
Submitted:
03 June 2024
Posted:
03 June 2024
You are already at the latest version
Abstract
Malignant melanoma (MM) is the most fatal form of skin cancer, accounting for 90% of all skin cancer deaths. Multiple dysregulated cellular pathways are involved in MM, including the MAPK/ERK and PI3K/PTEN/AKT pathways, which can be targeted with novel targeted inhibitors. However, these blockades can be overcome through the activation of alternative pathways, leading to treatment resistance. The high immunogenicity of MM has been exploited using checkpoint inhibitors resulting in improved survival outcomes of patients with advanced MM. However, their side effect profile and prohibitive cost remain a challenge and the survival outcomes for patients with metastatic MM remain poor. Treatment resistance has been attributed to the presence of cancer stem cells (CSCs), a small subpopulation of pluripotent, highly tumorigenic cells proposed to drive cancer progression, metastasis, and treatment resistance. CSCs reside within the MM tumor microenvironment (TME) regulated by the immune system, and the paracrine renin-angiotensin system (RAS), expressed in many cancer types, including MM. This article reviews the current treatment of advanced MM with targeted therapy and immune checkpoint blockers, highlights the emergence of mRNA vaccine. It also discusses the MM TME and its relationship with CSCs, and the local immune system and the paracrine RAS. It highlights the regulation of the MAPK/ERK and PI3K/AKT/mTOR pathways by the RAS via prorenin receptor and angiotensin II receptor 1, and how this relates to the CSCs and treatment resistance. This underscores the potential multi-modal therapeutic strategy by concurrent targeting of multiple components of the TME.
Keywords:
melanoma
; cancer vaccine
; mRNA vaccine
; renin-angiotensin system
; drug repurposing
; immunotherapy
; embryonic stem cells
; cancer stem cells
1. Introduction
Malignant melanoma (MM) is the most aggressive form of skin cancer accounting for 90% of all deaths from skin cancer [1]. Its global incidence has been increasing in recent decades, particularly among fair-skinned populations and those 60 years and older, with a projected ongoing rise [2]. MM is classified histopathologically into superficial spreading melanoma, lentigo maligna melanoma, nodular melanoma, and acral lentiginous melanoma subtypes. The etiopathogenesis of MM involves environmental, genetic, and epigenetic factors. MM originates from melanocytes, pigment-producing cells of neuroectodermal origin found in the basal layer of the epidermis of the skin, aerodigestive and anogenital tracts, eye, leptomeninges, and inner ear [1]. The incidence of new MM cases varies between countries, with rates of 50-60 per 100,000 in Australia, 35 per 100,000 in New Zealand, 20-30 per 100,000 in the USA, and 10-25 per 100,000 in Europe [3,4]. These geographic differences are attributed to the exposure to ultraviolet (UV) radiation – a significant modifiable risk factor influenced by latitude, altitude, and ozone depletion [3]. Most MMs are sporadic, with hereditary MM accounting for around 10% of cases. Mutations in high-risk genes such as cyclin-dependent kinase 2A (CDKN2A), POT1, TERT, and CDK4 are associated with hereditary MM. Moderate-risk genes involved in melanin synthesis include MC1R and MITF [5]. Other important heritable risk factors are melanocortin-1 receptor variants, large congenital nevi, increased number of common nevi, and multiple atypical nevi [6,7].
MM exhibits high metastatic potential and mortality rates. Recent advances in the treatment of metastatic MM include targeted therapy, and immunotherapy with checkpoint inhibitors. However, the emergence of drug resistance, severe adverse effects, and high costs necessitate exploration of more effective, better tolerated, and affordable treatment options. Poor outcomes of MM have been attributed to the presence of cancer stem cells (CSCs) found in both primary [8] and metastatic MM [9,10]. CSCs possess pluripotent and self-renewal capacities, generating identical CSCs and cancer cells by asymmetric cell division, giving rise to heterogenous tumor cell populations as proposed by the hierarchical model of cancer [11] (Figure 1). CSCs contribute to metastasis and resistance to chemotherapy, radiotherapy [12] and immunotherapy [13], leading to treatment failure [11]. This is in contrast to the stochastic model of cancer (also known as the clonal evolutional theory of cancer) which proposes that cancer arises from the stepwise accumulation of genetic and epigenetic changes that confer a heritable survival advantage. In this model cancer cells with advantageous survival traits undergo clonal expansion to form a tumor [14] (Figure 1).
MM involves multiple dysregulated cellular signaling pathways. Mutations in BRAF, NRAS, NF1, and KIT drive the overactivation of the MAPK/ERK and PI3K/PTEN/AKT pathways, fueling MM progression [16]. Targeting these pathways with agents such as dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) improve overall and progression-free survival in MM patients carrying BRAF V600E or V600K mutations [17,18]. However, drug resistance emerges in most patients within several months due to activation of alternative pathways involved in cellular proliferation. This inevitable development of drug resistance highlights the need for an effective multi-modal strategy [19] and has prompted the development of immunotherapy agents and small molecule inhibitors [20]. However, these treatments are associated with significant side effects, and their high-cost limits accessibility [21].
MM is an immunogenic tumor characterized by high tumor mutational burden and its association with UV exposure and vitiligo [22]. Additionally, MM exhibits brisk lymphocytic infiltration [23], with increased peritumoral lymphocyte infiltration being linked to a reduced risk of death [24]. This high immunogenicity has been exploited in the development of vaccines, albeit hitherto with limited outcomes. Vaccine trials for MM in the 1970s utilizing tumor lysate injections and pathogen adjuvants displayed promising but often short-lived responses [25]. Allogenic melanoma cells studies emerged in the 1980s, followed by attempts with ganglioside-based vaccines a decade later [26]. The oncolytic herpesvirus-based vaccine (T-VEC) demonstrates a response rate of 26% and a complete clinical response in 11% of patients with stage IIIB-IV melanoma [27]. Clinical trials involving cytokines such as interleukin-2 and interferon-α, along with genetically modified tumor cell vaccines secreting cytokines such as granulocyte-macrophage colony stimulating factor [28,29]. Until the recent development of mRNA vaccine for MM, these therapeutic approaches have largely been abandoned because of the lack of efficacy.
It has been over a decade since checkpoint inhibitors were introduced in the treatment of MM. The cytotoxic T-lymphocyte-associated protein (CTLA-4) blocking antibody, ipilimumab, approved by FDA in 2011, demonstrates prolonged survival in patients with metastatic MM [30]. Another crucial T cell immune checkpoint, programmed death 1 (PD-1), subsequently emerged as a viable treatment target. Phase III clinical trials show that PD-1 inhibitors, nivolumab and pembrolizumab, extend survival compared to ipilimumab and chemotherapy [31,32]. The EORTC1225-KEYNOTE-054 trial demonstrates pembrolizumab increases recurrence-free survival and distant metastasis-free survival of patients with resected stage III MM at a 3.5-year median follow-up, compared with placebo [33,34]. Additionally, pembrolizumab significantly reduces the risk of disease recurrence or death in patients with completely resected stage IIB or IIC MM [35].
The renin-angiotensin system (RAS) is a key regulatory pathway involved in regulation of CSCs through its impact on the tumor microenvironment (TME) [36,37]. Classically known for its crucial role in cardiovascular homeostasis, the paracrine function of the RAS in regulating CSCs within the TME is increasingly appreciated [15]. The TME consists of various components including blood vessels, extracellular matrix, signaling molecules, soluble molecules, inflammatory and immune cells, fibroblasts, keratinocytes, smooth muscle cells and endothelial cells [21,38]. The dynamic interplay between CSCs and the TME is influenced by the immune system and the RAS [36,38] with phenotypic CSCs in metastatic MM expressing components of the RAS [10,39]. This article discusses the role of CSCs and the RAS in the TME of MM, and underscores areas of future investigation.
Spurred by the advancements catalyzed by the COVID-19 pandemic, recent advances in mRNA technology, particularly mRNA vaccination encoding neoantigens for MM, have shown promise [40]. This article also explores initial findings involving mRNA-4157/V940 combined with pembrolizumab and discusses the potential enhancement of observed responses by targeting the RAS to augment local immune factors in the TME.
2. The Role of the Renin-Angiotensin System in Malignant Melanoma
The systemic RAS is a crucial endocrine system for maintaining cardiovascular homeostasis by controlling blood pressure, plasma volume, and electrolyte balance. Components of the RAS are locally expressed in many tissue sites and tumors, known as the paracrine or local RAS. The paracrine RAS plays key pathophysiological roles and is implicated in the cancer [41] by possessing a regulatory role in cellular proliferation, carcinogenesis, angiogenesis, and metastasis [42]. It is also involved in hematopoiesis in bone marrow [43]. Dysregulation of the RAS is common in various cancer types, including MM. Experimental models across different cancer types have shown that inhibition of the RAS reduces tumor growth, angiogenesis, and metastasis [44]. Despite its dysregulation in numerous cancer types, the role of the paracrine RAS in MM remains to be fully investigated [44].
Recent research reveals the complexity of the RAS and its downstream interactions (Figure 2). The classical view of the RAS is that of a cascade which begins with angiotensinogen (AGT), synthesized and secreted by the liver. When arterial pressure or plasma sodium levels decrease, the kidney releases renin. AGT is cleaved by renin to form angiotensin I (ATI) which is converted to angiotensin II (ATII) by angiotensin converting enzyme (ACE), also known as ACE1. ATI is also cleaved by angiotensin converting enzyme 2 (ACE2) to form angiotensin 1-7 (Ang1-7). ATII can be further converted into angiotensin III (ATIII) and angiotensin IV (ATIV) by aminopeptidases A and N. These angiotensin peptides can also be generated through alternative pathways involving enzymes such as cathepsins B, D and G, chymostatin-sensitive angiotensin II-generating enzyme, and chymase. Upon binding to their respective receptors, these angiotensin peptides trigger cellular signaling pathways, eliciting diverse physiological responses. Notably, ATII binds to ATII receptor 1 (AT1R) and ATII receptor 2 (AT2R), causing vascular smooth muscle contraction and stimulating aldosterone release, thus increasing blood pressure [44].
Apart from its role on cardiovascular function, signal transduction of ATII in the paracrine RAS is involved in a wide array of signaling pathways and multiple pathophysiological functions. These include inflammation, end-organ damage associated with aging, and metabolic dysfunction. It is also implicated in cancer, and heart, vascular, brain and kidney disease [45]. AT1R activation is involved in inflammation, as reactive oxygen species (ROS) produced via NADPH oxidase activation acts as a second messenger of AT1R, thereby imbuing ATII to function as an inflammatory cytokine [46]. Moreover, AT1R activation is pro-fibrotic, and induces hypertrophy and vasoconstriction [45]. Further, activation of pro-renin receptor (PRR), a key component of the RAS, also causes fibrosis and renal dysfunction [45]. The effects of AT2R activation are antagonistic to that of AT1R, which causes vasodilation, and anti-proliferative, anti-fibrotic, and anti-inflammatory effects [45]. MrgD and AT2R also cause vasodilatation, with the later causing sodium excretion [45]. The wide-ranging impacts of the paracrine RAS via ATII signaling highlights the potential of its inhibition in the treatment of MM.
We have observed expression of components of the RAS in metastatic head and neck MM to the regional lymph nodes, and MM primary-derived cells. The MM tissues express PRR, ACE, and AT2R with ACE localizing to the endothelium of the microvessels. Within metastatic head and neck MM to regional lymph nodes, SOX2+ and OCT4+ cells are found to express PRR and AT2R, demonstrating these phenotypic CSCs express components of the RAS [39]. Expression of components of the RAS by phenotypic CSCs has also been demonstrated in metastatic MM to the brain [10]. Further work is warranted to determine the regulatory impact the local RAS signaling on these CSCs, the proposed driver of MM and treatment resistance.
2.1. The Role of the Renin-Angiotensin System in Malignant Melanoma Tumor Microenvironment
The MM TME is complex and dynamic, comprising multiple interacting components through various signaling pathways. Of particular interest in this TME, is the paracrine RAS. PRR, a key component of the RAS, is known to interact with several overactive signaling pathways in MM, such as the MAPK/ERK pathway involving BRAF and the PI3K/AKT/mTOR pathways [47].
The observed heightened CpG island methylation of AGTR1, the gene encoding AT1R, and its reduced expression in metastatic MM compared to primary MM, suggests its role as a tumor suppressor in MM [48]. Further supporting a possible role of the RAS in MM is that antagonizing AT1R with an angiotensin receptor blocker (ARB), or shRNA-mediated knockdown in MM cell lines, facilitates melanoma cell proliferation in a serum-free environment [48]. Interestingly, an ATII-independent mechanism for AT1R exists, wherein ectopic expression of AGTR1 induces cellular proliferation in the absence of ATII ligand. This study also demonstrates that RAS inhibitors potentiate the action of BRAF and MEK inhibitors in BRAF V600 mutated melanoma cells. Treating melanoma cells with AT2R antagonists EMA401 and PD123319 potentiates the action of BRAF and MEK inhibitors while inhibiting angiogenesis and melanoma growth [48]. Moreover, treatment of melanoma cells with an AT2R agonist induces cellular proliferation under serum-free conditions, while its blockade inhibits angiogenesis [48].
2.2. The Renin-Angiotensin System and Cancer Metastasis
There is increasing evidence of the role of the RAS in cancer metastasis. A study by Martínez-Meza et al [49] investigating the role of AT2R in MM metastasis demonstrates that AT2R activation leads to inhibition of transendothelial migration, cellular migration, and metastasis of B16F10 murine melanoma cells. This finding is corroborated by the complete abrogation of these effects upon silencing AT2R using shRNA. Activation of AT2R notably reduces lung metastasis of murine melanoma cells in C57BL/6 mice, attributable to increased non-receptor protein tyrosine phosphatase 1B activity [49].
Upon injection of B16/F10 mouse melanoma cells into C57BL/6 mice, Ishikane et al, [50] observe a greater number of lung metastases in the ATII-treated group compared to the vehicle-treated group. Additionally, the effect of ATII is inhibited by the ARB valsartan. Notably, in AGTR1α knockout mice, melanoma lung metastases mediated by ATII are significantly reduced. Furthermore, ATII upregulates mRNA expression of E-selectin, thereby promoting adherence of melanoma cells to the vascular endothelium [50].
There is a paucity of investigations into the utilization of RAS inhibitors for treatment of metastatic MM. Treatment with RAS inhibitors may not only result from the downstream effects of AT1R and AT2R signaling but also from interactions with the overactive MAPK/ERK and PI3K/AKT/mTOR pathways in MM. The observation that AT2R antagonists potentiate the effects of BRAF and MEK inhibitors highlights the potential benefits of combing RAS modulators with targeted inhibitors in the treatment of MM. Further research is needed to investigate the effects of inhibiting or modulating these pathways with current targeted inhibitors and examine the effect of RAS inhibition on patient outcomes. Further research is warranted to explore the relationship between the paracrine RAS and the CSCs within the TME.
3. Immunotherapy and Cancer Vaccines for Metastatic Malignant Melanoma
3.1. Immunotherapy
Targeting the immune system in the management of advanced MM has achieved significant progress, as evidenced by the approximately 50% 5-year survival rate in patients treated with combination immunotherapy [51]. Specifically, patients with stage III or stage IV MM receiving a combination of nivolumab and ipilimumab have a 52% 5-year overall survival [52]. However, these treatments are associated with significant side effects.
The effectiveness of immunotherapy in MM is partly based on the tumor-induced immunosuppression seen in patients with metastatic MM. This immunosuppression is driven by persistent antigenic stimulation of the immune system and the elevated expression of inhibitory receptors on tumor antigen-specific T cells [51]. However, development of treatment resistance, recurrence, and the adverse effects of immunotherapy in a large proportion of patients underscore the need for better therapies.
Treatment with anti-CTLA4 monoclonal antibody ipilimumab that targets CTLA-4 significantly improves overall survival of patients with inoperable metastatic MM [30]. Additionally, monoclonal antibodies against PD-1 and its ligand PD-L1 have been developed. PD-1, an antigen present on APCs and MM cells, acts as a checkpoint inhibitor, preventing T-cell activation. Targeting these checkpoints with monoclonal antibodies allows T cells to remain active, causing anti-tumor cytotoxic effects [51]. Administration of nivolumab and pembrolizumab leads to increased survival when compared to chemotherapy or ipilimumab [31,32]. A combination of nivolumab and ipilimumab is currently considered the standard treatment for patients with inoperable advanced MM.
A recent phase II clinical trial demonstrates the use of pembrolizumab as a neoadjuvant treatment in patients with advanced MM prolongs event-free survival at 2 years (72%, 95% CI, 64 to 80), compared to those receiving adjuvant pembrolizumab alone (49%, 95% CI, 41 to 59) [53]. The cause of treatment failure in individual or combination immunotherapy treatment remains unclear. It has been attributed to the expression of alternative immune checkpoints, including lymphocyte-activation gene 3 (LAG-3), found on the surface of CD4+ T cells. A recent phase II/III clinical trial (RELATIVITY-047) demonstrates that targeting LAG-3 and PD-1 together with relatimab and nivolumab, respectively, provides greater survival benefit compared to targeting PD-1 alone [54]. Notwithstanding the success seen with immunotherapy, significant adverse effects are observed in both mono- and combination therapy. In the RELATIVITY-047 trial, grades three or four treatment-related adverse effects are observed in 18.9% of patients in the combination treatment group, compared to 9.7% in the those receiving nivolumab alone [54].
It is interesting to hypothesize whether targeting other components of the TME, such as the paracrine RAS which inhibits tumor immunity through its effect on myeloid cells and fibroblasts [41], could enhance the effects of immunotherapy. Nakamura et al demonstrate that inhibition of the RAS with ARBs increases the efficacy of anti-PD-1 immunotherapy in a MM mouse model [55]. This effect is achieved as ARB administration decreases RAS-stimulated fibroblast CC motif chemokine ligand 5 (CCL5) production in melanoma-transplanted mice. Subsequently, co-administration of anti-PD1 antibodies and an ARB is associated with greater inhibition of growth compared to monotherapy [55]. The OpACIN trial demonstrates significant survival advantages in stage III MM patients who received neoadjuvant treatment compared to adjuvant treatment alone, using ipilimumab and nivolumab. The 5-year overall survival was 90% (95% CI 73 to >99%) in the neoadjuvant arm, and 70% (95% CI 47% to >99%) in the adjuvant-only arm [56].
Cancer stemness refers to the extent to which a cancer cell exhibits properties of stem cells, including the ability to undergo self-renewal, multilineage differentiation potential, and proliferative ability. A recent study investigating the association between cancer stemness and scRNA-Seq datasets from patients treated with checkpoint inhibitors, shows that cancer stemness is significantly associated with resistance to checkpoint inhibitors in patients with MM. Using a large publicly available dataset, a signature Stem.Sig has been developed to predict which patients will benefit from immunotherapy [57]. This study demonstrated that cells with cancer stemness are less likely to respond to checkpoint inhibitors. This finding supports the notion that immunotherapy resistance in MM is attributable to resident CSC populations. Therefore, specifically targeting these cells may lead to improved survival outcomes. Further, selective targeting of CSCs using personalized neoantigens only found in those CSCs may spare other normal somatic and stem cells and thus reducing treatment side effects.
3.2. Cancer Vaccines
mRNA-4157 is an mRNA vaccine encoding 34 personalized tumor neoantigens. Results of the mRNA-4157-P201KEYNOTE-942 phase II open-labeled randomized clinical trial presented at the 2023 ASCO Annual Meeting II, shows clinically significant improvement in recurrence-free survival in patients with resected high-risk stage IIIB/C/D and stage IV MM, compared to pembrolizumab monotherapy. This combination treatment approach reduces the risk of recurrence or death by 44% (HR = 0.561; 95% CI 0.30 to 1.017) [58]. It is interesting to speculate whether co-administration of RAS inhibitors would further impact this result given their suppressive effect on the TME immune system, and possible regulatory role for resident CSCs.
CSCs resist apoptosis and toxins, potentially accumulating a higher mutation burden compared to other melanoma cells [59]. It is interesting to speculate whether mRNA vaccines based on personalized neoantigens derived from CSCs could improve survival outcomes.
4. Concurrent Targeting of the Renin-Angiotensin System and Other Dysregulated Pathways
Primary [60] and metastatic MM to the head and neck exhibits increased expression of PRR, a key component of the RAS [39]. PRR induces activation of the MAPK/ERK pathway upon binding to its ligands, renin and (pro)renin. In certain cell types, PRR-induced activation of MAPK/ERK signaling up-regulates extracellular signal-regulated kinase 1/2 (ERK 1/2), leading to the increased production of transforming growth factor-β and cellular proliferation, thereby promoting cancer progression and metastasis [60,61]. Additionally, through an ATII-independent mechanism, PRR induces the formation of ROS, which further increases PI3K/AKT/mTOR and MAPK/ERK signaling [60,62]. Further research is needed to determine whether PRR expression in MM contributes to over-activation of MAPK/ERK and PI3K/AKT/mTOR pathways through the production of ROS. In health these signaling pathways are integral to multiple processes of cellular function, including cell growth, cell survival, and nutrient intake [63]. Targeting mTOR signaling in MM to overcome the inevitable acquired resistance to BRAF and MEK inhibition has been investigated. The mTOR inhibitor rapamycin (sirolimus) and NVP-BEZ235 induce apoptosis and cell cycle arrest in BRAF mutant melanoma cell lines [64]. Metformin, the widely used anti-diabetic medication, targets AMPK/mTOR signaling, reducing cellular proliferation, and displaying anti-tumor properties across multiple cancer types [65]. In-vivo, metformin inhibits melanoma tumor development, with three of seven metformin-treated mice not demonstrating measurable tumors [65]. PI3K, mTOR and AKT have been shown to be overactive across a range of cancer types. These components are fundamental in embryogenesis with their absence often resulting in failure in the development of an embryo [66,67,68]. Multiple studies have demonstrated that PI3K/AKT/mTOR signaling preserves the ability for pluripotent stem cells to self-renew and differentiate. Human embryonic stem cells require PI3K/AKT signaling to maintain an undifferentiated state, whilst inhibition of PI3K leads to the loss of pluripotency [63]. Given the presence of CSCs within MM, it is interesting to consider what effects inhibition of PI3K/AKT/mTOR pathways may have on the differentiation and behavior of resident CSCs [39]. This is relevant as PRR is expressed on the phenotypic CSCs expressing SOX2, a CSC marker, in metastatic MM [39].
Figure 3.
A schema showing how the MAPK/ERK and PI3K/AKT/mTOR pathways can be inhibited in malignant melanoma. BRAF can be targeted by BRAF inhibitors, and MEK by MEK inhibitors. Pro-renin receptor (PRR) and angiotensin II receptor 1 which activates Ras, can be inhibited through inhibition of the renin-angiotensin system (RAS). PRR also increases the production of reactive oxygen species (ROS), which contributes to overactivation of the MAPK/ERK and PI3K/AKT/mTOR pathways. mTORC1 signaling can be inhibited by rapamycin (sirolimus) and metformin. Figure created using Biorender.
Figure 3.
A schema showing how the MAPK/ERK and PI3K/AKT/mTOR pathways can be inhibited in malignant melanoma. BRAF can be targeted by BRAF inhibitors, and MEK by MEK inhibitors. Pro-renin receptor (PRR) and angiotensin II receptor 1 which activates Ras, can be inhibited through inhibition of the renin-angiotensin system (RAS). PRR also increases the production of reactive oxygen species (ROS), which contributes to overactivation of the MAPK/ERK and PI3K/AKT/mTOR pathways. mTORC1 signaling can be inhibited by rapamycin (sirolimus) and metformin. Figure created using Biorender.
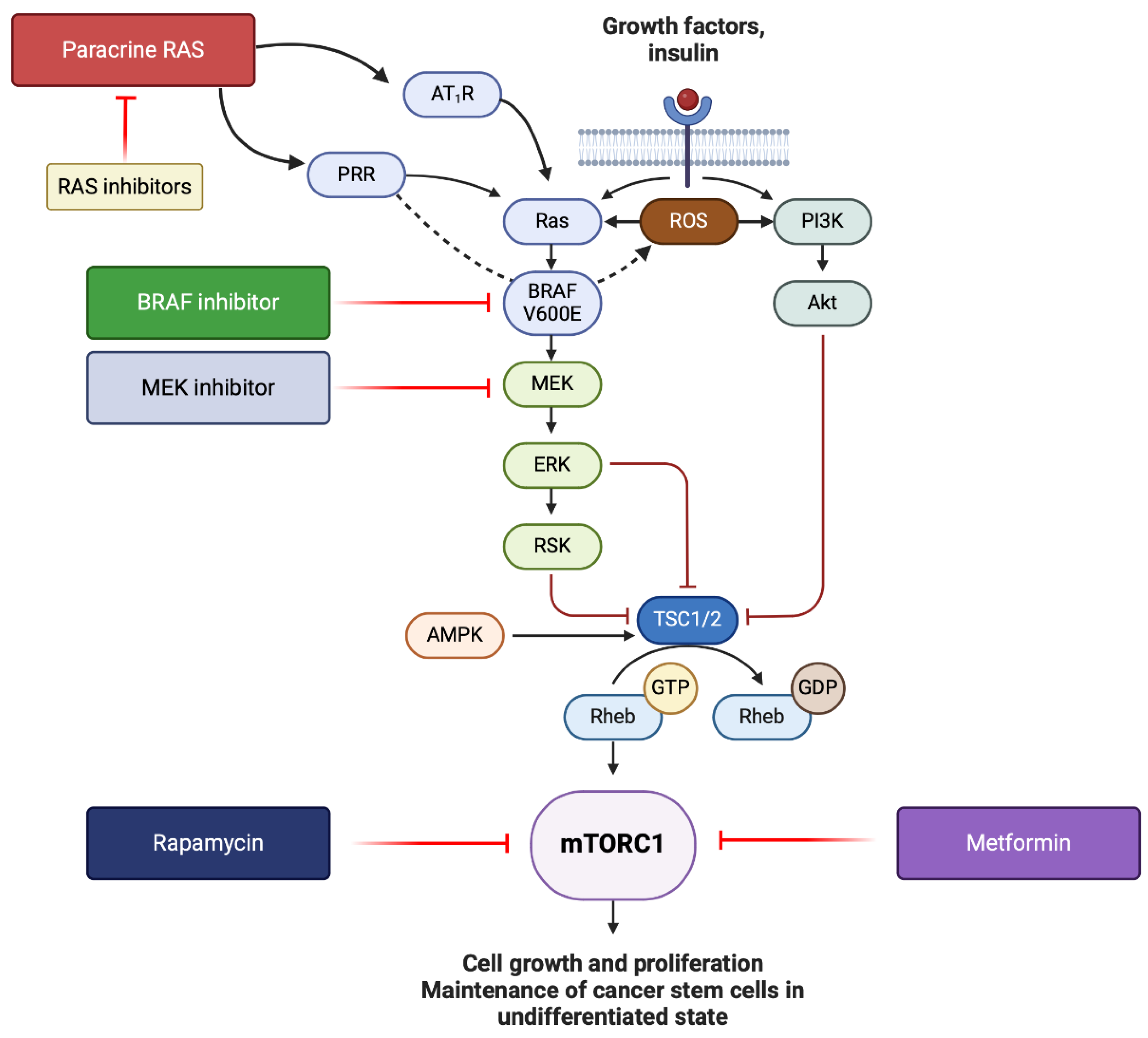
These findings suggest that PRR may contribute to carcinogenesis through its interaction with the MAPK/ERK and PI3K/AKT/mTOR pathways, leading to aberrant cellular proliferation and maintenance of cells in the undifferentiated state. PRR is also implicated in cancer progression through its involvement in other signaling pathways, namely, the vacuolar H+ ATPase and Wnt/β-catenin signaling pathways [60]. Given the overactivity of these pathways in MM, it is interesting to speculate whether PRR contributes to the progression of MM, considering its expression alongside other components of the RAS. Furthermore, ATII induces phosphorylation of PI3K/AKT, resulting in increased NF-κB activity, which promotes cancer progression, metastasis, and chemoradiotherapy resistance [60,69]. Considering the somewhat successful targeting of these overactive pathways in MM, we hypothesize that inhibition of the RAS as a whole or directly targeting PRR, in combination with a targeted therapy such as a BRAF inhibitor, would further suppress the MAPK/ERK and PI3K/AKT/mTOR pathways. Concurrent administration of personalized mRNA vaccines against CSC neoantigens may confer further survival advantage.
Current treatment of advanced MM usually consists of single agents. Further research into a multimodal targeting is warranted, given the numerous inter-related treatment targets. A combined approach may lead to a more durable treatment outcomes for advanced MM.
5. Conclusions
MM is the most fatal form of skin cancer. Future treatment of MM. Historically, vaccine approaches to MM have shown limited efficacy. Recent development of a melanoma mRNA vaccine shows promise. Advances in targeted therapies targeting the MAPK/ERK and PI3K/AKT/mTOR pathways, and immunotherapy, specifically checkpoint inhibitors, have led to improved survival outcomes for Stage III and stage IV disease. However, this is associated with a high incidence of side effects and treatment resistance, and the treatment costs remain prohibitive. Treatment resistance of MM has been attributed to the presence of CSCs, a small subpopulation of pluripotent, highly tumorigenic cells proposed to drive cancer progression, metastasis, and treatment resistance. These CSCs reside within the TME regulated by the immune system, and the paracrine RAS. This article explores regulation of the overactive MAPK/ERK and PI3K/AKT/mTOR pathways in MM, by the RAS via PRR and AT1R, and how this relates to CSCs and the TME. Future treatment strategies for MM will likely continue to exploit its high immunogenicity and involve multi-modal approach that targets CSCs and the TME. The use of combination therapies that inhibit the MAPK/ERK and PI3K/AKT/mTOR pathways using repurposed drugs that inhibits the RAS, targeted therapies, immunotherapy, and personalized mRNA vaccination, may improve survival outcomes of MM patients.
6. Patents
STT is an inventor of the patents Cancer Diagnosis and Therapy (PCT/NZ2015/050108, AUS/2012302419, JAP/2017528398, and US/0281472), Cancer Therapeutic (PCT/NZ2018/050006), and Novel Pharmaceutical Compositions for Cancer Therapy (PCT/NZ2019/050087).
Author Contributions
EJK drafted the manuscript and STT critically reviewed and commented on the manuscript. The authors approved the final manuscript.
Funding
None.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
ST is an inventor of the patents Cancer Diagnosis and Therapy (PCT/NZ2015/050108, AUS/2012302419, JAP/2017528398, and US/0281472), Cancer Therapeutic (PCT/NZ2018/050006), and Novel Pharmaceutical Compositions for Cancer Therapy (PCT/NZ2019/050087). The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; et al. European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 1: Diagnostics: Update 2022. Eur J Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J Invest Dermatol 2016, 136, 1161–1171. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Review: Ultraviolet Radiation and Skin Cancer. Int J Dermatol 2010, 49, 978–986. [Google Scholar] [CrossRef]
- https://www.tewhatuora.govt.nz/our-health-system/data-and-statistics/historical-cancer (Accessed on 25 June 2023) Historical Cancer Data.
- Serman, N.; Vranic, S.; Glibo, M.; Serman, L.; Bukvic Mokos, Z. Genetic Risk Factors in Melanoma Etiopathogenesis and the Role of Genetic Counseling: A Concise Review. Bosn J Basic Med Sci 2022, 22, 673–682. [Google Scholar] [CrossRef]
- Bauer, J.; Garbe, C. Acquired Melanocytic Nevi as Risk Factor for Melanoma Development. A Comprehensive Review of Epidemiological Data. Pigment Cell Res 2003, 16, 297–306. [Google Scholar] [CrossRef]
- Grob, J.J.; Gouvernet, J.; Aymar, D.; Mostaque, A.; Romano, M.H.; Collet, A.M.; Noe, M.C.; Diconstanzo, M.P.; Bonerandi, J.J. Count of Benign Melanocytic Nevi as a Major Indicator of Risk for Nonfamilial Nodular and Superficial Spreading Melanoma. Cancer 1990, 66, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Yoganandarajah, V.; Patel, J.; Schaijik, B. van; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Identification of Cancer Stem Cell Subpopulations in Head and Neck Metastatic Malignant Melanoma. Cells 2020, Vol. 9, Page 324 2020, 9, 324. [Google Scholar] [CrossRef]
- Sangster, A.B.; Chang-Mcdonald, B.; Patel, J.; Bockett, N.; Paterson, E.; Davis, P.F.; Tan, S.T. Expression of Cathepsins B and D by Cancer Stem Cells in Head and Neck Metastatic Malignant Melanoma. Melanoma Res 2021, 31, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Wickremesekera, A.C.; Brasch, H.D.; Lee, V.M.; Davis, P.F.; Woon, K.; Johnson, R.; Tan, S.T.; Itinteang, T. Expression of Cancer Stem Cell Markers in Metastatic Melanoma to the Brain. J Clin Neurosci 2019, 60, 112–116. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat Med 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Eyler, C.E.; Rich, J.N. Survival of the Fittest: Cancer Stem Cells in Therapeutic Resistance and Angiogenesis. J Clin Oncol 2008, 26, 2839–2845. [Google Scholar] [CrossRef]
- Gourab, G.; George, M.; Shakthika, S.; Hexin, C. Cancer Resistance to Immunotherapy: What Is the Role of Cancer Stem Cells? Cancer Drug Resist 2022, 5, 981–994. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Kilmister, E.J.; Koh, S.P.; Weth, F.R.; Gray, C.; Tan, S.T. Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment. Biomedicines 2022, 10, 2988. [Google Scholar] [CrossRef]
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int J Mol Sci 2021, 22, 6395. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Eng J Med 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N Eng J Med 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Ombra, M.N.; Colombino, M.; Casula, M.; Sini, M.C.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front Oncol 2015, 5, 151142. [Google Scholar] [CrossRef]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers (Basel) 2020, 12, 482. [Google Scholar] [CrossRef]
- Zhou, S.; Lu, J.; Liu, S.; Shao, J.; Liu, Z.; Li, J.; Xiao, W. Role of the Tumor Microenvironment in Malignant Melanoma Organoids during the Development and Metastasis of Tumors. Front Cell Dev Biol 2023, 11, 1166916. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A Decade of Checkpoint Blockade Immunotherapy in Melanoma: Understanding the Molecular Basis for Immune Sensitivity and Resistance. Nat Immunol 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.H.; From, L.; Bernardino, E.; Mihm, M. The Histogenesis and Biologic Behavior of Primary Human Malignant Melanomas of the Skin. Cancer Res 1969, 29, 705–727. [Google Scholar] [PubMed]
- Thomas, N.E.; Busam, K.J.; From, L.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; Gallagher, R.P.; Zanetti, R.; Rosso, S.; et al. Tumor-Infiltrating Lymphocyte Grade in Primary Melanomas Is Independently Associated with Melanoma-Specific Survival in the Population-Based Genes, Environment and Melanoma Study. J Clin Oncol 2013, 31, 4252–4259. [Google Scholar] [CrossRef] [PubMed]
- Maurer, D.M.; Butterfield, L.H.; Vujanovic, L. Melanoma Vaccines: Clinical Status and Immune Endpoints. Melanoma Res 2019, 29, 109–118. [Google Scholar] [CrossRef]
- Livingston, P.O.; Adluri, S.; Helling, F.; Yao, T.J.; Kensil, C.R.; Newman, M.J.; Marciani, D. Phase 1 Trial of Immunological Adjuvant QS-21 with a GM2 Ganglioside-Keyhole Limpet Haemocyanin Conjugate Vaccine in Patients with Malignant Melanoma. Vaccine 1994, 12, 1275–1280. [Google Scholar] [CrossRef]
- Ross, M.I.; Andtbacka, R.H.I.; Puzanov, I.; Milhem, M.M.; Collichio, F.A.; Delman, K.A.; Noyes, R.D.; Zager, J.S.; Cranmer, L.D.; Spitler, L.E.; et al. Patterns of Durable Response with Intralesional Talimogene Laherparepvec (T-VEC): Results from a Phase III Trial in Patients with Stage IIIb-IV Melanoma. J Clin Oncol 2014, 32, 9026–9026. [Google Scholar] [CrossRef]
- Stingl, G.; Brŏcker, E.B.; Mertelsmann, R.; Wolff, K.; Schreiber, S.; Kămpgen, E.; Schneeberger, A.; Dummer, W.; Brennscheid, U.; Veelken, H.; et al. Phase I Study to the Immunotherapy of Metastatic Malignant Melanoma by a Cancer Vaccine Consisting of Autologous Cancer Cells Transfected with the Human IL-2 Gene. Hum Gene Ther 1996, 7, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.E.; Sondak, V.K.; Bishop, D.K.; Nickoloff, B.J.; Mulligan, R.C.; Mulé, J.J. Adoptive Immunotherapy of Cancer with Activated Lymph Node Cells Primed in Vivo with Autologous Tumor Cells Transduced with the GM-CSF Gene. Hum Gene Ther 1996, 7, 773–792. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Eng J Med 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results from an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Jacques Grob, J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Eng J Med 2015, 372, 2521–2553. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma (EORTC 1325-MG/KEYNOTE-054): Distant Metastasis-Free Survival Results from a Double-Blind, Randomised, Controlled, Phase 3 Trial. Lancet Oncol 2021, 22, 643–654. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Longer Follow-up Confirms Recurrence-Free Survival Benefit of Adjuvant Pembrolizumab in High-Risk Stage III Melanoma: Updated Results from the EORTC 1325-MG/KEYNOTE-054 Trial. J Clin Oncol 2020, 38, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; de la Cruz Merino, L.; Khattak, M.A.; Schadendorf, D.; Long, G. V; et al. Pembrolizumab versus Placebo as Adjuvant Therapy in Completely Resected Stage IIB or IIC Melanoma (KEYNOTE-716): A Randomised, Double-Blind, Phase 3 Trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Tan, S.T. The Role of the Renin–Angiotensin System in the Cancer Stem Cell Niche. J Histochem Cytochem 2021, 69, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Robalo Cordeiro, C.; Medeiros, R. Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers (Basel) 2020, 12, 1457. [Google Scholar] [CrossRef]
- Kapoor-Narula, U.; Lenka, N. Cancer Stem Cells and Tumor Heterogeneity: Deciphering the Role in Tumor Progression and Metastasis. Cytokine 2022, 157, 155968. [Google Scholar] [CrossRef]
- Siljee, S.; Pilkington, T.; Brasch, H.D.; Bockett, N.; Patel, J.; Paterson, E.; Davis, P.F.; Tan, S.T. Cancer Stem Cells in Head and Neck Metastatic Malignant Melanoma Express Components of the Renin-Angiotensin System. Life 2020, 10, 268. [Google Scholar] [CrossRef]
- Bafaloukos, D.; Gazouli, I.; Koutserimpas, C.; Samonis, G. Evolution and Progress of MRNA Vaccines in the Treatment of Melanoma: Future Prospects. Vaccines (Basel) 2023, 11, 636. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yaguchi, T.; Ohmura, G.; Kobayashi, A.; Kawamura, N.; Iwata, T.; Kiniwa, Y.; Okuyama, R.; Kawakami, Y. Involvement of Local Renin-Angiotensin System in Immunosuppression of Tumor Microenvironment. Cancer Sci 2018, 109, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Almutlaq, M.; Alamro, A.A.; Alamri, H.S.; Alghamdi, A.A.; Barhoumi, T. The Effect of Local Renin Angiotensin System in the Common Types of Cancer. Front Endocrinol (Lausanne) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Durik, M.; Sevá Pessôa, B.; Roks, A.J.M. The Renin–Angiotensin System, Bone Marrow and Progenitor Cells. Clin Sci 2012, 123, 205–223. [Google Scholar] [CrossRef]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The Renin–Angiotensin System and Cancer: Old Dog, New Tricks. Nat Rev Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol Rev 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; Martin, A.S.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 Overexpression Potentiates Angiotensin II-Induced Hypertension and Vascular Smooth Muscle Hypertrophy in Transgenic Mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Tehranian, C.; Fankhauser, L.; Harter, P.N.; Ratcliffe, C.D.H.; Zeiner, P.S.; Messmer, J.M.; Hoffmann, D.C.; Frey, K.; Westphal, D.; Ronellenfitsch, M.W.; et al. The PI3K/Akt/MTOR Pathway as a Preventive Target in Melanoma Brain Metastasis. Neuro-Oncol 2022, 24, 213–225. [Google Scholar] [CrossRef]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C. Lo; Lattanzio, L.; Merlano, M.; Vega-Rioja, A.; del Carmen Fernandez-Carranco, M.; Hajji, N.; et al. The Renin Angiotensin System (RAS) Mediates Bifunctional Growth Regulation in Melanoma and Is a Novel Target for Therapeutic Intervention. Oncogene 2019, 38, 2320–2336. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Meza, S.; Díaz, J.; Sandoval-Bórquez, A.; Valenzuela-Valderrama, M.; Díaz-Valdivia, N.; Rojas-Celis, V.; Contreras, P.; Huilcaman, R.; Ocaranza, M.P.; Chiong, M.; et al. AT2 Receptor Mediated Activation of the Tyrosine Phosphatase PTP1B Blocks Caveolin-1 Enhanced Migration, Invasion and Metastasis of Cancer Cells. Cancers (Basel) 2019, 11, 1299. [Google Scholar] [CrossRef]
- Ishikane, S.; Hosoda, H.; Nojiri, T.; Tokudome, T.; Mizutani, T.; Miura, K.; Akitake, Y.; Kimura, T.; Imamichi, Y.; Kawabe, S.; et al. Angiotensin II Promotes Pulmonary Metastasis of Melanoma through the Activation of Adhesion Molecules in Vascular Endothelial Cells. Biochem Pharmacol 2018, 154, 136–147. [Google Scholar] [CrossRef]
- Frampton, A.E.; Sivakumar, S. A New Combination Immunotherapy in Advanced Melanoma. N Eng J Med 2022, 386, 91–92. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Eng J Med 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.-G.; et al. Neoadjuvant–Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N Eng J Med 2023, 388, 813–823. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Eng J Med 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Nakamura, K.; Kiniwa, Y.; Okuyama, R. CCL5 Production by Fibroblasts through a Local Renin–Angiotensin System in Malignant Melanoma Affects Tumor Immune Responses. J Cancer Res Clin Oncol 2021, 147, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.M.; Menzies, A.M.; Sikorska, K.; Rozeman, E.A.; Saw, R.P.M.; van Houdt, W.J.; Eriksson, H.; Klop, W.M.C.; Ch’ng, S.; van Thienen, J.V.; et al. Survival Update of Neoadjuvant Ipilimumab plus Nivolumab in Macroscopic Stage III Melanoma in the OpACIN and OpACIN-Neo Trials. Ann Oncol 2023, 34, 420–430. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.X.; Chen, Y.X.; Wu, H.X.; Yin, L.; Zhao, Q.; Luo, H.Y.; Zeng, Z.L.; Qiu, M.Z.; Xu, R.H. Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals a Pan-Cancer Stemness Signature Predicting Immunotherapy Response. Genome Med 2022, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Khattak, A.; Weber, J.S.; Meniawy, T.; Taylor, M.H.; Ansstas, G.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; Faries, M.B.; et al. Distant Metastasis-Free Survival Results from the Randomized, Phase 2 MRNA-4157-P201/KEYNOTE-942 Trial. J Clin Oncol 2023, 41, LBA9503–LBA9503. [Google Scholar] [CrossRef]
- Dillman, R.O.; Cornforth, A.N.; Nistor, G. Cancer Stem Cell Antigen-Based Vaccines: The Preferred Strategy for Active Specific Immunotherapy of Metastatic Melanoma? Expert Opin Biol Ther 2013, 13, 643–656. [Google Scholar] [CrossRef]
- Wang, J.; Nishiyama, A.; Matsuyama, M.; Wang, Z.; Yuan, Y. The (pro)Renin Receptor: A Novel Biomarker and Potential Therapeutic Target for Various Cancers. Cell Commun Signal 2020, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Noble, N.A.; Zhang, J.; Xu, C.; Border, W.A. Renin-Stimulated TGF-Beta1 Expression Is Regulated by a Mitogen-Activated Protein Kinase in Mesangial Cells. Kidney Int 2007, 72, 45–52. [Google Scholar] [CrossRef]
- Cardinale, J.P.; Sriramula, S.; Mariappan, N.; Agarwal, D.; Francis, J. Angiotensin II-Induced Hypertension Is Modulated by Nuclear Factor-ΚB in the Paraventricular Nucleus. Hypertension 2012, 59, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Cui, W. Proliferation, Survival and Metabolism: The Role of PI3K/AKT/MTOR Signalling in Pluripotency and Cell Fate Determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting MTOR Signaling Overcomes Acquired Resistance to Combined BRAF and MEK Inhibition in BRAF-Mutant Melanoma. Oncogene 2021 40:37 2021, 40, 5590–5599. [Google Scholar] [CrossRef]
- Tomic, T.; Botton, T.; Cerezo, M.; Robert, G.; Luciano, F.; Puissant, A.; Gounon, P.; Allegra, M.; Bertolotto, C.; Bereder, J.M.; et al. Metformin Inhibits Melanoma Development through Autophagy and Apoptosis Mechanisms. Cell Death Dis 2011, 2, e199. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. MTOR Is Essential for Growth and Proliferation in Early Mouse Embryos and Embryonic Stem Cells. Mol Cell Biol 2004, 24, 6710–6718. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Okabe, I.; Bernard, D.J.; Wynshaw-Boris, A.; Nussbaum, R.L. Proliferative Defect and Embryonic Lethality in Mice Homozygous for a Deletion in the P110α Subunit of Phosphoinositide 3-Kinase. J Biol Biochem 1999, 274, 10963–10968. [Google Scholar] [CrossRef]
- Peng, X.-D.; Xu, P.-Z.; Chen, M.-L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, Impaired Skin Development, Skeletal Muscle Atrophy, Delayed Bone Development, and Impeded Adipogenesis in Mice Lacking Akt1 and Akt2. Genes Dev 2003, 17, 1352–1365. [Google Scholar] [CrossRef]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-ΚB Addiction and Its Role in Cancer: ‘One Size Does Not Fit All. ’ Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef]
Figure 1.
(A) The stochastic model of cancer proposes that a normal somatic cell accumulates oncogenic mutations in a stepwise manner and becomes a cancer cell that undergoes clonal expansion to form a tumor. (B) The hierarchical model of cancer proposes the presence of a highly tumorigenic cancer stem cell (CSC) sitting atop the tumor cellular hierarchy and divides asymmetrically to form-non-tumorigenic cancer cells that form the bulk of the tumor, and identical CSCs that form new tumors like the original tumor. Figure modified and reproduced with permission from Biomedicines [15].
Figure 1.
(A) The stochastic model of cancer proposes that a normal somatic cell accumulates oncogenic mutations in a stepwise manner and becomes a cancer cell that undergoes clonal expansion to form a tumor. (B) The hierarchical model of cancer proposes the presence of a highly tumorigenic cancer stem cell (CSC) sitting atop the tumor cellular hierarchy and divides asymmetrically to form-non-tumorigenic cancer cells that form the bulk of the tumor, and identical CSCs that form new tumors like the original tumor. Figure modified and reproduced with permission from Biomedicines [15].
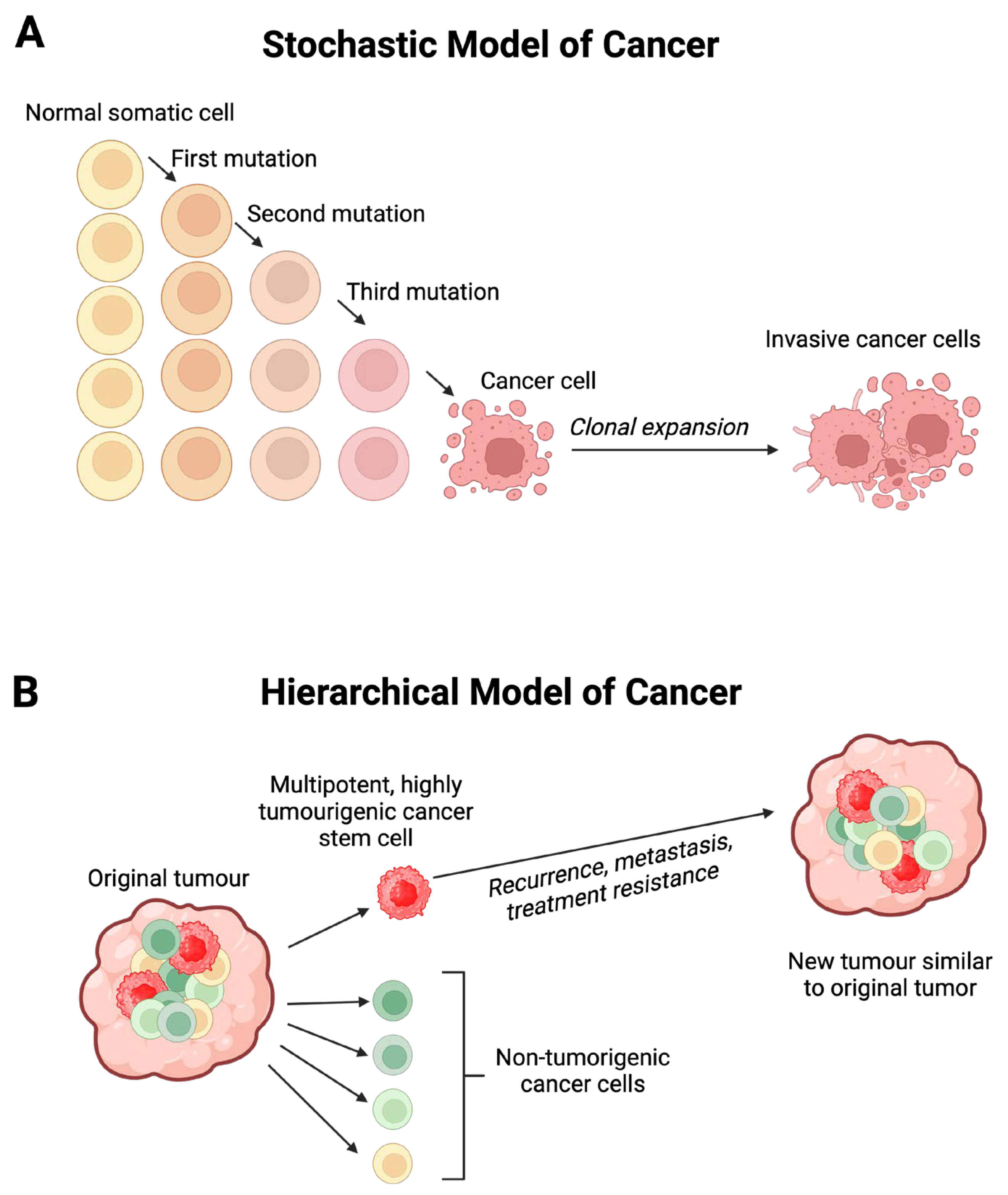
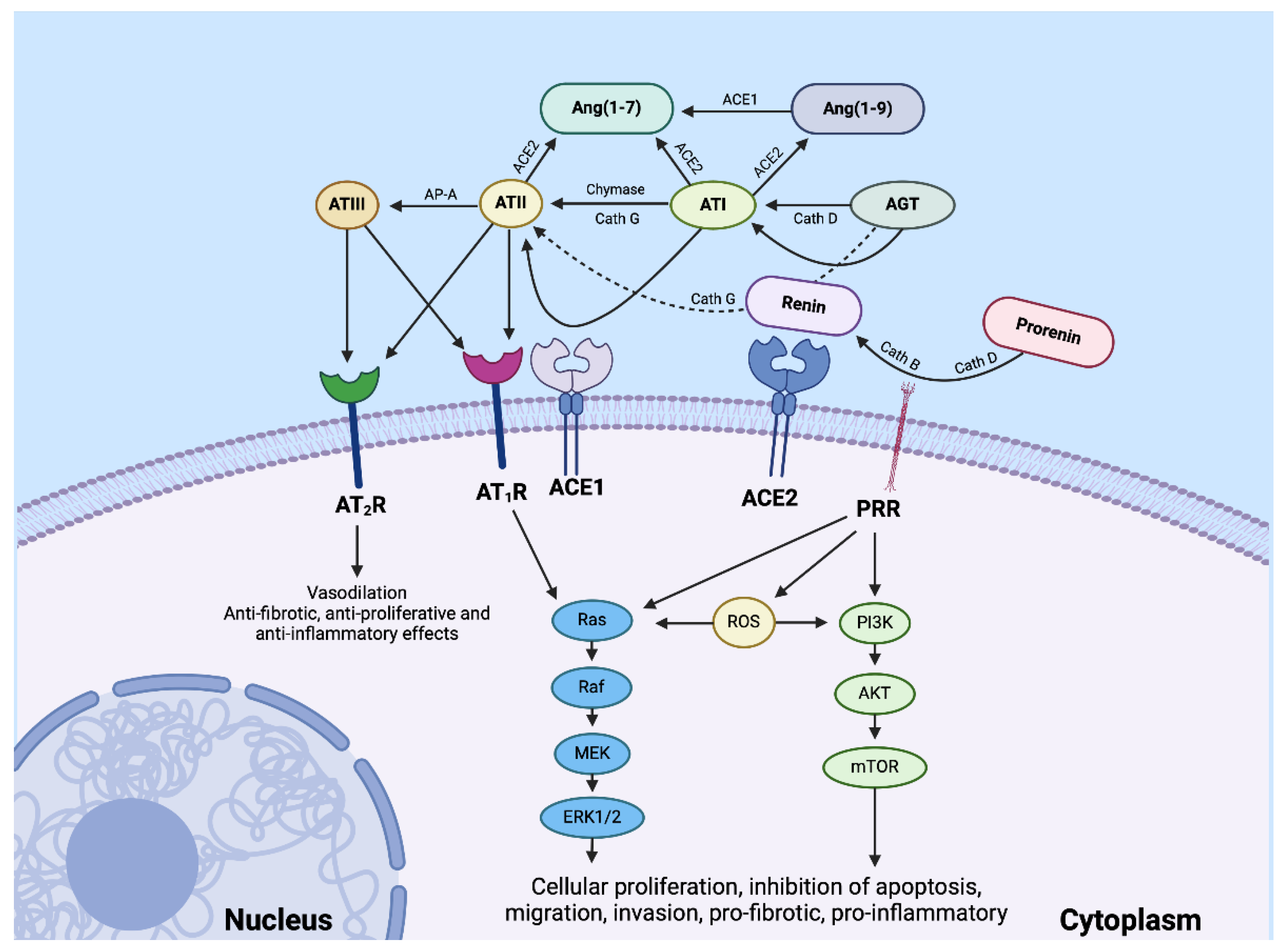
Figure 2.
A schema showing the effect of the paracrine renin–angiotensin system (RAS) and its convergent signaling pathways on the tumor microenvironment to influence cellular proliferation, invasiveness, and cell survival in cancer development. The RAS interacts with downstream pathways, such as the Ras/RAF/MEK/ERK (light blue) pathway and the PI3K/AKT/mTOR (light green) pathway, that influence cellular proliferation, migration, inhibition of apoptosis, migration, and invasion (see text). PRR, pro-renin receptor; Cath G, cathepsin G; Cath B, cathepsin B; Cath D, cathepsin D; ACE1, angiotensin-converting enzyme 1; ACE2,angiotensin-converting enzyme 2; AGT, angiotensinogen; ATP, adenosine triphosphate; Ang(1–7), angiotensin (1–7); Ang(1–9), angiotensin (1–9); AP-A, aminopeptidase-A; ATI, angiotensin I; ATII, angiotensin II; ATIII, angiotensin III; AT-eIV, angiotensin IV; AT1R, angiotensin II receptor 1; AT2R, angiotensin II receptor 2; mTOR, mammalian target of rapamycin. Reproduced and adapted with permission from Biomedicines [15].
Figure 2.
A schema showing the effect of the paracrine renin–angiotensin system (RAS) and its convergent signaling pathways on the tumor microenvironment to influence cellular proliferation, invasiveness, and cell survival in cancer development. The RAS interacts with downstream pathways, such as the Ras/RAF/MEK/ERK (light blue) pathway and the PI3K/AKT/mTOR (light green) pathway, that influence cellular proliferation, migration, inhibition of apoptosis, migration, and invasion (see text). PRR, pro-renin receptor; Cath G, cathepsin G; Cath B, cathepsin B; Cath D, cathepsin D; ACE1, angiotensin-converting enzyme 1; ACE2,angiotensin-converting enzyme 2; AGT, angiotensinogen; ATP, adenosine triphosphate; Ang(1–7), angiotensin (1–7); Ang(1–9), angiotensin (1–9); AP-A, aminopeptidase-A; ATI, angiotensin I; ATII, angiotensin II; ATIII, angiotensin III; AT-eIV, angiotensin IV; AT1R, angiotensin II receptor 1; AT2R, angiotensin II receptor 2; mTOR, mammalian target of rapamycin. Reproduced and adapted with permission from Biomedicines [15].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



