
Submitted:
31 May 2024
Posted:
03 June 2024
You are already at the latest version
Abstract
The ribonuclease H (RNase H) active site of HIV-1 reverse transcriptase (RT) is the only viral enzyme not targeted by approved antiretroviral drugs. Using a fluorescence-based in vitro assay, we screened 65,239 compounds at a final concentration of 10 µM to identify inhibitors of RT RNase H activity. We identified 41 compounds that exhibited 50% inhibitory concentration (i.e., IC50) values < 1.0 µM. Two of these compounds, 2-(4-methyl-3-(piperidin-1-ylsulfonyl)phenyl)benzo[d]isothiazol-3(2H)-one (1) and ethyl 2-(2-(3-oxobenzo[d]isothiazol-2(3H)-yl)thiazol-4-yl)acetate (2), which both share the same benzisothiazolone pharmacophore, demonstrate robust antiviral activity (50% effective concentrations of 1.68 ± 0.94 µM and 2.68 ± 0.54, respectively) in the absence of cellular toxicity. A limited structure-activity relationship analysis identified two additional benzisothiazolone analogs, 2-methylbenzo[d]isothiazol-3(2H)-one (3) and N,N-diethyl-3-(3-oxobenzo[d]isothiazol-2(3H)-yl)benzenesulfonamide (4), which also resulted in inhibition of RT RNase H activity and virus replication. Compounds 1, 2 and 4, but not 3, inhibited the DNA polymerase activity of RT (IC50 values ~ 1 to 6 µM). In conclusion, benzisothiazolone derivatives represent a new class of multifunctional RT inhibitors that warrants further assessment for the treatment of HIV-1 infection.
Keywords:
HIV-1
; reverse transcriptase
; polymerase
; ribonuclease H
; inhibitor
; benzisothiazolone
1. Introduction
The human immunodeficiency virus type-1 (HIV-1) encodes for the enzyme reverse transcriptase (RT) which catalyzes the conversion of the single-stranded viral RNA genome into double-stranded DNA. RT is multifunctional and exhibits both DNA polymerase and ribonuclease H (RNase H) activities [1,2]. Due to its essential role in virus replication, HIV-1 RT is a primary target for drug discovery [3,4,5]. Two therapeutic classes of RT inhibitors have been approved by the U.S. Food and Drug Administration for prevention and treatment of HIV-1 infection. These include the nucleoside/nucleotide RT inhibitors (NRTIs; e.g., abacavir, emtricitabine, lamivudine, tenofovir disoproxil fumarate/tenofovir alafenamide and zidovudine) and the nonnucleoside RT inhibitors (NNRTIs; e.g., nevirapine (NVP; Figure 1a), efavirenz, rilpivirine, etravirine and doravirine). Within the cell, NRTIs are metabolized to their active diphosphate or triphosphate forms and compete with the natural dNTP substrate for binding and incorporation into the nascent viral DNA strand. Once incorporated, they promote chain-termination of HIV-1 DNA synthesis. NNRTIs are allosteric inhibitors of reverse transcription and bind to a hydrophobic pocket, termed the NNRTI binding pocket. that is adjacent to, but separate from, the DNA polymerase active site of RT (Figure 1b). The NNRTI binding pocket does not exist in the absence of the inhibitor, and this binding pocket is not directly involved in substrate binding or viral DNA synthesis. As a consequence, point mutations in the NNRTI binding pocket confer inhibitor resistance but do not typically impact enzyme function.
Although the RT RNase H activity is critical for HIV-1 replication, the RNase H domain is the only viral enzyme not targeted by approved antiviral drugs. Several RNase H pharmacophores have been described including the diketo acid, N-hydroxyimide, hydroxytropolone (Figure 1a), pyrimidinol carboxylic acid, N-hydroxy naphthyridinone, pyrido-pyrimidinone, nitrofuran-2-carboxylic acid and thiocarbamates [6]. Most of these pharmacophores co-ordinate the metal ions at the RNase H active site (Figure 1b), and the bound and intact DNA/RNA template/primer (T/P) substrate can restrict access of RNase H active site inhibitors, thus creating a major obstacle to RNase H inibitor design [7]. In addition to active site inhibitors, several classes of allosteric RT RNase H inhibitors have also been identified including the acylhydrazones, 1,2,4-triazoles, thiocarbamates and vinylogous urea [6]. Of note, NNRTI binding to RT can also modulate the RNase H activity of the enzyme depending on the T/P substrate; in particular, NNRTIs accelerate the rates of polymerase-independent RNase H cleavages [8,9,10,11].
In this study, we describe the discovery of benzisothiazolone derivatives as inhibitors of both RT DNA polymerase and RNase H activity. Importantly, the active compounds demonstrate robust anti-HIV-1 activity in cell culture with minimal cytotoxicity. Consequently, benzisothiazolone derivatives are a new class of RT inhibitor that warrants further assessment for the treatment of HIV-1 infection.
2. Results
2.1. High Throughput Screen (HTS) to Identify HIV-1 RT RNase H Inhibitors
We implemented a rapid, robust, and inexpensive fluorescence-based 384-well assay for the measurement of RT RNase H activity, previously described by Parniak et al [12]. The substrate comprises an 18-nucleotide 3′-fluorescein-labeled heteropolymeric RNA annealed to a complementary 5′-dabcyl-modified DNA. RT cleaves the RNA strand of this substrate close to 3′ end, promoting dissociation of the fluorescein-labeled RNA fragment generating a detectable fluorescent signal. We used this HTS assay to screen 65,239 compounds at a final concentration of 10 µM. The intra-plate Z factor ranged from 0.83 - 0.86 and the signal-to-background ratio ranged from 13.0- to 14.0-fold. Intra-plate coefficients of variation ranged between 3.7 - 5.5 %, and the inter-plate coefficients of variation ranged from 4.1 -6.1 %. The screen yielded 1190 compounds with ≥ 50 % inhibition (1.82 % active rate; Figure 2). We cherry picked and reordered 1042 compounds that were re-tested in the primary HTS. Of these, 390 (37.4 %) were confirmed as active. We then counter-screened these compounds against fluorescein isothiocyanate (FTIC) to confirm that they did not simply quench the fluorescence signal. None of the compounds quenched FITC fluorescence significantly (≥ 20 %). Finally, we conducted 10-point concentration-response assays starting at 50 µM to calculate 50 % inhibitory concentrations (i.e., IC50). Of the 390 compounds, 372 (95.4 %) exhibited IC50 values less than 50 µM: 41 exhibited IC50 values < 1.0 µM; 257 exhibited IC50 values in the range of 1.0 to 10.0 µM; and 74 exhibited IC50 values in the range of 10.0 to 50.0 µM. Leadscope classification and clustering by recursive partitioning identified 65 singletons and 64 clusters ranging from 2 to 24 stuctures/cluster. Data from the HTS campaign were uploaded and deposited in PubChem under the indicated assay identifier (AID) numbers: primary HTS (AID 565), active confirmation and fluorescense quenching counter screen (AID 651), and IC50 determinations of hits (AID 652).
2.2. Identification of RT RNase H Inhibitors with Antiviral Activity
Next, we re-purchased hit compounds with IC50 values < 1.0 µM for inhibition of RT RNase H activity that were available in sufficient quantities for further analysis. We assessed their antiviral activity and cytotoxicty in TZM-bl cells, as described previously [13]. Of these compounds, only two (2-(4-methyl-3-(piperidin-1-ylsulfonyl)phenyl)benzo[d]isothiazol-3(2H)-one (1) and ethyl 2-(2-(3-oxobenzo[d]isothiazol-2(3H)-yl)thiazol-4-yl)acetate (2) (Figure 3)), which both shared the same benzisothiazolone pharmacophore (Figure 3, red), demonstrated robust antiviral activity in the absence of cellular toxicity (Table 1). The effective concentration of compound required to inhibit 50% of HIV-1 replication in TZM-bl cells (i.e., EC50) and the concentrations of inhibitor that resulted in 50% TZM-bl cell death (i.e., CC50) were calculated to be 1.68 ± 0.94 µM and ≥ 100 µM for 1, and 2.68 ± 0.54 and ≥ 100 µM for 2, respectively.
2.3. Structure Activity Relationships (SAR)
We conducted a limited SAR by purchasing commercially available compounds that were structurally related to 1 and 2 (Figure 4). This included four benzisothiazolone analogs (compounds 3-6), two compounds in which the benzisothiazolone was replaced with an imidazo(1,2a)pyridine moiety (blue, compounds 7 and 8), and three compounds in which the benzisothiazolone was replaced with a 1,3-benzothiazole moiety (purple, compounds 9-11). We assessed the capacity for all of these compounds to inhibit the in vitro RNase H and DNA-dependent DNA polymerase activity of WT RT (Table 1). Of the benzisothiazolone analogs, compound 4 exhibited IC50 values for inhibition of RT RNase H (IC50 = 0.26 ± 0.08) and DNA polymerase activity (IC50 = 1.1 ± 0.3) that were similar to 1 and 2 (Table 2). Compound 3, which largely comprised the benzothiazolone scaffold, inhibited RT RNase H activity with an IC50 of 2.5 ± 0.2 µM but was completely inactive in the RT DNA polymerase assay. Compounds 3 and 4 also exhibited anti-HIV-1 activity in the absence of toxicity in TZM-bl cells (Table 1). A further two analogs, 5 and 6, exhibited modest activity in both in vitro assays. All of the other compounds tested were inactive in both the RT RNase H and DNA polymerase assays.
2.4. Inhibitory Activity of 1, 2, 3 and 4 against Wild-Type (WT) HIV-1 RT and HIV-1 RT Containing NNRTI Resistance Mutations
Because NNRTI binding to RT can enhance or inhibit the RNase H activity of the enzyme depending on the T/P substrate [8,9,10,11], we assessed the capacity for 1, 2, 3 and 4 to inhibit the RNase H and DNA polymerase activity of WT RT and RT containing the NNRTI resistance mutations K101E, K103N, Y181C, Y188C, G190A and P236L (Table 2). Compound 19619 (Figure 1), a hydroxytroplone analog of β-thujaplicinol [14,15], was used as a postive control in the RNase H cleavage assay, and NVP (Figure 1) was used as a control in the DNA polymerase assay. Consistent with a previous study, the IC50 for 19619 was calculated to be 8.7 ± 3.0 µM [8]. A similar IC50 value was determined for all the RTs containing NNRTI resistant mutations. NVP did not inhibit the RT RNase H cleavage activity. All 4 benzisothiazolone analogs inhibited the RNase H activity of WT and NNRTI-resistant RT, but small fold-changes in IC50 (< 5-fold) were noted for Y188C. Y181C RT conferred decreased susceptibility to 1 and 2, but not 3 and 4. K103N RT displayed 2-fold decreased susceptibility to 1 and 3.2-fold decreased susceptibility to 4. In the DNA polymerase assay, compounds 1, 2 and 4 retained activity against all RTs containing NNRTI resistance mutations. Interestingly, Y181C and Y188C RT exhibited marked increased susceptibility to 1, 2 and 4, a result that contrasts with the data for inhibition of RNase H cleavage. As expected, NVP was inactive against all NNRTI-resistant RTs.
3. Discussion
HIV-1 RT is a primary target for antiretroviral drugs, and two distinct therapeutic classes of inhibitors, the NRTIs and NNRTIs, have been approved by the U.S. Food and Drug Administration for treatment of HIV-1 infection. Both these drug classes, however, target the DNA polymerase activity of the enzyme. Although RT RNase H activity is critical for virus replication, the RNase H domain is the only viral enzyme not targeted by approved antiviral drugs. In this study, we describe the discovery of benzisothiazolone derivatives as inhibitors of RT RNase H cleavage.
The benzisothiazolone derivatives 1 and 2 were discovered in a high throughput screen designed to identify inhibitors of RT RNase H cleavage. Both 1 and 2 display potent inhibition of RT RNase H activity (IC50 values of 160 ± 30 nM and 130 ± 40 nM, respectively) but were also found to inhibit the enzyme’s DNA polymerase activity, albeit with IC50 values in the micromolar range (5.97 ± 3.10 µM and 2.64 ± 1.85 µM for 1 and 2, respectively). Typically, RNase H active site inhibitors, such as the hydroxytroplones [14,15], inhibit only the RNase H activity of RT. However, allosteric inhibitors have been reported to block both RT functions. For example, the thiocarbamates and 1,2,4-triazoles were identified in a high throughput screen for RT RNase H inhibitors [16], but were found to also inhibit the enzyme’s DNA polymerase activity. Interestingly, computational studies and crystallography show that triazoles bind in the NNRTI binding pocket in the RT DNA polymerase domain [17]. Acylhydrazones also act as dual DNA polymerase and RNase H inhibitors. The crystal structure of dihydroxybenzoyl naphthyl hydrazone in complex with HIV-1 RT showed the inhibitor binds to RT in the polymerase domain, near to but not within the NNRTI binding pocket [18]. Vinylogous urea compounds, also identified in a screen for RNase H inhibitors, inhibit both the DNA polymerase and RNase H activities of RT [19]. Protein footprinting and mutagenesis approaches showed that they interact with residues in the RT p51 thumb at the interface with the p66 RNase H domain [19,20]. Currently, we do not have insight into the binding site(s) or binding stoichiometry of the benzisothiazolone derivatives to RT HIV-1 RT. However, our SAR analysis revealed that compound 3 demonstrated specificity for the RNase H active site, suggesting that it may be possible to uncouple inhibition of the DNA polymerase and RNase H activities.
There is considerable evidence that both inhibitor binding and mutations in the NNRTI-binding pocket in the RT DNA polymerase domain impact the activity of the spatially remote RT RNase H [8,9,10,11,21,22]. The mechanisms involved in this long-range alteration of RNase H activity are not entirely clear but likely involve changes in the positioning of the RNA/DNA duplex nucleic acid due to protein conformation changes in the polymerase domain following NNRTI binding [8]. The effect of NNRTIs on RT RNase H activity, however, is typically much less than on RT DNA polymerase activity. In this study, we assessed the activity of the benzisothiazolone derivatives against RTs containing NNRTIs resistance mutations. The hydroxytroplone 19619, which binds at the RNase H active site of RT, retained activity against all NNRTI-resistant RTs. In general, the benzisothiazolone derivatives inhibited the RNase H activity of all RTs tested, but we did observe that some mutations, such as Y188C, decreased inhibitor potency. Of note, the activity of the benzisothiazolone derivatives against the DNA polymerase activity of the NNRTI-resistant RTs was not reduced by any of the mutations tested. Instead, we report that Y188C RT was hyper-susceptible to all of the analogs tested in the polymerase assay. While these data highlight that mutations in the NNRTI-binding pocket modulate activity of the benzisothiazolone analogs, they do not provide definitive proof that they bind to this pocket. We are currently trying to identify the benzisothiazolone analog binding site in RT using structural biology approaches.
In conclusion, we identified benzisothiazolone derivatives as inhibitors of the RNase H activity of HIV-1 RT, but they also block the enzyme’s DNA polymerase activity. Importantly, these benzisothiazolone derivatives demonstrate robust anti-HIV-1 activity in cell culture with minimal cytotoxicity. Consequently, benzisothiazolone derivatives are a new class of RT inhibitor that warrants further assessment for the prevention and/or treatment of HIV-1 infection.
4. Materials and Methods
- Materials
WT (wild-type) RT, and RTs harboring the NNRTI-resistant mutations K101E, K103N, Y181C, Y188C, G190A and P236L were overexpressed and purified to homogeneity as described previously [23]. RT concentration was determined spectrophotometrically at 280 nm using an absorbance coefficient of 260450 M−1·cm−1. The 7-hydroxy tropolone derivative 19616 (2,7-dihydroxy 2,4,6-cyclo-heptatrien-1-one) was purchased from Molecular Diversity Preservation International. Nevirapine was purchased from Selleckchem (Houston, TX, USA). The benzisothiazolone derivatives were procured from MolPort (New York, NY, USA) or Enamine (Kyiv, Ukraine). The DNA and RNA oligonucleotides were synthesized and purified by Integrated DNA Technologies (Coralville, IA, USA). All other reagents were of the highest quality available and were used without further purification.
- Compound Library
The NIH Molecular Library Screening Center Network (MLSCN) library of 65,239 compounds was arrayed at 10 mM concentration in DMSO into 384-well microtiter master plates and distributed to the University of Pittsburgh Molecular Library Screening Center (PMLSC) by the small molecule repository (SMR), Biofocus-DPI (A Galapagos Company, San Francisco, CA). Compounds were identified by their PubChem substance identity numbers (SIDs). Daughter plates containing 2 µL of 1 mM compounds in DMSO were prepared and replicated from the MLSCN master plates using the Velocity11 Vprep (Velocity11, Menlo Park, CA) outfitted with a 384-well transfer head. Aluminum adhesive plate seals were applied with an Abgene plate sealer and plates were stored at -20ºC in a Matrical MatriMinistore (Spokane, WA) automated compound storage and retrieval system. Compound daughter plates were withdrawn from -20ºC storage, thawed at ambient temperature, centrifuged 1-2 min at 50 x g, and plate seals were removed prior to the transfer of 18 µL of 50 mM Tris pH 8.0, 60 mM KCl, 5 mM MgCl2 assay buffer to prepare an intermediate compound library concentration of 100 µM in 1% DMSO using the Velocity11 Vprep outfitted with a 384-well transfer head. Diluted compounds were mixed by repeated aspiration and dispensation using the 384-well transfer head of the Velocity11 Vprep and 10 µL were transferred to the compound wells of 384-well assay plates.
- Primary HTS Assay.
Compounds were screened at a final concentration of 10 µM in 1x assay buffer (50 mM Tris pH 8.0, 60 mM KCl, 5 mM MgCl2) containing 1% DMSO in 384-well Greiner black polystyrene plates (VWR catalog # 82051-272). 10 µl of intermediate diluted compounds (100 µM in 10% DMSO) were transferred into assay plates using the Evolution-3 (EP3) (Perkin Elmer, Waltham, MA) automated liquid handling platform outfitted with a 384-well transfer head. 40 µL of RT-RNase H enzyme (60 ng per well) were then dispensed into each well using the Titertek MAP-C2 reagent dispenser platform (Huntsville, AL). 50 µl of the RNA/DNA duplex (140 nM) substrate was then dispensed into each well using the MAP-C2 dispenser, and assay plates were then incubated at room temperature for 20 minutes. After 20 min, the assay was terminated by the addition of 10 µl of 1 M EDTA pH 9.0 stop buffer. Relative fluorescent unit (RFU) signals, Excitation 490 nm and Emission: 528 nm (Cutoff 515 nm) were captured on the Spectromax M5e plate reader (Molecular Devices, Menlo, CA). To analyze the percent inhibition of RT-RNase H by library compounds in the primary screen, we constructed an ActivityBase™ HTS template to associate compound SIDs with the calculated percent inhibition, based on the mean maximum (1% DMSO, n=32) and mean minimum (100 µM EDTA, n=24) plate controls. The template also calculated plate signal to background (S:B) ratios (mean maximum signal/mean minimum signal) and Z’-factor coefficient HTS performance statistics.
- T/P substrates
The following RNA/DNA T/P substrate was used in the present study: an 18 nucleotide RNA template (RNA-18T: 5’- GAUCUGAGCCUGGGAGCU-3’) was annealed to an 18 nucleotide primer (DNA-18T: 5’-AGCTCCCAGGCTCAGATC-3’), as described previously [12]. Fluorescein and IowaBlack FQTM were covalently attached to the 3’- and 5’-ter-mini of the RNA and DNA oligonucleotides respectively.
- FRET-based RNase H cleavage assays
The FRET-based RNase H cleavage assays were carried out as described previously [12]. Fluorescence was measured with a SpectraMax M2 microplate spectrofluorometer (Molecular Devices) using an excitation wave-length of 490 nm and an emission wavelength of 528 nm. Data were analysed using SoftMax Pro software (Molecular Devices). The concentration of each 7-hyroxy-tropo-lone analogue required to inhibit RNase H cleavage by 50% (i.e. IC50 ) was calculated using non-linear regression analyses (SigmaPlot Software Version 11, Systat Soft-ware) from at least three independent experiments.
- Fluorescence-based DDDP Assay
DDDP assays were also performed using a QuantiFluor ONE based spectrophotometric assay to detect double-stranded DNA synthesized by recombinant HIV-1 RT [24]. The reaction contained 40 nM of a template/primer comprising a 200 nt DNA template (5′-TCTCTCTGGTTAGACCAGATCTGAGCCTGGGAGCTCTCTGGCTGACTGGGACCCA CTGCTTAA-GCCTCAATAAA-GCTTGCCTTGAGTGCTTAAAGTAGATGTGTGTGCC CGTCTGTTGTGTGACTCTGGTAACTAGAGATCCCTCAGACCCTTTTAGTCAGTGTG GAATATCTCATAGCTTGGTGCTCGAACAGTGAC-3′) annealed to an 18 nucleotide DNA primer (5′-GTCACTGTTCGAGCACCA-3′), 50 mM Tris-HCl pH 7.8, 50 mM NaCl, 10 mM MgCl2 and 50 µM dNTP mix. Reactions were stopped after 45 minutes by adding the detection reagent containing 1 × Quant-Fluor One (Promega) and 12.5 mM ethylenediaminetet-raacetic acid; samples were transferred to a 96-well black plate, and fluorescence was measured at excitation/emission (490/535 nm).
- HIV-1 drug susceptibility assays.
The genes for Y181C, Y181I, Y181V, and Y181F were cloned into HIV-1LAI. NNRTI susceptibility was determined in TZM-bl cells, as described previously [25].
Author Contributions
Conceptualization, P.A.J, J.S.L.; M.A.P.; and N.S-C.; methodology, P.A.J.; J.J.S.; A.V.R.; H.D.; M.A.; formal analysis, P.A.J.; A.V.R.; H.D.; N.S-C..; investigation, A.V.R; H.D.; J.J.S.; M.A.; writing—original draft preparation, N.S-C.; writing—review and editing, all authors.; funding acquisition, P.A.J; N.S-C; J.S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health; Molecular Library Screening Center Network U54MH074411 (Lazo, PI), XO1 MH077605 (Parniak, PI)) and R01 AI175067 (Sluis-Cremer, MPI).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All raw data are available from the authors upon request.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xavier Ruiz, F.; Arnold, E. Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr Opin Struct Biol. 2020, 61, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N. Retroviral reverse transcriptase: Structure, function and inhibition. Enzymes 2021, 50, 179–194. [Google Scholar] [CrossRef]
- Singh, A.K.; Das, K. Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies. Viruses 2022, 14, 1027. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Pannecouque, C.; De Clercq, E.; Chen, F. Development of non-nucleoside reverse transcriptase inhibitors (NNRTIs): our past twenty years. Acta Pharm Sin B. 2020, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Honma, M.; Kimura, Y.; Abe, H. Structure, Synthesis and Inhibition Mechanism of Nucleoside Analogues as HIV-1 Reverse Transcriptase Inhibitors (NRTIs). ChemMedChem. 2021, 16, 743–766. [Google Scholar] [CrossRef] [PubMed]
- Ilina, T.V.; Brosenitsch, T.; Sluis-Cremer, N.; Ishima, R. Retroviral RNase H: Structure, mechanism, and inhibition. Enzymes. 2021, 50, 227–247. [Google Scholar] [CrossRef] [PubMed]
- Beilhartz, G.L.; Wendeler, M.; Baichoo, N.; Rausch, J.; Le Grice, S.; Götte, M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol. 2009, 388, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Herman BD, Sluis-Cremer N. Transient kinetic analyses of the ribonuclease H cleavage activity of HIV-1 reverse transcriptase in complex with efavirenz and/or a beta-thujaplicinol analogue. Biochem J. 2013, 455, 179–184. [CrossRef]
- Hang, J.Q.; Li, Y.; Yang, Y.; Cammack, N.; Mirzadegan, T.; Klumpp, K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biochem Biophys Res Commun. 2007, 352, 341–350. [Google Scholar] [CrossRef]
- Shaw-Reid, C.A.; Feuston, B.; Munshi, V.; Getty, K.; Krueger, J.; Hazuda, D.J.; Parniak, M.A.; Miller, M.D.; Lewis, D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry. 2005, 44, 1595–1606. [Google Scholar] [CrossRef]
- Radzio, J.; Sluis-Cremer, N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol Pharmacol. 2008, 73, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Parniak, M.A.; Min, K.L.; Budihas, S.R.; Le Grice, S.F.; Beutler, .JA. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal Biochem. 2003, 322, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.P.; Huber, K.D.; Sluis-Cremer, N. Nonnucleoside Reverse Transcriptase Inhibitor Hypersusceptibility and Resistance by Mutation of Residue 181 in HIV-1 Reverse Transcriptase. Antimicrob Agents Chemother. 2019, 63, e00676–19. [Google Scholar] [CrossRef] [PubMed]
- Budihas, S.R.; Gorshkova, I.; Gaidamakov, S.; Wamiru, A.; Bona, M.K.; Parniak, M.A.; Crouch, R.J.; McMahon, J.B.; Beutler, J.A.; Le Grice, S.F. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005, 33, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Maegley, K.A.; Pauly, T.A.; Bauman, J.D.; Das, K.; Dharia, C.; Clark, A.D. Jr.; Ryan, K.; Hickey, M.J.; Love, R.A.; Hughes, S.H.; Bergqvist, S.; Arnold, E. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009, 17, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Di Grandi, M.; Olson, M.; Prashad, A.S.; Bebernitz, G.; Luckay, A.; Mullen, S.; Hu, Y.; Krishnamurthy, G.; Pitts, K.; O'Connell, J. Small molecule inhibitors of HIV RT Ribonuclease H. Bioorg Med Chem Lett. 2010, 20, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Kirschberg, T.A.; Balakrishnan, M.; Huang, W.; Hluhanich, R.; Kutty, N.; Liclican, A.C.; McColl, D.J.; Squires, N.H.; Lansdon, E.B. Triazole derivatives as non-nucleoside inhibitors of HIV-1 reverse transcriptase-structure-activity relationships and crystallographic analysis. Bioorg. Med. Chem. Lett. 2008, 18, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D., Jr.; Knight, J.L.; Julias, J.G.; Clark, P.K.; Krogh-Jespersen, K.; Levy, R.M.; Hughes, S.H.; Parniak, M.A.; Arnold, E. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef]
- Wendeler, M.; Lee, H.-F.; Bermingham, A.; Miller, J.T.; Chertov, O.; Bona, M.K.; Baichoo, N.S.; Ehtesham, M.; Beutler, J.A.; O'Keefe, B.R.; Gotte, M.; Kvaratskhelia, M.; Le Grice, S.F.J. Vinylogous ureas as a novel class of reverse transcriptase-associated ribonuclease H activity. ACS Chem. Biol. 2008, 3, 635–644. [Google Scholar] [CrossRef]
- Chung, S.; Miller, J.T.; Johnson, B.C.; Hughes, S.H.; Le Grice, S.F. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J. Biol. Chem. 2012, 287, 4066–4075. [Google Scholar] [CrossRef]
- Archer, R.H.; Dykes, C.; Gerondelis, P.; Lloyd, A.; Fay, P.; Reichman, R.C.; Bambara, R.A.; Demeter, L.M. Mutants of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors demonstrate altered rates of RNase H cleavage that correlate with HIV-1 replication fitness in cell culture. J. Virol. 2000, 74, 8390–8401. [Google Scholar] [CrossRef] [PubMed]
- Archer, R.H.; Wisniewski, M.; Bambara, R.A.; Demeter, L.M. The Y181C mutant of HIV-1 reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors alters the size distribution of RNase H cleavages. Biochemistry 2001, 40, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Le Grice, S.F.; Grüninger-Leitch, F. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur J Biochem. 1990, 187, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Rumrill, S.; Dawson, S.; Johnson, A.; Pinson, J.A.; Gunzburg, M.J.; Latham, C.F.; Barlow, N.; Mbogo, G.W.; Ellenberg, P.; Headey, S.J.; Sluis-Cremer, N.; Tyssen, D.; Bauman, J.D.; Ruiz, F.X.; Arnold, E.; Chalmers, D.K.; Tachedjian, G. Targeting HIV-1 Reverse Transcriptase Using a Fragment-Based Approach. Molecules. 2023, 28, 3103. [Google Scholar] [CrossRef]
- Giacobbi, N.S.; Sluis-Cremer, N. In Vitro Cross-Resistance Profiles of Rilpivirine, Dapivirine, and MIV-150, Nonnucleoside Reverse Transcriptase Inhibitor Microbicides in Clinical Development for the Prevention of HIV-1 Infection. Antimicrob Agents Chemother. 2017, 61, e00277–17. [Google Scholar] [CrossRef]
Figure 1.
(a) Structures of the NNRTI nevirapine and the RNase H active site inhibitor β-thujaplicinol, a hydroxytroplone analog. (b) Three dimensional structure of the p66 kDa subunit of HIV-1 RT in complex with β-thujaplicinol (pdb 3IG1).
Figure 1.
(a) Structures of the NNRTI nevirapine and the RNase H active site inhibitor β-thujaplicinol, a hydroxytroplone analog. (b) Three dimensional structure of the p66 kDa subunit of HIV-1 RT in complex with β-thujaplicinol (pdb 3IG1).
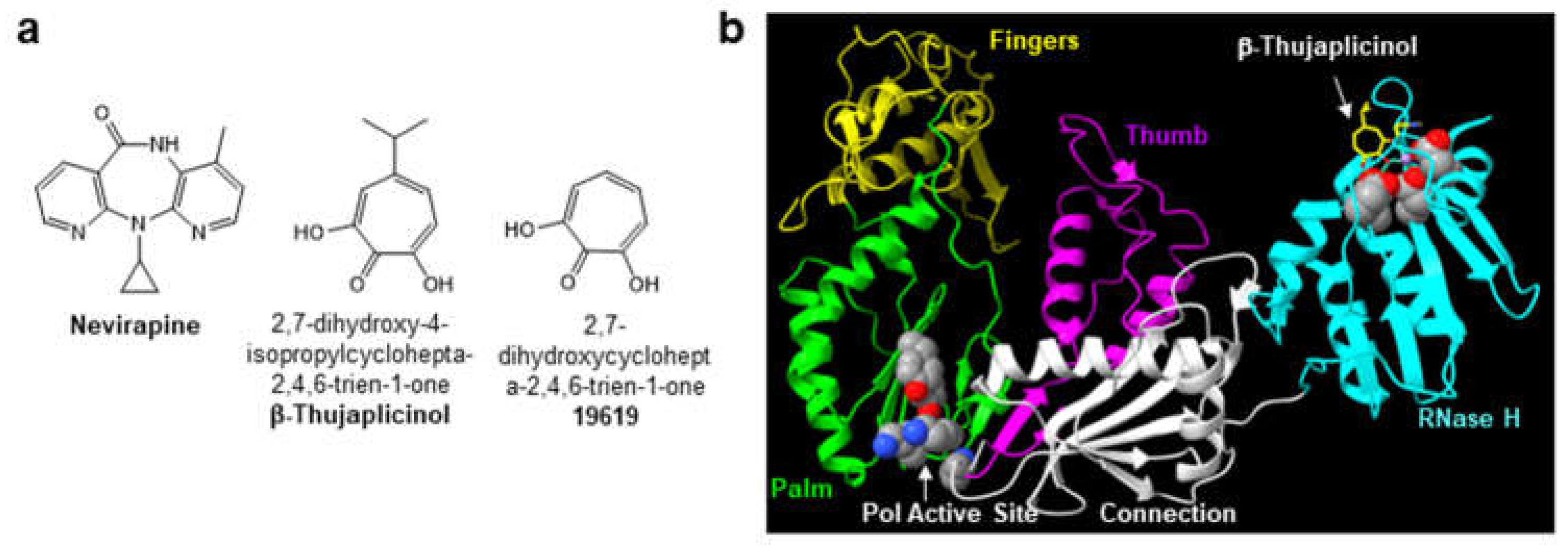
Figure 2.
Overview of the HTS campaign to identify RT RNase H inhibitors.
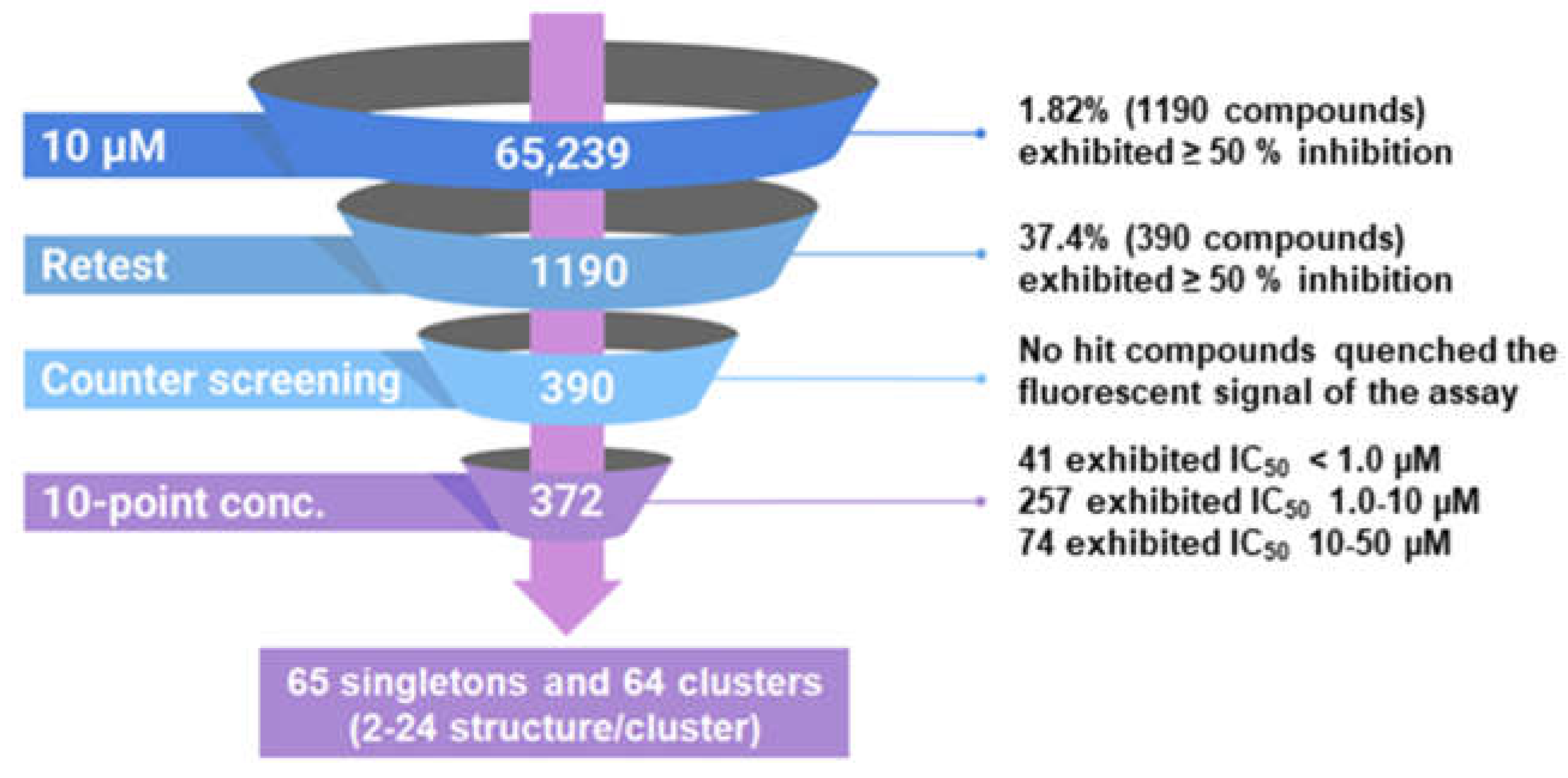
Figure 3.
Chemical structures of compounds 1 and 2.
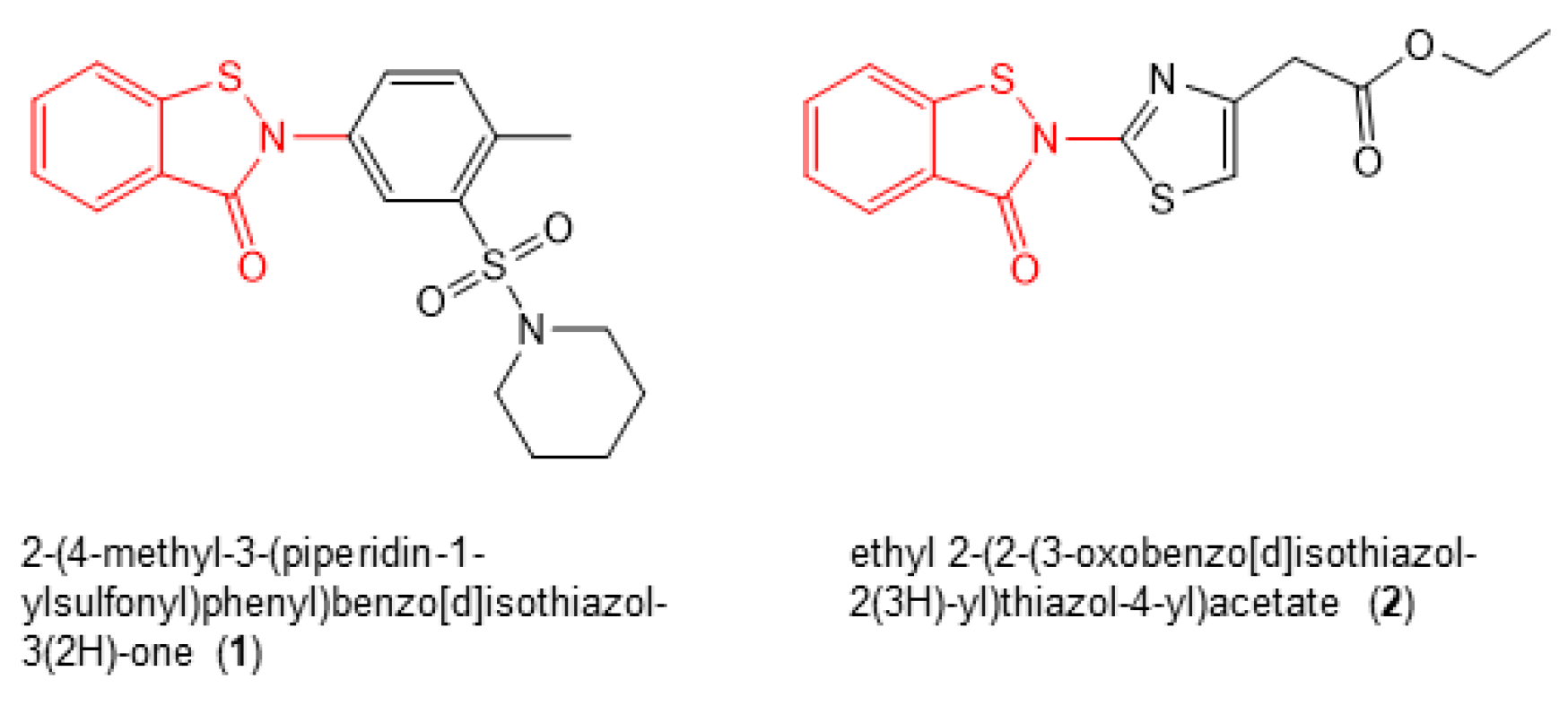
Figure 4.
Chemical structures of compounds 4 - 11.
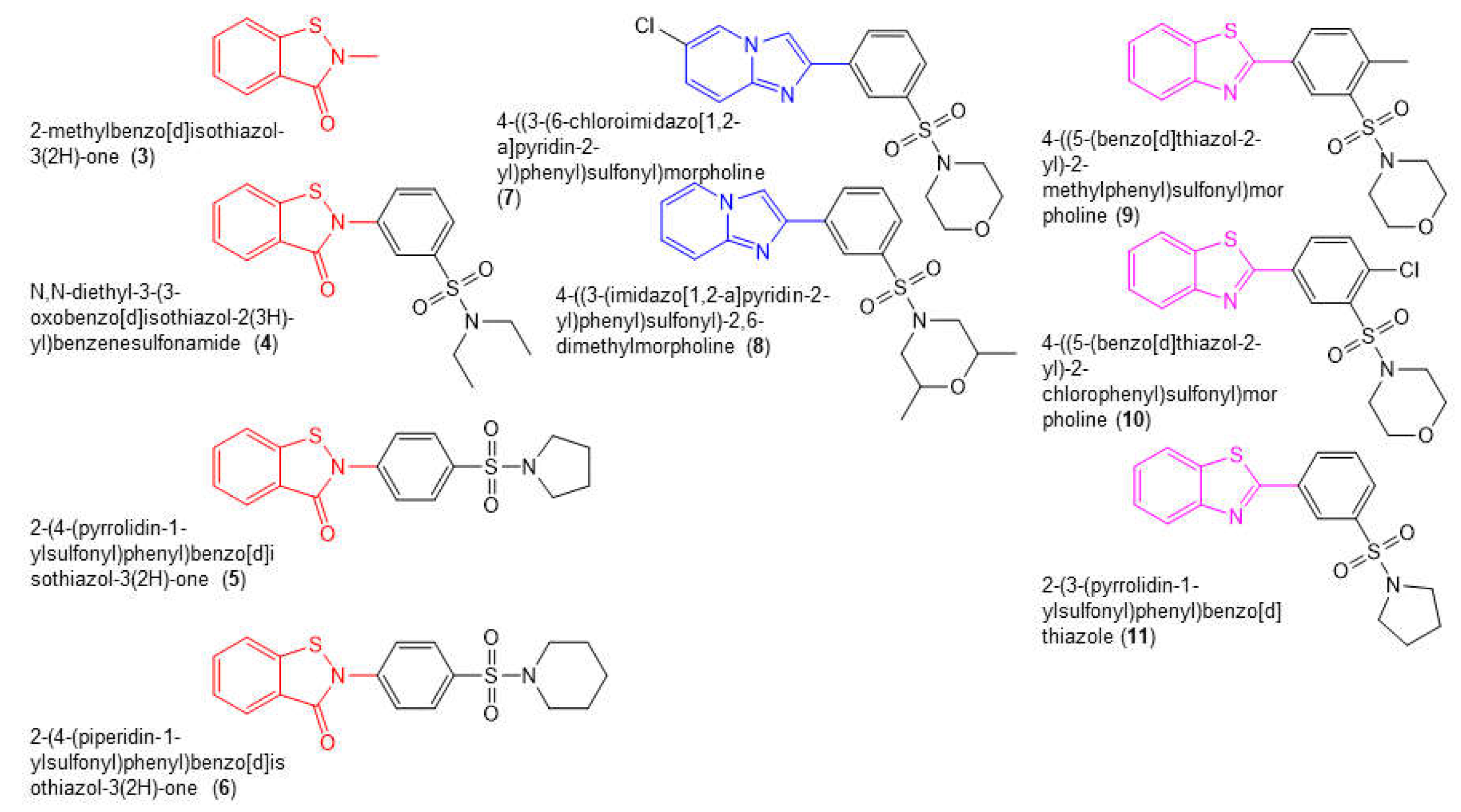

Table 1.
Activity of 1 – 11 against purified RT and HIV-1.
| RNase H Activity | Polymerase Activity | Antiviral Activity | Cytotoxicity | |
| IC50 (µM) | IC50 (µM) | EC50 (µM) | CC50 (µM) | |
| RNase H | DNA Polymerase | HIV-1 | ||
| 1 | 0.16 ± 0.03 | 5.97 ± 3.10 | 1.68 ± 0.94 | ≥ 100 |
| 2 | 0.13 ± 0.04 | 2.64 ± 1.85 | 2.68 ± 0.54 | ≥ 100 |
| 3 | 2.5±0.2 | > 81 | 28.45 ± 12.26 | ≥ 100 |
| 4 | 0.26±0.08 | 1.1±0.3 | 3.27 ± 0.49 | ≥ 100 |
| 5 | 15.41±9.2 | 25±25 | ||
| 6 | 39.21±20.99 | 12.2±3.7 | ||
| 7 | > 81 | > 81 | ||
| 8 | > 81 | > 81 | ||
| 9 | > 81 | > 81 | ||
| 10 | > 81 | > 81 | ||
| 11 | > 81 | 62±38 |
Table 2.
Inhibition of WT and NNRTI-resistant HIV-1 RT RNase H and DNA-dependent DNA polymerase activity by 1 and 2.
Table 2.
Inhibition of WT and NNRTI-resistant HIV-1 RT RNase H and DNA-dependent DNA polymerase activity by 1 and 2.
| Inhibition of RT RNase H Activity | ||||||||||
| RT | 19619 | 1 | 2 | 3 | 4 | |||||
| IC50 (µM) | Fold-R1 | IC50 (µM) | Fold-R | IC50 (µM) | Fold-R | IC50 (µM) | Fold-R | IC50 (µM) | Fold-R | |
| WT | 8.7±3.0 | - | 0.16±0.03 | - | 0.13±0.04 | - | 2.5±0.2 | 0.26±0.08 | ||
| Y181C | 12.1±1.6 | 1.4 | 0.47±0.05 | 2.9 | 0.55±0.02 | 4.2 | 2.1±0.3 | 0.8 | 0.27±0.03 | 1.0 |
| K103N | 9.4±0.9 | 1.1 | 0.34±0.04 | 2.1 | 0.24±0.12 | 1.8 | 2.4±0.2 | 1.0 | 0.83±0.23 | 3.2 |
| G190A | 7.1±1.9 | 0.8 | 0.21±0.02 | 1.3 | 0.23±0.02 | 1.8 | 2.4±0.4 | 1.0 | 0.31±0.04 | 1.2 |
| K101E | 8.1±1.7 | 0.9 | 0.25±0.06 | 1.6 | 0.28±0.02 | 2.2 | 2.2±0.3 | 0.9 | 0.09±0.03 | 0.3 |
| Y188C | 5.5±2.4 | 0.6 | 0.32±0.01 | 2.0 | 0.59±0.04 | 4.5 | 5.7±1.2 | 2.3 | 1.10±0.32 | 4.2 |
| P236L | 8.9±2.5 | 1.0 | 0.22±0.01 | 1.4 | 0.21±0.01 | 1.6 | 2.2±0.2 | 0.9 | 0.31±0.04 | 1.2 |
| Inhibition of RT DNA polymerase activity | ||||||||||
| NVP | 1 | 2 | 3 | 4 | ||||||
| WT | 4.96±3.19 | - | 5.97±3.10 | - | 2.64±1.85 | - | - | 1.1±0.3 | - | |
| Y181C | > 81 | > 16 | 0.61±0.06 | 0.1 | 0.71±0.10 | 0.3 | - | 0.5±0.2 | 0.45 | |
| K103N | > 81 | > 16 | 3.66±1.84 | 0.6 | 3.28±1.53 | 1.2 | - | 2.1±0.7 | 1.9 | |
| G190A | > 81 | > 16 | 1.60±0.88 | 0.2 | 1.93±0.49 | 0.7 | - | 1.7±1.0 | 1.5 | |
| K101E | > 81 | > 16 | 7.33±7.05 | 1.2 | 4.88±1.56 | 1.8 | - | 1.8±0.7 | 1.6 | |
| Y188C | > 81 | > 16 | 0.14±0.01 | 0.02 | 0.53±0.08 | 0.2 | - | 0.3±0.1 | 0.3 | |
| P236L | > 81 | > 16 | 2.22±0.18 | 0.4 | 5.13±1.68 | 1.9 | - | 1.6±0.9 | 1.5 | |
1 Fold-R; fold resistance; IC50(mutant)/IC50(WT).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



