
Submitted:
30 May 2024
Posted:
03 June 2024
You are already at the latest version
Abstract
We present a case of adult-onset systemic chronic active EBV disease (CAEBV) in a 40-year-old woman with chronic HBV hepatitis. Initial symptoms resembled a viral illness, progressing to recurrent fever, transaminitis, and anasarca. Investigations revealed high-level EBV viremia and an abnormal T-cell population in liver and bone marrow, indicative of CAEBV. Liver biopsy showed CD3+ T-cells lacking TCRbeta and displaying dim/negative CD5, with elevated EBV-infected T-cells. Next-generation sequencing identified rare variants in CREBBP, SPEN, TP73, and PLCG2, suggesting potential contributions to disease pathogenesis. This case underscores diagnostic challenges and management complexities of adult-onset CAEBV, particularly with underlying chronic HBV infection. Genomic profiling offers crucial insights into the molecular landscape of rare lymphoid malignancies, highlighting the importance of personalized treatment strategies. The distinct immunophenotypic features underscore the heterogeneity in EBV-associated T-cell LPDs, urging further research for optimized clinical management
Keywords:
chronic EBV infection
; T cell lymphoproliferative disorder
; mutational profile
1. Introduction
The Epstein-Barr virus (EBV) is a prevalent member of the human herpesvirus family. It infects over 90% of adults globally. While its primary target is B-cells, EBV can sporadically infect T- and natural killer (NK)- cells. Following an often asymptomatic initial infection, EBV establishes latency within B-cells. Reactivation of the virus, influenced by host immune response and viral properties, can lead to lymphoproliferation and a diverse range of clinical manifestations. (1, 2)
EBV-positive T-cell and NK-cell lymphoproliferative disorders represent a spectrum of rare conditions, predominantly observed in individuals of East Asian and Native American descent from Central and South America. Clinical manifestations encompass a broad spectrum, ranging from non-neoplastic entities like EBV-associated hemophagocytic lymphohistiocytosis to more persistent conditions such as chronic active EBV disease (CAEBV) affecting T- and NK-cell lineages. Overtly malignant presentations include systemic EBV-positive T-cell lymphoma in children and adults, aggressive NK-cell leukemia, extranodal NK/T-cell lymphoma of nasal type, and primary EBV-positive nodal T/NK-cell lymphoma. (3, 4)
Following an acute episode of infectious mononucleosis, the majority of patients spontaneously recover upon the establishment of EBV-specific immunity. However, a subset of rare immunocompetent hosts may develop a chronic trajectory characterized by persistent EBV infection within T and/or NK cells, alongside elevated EBV levels in blood and tissues. This condition, primarily observed in children, is delineated as systemic chronic active EBV disease (CAEBV) in the 5th edition of the World Health Organization (WHO) classification and International Consensus Classification. A hallmark feature of CAEBV is the polyclonal, oligoclonal, or frequently monoclonal proliferation of EBV-infected T or NK cells, which infiltrate tissues, eliciting end-organ damage. (5-9)
In this report, we describe a case of adult-onset systemic chronic active EBV disease (CAEBV) with a distinctive immunophenotype in a 40-year-old woman of Chinese descent with chronic HBV hepatitis.
2. Case Presentation
The patient initially presented with a 2-week history of fever, cough, myalgia, and periorbital edema. Laboratory findings revealed mild pancytopenia (hemoglobin: 118 g/L, white blood cell count: 3.5 x 10^9/L, platelet count: 120 x 10^9/L) and mildly elevated liver enzymes, with normal bilirubin levels (aspartate transaminase [AST] 49 U/L, alanine transaminase [ALT] 88 U/L, alkaline phosphatase [ALP] 313 U/L, bilirubin 11 µmol/L). Symptomatic management was provided for what was considered a viral illness.
Five months later, she presented with a two-week history of fever accompanied by chills and tachycardia. Her liver enzyme levels were found to be elevated (AST 701 U/L, ALT 662 U/L, ALP 1130 U/L, and bilirubin 36 µmol/L). Further investigation unveiled her positivity for Hepatitis B surface antigen (HBsAg) with HBcAb positivity (IgM negative), and a viral DNA count of 1.48 x 10^1 copies, prompting initiation of antiviral therapy (Tenofovir, later switched to Entecavir). EBV titers were also notably elevated. Laboratory analyses including ceruloplasmin, alpha-1 antitrypsin, and quantitative immunoglobulin yielded unremarkable results. ANA testing demonstrated a borderline positive result (1:160) with a speckled pattern, while anti-mitochondrial antibody (AMA) and anti-smooth muscle antibody (ASMA) tests were negative. Pelvic MRI revealed a small amount of fluid and an ovarian lesion, with negative findings on cytology following paracentesis.
Nine months following the initial presentation, she was admitted yet again with fever, night sweats, tachycardia, and bilateral pedal edema, which subsequently advanced to anasarca. Comprehensive evaluation revealed no indications of congestive heart failure, nephrotic syndrome, or cirrhosis. Laboratory investigations upon admission revealed anemia (hemoglobin: 92 g/L, WBC: 5.0 x 10^9/L, platelet count: 174 x 10^9/L), along with elevated liver enzymes (AST 143 U/L, ALT 273 U/L, ALP 672 U/L, bilirubin 74 µmol/L), low albumin (25 g/L), and normal INR (0.9) and creatinine (38 µmol/L). High-level EBV viremia was detected (EBV PCR on peripheral blood 4.41 x 10^7 IU/ml) with positive viral capsid antigen (VCA) IgG and Epstein-Barr nuclear antigen (EBNA) IgG, and negative VCA IgM, indicative of a late-stage infection. HBV DNA was, however, undetectable at this juncture. Testing for other viral pathogens, including HIV, Delta virus, COVID-19, respiratory swab pathogens, and human T-cell lymphotropic virus (HTLV-1/2), returned negative results. Notably, criteria for hemophagocytic lymphohistiocytosis (HLH) were not met. Imaging via CT abdomen revealed marked hepatomegaly and moderate splenomegaly, without significantly enlarged abdominal or pelvic lymph nodes or signs of cirrhosis. CT brain and neck scans yielded unremarkable findings. Additionally, a positron emission tomography (PET) scan demonstrated no definitive evidence of fluorodeoxyglucose (FDG)-avid nodal or extranodal sites suggestive of a lymphoproliferative disorder. Subsequently, the patient underwent liver biopsy and bone marrow examination.
3. Results
The liver biopsy revealed moderate periportal and intrasinusoidal lymphoid infiltrates, alongside moderate macrovesicular steatosis, no apparent steatohepatitis and minimal fibrosis. Notably, a large subset of lymphoid infiltrates comprised CD3+ T-cells exhibiting a distinct immunophenotype characterized by the absence of T-cell receptor beta (TCRbeta), dim expression of CD8, and dim/negative expression of CD5, while displaying bright expression of CD2 and CD7. Ki-67 staining assessment indicated a low proliferation rate of less than 10%. Cytotoxic markers such as Gran B and TIA-1 as well as CD4, CD10, CD30, and BCL6 were predominantly negative in these cells. In situ hybridization for Epstein-Barr virus-encoded small RNA (EBER) revealed numerous positive T-cells. Additionally, only scattered hepatocytes, and not T cells, exhibited positive staining with hepatitis B surface antigen (HBsAg) (Figure 1). Unequivocal features of lymphoma were not appreciated in the biopsy specimen. The culmination of these findings led to the collective interpretation of an immune-mediated hepatic pathology characterized by significant T-cell involvement and indicative of chronic Epstein-Barr virus (EBV) infection, without definitive evidence of overt lymphoma.
The bone marrow aspirate showed T-cell lymphocytosis (~20%), characterized by small-sized lymphoid cells lacking significant atypia. EBER staining was positive within the T cells. Immunohistochemistry and flow cytometry analyses demonstrated an immunophenotypically abnormal T cells, characterized by positivity for CD2 (bright/homogeneous), CD3, CD7 (bright/homogeneous), CD8 (dim, partial), CD16 (partial, ~40%), CD26, CD28, CD38, CD94 (bright/homogeneous), HLA-DR, and T-cell receptor gamma delta (TCR γδ), and negativity for CD4, CD5, CD10, CD20, CD25, CD56, CD57, and PD-1 (CD279). Furthermore, T-cell clonality analysis by polymerase chain reaction (PCR) demonstrated a single clonal peak in TCRβ and a single clonal peak in TCRγ primer mixes.
The small size of the T-cells, their immunophenotype, and absence of cellular atypia did not favor a diagnosis of extranodal NK/T-cell lymphoma. The clinical presentation of several episodes of fever and general malaise over 9 months, with hepatosplenomegaly and without evidence of nasal involvement was more consistent with a diagnosis of systemic chronic active EBV-associated lymphoproliferative disorder. She was started on Dexamethasone, currently on tapering doses, and has been stable after discharge. She was initiated on Nivolumab and planned for an allogenic stem cell transplantation in the future when suitable donor becomes available.
Next-generation sequencing was conducted on the liver biopsy sample using a comprehensive gene panel focused on lymphoid-related genes, comprising 120 genes. After filtering out variants found in reference population database (gnomAD v2.1.1), four variants were detected in CREBBP, SPEN, TP73 and PLGC2 with no known functional evidence (Table 1). In silico prediction tools predict the CREBBP variant to be disease causing with a REVEL score of 0.559 (PMID: 27666373). The SPEN, TP73 and PLGC2 variants are not predicted to be disease causing with REVEL scores well below 0.5. Both TP73 and PLCG2 variants are found in the reference population database (gnomADv2.1.1) at very low minor allele frequencies (0.000077 and 0.00004274, respectively).3.1. Subsection
4. Discussion
According to the International Agency for Research on Cancer (IARC), EBV is recognized as one of the seven major oncogenic viruses.(10) In addition to B cells, EBV has demonstrated the capacity to infect T-cells and NK-cells, although the precise mechanism remains incompletely understood. The proliferation of EBV within T and NK cells has been implicated in the pathogenesis of EBV-associated T and NK cell lymphoproliferative disorders. The latest World Health Organization (WHO) classification recognizes two primary categories within this spectrum: 1) EBV-positive NK-cell and T-cell lymphomas, encompassing EBV-positive nodal T-and NK-cell lymphoma and extranodal NK/T-cell lymphoma, and 2) EBV-positive T-cell and NK-cell lymphoid proliferations and lymphomas of childhood.(7)
EBV-associated lymphoproliferative diseases in pediatric populations are overall rare conditions, with higher incidence in Asian populations and Native Americans from Central and South America. Systemic CAEBV presents symptoms resembling to infectious mononucleosis for over 3 months, diagnosed via increased EBV DNA or EBER-positive T/NK-cells, excluding other conditions. Kawada et al recommend a threshold of ≥ 10,000 IU/ml EBV DNA for diagnosis. (5) Initially seen as a childhood disease, CAEBV is now recognized in adults, as also highlighted by our 40-year-old patient. Recent data from Japan indicate a median age of 21 years among CAEBV patients, suggesting a shift towards older age groups. (11, 12) Adult-onset CAEBV differs clinically, with lower fever frequency but higher skin lesion incidence, particularly erythema. Notably, adult cases have less mosquito bite hypersensitivity and hydroa vacciniforme. Laboratory-wise, adult-onset CAEBV often involves elevated liver enzymes and hemophagocytic syndrome, as seen in our patient presenting with recurrent fever and transaminitis.
As opposed to the systemic CAEBV disease, systemic EBV-positive T-cell lymphoma of childhood typically manifests with an acute and fulminant clinical course, characterized by symptom duration of less than 3 months. This condition commonly presents with hemophagocytic lymphohistiocytosis (HLH), which was not observed in our case. Histologically, the lymphoid cells in the affected tissue exhibit a broad cytological spectrum, ranging from small to medium-sized and large atypical lymphoid cells.
The most appropriate diagnosis in our case, therefore, after exclusion of other differentials was systemic CAEBV disease. The EBV infected lymphocytes in CAEBV is most commonly of T lineage than NK cell. Among the T-cells, CD4+ T cells are more commonly seen. As opposed to systemic EBV-positive T-cell lymphoma, only minority of cases show a cytotoxic CD8+ phenotype. The gamma delta T-cell type usually comprise only a small proportion of CAEBV cases.(8)
In recent genomic analyses DDX3X mutations were found to be prevalent in EBV-infected lymphoid progenitor cells in CAEBV.(13, 14) Additionally, frequent intragenic deletions, particularly involving the BamHI-A Rightward transcript microRNA clusters, were identified in CAEBV and other EBV-associated neoplasms. Interestingly, these deletions were absent in infectious mononucleosis or post transplant lymphoproliferative disorders, suggesting a unique pathogenic mechanism underlying lymphomagenesis. (13, 14) Other mutations identified in CAEBV disease involved tumor suppressors (TP53 and MGA), JAK-STAT-pathway (STAT3 and STAT5B) and epigenetic modifiers (MLL2, ARID1A, EP300 and ASXL3).(15)
TP73, a homologue of TP53, is rarely found mutated in cancer, previously accounting for no more than 0.6% of primary tumors. However, its deletion (1p36) is much more commonly observed in various cancers, including lymphomas.(16, 17) Interestingly, TP73 mutation has been reported in HBV-associated B cell lymphomas but not in T cell lymphomas, despite chronic HBV infection being linked to an increased risk of NK/T cell lymphomas.(18, 19) While our patient harbored HBV infection with focal hepatocyte involvement, overall control of the infection was evident. However, T cells did not show signs of HBV infection. The significance of the TP73 variant is unclear in this patient, but noted a low variant allele fraction at 10.4% that may suggest a somatic origin.
CREBBP mutations are frequently observed in B cell lymphomas originating from the germinal center, but they are only rarely reported in T cell or NK cell lymphomas.(20-23) EP300, a prologue of CREBBP, is, however, relatively commonly mutated in the PTCL including extra-nodal NK/T cell lymphoma and adult T cell leukemia/lymphoma.(24) CREBBP belongs to the KAT3 family of histone acetyltransferases and functions as a transcriptional co-activator through H3K27 acetylation.(25) With the emergence of potential acetyltransferase inhibitors, targeting CREBBP can be therapeutically considered in lymphomas with epigenetic alterations.(26, 27) The clinical significance of the CREBBP variant is unclear; however, is predicted to be deleterious and could be contributing to the oncogenesis of the tumor.
SPEN and PLCG2 mutations were detected at a high VAF of close to 50% indicating either germline alterations or loss of heterozygosity (LOH). SPEN is located at 1p36 adjacent to TP73 making both alterations possibly disease related. SPEN mutations have been reported in B cell lymphoma but not in T cell lymphoma.(28) SPEN is a nuclear protein with crucial roles in gene silencing and transcriptional regulation.(29) PLCG2 is located at 16q23 and is commonly mutated in chronic lymphocytic leukemia (CLL) and rare diffuse large B cell lymphomas.(30, 31) While PLCG1 mutation was reported in T cell lymphomas, PLCG2 is rarely detected and is likely not contributory to the patient’s phenotype.(32, 33)
5. Conclusions
This case sheds light on the intricate interplay between chronic viral infections, immune dysregulation, and lymphomagenesis. The presentation of adult-onset chronic Epstein-Barr virus (EBV)-associated T-cell lymphoproliferative disorder (LPD) within the backdrop of chronic HBV hepatitis underscores the complexity of viral interactions within the immune system.
Among four detected mutational changes in this case only CREBBP is relatively uncommonly seen in T cell lymphomas.(24) The identification of novel TP73, SPEN and PLCG2 mutations adds a layer of complexity to the understanding of the molecular mechanisms underlying EBV-associated lymphoproliferative disorders. TP73 mutations, although rare in cancer, have been implicated in HBV-associated B-cell lymphomas, suggesting a potential link between HBV infection and T-cell lymphomagenesis. (34)
This case underscores the importance of comprehensive genomic profiling in elucidating the molecular landscape of rare lymphoid malignancies, paving the way for personalized treatment strategies. Moreover, the distinctive immunophenotypic features observed in this case highlight the heterogeneity of EBV-associated T-cell LPDs and underscore the need for further research to delineate their clinical behavior and optimal management approaches.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table S1: title; Video S1: title.
Author Contributions
AS: study design, acquisition, assembly, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content. TG: acquisition and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content. PJBS and TZ: acquisition, analysis, and interpretation of molecular data, drafting of the manuscript, and critical revision of the manuscript for important intellectual content.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by the “Research Ethics Board” of the University Health Network (REB#22-5613) and conducted in compliance with the Declaration of Helsinki.
Informed Consent Statement
Patient consent was waived by the institutional ethics board as the study does not entail the disclosure of personal Health Information (PHI) of the patient.
Data Availability Statement
The dataset generated during and/or analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cohen, J.I.; Kimura, H.; Nakamura, S.; Ko, Y.H.; Jaffe, E.S. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009, 20, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Afzali, B.; Kazemian, M. EBV-associated diseases: Current therapeutics and emerging technologies. Front. Immunol. 2022, 13, 1059133. [Google Scholar] [CrossRef]
- Montes-Mojarro, I.A.; Kim, W.Y.; Fend, F.; Quintanilla-Martinez, L. Epstein - Barr virus positive T and NK-cell lymphoproliferations: Morphological features and differential diagnosis. Semin. Diagn. Pathol. 2020, 37, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Swerdlow, S.H.; Tousseyn, T.; Barrionuevo, C.; Nakamura, S.; Jaffe, E.S. New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders. Virchows Arch. 2022, 482, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Kawada, J.-I.; Ito, Y.; Ohshima, K.; Yamada, M.; Kataoka, S.; Muramatsu, H.; Sawada, A.; Wada, T.; Imadome, K.-I.; Arai, A.; et al. Updated guidelines for chronic active Epstein–Barr virus disease. Int. J. Hematol. 2023, 118, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (Eds):. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). Lyon: IARC; 2017.
- WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet]. International Agency for Research on Cancer; 2024. (WHO classification of tumours series t. 2024.
- Kimura, H.; Ito, Y.; Kawabe, S.; Gotoh, K.; Takahashi, Y.; Kojima, S.; Naoe, T.; Esaki, S.; Kikuta, A.; Sawada, A.; et al. EBV-associated T/NK–cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 2012, 119, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. Virus infection and human cancer: an overview. Recent Results Cancer Res. 2014, 193, 1–10. [Google Scholar] [PubMed]
- Kimura, H.; Morishima, T.; Kanegane, H.; Ohga, S.; Hoshino, Y.; Maeda, A.; Imai, S.; Okano, M.; Morio, T.; Yokota, S.; et al. Prognostic Factors for Chronic Active Epstein-Barr Virus Infection. J. Infect. Dis. 2003, 187, 527–533. [Google Scholar] [CrossRef]
- Yonese, I.; Sakashita, C.; Imadome, K.-I.; Kobayashi, T.; Yamamoto, M.; Sawada, A.; Ito, Y.; Fukuhara, N.; Hirose, A.; Takeda, Y.; et al. Nationwide survey of systemic chronic active EBV infection in Japan in accordance with the new WHO classification. Blood Adv. 2020, 4, 2918–2926. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2018, 4, 404–413, Erratum in Nat Microbiol. 2018, 4, 544. [Google Scholar] [CrossRef]
- Murata, T.; Okuno, Y.; Sato, Y.; Watanabe, T.; Kimura, H. Oncogenesis of CAEBV revealed: Intragenic deletions in the viral genome and leaky expression of lytic genes. Rev. Med Virol. 2019, 30, e2095. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gu, Z.-H.; Yan, Z.-X.; Zhao, X.; Xie, Y.-Y.; Zhang, Z.-G.; Pan, C.-M.; Hu, Y.; Cai, C.-P.; Dong, Y.; et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.Z.L.; Yokomori, R.; Wong, R.W.J.; Tan, T.K.; Ueda, R.; Ishida, T.; Iida, S.; Sanda, T. Requirement for TP73 and genetic alterations originating from its intragenic super-enhancer in adult T-cell leukemia. Leukemia 2022, 36, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Dave, B.; Singh, R. TP73, an under-appreciated player in non-Hodgkin lymphoma pathogenesis and management. Curr. Mol. Med. 2014, 14, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, H.; He, W.; Xia, Y.; Xia, Z.; Li, S.; Huang, H.; Li, Z.; Liu, P.; Jiang, W. Association between extranodal natural killer/T-cell lymphoma and hepatitis B viral infection: a case-control study. J. Cancer 2017, 8, 2676–2683. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Poluch, M.; Thomas, C.; Sindaco, P.; Khoo, A.; Porcu, P. Hepatitis B Virus and B-cell lymphoma: evidence, unmet need, clinical impact, and opportunities. Front. Oncol. 2023, 13, 1275800. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.N.; Scuoppo, C.; Vlasevska, S.; Bal, E.; Holmes, A.B.; Holloman, M.; Garcia-Ibanez, L.; Nataraj, S.; Duval, R.; Vantrimpont, T.; et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 2019, 51, 535–547. [Google Scholar] [CrossRef]
- Almeida, A.C.d.S.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef]
- Schatz, J.H.; Horwitz, S.M.; Teruya-Feldstein, J.; A Lunning, M.; Viale, A.; Huberman, K.; Socci, N.D.; Lailler, N.; Heguy, A.; Dolgalev, I.; et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia 2014, 29, 237–241. [Google Scholar] [CrossRef]
- Hathuc, V.; Kreisel, F. Genetic Landscape of Peripheral T-Cell Lymphoma. Life 2022, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.A.; Chung, E.Y.; Giricz, O.; Pradhan, K.; Kataoka, K.; Gordon-Mitchell, S.; Bhagat, T.D.; Mai, Y.; Wei, Y.; Ishida, E.; et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood 2018, 132, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Z.; Li, Y.; Peng, H.; Liu, J.; Zhang, J.; Xiao, X. The Role of CREBBP/EP300 and Its Therapeutic Implications in Hematological Malignancies. Cancers 2023, 15, 1219. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Cheng, D.; Lin, K.; Villarreal, O.D.; Gao, G.; Perez-Oquendo, M.; Van, H.T.; Stratton, S.A.; Green, M.; Xu, H.; et al. CARM1 inhibition reduces histone acetyltransferase activity causing synthetic lethality in CREBBP/EP300-mutated lymphomas. Leukemia 2020, 34, 3269–3285. [Google Scholar] [CrossRef] [PubMed]
- Hartert, K.T.; Wenzl, K.; Krull, J.E.; Manske, M.; Sarangi, V.; Asmann, Y.; Larson, M.C.; Maurer, M.J.; Slager, S.; Macon, W.R.; et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia 2020, 35, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, M.; Schwabe, J.W. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev. 2003, 17, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Quinquenel, A.; Fornecker, L.-M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.-S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef]
- Symes, E.; Wang, P.; Lager, A.M.; Bishop, M.R.; Aqil, B.; Venkataraman, G. TP53/PLCG2-mutated diffuse large B-cell lymphoma richter transformation (DLBCL-RT) of CLL with unusual CD2 and PD-1 expression. Leuk Lymphoma 2022, 63, 2735–2738. [Google Scholar] [CrossRef]
- Manso, R.; Rodríguez-Pinilla, S.M.; González-Rincón, J.; Gómez, S.; Monsalvo, S.; Llamas, P.; Rojo, F.; Pérez-Callejo, D.; Cereceda, L.; Limeres, M.A.; et al. Recurrent presence of the PLCG1 S345F mutation in nodal peripheral T-cell lymphomas. Haematologica 2014, 100, e25–e27. [Google Scholar] [CrossRef]
- Vaqué, J.P.; Gómez-López, G.; Monsálvez, V.; Varela, I.; Martínez, N.; Pérez, C.; Domínguez, O.; Graña, O.; Rodríguez-Peralto, J.L.; Rodríguez-Pinilla, S.M.; et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014, 123, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Ye, X.; Su, H.; Li, W.; Liu, D.; Pirmoradian, M.; Wang, X.; Zhang, B.; Zhang, Q.; Chen, L.; et al. Genetic landscape of hepatitis B virus–associated diffuse large B-cell lymphoma. Blood 2018, 131, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Morphological and immunophenotypic characteristics of systemic chronic active EBV (CAEBV) disease. Focal infiltration of hepatic parenchyma by small lymphocytes with portal accentuation. (A, B, C, Hematoxylin and Eosin, 10x, 20x and 40x). No architectural alteration, epithelial destruction or necrosis is present. Atypical lymphocytes are positive for CD3 (D, E, 20x, and 40x), CD2 (F, 40x), and CD7 (G, 40x). The same cells are negative for CD5 (G, inlet, 40x), CD4 (H, 40x), and TCRb (D, inlet, 40x). The cells are partially but dimly positive for CD8 (I, 40x). EBV is diffusely positive in lymphocytes (J, 20x) while only hepatocytes (focally) are positive for HBsAg (K, 20X). Proliferation index is approximately 5% (not shown).
Figure 1.
Morphological and immunophenotypic characteristics of systemic chronic active EBV (CAEBV) disease. Focal infiltration of hepatic parenchyma by small lymphocytes with portal accentuation. (A, B, C, Hematoxylin and Eosin, 10x, 20x and 40x). No architectural alteration, epithelial destruction or necrosis is present. Atypical lymphocytes are positive for CD3 (D, E, 20x, and 40x), CD2 (F, 40x), and CD7 (G, 40x). The same cells are negative for CD5 (G, inlet, 40x), CD4 (H, 40x), and TCRb (D, inlet, 40x). The cells are partially but dimly positive for CD8 (I, 40x). EBV is diffusely positive in lymphocytes (J, 20x) while only hepatocytes (focally) are positive for HBsAg (K, 20X). Proliferation index is approximately 5% (not shown).
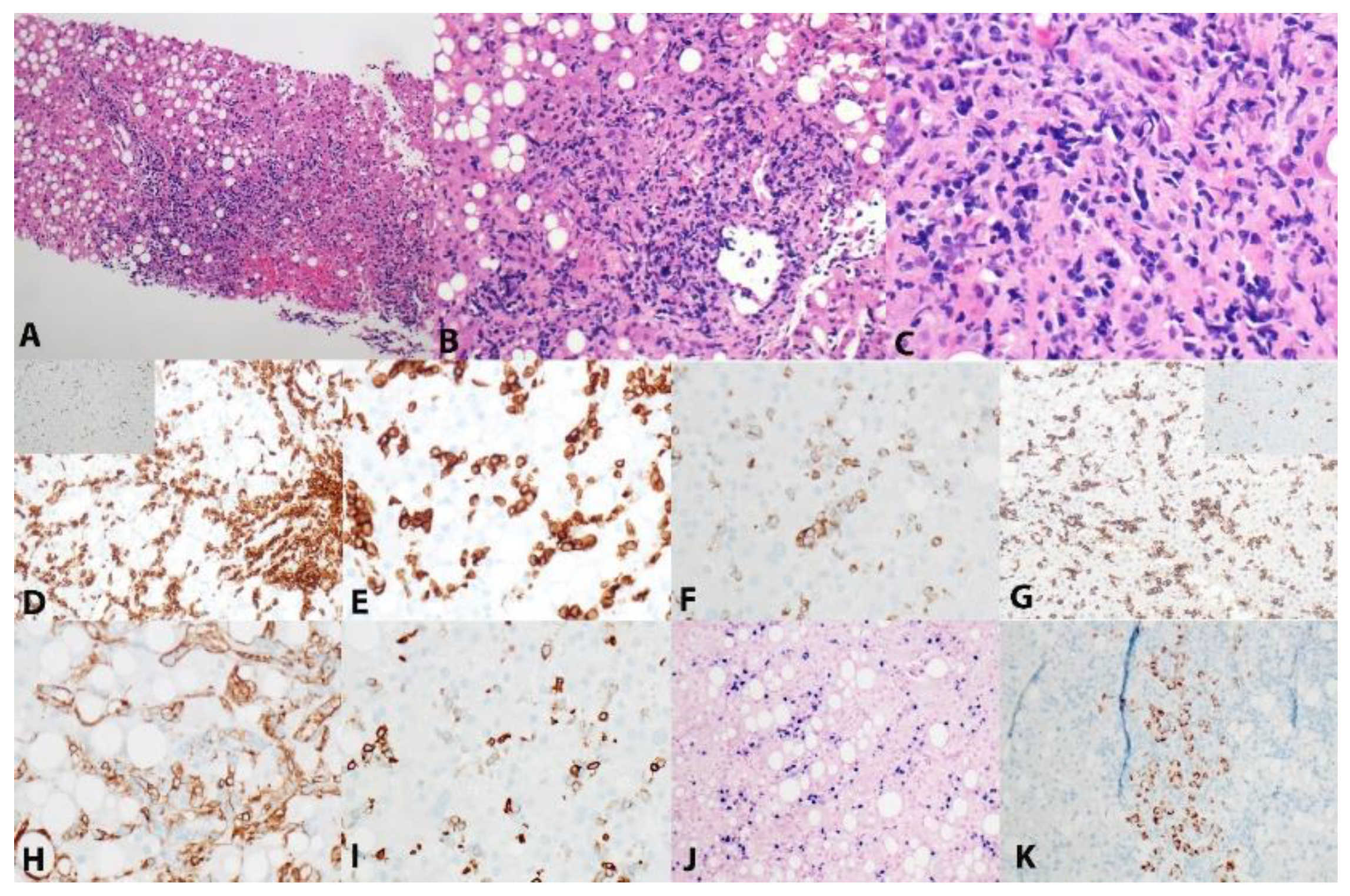

Table 1.
The possible oncogenic variants detected in the case of systemic CAEBV.
| Gene | Transcript | cDNA | Protein | VAF |
|---|---|---|---|---|
| CREBBP | NM_004380.2 | c.2447A>T | p.Gln816Leu | 9.6% |
| PLCG2 | NM_002661.3 | c.202A>T | p.Met68Leu | 48.5% |
| SPEN | NM_015001.2 | c.2318G>A | p.Arg773Lys | 47.6% |
| TP73 | NM_005427.3 | c.958G>A | p.Ala320Thr | 10.4% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



