
Submitted:
30 May 2024
Posted:
31 May 2024
You are already at the latest version
Abstract
Polyalcohol liquefaction can be done by acid or basic catalysis producing polyols with different properties. This study compared the mechanical properties of foams produced using polyols from liquefied Cytisus scoparius obtained by acid and basic catalysis and using two different foam catalysts. The differences were monitored using FTIR analysis. Acid-catalyzed liquefaction yielded 95.1 %, with the resultant polyol having an OH index of 1081 mg KOH/g while basic catalysis yielded 82.5 %, with a similar OH index of 1070 mg KOH/g. Generally, compressive strength with dibutyltin dilaurate (DBTDL) ranged from 16 - 31 kPa (acid-liquefied polyol) and 12 -21 kPa (base-liquefied polyol), while with stannous octoate (TIN), it ranged from 17 - 42 kPa (acid) and 29 - 68 kPa (base). Increasing water content generally decreased the compressive modulus and strength of foams. Higher water content led to a higher absorption at 1670 cm-1 in the FTIR spectrum due to the formation of urea. Higher isocyanate indices generally improved compressive strength, but high amounts led to unreacted isocyanate that could be seen by a higher absorption at 2265 cm-1 and 3290 cm-1. DBTL was shown to be the best foam catalyst due to higher trimer conversion seen in the spectra by a higher absorption at 1410 cm-1. Acid and base-derived polyols lead to different polyurethane foams with different FTIR spectra, particularly with a higher absorption at 1670 cm-1 for foams from acid-derived liquefaction.
Keywords:
Acid liquefaction
; basic liquefaction
; compressive strength
; FTIR
; polyurethane foams
1. Introduction
Polyurethane foams (PUF) derived from lignocellulosic materials represent an environmentally friendly and sustainable alternative to traditional petroleum-based polyurethane foams [1]. Lignocellulosic materials, such as wood, agricultural residues, and other plant-based sources, can be converted into liquid form through various processes like liquefaction or hydrothermal treatment. The resulting liquefied lignocellulosic materials can then be used as a renewable feedstock for the synthesis of polyurethane foams. Among the lignocellulosic materials used for the production of polyurethane foams, wood is probably the most used [2,3,4,5,6], and several agro-industrial residues are also used [7]. The use of renewable lignocellulosic materials reduces dependence on fossil fuels, making the process more sustainable. These bio-based polyurethane foams' carbon footprint is generally lower than traditional petroleum-based foams. Research and development in this field aim to optimize the production process, enhance foam properties, and make the technology economically viable on a larger scale. Using agricultural residues and waste biomass for polyurethane foam production aligns with the circular economy and sustainable development principles.
There are two main procedures for producing polyols from lignocellulosic materials to be used in polyurethane foam production: oxypropylation and liquefaction. Oxypropylation of biomass involves the addition of propylene oxide to biomass-derived compounds containing hydroxyl groups. This process is a way to modify the structure of biomass-derived materials, making them more suitable for various applications, including the production of bio-based polyols for polyurethane foams. It is generally done with high pressure and temperature using KOH as a catalyst [8]. This process has been used for several lignocellulosic materials such as cork [9,10]. The liquefaction process with polyalcohols offers several advantages, such as promoting the solubility of biomass components and facilitating solvolytic reactions that lead to the breakdown of complex structures. Polyalcohols, which are compounds with multiple hydroxyl (OH) groups, can be used as solvents or reactants in the liquefaction process. Common polyalcohols used in liquefaction include glycerol, ethylene glycol or polyethylene glycols (PEGs) [11,12,13,14]. This process can have acid [15,16,17], or basic catalysis [11,12,18], and the choice of catalyst depends on the initial material. Generally, acid catalysts lead to higher liquefaction yields, but basic catalysts are preferred for materials with suberin content, like cork barks [19].
The selection of liquefaction solvents depends on the intended properties of the liquefied material. The most important properties are hydroxyl number, acid number, viscosity and molecular weight. According to Hu et al [8] polyols obtained by polyalcohol liquefaction generally exhibit hydroxyl numbers between 100 to 600 mg KOH/g, acid numbers from 0 to 40 mgKOH/g, viscosities from 300 to 4500 cps, and molecular weight (MW) from 250 to over 7000 gmol−1. Nevertheless, these parameters depend on the type and amount of solvent used in the liquefaction procedure. For example, glycerol has been reported to have a hydroxyl value of around 1800 mg KOH/g [20], while PEG200, PEG400 and PEG600 have significantly lower hydroxyl values of respectively 534–590, 268–294 and 178–196 mg KOH/g [21]. Therefore, the amount of polyalcohols used in the liquefaction process influences the final hydroxyl value. For instance, the liquefaction of corn stover with different glycerol/corn stover ratios led to different hydroxyl values, ranging from 267 mg KOH/g to 346 mg KOH/g for 1:2 and 1:5 corn stover/glycerol ratios [22]. The hydroxyl number is important for polyurethane (PU) production and is known to decrease along the liquefaction, which has been considered to be due to the consumption of hydroxyl in oxidation and dehydration reactions [8,23,24]. Another important property, viscosity, usually also decreases with the progress of the liquefaction process [8]. However, D’Souza and Yan [25], who studied the effect of temperature on the production of bark-based polyols through liquefaction, stated that for higher liquefaction temperatures, the polyols exhibited an elevated viscosity, accompanied by an increase of the MW distributions.
In PU production, polyols can have from 2 to 8 reactive hydroxyl (OH) groups present in a polyol molecule and molecular weight from 200 to 8000 gmol-1. Therefore, polyurethane properties can be adjusted according to the needs [8]. The formation of polyurethane foams involves a complex reaction known as polyurethane synthesis, which typically consists of two main reactions: the polyol-isocyanate reaction (Foam formation) and the polyol-water reaction (Blowing Reaction). These reactions occur simultaneously and form a three-dimensional polymer network with the characteristic properties of polyurethane foam. Catalysts facilitate the polymerization reaction between polyols and isocyanates to form the polyurethane matrix. Common catalysts used in the production of polyurethane foams include tertiary amine compounds, such as triethylenediamine (TEDA) or dimethylcyclohexylamine (DMCHA) or organotin compounds, such as stannous octoate (Tin(II) 2-ethylhexanoate, TIN) and dibutyltin dilaurate (DBTDL), are also widely employed [26]. Tertiary amines catalyze both formation and expansion reactions, but they exhibit a distinctive characteristic wherein they demonstrate significantly higher efficacy in the isocyanate–hydroxyl reaction when employed with aliphatic isocyanates, contrasting to the combination with aromatic isocyanates [26].
Blowing agents, such as water or certain hydrocarbons, are used to generate gas during the reaction, leading to the formation of foam [1]. Water, in particular, reacts with isocyanate to release carbon dioxide, contributing to foam expansion. The choice of blowing agent significantly affects the foam properties. For example, Kurańska et al. [27] studied the effects of blowing agent type on the foaming process, cellular structure, mechanical properties, and changes in thermal conductivity during one year of ageing and concluded that carbon dioxide exhibited the highest reactivity. Additionally, foams blown with carbon dioxide displayed a cellular structure characterized by smaller cell sizes compared to those using physical blowing agents. The lowest thermal conductivity was however observed in polyurethane systems foamed with isopentane and a mixture of isopentane and cyclopentane.
Silicone surfactants are often added to improve the cell structure and overall foam properties. They help in controlling the size and distribution of cells in the foam. Zhang et al. [28] studied the role of silicone surfactant in flexible polyurethane foams using different siloxane-to-polyether ratios and concluded that silicone surfactants with a higher silicone content had lower surface tension, resulting in smaller bubble size and an increased bubble generation rate but leading to unstable foams. On the other hand, surfactants with a siloxane backbone to polyether ratio ranging from 0.32 to 0.5 demonstrated a balanced performance, exhibiting a moderate equilibrium between surface tension and lamella elasticity. Lower surface tension generally leads to lower cell sizes and higher closed cell content [29]. These authors also stated that the reduction in cell size also leads to lower thermal conductivity of foams, showing a linear relationship between the two variables across a broad range of cell sizes [29]. Similar results were presented for rigid polyurethane foams, where a smaller cell size enhanced the thermal insulation properties of rigid PUF and was considered to be a crucial factor in decreasing the thermal conductivity of the foams [30].
Acid or basic liquefaction yield depends on the type of the lignocellulosic material. Generally, alkaline hydrolysis is better for barks with higher cork content since an alkaline pH is needed for the saponification of suberin, which is the main content of cork. This was proven before, for example, by Yona et al. [19] for the liquefaction of Quercus suber bark that yielded around 61–85 % for alkaline catalysis and 43–50 % for acid catalysis or for Douglas-fir bark which also has a high suberin content and yield around 80 % for alkaline hydrolysis and 30 % for acid hydrolysis [11]. For lignocellulosic materials without suberin acid, catalysis is more efficient [18].
In this study, a comparison was made between the mechanical properties of the foams with varying percentages of two different catalyzers, blowing agents and isocyanate for both acid and base-catalyzed liquefaction. The changes were monitored by FTIR.
2. Materials and Methods
2.1. Sample Liquefaction
The Cytisus scoparius samples underwent a drying process in an oven at 100 °C and were finely ground to enhance their surface area. A precisely measured 10 g of sample was then weighed and transferred into the reactor. Subsequently, a mixture of glycerol and ethylene glycol in a 50:50 ratio, along with 3 % sulfuric acid based on the sample weight (used as a catalyst), was introduced. The glycerol-ethylene glycol mixture was added to fully cover the wood sample, and the Parr reactor was tightly sealed to prevent any potential leaks. The agitator was turned on at 75 rpm to ensure a complete mixture of the sample with the solvents. The temperature was gradually increased to 180 °C and maintained at this level for 60 minutes. After the designated time, the reactor was cooled to room temperature. Upon opening the reactor, the liquefied wood product was collected. This product, dissolved in 100 mL of methanol, underwent filtration for further processing. Samples of Cytisus scoparius were also liquefied with alkaline catalysis. In this case, potassium hydroxide (KOH) was used as the catalyst, with a maximum of around 6 %.
2.2. Determination of Hydroxyl Value
The OH index was determined through potentiometric titration of the residual acetic acid present after the esterification of free OH groups. An approximate weight of 20 mg of the sample was placed in a screw-cap tube. Subsequently, 0.1 mL of acetylation mixture was added, which had been prepared just prior to the analysis by mixing 4.7 mL of acetic anhydride with 4 mL of pyridine. The tube's content was then homogenized and kept for 24 hours in an oven set at 50 °C. Following cooling to room temperature, the mixture was transferred quantitatively to a 100 mL beaker using 10 mL of acetone and an equal amount of distilled water was added. The mixture was then titrated using a potentiometric method with standardized 0.1 N LiOH. The average value of three replicates was obtained, and the number of milligrams of KOH required to neutralize one gram of the sample was calculated using the following equations:
where V is the volume of LiOH solution required for the titration of the sample (mL); Vb is the volume of LiOH solution required for the titration of the blank (mL); ms is the acetylating mixture of the sample (mg); mb is the blank ( acetic anhydride and pyridine) in mg; f is the standardized titer of LiOH solution; W is the weight of the sample (mg); 1.7 is the mass, in mg, of OH groups equivalent to 1 mL of 0.1 M LiOH.
2.3. Foam Preparation
Approximately 4 g of neutralized and dried polyol was weighed and placed in a polypropylene container on a stable surface. The measured isocyanate was added to the polyol in a cylindrical container with a dimension of 60 × 120 mm3 (diameter × height) using a syringe. The surfactant was introduced into the mixture to stabilize the foam by controlling the size and distribution of bubbles. The measured amount of water was then added as the blowing agent, reacting with isocyanate to release carbon dioxide, contributing to the expansion of the foam. The mixture underwent mixing at 2000 rpm for 1 or 2 minutes, ensuring homogeneity and initiating reactions effectively. Subsequently, the catalyst (DBTDL or TIN) was added to the mixture to facilitate and accelerate the reaction between polyol and isocyanate, promoting the foaming process. Further mixing at 2000 rpm for 1 or 2 minutes followed until the foam started to rise. It was allowed to rise freely at room conditions.
2.4. Foam Testing
The polyurethane foam sample was prepared by cutting it into a cylinder with approximately 60 mm diameter and 30 mm high. The polyurethane foam sample was placed between the compression platens. The testing parameters, including compression speed and limit, were adjusted. The compression speed was 5 mm/min. The Universal Test Machine was started, applying a gradual and uniform compression force to the polyurethane foam.
During the test, data on applied force and corresponding deformation were recorded in real time using testing software. The compression continued until the sample underwent deformation, and the force stabilized, indicating a significant portion of the foam had compressed.
2.5. FTIR Analysis
Foams were dried overnight in an oven at 102 ± 2 °C and ground in a mortar. A Perkin Elmer UATR Two FT-IR Spectrometer (Beaconsfield, UK) was used with a resolution of 4.0 cm⁻¹, recording 72 scans in the range of 4000 - 400 cm⁻¹. The powder was placed directly on the crystal, completely covering the surface. Three spectra were collected for each sample.
3. Results and Discussion
3.1. Mechanical Properties
Acid catalysis led to a 95.1 % yield, and the resultant polyol presented an OH index of 1081 mg KOH/g and an acid number of 2.56 mg KOH/g (ethanol blank), while basic catalysis had an 82.5 % yield and an OH index of 1070 mg KOH/g and an acid number of 2.85 mg KOH/g (ethanol blank). Therefore, no significant difference has been observed for base or acid-liquefied polyols, although acid liquefaction was more efficient, leading to a higher amount of polyol. The high hydroxyl value of the polyols is due to the high amount of solvent used in the liquefaction procedure with 1:10 ratio sample:solvent. The solvent used was glycerol/ethylene glycol (50:50), and both have high OH index, ethylene glycol (IOH = 1808 mg KOH/g) and glycerol (IOH = 1827 mg KOH/g) in accordance to Chajęcka [31].
Water was chosen as the blowing agent. Figure 1 shows the variation of compressive modulus and compressive strength for increasing water content using DBTDL and TIN catalyzers. Overall, under the same conditions, PUF made with a TIN catalyzer presented higher compressive strength and compressive modulus.
The compressive strength of the foams using DBTDL catalyzer varied between 16 kPa and 31 kPa for the acid-liquefied polyol and 12 - 21 kPa for the base-liquefied polyol. In relation to the foams made with TIN catalyst, compressive strength varied between 17 kPa and 42 kPa and 29 kPa to 32 kPa for base-liquefied polyol. Therefore, results show that the compressive strength of foams with TIN catalyst is higher than the ones using DBTDL. Nevertheless, all of them are smaller than the compressive strengths of 80 – 150 kPa obtained for PU foams produced with polyols with high biomass content (ca. 50 %) produced by the combined liquefaction of wood and starch by Yao et al. [35] or PUF with polyols obtained by liquefaction of several waste papers (68 to 195 kPa) by Hu et al. [8]. The maximum compressive strength of about 21 kPa (DBTDL) and 42 kPA were obtained for 5 % water content. The compressive strength reported by Li et al. [33] for 7 % water content was 147 kPa; however, the polyether polyol used was prepared from a mixture of sugar and glycerol catalyzed with potassium hydroxide at low temperature (90 °C).
Compressive modulus seems to follow a similar trend for foams using DBTDL catalyst, varying between 233 kPa and 371 kPa for acid-liquefied polyol and between 197 kPa and 319 kPa for the base-liquefied one. These values are also lower than the ones reported before, for example, by Lee et al. [36], who reported 800 to 3400 kPa for a biodegradable polyurethane foam produced from liquefied waste paper. Nevertheless, mechanical properties depend on the equilibrium between the blowing agent, catalyzer and isocyanate and higher mechanical properties can be achieved, as reported in Figure 2 and Figure 3.
Results show that with the increase in water content, there was a decrease in both compressive modulus and compressive strength for the foams using DBTDL catalyzer. This decrease was observed for both acid and base-liquefied derived polyols. The results using TIN catalyzer are less consistent with high standard deviation but generally have a similar trend, although the highest values are obtained for 10.75 % water content. Similar results were presented before for other lignocellulosic polyols, for instance, for polyurethane foams from liquefied Eucalyptus globulus branches [6] or orange peel wastes [32] using tertiary amines catalyzers. The decrease can be associated with the higher expansion of the foam, producing a less densified structure and, therefore, a more resistant matrix. The same was observed before, for example, by Li et al. [33], who studied the influence of the water content on the apparent density and compressive strength of rigid foams reaching a compressive strength from 147 kPa (7% water content) and 401 kPa (3 % water content) or Hakim et al. [34] who reported the compressive strength variation of rigid polyurethane foam prepared from sugar-cane bagasse polyol.
Figure 2 presents the variation of compressive strength and compressive modulus for different NCO/OH ratios (isocyanate index). Compressive strength for index 1 was around 18 kPa for acid-liquefied derived polyol and under 10 kPa for base-liquefied polyol using DBTDL as the catalyst. With the increase in NCO/OH ratio, compressive strength increased, reaching a maximum of 31 kPa for acid-liquefied polyol. In relation to base-liquefied polyol, there was an increase from 1 to 1.2 isocyanate index followed by a decrease for 1.5 index. This decrease for 1.5 index may be due to unreacted materials. Higher isocyanate content usually leads to higher mechanical properties, which have been attributed to the increased hard segment content and crosslinking densities in polymer networks [8,37]. Nevertheless, if the amount of isocyanate is too high, the mechanical properties are affected due to incomplete curing of the isocyanate has been reported before [8,37]. This is confirmed by the FTIR analysis presented in section 3.2.
Using TIN catalyst, there was an increase followed by a decrease in both acid-liquefied and base-liquefied polyols' compressive strength. The highest compressive strength was attained for base-liquefied polyol using TIN catalyst, nevertheless, this is probably due to the lower grown of these foams. Generally, compressive modulus followed a similar trend. This means that with TIN catalyst, an isocyanate index higher than 1.2 is detrimental to the mechanical properties of the foams. Similar results were reported by Hakim et al. [34], who attributed this behaviour to an increased blowing effect by the CO2 created in extra condensation reactions between the isocyanate groups.
Figure 3 presents the variation of mechanical properties with varying amounts of catalyzers. Higher amounts of catalyst appear to increase compressive strength, which is particularly noticeable with a TIN catalyst. This effect could be caused by the rapid gelling reaction outpacing the expansion reaction. The faster gelling process might hinder the generation of sufficient CO2 to facilitate foam expansion. Additionally, the substantial rise in temperature induced by higher catalyst concentrations could enhance the reaction between the hydrogen-bonded with the nitrogen atom in the urethane group and the additional isocyanate, leading to the formation of allophanate [38]. Using DBTDL catalyst, there is no significant difference between acid and base-derived polyols, but for TIN catalyst, there is a significantly higher compressive strength for base-derived polyol. The highest compressive strength of around 69 kPa was achieved for base-derived polyol using 10% TIN catalyst. Compressive modulus also increases for higher amounts of catalyst. The highest compressive modulus was also achieved for TIN catalyst using basic-derived polyol with 1100 kPa.
3.2. FTIR Analysis
Figure 4 and Figure 5 present the FTIR spectra of foams made from acid and basic liquefied polyols and two different catalysts. For visualization purposes, the spectra are only presented in the 2400-1000 cm-1 range. The full spectra can be found in the supplementary material. The main peak assignments are presented in Table 1.
Overall, all the spectra presented the usual peaks of polyurethane foams.
All the foams exhibit a peak at 3290 cm-1 which has been assigned to N-H stretching vibration of urethane groups. No significant differences are observed in this peak at this wavelength. The peaks of unreacted OH and unreacted NCO groups can be found at around 3400 cm-1 and 2265 cm-1 [39]. Some of the foams still have the peak at 2265 cm-1 in conjunction with a higher absorption rate at 3400 cm-1, which indicates that there were some unreacted NCO and OH groups. When comparing the foams with higher isocyanate content (nº 7) with foams with lower isocyanate content (nº 6), a higher absorption is visible. Increasing isocyanate content above this point would result in a higher amount of unreacted materials. This might be the reason for the decrease in mechanical strength observed for base-liquefied polyol catalyzed by DBTL and for both acid and base-derived polyols catalyzed with TIN.
The carbonyl peak has its maximum absorbance at 1710 cm-1, which is associated to hydrogen-bonded urethane carbonyl, while free urethane absorbs at 1730 cm-1 [40,41,42]. The shoulder at 1670 cm-1 is due to urea carbonyl [43]. The foams with higher water content (nº 3) exhibit a higher urea carbonyl peak, except for base TIN, which has been stated to be due to the formation of more urea as a result of the reaction of isocyanate with water [40]. Similarly, the foams with less water content have a lower urea carbonyl peak (nº 2). Generally, the foams using the acid-liquefied polyols have a higher absorption at 1670 cm-1 compared to the absorption at 1710 cm-1, and this is observed for both catalysts.
The 1593 cm−1 band corresponds to the C-C stretching of the aromatic ring which is present in MDI [44]. No significant difference has been found in this peak for both polyols and for different catalysts. The band at around 1500 cm-1, with a maximum at 1507 cm-1 is generally attributed to bending vibrations of polyurethane N-H groups [45] but can also have some collaboration of C-N stretching. The main difference in this band is observed between acid and base-derived polyols since the shoulder at around 1530 cm-1 seems to be higher for base liquefied polyols.
The peak at 1410 cm-1 is associated with the isocyanurate C-N stretching [46,47]. This peak demonstrates the catalysts' potency to achieve high trimer conversion, which is crucial for enhancing fire retardancy in rigid foams, as stated before [46]. Overall, this peak is slightly higher for DBTL-catalyzed foams, which might indicate that this catalyst is better when compared to TIN with respect to fire retardancy. Uretidinedione also presents a similar band, nevertheless, it is accompanied by a band in the region 1700-1800 cm-1, which is not present in this case [47].
The peak at 1308 cm-1 has been assigned to aliphatic C-H bending vibrations [48], while the band around 1220 cm-1 can have several contributions. In this case the maximum is around 1200 cm-1.
5. Conclusions
Polyalcohol liquefaction of Cytisus scoparius was done using acid or basic catalysis, resulting in polyols with varying properties. Although with a higher liquefaction percentage for acid-catalyzed liquefaction, the OH index was similar for both polyols. The compressive strength and compressive modulus of foams made with DBTDL catalyst were similar for both acid and base-derived polyols, while with TIN catalyst, mechanical properties were higher for base-liquefied polyol. Results indicated that increasing water content generally decreased the compressive modulus and strength of the foams. Higher water content was associated with increased absorption at 1670 cm-1 due to urea formation. Higher isocyanate indices typically improved compressive strength, but excessive amounts led to unreacted isocyanate, as indicated by higher absorption at 2265 cm-1 and 3290 cm-1. The higher absorption at 1410 cm-1 identified DBTL as the best foam catalyst due to higher trimer conversion, which is responsible for the increase of fire retardancy in polyurethane foams. Additionally, polyols derived from acid and base catalysis resulted in different polyurethane foams with distinct FTIR spectra, generally showing higher absorption at 1670 cm-1 for foams derived from acid-catalyzed liquefaction.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: FTIR spectra of foams using acid liquefied polyol and DBTDL as catalyzer; Figure S2: FTIR spectra of foams using base liquefied polyol and DBTDL as catalyzer; Figure S3: FTIR spectra of foams using acid liquefied polyol and TIN as catalyzer; Figure S4: FTIR spectra of foams using base liquefied polyol and TIN as catalyzer;.
Author Contributions
Conceptualization, Y.D, B.E., I.D., L.P.C.-L. and R.P.F.G;; formal analysis, Y.D., I.D.,B:E. and L.P.C.-L.; investigation, Y.D., I.D.; resources, Y.D, B.E., I.D., L.P.C.-L. and R.P.F.G.; writing—original draft preparation, B.E.; writing—review and editing, B.E., Y.D., I.D., R.P.F.G. and L.P.C.-L.; funding acquisition, Y.D, B.E., I.D., L.P.C.-L. and R.P.F.G All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Funds through the FCT – Foundation for Science & Technology through (Proj. UIDB/00681/2020 (CERNAS) DOI: 10.54499/UIDB/00681/2020) and Polytechnic University of Viseu.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eling, B.; Tomović, Ž.; Schädler, V. Current and Future Trends in Polyurethanes: An Industrial Perspective. Macro Chemistry & Physics 2020, 221, 2000114. [Google Scholar] [CrossRef]
- Ertaş, M.; Fidan, M.S.; Alma, M.H. Preparation and Characterization of Biodegradable Rigid Polyurethane Foams from the Liquefied Eucalyptus and Pine Woods. Wood Res.-Slovakia 2014, 59, 97–108. [Google Scholar]
- Pan, H.; Zheng, Z.; Hse, C.Y. Microwave-Assisted Liquefaction of Wood with Polyhydric Alcohols and Its Application in Preparation of Polyurethane (PU) Foams. European Journal of Wood and Wood Products 2012, 70, 461–470. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, J.; Hse, C.-Y.; Shupe, T.F. Preparation of Polyurethane Foams Using Fractionated Products in Liquefied Wood. Journal of Applied Polymer Science 2014, 131, n/a-n/a. [Google Scholar] [CrossRef]
- Zheng, Z.; Pan, H.; Huang, Y.; Chung, Yh.; Zhang, X.; Feng, H. Rapid Liquefaction of Wood in Polyhydric Alcohols under Microwave Heating and Its Liquefied Products for Preparation of Rigid Polyurethane Foam. Open Materials Science Journal 2011, 5, 1–8. [Google Scholar] [CrossRef]
- Domingos, I.; Fernandes, A.P.; Ferreira, J.; Cruz-Lopes, L.; Esteves, B.M. Polyurethane Foams from Liquefied Eucalyptus Globulus Branches. BioResources 2019, 14, 31–43. [Google Scholar] [CrossRef]
- Zhang, J.; Hori, N.; Takemura, A. Optimization of Agricultural Wastes Liquefaction Process and Preparing Bio-Based Polyurethane Foams by the Obtained Polyols. Industrial Crops and Products 2019, 138, 111455. [Google Scholar] [CrossRef]
- Hu, S.; Luo, X.; Li, Y. Polyols and Polyurethanes from the Liquefaction of Lignocellulosic Biomass. ChemSusChem 2014, 7, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Evtiouguina, M.; Barros-Timmons, A.; Cruz-Pinto, J.J.; Neto, C.P.; Belgacem, M.N.; Gandini, A. Oxypropylation of Cork and the Use of the Ensuing Polyols in Polyurethane Formulations. Biomacromolecules 2002, 3, 57–62. [Google Scholar] [CrossRef]
- Evtiouguina, M.; Margarida Barros, A.; Cruz-Pinto, J.J.; Pascoal Neto, C.; Belgacem, N.; Pavier, C.; Gandini, A. The Oxypropylation of Cork Residues: Preliminary Results. Bioresource Technology 2000, 73, 187–189. [Google Scholar] [CrossRef]
- Esteves, B.; Cruz-Lopes, L.; Ferreira, J.; Domingos, I.; Nunes, L.; Pereira, H. Optimizing Douglas-Fir Bark Liquefaction in Mixtures of Glycerol and Polyethylene Glycol and KOH. Holzforschung 2018, 72, 25–30. [Google Scholar] [CrossRef]
- Hu, S.; Li, Y. Polyols and Polyurethane Foams from Base-Catalyzed Liquefaction of Lignocellulosic Biomass by Crude Glycerol: Effects of Crude Glycerol Impurities. Industrial Crops and Products 2014, 57, 188–194. [Google Scholar] [CrossRef]
- Jasiukaitytė-Grojzdek, E.; Kunaver, M.; Crestini, C. Lignin Structural Changes During Liquefaction in Acidified Ethylene Glycol. Journal of Wood Chemistry and Technology 2012, 32, 342–360. [Google Scholar] [CrossRef]
- Jin, Y.; Ruan, X.; Cheng, X.; Lü, Q. Liquefaction of Lignin by Polyethyleneglycol and Glycerol. Bioresource Technology 2011, 102, 3581–3583. [Google Scholar] [CrossRef] [PubMed]
- Soares, B.; Gama, N.; Freire, C.; Barros-Timmons, A.; Brandão, I.; Silva, R.; Pascoal Neto, C.; Ferreira, A. Ecopolyol Production from Industrial Cork Powder via Acid Liquefaction Using Polyhydric Alcohols. ACS Sustainable Chemistry & Engineering 2014, 2, 846–854. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, F.; Luo, C.; Xiong, L.; Chen, X. Liquefaction and Characterization of Acid Hydrolysis Residue of Corncob in Polyhydric Alcohols. Industrial Crops and Products 2012, 39, 47–51. [Google Scholar] [CrossRef]
- Mateus, M.M.; Guerreiro, D.; Ferreira, O.; Bordado, J.C.; Santos, R.G. dos Heuristic Analysis of Eucalyptus Globulus Bark Depolymerization via Acid-Liquefaction. Cellulose 2017, 24, 659–668. [Google Scholar] [CrossRef]
- Maldas, D.; Shiraishi, N. Liquefaction of Wood in the Presence of Polyol Using NaOH as a Catalyst and Its Application to Polyurethane Foams. International Journal of Polymeric Materials 1996, 33, 61–71. [Google Scholar] [CrossRef]
- Yona, A.M.C.; Budija, F.; Kričej, B.; Kutnar, A.; Pavlič, M.; Pori, P.; Tavzes, Č.; Petrič, M. Production of Biomaterials from Cork: Liquefaction in Polyhydric Alcohols at Moderate Temperatures. Industrial Crops and Products 2014, 54, 296–301. [Google Scholar] [CrossRef]
- Mamiński, M.; Szymański, R.; Parzuchowski, P.; Antczak, A.; Szymona, K. Hyperbranched Polyglycerols with Bisphenol A Core as Glycerol-Derived Components of Polyurethane Wood Adhesives. BioResources 2012, 7. [Google Scholar] [CrossRef]
- Liang, Y.; Ding, X.; Du, Z.; Wang, J.; Zhao, M.; Dan, Y.; Jiang, L.; Chen, Y. Low-Temperature Performance Controlled by Hydroxyl Value in Polyethylene Glycol Enveloping Pt-Based Catalyst for CO/C3H6/NO Oxidation. Molecular Catalysis 2020, 484, 110740. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Wan, Y.; Lei, H.; Yu, F.; Chen, P.; Lin, X.; Liu, Y.; Ruan, R. Liquefaction of Corn Stover Using Industrial Biodiesel Glycerol. International Journal of Agricultural and Biological Engineering 2009, 2, 32–40. [Google Scholar]
- Yamada, T.; Ono, H. Characterization of the Products Resulting from Ethylene Glycol Liquefaction of Cellulose. Journal of Wood Science 2001, 47, 458–464. [Google Scholar] [CrossRef]
- Yamada, T.; Aratani, M.; Kubo, S.; Ono, H. Chemical Analysis of the Product in Acid-Catalyzed Solvolysis of Cellulose Using Polyethylene Glycol and Ethylene Carbonate. J Wood Sci 2007, 53, 487–493. [Google Scholar] [CrossRef]
- D’Souza, J.; Yan, N. Producing Bark-Based Polyols through Liquefaction: Effect of Liquefaction Temperature. ACS Sustainable Chemistry & Engineering 2013, 1, 534–540. [Google Scholar] [CrossRef]
- Silva, A.L.; Bordado, J.C. Recent Developments in Polyurethane Catalysis: Catalytic Mechanisms Review. Catalysis Reviews 2004, 46, 31–51. [Google Scholar] [CrossRef]
- Kurańska, M.; Prociak, A.; Michalowski, S.; Zawadzińska, A. The Influence of Blowing Agents Type on Foaming Process and Properties of Rigid Polyurethane Foams. Polimery 2018, 63. [Google Scholar] [CrossRef]
- Zhang, X.D.; Macosko, C.W.; Davis, H.T.; Nikolov, A.D.; Wasan, D.T. Role of Silicone Surfactant in Flexible Polyurethane Foam. Journal of Colloid and Interface Science 1999, 215, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Kim, S.H.; Kim, B.K. Effects of Silicon Surfactant in Rigid Polyurethane Foams. Express Polymer Letters 2008, 2, 194–200. [Google Scholar] [CrossRef]
- Han, M.S.; Choi, S.J.; Kim, J.M.; Kim, Y.H.; Kim, W.N.; Lee, H.S.; Sung, J.Y. Effects of Silicone Surfactant on the Cell Size and Thermal Conductivity of Rigid Polyurethane Foams by Environmentally Friendly Blowing Agents. Macromol. Res. 2009, 17, 44–50. [Google Scholar] [CrossRef]
- Chajęcka, J.M. Synthesis of Biodegradable and Biocompostable Polyesters. PhD Thesis, Dissertation, Instituo Superior Tecnico, Universide Tecnica de Lisboa, Lisboa, 2011. [Google Scholar]
- Domingos, I.; Ferreira, J.; Cruz-Lopes, L.; Esteves, B. Polyurethane Foams from Liquefied Orange Peel Wastes. Food and Bioproducts Processing 2019, 115, 223–229. [Google Scholar] [CrossRef]
- Li, X.; Cao, H.; Zhang, Y. Structures and Physical Properties of Rigid Polyurethane Foams with Water as the Sole Blowing Agent. Science in China Series B: Chemistry 2006, 49, 363–370. [Google Scholar] [CrossRef]
- Hakim, A.A.; Nassar, M.; Emam, A.; Sultan, M. Preparation and Characterization of Rigid Polyurethane Foam Prepared from Sugar-Cane Bagasse Polyol. Materials Chemistry and Physics 2011, 129, 301–307. [Google Scholar] [CrossRef]
- Yao, Y.; Yoshioka, M.; Shiraishi, N. Combined Liquefaction of Wood and Starch in a Polyethylene Glycol/Glycerin Blended Solvent. Mokuzai Gakkaishi 1993, 39, 930–938. [Google Scholar]
- Lee, S.-H.; Teramoto, Y.; Shiraishi, N. Biodegradable Polyurethane Foam from Liquefied Waste Paper and Its Thermal Stability, Biodegradability, and Genotoxicity. Journal of Applied Polymer Science 2002, 83, 1482–1489. [Google Scholar] [CrossRef]
- Yan, Y.; Pang, H.; Yang, X.; Zhang, R.; Liao, B. Preparation and Characterization of Water-blown Polyurethane Foams from Liquefied Cornstalk Polyol. Journal of applied polymer science 2008, 110, 1099–1111. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C. (Charles) Depolymerization of Lignins and Their Applications for the Preparation of Polyols and Rigid Polyurethane Foams: A Review. Renewable and Sustainable Energy Reviews 2016, 60, 317–329. [Google Scholar] [CrossRef]
- Kwon, O.-J.; Yang, S.-R.; Kim, D.-H.; Park, J.-S. Characterization of Polyurethane Foam Prepared by Using Starch as Polyol. Journal of Applied Polymer Science 2007, 103, 1544–1553. [Google Scholar] [CrossRef]
- Oka, H.; Tokunaga, Y.; Masuda, T.; Kiso, H.; Yoshimura, H. Characterization of Local Structures in Flexible Polyurethane Foams by Solid-State NMR and FTIR Spectroscopy. Journal of Cellular Plastics 2006, 42, 307–323. [Google Scholar] [CrossRef]
- Coleman, M.M.; Skrovanek, D.J.; Hu, J.; Painter, P.C. Hydrogen Bonding in Polymer Blends. 1. FTIR Studies of Urethane-Ether Blends. Macromolecules 1988, 21, 59–65. [Google Scholar] [CrossRef]
- Kardeş, M.; Yatmaz, H.C.; Öztürk, K. ZnO Nanorods Grown on Flexible Polyurethane Foam Surfaces for Photocatalytic Azo Dye Treatment. ACS Appl. Nano Mater. 2023, 6, 6605–6613. [Google Scholar] [CrossRef]
- Luo; Wang; Ying Hydrogen-Bonding Properties of Segmented Polyether Poly (Urethane Urea) Copolymer. Macromolecules 1997, 30, 4405–4409. [CrossRef]
- Węgrzyk, G.; Grzęda, D.; Ryszkowska, J. The Effect of Mixing Pressure in a High-Pressure Machine on Morphological and Physical Properties of Free-Rising Rigid Polyurethane Foams—A Case Study. Materials 2023, 16, 857. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Kumagai, S.; Motokucho, S.; Kameda, T.; Saito, Y.; Watanabe, A.; Nakatani, H.; Yoshioka, T. Temperature-Dependent Pyrolysis Behavior of Polyurethane Elastomers with Different Hard- and Soft-Segment Compositions. Journal of Analytical and Applied Pyrolysis 2020, 145, 104754. [Google Scholar] [CrossRef]
- Chaffanjon, P.; Grisgby, R.A.; Rister, E.L.; Zimmerman, R.L. Use of Real-Time FTIR to Characterize Kinetics of Amine Catalysts and to Develop New Grades for Various Polyurethane Applications, Including Low Emission Catalysts. Journal of Cellular Plastics 2003, 39, 187–210. [Google Scholar] [CrossRef]
- Reignier, J.; Méchin, F.; Sarbu, A. Chemical Gradients in PIR Foams as Probed by ATR-FTIR Analysis and Consequences on Fire Resistance. Polymer Testing 2021, 93, 106972. [Google Scholar] [CrossRef]
- 48. Yi, S.; Cho, S.; Lee, C.-K.; Cho, Y.-J. Thermal Resistance of Polyurethane Adhesives Containing Aluminum Hydroxide and Dealkaline or Alkaline Lignin. Journal of Applied Polymer Science 2024, 141, e55120. [Google Scholar] [CrossRef]
Figure 1.
Compressive strength and compressive modulus of PUF made with different amounts of blowing agent (water).
Figure 1.
Compressive strength and compressive modulus of PUF made with different amounts of blowing agent (water).
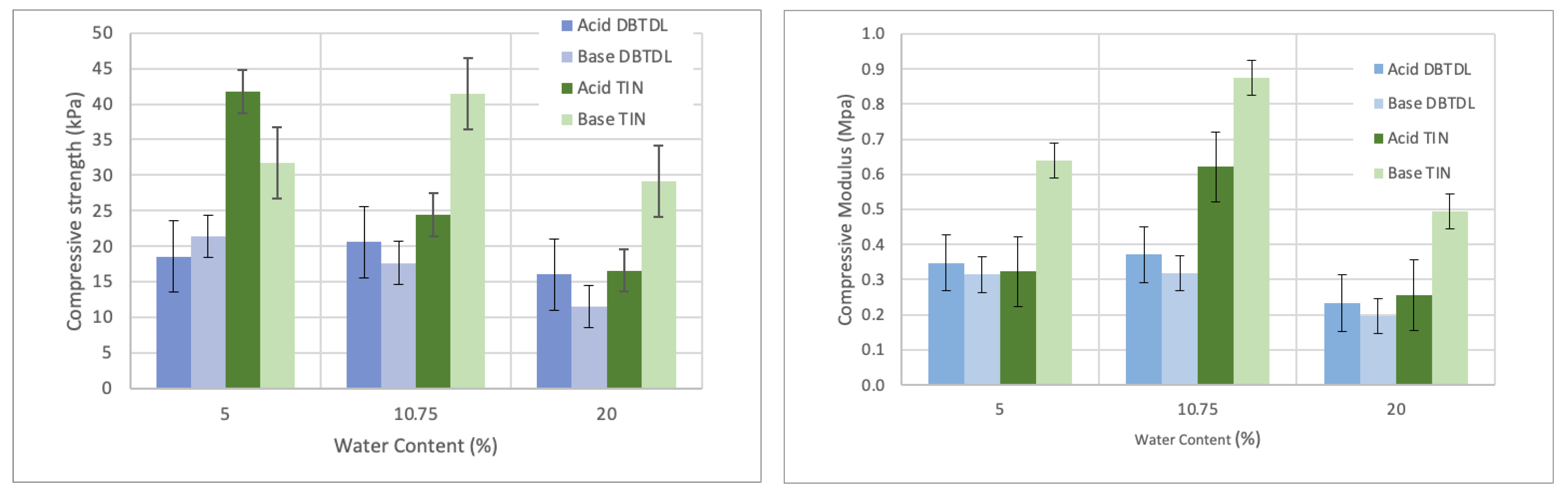
Figure 2.
Compressive strength and compressive modulus of PUF made with different isocyanate index.
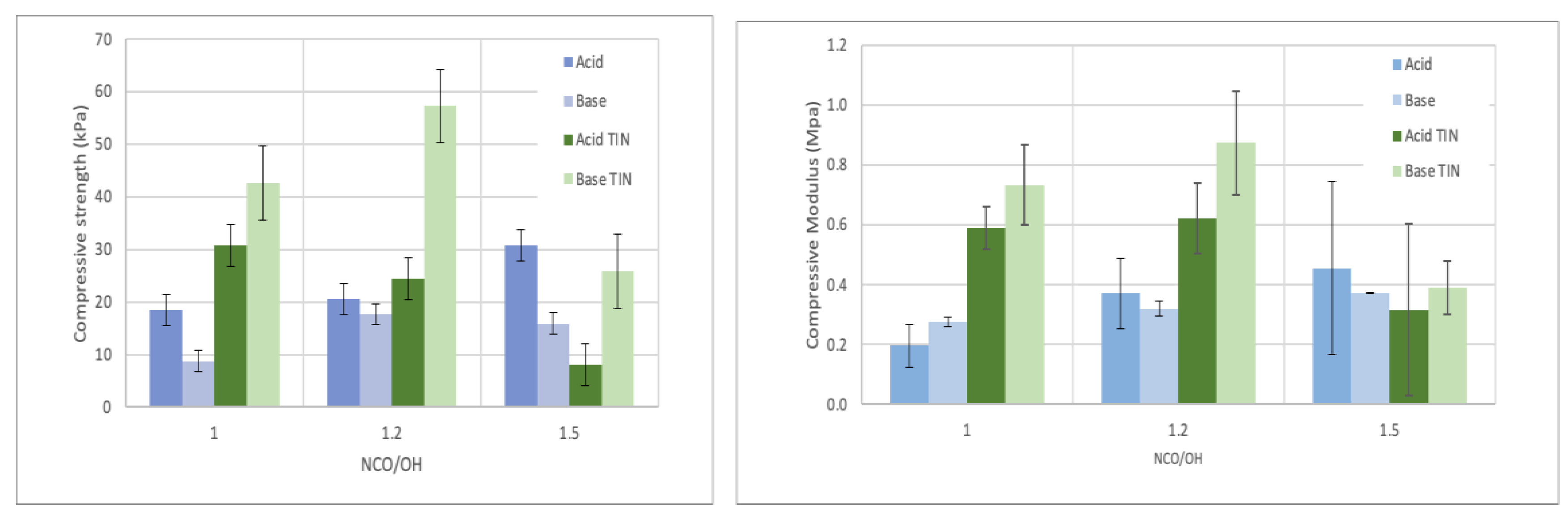
Figure 3.
Compressive modulus and compressive strength of PUF made with different amounts of catalyzer (DBTDL) for TIN catalyst.
Figure 3.
Compressive modulus and compressive strength of PUF made with different amounts of catalyzer (DBTDL) for TIN catalyst.

Figure 4.
FTIR spectra of foams using acid liquefied polyol(left) and base liquefied polyol(right) and DBTDL as catalyzer.
Figure 4.
FTIR spectra of foams using acid liquefied polyol(left) and base liquefied polyol(right) and DBTDL as catalyzer.
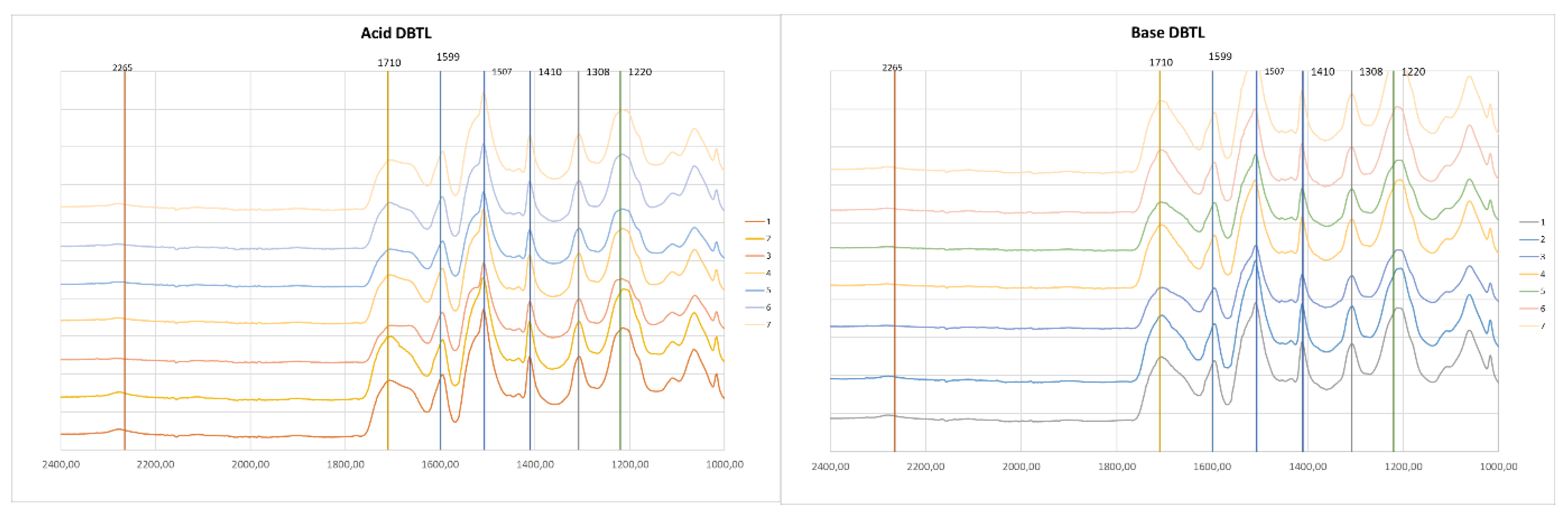
Figure 5.
FTIR spectra of foams using acid-liquefied polyol(left) and base-liquefied polyol(right) and TIN as catalyzer.
Figure 5.
FTIR spectra of foams using acid-liquefied polyol(left) and base-liquefied polyol(right) and TIN as catalyzer.
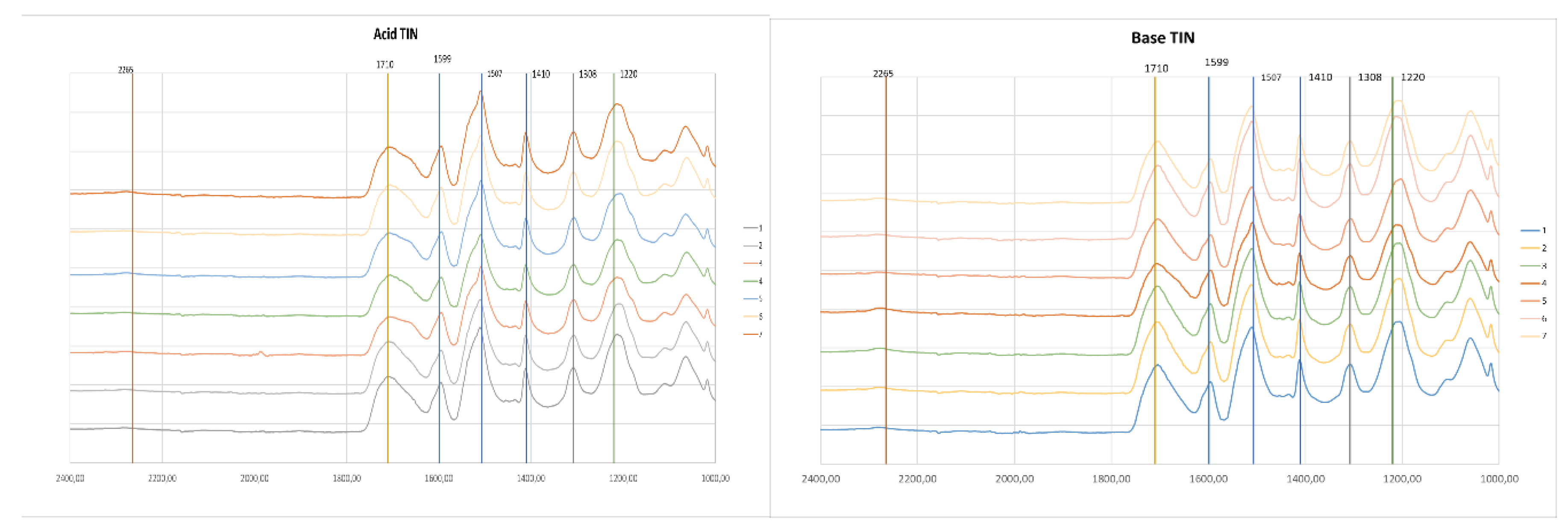

Table 1.
FTIR spectra assignment of bands in PU Foams.
| Wavenumber (cm-1) | Peak assignment |
| 3400 | O-H stretching |
| 3290 | N-H stretching vibration of urethane groups |
| 2265 | Antisymmetric stretching vibration of NCO |
| 1730 | C=O stretching (free urethane) |
| 1710 | C=O stretching (hydrogen-bonded urethane) |
| 1670 | C=O stretching (urea) |
| 1593 | C-C stretching of the aromatic ring |
| 1530 | C-N stretching of urethane group |
| 1507 | N-H bending vibration |
| 1410 | C-N stretching of the aromatic ring |
| 1308 | Aliphatic C-H bending vibrations |
| 1200-1230 | C-N stretching of urethane group |
| 1096 | C-O-C stretching |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



