
Submitted:
31 May 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
As precision medicine such as targeted therapy and immunotherapy often have limited accessibility, low response rate and evolved resistance, it is urgent to develop simple, low-cost, and quick-turnaround personalized diagnostic technologies for drug response prediction with high sensitivity, speed, and accuracy. Major challenges of drug response prediction strategies employing digital database modeling are the scarcity of labeled clinical data, applicable only to a few classes of drugs, and losing the resolution at the individual patient level. Although these challenges have been partially addressed by large-scale cancer cell line datasets and more patient-relevant cell-based systems, the integration of different data types and data translation from pre-clinical to clinical utilities are still far-fetched. To overcome current limitations of precision medicine with a clinically proven drug response prediction assay, we have developed an innovative and proprietary technology based on in vitro patient testing and in silico data analytics. First, a patient-derived gene expression signature was established via transcriptomic profiling of cell-free mRNA (cfmRNA) from the patient’s blood. Second, a gene-to-drug data fusion and overlaying mechanism to transfer data was performed. Finally, a semi-supervised method was used for database searching, matching, annotation and ranking of drug efficacies from a pool of ~700 approved, investigational or clinical trial drug candidates. A personalized drug response report can be delivered to inform clinical decision within a week. The PGA (Patient-derived Gene expression-informed Anticancer drug efficacy) test has significantly improved patient outcomes when compared to the treatment plans without PGA support. Implementation of PGA, which combines patient-unique cfmRNA fingerprints with drug mapping power, has the potential to identify treatment options when patients are no longer responding to therapy and when standard-of-care is exhausted.
Keywords:
precision medicine
; drug response prediction
; cell-free mRNA
; transcriptomic profiling
; gene expression signature
1. Introduction
The deciphering of human genome sequence has expedited genetic data-driven transformation in medicine and healthcare. This revolution, now recognized as precision medicine, has provided better diagnoses, more targeted and effective treatment, and early intervention of disease. Precision medicine promises improved health outcomes by providing the right therapy to the right patient, at the “first” time without delay [1]. Current standard of care is to utilize genomic alteration data to tailor therapy for an individual cancer patient. However, the reality of today’s precision medicine is only 5-10% of cancer patients experience a clinical benefit from treatments matched to tumor DNA mutations via biomarker testing [2,3,4]. Although there are many factors underlying this modest success rate, improved drug response prediction will significantly benefit more patients, especially for those non-responders to targeted therapy or immunotherapy [4,5,6].
Mega pre-clinical databases such as Genomics of Drug Sensitivity in Cancer (GDSC) [7,8] and Cancer Cell Line Encyclopedia (CCLE) [9] provide plentiful genomic profiles consisting of somatic mutation, copy number aberration, structural variant, transcriptomic, and methylomic data, together with the response to a large number of targeted and chemotherapy drugs. However, these are different from clinical datasets which register the response only to monotherapy or combination therapy that have been administered to a patient, e.g., The Cancer Genome Atlas (TCGA) and ClinicalTrials.gov. Nevertheless, the pre-clinical datasets enable drug response modeling, training and prediction, in particular for many drugs, from various types of pre-clinical systems to patients [8,9]. Working on pre-clinical big data, current in silico analyses usually aimed at building computational deep-learning, deep neural network methods to predict drug response [10,11]. However, it remains challenging to integrate and interpret the diverse and large number of the high-dimensional multiomics data points in a clinically relevant manner. Further, the complex cellular signaling networks that regulate the anticancer drug response are largely overlooked in digital computation and simulation, thereby losing the translatability to real-world patients [12,13]. A computational approach should be trained on relevant and standardized clinical data to achieve translatability, unfortunately the available clinical datasets such as TCGA do not have sufficient patient records with drug response information.
Extensive studies have suggested that gene expression data is the most effective data type for drug response prediction [11,14,15]. Although gene expression profiles provide a machine learning model with deeper insight of the same sample and promise better characterization of biological processes, this approach has several limitations. First, it will miss much-needed resolution at the individual patient level, which may limit its ability to predict personalized drug response. Second, the non-realtime gene expression patterns will misrepresent the dynamics of input genes and data. Third, sample-specific gene expression data were not deployed yet. The digital modeling was used to show gene-relatedness in the context of cancer, but not for sample-specific prediction tasks (e.g., drug response prediction). Therefore, it is not appropriate to use most important input gene sets for each sample.
In the clinical application of drug response prediction, our goal is to predict which drugs will most likely to benefit the patient based on the patient’s own gene expression signature. Since clinical gene-drug datasets are small and hard to obtain, many studies have focused on large pre-clinical pharmacogenomics datasets such as cancer cell lines as a proxy to patients. A majority of the digital computation methods are trained on cell line datasets and then tested on patient datasets [16,17]. However, cell lines even with the same set of genes often do not recapitulate patient’s drug response due to the lack of an immune system and tumor microenvironment (TME) [18]. Moreover, in cell lines, the drug response is often measured by the IC50 or AUC (Area Under Curve), whereas in patients, it is often based on changes in the size of the tumor and measured by metrics such as response evaluation criteria in solid tumors (RECIST) [19]. This means that drug response prediction is a regression problem in cell lines but a classification problem in patients. Therefore, discrepancies exist in both the input and output pharmacogenomics datasets and an urgent need exists for a translational technology to bridge this gap.
In this study, we developed an innovative liquid biopsy cell-free mRNA (cfmRNA) based technology, called patient-derived gene expression-informed anticancer drug efficacy (PGA), for predicting cancer drug responses. It applied cfmRNA profiling to measure gene expression and established a cancer type-specific, patient-unique gene expression signature. The signature was then used to digitally query, search, match, categorize and rank drug efficacies from a library of more than 700 anticancer drugs to identify the most effective drugs for the patient. Importantly, PGA was further prospectively and clinically tested on gene expression data from a real-life group of patients with refractory or relapsed non-small cell lung cancer (NSCLC) to identify potentially effective drugs. Our results demonstrated that the first-ever PGA platform, combining in vitro patient testing with in silico data computation, enabled us to analyze each patient’s cfmRNA data in real time to better match them with tailored treatments and drug combinations. These findings underscored the clinical utility of PGA and contribute to the advancement of drug response prediction.
2. Materials and Methods
2.1. Sample Processing and RNA Isolation
All paired tissue and blood samples were purchased from iSpecimen (Lexington, MA, USA), Discovery Life Sciences (Huntsville, AL, USA) or Precision for Medicine (Carlsbad, CA, USA). Blood samples (approximately 8 mL) were collected and centrifuged within one hour at 1100× g for 10 min. The plasma was additionally centrifuged at 16,000× g for 10 min, transferred to cryogenic vials, and stored at −80 °C until processed. Archival RNA were isolated from formalin-fixed, paraffin-embedded (FFPE) samples. Most tumor tissue samples contained more than 50% tumor cells and the median tumor cell percentage was 77.5%. All plasma samples underwent one freeze-thaw cycle. Circulating cell-free RNA was extracted from 400 μL double-spun plasma using the MagMAX™ Viral/Pathogen Nucleic Acid Isolation Kit. Tumor and adjacent normal tissue RNA were each extracted from 5 consecutive 10 μm FFPE sections by MagMAX™ FFPE DNA/RNA Ultra Kit (Applied Biosystems, Foster City, CA, USA). Both extractions were performed on the KingFisher™ Duo Prime Purification System (Thermo Fisher Scientific, Waltham, MA, USA). RNA isolated was quantified using Qubit RNA HS Assay Kit and Qubit 2.0 Fluorometer (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). The Qubit working solution was made according to manufacturer’s instructions. The size distribution of RNA fragments within the extracts was assessed using the RNA 6000 Pico kit on a 2100 Bioanalyzer Lab-on-a-Chip platform (Agilent Technologies, Santa Clara, CA, USA), and expressed as the percentage of fragments greater than 200 base pairs (DV200).
2.2. Reverse Transcriptase Quantitative PCR (RT-qPCR)
Double-stranded cDNA was synthesized from 1 μg of total RNA using NEBNext RNA First Strand Synthesis Module and NEBNext Ultra™ II Non-Directional RNA Second Strand Synthesis Module (New England Biolabs, Ipswich, MA) according to manufacturer’s instruction. For targeted plasma transcriptomic profiling, 9 TaqMan Gene Expression Arrays (Applied Biosystems, Foster City, CA, USA) covering 9 major signaling pathways of about 750 cancer-associated genes were employed. They were TaqMan Array Immune Response #4414073, Cell Surface Markers #4418754, DNA Repair Mechanism #4418773, DNA Methylation #4414127, Transcription Factors #4418784, p53 Signaling #4414168, MAPK Pathways #4414093, Molecular Mechanisms of Cancer #4418806, Tumor Metastasis #4418743. Real-time qRT-PCR amplification and detection were performed with TaqMan Gene Expression Assay reagents in an QuantStudio 12K Flex System (Applied Biosystems, Foster City, CA, USA) using standard settings and cycling parameters. The 20-μL reactions were carried out containing 10 μL TaqMan Fast Advanced Master Mix and 10 ng of a cDNA template per well in a 96-well format.
The expression of each of the genes in all specimens was normalized to its expression in a reference RNA pool (made by pooling equal amounts of total RNA from each of the tumor-free specimens) used as a calibrator. The ribosomal 18S RNA was used as the internal control. The relative changes in gene expression were determined by the ΔΔCt method using the Sequence Detection System (SDS) 2.1 software (Applied Biosystems, Foster City, CA, USA). The ΔΔCt method gives the amount of target normalized to an endogenous reference and relative to a calibrator. We chose this method to allow comparison with the RNA-Seq study, because most sequencing data provide relative counts of genes, whereas relative quantification can be used to analyze data obtained through qRT-PCR.
2.3. FFPE Tissue RNA Sequencing
The cDNA fragments of preferentially 250–300 bp in length were selected for sequencing library construction. The libraries were sequenced on an Illumina HiSeq platform (Illumina, San Diego, CA, USA) and 125 bp/150 bp paired-end reads were generated. Raw data were cleaned with Cutadapt 4.0 to remove reads that were of low quality, low-read and those containing adapters and sequencing artifacts. The clean reads were aligned to the reference genome using TopHat v2.0.12. HTSeq v0.6.1 was used to count the number of reads mapped to each gene. Subsequent normalization to correct for larger genes having higher read counts include Transcripts Per Million (TPM) or Reads/Fragments Per Kilo-base per Million mapped reads (RPKM/FPKM), which was calculated to determine relative gene expression levels [20]. Differential expression analysis was performed using the DESeq R package v1.18.0, which provided statistical methods for determining differential expression in digital gene expression data using a model based on negative binomial distribution. Top high-confidence differentially expressed genes were identified by comparing the expression levels of all transcripts in the TUMOR groups with those in the NORMAL group. Genes with |log2 (fold change)| > 1 and an adjusted P-value <0.05 were considered to be differentially expressed.
2.4. Single Cell Gene Expression Profiling by RNA-Seq
Tumor tissues were digested with a human tumor dissociation kit (Miltenyi Biotec, Gaithersburg, MD, USA) following the manufacturer’s protocol. If more than 5% of dead cells in cell suspensions were indicated by trypan blue staining, dead cells were filtered out using dead cell removal kit (Miltenyi Biotec, Gaithersburg, MD, USA). Purified cells were directly used for single cell RNA sequencing or frozen in 90% fetal bovine serum supplied with 10% dimethyl sulfoxide at −80°C with cell concentration within 100-2,000/μL.
Single cells were captured and barcoded using the 10X Chromium platform (10X Genomics, Pleasanton, CA, USA). RNA-seq libraries were prepared following the instructions from the Chromium Single Cell 3ʹ Reagent v3 Kits. Approximately 5,000 cells were loaded into each lane of a 10X chip. Cells were partitioned into single-cell gel beads in emulsions (GEMs) inside the Chromium instrument, where full-length cDNA synthesis occurred. After reverse transcription and cleanup, the cDNA from barcoded single-cell RNAs were amplified, and the 3’ gene expression libraries were constructed. The cDNA pool corresponding to an insertion size of ∼350-400 bp was selected. Sequencing libraries were quantified using Agilent Bioanalyzer High Sensitivity DNA chips (Agilent Technologies, Santa Clara, CA, USA) and pooled together to get similar numbers of reads from each single cell before sequencing on the NovaSeq 6000 S4 (Illumina, San Diego, CA, USA).
2.5. Single Cell Spatial Transcriptomics Analysis
Single-Cell RNA-Seq reads were mapped to the human genome (GRCh38) using Cell Ranger v1.1.0 pipeline with default settings. The resulting gene-cell matrixes were subsequently imported into the Seurat (v3.1.5) R toolkit for further quality control and downstream analysis [21,22]. To search for clinically relevant genes co-localized and co-expressed with the selected PGA Lung biomarkers for drug efficacy prediction, we identified cell clusters using Seurat graph-based clustering methods at increasing resolutions to identify major cell types within a single cell RNA-Seq dataset. We used marker genes – EGFR, KRAS, BRAF, MET, HER2, ALK, ROS1, RET – to identify lung tumor cells; CD8, CD25, CD69, PD-1, CTLA-4 and B cell markers for immune cell population; KI67 and PCNA for highly proliferative cells; and SOX2, OCT4, KLF4, and MYC for cancer stem cells. Mesenchymal, stromal, vascular endothelial and other cell-of-interest clusters can be further annotated based on canonical markers for further dimensionality reduction using the FindClusters function in the Seurat package. Cell clusters were identified at resolution 0.3 and annotated based on prior knowledge. We recognized a cluster with a minimum of 5% percent of cells from the total cell population. If the marker genes are not our primary interest, we would leave the original clusters unchanged.
2.6. Correlation Between Gene Expression and Drug Efficacy
A method of gene pathway/network generalization for drug response prediction needed to take both pre-clinical or clinical samples during deployment. Therefore, we selected datasets and developed gene-drug correlation based on cancer cell lines, archived tumors, single cell transcriptomics and real-world patients. We employed the following resources for gene pathway generalization: Cancer Cell Line Encyclopedia (CCLE), The Genomics of Drug Sensitivity in Cancer (GDSCv1/2); The Cancer Therapeutics Response Portal (CTRPv2), EMBL-EBI Single Cell Expression Atlas, cBioPortal for Cancer Genomics, CREAMMIST database, Cancer Treatment Response Gene Signature database (CTR-DB), and The Cancer Genomic Atlas (TCGA). All datasets were downloaded from ORCESTRA platform [23]. We focused on bridging cell line datasets to patient tumors because they are the missing link for translation from pre-clinical to clinical [24].
Correlation at the gene level: The correlation between cell lines and the corresponding TCGA cohorts was measured by two computational analytics: (i) Spearman’s correlation coefficient (ρ) between every cancer cell line and its corresponding TCGA cohort was determined at the gene level. For this, for each gene in a TCGA cohort, the TPM values were averaged per cohort. Then, for each TCGA cohort, Spearman’s ρ was calculated between the averaged TPM values and those of the disease-matched cell lines based on the common 20,053 protein-coding genes. (ii) The enrichment of the TCGA cohort overexpressed genes (i.e., the union of enriched, group enriched, and enhanced genes in the TCGA cohort) in cell lines was analyzed by gene set enrichment analysis (GSEA). The concept is that genes that have an upregulated expression in a TCGA cohort can be considered as the cohort signature, and their high expression should be reflected by cell line models. For every cell line, we calculated the fold change of every gene relative to the disease baseline expression, followed by the log2 transformation. The gene log2 fold changes were sorted from high to low, followed by the GSEA of the TCGA cohort overexpressed genes against the sorted gene list. The correlation results were represented as the normalized enrichment score (NES), with a positive value showing high correlation between a cell line and a disease-matched TCGA cohort.
Correlation at the pathway level: The activity of a total of 14 cancer-related pathways were interrogated using PROGENy, a pathway-response signature based approach that is capable of deep data mining to obtain cancer-related pathway responsive genes [25], together with the CytoSig program which analyzes 43 cytokines gene expression profiles [26]. Both results were presented as z-scores to represent the relative activities, with a p-value < 0.05 as significant.
In the output, the latent representations of the patient’s tumor molecular profile must then be matched with each drug’s latent representation through cell line data links. Low-rank multimodal fusion (LMF) is a technique for combining multiple modalities in a neural network such that the latent representations of different features are forced to “interact” with each other [27]. LMF has shown higher performance than other fusion methods. It is especially important for modeling biology since it is known that various biomolecules in the cell interact with each other and thus must also be allowed to interact when modeling biology in silico. LMF was employed as the fusion method, and the output from this fusion is then passed to the final module which predicted the drug efficacy.
2.7. Statistical Analysis
Data on patients were analyzed from the date of therapy with or without PGA guidance to the time of the death or the date on which data were censored. All statistical analyses were performed using SPSS software v20.0 (SPSS Inc., Chicago, IL, USA). Progression-free survival (PFS) and overall survival (OS) were obtained with the Kaplan-Meier product limit method for each cohort. Comparisons were made with the bilateral log-rank test. In addition, the combined effects of those variables on both PFS and OS were examined in multivariate analysis using Cox proportional hazards regression models. P<0.05 was considered to indicate a statistically significant difference.
3. Results
3.1. Cancer Type-Specific and Patient-Derived Gene Expression Profiles
Although RNA-Seq and microarrays were standard methods benchmarked for differential gene expression and prediction model development, the challenge of quantifying low-abundance, short half-life cfmRNA species is compounded by the time-consuming and labor-intensive workflows. The requirement of high-quality and sufficient quantity of cfmRNA also imposes a technical barrier on top of the interference from high and variable levels of globin mRNA and ribosomal RNA (rRNA). Although rRNA depletion and globin reduction have been shown to mitigate some of these issues, they require a large amount of total cfRNA pool and may induce biases in the quantification of gene expression. To overcome these limitations, we applied targeted transcriptomic profiling based on multiplex RT-qPCR amplification followed by quantitative analysis of cfmRNA abundance by ΔΔCt, the difference of Ct values between reference gene (18S) and target gene, and normalized to the control samples.
Plasma circulating cfmRNA was extracted from pooled plasma cohorts of patients with lung, pancreatic or breast cancer. Approximately 750 well-established cancer-associated genes belong to 9 major cancer signaling pathways were profiled: immune response (IR), cell surface markers (CSM), transcription factors (TF), DNA repair (DR), DNA methylation (DM), oncogenesis (ONC), tumor metastasis (TM), TP53 signaling (TS), MAP kinases (MK). The distribution of detected cfmRNA species from the lung cancer cohort was demonstrated in Figure 1A. We identified same percentage of genes belonged to cell surface markers and TP53 signaling pathway (21%), 17% of detected transcripts were members of the MAP kinase family, 13% involved in DNA repair, 8% correlated with oncogenesis, 7% associated with tumor metastasis, 6% involved in immune response, 5% are transcription factors and 2% related to DNA methylation.
Figure 1B illustrated a global cfmRNA expression and functional landscape in lung, breast and pancreatic cancers. The circulating cell-free transcriptome composition of TP53 signaling and MAP kinases were particularly dominant in these three cancer types. For quantification of cfmRNA expression levels, genes with ΔCt values between 0-15 was classified as “high expression” (blue); ΔCt values between 15-20 was interpreted as “medium expression” (green); and ΔCt values of 20-30 was called “low expression” (red) after normalization with the control samples. The genes with ΔCt values >30 were not color coded. From the representative cfmRNA expression heatmaps, differentially expressed cancer type-specific genes can be easily identified in the same functional cluster, for example, ERCC2, MDM2, POLR2B, PSMB10 in DNA repair cluster are highly expressed and are pancreatic cancer-specific genes; whereas FANCG is breast cancer-specific gene; and POLH, RPA2 are strongly expressed as lung cancer-specific genes. Among cell surface markers, C5AR1, CD24, CD28, SELP are highly expressed as pancreatic cancer-specific genes; whereas CD7, CD8A, FAS are breast cancer-specific genes; and CD79 and MS4A1 are strongly expressed as lung cancer-specific genes.
Here, we have obtained highly distinct cfmRNA expression profiles and functional clusters specific for lung, breast and pancreatic cancers. Pancreatic cancer revealed the largest heterogeneity of gene expression as a wide spectrum of cfmRNA transcripts are produced by its transcriptional machinery. In contrast, lung cancer has relatively low cfmRNA heterogeneity and fewer specific genes that contribute to the total cfmRNA composition. Multiple pathways are responsible for transducing mechanical and growth stimuli into changes in gene expression during cancer development and association with drug susceptibility. In this work we have established an unprecedented functional cfmRNA database which will guide for (i) the illustration of a comprehensive landscape of cfmRNA in circulation, (ii) the classification of cfmRNA species by their functions, (iii) the identification of differentially expressed cfmRNA in a particular cancer type, and (iv) the establishment of specific cfmRNA expression signatures for different cancer types. The cfmRNA expression profiles identified in this study represented the functional genomic fingerprints in circulation for specific cancer types, thus offering the exciting opportunity of personalized drug efficacy prediction.
3.2. Selection and Validation of PGA Lung cfmRNA Biomarkers
Genomic features are regarded as the state-of-the-art method for drug response prediction. Numerous studies have shown that measurement of gene expression is a potent and still under-utilized method for identifying cell vulnerabilities, with superior performance over genomic features in genetic and compound response prediction [28,29,30]. The advantage of expression-based profiles over DNA-based alterations held consistently across multiple experimental platforms, models and databases [31].
Most importantly, contrary to the common perception in the literature, the most accurate expression-based models depended on only few features and are amenable to biological interpretation, suggesting that a full RNA-Seq profile of tumors is not necessary to gain powerful prediction for precision therapy [31]. Since many new or established cell vulnerabilities can be identified with just one or two expression features, cost- and time-efficient technologies such as RT-qPCR with identified biomarkers would still offer considerable benefits. Specifically, genes exhibiting bimodal expression and covering important cancer-associated pathways can be used to robustly predict drug response across datasets [32]. These bimodal predictive biomarkers have a high potential of clinical translatability given the clear separation they would provide between patient responder and non-responder cohorts, and the practicality of measuring a few genes for treatment planning using various targeted assays instead of whole-transcriptome sequencing.
Consistently, another line of evidence, essentially by guided trial-and-error, have demonstrated that it is possible to “reprogram” cell type by manipulating only a handful of genes [33]. It has been delicately proven that forced expression of only 4 genes SOX2, OCT4, KLF4, MYC was able to turn adult cells back into pluripotent or embryonic-like stem cells [34]. Overall, it was estimated that 10-200 meta-analytic genes are required to provide optimal downstream performance and make available replicable marker lists for the 85 BICCN cell types [33]. Even modern precision medicine supports this notion that single hotspot mutation in a single gene, e.g., EGFR, KRAS, BRAF, ABL1, JAK2, is sufficient to predict effective targeted therapy.
We thus set up to select dozens of lung cancer-specific cfmRNA biomarkers based on four criteria: (i) tumor-specific, highly expressed and readily detectable biomarkers, (ii) biomarkers involved in 9 major cancer functional clusters, directly affecting more than 10,000 genes, (iii) biomarkers that were retrospectively verified in tumor tissues as overactive, (iv) biomarkers associated with drug efficacy, e.g., cell death, proliferation, survival, hypoxia and microsatellite instability (MSI). The selected biomarkers were next interrogated through TCGA database to assess their expression in patient tumors. As expected, these PGA Lung biomarkers were found to be overexpressed in 60-70% of 1,145 lung cancer patient tumors (Figure 2A). Further, the overexpression of these selected biomarkers significantly correlated with hypoxia (p=0.0177, Figure 2B) and MSI scores (p=0.0143, Figure 2C) in TCGA PanCancer Database of 510 LUAD samples.
In parallel, transcriptome-wide characterization of tumor tissue of lung cancer was also conducted using RNA-Seq technology. Of 17,780 detected and annotated transcripts, 5,185 (29%) displayed at least 1.2-fold higher expression over non-cancer samples. Within those low-variation genes, we identified lung cancer-specific transcripts that are recurrently detected in both plasma and tissue. These transcripts met our set criteria: (i) they were not detected in non-cancer or other cancer plasma, (ii) they were upregulated in the cancer group compared to the non-cancer group, and (iii) they were detected in more than one cancer sample in our cohort. Our results showed strong correlation between cfmRNA levels in plasma and mRNA expression in tissue, suggesting that these biomarkers with relatively high expression in tumor tissue could enhance cancer detection in patients with circulating cfmRNA (Figure 3). Overall, our data validated the clinical relevance of the selected PGA Lung biomarkers for informed drug efficacy.
3.3. Single Cell Spatial Transcriptomics Analyses
Obtaining quantitative information on gene expression changes within cells can be laborious and challenging. Spatial transcriptomes, an emerging technique that utilizes spatially barcoded, complementary DNA primers for full-transcriptome capture on tissue sections can be added to RNA-seq data to transform our understanding of tissue functional organization and cell-to-cell interactions in situ. Analysis of single-cell RNA expression in their spatial context provides critical insight about tumor, immune cells and their microenvironment. This also helps to decipher subcellular co-localization and co-expression of target RNA biomarkers, leading to an unprecedented resolution for drug efficacy prediction.
To characterize the phenotypic and functional interaction of tumor cells and their microenvironment in lung cancer, we first performed single cell RNA-Seq with spatial transcriptomics followed by graph-based clustering analyses to distinguish EGFR-expressing tumor cells in three lung carcinomas in a total of 32,341 cells (Figure 4). Distinct from other tumor clusters, EGFR-expressing tumor cells only made up a fraction (less than 30%) of entire tumor population. Interestingly, EGFR+ staining was highly overlapping with MET, HER2 and ROS1 expression, whereas very few ALK- and RET-expressing cells were detected, and distinct from EGFR+ cells. The high expression level of KRAS was detected in over 50% of tumor population, and its distribution was more consistent with BRAF expression. The single cell spatial analyses have distinguished diverse cell types in lung cancer: EGFR/MET/HER2/ROS1-expressing, KRAS/BRAF-positive, and ALK+ or RET+ cells. Our novel observations here were somehow surprising in terms of the high and complex heterogeneity at the single cell level, and will provide potential guidance on target-tailored therapy strategy in lung cancer.
Next, we took a closer look at the expression distribution of the selected PGA Lung biomarkers, to infer their roles in regulating drug responses. Most of the selected PGA Lung biomarkers were similar in expression patterns, resemble to KRAS/BRAF, and they may constitute a relatively homogeneous population (Figure 5A). We also discovered a concurrent expression of PCNA in this population, indicative of highly proliferative activities. PCNA is recognized as an important prognostic indicator of cancer. Its expression has been found to be significantly elevated in various malignant tumors. PCNA expression thus can reflect cell dynamics and represent the proliferative potentials of cells, and can be used as a marker for chemotherapy efficacy [35].
The immune cells existed in other clusters were also examined by the following markers: CD4, CD8, CD25, CD69, CD19, CD20, PD-1, CTLA-4. However, these immune cells were not in close proximity to EGFR/MET/HER2/ROS1-expressing cells suggesting that they might not be the infiltrated immune cells (Supplementary Data; Figure S1). It will be of great interest to assess markers closely related to the pro-invasive or immunosuppressive tumor microenvironment to predict immunotherapy response.
To confirm what we have observed in lung carcinoma tissues, we set out to conduct single cell spatial transcriptomics of tumor cells obtained from pleural effusion of lung adenocarcinoma patients with a total of 7,511 cells (Figure 5B). As expected and consistent with the tumor tissue results, dissociated tumor cells in pleural effusion showed similar expression distribution among the selected PGA Lung biomarkers, and most also co-expressed PCNA. Together, we have demonstrated the coexpression of the selected PGA Lung biomarkers with PCNA in two different sample types from different lung cancer patients.
3.4. From Patient’s Gene Expression Signature to Drug Efficacy Prediction
Cancer cell lines with pharmacological, genomic, transcriptomic characteristics are the most important resource available today for drug response study. These datasets can be pooled, analyzed and trained from Cancer Cell Line Encyclopedia (CCLE), The Genomics of Drug Sensitivity in Cancer (GDSCv1/2), and The Cancer Therapeutics Response Portal (CTRPv2). Total 232 lung cancer cell lines were analyzed for their representability of the corresponding TCGA lung cancer cohorts (total 1,089 patient tumors). Considering that tumor samples also harbor immune and other cells, TCGA samples with a tumor purity score lower than 0.7 were excluded from the analysis. The similarity between cell lines and the corresponding TCGA cohort was estimated by Spearman’s correlation coefficient (ρ) and the normalized enrichment score (NES). A positive value indicated high consistency between a cell line and a disease-matched TCGA cohort. The cell lines were then ranked combining Spearman’s ρ with NES. Overall, we found strong genetic similarity between lung cancer cell lines and lung cancer patient tumors (Figure 6). These cell lines faithfully recapitulate gene expression profiles and major cancer pathway activities in tumors, many of these associated with drug sensitivity/resistance.
We next took advantage of the high-degree representation of tumor functional activities in cancer cell lines for pharmacogenomic prediction of drug sensitivity. Publicly available gene expression datasets for a large cohort of cell lines (CCLE), single cells (EMBL) and primary tumors (TCGA) were retrospectively pooled and merged to identify clinically relevant features called cancer consensus modules (CCM). Prospectively collected cancer type-specific, patient-derived gene expression signatures were then used to align, filter, homogenize and map with CCM. The resultant datasets were applied to predict in vivo drug efficacies (Figure 7). We have identified significant gene-drug interactions for the majority of 700+ anticancer drugs (approved, investigational or clinical trial) via PGA. A pathway-centric approach highlighted the power of drug efficacy prediction by those PGA Lung biomarkers involved in cancer pathways. For example, MEK and PARP inhibitors have been identified by the PGA test to be effective for a number of refractory or recurrent lung cancer patients. Together, we have discovered and established a translational linkage demonstrating that lung cancer patient-derived gene expression signatures can be mapped onto molecularly annotated human cancer cell lines and correlated with sensitivity to more than 700 anticancer drugs. Our data fusion and mapping analytics ensured accurate translation from functional genotypes to cellular phenotypes and to identify effective therapeutics to benefit lung cancer patients.
3.5. Clinical Utility and Validity of the PGA Lung Test
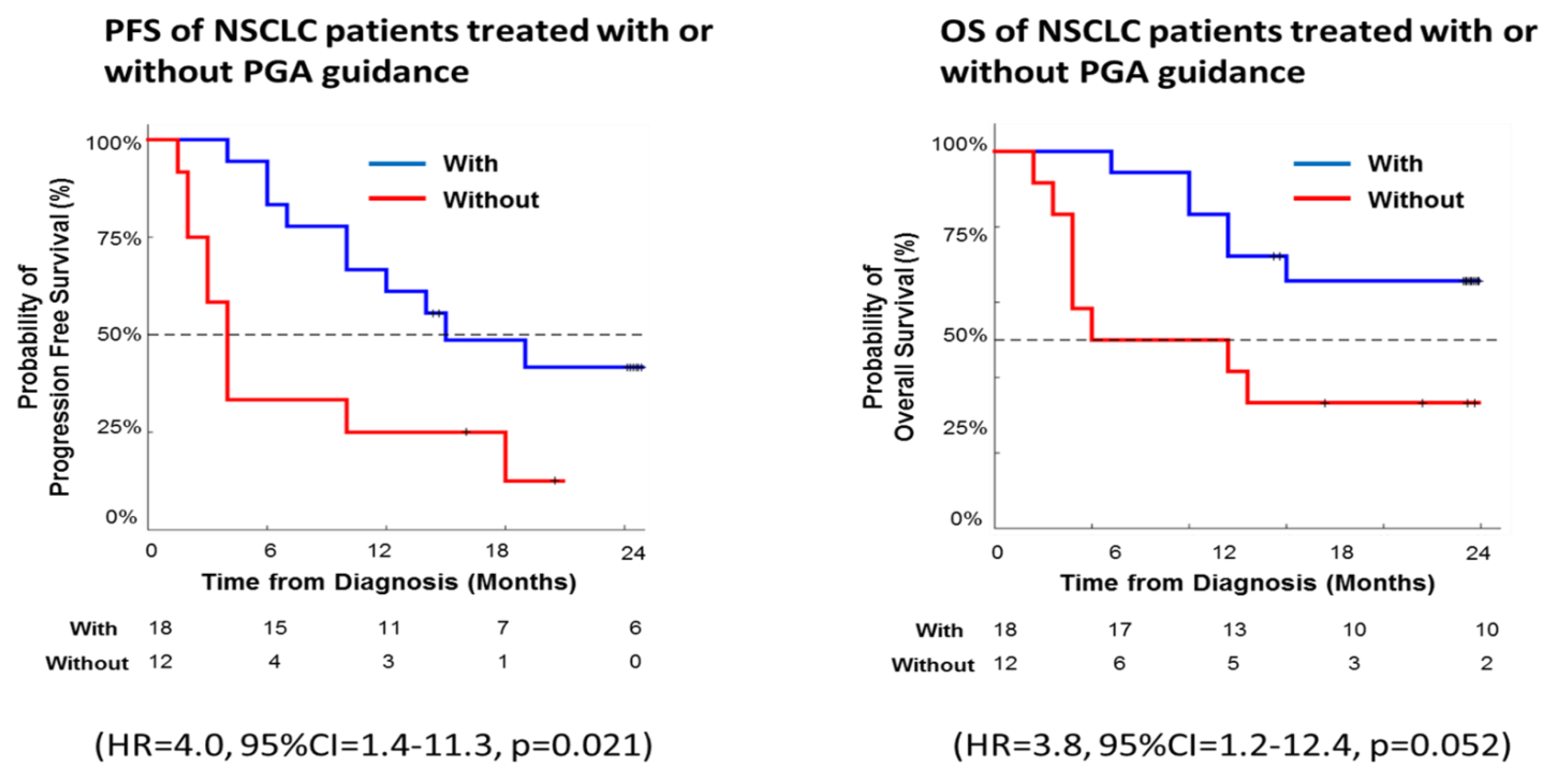
As a proof of principle, we further evaluated PGA clinical validity on a small cohort of 30 patients with recurrent or progressive lung cancer. To ensure the cross-group comparison of the trial, we divided patients into two groups each with the indicated numbers of age-, gender- and stage-matched subjects. In the placebo group of 12 patients, clinicians treated these patients according to current medical guidelines without PGA test; while in the experimental group of 18 patients, patients went through PGA test and clinicians treated these patients with PGA’s drug efficacy information. Tumor response was evaluated by a computed tomography scan based on the Response Evaluation Criteria in Solid Tumors (RECIST). The Kaplan-Meier method and a log-rank test were used to analyze the univariate discrimination of progression-free survival (PFS) and overall survival (OS) by demographic data, baseline clinical information and toxicities. Kaplan-Meier curve is one of the best analyses to be used to measure the fraction of subjects living for a certain amount of time with or without treatment. In clinical trials or community trials, the effect of an intervention is assessed by measuring the number of subjects survived or saved after that intervention over a period of time. The log-rank test is used to test whether the difference between survival times between two groups is statistically different or not.
In our pilot trial, Kaplan-Meier survival analysis revealed significantly longer PFS and OS among PGA-guided patients compared with patients without PGA support (PFS: hazard ratio, 4.0; 95% CI, 1.4-11.3; p = 0.021; OS: hazard ratio, 3.8; 95% CI, 1.2-12.4; p = 0.052) (Figure 8). Thus, the real-world data here demonstrated PGA’s clinical utility and validity with a significant effect on the long-term survival in our cohort of lung cancer patients.
4. Discussion
The advantage of precision oncology is the ability to obtain early human evidence of whether the medicine is working and use that to inform clinical decisions. All too often, the actual benefits of targeted therapy to patients are short-lived because tumors are heterogeneous and drug resistance emerges quickly. The harsh reality is that only 20-30% of cancer patients are eligible, and in the qualified population about one-fourth actually responds to targeted treatment. As a result, a small fraction of cancer patients (5-10%) experience a clinical benefit from treatments matched to tumor DNA mutations (via biomarker testing). Finding reliable and interpretable biomarkers that can predict non-responder patients’ response to anticancer drugs thus remain a huge unmet clinical need. In this study, we have invented and employed cutting-edge functional genomics to translate patient’s genetic prolife into drug response to benefit more patients. The PGA classifier was designed to categorize patient’s cfmRNA expression data into “responder” or “not responder”—in reality, PGA doesn’t provide a binary answer but instead generates drug efficacy prediction (or drug response prediction) to more than 700 anticancer drugs.
Gene expression profiling is a novel functional genomics for identifying tumor vulnerabilities, with superior performance over genomic features in both genetic and drug response prediction. Studies have shown the advantage of expression-based features over DNA-based alterations held consistently across multiple experimental platforms using different perturbation technologies. It has been suggested that the expression of gene panels, such as pathway clusters or transcription factor classes, are more robust and reliable predictors than expression of individual genes [36,37]. Moreover, it was able to “reprogram” cell type by manipulating only a handful of genes, and it was estimated that 10-200 genes are sufficient to robustly determine a cell's type. Based on these findings, we have conducted plasma cfmRNA profiling to identify cancer type-specific, patient-unique gene expression signature for drug efficacy prediction. We have selected dozens of tumor-overexpressed biomarkers involved in 9 cancer pathways, i.e., immune response, cell surface markers, DNA repair, DNA methylation, oncogenesis, tumor metastasis, transcription factors, TP53 signaling, and MAPK pathways to be broadly representative and ensured capture of tumor and non-tumor signals. These selected PGA Lung biomarkers are capable of directly affecting more than 10,000 genes, and their over-activation have been retrospectively verified in tumor tissues and TCGA cohorts. Most significantly, these biomarkers were implicated in drug response, e.g., cell growth, survival, death, hypoxia and microsatellite instability (MSI).
We further profiled the cell subtypes expressing PGA Lung biomarkers and their spatial distribution in lung tumor tissues as well as dissociated tumor cells from pleural effusion by single cell RNA-Seq and spatial transcriptome. As a result, we created an atlas of PGA Lung biomarker-expressing cells in lung cancer. We defined these cell clusters using representative PGA Lung biomarkers, key lung cancer driver genes, immune cell markers, and identified their spatial distribution. We found that EGFR/MET/HER2/ROS1-expressing tumor cells constituted only a small fraction of tumor population, while KRAS/BRAF-positive cell clusters were distributed over the entire tumor section. Most cells expressing PGA Lung biomarkers were also KRAS+/BRAF+, suggesting this relatively homogeneous population could be a more effective target for therapeutic intervention than EGFR/MET/HER2/ROS1-positive cells. Interestingly, immune T- and B-cells were found to be in distinct cell clusters and distant from EGFR/MET/HER2/ROS1-expressing tumor cells. The PGA Lung biomarker-expressing cells were also enriched with PCNA, indicative of high proliferation potential. The spatial patterns were reproducible in tumor cells from pleural effusion. Therefore, our data identified, for the first time, the PGA Lung/KRAS/PCNA-coexpressing cells as the dominant and representative subtype in lung tumors which will serve as an important cell atlas in illustrating the complex transcriptomics and potential therapeutic targets for lung cancer. Moreover, treatment strategies targeting EGFR/MET/HER2/ROS1 may not be sufficient. Tumor cell subtypes, immune cell proximity and gene expression in individual cell types in lung cancer could partly explain the failure of targeted therapy and immunotherapy. The single cell spatial transcriptome also revealed that the relatively homogeneous coexpression of PGA Lung biomarkers in the same population, instead of heterogeneous expression in different cell clusters, would make PGA Lung assay more accurate and consistent for drug efficacy prediction. Overall, the spatial atlas of the transcriptional profiles of PGA Lung biomarkers in the tumor cell subtypes further validated their predictive power for drug efficacy.
To date, multiple cellular and molecular changes in lung cancer, including those mutations in the driver genes and interaction between tumor and immune cells, are thought to contribute to the pathological state. However, it is a great challenge to transfer experimental results from cells directly to humans. A number of computational drug response predictions are developed on preclinical datasets and subsequently “humanized” to focus on the similarities between preclinical models and human tumors. Most approaches applied molecular profiles and drug screens from large-scale databases of preclinical models with advanced machine learning and training, e.g., transfer learning or deep neural network learning, to correct for differences between preclinical models and human tumors [38,39,40]. Although promising, these approaches either do not take into account the real-time, real-world patient data and dynamic tumor evolution or only model these differences as a technical batch effect, leading to “one-size-fits-all” generalized software packages. To reach accuracies that are acceptable for clinical application, existing databases just can’t provide the required training samples sizes. In this study, we have correlated gene overexpression patterns and pathway activities of more than 200 lung cancer cell lines with the corresponding TCGA tumor cohorts. Our results of cell-tumor comparisons demonstrated substantial similarities in the gene/pathway functional profiles across preclinical and clinical barrier. Our work established the first-ever molecular algorithm for data fusion, translation and extrapolation combining in vitro patient testing and in silico analytics, providing a quantum leap for drug efficacy prediction in lung cancer. In the long term, PGA technology could serve as a powerful tool to advance our understanding of the molecular mechanisms in cancer that mediate vulnerability or drug sensitivity.
Our analysis of >1,000 patient tumor samples, ~40,000 single cells and the subsequent superimposing of consensus genomic features onto cell lines exemplifies how gene expression signatures can be used to reliably predict drug efficacy at individual patient level, and maximizes the clinical utility of the PGA Lung test reported. The majority of cancer consensus modules (CCM) identified from TCGA tumors and single cell transcriptomics are captured within a large number of lung cancer cell lines and often at a similar extent to those observed in patient cohorts. Pharmacological datasets in cancer cell lines also offer an unbiased and plug-and-play resource for potential leverage on drug efficacy.
We introduced PGA Lung test to integrate preclinical and clinical data in a semi-supervised way. Our approach functionally aligned cell-to-tumor similarity matrices and extracted relevant CCM for mapping drug efficacy. By performing a functional gene/pathway alignment instead of a direct database comparison, CCM limited the effect of sample selection bias and filtered out variables. Although we restricted ourselves to dozens of PGA Lung biomarkers, deploying CCM that incorporate patient-derived gene expression signatures, specifically tailored for personalized drug efficacy prediction, is a potentially revolutionary avenue. The identified and defined CCM was present in real-world patients at a frequency that would make PGA Lung testing in a clinical setting feasible. We have found that more than 90% of primary tumor samples harbor at least one CCM associated with increased drug response. Hence, prioritizing molecular diagnostics that deliver real-time gene expression profiles could be the most cost and time effective means to stratify patients for cancer treatment.
Today, the vast majority of cancer patients have no detectable biomarkers for precision medicine. Therefore, expanding our arsenal of accurate theranostics would pave the way for personalized medicine by identifying the most effective drug for each patient. PGA Lung test was able to predict drug efficacies for patients, either as monotherapy or combination therapy. We convincingly demonstrated that its performance was substantially better than educated guess for a number of therapies of high clinical importance, such as platinum-based chemotherapies, gemcitabine and paclitaxel. PGA Lung assay is versatile, generalizable, scalable, and can be implemented to provide guidance in alternative treatment options (e.g., drug repurposing) for patients with refractory or relapsed disease or when standard-of-care treatments are exhausted.
PGA Lung technology still had room for improvement. First, few CCM are not well represented by a single cell line or not at all, and coverage by individual patient is variable. As we are in an era of precision oncology, where many drugs are active in small molecularly defined subgroups of patients, the broadness of CCM for different tumor genotypes could be further improved. As the preclinical and clinical databases keep expanding, they will make CCM encompassing the molecular diversity of cancer a realistic possibility. Second, our ability to validate some pharmacogenomic associations was restricted by the limited number of overlapping cell lines and drugs between these studies. The consistency between datasets is not perfect, and efforts toward standardization to reduce methodological and biological differences across the different studies are likely to improve future CCM representation between datasets. Third, we focused on cfmRNA expression. Integration of other genomic features—for example, mutations, copy number, methylation, and chromatin accessibility—may help refine drug efficacy prediction by providing additional signals. Finally, we do assume the functional clustering from CCM follow the same monotonicity in preclinical models and human tumors. This assumption, albeit reasonable, might be debatable.
Functional precision medicine opens new paths to understanding how cancer drugs can be better matched to patients. The breakthrough PGA technology enables us to analyze each patient’s data to better match them with tailored treatments and drug combinations. PGA Lung test also allows us to understand the complex relationships between gene activity within tumors and how different treatments will affect them.
5. Conclusions
PGA-based drug efficacy predictions, for the first time, revealed clinically and biologically strong interactions of drugs and gene pathways in the context of treatment response. Our study connected a systematic drug efficacy prediction pipeline with layered in vitro and in silico analyses involving plasma cfmRNA profiling, cancer type-specific biomarkers, individualized gene expression signatures, and anticancer drug database, which are the most important prerequisite for the clinical implementation of the PGA Lung platform.
Owing to the explicit use of cfmRNA biomarkers, PGA Lung highlights the underpinning biological mechanisms contributing to drug efficacy. The plasma gene expression-based prediction approach allowed us to capture novel signals from non-tumor environment, immune cell communication and interaction in real-time. This can enable drug efficacy prediction at cellular resolution from both tumor and non-tumor tissues, thus providing high degree of specificity much more so than using tumor DNA sequencing data alone.
The number of tumor mutations can sometimes help doctors identify the patients most likely to benefit from targeted therapy but unfortunately, most cancer patients (70-80%) carry no actionable mutations and don’t respond to targeted therapy. Even in those responders, drug resistance will inevitably develop. Treatment options for progressive disease continue to dwindle as mortality rates are rising. The one-of-a-kind PGA Lung technology is able to nominate existing drugs for further consideration to meet the unmet demands of enabling personalized treatments for “non-responder” patients based on tumor molecular profiles, thereby fulfilling the precision medicine promise.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Distinct immune cell clusters in tumor microenvironment by single cell RNA-Seq spatial transcriptomic analysis in lung carcinoma tissues (32,341 cells).
Author Contributions
Conceptualization, C.Y.; Data curation, S-T.L. and H-C.L.; Formal analysis, C.Y.; Funding acquisition, C.Y.; Investigation, C.Y., S-T.L. and H-C.L.; Methodology, C.Y.; Project administration, S-T.L.; Resources, C.Y.; Software, S-T.L.; Supervision, C.Y.; Validation, S-T.L. and H-C.L.; Visualization, C.Y.; Writing—original draft, C.Y.; Writing—review and editing, C.Y. S-T.L. and H-C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by OncoDxRx.
Institutional Review Board Statement
The study was conducted according to the guidelines of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Guideline for Good Clinical Practice (ICH-GCP), and approved by the Institutional Research Board of Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan, IRB 20180804Rv2.
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study for the use and publication of data (2018-08-28 version 2). All experiments were carried out in accordance with the ICH-GCP in its last revised version.
Data Availability Statement
The data presented in this study are available within the article.
Acknowledgments
We would like to thank Mr. Daniel Lin and Ms. Sharon Yeh for their project management, experimental and logistic support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Denny, J.C.; Collins, F.S. , “Precision medicine in 2030—seven ways to transform healthcare,” Cell, vol. 184, no. 6, pp. 1415–1419, 2021. [CrossRef]
- Acanda De La Rocha, A.M., Berlow, N.E., Fader, M. et al. Feasibility of functional precision medicine for guiding treatment of relapsed or refractory pediatric cancers. Nat Med 30, 990–1000 (2024). [CrossRef]
- Cheng, M.L.; Berger, M.F.; Hyman, D.M.; Solit, D.B. , “Clinical tumour sequencing for precision oncology: time for a universal strategy,” Nature Reviews Cancer, vol. 18, no. 9, p. 527, 2018. [CrossRef]
- Marquart, J.; Chen, E.Y.; Prasad, V. , “Estimation of the percentage of us patients with cancer who benefit from genome-driven oncology,” JAMA oncology, 2018. [CrossRef]
- Gavan, S.P.; Thompson, A.J.; Payne, K. , “The economic case for precision medicine,” Expert review of precision medicine and drug development, vol. 3, no. 1, pp. 1–9, 2018. [CrossRef]
- Mishra, A.; Verma, M. , “Cancer biomarkers: are we ready for the prime time?,” Cancers, vol. 2, no. 1, pp. 190–208, 2010. [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W. , et al. “Systematic identification of genomic markers of drug sensitivity in cancer cells,” Nature, vol. 483, pp. 570–575, Mar. 2012. [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M. , et al., “A landscape of pharmacogenomic interactions in cancer,” Cell, vol. 166, no. 3, pp. 740–754, 2016. [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S. , et al., “The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity,” Nature, vol. 483, no. 7391, p. 603, 2012. [CrossRef]
- Partin, A; TS, B.; Zhu, Y; Narykov, O; Clyde, A; Overbeek, J; RL, S. Partin A; TS, B.; Zhu Y; Narykov O; Clyde A; Overbeek J; RL, S. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front Med (Lausanne). 2023 Feb 15;10:1086097. [CrossRef]
- Chen, H., King, F.J., Zhou, B. et al. Drug target prediction through deep learning functional representation of gene signatures. Nat Commun 15, 1853 (2024). [CrossRef]
- Farzan Taj, Lincoln D Stein, MMDRP: drug response prediction and biomarker discovery using multi-modal deep learning, Bioinformatics Advances, Volume 4, Issue 1, 2024, vbae010. [CrossRef]
- He, D., Liu, Q., Wu, Y. et al. A context-aware deconfounding autoencoder for robust prediction of personalized clinical drug response from cell-line compound screening. Nat Mach Intell 4, 879–892 (2022). [CrossRef]
- Chawla, S., Rockstroh, A., Lehman, M. et al. Gene expression based inference of cancer drug sensitivity. Nat Commun 13, 5680 (2022). [CrossRef]
- Park, A., Lee, Y. & Nam, S. A performance evaluation of drug response prediction models for individual drugs. Sci Rep 13, 11911 (2023). [CrossRef]
- Tang, YC., Powell, R.T. & Gottlieb, A. Molecular pathways enhance drug response prediction using transfer learning from cell lines to tumors and patient-derived xenografts. Sci Rep 12, 16109 (2022). [CrossRef]
- Partin, A., Brettin, T., Evrard, Y.A. et al. Learning curves for drug response prediction in cancer cell lines. BMC Bioinformatics 22, 252 (2021). [CrossRef]
- Mourragui, S.; Loog, M.; van de Wiel, M.A.; Reinders, M.J.; Wessels, L.F. , “Precise: a domain adaptation approach to transfer predictors of drug response from pre-clinical models to tumors,” Bioinformatics, vol. 35, no. 14, pp. i510–i519, 2019. [CrossRef]
- Schwartz, L.H.; Litiè, *!!! REPLACE !!!*; re, S.; de Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S. , et al., “Recist 1.1—update and clarification: From the recist committee,” European journal of cancer, vol. 62, pp. 132–137, 2016. [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 5 (7) (2008), pp. 621-628. [CrossRef]
- Stuart T, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e1821. [CrossRef]
- Su Z, Ho JWK, Yau RCH, Lam YL, Shek TWH, Yeung MCF, et al. A single-cell atlas of conventional central chondrosarcoma reveals the role of endoplasmic reticulum stress in malignant transformation. Commun Biol. 2024 Jan 24;7(1):124. [CrossRef]
- Mammoliti, A.; Smirnov, P.; Nakano, M.; Safikhani, Z.; Ho, C.; Beri, G.; Kains, B.H. , “ORCESTRA: a platform for orchestrating and sharing high-throughput pharmacogenomic analyses.” Sept. 2020. [CrossRef]
- Jin H, Zhang C, Zwahlen M, von Feilitzen K, Karlsson M, Shi M, Yuan M, Song X, Li X, Yang H, Turkez H, Fagerberg L, Uhlén M, Mardinoglu A. Systematic transcriptional analysis of human cell lines for gene expression landscape and tumor representation. Nat Commun. 2023 Sep 5;14(1):5417. [CrossRef]
- Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N, Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun. 2018 Jan 2;9(1):20. [CrossRef]
- Jiang P, Zhang Y, Ru B, Yang Y, Vu T, Paul R, Mirza A, Altan-Bonnet G, Liu L, Ruppin E, Wakefield L, Wucherpfennig KW. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat Methods. 2021 Oct;18(10):1181-1191. [CrossRef]
- Liu Z, Shen Y, Lakshminarasimha VB et al. Efficient low-rank multimodal fusion with modality-specific factors. In: ACL 2018—56th Annual Meeting of the Association for Computational Linguistics, Proceedings of the Conference (Long Papers), Melbourne, Australia, Vol. 1. Association for Computational Linguistics, 2018, 2247–2256. [CrossRef]
- Ali, M. & Aittokallio, T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys. Rev. 11, 31–39 (2019). [CrossRef]
- Ding Z, Zu S, Gu J. Evaluating the molecule-based prediction of clinical drug responses in cancer. Bioinformatics. 2016;32: 2891–2895. [CrossRef]
- Costello JC, NCI DREAM Community, Heiser LM, Georgii E, Gönen M, Menden MP, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nature Biotechnology. 2014. pp. 1202–1212. [CrossRef] [PubMed]
- Joshua M. Dempster, John M. Krill-Burger, James M. McFarland, Allison Warren, Jesse S. Boehm, Francisca Vazquez, et al. Gene expression has more power for predicting in vitro cancer cell vulnerabilities than genomics. bioRxiv 2020.02.21.959627. [CrossRef]
- Wail Ba-Alawi, Sisira Kadambat Nair, Bo Li, Anthony Mammoliti, Petr Smirnov, Arvind Singh Mer, Linda Z. Penn, Benjamin Haibe-Kains; Bimodal Gene Expression in Patients with Cancer Provides Interpretable Biomarkers for Drug Sensitivity. Cancer Res. 2022; 82 (13): 2378–2387. [CrossRef]
- Fischer S, Gillis J. How many markers are needed to robustly determine a cell's type? iScience. 2021 Oct 14;24(11):103292. [CrossRef]
- Takahashi, K. , Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. [CrossRef]
- Ye, Xiaolan; Ling, Bai; Xu, Hanrong; Li, Gongqi; Zhao, Xinguo; Xu, Jiangyan, et al. Clinical significance of high expression of proliferating cell nuclear antigen in non-small cell lung cancer. Medicine 99(16):p e19755, April 2020. [CrossRef]
- Wang X, Sun Z, Zimmermann MT, Bugrim A, Kocher J-P. Predict drug sensitivity of cancer cells with pathway activity inference. BMC Med Genomics. 2019;12: 15. [CrossRef]
- Rydenfelt M, Wongchenko M, Klinger B, Yan Y, Blüthgen N. The cancer cell proteome and transcriptome predicts sensitivity to targeted and cytotoxic drugs. Life Sci Alliance. 2019;2. [CrossRef]
- Park A, Lee Y, Nam S. A performance evaluation of drug response prediction models for individual drugs. Sci Rep. 2023 Jul 24;13(1):11911. [CrossRef]
- Partin A, Brettin TS, Zhu Y, Narykov O, Clyde A, Overbeek J, Stevens RL. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front Med (Lausanne). 2023 Feb 15;10:1086097. [CrossRef]
- Farzan Taj, Lincoln D Stein, MMDRP: drug response prediction and biomarker discovery using multi-modal deep learning, Bioinformatics Advances, Volume 4, Issue 1, 2024, vbae010. [CrossRef]
Figure 1.
Plasma cfmRNA profiling by cancer type, functional cluster and expression level. (A) The pie chart displayed distribution of various functional classes of cfmRNA in lung cancer; (B) Representative gene expression heatmaps showing high-, medium- and low-expressing transcripts involved in different pathways from different cancer types.
Figure 1.
Plasma cfmRNA profiling by cancer type, functional cluster and expression level. (A) The pie chart displayed distribution of various functional classes of cfmRNA in lung cancer; (B) Representative gene expression heatmaps showing high-, medium- and low-expressing transcripts involved in different pathways from different cancer types.
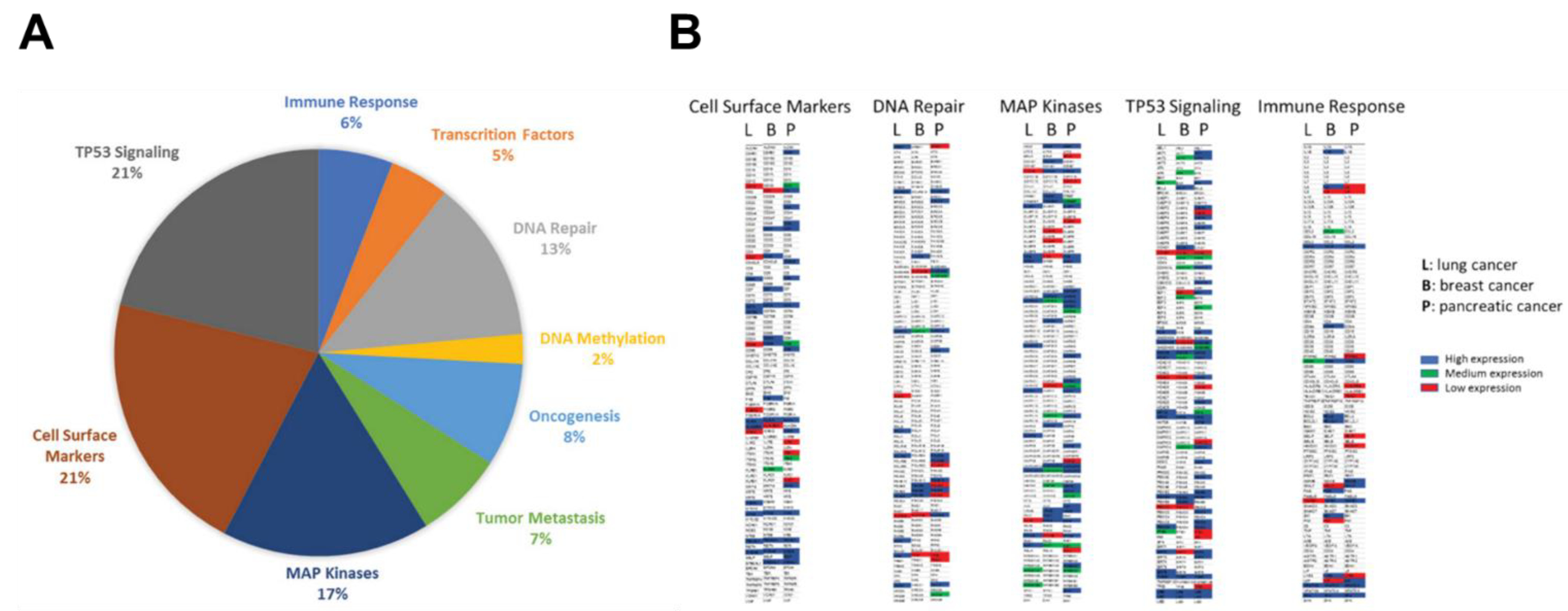
Figure 2.
Validation of the selected PGA Lung biomarkers for drug efficacy prediction. (A) Overexpression of PGA Lung biomarkers in most lung tumor tissues from the TCGA database (1,145 samples). Significant association of PGA Lung biomarkers with hypoxia (B) and MSI scores (C) in TCGA PanCancer database (510 LUAD samples).
Figure 2.
Validation of the selected PGA Lung biomarkers for drug efficacy prediction. (A) Overexpression of PGA Lung biomarkers in most lung tumor tissues from the TCGA database (1,145 samples). Significant association of PGA Lung biomarkers with hypoxia (B) and MSI scores (C) in TCGA PanCancer database (510 LUAD samples).
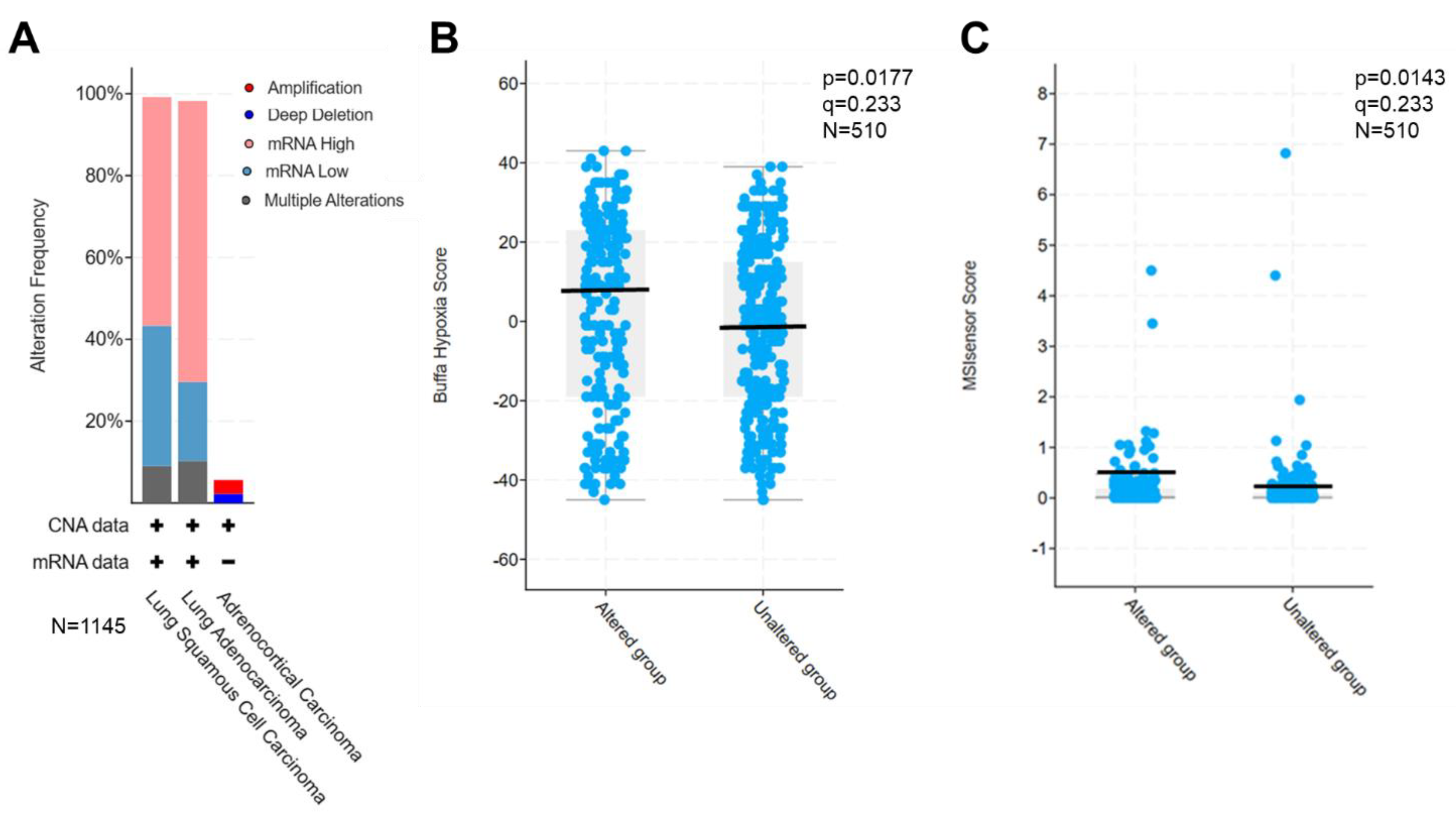
Figure 3.
Strong correlation of the PGA Lung biomarker expression levels between plasma and tissue samples. Relative cfmRNA levels were expressed as delta Ct values, whereas tissue mRNA expression was normalized as fold expression. The data showed a positive correlation between cfmRNA and tissue mRNA expression (i.e., an inverse relationship between delta Ct and fold expression).
Figure 3.
Strong correlation of the PGA Lung biomarker expression levels between plasma and tissue samples. Relative cfmRNA levels were expressed as delta Ct values, whereas tissue mRNA expression was normalized as fold expression. The data showed a positive correlation between cfmRNA and tissue mRNA expression (i.e., an inverse relationship between delta Ct and fold expression).
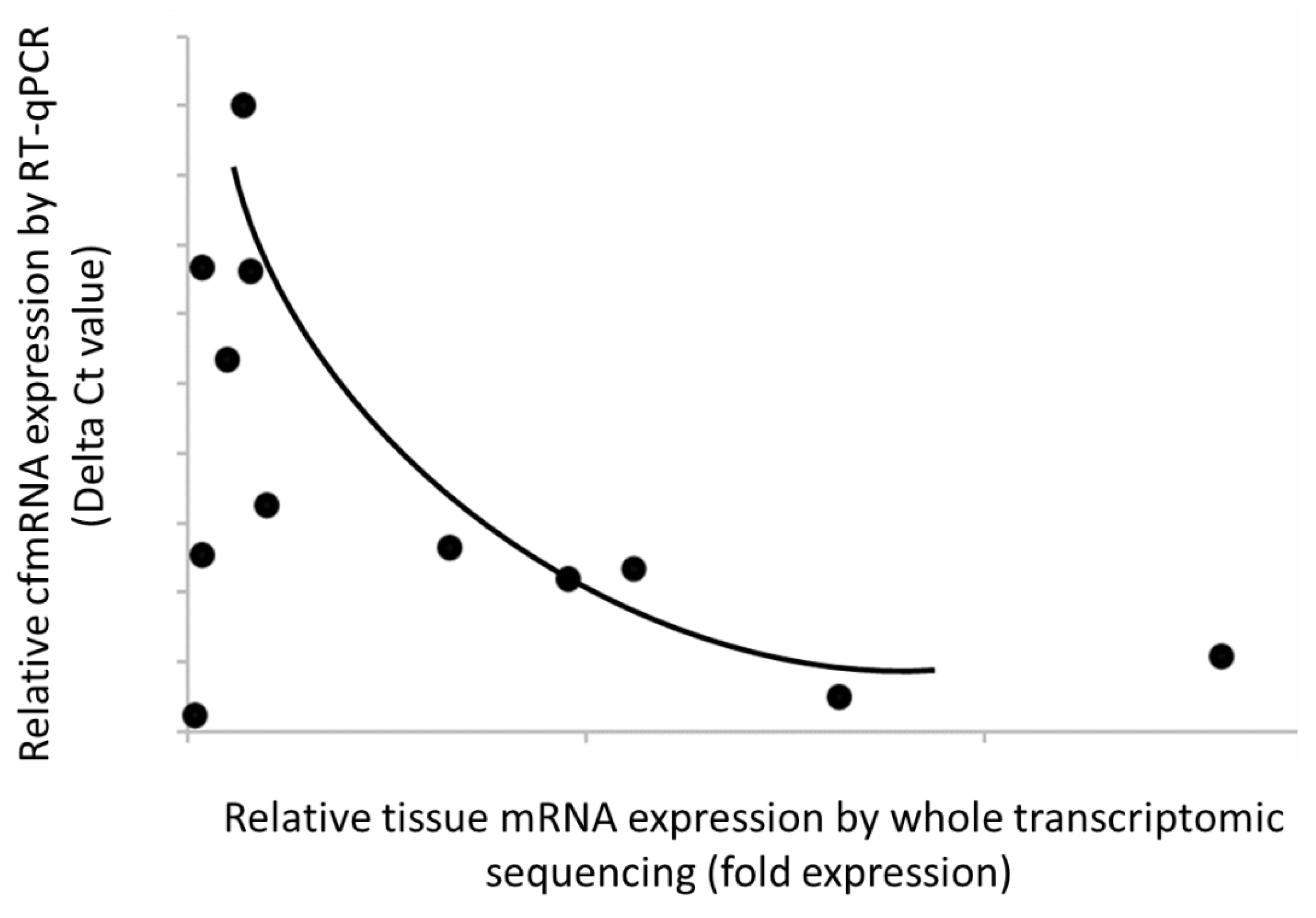
Figure 4.
Single cell RNA-Seq spatial transcriptomic analysis in lung carcinoma tissues (32,341 cells). Visualization of tumor cells expressing the key lung cancer driver genes EGFR, KRAS, BRAF, MET, HER2, ALK, ROS1, or RET. The expression patterns of EGFR, MET, HER2 and ROS1 were highly overlapped, and these EGFR/MET/HER2/ROS1-coexpressing cells only constituted a small fraction of entire tumor population. By contrast, the expression profiles of KRAS and BRAF were similar and distributed across the entire section.
Figure 4.
Single cell RNA-Seq spatial transcriptomic analysis in lung carcinoma tissues (32,341 cells). Visualization of tumor cells expressing the key lung cancer driver genes EGFR, KRAS, BRAF, MET, HER2, ALK, ROS1, or RET. The expression patterns of EGFR, MET, HER2 and ROS1 were highly overlapped, and these EGFR/MET/HER2/ROS1-coexpressing cells only constituted a small fraction of entire tumor population. By contrast, the expression profiles of KRAS and BRAF were similar and distributed across the entire section.
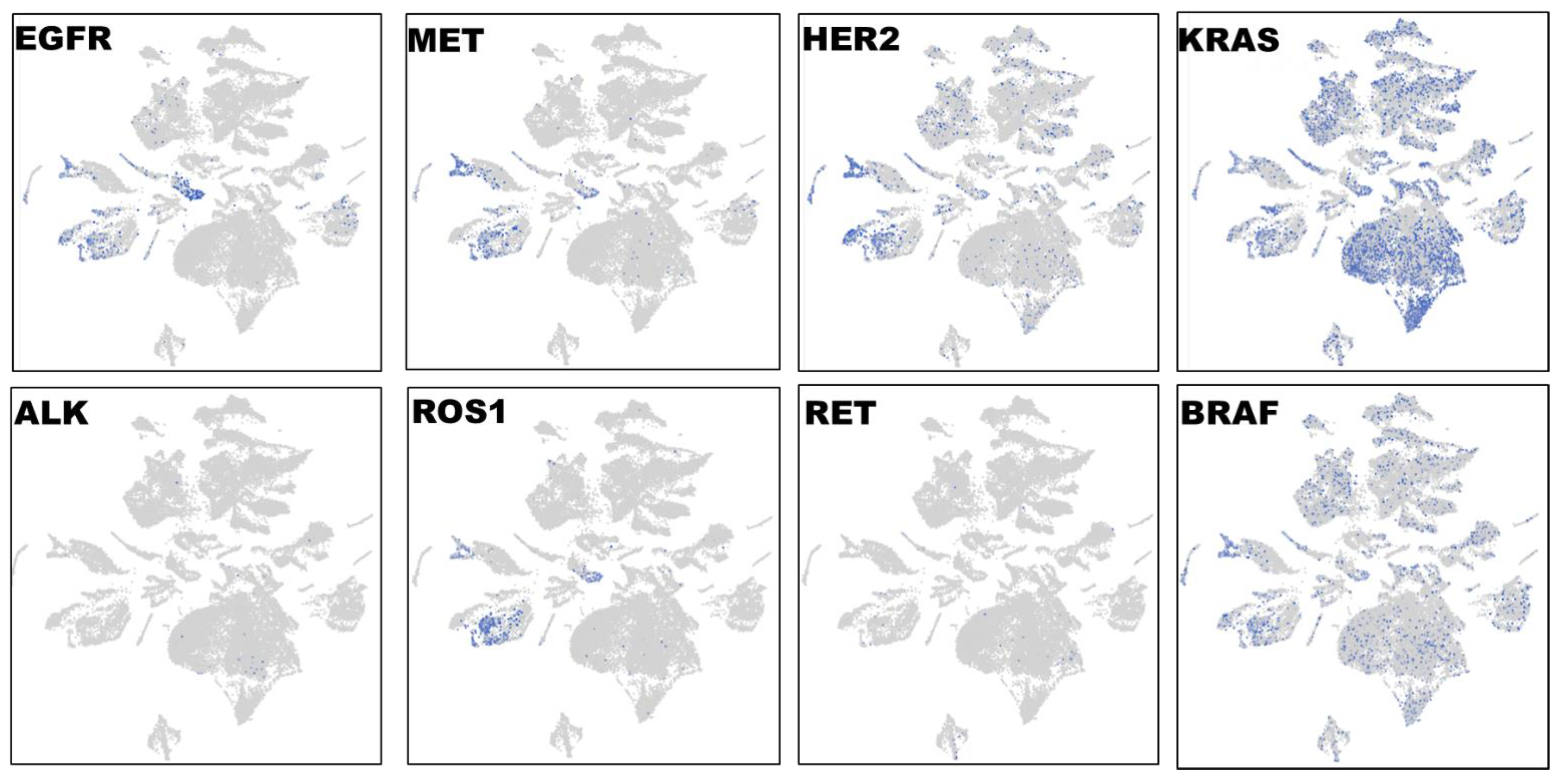
Figure 5.
Single cell RNA-Seq spatial transcriptomic analysis of PGA Lung biomarkers in (A) lung carcinoma tissues (32,341 cells) and (B) dissociated tumor cells from pleural effusion of lung adenocarcinoma patients (7,511 cells). Visualization of tumor cells expressing the representative PGA Lung biomarkers 1-8. The expression patterns of these PGA Lung genes were highly similar and distributed across the entire section, resembling to those of KRAS and BRAF. Most significantly, the population of tumor cells expressing PGA Lung biomarkers were found to be PCNA positive, indicative of high proliferation potential.
Figure 5.
Single cell RNA-Seq spatial transcriptomic analysis of PGA Lung biomarkers in (A) lung carcinoma tissues (32,341 cells) and (B) dissociated tumor cells from pleural effusion of lung adenocarcinoma patients (7,511 cells). Visualization of tumor cells expressing the representative PGA Lung biomarkers 1-8. The expression patterns of these PGA Lung genes were highly similar and distributed across the entire section, resembling to those of KRAS and BRAF. Most significantly, the population of tumor cells expressing PGA Lung biomarkers were found to be PCNA positive, indicative of high proliferation potential.
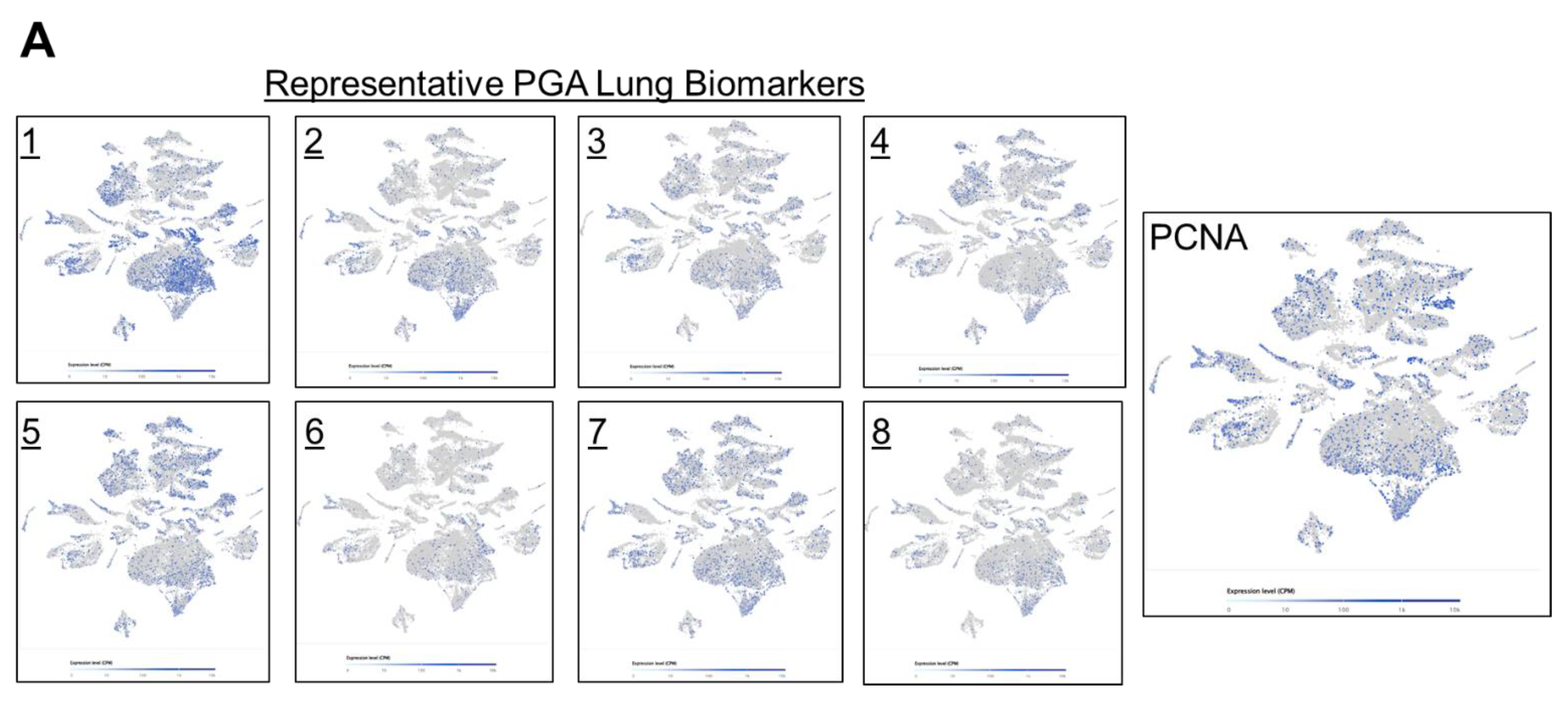
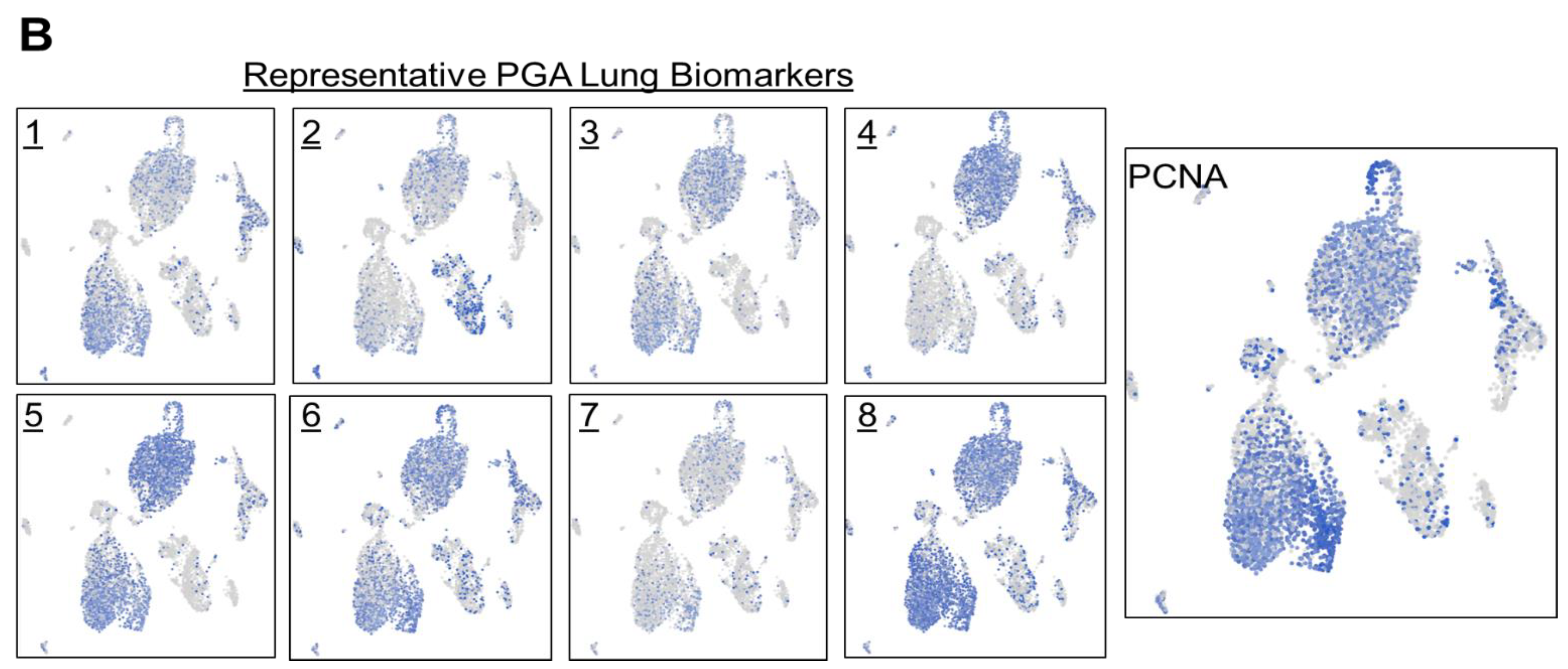
Figure 6.
Strong functional genomics similarity between TCGA lung tumors and lung cancer cell lines. Spearman correlation and normalized enrichment score (NES) were derived from expression patterns of overactive genes and the activities of cancer-related pathways.
Figure 6.
Strong functional genomics similarity between TCGA lung tumors and lung cancer cell lines. Spearman correlation and normalized enrichment score (NES) were derived from expression patterns of overactive genes and the activities of cancer-related pathways.
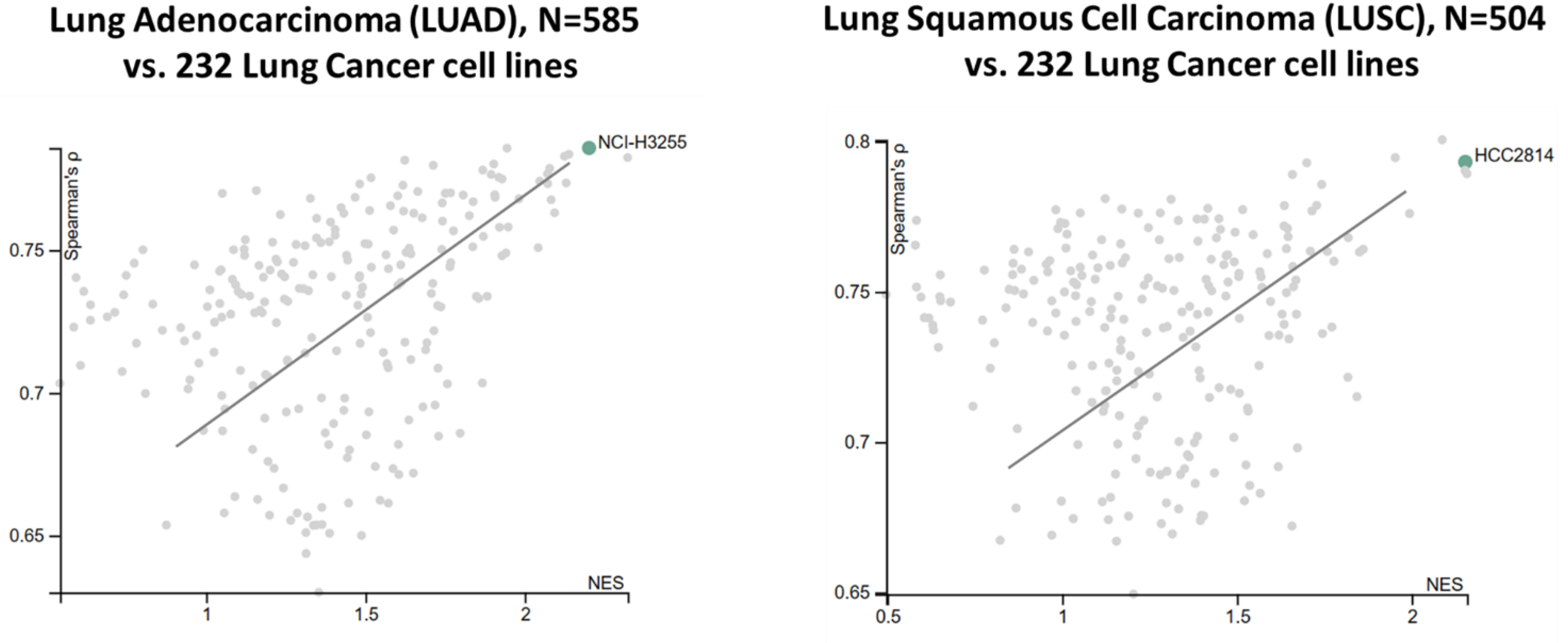
Figure 7.
Overview of in silico data fusion, annotation, mapping and analyses in the PGA Lung test.
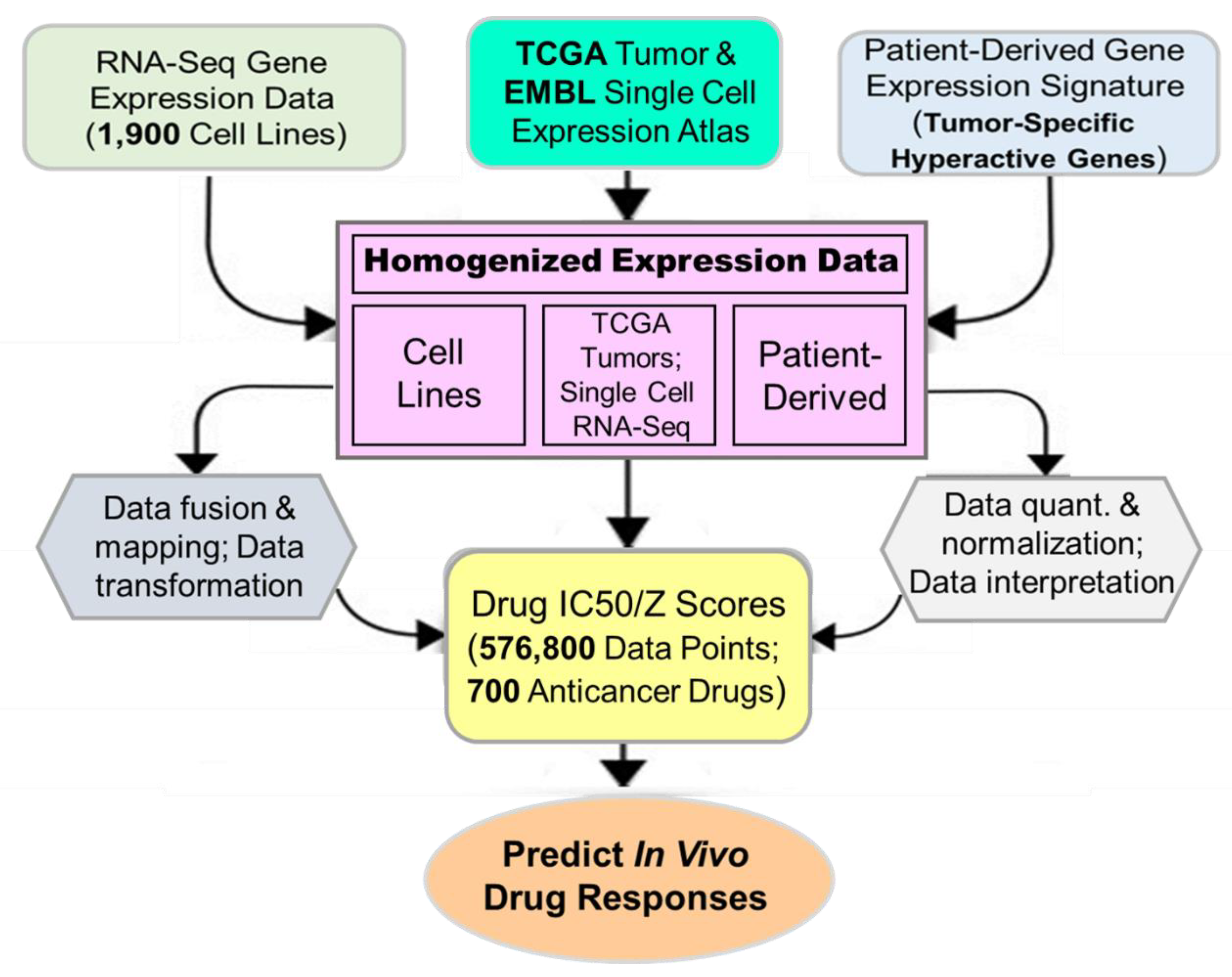
Figure 8.
Kaplan-Meier analysis of progression-free survival (PFS) and overall survival (OS) for the treatment of real-world lung cancer patients with or without the support from the PGA Lung test.
Figure 8.
Kaplan-Meier analysis of progression-free survival (PFS) and overall survival (OS) for the treatment of real-world lung cancer patients with or without the support from the PGA Lung test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



