
Preprint
Article
Mitochondrial Control Region Database in Hungarian Fallow Deer (Dama dama) Populations for Forensic Use
Altmetrics
Downloads
93
Views
50
Comments
0
A peer-reviewed article of this preprint also exists.


This version is not peer-reviewed
Submitted:
03 June 2024
Posted:
04 June 2024
You are already at the latest version
Alerts
Abstract
The evidential value of an mtDNA match between biological remains and its potential donor is determined by the random match probability of the haplotype. This probability is based on the haplotype’s population frequency estimate. Consequently, implementing a population study representative of the population relevant to the forensic case is vital to correctly evaluating the evidence. The emerging number of poaching cases and the limited availability of such data emphasizes the need for improved fallow deer mtDNA population databank for forensic purposes, including targeting the entire mitochondrial control region. By sequencing the whole mitochondrial control region in 138 animals from five populations in Hungary we found four different haplotypes, including one which has not yet been described. Our results, supplemented with data already available from previous research do not support the possibility of determining the population of origin, although some patterns of geographical separation can be distinguished. Estimates of molecular diversity indicate approximately similarly low mtDNA diversity (Hd=0.565 and π = 0.002), compared with data from other countries. The calculated random match probability of 0.547 shows a high probability of coincidence and, therefore, a limited capacity for exclusion. Consequently, it may be necessary to search for polymorphic sites on other mitochondrial gene sections or to sequence the entire mitogenome of fallow deer.
Keywords:
Subject: Biology and Life Sciences - Biochemistry and Molecular Biology
1. Introduction
The fallow deer (Dama dama) has a cosmopolitan distribution across nearly every continent worldwide, facilitated by human intervention, and boasts a significant population of more than 40,000 individuals in Hungary [1,2]. Almost 45% of this population is harvested annually, underscoring the species' substantial game management value. This is due to the infrastructure established to support their hunting, as well as the meat (venison) and trophies obtained from hunting, which also hold cultural and nature conservation significance [3]. Although hunting of fallow deer in Hungary operates within the legal framework (e.g., Act LV of 1996) [4], instances of poaching still occur. Hunting is only permitted for individuals authorized to do so, using approved hunting tools, and strictly during the designated hunting season. Failure to meet any of these conditions constitutes illegal hunting, a trend highlighted by several studies conducted in Hungary [5,6].
In cases with significant legal implications, proving the suspect's guilt poses a challenge. However, genetic identification can be employed to compare biological remains (e.g., hairs, blood contamination) found at the scene with those on the suspect’s belongings, as well as identification and selection of out portioned, vast number of uncontrolled or illegal meats from private or restaurant freezers. If the test results reveal differences between these samples, the genetic evidence cannot support the suspect's involvement in the crime. Conversely, if there is a match, it strengthens this assumption. The representativeness of the reference samples in the database is crucial in assessing the rarity of DNA profiles associated with evidence from a crime scene when both reference and evidentiary samples originate from the same geographic populations. Hence, a more precise understanding of the genetic structure of local and global deer populations is necessary to interpret matching DNA haplotypes or genotypes accurately. While a genetic method capable of identifying individual fallow deer is available, it is not universally applicable. Many crime scenes are outdoors, leading to environmental breakdown of evidential samples and significant DNA degradation over time due to factors like UV exposure, moisture, and bacterial activity.
The advantage of mitochondrial DNA (mtDNA) with a circular structure is that it is surrounded by a strong protein coat and can be found in multiple copies per cell, thus it can usually be successfully detected even in small amounts of degraded samples [7]. Therefore, mitochondrial DNA is useful to assist with the identification of the source of a biological sample (such as species- and subspecies determination) or to confirm matrilineal relatedness in phylogenetic studies [8,9,10,11]. The examination of mtDNA can be important for differentiating populations [12,13] and for determining the geographical origin, as a haplotype may become fixed in a particular region, the place of origin of an individual can be determined based on the haplotype of a biological sample of unknown origin [8]. The mutation rate of the mitochondrial non-coding control region (CR or D-loop) is five to ten times higher than the average rate of synonymous substitutions of nuclear genes and therefore more polymorphic, making it widely researched in various Cervidae species [12,13,14,15,16,17,18,19]. Although mitochondrial DNA is not a tool for individualization purposes owing to its matrilineal inheritance, mtDNA directly links maternal relatives, which can be used as match references where two or more nucleotide discrepancies are needed for a mismatch or exclusion [20], capable of excluding many potential sources despite lower discriminating power than nuclear DNA. For this marker type, it is also necessary to create databases to assess the frequency of alleles or haplotypes within a relevant population [21]. For haploid markers such as mtDNA, where profiles are expected to be shared by many matrilineally related individuals, the strength of the evidence is determined not only by the variability of the sequence but also by the size of the geographically relevant genetic database, which should be large enough to accurately reflect the local diversity [22]. It is well known from previous studies that, especially in fallow deer, the founder effect and the relatively low mutation rate of the mitochondrial genome (mitogenome) mean that there can be large sets of matrilineally related individuals sharing a common mitogenome [8,9,10,11]. While CR is widely used in various fields, the primers and consequently the length of the amplicon are not standardized. Although shorter sequences are easier to amplify, information loss can occur, therefore we plan to assess the most informative region. Since many articles support the fact that polymorphic sites may occur outside of the conventionally examined shorter, 'mutation hotspot' section, we also aimed to examine the sequence of the entire control region in the samples.
Assessment of the diversity of fallow deer populations based on the mitochondrial CR has already begun in the peripheral regions of Hungary. However, these studies examined only shorter sections of the control region: 708 bp in Southern Hungary (n=13) and 450 bp in Northeastern Hungary (n=41) [8,9] and no such data are available from other parts of the country. It was important to determine to what extent the control region can be both sufficient and efficient for the regional differentiation of the domestic fallow deer populations, and thus, to what extent it can assist in cases with legal consequences. For this reason, based on the aggregated (newly defined plus existing) haplotype data, we assessed the probability of matching and whether this extended section of CR is suitable for the regional separation of domestic fallow deer herds.
2. Materials and Methods
2.1. Sampling and DNA Extraction
Muscle or hide samples (n=138) were collected from registered shootings by hunters with a license between 2019 and 2024 from five regions in Hungary, focusing on those regions from which investigations have not been carried out so far and where the occurrence and hunting of fallow deer are common (Figure 2). Genomic DNA was isolated using a FavorPrepTM Tissue Genomic DNA Extraction Mini Kit (Favorgen Biotech, Ping-Tung, Taiwan) following the procedural guidelines. The quality of the extracted DNA was tested using a 1% agarose gel stained with GelRedTM Nucleic Acid Gel Stain (Biotium, Fremont, CA, USA), and the concentration was measured using a Qubit 2.0 Fluorometer (Life Technologies Corporation, Carlsbad, CA, USA). Isolated DNA from the tissue samples was stored at −20°C until subsequent analysis.
2.2. Mitochondrial Control Region Amplification, Sequencing, and Haplotype Determination
The most informative length and variable sites for the fallow deer CR sequences were determined by analysis of previous studies [8,9,10,11]. Based on the mitogenome sequence found in GenBank (Accession Number: NC_020700), the entire mitochondrial control region (15,400-16,146 base pair) was decided to be amplified using newly designed primers (Primer Designer 4 software [http://www.scied.com]), positioned outside the CR. A primer naming convention was used, where the primer name indicates the position of the 5’ base. The forward primer (5’-ACCCCACTATCAACACCC-3’) was defined as F15,386, and the reverse primer (5’-TATGCATAATTAGAGAAAAATTGG-3’) was defined as R16,330.
Amplification was performed in a 25 μl reaction volume containing 12.5 μl DreamTaq™ Green DNA Polymerase (ThermoFisher Scientific), 0.5 μM forward and reverse primer, 1-10 ng DNA template and PCR grade H2O to volume. PCR was carried out on 2720 Thermal Cyclers (Applied Biosystems) using the following conditions: 36 cycles of 40 sec at 94ºC, 40 sec at 56ºC, 60 sec at 72ºC, and a final extension for 2 m at 60ºC. Sequencing reactions of the purified DNA fragments (GenElute™ PCR Clean-Up Kit, Sigma-Aldrich) were carried out with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and on an ABI3500 genetic analyzer (ThermoFisher Scientific). Sequence data were analyzed by Sequence Analysis 3.4.1 (Applied Biosystems) and aligned against a reference sequence (GenBank Acc. No: NC_020700) by SequencherTM 5.4.6 software (Gene Codes Corp) for unique haplotypes identification.
Our results were supplemented with the previous control region sequences of 54 Hungarian fallow deer [8,9], downloaded from the NCBI (National Center for Biotechnology Information) GenBank database. The downloaded (n=54) and our new sequences (n=138) were aligned using MEGA11 software with ClustalW default settings [23]. Statistical tests were performed with a combined examination of a total of these 192 sequences. These sequences were also analyzed separately into six populations, mainly divided based on natural barriers such as rivers and lakes (Figure 2).
Wright's F-statistic was calculated using the DNA Sequence Polymorphism (DnaSP) software [24], and the Fst value was calculated per population pair. Additionally, we determined the number of polymorphic sites and haplotypes found in the sequence for each sampling site and all samples, as well as the haplotype and nucleotide diversity. To estimate the overall haplotype match probability for fallow deer sampled at random within the Hungarian population, we used RMP=∑pi2 where p is the frequency of the observed haplotype. The Random Match Probability, or probability of matching (PM), is defined as the probability of observing a random match between two unrelated individuals [25] and conveying the significance of a statistical match between reference and evidential sequences in forensic cases.
3. Results
Based on previous research and sequences downloaded from GenBank, we determined the investigated section of the control region and the frequency of polymorphic sites in fallow deer introduced to different countries (Figure 1). Sequences from original populations, such as Rhodes or Turkey, were not included in this examination, as they have many more variable sites, indicating that they still possess some of the original genetic variety of fallow deer not found in the current introduced populations thus excluding them from the study’s main target [8,9,10,11]. Figure 1. shows that the entire length of the mitochondrial control region, as well as the section that follows, contains variable SNP (Single Nucleotide Polymorphism) sites. Based on this information, a 920 bp long section containing the whole CR was amplified with the designed primer pair, between positions 15,400 and 16,320. Due to our selected CR section, four haplotypes were detected for a total of 138 fallow deer samples from the five sampling sites, three of which were already described in previous research [8,9]. A new haplotype sequence was detected from the NM region (Figure 1 and Figure 2), which has been uploaded to GenBank (Accession Number: PP558272). Based on the sequence alignments, we standardized the names of the haplotypes for the current and previous results obtained from Hungarian fallow deer (Figure 1), and henceforth we used these names.
We analyzed the observed haplotypes and their frequencies, supplemented with domestic fallow deer sequences (n=54) from previous research [8,9]. This process resulted in the creation of six populations, within each of which three haplotypes were detected, except for the North-West (NW) region, where only two haplotypes occurred (Figure 2). Altogether, six haplotypes were identified, including six variable sites among the 192 individuals (Figure 1). All polymorphic positions were the result of substitutions, with no indels observed. Regarding haplotype frequencies, 66 individuals had Hun1 (34%, previously named H1 or Hap17 [8,9], and 108 had Hun2 (56%, previously named H2 [8]). One individual each had Hun3 (previously named H3 [9]), and Hun4 (0.5% each). In comparison, nine individuals had Hun5 (4.7%, previously named Hap5), and seven individuals had Hun6 (3.6%, previously named Hap9) haplotypes among the 192 Hungarian samples examined so far (Figure 2). Indices of molecular diversity for each group are presented in Table 1. Based on our calculation, F statistical tests showed that only two southern regions (SM and SE) differ significantly (Fst values ranged between 0.177 and 0.359) from all other sampling locations in Hungary. No significant differences (<0.15) were observed between the other sampling sites, and the negative Fst value (effectively seen as zero) was calculated between NM and NE, indicating that there is no genetic subdivision among these populations.
The aggregated frequencies from recent and previous studies provided a random match probability between the six areas ranging from 0.429 (SE) to 0.677 (SM).
4. Discussion
The importance or legal concern of representatives of the wild animal kingdom can be different by country. Fallow deer from Hungary have some of the finest known antlers for the species, and this is one of the reasons why the species frequently falls victim to poaching. In forensic cases, there is a need to obtain the most accurate and informative data possible. Based on the comparative analysis of fallow deer sequence data from several countries, differences are found in the location of the polymorphic positions of the mitochondrial control region in introduced populations with very low diversity due to the founder effect. The number and location of these variations strongly depend on the region (country) of origin of the sequences. This means that sequence regions showing mutational hotspots in one country show no diversity at all in another geographic area. At the same time, it can also be seen that the pattern of sections frequented by mutations is similar in sequences with different origins. The length and position of the corresponding genetic section to be examined must therefore be assessed based on the population study of the given country.
Mitochondrial control region databases are highly valuable for the analysis of minimal amounts and/or degraded samples (such as hair, feces, and processed samples). These databases have been developed for several species besides humans for forensic use [26,27,28,29,30]. For this reason, we aimed to assess the accuracy of the CR sequences of different lengths and whether the implementation of longer sections would be beneficial for forensic investigations. With the help of the 127 fallow deer sequences worldwide downloaded from GenBank, we showed that mutation hot spots can occur in the entire length of the CR. The examination of the Hungarian fallow deer population revealed that mutations within the CR are concentrated in a shorter section, resulting in all six SNP positions being found within the shorter (450 bp) region previously examined in Hungary [9]. However, further surveys in herds from other countries can result in different patterns.
Since fallow deer, except for the native populations in Turkey and Rhodes, have a very limited genetic diversity [8,9,10,11], every opportunity should be explored to examine potential polymorphic sites, thus detecting possible deviations. The variation reported herein was compared with the published fallow deer genetic data to determine whether our database was typical of fallow deer populations in other countries. Haplotype diversity (Hd) is the probability that the haplotypes of two randomly selected individuals differ, while nucleotide diversity (π) is the average number of nucleotide differences between sequences per base position [31]. The investigated Hungarian fallow deer populations show approximately similarly low average mtDNA diversity (Hd=0.565) and nucleotide diversity (π=0.002), compared with data from other countries (Hd=0-0.902; π=0-0.01029) [8,10] and previous data from Hungary [9].
While previous studies rarely examined more than 60 individuals per country [8,9] the current study includes broader sampling within the country to uncover additional variations. In our study, a new haplotype was registered; therefore, altogether six haplotypes have been detected so far in Hungary from 192 samples. A similar degree of polymorphism was detected in Germany, where 10 haplotypes were detected by surveying a similarly large number of samples (n=365) [10]. In other European countries, the number of haplotypes ranged between 3 and 15, investigating a few dozen samples. Determination of when sufficient samples have been ascertained to adequately represent a population depends on the population size and the mitotype diversity observed within a population. An ideal sample set would be considered saturated when sampling additional individuals from the population no longer increases the absolute number of observed types [32]. Generally, the number of observed haplotypes increases with sample size, while the proportion of rare haplotypes (i.e., encountered only once or twice) goes down [33]. Our results in most populations investigated support these statements; and as a result, the exclusion probability largely remained the same with sample size expansion [34]. In one of the regions investigated in this study, only a limited sample size was available (SW=7), thus tending to overestimate haplotype frequencies, which decreases the evidential value of an mtDNA match. Since overestimations do not increase the risk of incriminating a false suspect, under-sampling can be considered a conservative error [35].
In general, genetic subdivisions and inbreeding must be considered in wildlife forensic DNA analysis, particularly in the case of fallow deer. DNA databases that reflect the genetic composition and geographic structure are important for accurately calculating the rarity of allele frequencies and mtDNA haplotypes to determine exclusion probabilities [20]. Therefore, we examined the possibilities of the mitochondrial DNA control region to determine whether there is geographical separation of the haplotypes, which can also help solve cases with legal consequences. To evaluate the significance of a haplotype match between a biological trace and its suspected donor, a population sample should reliably represent the population to which the donor of the trace is supposed to belong. Regarding the distribution of domestic haplotypes, Hun3 and Hun4 have so far only occurred in one individual each in the north-eastern (NE) and north-middle (NM) regions of Hungary, respectively; while Hun5 and Hun6 only in Southern Hungary, in nine and seven individuals, respectively. The other two haplotypes (Hun1 and Hun2) were far more common with 90.6%, occurring almost throughout the country. Although earlier genetic research conducted in European fallow deer showed a high degree of nuclear and mitochondrial DNA diversity, only a small degree of variation is present per country due to the fixation of local allelic variants [8,9,12,17,36,37,38,39,40,41,42]. The Fst (fixation index) value provides information on the segmentation of the populations, as it shows how much the proportion of haplotypes decreases because the metapopulation consists of two or more subpopulations with different haplotype frequencies. The higher the fixation index, the more suitable the genetic marker is for separating populations. Fst ≥ 0.15 already indicates a significant genetic difference between subpopulations [43]. Our results indicate that despite the overall low genetic mtDNA diversity within the Hungarian fallow deer samples, a pattern of differentiation among the regions is present, which can have relevance from a forensic point of view.
We are aware of the limitations of our dataset in identifying individuals derived from the study of autosomal microsatellites [44]; however, these tests can be conducted faster and more easily. Furthermore, in case a nuclear DNA test is unsuccessful, this type of extranuclear genome can likely be used primarily as an exclusionary tool. Nonetheless, using these techniques without reference data for comparison may lead to incompatible case reporting, therefore comprehensive domestic genetic databases are needed. These can help investigators trace samples to their source of origin (population, species, geographic region), and aid in the arrest, conviction, and subsequent sentencing of perpetrators for smuggling, poaching, or possession [45]. Despite that our calculated random match probability (RMP) of 0.547 shows a high probability of coincidence and, therefore, a limited capacity of exclusionary potential, this work helps interpret the strength of evidence in forensic cases. As the power of genetic evidence is directly correlated to the exclusionary power generated by the local haplotype frequencies available [32], this dataset provides an additional forensic asset for a fallow deer mtDNA CR database.
5. Conclusions
Country-by-country differences in the location of polymorphic positions of mtDNA control region necessitate the assessment of local populations in connection with the examination of the most informative sections. By increasing the number of tested sample elements, although it is possible to detect new haplotypes, the probability of exclusion remains largely the same. Based on the detected uneven haplotype frequencies, there can be a partial geographical separation between the surveyed stocks. However, the probability of random matching within the populations is still very high in Hungary. Therefore, additional genetic tests may be necessary for the exclusion.
Author Contributions
O.K.Z. and P.Z. developed the methodology and made an effort to write the original draft. O.K.Z. dealt with DNA purification and PCR amplification and had a major role in data analysis. Z.W. provided the resources for the implementation of the investigation. P.L. conducted the sample taking. P.Z. and Z.P. conceived and supervised the investigation, and had substantial input into the manuscript's completion.
Funding
This study was supported by the strategic research fund of the University of Veterinary Medicine Budapest (Grant No. SRF-001.).
Institutional Review Board Statement
None of the tests described in the manuscript titled ‘Mitochondrial control region database in Hungarian fallow deer (Dama dama) populations for forensic use’ required the approval of the Ethics Committee or Institutional Review Board. There were not any experimental animals involved in the study. The samples required for the tests were obtained from animals that were shot in compliance with the current national and international laws in force. Hunting licenses are personal documents owned by professional hunters and cannot be disclosed due to the General Data Protection Regulations (GDPR). The authors do not have access to any such documents.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data will be available from the corresponding author upon request.
Conflicts of Interest
The authors declare that they have no interests that could have appeared to influence the work reported in this paper.
References
- Chapman NG, Chapman DI (1980) The distribution of fallow deer: a worldwide review. Mammal Review 10:61–138. [CrossRef]
- Pemberton J, Smith R (1985) Lack of biochemical polymorphism in British fallow deer. Heredity 55:199–207. [CrossRef]
- Faragó S, Köller J, Zoltán A (2009) Természeti-vadászati örökségünk. A legkiválóbb magyar vadásztrófeák.
- évi LV. törvény - Nemzeti Jogszabálytár. Available online: https://njt.hu/jogszabaly/1996-55-00-00.49 (accessed on 23 September 2023).
- Elek, BS. The criminalization of poaching in Hungary. Zb. Rad. 2019, 53, 653. [Google Scholar] [CrossRef]
- Szabolcsi Z, Egyed B, Zenke P, Padar Z, Borsy A, Steger V, Pasztor E, Csanyi S, Buzas Z, Orosz L (2014) Constructing STR multiplexes for individual identification of Hungarian red deer. Journal of Forensic Sciences 59:1090–1099. [CrossRef]
- Alacs EA, Georges A, FitzSimmons NN, Robertson J (2010) DNA detective: a review of molecular approaches to wildlife forensics. Forensic Science, Medicine, and Pathology 6:180–194. [CrossRef]
- Baker K, Gray H, Ramovs V, Mertzanidou D, Akın Pekşen Ç, Bilgin CC, Sykes N, Hoelzel A (2017) Strong population structure in a species manipulated by humans since the Neolithic: the European fallow deer (Dama dama dama). Heredity 119:16–26. [CrossRef]
- Kusza S, Ashrafzadeh MR, Tóth B, Jávor A (2018) Maternal genetic variation in the northeastern Hungarian fallow deer (Dama dama) population. Mammalian Biology 93:21–28. [CrossRef]
- Ludwig A, Vernesi C, Lieckfeldt D, Lattenkamp EZ, Wiethölter A, Lutz W (2012) Origin and patterns of genetic diversity of German fallow deer as inferred from mitochondrial DNA. European Journal of Wildlife Research 58:495–501. [CrossRef]
- Masseti M, Pecchioli E, Vernesi C (2008) Phylogeography of the last surviving populations of Rhodian and Anatolian fallow deer (Dama dama dama L., 1758). Biological Journal of the Linnean Society 93:835–844. [CrossRef]
- Randi E, Apollonio M (1988) Low biochemical variability in European fallow deer (Dama dama L.): natural bottlenecks and the effects of domestication. Heredity 61:405–410. [CrossRef]
- Markov GG, Kuznetsova MV, Danilkin AA, Kholodova MV, Sugár L, Heltai M (2015) Genetic diversity of the red deer (Cervus elphus L.) in Hungary revealed by cytochrome b gene. 1: Acta Zoologica Bulgarica 67.
- Fajardo V, González I, López-Calleja I, Martín I, Rojas M, Hernández P, García T, Martín R (2007) Identification of meats from red deer (Cervus elaphus), fallow deer (Dama dama), and roe deer (Capreolus capreolus) using polymerase chain reaction targeting specific sequences from the mitochondrial 12S rRNA gene. Meat Science 76:234–240. [CrossRef]
- Brodmann P, Nicholas G, Schaltenbrand P, Ilg E (2001) Identifying unknown game species: experience with nucleotide sequencing of the mitochondrial cytochrome b gene and a subsequent basic local alignment search tool search. European Food Research and Technology 212:491–496. [CrossRef]
- Parkanyi V, Ondruska L, Vasicek D, Slamecka J (2014) Multilevel D-loop PCR identification of hunting game. Applied & Translational Genomics 3:1–7. [CrossRef]
- Douzery E, Randi E (1997) The mitochondrial control region of Cervidae: evolutionary patterns and phylogenetic content. Molecular Biology and Evolution 14:1154–1166. [CrossRef]
- Polziehn R, Strobeck C (1998) Phylogeny of wapiti, red deer, sika deer, and other North American cervids as determined from mitochondrial DNA. Molecular Phylogenetics and Evolution 10:249–258. [CrossRef]
- Polziehn RO, Strobeck C (2002) A phylogenetic comparison of red deer and wapiti using mitochondrial DNA. Molecular Phylogenetics and Evolution 22:342–356. [CrossRef]
- Kanthaswamy S (2015) domestic animal forensic genetics–biological evidence, genetic markers, analytical approaches and challenges. Animal Genetics 46:473–484. [CrossRef]
- Tully G, Wetton JH (2009) Interpretation of mitochondrial DNA evidence. Wiley Encyclopedia of Forensic Science 1–10. [CrossRef]
- Khademi TG (2014) A review of the biological status of Persian fallow deer (Dama mesopotamica), a precious and endangered animal species in Iran.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution 35:1547. [CrossRef]
- Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution 34:3299–3302. [CrossRef]
- Balding DJ, Nichols RA (1994) DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Science International 64:125–140. [CrossRef]
- Johnson RN, Wilson-Wilde L, Linacre A (2014) Current and future directions of DNA in wildlife forensic science. Forensic Science International: Genetics 10:1–11. [CrossRef]
- Duleba A, Skonieczna K, Bogdanowicz W, Malyarchuk B, Grzybowski T (2015) Complete mitochondrial genome database and standardized classification system for Canis lupus familiaris. Forensic Science International: Genetics 19:123–129. [CrossRef]
- Ottolini B, Lall GM, Sacchini F, Jobling MA, Wetton JH (2017) Application of a mitochondrial DNA control region frequency database for UK domestic cats. Forensic Science International: Genetics 27:149–155. [CrossRef]
- Zhao K, Ishida Y, Green CE, Davidson AG, Sitam FA, Donnelly CL, De Flamingh A, Perrin-Stowe TI, Bourgeois S, Brandt AL, others (2019) Loxodonta Localizer: a software tool for inferring the provenance of African elephants and their ivory using mitochondrial DNA. Journal of Heredity 110:761–768. [CrossRef]
- Syndercombe Court D (2021) Mitochondrial DNA in forensic use. Emerging Topics in Life Sciences 5:415–426. [CrossRef]
- Nei M (1987) Molecular evolutionary genetics.
- Grahn RA, Alhaddad H, Alves PC, Randi E, Waly NE, Lyons LA (2015) Feline mitochondrial DNA sampling for forensic analysis: when enough is enough! Forensic Science International: Genetics 16:52–57. [CrossRef]
- Verscheure S, Backeljau T, Desmyter S (2013) Reviewing population studies for forensic purposes: Dog mitochondrial DNA. ZooKeys 381. [CrossRef]
- Webb K, Allard M (2010) Assessment of minimum sample sizes required to adequately represent diversity reveals inadequacies in datasets of domestic dog mitochondrial DNA. Mitochondrial DNA 21:19–31. [CrossRef]
- Salas A, Bandelt H-J, Macaulay V, Richards MB (2007) Phylogeographic investigations: the role of trees in forensic genetics. Forensic Science International 168:1–13. [CrossRef]
- Say L, Naulty F, Hayden T (2003) Genetic and behavioural estimates of reproductive skew in male fallow deer. Molecular Ecology 12:2793–2800. [CrossRef]
- Pitarch JL, Raksa HC, Arnal MC, Revilla M, Martínez D, Fernández de Luco D, Badiola JJ, Goldmann W, Acín C (2018) Low sequence diversity of the prion protein gene (PRNP) in wild deer and goat species from Spain. Veterinary Research 49:1–7. [CrossRef]
- Webley LS, Zenger KR, Hall GP, Cooper DW (2007) Genetic structure of introduced European fallow deer (Dama dama dama) in Tasmania, Australia. European Journal of Wildlife Research 53:40–46. [CrossRef]
- Luikart G, Allendorf F, Cornuet J, Sherwin W (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. Journal of Heredity 89:238–247. [CrossRef]
- Hartl G, Schleger A, Slowak M (1986) Genetic variability in fallow deer, Dama dama L. Animal Genetics 17:335–341. [CrossRef]
- Scandura M, Tiedemann R, Apollonio M, Hartl GB (1998) Genetic variation in an isolated Italian population of fallow deer Dama dama as revealed by RAPD-PCR. 1: Acta Theriologica 43.
- Fadhil A, others (2023) Documentation of Genetic Diversity by Insulin-Like Growth Factor1 Receptor (Exon2) Gene for Fallow Deer (Dama dama) in Iraq. 6: Archives of Razi Institute 78.
- Frankham R, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics.
- Zorkóczy OK, Turi O, Wagenhoffer Z, Ózsvári L, Lehotzky P, Pádár Z, Zenke P (2023) A Selection of 14 Tetrameric Microsatellite Markers for Genetic Investigations in Fallow Deer (Dama dama). 2: Animals 13, 2083.
- Kanthaswamy S (2023) Wildlife forensic genetics—Biological evidence, DNA markers, analytical approaches, and challenges.
Figure 1.
Comparison of the polymorphic sites of the mitochondrial control region (CR) to the reference sequence (NC_020700) based on the length of the investigated segment in introduced fallow deer stocks. Ref: previous studies and sequences downloaded from GenBank (GB). Colour codes: 

 polymorphic sites per 10 base pairs; N: number of sequences, O: number of countries the samples originated from, S: number of variable sites, k: number of haplotypes, GB': MK473448-50; GB'': ON321840-41; GB''': OQ535577-79; GB'''': OR220344-89; GB''''': OR232305-17; ni: no information.
polymorphic sites per 10 base pairs; N: number of sequences, O: number of countries the samples originated from, S: number of variable sites, k: number of haplotypes, GB': MK473448-50; GB'': ON321840-41; GB''': OQ535577-79; GB'''': OR220344-89; GB''''': OR232305-17; ni: no information.
polymorphic sites per 10 base pairs; N: number of sequences, O: number of countries the samples originated from, S: number of variable sites, k: number of haplotypes, GB': MK473448-50; GB'': ON321840-41; GB''': OQ535577-79; GB'''': OR220344-89; GB''''': OR232305-17; ni: no information.
Figure 1.
Comparison of the polymorphic sites of the mitochondrial control region (CR) to the reference sequence (NC_020700) based on the length of the investigated segment in introduced fallow deer stocks. Ref: previous studies and sequences downloaded from GenBank (GB). Colour codes: polymorphic sites per 10 base pairs; N: number of sequences, O: number of countries the samples originated from, S: number of variable sites, k: number of haplotypes, GB': MK473448-50; GB'': ON321840-41; GB''': OQ535577-79; GB'''': OR220344-89; GB''''': OR232305-17; ni: no information.
polymorphic sites per 10 base pairs; N: number of sequences, O: number of countries the samples originated from, S: number of variable sites, k: number of haplotypes, GB': MK473448-50; GB'': ON321840-41; GB''': OQ535577-79; GB'''': OR220344-89; GB''''': OR232305-17; ni: no information.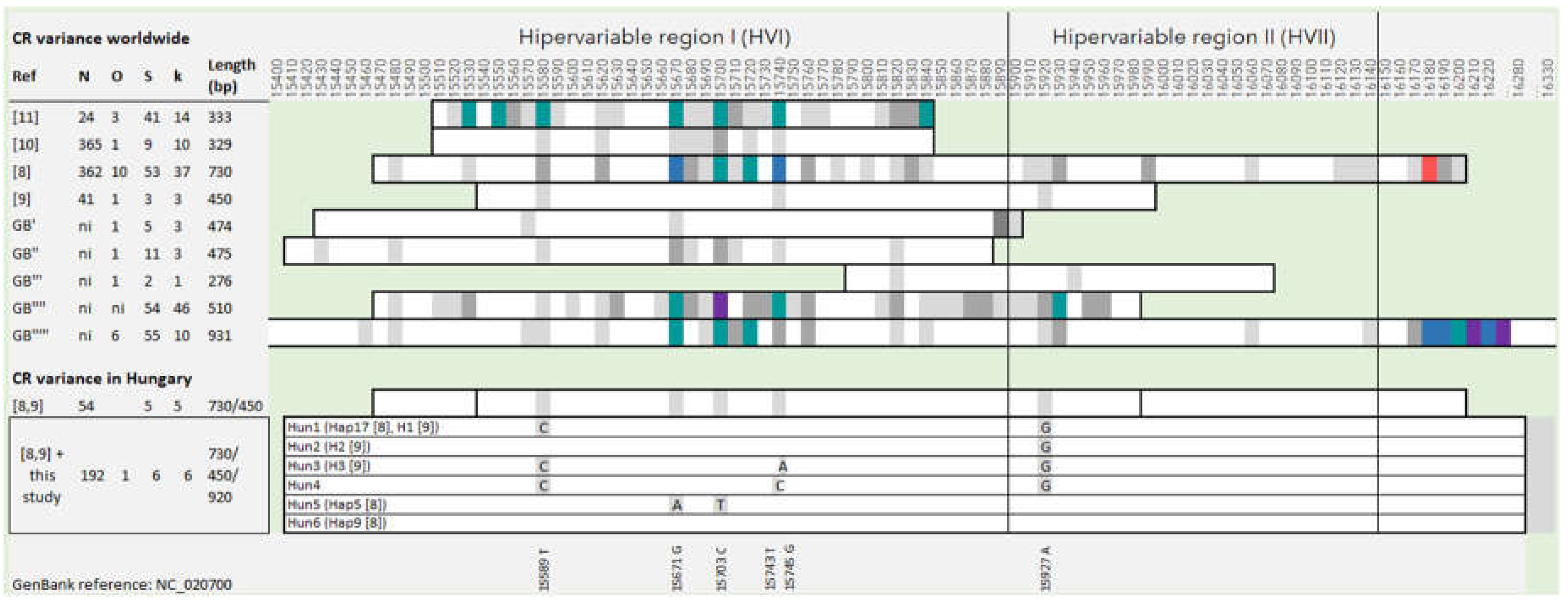
Figure 2.
The distribution of haplotypes Hun1-Hun6 of the fallow deer populations studied so far in Hungary in six regions. n: sample size, ⁎: seven and six sequences from populations SW and SE, respectively, originated from a previous study [8].
Figure 2.
The distribution of haplotypes Hun1-Hun6 of the fallow deer populations studied so far in Hungary in six regions. n: sample size, ⁎: seven and six sequences from populations SW and SE, respectively, originated from a previous study [8].
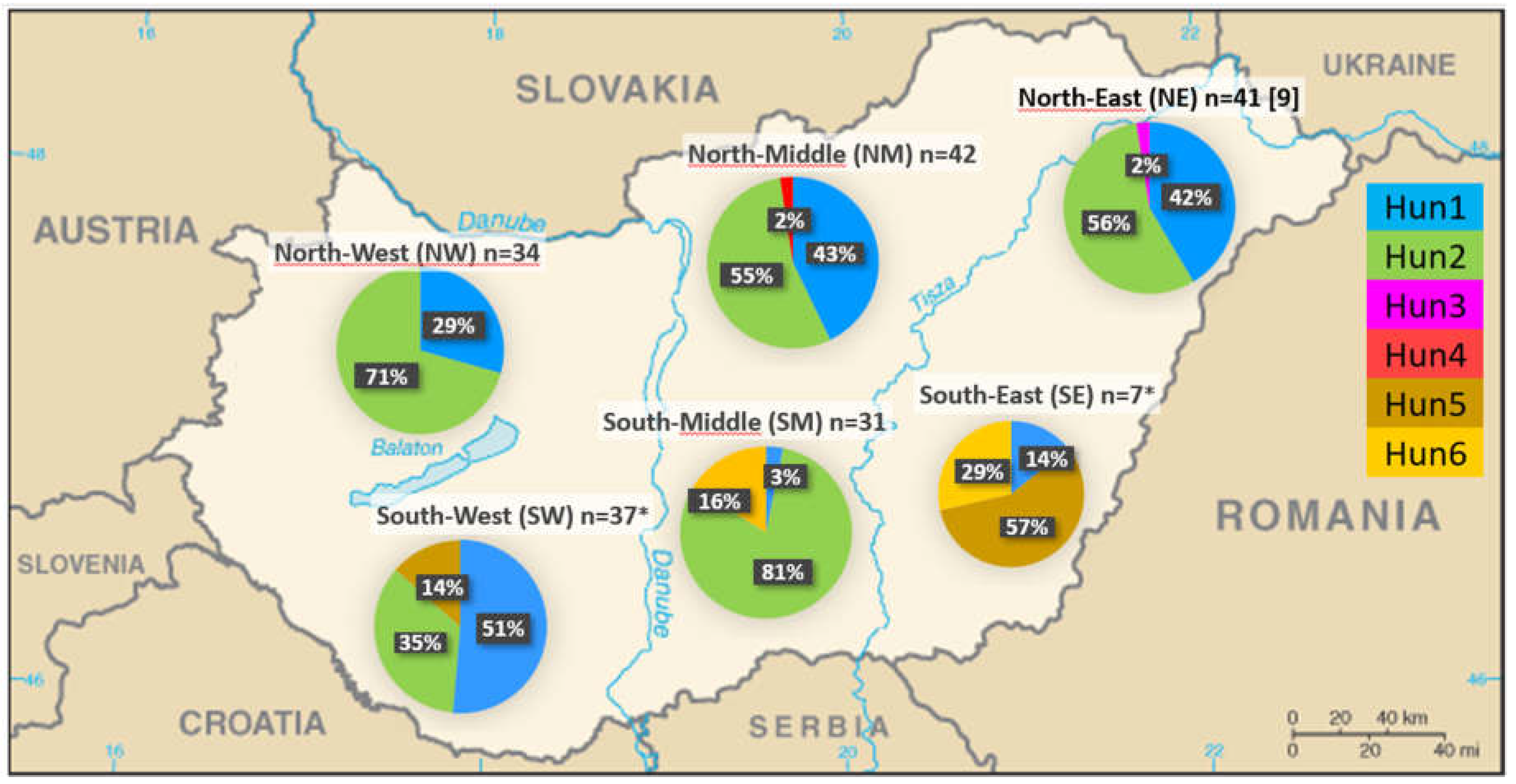

Table 1.
Parameters of the six sampling sites analyzed by the DnaSP program. NW=North-West, NM=North-Middle, NE=North-East, SW=South-West, SM=South-Middle, SE=South-East, n: sample size, S: number of variable sites, k: number of haplotypes, Hd: haplotype diversity, π: nucleotide diversity.
Table 1.
Parameters of the six sampling sites analyzed by the DnaSP program. NW=North-West, NM=North-Middle, NE=North-East, SW=South-West, SM=South-Middle, SE=South-East, n: sample size, S: number of variable sites, k: number of haplotypes, Hd: haplotype diversity, π: nucleotide diversity.
| Region | NW | NM | NE [9] | SW | SM | SE | Sum |
|---|---|---|---|---|---|---|---|
| n | 34 | 42 | 41 | 37 | 31 | 7 | 192 |
| S | 1 | 2 | 2 | 3 | 2 | 4 | 6 |
| k | 2 | 3 | 3 | 3 | 3 | 3 | 6 |
| Hd | 0.428 | 0.529 | 0.526 | 0.611 | 0.333 | 0.667 | 0.565 |
| π | 0.00095 | 0.00123 | 0.00123 | 0.00221 | 0.00076 | 0.00423 | 0.00162 |
| RMP | 0.585 | 0.484 | 0.487 | 0.617 | 0.677 | 0.429 | 0.547 |
| NW | 0.025 | 0.017 | 0.084 | 0.177 | 0.345 | Fst | |
| NM | -0.022 | 0.026 | 0.298 | 0.359 | |||
| NE | 0.028 | 0.286 | 0.356 | ||||
| SW | 0.291 | 0.237 | |||||
| SM | 0.324 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated