
Submitted:
04 June 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
The genus Amomum encompasses six medicinal species that are extensively utilized and have a significant historical background. Due to their morphological similarities, however, the presence of counterfeit and substandard products remains a challenge. Accurate plant identification is therefore essential to address these issues. This study utilized 11 newly sequenced samples along with extensive data from NCBI to perform molecular identification of these six species. The plastomes of Amomum displayed a typical quadripartite structure with conserved gene content, yet showed independent variations in the SC/IR boundary shifts at both inter- and intra-specific levels. Our approach incorporated ITS, ITS1, ITS2, complete plastomes, matK, rbcL, and psbA-trnH sequences for molecular identification, which effectively differentiated the six medicinal species within the genus Amomum, as confirmed by distance-based and phylogenetic tree analyses. Among these, the ITS, ITS1, and complete plastomes sequences demonstrated the highest identification success rate (3/6), followed by ITS2, matK, and psbA-trnH (1/6). In contrast, rbcL failed to identify any species. This research successfully established a reliable molecular identification method for Amomum plants, to protect wild plant resources and promote the sustainable use of medicinal plants and restrict the exploitation of these resources.
Keywords:
Amomum
; plastome
; DNA barcoding
; ITS
; medicinal plants
; species identification
1. Introduction
Species identification is crucial in field of biology and ecology, holding widespread importance [1]. It serves as the foundation for ecological research, enabling the understanding of species richness, diversity, and ecosystem health [2]. Additionally, it aids in identifying endangered, invasive, and keystone species within ecosystems, facilitating effective conservation and management strategies [3]. Moreover, species identification plays a crucial role in predicting and preventing infectious disease outbreaks by identifying potential disease hosts and transmitters among wild animal species [4]. In food production industries, species identification ensures authenticity, quality, and safety, preventing fraud and the circulation of substandard products [5]. Furthermore, it has relevance in criminal and forensic cases, aiding in identifying the origin of wildlife products. [6]. Traditional methods of species identification, relying on morphological characteristics, have limitations in discriminating taxa with minimal morphological differences or complex phylogeny. To overcome these challenges, DNA barcoding technology has emerged as an effective advancement.
DNA barcoding is a molecular biology technique for the identification of biological species by examining distinct DNA segments. It uses variations in short DNA sequences to provide rapid and reliable species identification [5,7,8,9,10,11]. DNA barcoding enables the analysis of specific gene regions, aiding in the identification and differentiation of morphologically similar species [5,10]. The concept of DNA barcoding was first proposed by Paul Hebert, who suggested using a small, highly conserved genetic sequence called the “ribosomal RNA gene region” to identify species [5]. Initially, DNA barcoding was widely used in animal, where the gene encoding cytochrome c oxidase I (COI) in mitochondria has a high species differentiation potential, especially in insects, birds and fish [12,13]. Therefore, the COI gene has become the preferred choice for universal DNA barcoding in animals due to its high level of accuracy in species identification [14]. However, in plant mitochondrial genomes, the COI gene shows a high degree of conservation and is not suitable as a DNA barcode selection [15]. In addition, complex evolutionary events such as hybridization, polyploidization, and lineage selection are more common in plants than in animals, further increasing the difficulty of screening fragments suitable for DNA barcoding [16]. Unlike the universal COI gene fragments in animals, DNA barcoding research in plants has undergone a screening process of a large number of fragments [17]. Currently, the internationally recognized universal plant DNA barcodes include four gene regions including ITS (internal transcribed spacer: internal transcribed spacer 1-5.8S-internal transcribed spacer 2), matK, rbcL and psbA-trnH [18]. The selection of these gene regions takes into account the genetic diversity and evolutionary history of the plant kingdom to improve the identification ability and applicability of plant DNA barcodes. But these fragments also have limitations, so Kane and Cronk proposed ultra-barcoding, which uses the complete plastomes for plant species identification [19]. DNA barcoding has significant success in plant species identification and classification and has provided a common standard for the international botanical community [20]. It has been widely used in diverse biological areas such as unveiling hidden species, identifying invasive ones, and elucidating food networks [21]. Moreover, it serves as a reliable method for verifying herbal medicinal products, detecting instances of product substitution, and contamination [22,23,24,25]. Distressingly, it’s not uncommon to find herbs that appear similar being used as adulterants in the commercial herbal arena. Although discerning closely related species using DNA barcoding can pose challenges, the technique excels in distinguishing between species that are morphologically indistinguishable but genetically distinct [26]. In conclusion, DNA barcoding is a valuable tool in biological research, enabling rapid and reliable species identification and classification in diverse organisms.
Amomum Roxb. is the second-largest genus in the Zingiberaceae family after Alpinia, which includes approximately 111 [27] to 150 [28,29] species distributed in tropical Asia and Australia, particularly in Southeast Asia, such as India, Malaysia and Indonesia [29]. In China, Amomum comprises 39 species (29 endemic, one introduced) [29], mainly distributed across provinces like Fujian, Guangdong, Guangxi, Guizhou, Yunnan and Tibet [28]. Among them, six species have been listed in the Chinese Pharmacopoeia [30]. These species encompass (1) A. compactum Solander ex Maton (synonyms: Wurfbainia compacta (Sol. ex Maton) Škorničk. & A.D.Poulsen [31]), (2) A. kravanh Pierre ex Gagnep. (synonyms: A. krervanh Pierre ex Gagnep [32], W. vera (Blackw.) Škorničk. & A.D.Poulsen and A. verum Blackw. [32,33]), (3) A. longiligulare T. L. Wu (synonyms: W. longiligularis (T.L.Wu) Škorničk. & A.D.Poulsen [34]), (4) A. tsao-ko Crevost et Lemarie(synonym: Lanxangia tsao-ko (Crevost & Lemarié) M.F.Newman & Škorničk [35]), (5) A. villosum Lour. (synonyms: W. villosa (Lour.) Škorničk. & A.D.Poulsen [36]) and (6) A. villosum var. xanthioides (Wall.ex Bak.) T.L.Wu & S.J.Chen (synonyms: W. villosa var. xanthioides (Wall. ex Baker) Škorničk. & A.D.Poulsen [37]). They exhibit a diverse range of characteristics and applications. For instance, A. compactum is a widely used culinary spice, and its fruits, leaves and seeds have a wide range of pharmacological activities in traditional medicine, such as antifungal, antibacterial, antioxidant, gastroprotective, anti-inflammatory, immunomodulatory, anticancer, antiasthmatic and acute renal failure [38]. Fruits of A. kravanh have showed antibacterial activity [39]. The active ingredients in A. longiligulare and A. villosum var. xanthioides have antibacterial activity [40,41]. Besides, powerful antioxidant properties of A. villosum var. xanthioides in the treatment of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [42]. A. tsao-ko has been found to contain antifungal active substances [43] and antioxidant ingredients [44], indicating its potential medicinal properties; recent research also suggests that it has the ability to relieve constipation and could be a promising candidate for developing laxatives in the future [45]. The total flavonoids extracted from A. villosum have shown promising potential for developing new drugs to treat gastric cancer [46]. Chemical components found in the seeds of A. villosum can enhance cellular antioxidant activity, as reported by [47]. Additionally, Chen et al. (2018) have confirmed the potential beneficial effects of A. villosum in the treatment of inflammatory bowel disease [48]. Moreover, Li et al. (2016) have demonstrated that the fresh stems and leaves of A. villosum can be used as high-quality feed for cattle, sheep, and other grass-eating livestock [49]. However, their morphological similarities make it easy to confuse these species with one another, and they are also prone to being replaced by other species within the same genus. Therefore, employing molecular identification through DNA barcoding is crucial for accurately identifying Amomum six species.
The ITS sequence is approximately 500-700 bp long. It exhibits a high degree of conservation, making them applicable across a wide spectrum of biological species, particularly in the case of plants and fungi. The sequencing and analysis of ITS are often characterized by their rapidity and cost-effectiveness, especially when compared to traditional morphological classification methods. Additionally, an extensive repository of ITS is available in public databases, providing researchers with a wealth of reference resources that facilitate expedited species identification and classification. The GenBank database at the National Center for Biotechnology Information (NCBI) hosts an extensive collection of ITS sequences for Amomum and its synonymous plants. As of April 11, 2024, it includes 572 sequences that represent 159 species. This extensive dataset serves as a valuable resource for our DNA barcoding research, providing comprehensive and diverse information. In the identification of medicinal plants and distinguishing them from counterfeits, Selvaraj et al. (2012) found that ITS and specifically ITS1 (internal transcribed spacer 1) are effective DNA barcodes for Boerhavia diffusa Linnaeus [50]. The ITS2 (internal transcribed spacer 2) region has been utilized for the identification of medicinal plants and their closely related species [51], such as within the Polygonaceae A. L. Jussieu family [52] and the Dendrobium Sw. genus [53]. The ITS2 region has been demonstrated to be the most promising universal DNA barcode for Zingiberaceae Martinov family [54]. The super-barcode complete plastomes, as well as the matK and rbcL genes, can effectively distinguish A. compactum, A. longiligulare, and A. villosum [55,56]. Additionally, the matK gene and the psbA-trnH intergenic spacer exhibit high identification efficiency for A. tsao-ko and other Amomum species [57]. Among them, the barcodes that are more effective for molecular identification of Amomum are ITS [57,58,59], ITS1 [60,61], and ITS2 [61,62,63]. These research findings demonstrate the promising potential application of DNA barcoding technology in species identification and classification within Amomum. By using DNA barcoding, researchers can accurately identify and classify different Amomum species, which helps us understand their diversity and evolutionary relationships, and provides effective tools and methods for the protection, sustainable utilization and medicinal value research of Amomum.
In this study, we employed a combination of newly sequenced data and sequences obtained from the NCBI database, including (1) ITS, (2) ITS1, (3) ITS2, (4) complete plastomes, (5) matK, (6) rbcL, and (7) psbA-trnH, to facilitate the calibration and precise identification of six medicinal plants within the Amomum genus. By utilizing DNA barcode technology, we were able to identify different Amomum medicinal species at the molecular level, thereby reducing the potential errors associated with traditional morphological methods. Our findings have the potential to enhance the sustainable utilization and conservation of Amomum resources, facilitate industry development and quality control, and ultimately provide significant scientific and societal benefits.
2. Results
2.1. Plastome Structural Variation, Sequence Divergences, and Hypervariable Regions
All 41 individuals from the six examined Amomum species exhibited a quadripartite structure (Figure 1) and showed limited intraspecific variation in plastome size (Table S1). The complete plastomes of these species ranged in size from 162,678 to 164,332 bp. The lengths of the Large Single Copy (LSC), Small Single Copy (SSC), and Inverted Repeat (IR) regions in the six Amomum species ranged from 87,632 to 89,067 bp, 14,895 to 15,754 bp, and 29,642 to 29,971 bp, respectively (Table S1). There was only slight variation in the total GC content, which ranged from 36.0% to 36.4% (Table S1). However, the GC content was higher in the IR regions (41.0–41.2%) compared to the LSC (33.7–34.1%) and SSC (29.6–30.3%) regions (Table S1). The Amomum plastomes are highly conserved and encode between 121 and 133 genes, including 82 to 87 protein-coding genes, eight rRNA genes, and 30 to 38 tRNA genes (Table S1).
We compared the contraction and expansion of IRs regions at four junctions between the two IRs (IRa and IRb) and the two single–copy regions (LSC and SSC) among six species of Amomum genus (Figure. 2). Figure 2 showed the boundary shifts in the plastomes of the studied Amomum species. Specifically, the LSC/IRb boundary is embedded in the rpl22-rps19 region (except for A. compactum YWB91902-1 and A. kravanh YWB91901-1, which are directly at the rpl22 gene); the IRb/SSC and SSC/IRa boundary is within the ycf1 gene; the IRa/LSC boundary is in the rps19–psbA region. These boundary shifts exhibit independent variations both between and within species.
The nucleotide diversity (Pi) values were calculated with DnaSP to test divergence level within different regions among the six Amomum complete plastomes. The average value of nucleotide diversity (Pi) was 0.00469. The nucleotide diversity (π) value ranged from 0 to 0.02354 across the plastomes, and the most hypervariable region was ycf1 (Figure 3).
2.2. Sequence Characteristics
The matrices characteristics of ITS, ITS1, ITS2, complete plastomes, matK, rbcL and psbA-trnH of six medicinal plants of Amomum were listed in Table 1. ITS2 had the highest percentage of variable sites, but complete plastomes had the most variable sites. The same was true for singleton sites (Table 1). ITS1 had the highest percentage of parsimony informative sites (Table 1).
2.3. Distance based Species Discrimination
Analyses of intra- and interspecific Kimura 2-parameter (K2P) distances identified varying barcoding gaps within six Amomum plants across different datasets. In barcoding gap analysis, the ITS1 and complete plastomes barcodes exhibited the highest discriminatory power, successfully identifying 50% of the species (3 out of 6 species; Table S2; Figure 4; Figure 7). The ITS barcode was the next most effective, identifying 33% of the species (2 out of 6 species; Table S2; Figure 4; Figure 7). The ITS2 and psbA-trnH barcodes could only identify one species each, accounting for 17% of the species (1 out of 6 species; Table S2; Figure 4; Figure 7). The matK and rbcL barcodes were unable to identify any species (Table S2; Figure 4; Figure 7). In ABGD analysis, ITS and ITS1 performed best (3/6; 50%; Table S3) whereas other five performed the least (1/6; 17%; Table S3). The number of generated OTUs varied across ABGD analysis with the different prior intraspecific divergence both in initial and recursive partitions (Table S3).
2.4. Tree based Species Discrimination
In the ITS dataset, due to the abundance of sequences for A. villosum (W. villosa), Maximum Likelihood (ML) and Bayesian Inference (BI) trees were initially constructed for all individuals (Figures S1-S2). Subsequently, three individuals were selected from the A. villosum (W. villosa) branch of the ML tree to participate in the construction of subsequent ITS, ITS1, and ITS2 trees. Similarly, in the matK (Figures S3-S4) and rbcL (Figures S5-S6) datasets, three individuals were selected from the same branch in the ML tree for the construction of subsequent matK and rbcL trees. In both cases, these individuals were chosen from the top, middle, and bottom of the branch to represent the full range of genetic diversity.
The ML and BI topologies derived from six of the seven datasets for the six species were congruent in showing which species were monophyletic (Figure 5; Figure 7 and Figure 8; Figures S7-S16), with the exception of the ITS1 dataset, which differed from the others (Figure 6; Figure 8; Figure S17). Across all datasets, including ITS, ITS1, ITS2, complete plastomes, matK, rbcL and psbA-trnH, A. tsao-ko and all its synonymous individuals formed a monophyletic group, demonstrating the successful identification of A. tsao-ko (Figure 5, Figure 6 and Figure 7; Figures S7-S17). Similarly, A. compactum, along with all synonymous individuals, formed a monophyletic group in both the ITS, ITS1 and complete plastomes datasets (Figure 5, Figure 6 and Figure 7; Figure S7; Figure S10). In the ITS, ITS1 and complete plastomes datasets, individuals of A. kravanh, along with all synonymous individuals, exhibited a monophyletic group (Figure 5, Figure 6 and Figure 7; Figure S7; Figure S10; Figure S17). Overall, the ITS, ITS1 and complete plastomes datasets can successfully identify A. compactum, A. kravanh and A. tsao-ko (3/6; Figure 5, Figure 6 and Figure 7; Figure S7; Figure S10); the ITS2, matK, psbA-trnH datasets can successfully identify A. tsao-ko (1/6; Figures S8-S9; Figures S11-S14); the rbcL dataset cannot identify any species (Figure 8; Figures S15-S16). However, A. longiligulare, A. villosum, A. villosum var. xanthioides and their synonymous individuals didn’t form monophyly in the four datasets (Figure 5, Figure 6 and Figure 7; Figures S7-S17).
3. Discussion
3.1. Plastome Characteristics and DNA Barcodes Performance
The plastomes of Amomum are highly conserved and exhibited a typical quadripartite structure, a characteristic shared with nine species within the subfamily Zingiberoideae [64], Zingiber Boehm. [65], various species of Curcuma L. [66], and other photosynthetic angiosperms [67,68,69]. In six medicinal Amomum plants, the maximum possible species discrimination was 3/6 because A. longiligulare, A. villosum, and A. villosum var. xanthioides were non-monophyletic for both ITS, ITS1 and plastome (Figure 5, Figure 6 and Figure 7; Figure S7; Figure S10).
Taxon-specific markers present a feasible alternative that balances the costs associated with comprehensive super-barcodes, such as whole plastomes, against the limited genetic variability often found in standard barcodes. For the genus Amomum, we pinpointed the most significant mutational hotspots in the ycf1 gene with a π value of 0.02354 (Figure 3), a pattern also observed in other members of the Zingiberaceae family [65,66]. While the four conventional barcodes (ITS2, matK, rbcL, and psbA-trnH) were each only able to reliably identify a single species at most, so the ycf1 gene region could serve as a viable alternative when these standard barcodes are inadequate. Given the financial and temporal demands of full plastome sequencing, this gene region offers a cost-effective and efficient method for future population genetic research on Amomum. This approach aids in the development of a growing database for taxon-specific barcodes.
3.2. Performance Comparison of Species Delimitation Methods
Consistent with previous research [70,71,72], species delimitation outcomes vary with the data and methodologies applied. Among the methods evaluated—ABGD, BG, BI, and ML—ML stands out as the most effective for species identification, closely followed by BI, as illustrated in Figure 8. Additionally, the topological structures produced by ML and BI are largely similar, suggesting that these methods consistently achieve the highest identification rates (Figure 8). Consequently, ML is recommended as the primary choice, with BI as a secondary option. While the identification rates for ABGD and BG differ, ABGD generally outperforms BG (Figure 8), leading to a method ranking of ML > BI > ABGD > BG. Given the demonstrated robustness and efficiency of ML and BI in this study, these methods are recommended as the preferred approaches for species delimitation in DNA barcode-based identification, particularly when employing super-barcodes.
3.3. DNA Barcoding in Six Medicinal Plants within Amomum
Previous studies have indicated that the single plastome fragment is not suitable for the identification of several medicinal plants within the genus Amomum [55,61,63,73,74,75,76]. The complete plastomes have demonstrated a strong capability to differentiate species of Amomum [55]. The results of this study have further validated these findings. The ability to distinguish species of Amomum is enhanced by the length of the complete plastomes sequence, which is approximately 160,000 bp long, and its inclusion of a multitude of informative sites. However, it is important to note that the sequencing and analysis of the complete plastomes are considerably more expensive and resource-intensive compared to ITS sequences. ITS sequencing is more cost-effective and demands fewer computational resources for analysis. Despite its relatively short length of approximately 600 bp, the informative sites within the ITS region can accurately distinguish between A. compactum, A. kravanh, and A. tsao-ko, similar to the capabilities of ITS1. ITS2 can only successfully identify A. tsao-ko. Although ITS2 contains the highest proportion of variable sites, the complete plastomes holds the greatest total number of variable sites (Table 1).
Previous studies have shown that the identification rate of ITS/ITS1/ITS2 is higher compared to the plastome fragments [59,61,77]. This may be because the plastome only contains maternal genetic information [78], while ITS/ITS1/ITS2 contains richer biparental genetic information [79]. ITS sequences typically have multiple copies, which may increase its variability and improve the accuracy of species identification. Conversely, plastome fragments may only have a single copy, limiting its identification capabilities in certain taxa. Compared to this, ITS sequences may have higher variability in these taxa. These taxa may contain hybrid species or show hybridization phenomena, leading to difficulties in species identification using plastome fragments. In this case, ITS sequences may better reflect the genetic differences between the six species, thereby improving the identification rate.
In the seven datasets, some individuals were placed within monophyletic groups (Figure 5, Figure 6 and Figure 7; Figures S1-S17), which may be due to misidentification. The inclusion of some non-target species individuals in the monophyletic branches might be due to errors in species identification, given that the NCBI database has a very wide range of sources. Previously, a number of studies solely relied on distance for species identification. However, subsequent research has indicated that the barcode gap may be a result of human error in under-sampled taxonomic groups [80]. Therefore, when carrying out species identification, we should incorporate other analyses. The relationship between the minimum interspecific distance and the maximum intraspecific distance among the six species, along with the consistency between the ABGD grouping results and the tree results, provides strong evidence to support the species identification and classification of these species.
3.4. Application of NCBI Database
NCBI database has provided comprehensive biological and biomedical information [81], offering a vast collection of genetic sequences, gene expression data, protein structures, and scientific literature. Its user-friendly interface and open access policy promote global scientific collaboration. However, challenges include navigating through the extensive data and ensuring the quality and accuracy of the information due to varying submissions from researchers and institutions. This research was conducted based on a large amount of NCBI data and obtained reliable results. The NCBI database provides great convenience for research.
ITS1 sequence is approximately 100-200 bp in length. In most cases, it does not require sequencing but can be obtained directly through Polymerase Chain Reaction (PCR) amplification. This process is cost-effective and can swiftly obtain the necessary data. Furthermore, abundant ITS sequences of the genus Amomum can be directly extracted from the NCBI database. Through multiple analyses in this study, it has been mutually verified that ITS1 has the highest identification rate. This suggests that in the future identification of these six medicinal plants within the genus Amomum, ITS1 should be considered first.
3.5. ITS vs. ITS1
ITS1 is a part of ITS, both of which can be directly obtained through PCR amplification. The ITS region and/or its subregion (ITS2) have been proposed as a standard DNA barcode marker in fungi [82] and plants [83]. In our study, the identification rate of ITS1 was higher than that of ITS2, which aligned with the view that ITS1 is a better barcode than ITS2 in eukaryotes [84]. Despite the evaluation of ITS1 and ITS2 as meta-barcode markers for fungi [85], their identification efficacy as DNA barcode markers varies across different taxa. In this study, the individuals used across the ITS, ITS1, and ITS2 datasets are consistent. Therefore, this research serves as a reference, suggesting that the ITS1 dataset might be considered first in practical applications when the experimental individuals are identical.
Although ITS is significantly longer than ITS1, there is no noticeable difference in the difficulty of amplification between the two. Even though the ITS dataset had more variable sites than ITS1, it didn’t necessarily mean that it surpasses ITS1 in identification rate. Importantly, the percentage of variable sites within the ITS1 sequence is higher than that of variable sites within the entire ITS sequence. There are many factors that can affect the identification rate, including: the presence of key variable sites that can significantly distinguish species, the amount of recognizable feature information within the dataset, and the size of the differences in the species being identified. In these aspects, the ITS1 dataset may be superior to ITS, and thus, have a higher identification rate.
4. Materials and Methods
4.1. Taxon Sampling
Based on the phylogenetic relationships of the genus Amomum established by Boer et al. [86], we selected close relatives of six target species for our study. Our data collection and analysis focused on six target species, their close relatives, and the synonyms associated with both the target and related species. We sampled 11 individuals from Amomum (Table 2), as well as numerous individuals represented by ITS, complete plastomes, matK, rbcL, and psbA-trnH sequences from both Amomum and its synonymous taxa available on NCBI (Table S4). To download the second-generation sequencing data encompassing these groups, we utilized the prefetch tool in SRA Toolkit v.3.1.0, accessible at https://github.com/ncbi/sra-tools, from the NCBI database. The cut-off date for downloading data from NCBI was April 11, 2024. Detailed species information that we sequenced is listed in Table 2, and all of them have been uploaded to the NCBI GenBank database. Alpinia nigra (Gaertn.) Burtt (MF076960) and Alpinia galanga (L.) Willd. (AF478715) were chosen as outgroups for constructing the matrices of ITS, ITS1 and ITS2 sequences. For the complete plastomes, matK, rbcL and psbA-trnH matrices, A. nigra (MK940826) and A. galanga (MK940825) were selected as outgroups. This selection of outgroups was informed by the research of Gong et al. [63]. We have downloaded 232, 31, 138, 224 and 53 sequences of ITS/ITS1/ITS2, complete plastomes, matK, rbcL and psbA-trnH respectively from NCBI (Table S4). The ITS, matK and rbcL dataset contains numerous instances of A. villosum (synonym: W. villosa). Initially, we constructed a phylogenetic tree using all available data. Subsequently, we selected three individuals from the A. villosum (W. villosa) clade within the tree of A. villosum (W. villosa) based on genetic distance. These individuals, downloaded from NCBI, were chosen for further analysis.
It is worth noting that A. krervanh was revised to A. kravanh [32]. The species name of the data downloaded by NCBI cannot be changed at will, so A. krervanh on NCBI is still A. krervanh, and our own data is named the revised A. kravanh.
4.2. DNA Extraction, Sequencing, Assembly and Annotation
We extracted total DNA from 0.2 g of the gel-dried leaves and herbarium samples using the modified 4 × CTAB method [87]. The quality of DNA was assessed using 1% agarose gel electrophoresis and a NanoDrop® ND-1000 spectrophotometer. We constructed a DNA library (300-500 bp) using the NEBNext UItra II DNA library prep kit for Illumina, and performed two-end sequencing (2×150 bp) on the DNBSEQ-T7 high-throughput platform, generating a total data amount of no less than 3 Gb. The length of single-ended sequencing reads was 150 bp (sequencing strategy PE150). To convert SRA files downloaded from NCBI into FASTQ format using fasterq-dump-orig from SRA Toolkit v.3.1.0 (https://github.com/ncbi/sra-tools). Then, compress the ‘fastq’ files into ‘fastq.gz’ format suitable for GetOrganelle assembly using the open-source tool pigz v. 2.2.5 (https://zlib.net/pigz/).
The ITS sequence, spanning approximately 600-700 bp, was first assembled utilizing GetOrganelle v.1.7.5.3 [88]. Following assembly, both the resultant FASTG file and the reference from Amomum sericeum Roxb. (KY438097.1) were aligned using the Map function in Geneious v.9.0.2 [89] to prepare the sequence for annotation. Subsequently, annotation was performed through Geneious v.9.0.2[89] with the reference to acquire the ITS sequence. ITS1/ITS2 sequences were then extracted based on annotation information using Geneious v.9.0.2 [89].
The plastome assembly and annotation methods of sequences were conducted following the protocol described by Li et al. [90]. The clean data obtained from high-throughput sequencing were directly assembled using GetOrganelle v.1.7.5.3 [88], and the complete circular plastid genome was automatically generated. In cases where the circular structure could not be obtained, results were visually inspected using Bandage v.0.8.1 [91]. Subsequently, reliable plastid genome contigs or scaffolds were identified by manually removing non-target contigs from the ‘fastg’ file. The selected sequences were then manually edited and spliced to obtain a complete plastid genome. Annotation of the plastid genome was performed using Geneious v.9.0.2 [89], with the published genome of A. krervanh (NC_036935.1) as the reference, and then combined with ORF (open reading frame) for correction. The matK, rbcL and psbA-trnH were extracted using Geneious v.9.0.2 [89] based on annotation information.
4.3. Data Analysis
4.3.1. Plastome Structural Variation, Divergence, and Mutational Hotspot Analyses
We analyzed the characteristics of 41 plastomes of six medicinal Amomum plants, focusing on aspects such as genome size, gene content (including protein-coding genes, tRNAs, and rRNAs), and GC content. We performed comparative analyses on the expansion and contraction of the Inverted Repeats (IR) at the four junctions of the plastomes using Geneious v.9.0.2 [89], and visualized the results with IRscope [92]. To pinpoint hypervariable regions, we carried out a sliding window analysis using DnaSP v.5 [93], with a step size of 200 bp and a window length of 600 bp, identifying the top three sequences as the most variable regions. Finally, we constructed a physical circular map of the plastome with OGDRAW v.1.3.1 [94].
4.3.2. Sequence-Based Analyses
We conducted distance-based analysis using matrices generated from a subset of target and closely related species individuals selected from all individuals of the Amomum genus and its synonyms for tree construction according to Boer et al. [86]. Two primary species delimitation approaches were employed: barcoding gaps (BG) [95] and automatic barcode gap discovery (ABGD) [96]. To investigate the existence of barcoding gaps within each dataset (ITS, ITS1, ITS2, complete plastomes, matK, rbcL and psbA-trnH), we conducted pairwise distance calculations implemented in MEGA-11 [97] using the K2P model. A scatter plot was employed to identify barcoding gaps by visualizing the relationship between the minimum interspecific distance and maximum intraspecific distance for the six species. A species is considered accurately identified when the minimum interspecific distance is larger than its maximum intraspecific distance [98]. The ABGD analysis was conducted using an online platform (https://bioinfo.mnhn.fr/abi/publi c/abgd/), employing three distinct distance models: Jukes-Cantor [JC69], Kimura [K80] TS/TV 2.0 and Simple Distance. The analysis was configured with the following parameters: Pmin = 0.001, Pmax = 0.1, Steps = 10, X = 1.5, Nb bins = 20. The best partition was identified as the one most closely aligning with the delimitation of nominal species among the partitions obtained.
4.3.3. Phylogenetic Tree-Based Analyses
We constructed phylogenetic trees based on ML and BI methods from seven datasets: (1) ITS, (2) ITS1, (3) ITS2, (4) complete plastomes, (5) matK, (6) rbcL, and (7) psbA-trnH sequences. The sequence matrices of each dataset was aligned using MAFFT implemented in Geneious v.9.0.2 [89]. The ML tree was constructed using RAxML v.8.2.11 [99] by the GTRGAMMAI model with 1000 rapid bootstrap replicates. MrBayes v.3.2.7 [100] was utilized for BI analyses runs with 1,000,000 generations, employing the best-fit model specified according to the optimal scheme selected by jModeltest v.2.1.7 [101] using the Akaike Information Criterion (AIC) criteria. Phylogenetic trees were then visualized by tvBOT [102]. When all individuals of the same species and its synonyms cluster into a single clade, we consider it to be successfully identified.
5. Conclusions
We examined plastome structural variations and investigated the efficacy of standard and super DNA barcodes for resolving species boundaries based on within and between species variation within six medicinal Amomum plants. In this study, six medicinal plants of the genus Amomum were molecularly identified using the ITS, ITS1, ITS2, complete plastomes, matK, rbcL, and psbA-trnH sequences. Among these seven sequences, ITS, ITS1 and complete plastomes were effective in identifying A. compactum, A. kravanh, and A. tsao-ko, while ITS2, matK, and psbA-trnH only can successfully identify A. tsao-ko. In contrast, rbcL failed to identify any species. In summary, ITS, ITS1 and complete plastomes demonstrates the highest identification rate, followed by ITS2, matK, and psbA-trnH, with rbcL having the lowest identification rates. In conclusion, considering factors such as cost, for the molecular identification of the six medicinal plants within the Amomum genus, the use of ITS1 is strongly recommended. This study developed reliable molecular identification methods for the genus Amomum, crucial for protecting wild plant resources, rational use of medicinal plants, and preventing resource misuse. In summary, it provided essential molecular tools for species identification and classification, enhancing our understanding of Amomum medicinal plants.
Supplementary Material: The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The Supplementary Material for this article as follows: Tables: Supplementary Table 1. Summary of significant characteristics of six medicinal Amomum plants plastomes, including aspects of genome size, G-C content, and gene number. Supplementary Table 2. Net differences between minimum interspecific and maximum intraspecific distances for six medicinal plants in the genus Amomum across seven datasets, derived from barcoding gap analysis. Supplementary Table 3. The number of putative species recognized by automatic barcode gap discovery (ABGD) analyses of seven datasets using three distance metrics. Supplementary Table 4. All samples of Amomum and their synonyms used in this study (those marked with “*” are individuals sequenced by ourselves, others are downloaded from NCBI). Figures: Supplementary Figure 1. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS set of all individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 2. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the ITS set of all individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 3. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the matK set of all individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 4. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the matK set of all individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 5. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the rbcL set of all individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 6. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the rbcL set of all individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 7. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the ITS set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 8. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS2 set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 9. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the ITS2 set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 10. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the complete plastomes set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 11. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the matK set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 12. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the matK set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 13. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the psbA-trnH set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 14. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the psbA-trnH set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 15. Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the rbcL set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate ML bootstrap values (BS). Supplementary Figure 16. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the rbcL set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP). Supplementary Figure 17. Phylogenetic tree was reconstructed based on the Bayesian Inference (BI) method with the ITS1 set of selected individuals of Amomum and its synonyms. The numbers at nodes indicate BI posterior probabilities (PP).
Author Contributions
JBY conceived the project and designed the research; YZ carried out data analysis and wrote the manuscript with input from all co-authors; AK corrected draft syntax; all authors contributed to revisions.
Funding
The study was supported by the Obtaining Super Barcodes of Important Wild Plants in Gaoligong Mountain (Grant No. 2021FY100204) to JBY.
Data Availability Statement
The datasets presented in this study can be accessed at NCBI GenBank; the list of accessions can be found in Table 2 and Supplementary Table 4.
Acknowledgments
The authors are grateful to the iFlora High Performance Computing Center of Germplasm Bank of Wild Species for providing a stable and fast computing environment and the Germplasm Bank of Wild Species for facilitating the laboratory work. Thanks to the NCBI database for providing us with a large amount of data for analysis. We are grateful to Prof. Wen Bin Yu (Xishuangbanna Tropical Botanical Garden, CAS) for kindly providing the samples. We also thank Jing Yang, Zheng Shan He, Chun Yan Lin, Ji Xiong Yang Wen-Bin Yuan and other supporting staff from the Molecular Biology Experiment Center of GBOWS.
References
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K. L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2006, 22, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Gotelli, N.J.; Colwell, R.K. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 2001, 4, 379–391. [Google Scholar] [CrossRef]
- Soulé, M.E.; Wilcox, B.A. , Conservation biology. An evolutionary-ecological perspective. Addison-Wesle: London, U.K., 1980; pp. 395.
- Smith, K.F.; Behrens, M.; Schloegel, L.M.; Marano, N.; Burgiel, S.; Daszak, P. Reducing the risks of the wildlife trade. Science 2009, 324, 594–595. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Linacre, A.; Gusmão, L.; Hecht, W.; Hellmann, A.P.; Mayr, W.R.; Parson, W.; Prinz, M.; Schneider, P.M.; Morling, N. ISFG: recommendations regarding the use of non-human (animal) DNA in forensic genetic investigations. Forensic Sci. Int.: Genet.
- Hebert, P.D.; Gregory, T.R. The promise of DNA barcoding for taxonomy. Syst. Biol. 2005, 54, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Erickson, D.L. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2007, 2, e508. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Van Alphen Stahl, J.; Kelly, L.J.; Wikström, N.; Hollingsworth, P.M.; Duff, R.J.; Hoot, S.B. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc. 2009, 159, 1–11. [Google Scholar] [CrossRef]
- Hollingsworth, P.M. F., L. L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S. A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 12794–12797. [Google Scholar]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS One 2011, 6, e19254. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of birds through DNA barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef]
- Ward, R.D.; Holmes, B.H.; O’Hara, T.D. DNA barcoding discriminates echinoderm species. Mol. Ecol. Resour. 2008, 8, 1202–11. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.S.; Eah, J.; Kim, J.S.; Kim, Y.; Min, M.; Paek, W.K.; Lee, H.; Kim, C. DNA barcoding Korean birds. Mol. Cells 2006, 22, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Erickson, D.L.; Jones, F.A.; Swenson, N.G.; Perez, R.; Sanjur, O.; Bermingham, E. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18621–18626. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Van Alphen Stahl, J.; Kelly, L.J.; Wikström, N.; Hollingsworth, P.M.; Duff, R.J.; Sarah, B. Hoot, R.S. C., Mark W. Chase, Mike J. Wilkinson. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc. 2009, 159, 1–11. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.Z.; van der Bank, M.; Twyford, A. Telling plant species apart with DNA: from barcodes to genomes. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 2015. [Google Scholar]
- Kane, N.C.; Cronk, Q. Botany without borders: barcoding in focus. Mol. Ecol. 2008, 17, 5175–5176. [Google Scholar] [CrossRef] [PubMed]
- von Cräutlein, M.; Korpelainen, H.; Pietiläinen, M.; Rikkinen, J. DNA barcoding: a tool for improved taxon identification and detection of species diversity. Biodivers. Conserv. 2011, 20, 373–389. [Google Scholar] [CrossRef]
- Vijayan, K.; Tsou, C.H. DNA barcoding in plants: taxonomy in a new perspective. Curr. Sci. 2010, 99, 1530–1541. [Google Scholar]
- Wang, H.; Kim, M.K.; Kwon, W.S.; Jin, H.; Liang, Z.; Yang, D.C. Molecular authentication of Panax ginseng and ginseng products using robust SNP markers in ribosomal external transcribed spacer region. J. Pharm. Biomed. Anal. 2011, 55, 972–976. [Google Scholar] [CrossRef]
- Vinitha, M.R.; Kumar, U.S.; Aishwarya, K.; Sabu, M.; Thomas, G. Prospects for discriminating Zingiberaceae species in India using DNA barcodes. J. Integr. Plant Biol. 2014, 56, 760–773. [Google Scholar] [CrossRef]
- Wang, H.; Kim, M.K.; Kim, Y.J.; Lee, H.N.; Jin, H.; Chen, J.; Yang, D.C. Molecular authentication of the oriental medicines Pericarpium Citri Reticulatae and Citri Unshius Pericarpium using SNP markers. Gene 2011, 494, 92–95. [Google Scholar] [CrossRef] [PubMed]
- JungHoon, K.J. K.; EuiJeong, D.E. D.; GuemSan, L.G. L. Evaluation of medicinal categorization of Atractylodes japonica Koidz. by using internal transcribed spacer sequencing analysis and HPLC fingerprinting combined with statistical tools. Evid. Based Complement. Alternat. Med. 2016, 2016. [Google Scholar]
- Mishra, P.; Kumar, A.; Nagireddy, A.; Mani, D.N.; Shukla, A.K.; Tiwari, R.; Sundaresan, V. DNA barcoding: an efficient tool to overcome authentication challenges in the herbal market. Plant Biotechnol. J. 2016, 14, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:327296-2 (archived on 2024). 22 February.
- Xu, H.Z., <italic>Amomum</italic> Roxb. In <italic>Flora of China</italic>, Xu, H.Z., Ed. Science Press; Missouri Botanical Garden Press: Beijing, China; St. Louis, USA, 1981; Vol. 16, pp 110-135.
- Yao, J.Y., <italic>Amomum</italic> Roxb. In <italic>Flora of China</italic>, Yao, J.Y., Ed. Science Press; Missouri Botanical Garden Press: Beijing, China; St. Louis, USA, 2000; Vol. 24, pp 347–356.
- China Pharmacopoeia Commission, Pharmacopoeia of the People’s Republic of China: Part One. China Medical Science and Technology Press: Beijing, China, 2020; pp. 175–264.
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:795489-1 (archived on February 22 2024).
- iPlant. http://www.iplant.cn/info/Amomum%20krervanh (archived on February 22 2023).
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:77178294-1#synonyms (archived on February 22 2024).
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:77178281-1#synonyms (archived on February 22 2024).
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:872172-1 (archived on February 22 2024).
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:77178295-1#synonyms (archived on February 22 2024).
- Plants of the World Online Kew Science. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:892796-1 (archived on February 22 2024).
- Alkandahri, M.Y.; Shafirany, M.Z.; Rusdin, A.; Agustina, L.S.; Pangaribuan, F.; Fitrianti, F.; Farhamzah; Kusumawati, A.H.; Sugiharta, S.; Arfania, M.; Mardiana, L.A. <italic>Amomum compactum</italic>: A review of pharmacological studies. Plant Cell Biotechnol. Mol. Biol. 2021, 22, 61–69. [Google Scholar]
- Diao, W.R.; Zhang, L.L.; Feng, S.S.; Xu, J.G. Chemical composition, antibacterial activity, and mechanism of action of the essential oil from Amomum kravanh. J. Food Prot. 2014, 77, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Thinh, B.B.; Chac, L.D.; Hanh, D.H.; Korneeva, A.A.; Hung, N.; Igoli, J.O. Effect of extraction method on yield, chemical composition and antimicrobial activity of essential oil from the fruits of Amomum villosum var. xanthioides . J. Essent. Oil-Bear. Plants 2022, 25, 25–25. [Google Scholar]
- Chau, L.T. M.; Thang, T.D.; Huong, L.T.; Ogunwande, I.A. Constituents of Essential Oils from Amomum longiligulare from Vietnam. Chem. Nat. Compd. 2015, 51, 1181–1183. [Google Scholar] [CrossRef]
- Cho, J.H.; Lee, J.S.; Kim, H.G.; Lee, H.W.; Fang, Z.; Kwon, H.H.; Kim, D.W.; Lee, C.M.; Jeong, J.W. Ethyl acetate fraction of Amomum villosum var. xanthioides attenuates hepatic endoplasmic reticulum stress-induced non-alcoholic steatohepatitis via improvement of antioxidant capacities. Antioxidants.
- Moon, S.S.; Lee, J.Y.; Cho, S.C. Isotsaokoin, an antifungal agent from Amomum tsao-ko. J. Nat. Prod. 2004, 67, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.S.; Kikuzaki, H.; Hisamoto, M.; Nakatani, N. Constituents of Amomum tsao-ko and their radical scavenging and antioxidant activities. J. Am. Oil Chem. Soc. 2000, 77, 667–673. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, X.; Zhao, Y.; Liu, S.; Luo, K.; Fu, X.; Li, J.; Sheng, J.; Tian, Y.; Fan, Y. Flavonoids in Amomum tsaoko crevost et lemarie ameliorate loperamide-induced constipation in mice by regulating gut microbiota and related metabolites. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Yue, J.; Zhang, S.; Zheng, B.; Raza, F.; Luo, Z.; Li, X.; Zhang, Y.; Nie, Q.; Qiu, M. Efficacy and mechanism of active fractions in fruit of Amomum villosum Lour. for gastric cancer. J. Cancer 2021, 12, 5991–5998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, S.; Xiong, Q.; Jiang, C.; Lai, X. Extraction, characterization and biological activities of polysaccharides from Amomum villosum. Carbohydr. Polym. 2013, 95, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ni, W.; Yang, C.; Zhang, T.; Lu, S.; Zhao, R.; Mao, X.; Yu, J. Therapeutic effect of Amomum villosum on inflammatory bowel disease in rats. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Li, Q.X.; Gao, G.; Ye, K.X.; Liu, J.Y.; Huang, B.Z.; Wang, A.K.; Wang, X.; Yang, G.R. Evaluation on feeding value of stems and leaves in fructus amomi. Anim. Husb. Vet. Med. 2016, 48, 61–63. [Google Scholar]
- Selvaraj, D.; Shanmughanandhan, D.; Sarma, R.K.; Joseph, J.C.; Srinivasan, R.V.; Ramalingam, S. DNA barcode its effectively distinguishes the medicinal plant Boerhavia diffusa from its adulterants. Genomics Proteomics Bioinformatics 2012, 10, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 2010, 5, e8613. [Google Scholar] [CrossRef] [PubMed]
- Youngbae, S.; Kim, S.; Park, C.W. A phylogenetic study of Polygonum sect. Tovara (Polygonaceae) based on ITS sequences of nuclear ribosomal DNA. J. Plant Biol.
- Yao, H.; Song, J.Y.; Ma, X.Y.; Liu, C.; Li, Y.; Xu, H.X.; Han, J.P.; Duan, L.S.; Chen, S.L. Identification of Dendrobium species by a candidate DNA barcode sequence: the chloroplast psbA-trnH intergenic region. Planta Med. 2009, 75, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Girenahalli, R.; Tavareakere Venkataravanappa, J.; Kumar, P.; Subbarayappa, K.; Nanjappa, L. DNA based identification of species of Zingiberaceae family plants using Bar-Hrm analysis. Int. J. Res. Anal. Rev. 2019, 6, 289–294. [Google Scholar]
- Cui, Y.; Chen, X.; Nie, L.; Sun, W.; Hu, H.; Lin, Y.; Li, H.; Zheng, X.; Song, J.; Yao, H. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- HU, Y.F. DNA barcoding sequence analysis of Amomum tsao-ko germplasm resources in Yunnan province. Chin. Tradit. Herb. Drugs 2019, 6091–6097. [Google Scholar]
- Hu, Y.F.; Zhang, X.M.; Shi, N.X.; Yang, Z.Q. DNA barcoding sequence analysis of Amomum tsao-ko germplasm resources in Yunnan province. Chin. Tradit. Herb. Drugs 2019, 50, 6091–6097. [Google Scholar]
- Doh, E.J.; Kim, J.H.; Lee, G. Identification and monitoring of Amomi fructus and its adulterants based on DNA barcoding analysis and designed DNA markers. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Sone, M.; Zhu, S.; Cheng, X.; Ketphanh, S.; Swe, S.; Tun, T.L.; Kawano, N.; Kawahara, N.; Komatsu, K. Genetic diversity of Amomum xanthioides and its related species from Southeast Asia and China. J. Nat. Med. 2021, 75, 798–812. [Google Scholar] [CrossRef]
- Pan, H.; Huang, F.; Wang, P.; Zhou, L.; Cao, L.; Liang, R. Identification of Amomum villosum, Amomum villosum var. xanthioides and Amomum longiligulare by ITS-1 sequencing. J. Chin. Med. Mater.
- Shi, L.; Song, J.; Chen, S.; Yao, H.; Han, J. Identification of Amomum (Zingiberaceae) through DNA Barcodes. World Sci. Techno. Mod. Tradit. Chin. Med. 2010, 12, 473–479. [Google Scholar]
- Han, J.P.; Li, M.N.; Shi, L.C.; Yao, H.; Song, J.Y. ITS2 sequence identification of cardamom and its adulterants. Global Tradit. Chin. Med. 2011, 4, 99–102. [Google Scholar]
- Gong, L.; Ding, X.; Guan, W.; Zhang, D.; Zhang, J.; Bai, J.; Xu, W.; Huang, J.; Qiu, X.; Zheng, X.; Zhang, D.; Li, S.; Huang, Z.; Su, H. Comparative chloroplast genome analyses of Amomum: insights into evolutionary history and species identification. BMC Plant Biol. 2022, 22. [Google Scholar] [CrossRef]
- Li, D.M.; Li, J.; Wang, D.R.; Xu, Y.C.; Zhu, G.F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae). BMC Plant Biol. 2021, 21, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.Z.; Cai, X.D.; Gong, M.; Xia, M.Q.; Xing, H.T.; Dong, S.S.; Tian, S.M.; Li, J.L.; Lin, J.Y.; Liu, Y.Q. Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genom. 2023, 24, 30. [Google Scholar]
- Liang, H.; Zhang, Y.; Deng, J.B.; Gao, G.; Ding, C.B.; Zhang, L.; Yang, R.W. The complete chloroplast genome sequences of 14 Curcuma species: insights into genome evolution and phylogenetic relationships within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Amenu, S.G.; Wei, N.; Wu, L.; Oyebanji, O.; Hu, G.W.; Zhou, Y.D.; Wang, Q. Phylogenomic and comparative analyses of Coffeeae alliance (Rubiaceae): deep insights into phylogenetic relationships and plastome evolution. BMC Plant Biol. 2022, 22, 88. [Google Scholar] [CrossRef]
- Yang, Q.; Fu, G.F.; Wu, Z.Q.; Li, L.; Zhao, J.L.; Li, Q.J. Chloroplast genome evolution in four montane Zingiberaceae taxa in China. Frontiers in Plant Science 2022, 12, 774482. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Zhu, G.F.; Xu, Y.C.; Ye, Y.J.; Liu, J.M. Complete chloroplast genomes of three medicinal Alpinia species: genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 2020, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Milne, R.I.; Mö, *!!! REPLACE !!!*; ller, M.; Zhu, G.F.; Ye, L.J.; Luo, Y.H.; Yang, J.B.; Wambulwa, M.C.; Wang, C.N.; Li, D.Z. Integrating a comprehensive DNA barcode reference library with a global map of yews (<italic>Taxus</italic> L.) for forensic identification. Mol. Ecol. Resour. 2018, 18, 1115–1115. [Google Scholar]
- Magoga, G.; Fontaneto, D.; Montagna, M. Factors affecting the efficiency of molecular species delimitation in a species-rich insect family. Mol. Ecol. Resour. 2021, 21, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.W.; Huang, J.L.; Ya, J.D.; Zhe, M.Q.; Zeng, C.X.; Zhang, Z.R.; Zhang, S.B.; Li, D.Z.; Li, H.T. DNA barcoding of Cymbidium by genome skimming: call for next-generation nuclear barcodes. Mol. Ecol. Resour. 2023, 23, 424–439. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Zhang, L. Screening potential DNA barcode regions in Amomum (Zingiberaceae). Acta Bot. Yunnanica 2010, 32, 393–400. [Google Scholar]
- Segersäll, M. DNA barcoding of commercialized plants; an examination of Amomum (Zingiberaceae) in South-East Asia. Uppsala University, Sweden, 2011.
- Leung, F.C. C.; Huang, Q.; Duan, Z.; Yang, J.; Ma, X.; Zhan, R.; Xu, H.; Chen, W. SNP typing for germplasm identification of Amomum villosum Lour. based on DNA barcoding markers. PLoS One 2014, 9. [Google Scholar]
- Gong, L.; Zhang, D.; Ding, X.; Huang, J.; Guan, W.; Qiu, X.; Huang, Z. DNA barcode reference library construction and genetic diversity and structure analysis of <italic>Amomum villosum</italic> Lour. (Zingiberaceae) populations in Guangdong province. PeerJ 2021, 9. [Google Scholar]
- Zhai, E.A.; Mi, W.J.; Cui, Y.; Hong, W.F.; Wang, Y.S.; Guo, X.Y.; Zou, H.Q.; Yan, Y.H. Comparative study of morphological identification and DNA barcoding for the authentication of medicinal Fructus Amomi. China J. Chin. Mater. Med. 2022, 47, 4600–4608. [Google Scholar]
- Kuroiwa, T.; Kawano, S.; Nishibayashi, S.; Sato, C. Epifluorescent microscopic evidence for maternal inheritance of chloroplast DNA. Nature 1982, 298, 481–483. [Google Scholar] [CrossRef]
- Smith, S.E. , Plant Breeding Reviews. Timber Press: Portland, Oregon, USA, 1989; Vol. 6.
- Wiemers, M.; Fiedler, K. Does the DNA barcoding gap exist? –a case study in blue butterflies (Lepidoptera: Lycaenidae). Front. Zool. 2007, 4, 1–16. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 35, D5–D12. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. ; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 6241–6241. [Google Scholar] [CrossRef] [PubMed]
- China Plant BOL Group, 1; Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L. 83. China Plant BOL Group 1; Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 19641–19646. [Google Scholar]
- Wang, X.C.; Liu, C.; Huang, L.; Bengtsson-Palme, J.; Chen, H.; Zhang, J.H.; Cai, D.; Li, J.Q. ITS1: a DNA barcode better than ITS2 in eukaryotes? Mol. Ecol. Resour. 2015, 15, 573–586. [Google Scholar] [CrossRef]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.; Kauserud, H. ITS 1 versus ITS 2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Boer, H. d.; Newman, M.; Poulsen, A.D.; Droop, A.J.; Fér, T.; Thu Hiền, L.T.; Hlavatá, K.; Lamxay, V.; Richardson, J.E.; Steffen, K.; Leong-Škorničková, J. Convergent morphology in Alpinieae (Zingiberaceae): Recircumscribing Amomum as a monophyletic genus. Taxon 2018, 67, 6–36. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; Thierer, T.; Ashton, B.; Meintjes, P.; Drummond, A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Li, R.Z.; Cai, J.; Yang, J.B.; Zhang, Z.R.; Li, D.Z.; Yu, W.B. Plastid phylogenomics resolving phylogenetic placement and genera phylogeny of Sterculioideae (Malvaceae s. l.). Guihaia 2022, 42, 42–38. [Google Scholar]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; OrganellarGenomeDRAW, R.B. A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Čandek, K.; Kuntner, M. DNA barcoding gap: reliable species identification over morphological and geographical scales. Mol. Ecol. Resour. 2015, 15, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Collins, R.; Cruickshank, R. The seven deadly sins of DNA barcoding. Mol. Ecol. Resour. 2013, 13, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
Figure 1.
Plastome gene map of Amomum compactum YWB91902-2 showing the typical structure organization in Amomum plastomes. Genes inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes in different functional groups are color-coded. The small and large single copy regions (SSC and LSC) and inverted repeat (IRa and IRb) regions are noted in the inner circle.
Figure 1.
Plastome gene map of Amomum compactum YWB91902-2 showing the typical structure organization in Amomum plastomes. Genes inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes in different functional groups are color-coded. The small and large single copy regions (SSC and LSC) and inverted repeat (IRa and IRb) regions are noted in the inner circle.
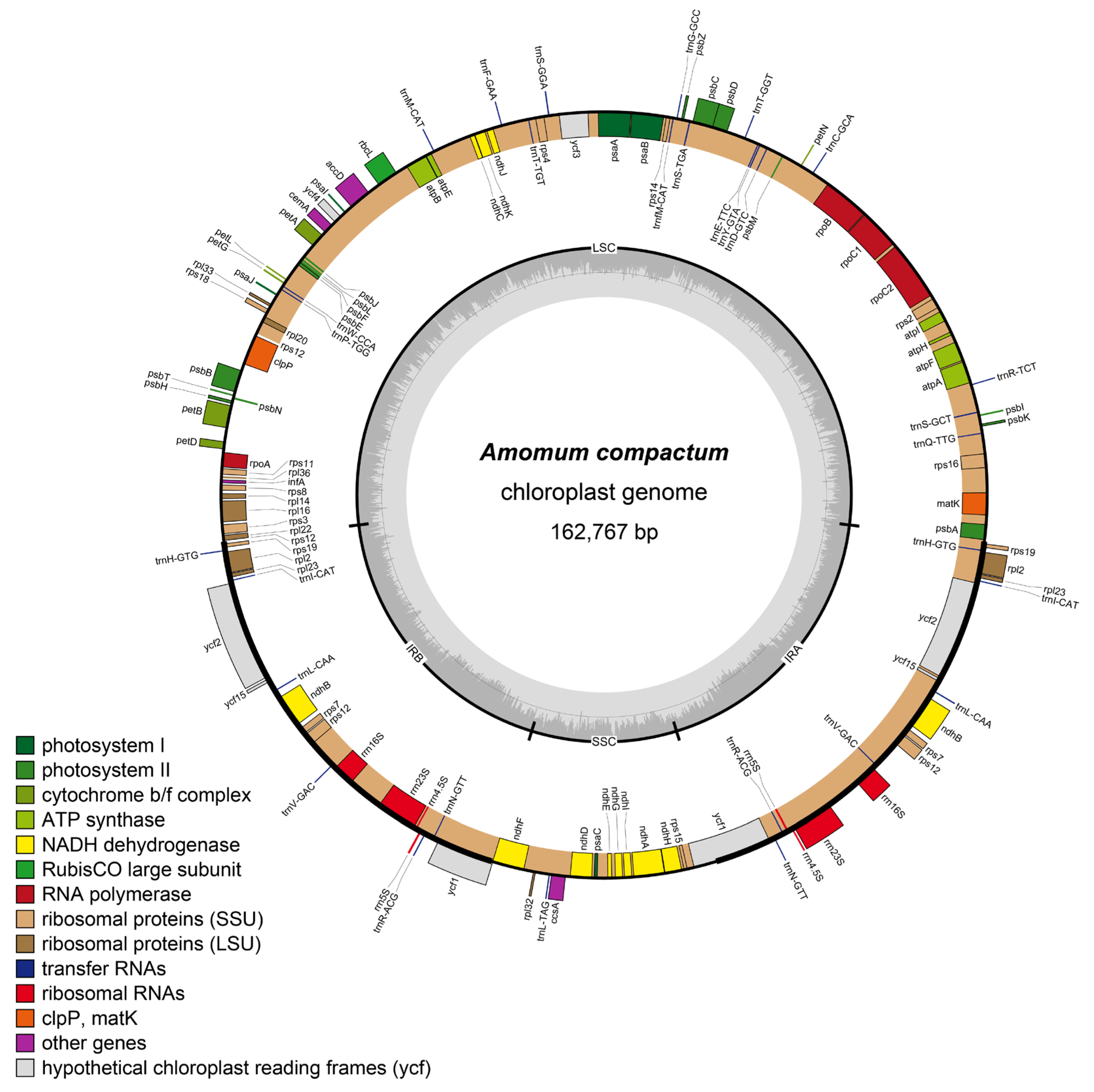
Figure 2.
Comparison of the borders of the LSC, SSC, and IR regions among six plastomes of Amomum. W. villosa var. xanthioides is synonymous with A. villosum var. xanthioides; L. tsao-ko is synonymous with A. tsao-ko; W. longiligularis is synonymous with A. longiligulare.
Figure 2.
Comparison of the borders of the LSC, SSC, and IR regions among six plastomes of Amomum. W. villosa var. xanthioides is synonymous with A. villosum var. xanthioides; L. tsao-ko is synonymous with A. tsao-ko; W. longiligularis is synonymous with A. longiligulare.
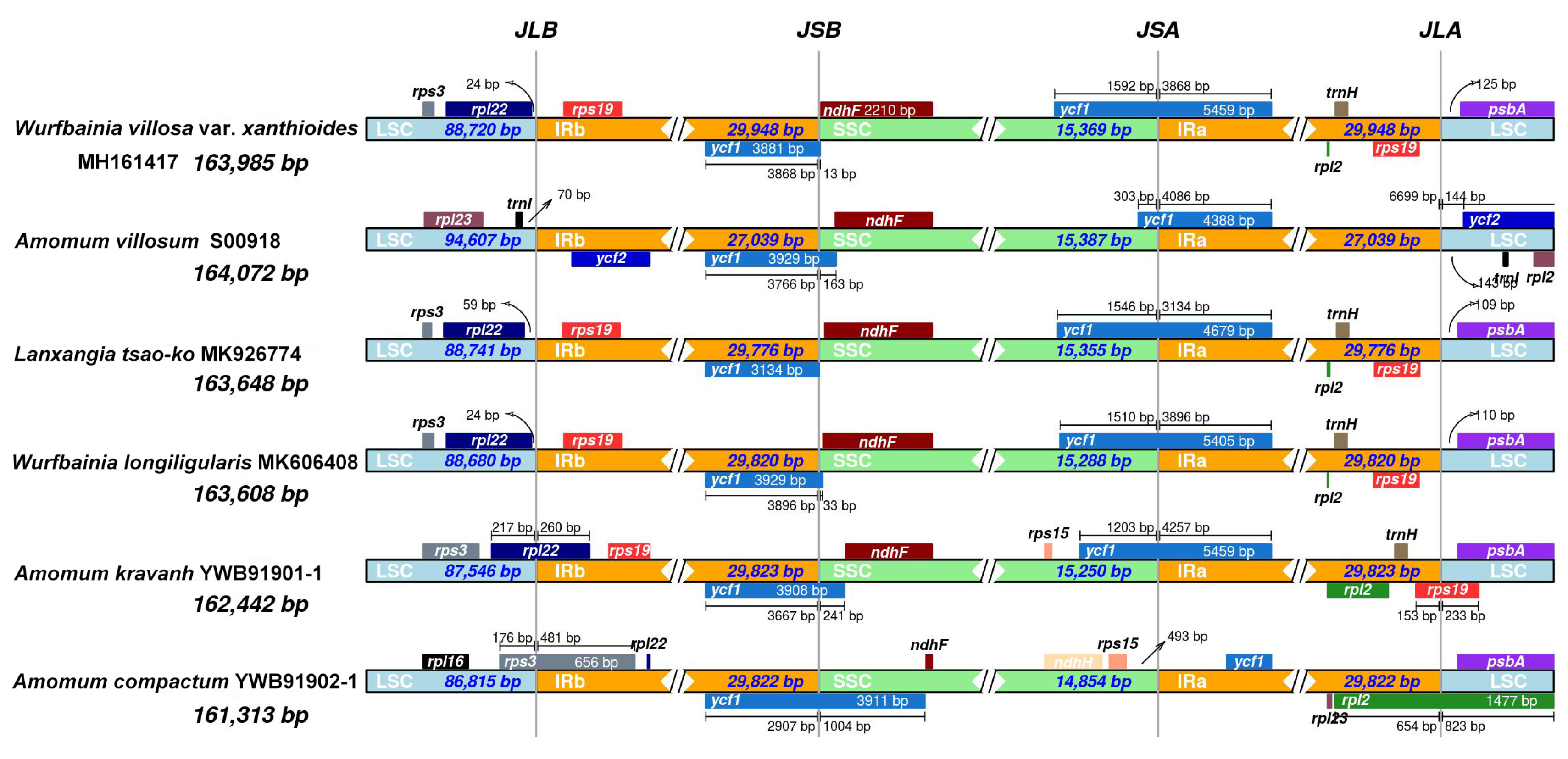
Figure 3.
The variable sites in the homologous regions of 41 Amomum plastomes. The y-axis represents the nucleotide diversity (Pi), and the x-axis indicates the nucleotide midpoints.
Figure 3.
The variable sites in the homologous regions of 41 Amomum plastomes. The y-axis represents the nucleotide diversity (Pi), and the x-axis indicates the nucleotide midpoints.
Figure 4.
Scatter plot of barcoding gap analysis of the seven datasets across the six Amomum species. The y-axis represents the genetic divergence, with the plots above the blue line of best fit representing successfully delimited species, and those along and below the line represent the overlap. “CP” represents complete plastomes.
Figure 4.
Scatter plot of barcoding gap analysis of the seven datasets across the six Amomum species. The y-axis represents the genetic divergence, with the plots above the blue line of best fit representing successfully delimited species, and those along and below the line represent the overlap. “CP” represents complete plastomes.
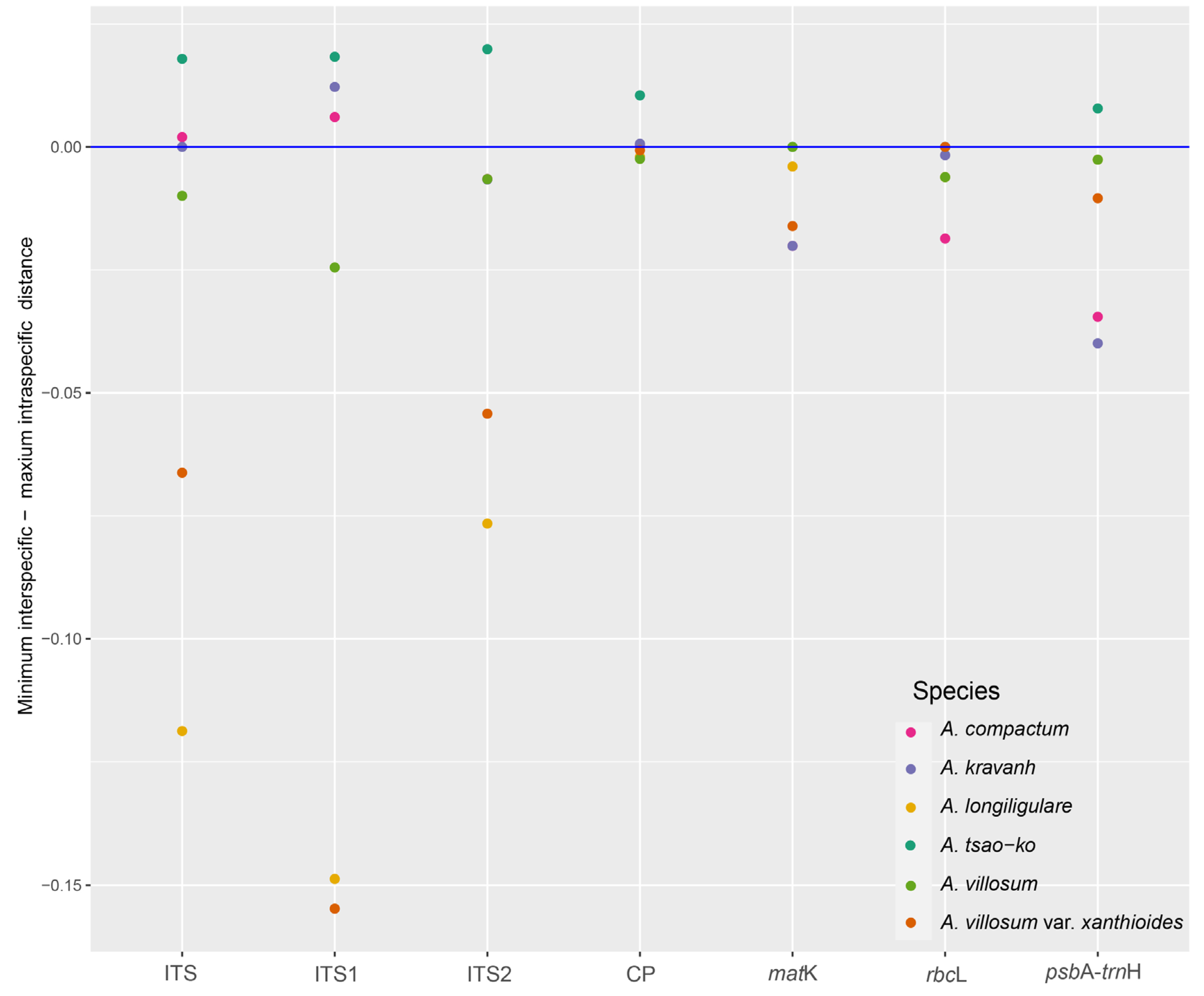
Figure 5.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
Figure 5.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
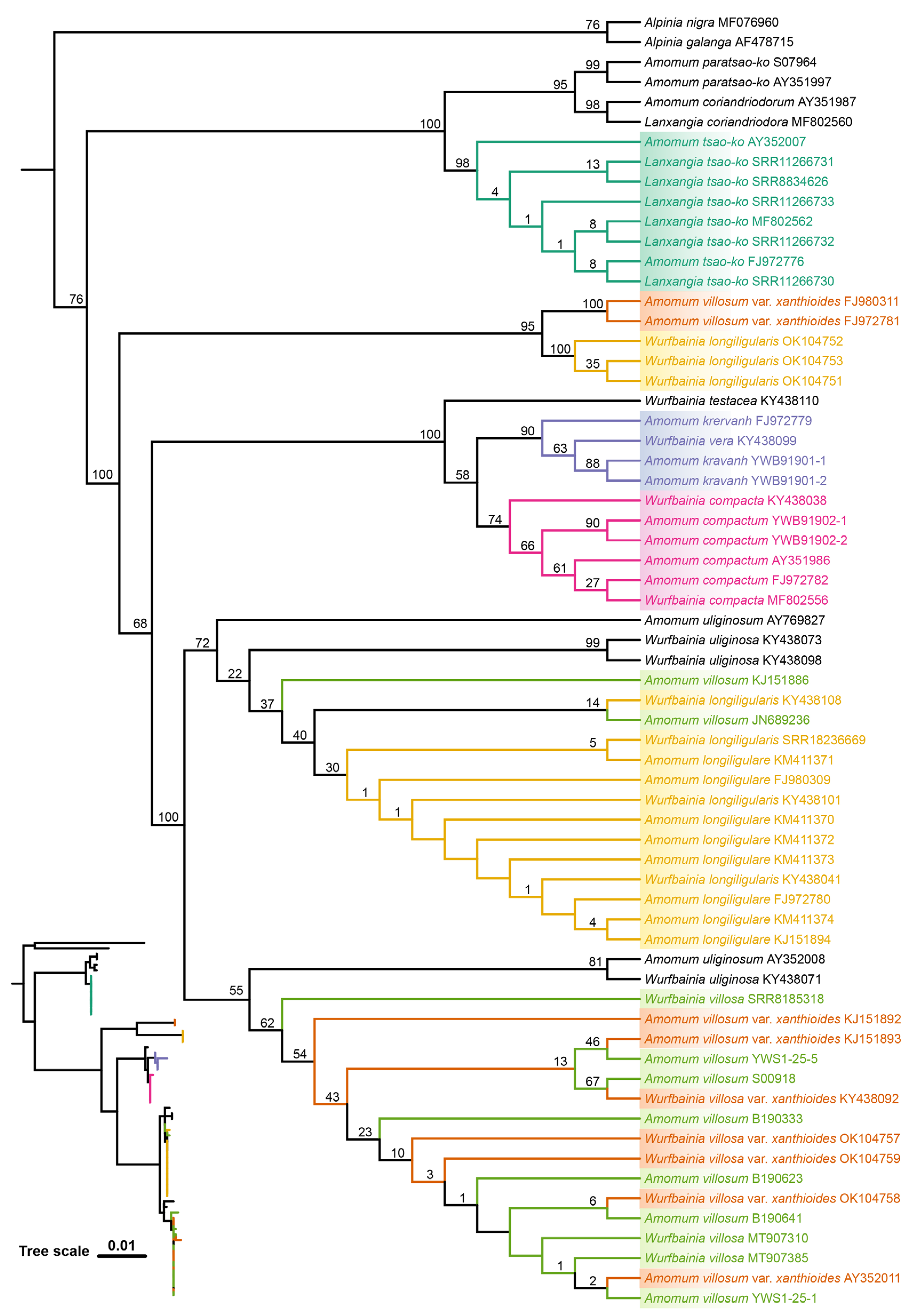
Figure 6.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS1 set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
Figure 6.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the ITS1 set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
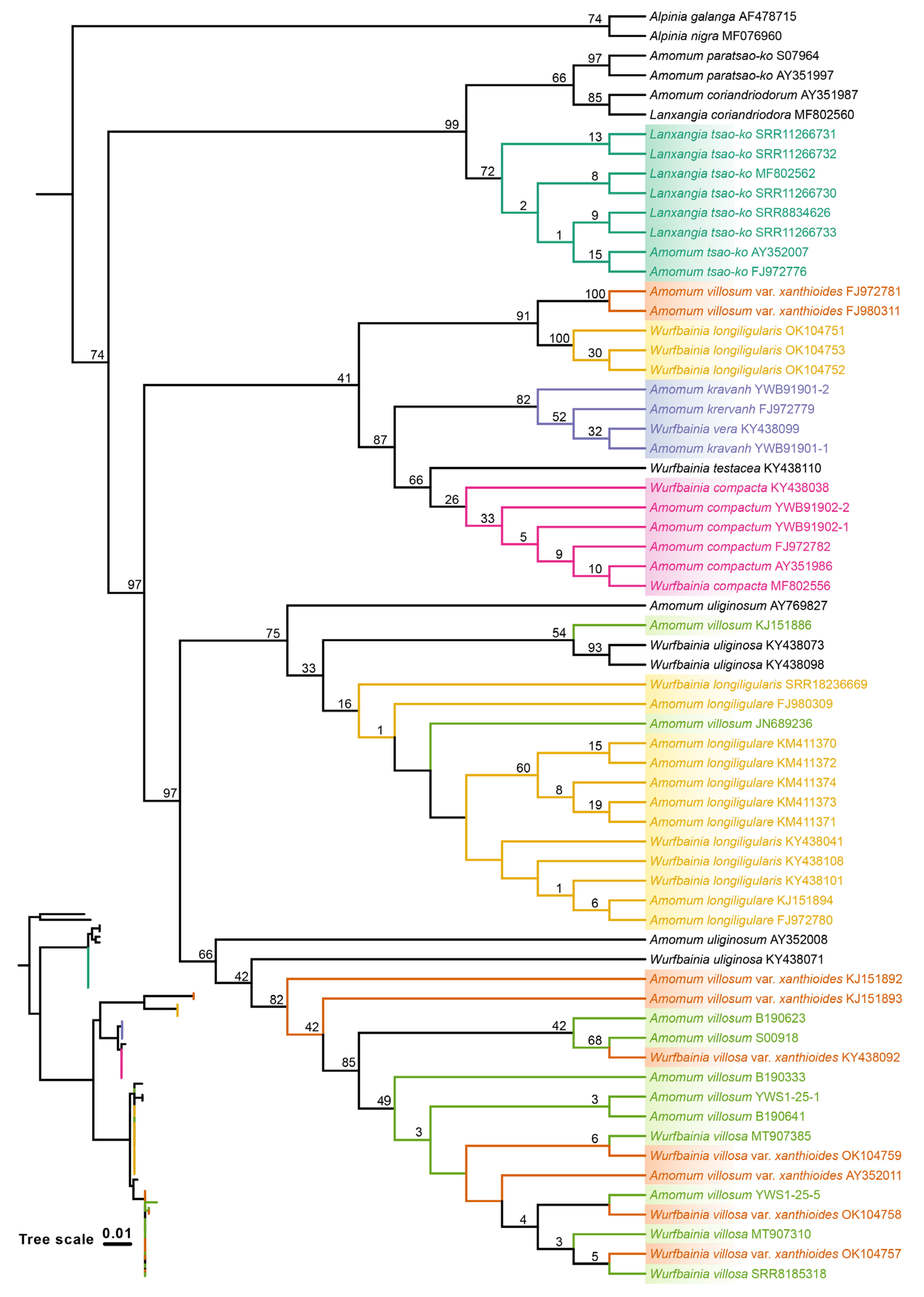
Figure 7.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the complete plastomes set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
Figure 7.
Phylogenetic tree was reconstructed based on the Maximum likelihood (ML) method with the complete plastomes set of six medicinal Amomum plants. The numbers at nodes indicate bootstrap values.
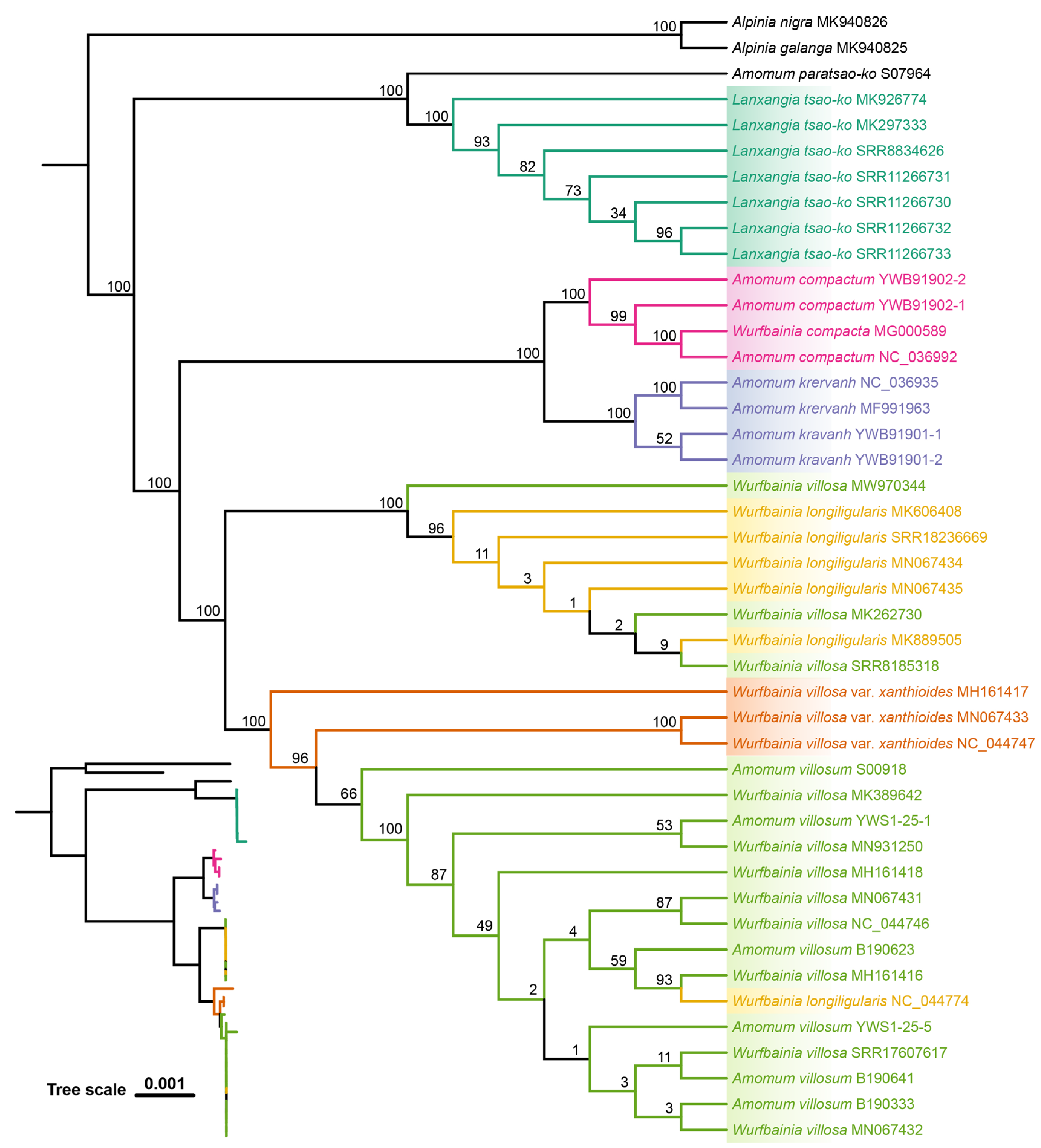
Figure 8.
The species discrimination success for candidate barcodes of six medicinal Amomum plants across different delimitation methods. The success rate is presented as the number of species successfully delimited to species in the different DNA markers. “CP” represents complete plastomes.
Figure 8.
The species discrimination success for candidate barcodes of six medicinal Amomum plants across different delimitation methods. The success rate is presented as the number of species successfully delimited to species in the different DNA markers. “CP” represents complete plastomes.
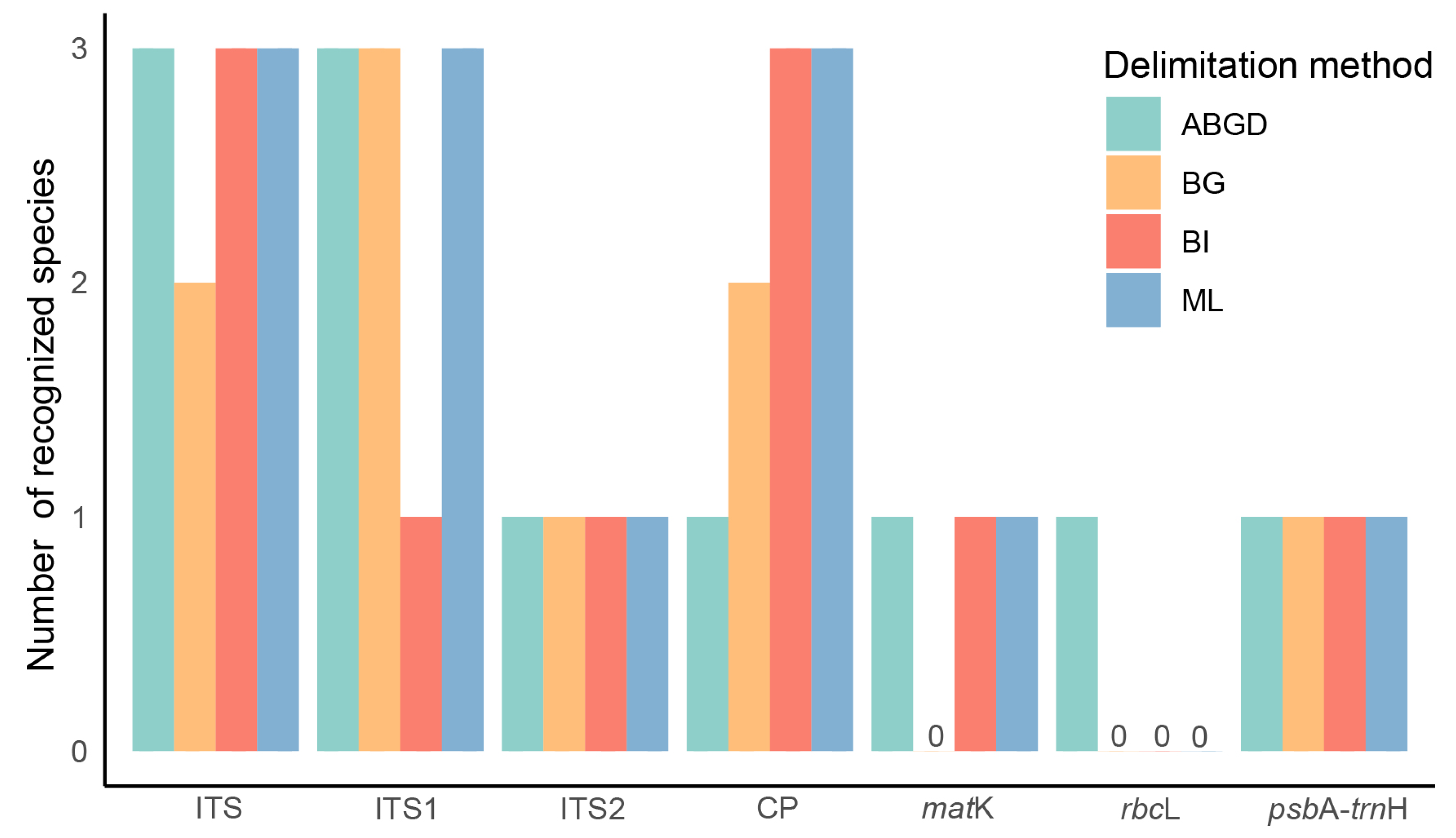

Table 1.
Comparison of characteristics of seven datasets in six medicinal Amomum plants.
| Dataset | No. of samples | Aligned length (bp) | No. of variable sites (% divergence) | No. of parsimony informative sites (% divergence) | GC content (%) | No. of conserved sites (% divergence) | No. of singleton sites (% divergence) |
|---|---|---|---|---|---|---|---|
| ITS | 65 | 609 | 164 (26.9) | 120 (19.7) | 56.2 | 422 (69.3) | 42 (6.9) |
| ITS1 | 65 | 194 | 70 (36.1) | 59 (30.4) | 56.5 | 111 (57.2) | 11 (5.7) |
| ITS2 | 65 | 222 | 83 (37.4) | 58 (26.1) | 60.1 | 130 (58.6) | 23 (10.4) |
| Complete plastomes | 44 | 168519 | 5299 (3.1) | 3280 (1.9) | 36.1 | 161202 (95.7) | 1980 (1.2) |
| matK | 82 | 716 | 44 (6.1) | 28 (3.9) | 28.7 | 672 (93.9) | 16 (2.2) |
| rbcL | 61 | 490 | 12 (2.4) | 9 (1.8) | 43.2 | 478 (97.6) | 3 (0.6) |
| psbA-trnH | 66 | 804 | 65 (8.1) | 35 (4.4) | 29.2 | 690 (85.8) | 30 (3.7) |
Table 2.
Detailed species individual collection information of Amomum which we sequenced.
| Sample number | Species | Country | Province | Region | County | GenBank accession numbers for each DNA region | |
|---|---|---|---|---|---|---|---|
| ITS/ITS1/ITS2 | CP/matK/rbcL/psbA-trnH | ||||||
| YWB91902-1 | Amomum compactum | China | Yunnan | Xishuangbanna Dai Autonomous Prefecture | Mengla County | OR801269 | PP826179 |
| YWB91902-2 | Amomum compactum | China | Yunnan | Xishuangbanna Dai Autonomous Prefecture | Mengla County | OR801270 | PP826180 |
| YWB91901-1 | Amomum kravanh | China | Yunnan | Xishuangbanna Dai Autonomous Prefecture | Mengla County | OR801267 | PP826177 |
| YWB91901-2 | Amomum kravanh | China | Yunnan | Xishuangbanna Dai Autonomous Prefecture | Mengla County | OR801268 | PP826178 |
| S07964 | Amomum paratsao-ko | China | Yunnan | Honghe Hani and Yi Autonomous Prefecture | Yuanyang County | OR801266 | PP826176 |
| S00918 | Amomum villosum | China | Guangxi | Fangchengang City | Shangsi County | OR801265 | PP826175 |
| B190333 | Amomum villosum | China | Yunnan | Kunming City | Xishan District | OR801256 | PP826171 |
| B190623 | Amomum villosum | China | Yunnan | Kunming City | Xishan District | OR801257 | PP826172 |
| B190641 | Amomum villosum | China | Yunnan | Kunming City | Xishan District | OR801258 | PP826173 |
| YWS1-25-1 | Amomum villosum | – | – | – | – | OR801271 | PP826181 |
| YWS1-25-5 | Amomum villosum | – | – | – | – | OR801272 | PP853448 |
Note: “CP” represents complete plastomes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



