDH is notably more expressed in bortezomib resistant MM cells, and a significantly higher percentage of the resistant cells express ALDH [145]. Inhibition of the metabolic enzyme offers a viable treatment application for MM. An in vitro and in vivo study observed that CuET can negatively affect stem cell qualities of MM cells, minimize tumor growth, and eliminate clonogenicity by inhibiting ALDH via the Hedgehog and ALDH1A1 pathways [146]. Furthermore, CuET successfully induced apoptosis in bortezomib and carfilzomib resistant MM cells and effected MM cells in ways similar to proteasome inhibitors as it induced unfolded protein response [147]. CuET’s mechanism of action and effects on MM cells in vitro and in vivo establish the drug as a promising target to be repurposed for MM treatment.
3.12. Albendazole
Albendazole (ABZ) is an antiparasitic agent used to treat intestinal worm infections and filarial infections [148]. ABZ prevents the uptake of glucose by parasites and mammalian cells through the inhibition of the microtubule systems [148]. However, the drug has applicable anti-cancer properties. In accordance with its mechanism in parasites, in leukemia cells, ABZ downregulates protein SIRT3 to cause upregulation of TNF-α which subsequently destabilizes microtubules within the cell, halting the cell cycle [149]. Additionally, ABZ treated leukemia cells showed evidence of ROS production, activation of death receptor-mediated pathway, and activation of p38 MAPK which ultimately resulted in apoptosis of the malignant cells [149]. Additionally, ABZ treated leukemia cells showed evidence of ROS production, activation of. These apoptotic effects are consistent in MM cells both in vitro and in vivo [150]. Among MMScs, ABZ reduced the number of ALDH-positive cells, which correlates with higher rates of tumorigenesis and acquired resistance, and in doing this, resistant MM cells were resensitized to bortezomib [150]. Furthermore, treatment of MM cells with ABZ inhibited the NF-κB pathway by diminishing transcription factor p65 [150]. Repurposing ABZ to be utilized in relapsed MM treatments offers a promising outlook given current evidence.
4. Conclusions and Future Prospectives
It has proven challenging to find a comprehensive treatment for MM, which is why there is a constant need for new medications. However, this demand requires a significant investment of both money and time. There has been a growing interest among physicians and researchers in recent decades to explore new applications for drugs that have already been approved for use in patients because repositioning drugs bypasses various obstacles. Drug repurposing plays a crucial role in the development of groundbreaking anti-cancer treatments. This study discusses various drugs used as anti-cancer agents and potential pharmaceuticals for MM treatment. Thalidomide's successful repurposing is a rare achievement in the field of cancer therapy, and successful repurposing of a limited number of non-cancer drugs for cancer treatment has sparked extensive study. This approach has the potential to save both time and money that would otherwise be needed to develop new anti-cancer drugs. However, it is crucial to exercise utmost caution when conducting tests on patients with non-cancer drugs. In doing so, it is crucial to establish the drug's effectiveness and mechanism of action before proceeding with clinical trials. Appropriate dosing of repurposed drugs needs to be explored in the context of MM pateints. Additionally, the potential synergistic effects of the repurposed drug alongside more traditional MM treatments should be evaluated and the most advantagoues treatment regimine must be determined. Therefore, it is crucial to thoroughly understand the mechanisms of drugs before subjecting cancer patients to testing. Using cancer and medication databases, like Table 1 details, can help expedite this process. The core concerns surrounding drug effectiveness and safety in clinical trials have remained consistent over time. There is room for discussion regarding the potential repurposing of non-cancer drugs that did not succeed in Phase II or Phase III as this opens up a large umber of drugs for exploration within MM treatment.This strategy has the potential to save a significant amount of time and money, it a more favorable option for repurposing approved non-cancer medications for MM. Approximately 48 percent of the total cost of drug development is dedicated to preclinical development and Phase I clinical trials of a new chemical entity (NCE). These crucial stages typically last for an average of seven years [9]. During the transition from Phase I to Phase II clinical trials, most medications fail due to their lack of effectiveness or toxicity. Therefore, medicines that do not pass Phase II can be considered as potential candidates for repurposing if they show anticancer activity. Several non-cancer medications demonstrated promising results in preclinical tumor models. However, it is still unclear whether their translation has any therapeutic applications.
- Practice points
- Although there have been numerous clinical trials conducted to evaluate different approaches for treating cancer, the 5-year survival rate for individuals with MM in the US remains at a modest 55%.
- Myeloma remains a challenging malignancy to treat due to the development of drug resistance, resulting in relapse for all patients.
- There is an ongoing demand for new medications. However, the process of finding a new treatment can often be quite time-consuming. Therefore, repurposing already approved non-cancer medication for MM can aid in the discovery of new effective drugs.
- The potential for repurposing approved drugs is promising, although a thorough analysis of these agents is necessary before they can be considered for clinical trials.
Research agenda
- The potential of various non anti-cancer drugs as an anti-myeloma treatment was discussed.
- Thalidomide stands out as an exemplary repurposed agent for treating MM.
- There is encouraging evidence that statins, rapamycin, clarithromycin, and leflunomide can inhibit MM.
- Extensive animal studies using the MM animal model, along with phase 1 clinical studies, are necessary to thoroughly investigate these agents as potential MM therapies.
Author Contributions
MKP, TBA, and SCJ collaborated on the idea, layout, literature search, and editing of the article. The OSA and EN conducted a thorough literature search, authored the manuscript, organized all sections, and created the necessary figures and tables. Figures were generated using BioRender.com.
Funding
The work received support from various sources including the Camden Research Initiative fund (M.K.P., T.B.A., and S.J.), the New Jersey Health Foundation (S.J., M.K.P.), and an interdepartmental fund from Cooper Medical School of Rowan University (M.K.P.) in Camden, NJ.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Authors do not report any conflict of interest.
Consent for publication
Not applicable.
References
- Yang, J., et al., Disparities in mortality risk after diagnosis of hematological malignancies in 185 countries: A global data analysis. Cancer Letters, 2024: p. 216793.
- Shameer, K., et al., Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Briefings in bioinformatics, 2018. 19(4): p. 656-678.
- Ashburn, T.T. and K.B. Thor, Drug repositioning: identifying and developing new uses for existing drugs. Nature reviews Drug discovery, 2004. 3(8): p. 673-683.
- Scannell, J.W., et al., Diagnosing the decline in pharmaceutical R&D efficiency. Nature reviews Drug discovery, 2012. 11(3): p. 191-200.
- Pammolli, F., L. Magazzini, and M. Riccaboni, The productivity crisis in pharmaceutical R&D. Nature reviews Drug discovery, 2011. 10(6): p. 428-438.
- Waring, M.J., et al., An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nature reviews Drug discovery, 2015. 14(7): p. 475-486.
- Beachy, S.H., et al., Drug repurposing and repositioning: workshop summary. 2014: National Academies Press Washington, DC.
- DiMasi, J.A., H.G. Grabowski, and R.W. Hansen, Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of health economics, 2016. 47: p. 20-33.
- Paul, S.M., et al., How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature reviews Drug discovery, 2010. 9(3): p. 203-214.
- Papapetropoulos, A.
- Hernandez, J.J., et al., Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as cancer therapeutics. Frontiers in oncology, 2017. 7: p. 273.
- Nosengo, N., Can you teach old drugs new tricks? Nature, 2016. 534(7607): p. 314-316.
- Ishida, J., et al., Repurposing of approved cardiovascular drugs. Journal of translational medicine, 2016. 14: p. 1-15.
- Cheng, F., et al., A genome-wide positioning systems network algorithm for in silico drug repurposing. Nature communications, 2019. 10(1): p. 3476.
- Rudrapal, M., S.J. Khairnar, and A.G. Jadhav, Drug repurposing (DR): an emerging approach in drug discovery. Drug repurposing-hypothesis, molecular aspects and therapeutic applications, 2020. 10.
- Rajkumar, S.V. and S. Kumar, Multiple myeloma current treatment algorithms. Blood cancer journal, 2020. 10(9): p. 1-10.
- Dehghanifard, A., et al., Various signaling pathways in multiple myeloma cells and effects of treatment on these pathways. Clinical Lymphoma Myeloma and Leukemia, 2018. 18(5): p. 311-320.
- Al-Odat, O.S., et al., Mcl-1 inhibition: managing malignancy in multiple myeloma. Frontiers in Pharmacology, 2021. 12: p. 699629.
- Hurle, M.R., et al., Computational drug repositioning: from data to therapeutics. Clinical Pharmacology & Therapeutics, 2013. 93(4): p. 335-341.
- Keiser, M.J., et al., Predicting new molecular targets for known drugs. Nature, 2009. 462(7270): p. 175-181.
- Hieronymus, H., et al., Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer cell, 2006. 10(4): p. 321-330.
- Data, O., Gross domestic spending on R&D. Retrieved Mai, 2017. 14.
- Cheng, F., et al., Network-based approach to prediction and population-based validation of in silico drug repurposing. Nature communications, 2018. 9(1): p. 2691.
- Nam, Y., et al., Drug repurposing with network reinforcement. BMC bioinformatics, 2019. 20: p. 1-10.
- Zickenrott, S., et al., Prediction of disease–gene–drug relationships following a differential network analysis. Cell death & disease, 2016. 7(1): p. e2040-e2040.
- Hu, G. and P. Agarwal, Human disease-drug network based on genomic expression profiles. PloS one, 2009. 4(8): p. e6536.
- Peyvandipour, A., et al., A novel computational approach for drug repurposing using systems biology. Bioinformatics, 2018. 34(16): p. 2817-2825.
- Naylor, D.M., D. Kauppi, and J. Schonfeld, Therapeutic drug repurposing, repositioning and rescue. Drug Discovery, 2015. 57.
- Chatr-Aryamontri, A., et al., The BioGRID interaction database: 2013 update. Nucleic acids research, 2012. 41(D1): p. D816-D823.
- Szklarczyk, D., et al., STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic acids research, 2015. 43(D1): p. D447-D452.
- Chen, J.Y., R. Pandey, and T.M. Nguyen, HAPPI-2: a comprehensive and high-quality map of human annotated and predicted protein interactions. BMC genomics, 2017. 18(1): p. 1-16.
- Kanehisa, M., et al., KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic acids research, 2017. 45(D1): p. D353-D361.
- Croft, D., et al., Nucleic Acids Research 39 (Database issue)(2011) D691–D697. URL:< http://dx. doi. org/10.1093/nar/gkq1018.
- Wang, Z., M. Jensen, and J. Zenklusen, A Practical Guide to the Cancer Genome Atlas (TCGA). Statistical Genomics. Methods and Protocols, 2016: p. 111-141.
- Uhlén, M., et al., Tissue-based map of the human proteome. Science, 2015. 347(6220): p. 1260419.
- Uhlen, M., et al., Towards a knowledge-based human protein atlas. Nature biotechnology, 2010. 28(12): p. 1248-1250.
- Uhlén, M., et al., A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & cellular proteomics, 2005. 4(12): p. 1920-1932.
- Barretina, J., et al., The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603-607.
- Lamb, J., et al., The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. science, 2006. 313(5795): p. 1929-1935.
- Lamb, J., The Connectivity Map: a new tool for biomedical research. Nature reviews cancer, 2007. 7(1): p. 54-60.
- Borate, B. and A.D. Baxevanis, Searching Online Mendelian Inheritance in Man (OMIM) for information on genetic loci involved in human disease. Current protocols in bioinformatics, 2009. 27(1): p. 1.2. 1-1.2. 13.
- Goh, K.-I., et al., The human disease network. Proceedings of the National Academy of Sciences, 2007. 104(21): p. 8685-8690.
- Yang, J., et al., DNetDB: The human disease network database based on dysfunctional regulation mechanism. BMC systems biology, 2016. 10(1): p. 1-8.
- Law, V., et al., DrugBank 4.0: shedding new light on drug metabolism. Nucleic acids research, 2014. 42(D1): p. D1091-D1097.
- Andersson, M.L., et al., Evaluation of usage patterns and user perception of the drug–drug interaction database SFINX. International Journal of Medical Informatics, 2015. 84(5): p. 327-333.
- Kale, V.P., et al. Old drugs, new uses: Drug repurposing in hematological malignancies. in Seminars in cancer biology. 2021. Elsevier.
- Kuhn, M., et al., The SIDER database of drugs and side effects. Nucleic acids research, 2016. 44(D1): p. D1075-D1079.
- Siramshetty, V.B., et al., WITHDRAWN—a resource for withdrawn and discontinued drugs. Nucleic acids research, 2016. 44(D1): p. D1080-D1086.
- Kuhn, M., et al., STITCH 3: zooming in on protein–chemical interactions. Nucleic acids research, 2012. 40(D1): p. D876-D880.
- Garnett, M.J., et al., Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature, 2012. 483(7391): p. 570-575.
- Lee, B.K.B., et al., DeSigN: connecting gene expression with therapeutics for drug repurposing and development. BMC genomics, 2017. 18(1): p. 1-11.
- Kumar, S., T. Witzig, and S.V. Rajkumar, Thalidomide as an anti-cancer agent. Journal of cellular and molecular medicine, 2002. 6(2): p. 160-174.
- Miller, M.T., Thalidomide embryopathy: a model for the study of congenital incomitant horizontal strabismus. Transactions of the American Ophthalmological Society, 1991. 89: p. 623.
- Grover, J., et al., Thalidomide: a re-look. The National Medical Journal of India, 2000. 13(3): p. 132-141.
- Perri III, A.J. and S. Hsu, A review of thalidomide's history and current dermatological applications. Dermatology online journal, 2003. 9(3).
- Gordon, J. and P. Goggin, Thalidomide and its derivatives: emerging from the wilderness. Postgraduate medical journal, 2003. 79(929): p. 127-132.
- Holmes, A., et al., Thalidomide Therapy for the Treatment of Hypertrophic Herpes Simplex Virus—Related Genitalis in HIV-Infected Individuals. Clinical Infectious Diseases, 2007. 44(11): p. e96-e99.
- Holland, S., Cytokine therapy of mycobacterial infections. Advances in internal medicine, 2000. 45: p. 431-452.
- Gupta, S.C., et al., Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends in pharmacological sciences, 2013. 34(9): p. 508-517.
- Locksley, R.M., N. Killeen, and M.J. Lenardo, The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell, 2001. 104(4): p. 487-501.
- Hideshima, T., et al., NF-κB as a therapeutic target in multiple myeloma. Journal of Biological Chemistry, 2002. 277(19): p. 16639-16647.
- Singhal, S. and J. Mehta, Thalidomide in cancer. Biomedicine & Pharmacotherapy, 2002. 56(1): p. 4-12.
- Kumar, V. and S. Chhibber, Thalidomide: an old drug with new action. Journal of Chemotherapy, 2011. 23(6): p. 326-334.
- Solèr, R.A., et al., Regression of AIDS-related Kaposi's sarcoma during therapy with thalidomide. Clinical infectious diseases, 1996. 23(3): p. 501-503.
- Tunio, M.A., et al., Low-dose thalidomide in patients with metastatic renal cell carcinoma. Brain, 2012. 3: p. 3.75.
- Franks, M.E., G.R. Macpherson, and W.D. Figg, Thalidomide. The Lancet, 2004. 363(9423): p. 1802-1811.
- Figg, W.D., et al., A double-blind randomized crossover study of oral thalidomide versus placebo for androgen dependent prostate cancer treated with intermittent androgen ablation. The Journal of urology, 2009. 181(3): p. 1104-1113.
- Ghobrial, I.M. and S.V. Rajkumar, Management of thalidomide toxicity. The journal of supportive oncology, 2003. 1(3): p. 194.
- Fadul, C.E., et al., A phase II study of thalidomide and irinotecan for treatment of glioblastoma multiforme. Journal of Neuro-oncology, 2008. 90: p. 229-235.
- Penas-Prado, M., et al., Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro-oncology, 2015. 17(2): p. 266-273.
- Chapman-Shimshoni, D., et al., Simvastatin induces apoptosis of B-CLL cells by activation of mitochondrial caspase 9. Experimental hematology, 2003. 31(9): p. 779-783.
- Pradelli, D., et al., Statins use and the risk of all and subtype hematological malignancies: a meta-analysis of observational studies. Cancer Medicine, 2015. 4(5): p. 770-780.
- Yi, X., et al., Statin use is associated with reduced risk of haematological malignancies: evidence from a meta-analysis. PloS one, 2014. 9(1): p. e87019.
- Afzal, A., et al., Statins reduce mortality in multiple myeloma: a population-based US study. Clinical Lymphoma Myeloma and Leukemia, 2020. 20(12): p. e937-e943.
- Brånvall, E., et al., Statin use is associated with improved survival in multiple myeloma: A Swedish population-based study of 4315 patients. American Journal of Hematology, 2020. 95(6): p. 652-661.
- Sanfilippo, K.M., et al., Statins are associated with reduced mortality in multiple myeloma. Journal of Clinical Oncology, 2016. 34(33): p. 4008.
- Epstein, M.M., et al., Statin use and risk of multiple myeloma: An analysis from the cancer research network. International journal of cancer, 2017. 141(3): p. 480-487.
- Zhang, P. and B. Liu, Statin use and the risk of multiple myeloma: A PRISMA-compliant meta-analysis. Annals of Hematology, 2020. 99: p. 1805-1812.
- Longo, J., et al., The mevalonate pathway is an actionable vulnerability of t (4; 14)-positive multiple myeloma. Leukemia, 2021. 35(3): p. 796-808.
- Juarez, D., et al., Statin-induced mitochondrial priming sensitizes multiple myeloma cells to BCL2 and MCL1 inhibitors. Cancer Research Communications, 2023.
- Cetin, M., et al., Overexpression of cyclooxygenase-2 in multiple myeloma: association with reduced survival. American journal of hematology, 2005. 80(3): p. 169-173.
- Ladetto, M., et al., Cyclooxygenase-2 (COX-2) is frequently expressed in multiple myeloma and is an independent predictor of poor outcome. Blood, 2005. 105(12): p. 4784-4791.
- Bernard, M., et al., Targeting cyclooxygenase-2 in hematological malignancies: rationale and promise. Current pharmaceutical design, 2008. 14(21): p. 2051-2060.
- Trojan, A., et al., Clinical significance of cyclooxygenase-2 (COX-2) in multiple myeloma. Swiss medical weekly, 2006. 136(25-26): p. 400-403.
- Tołoczko-Iwaniuk, N., et al., Celecoxib in cancer therapy and prevention–review. Current Drug Targets, 2019. 20(3): p. 302-315.
- Jendrossek, V., Targeting apoptosis pathways by Celecoxib in cancer. Cancer letters, 2013. 332(2): p. 313-324.
- Steinbach, G., et al., The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. New England Journal of Medicine, 2000. 342(26): p. 1946-1952.
- Scilimati, A., Patient Bone Marrow Aspiration to Explore the Cyclooxygenases (COXs) Involvement in Multiple Myeloma. J. Cancer Res. Therap. Oncol, 2021. 9: p. 1-19.
- Roy, P., U.A. Sarkar, and S. Basak, The NF-κB activating pathways in multiple myeloma. Biomedicines, 2018. 6(2): p. 59.
- Fan, L., et al., High expression of phosphorylated extracellular signal-regulated kinase (ERK1/2) is associated with poor prognosis in newly diagnosed patients with multiple myeloma. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research, 2017. 23: p. 2636.
- Liu, H., et al., Aspirin exerts anti-tumor effect through inhibiting Blimp1 and activating ATF4/CHOP pathway in multiple myeloma. Biomedicine & Pharmacotherapy, 2020. 125: p. 110005.
- Marinac, C.R., et al., Aspirin Use and Survival in Multiple Myeloma Patients. Blood, 2018. 132: p. 3250.
- Birmann, B.M., et al., Regular aspirin use and risk of multiple myeloma: a prospective analysis in the health professionals follow-up study and nurses' health study. Cancer prevention research, 2014. 7(1): p. 33-41.
- Sakamoto, M., et al., Effect of clarithromycin treatment of natural killer cell activity in patients with advanced non-small cell lung cancer. Gan to kagaku ryoho. Cancer & chemotherapy, 1998. 25(14): p. 2259-2266.
- Van Nuffel, A.M., et al., Repurposing Drugs in Oncology (ReDO)—clarithromycin as an anti-cancer agent. ecancermedicalscience, 2015. 9.
- Musto, P., et al., Inefficacy of clarithromycin in advanced multiple myeloma: a definitive report. Haematologica, 2002. 87(6): p. 658-659.
- Durie, B., Clarithromycin (Biaxin) as primary treatment for myeloma. Blood, 1997. 90: p. 579.
- Stewart, A., et al., Lack of response to short-term use of clarithromycin (BIAXIN) in multiple myeloma. Blood, The Journal of the American Society of Hematology, 1999. 93(12): p. 4441-4442.
- Moreau, P., et al., Lack of efficacy of clarithromycin in advanced multiple myeloma. Leukemia, 1999. 13(3): p. 490-491.
- Morris, T., et al., Phase II trial of clarithromycin and pamidronate therapy in myeloma. Medical Oncology, 2001. 18: p. 79-84.
- Coleman, M., et al., BLT-D (clarithromycin [Biaxin], low-dose thalidomide, and dexamethasone) for the treatment of myeloma and Waldenström's macroglobulinemia. Leukemia & lymphoma, 2002. 43(9): p. 1777-1782.
- Mark, T.M., et al., Thalidomide, clarithromycin, lenalidomide and dexamethasone therapy in newly diagnosed, symptomatic multiple myeloma. Leukemia & Lymphoma, 2014. 55(12): p. 2842-2849.
- Morris, T., et al., Clarithromycin with low dose dexamethasone and thalidomide is effective therapy in relapsed/refractory myeloma. British journal of haematology, 2008. 143(3): p. 349-354.
- Puig, N., et al., Lenalidomide and dexamethasone with or without clarithromycin in patients with multiple myeloma ineligible for autologous transplant: a randomized trial. Blood Cancer Journal, 2021. 11(5): p. 101.
- Klein, B., et al., Interleukin-6 in human multiple myeloma. 1995.
- Khan, A., et al., Effect of clarithromycin and azithromycin on production of cytokines by human monocytes. International journal of antimicrobial agents, 1999. 11(2): p. 121-132.
- Moriya, S., et al., Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress-mediated cell death in multiple myeloma cells. International journal of oncology, 2015. 46(2): p. 474-486.
- Takemori, N., et al., Possible mechanisms of action of clarithromycin and its clinical application as a repurposing drug for treating multiple myeloma. ecancermedicalscience, 2020. 14.
- Sehgal, S., H. Baker, and C. Vézina, Rapamycin (AY-22, 989), a new antifungal antibiotic II. Fermentation, isolation and characterization. The Journal of antibiotics, 1975. 28(10): p. 727-732.
- Saunders, R.N., M.S. Metcalfe, and M.L. Nicholson, Rapamycin in transplantation: a review of the evidence. Kidney international, 2001. 59(1): p. 3-16.
- 1. Laplante, M. and D.M. Sabatini, mTOR signaling at a glance. Journal of cell science, 2009. 122(20): p. 3589-3594.
- Strömberg, T., et al., Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood, 2004. 103(8): p. 3138-3147.
- Gera, J. and A. Lichtenstein, The mammalian target of rapamycin pathway as a therapeutic target in multiple myeloma. Leukemia & lymphoma, 2011. 52(10): p. 1857-1866.
- Raje, N., et al., Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood, 2004. 104(13): p. 4188-4193.
- Kikuchi, J., et al., Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood, The Journal of the American Society of Hematology, 2010. 116(3): p. 406-417.
- Ichiyama, T., et al., Sodium valproate inhibits production of TNF-α and IL-6 and activation of NF-κB. Brain research, 2000. 857(1-2): p. 246-251.
- Wang, Y., et al., Valproic acid increased autophagic flux in human multiple myeloma cells in vitro. Biomedicine & Pharmacotherapy, 2020. 127: p. 110167.
- Zhang, Y., et al., Valproic acid activates autophagy in multiple myeloma cell lines RPMI8226 and U266. Zhonghua xue ye xue za zhi= Zhonghua Xueyexue Zazhi, 2016. 37(6): p. 478-483.
- Kitazoe, K.-i., et al., Valproic acid exerts anti-tumor as well as anti-angiogenic effects on myeloma. International journal of hematology, 2009. 89: p. 45-57.
- Yang, Y., et al., HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer science, 2005. 96(7): p. 425-433.
- 1. Bono, C., et al., The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica, 2012. 97(7): p. 1101.
- 2. Kawabata, S., et al., Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell death & disease, 2012. 3(7): p. e353-e353.
- Kraus, M., et al., Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood cancer journal, 2013. 3(3): p. e103-e103.
- Kalender, A., et al., Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell metabolism, 2010. 11(5): p. 390-401.
- Del Barco, S., et al., Metformin: multi-faceted protection against cancer. Oncotarget, 2011. 2(12): p. 896.
- Greaves, D. and Y. Calle, Epithelial Mesenchymal Transition (EMT) and Associated Invasive Adhesions in Solid and Haematological Tumours. Cells, 2022. 11(4): p. 649.
- Noto, H., et al., Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PloS one, 2012. 7(3): p. e33411.
- Bodmer, M., et al., Long-term metformin use is associated with decreased risk of breast cancer. Diabetes care, 2010. 33(6): p. 1304-1308.
- Machado-Neto, J.A., et al., Metformin exerts multitarget antileukemia activity in JAK2V617F-positive myeloproliferative neoplasms. Cell death & disease, 2018. 9(3): p. 311.
- Mishra, A.K. and D. Dingli, Metformin inhibits IL-6 signaling by decreasing IL-6R expression on multiple myeloma cells. Leukemia, 2019. 33(11): p. 2695-2709.
- 1. Chang, S.-H., et al., Association between metformin use and progression of monoclonal gammopathy of undetermined significance to multiple myeloma in US veterans with diabetes mellitus: a population-based retrospective cohort study. The Lancet Haematology, 2015. 2(1): p. e30-e36.
- 2. Boursi, B., et al., Impact of metformin on the progression of MGUS to multiple myeloma. Leukemia & lymphoma, 2017. 58(5): p. 1265-1267.
- 3. LeGrand, J., et al., Global gene expression profiling in mouse plasma cell tumor precursor and bystander cells reveals potential intervention targets for plasma cell neoplasia. Blood, The Journal of the American Society of Hematology, 2012. 119(4): p. 1018-1028.
- Dalva-Aydemir, S., et al., Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clinical Cancer Research, 2015. 21(5): p. 1161-1171.
- Mishra, R.K., et al., In silico modeling-based identification of glucose transporter 4 (GLUT4)-selective inhibitors for cancer therapy. Journal of Biological Chemistry, 2015. 290(23): p. 14441-14453.
- Zhao, Y., et al., Metformin and FTY720 synergistically induce apoptosis in multiple myeloma cells. Cellular Physiology and Biochemistry, 2018. 48(2): p. 785-800.
- Jagannathan, S., et al., Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia, 2015. 29(11): p. 2184-2191.
- Berenson, J.R., Antitumor effects of bisphosphonates: from the laboratory to the clinic. Current opinion in supportive and palliative care, 2011. 5(3): p. 233-240.
- Weinstein, R.S., P.K. Roberson, and S.C. Manolagas, Giant osteoclast formation and long-term oral bisphosphonate therapy. New England Journal of Medicine, 2009. 360(1): p. 53-62.
- Mansour, A., A. Wakkach, and C. Blin-Wakkach, Emerging roles of osteoclasts in the modulation of bone microenvironment and immune suppression in multiple myeloma. Frontiers in immunology, 2017. 8: p. 954.
- 1. Tai, Y.-T., S.-F. Cho, and K.C. Anderson, Osteoclast immunosuppressive effects in multiple myeloma: role of programmed cell death ligand 1. Frontiers in immunology, 2018. 9: p. 1822.
- 2. Huang, X., et al., Diethyldithiocarbamate-copper complex (CuET) inhibits colorectal cancer progression via miR-16-5p and 15b-5p/ALDH1A3/PKM2 axis-mediated aerobic glycolysis pathway. Oncogenesis, 2021. 10(1): p. 4.
- 3. Suh, J.J., et al., The status of disulfiram: a half of a century later. Journal of clinical psychopharmacology, 2006. 26(3): p. 290-302.
- 4. Kannappan, V., et al., Recent advances in repurposing disulfiram and disulfiram derivatives as copper-dependent anticancer agents. Frontiers in Molecular Biosciences, 2021. 8: p. 741316.
- Guo, W., et al., Identification and characterization of multiple myeloma stem cell-like cells. Cancers, 2021. 13(14): p. 3523.
- Jin, N., et al., Disulfiram/copper targets stem cell-like ALDH+ population of multiple myeloma by inhibition of ALDH1A1 and Hedgehog pathway. Journal of cellular biochemistry, 2018. 119(8): p. 6882-6893.
- Chroma, K., et al., A drug repurposing strategy for overcoming human multiple myeloma resistance to standard-of-care treatment. Cell Death & Disease, 2022. 13(3): p. 203.
- Chai, J.-Y., B.-K. Jung, and S.-J. Hong, Albendazole and mebendazole as anti-parasitic and anti-cancer agents: an update. The Korean Journal of Parasitology, 2021. 59(3): p. 189.
- Wang, L.-J., et al., Non-mitotic effect of albendazole triggers apoptosis of human leukemia cells via SIRT3/ROS/p38 MAPK/TTP axis-mediated TNF-α upregulation. Biochemical Pharmacology, 2019. 162: p. 154-168.
- Yi, H., et al., Albendazole inhibits NF-κB signaling pathway to overcome tumor stemness and bortezomib resistance in multiple myeloma. Cancer Letters, 2021. 520: p. 307-320.
- Singhal, S., et al., Antitumor activity of thalidomide in refractory multiple myeloma. New England Journal of Medicine, 1999. 341(21): p. 1565-1571.
- Rajkumar, S.V., et al., Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. Journal of clinical oncology, 2006. 24(3): p. 431-436.
- Kardosh, A., et al., Multitarget inhibition of drug-resistant multiple myeloma cell lines by dimethyl-celecoxib (DMC), a non–COX-2 inhibitory analog of celecoxib. Blood, 2005. 106(13): p. 4330-4338.
- Holien, T., et al., Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. European Journal of Haematology, 2013. 91(4): p. 339-346.
- Papanikolaou, X., et al., Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget, 2014. 5(12): p. 4118.
- Li, S., et al., Effect of artesunate on inhibiting proliferation and inducing apoptosis of SP2/0 myeloma cells through affecting NFκB p65. International journal of hematology, 2009. 90: p. 513-521.
- Hu, G.-F., et al., Effects of Artesunate Combined with Arsenious Acid on Proliferation and Apoptosis of Multiple Myeloma Cells via PI3K/AKT Signaling Pathway. Zhongguo shi yan xue ye xue za zhi, 2021. 29(6): p. 1819-1824.
- Baumann, P., et al., Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Molecular cancer therapeutics, 2009. 8(2): p. 366-375.
- Rosenzweig, M., et al., Repurposing leflunomide for relapsed/refractory multiple myeloma: a phase 1 study. Leukemia & lymphoma, 2020. 61(7): p. 1669-1677.
- Yee, A.J., et al., Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. British journal of haematology, 2014. 166(3): p. 401-409.
- Koltai, T., Nelfinavir and other protease inhibitors in cancer: mechanisms involved in anticancer activity. F1000Research, 2015. 4.
- Alodhaibi, I., et al., An Open-Label Phase I Study of Metformin and Nelfinavir in Combination With Bortezomib in Patients With Relapsed and Refractory Multiple Myeloma. Clinical Lymphoma Myeloma and Leukemia, 2024.
Figure 1.
Drug discovery and development stages and process.
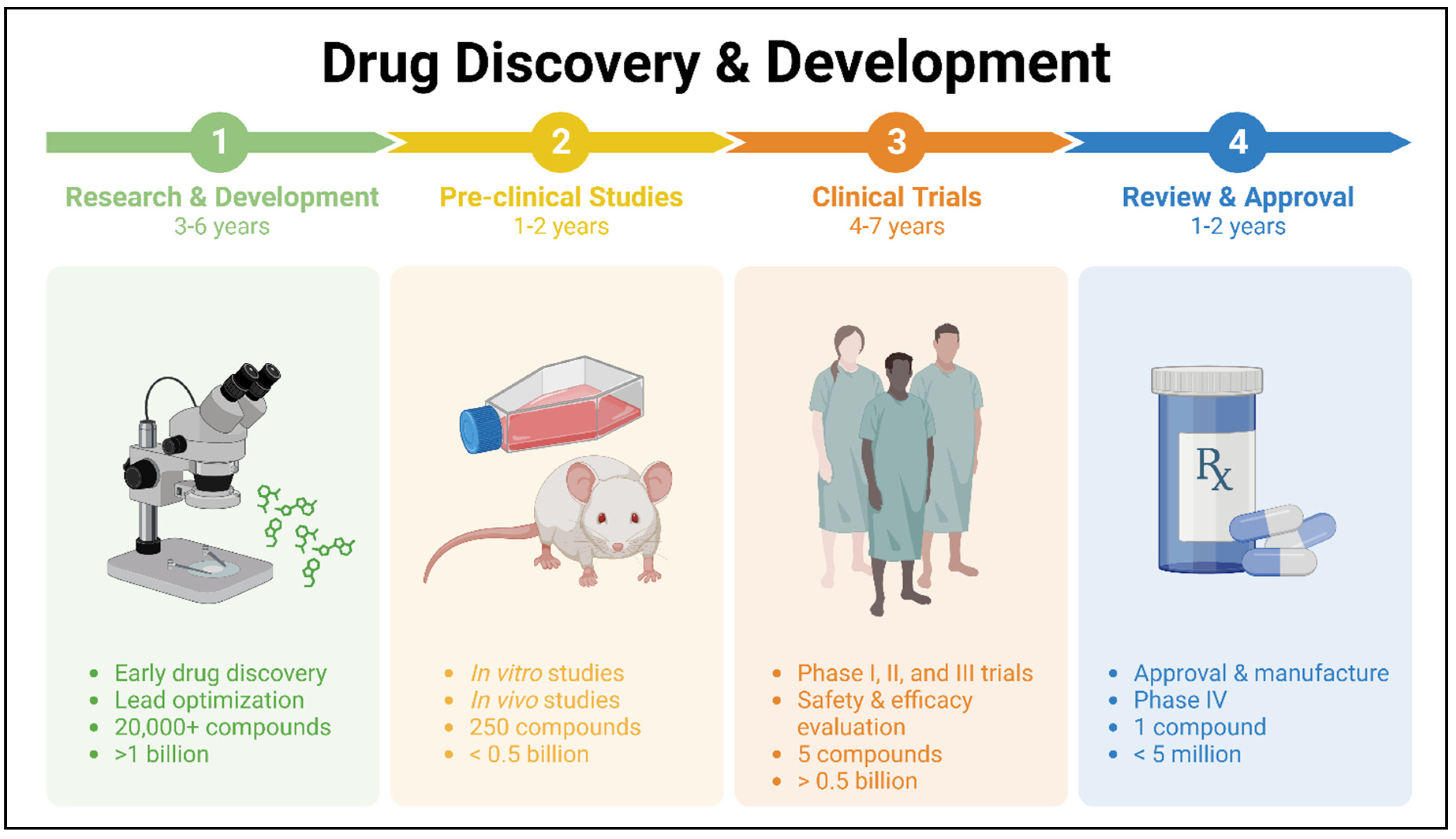
Figure 2.
Drug repurposing employs diverse computational approaches, either independently or in concert, to methodically scrutinize various large-scale datasets for insightful interpretations regarding repurposing hypotheses. Additionally, experimental approaches can uncover repurposing prospects. EHR stands for electronic health record.
Figure 2.
Drug repurposing employs diverse computational approaches, either independently or in concert, to methodically scrutinize various large-scale datasets for insightful interpretations regarding repurposing hypotheses. Additionally, experimental approaches can uncover repurposing prospects. EHR stands for electronic health record.
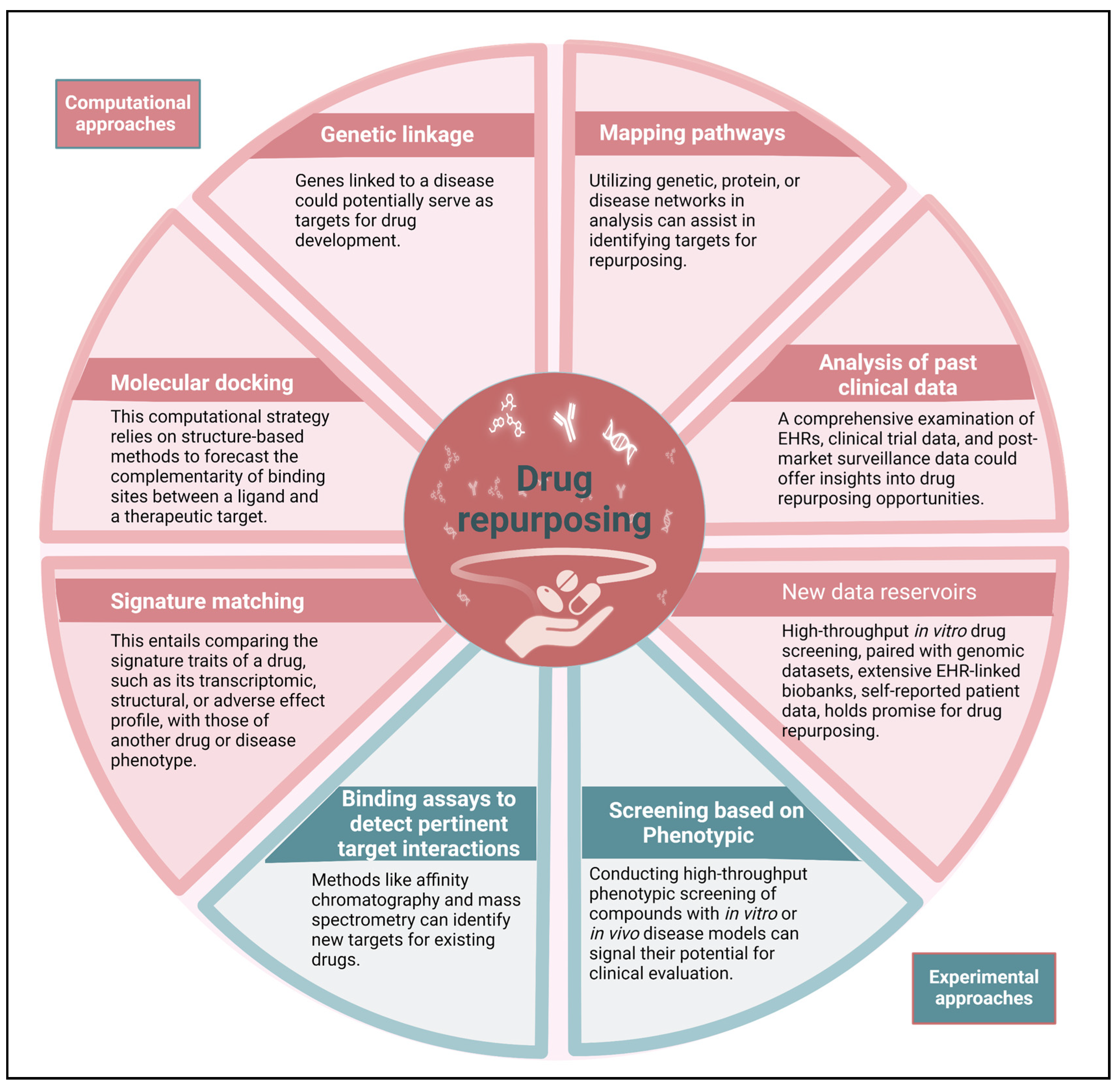
Figure 3.
Drug candidates for repurposing to treat multiple myeloma (MM).
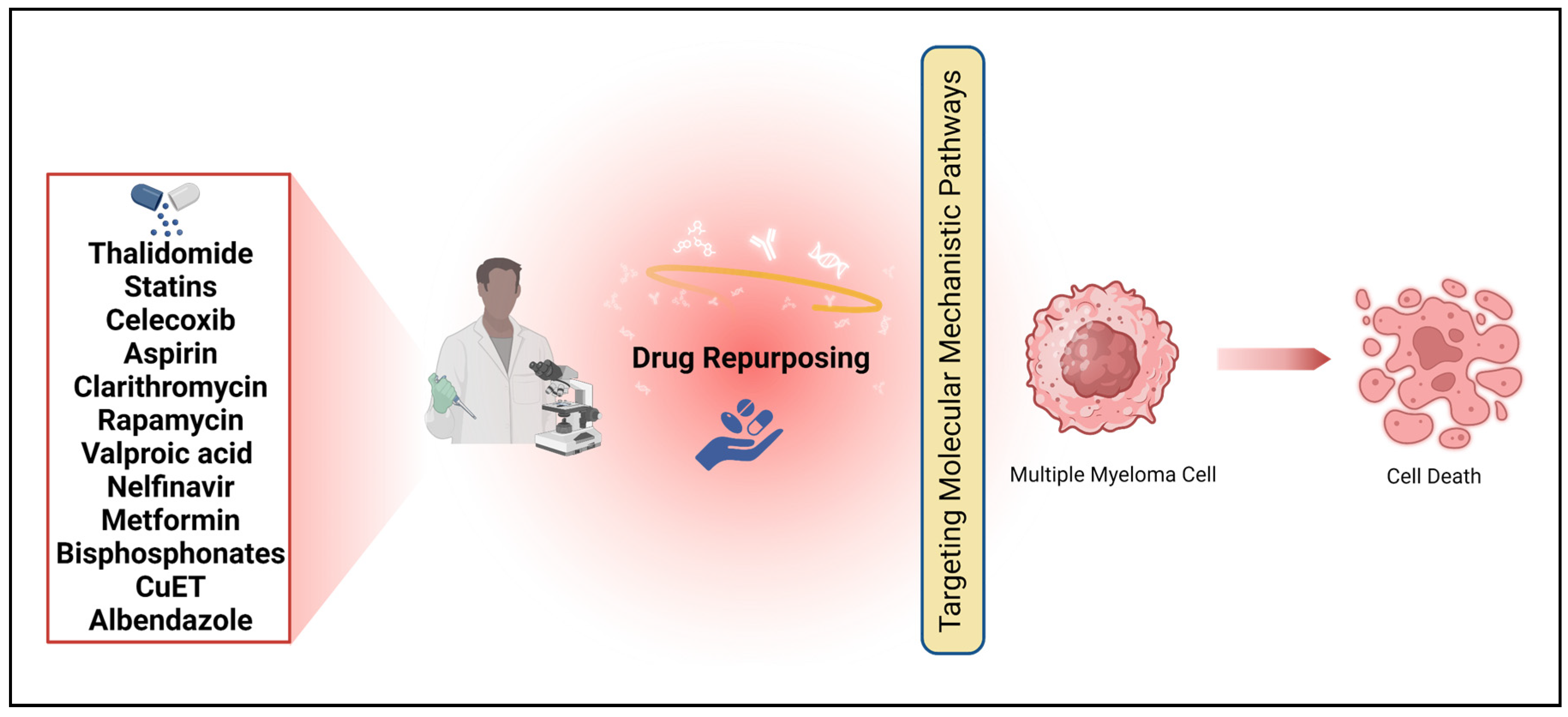

Table 1.
Resources for identifying drug interactions that may possibly be employed in drug repurposing.
Table 1.
Resources for identifying drug interactions that may possibly be employed in drug repurposing.
| Purpose | Resource | Ref. |
|---|---|---|
| Human pathways and protein-protein interaction (PPI) | BiGRID, STRING, HAPPI, KEGG, Reactome | [29,30,31,32,33] |
| Molecular classification of more than 20,000 main cancer matched normal tissue from 33 types of cancer | Cancer Genome Atlas | [34] |
| Protein expression in cancer, matched normal tissues, and cancer human cell lines | The Human Protein Atlas | [35,36,37] |
| Drug sensitivity, gene expression, and genotype for human cancer cell lines | Cancer Cell Line Encyclopedia | [38] |
| Data of genome-wide transcription expression from cultured human cancer cells with many small compounds | Connectivity Map 02 (CMap) | [39,40] |
| Disease-specific gene curation and analysis | OMIM, GEO | [41,42] |
| Disease-disease connectivity; connectivity of two genes elaborated within the same disease | The human disease network | [42] |
| Disease similarities as seen through the lens of gene regulatory mechanisms; comprehension of disease etiology and pathophysiology | Human Disease Network Database (DNetDB) | [43] |
| Drug-drug interaction; comprehensive drug-target information on tens of thousands of drugs and targets | DrugBank | [44] |
| Drug-drug interaction | SFINX | [45] |
| Database of more than 270 non-cancer drugs for potential repurposing for anti-cancer therapy | Repurposing Drugs in Oncology (ReDO) | [46] |
| Database of drugs and adverse drug reactions (ADRs) | Side Effect Resource (SIDER) | [47] |
| Withdrawn or discontinued drugs | WITHDRAWN | [48,49] |
| An inventory of main and secondary uses for repurposed pharmaceuticals | RepurposeDB | [2] |
| Chemical (including drugs)-protein interaction network | STITCH | [49] |
| Data on the sensitivity of hundreds of compounds and over a thousand cancer cell lines | Genomics of Drug Sensitivity in Cancer (GDSC) | [50] |
| Gene expression pattern-based prediction of drug effectiveness against cancer | DeSigN | [51] |
Table 2.
Drugs being repurposed for multiple myeloma (MM). .
| Drug Name | Old-Indication | New-Indication | Mechanism of Action | Clinical Trials Status | Ref. |
|---|---|---|---|---|---|
| Thalidomide | Sedative, anti-nausea | MM | Inhibits IKK (also NF-κB); inhibits TNF; inhibits IL-1, IL-6, IL-12, VEGF | Approved in combination with dexamethasone | [151,152] |
| Statins | High Cholesterol | MM | HMG-CoA reductase inhibitors, upregulation of PUMA and NOXA | Smouldering MM, phase II | [74,75,76,77] |
| Celecoxib | Anti- inflammatory | MM and drug resistant MM | Inhibits COX-2, inhibits Mcl-1, Bcl-2, survivin, Akt | Not for MM, approved for FAP | [83,85,153] |
| Aspirin | Anti- inflammatory | MM | Inhibits COX-1 and COX-2, suppresses cytokines and NF-κB, inhibits EKR and Blimp1, activates ATF4/CHOP | Preclinical | [92,93] |
| Artesunate | Malaria | MM and drug resistant MM | Decreased expression of MYC and Bcl-2, triggers cleavage of caspase-3 | Preclinical | [154,155,156,157] |
| Leflunomide | Rheumatism | MM | DHODH inhibitor, cyclin D2 and pRb inhibition | Phase II NCT04508790 |
[158,159] |
| Clarithromycin | Antibiotic | MM and drug resistant MM | Inhibits IL-6 and MGFs | Phase II | [95,102] |
| Rapamycin | Fungal infections | MM | Antagonist of mTOR | Phase I | [112,113,114,160] |
| Valproic acid | Seizures, migraine, and epilepsy | MM | Block HDAC, inhibits NF-κB and cytokines | Preclinical | [115] |
| Nelfinavir | HIV Infection | MM and drug resistant MM | Inhibits 26S proteasome- disrupts Akt and STAT3, ERK1/2 | MM phase I | [121,123,161,162] |
| Metformin | Diabetes mellitus type 2 | MM | Activates AMPK (suppresses mTORC1, activates p53), inhibits EMT, regulates cell cycle proteins (ERK1/2, JAK2/STAT), IL-6 suppression | Smouldering Myeloma and Monoclonal gammopathy of undetermined significance phase II, MM phase I | [134,135,137,162] |
| Bisphosphonates | Osteoporosis | MM | HMG-CoA pathway suppression, osteoclast apoptosis | Preclinical | [140,141] |
| CuET | Alcohol-abuse drug disulfiram (DSF) |
Drug resistant MM | ALDH inhibition | Preclinical | [147] |
| Albendazole | Parasitic infections | Drug resistant MM | Microtubule system interference, p65/NF-κB pathway inhibtion | Preclinical | [150] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



