
Submitted:
05 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
. Protein cysteine S-glycosylation is a relatively rare and less well characterized post-translational modification (PTM). Creating reliable model proteins that carry this modification is challenging. The lack of available model or natural S-glycosylated proteins significantly hampers the development of MS-based methodologies for detecting protein cysteine S-glycosylation in real-world proteomic studies. There is also limited MS-sequencing data describing easier to create synthetic S-glycopeptides. Here, we present the results of an in-depth manual analysis of automatically annotated CID/HCD spectra for model S-glucopeptides. The CID spectra show a long series of y/b-fragment ions with retained S-glucosylation, regardless of the dominant m/z signals corresponding to neutral loss of 1,2-anhydroglucose from the precursor ions. In addition, the spectra show signals manifesting glucosyl transfer from the cysteine position onto Lys, Arg side chains, and a peptide N-terminus. Other spectral evidence indicates that the N-glucosylated initial products of transfer are converted into N-furanosylated (i.e., glycated) structures due to Amadori rearrangement. We discuss the peculiar transfer of the glucose oxocarbenium ion (Glc+) to positively charged guanidinium residue (ArgH+) and propose a mechanism for the gas-phase Amadori rearrangement involving a hydride ion shift.
Keywords:
glycoproteomics
; S-glycosylation
; gas phase glycosyl transfer
; glycation
; Amadori rearrangement
1. Introduction
Cellular glycosylation of proteins is an enzyme-directed process that leads to diverse protein-glycan structures, which are responsible for driving particular biological functions [1]. Most glycans are assembled from a limited set of reducing monosaccharides linked by O, O-acetal bridges, which are themselves attached through O- or N-glycosidic bonds to side chains of serine/threonine (Ser/Thr) or asparagine (Asn) residues at specific locations on the polypeptide chains [2]. These major types of glycosidic bonds have distinctive properties, which can be analyzed using well-established MS-based technologies [3].
The chemical synthesis of glycans and glycoconjugates has enabled the development of modern glycoproteomics, by providing reference samples for systematic biological studies [4]. In general, the synthesis of complex glycan intermediates relies on the transfer of the glycosyl residue from a selected, activated donor to a saccharide serving as an acceptor molecule. In classic chemical strategies, the acceptor has a target hydroxyl group accessible for the formation of a glycosidic linkage, whereas the reducing end of the growing glycan chain is orthogonally protected to permit the eventual assembly of final protein glycoconjugates [5]. Another original approach achieves control over the regio- and stereochemistry of glycan synthesis by intramolecular glycosyl transfer. However, this strategy is not frequently used due to the great difficulty of acquiring sophisticated molecules concurrently exhibiting glycosyl donor and glycosyl acceptor functionalities [6].
Thioglycosides are favorable glycosyl donors, due to their efficient activation by various thiophilic promoter systems [7]. Chemical glycosylation is accomplished in extra-dry organic solvents to protect the activated donors from hydrolysis. In addition to the desired glycosylation product, the reaction typically generates a byproduct with no nucleophilic activity—the thioaglycon. The thioaglycon often remains tightly bound to the promoter used for activation and should be removed from the reaction mixture.
In contrast to extensively studied, structurally complex N- and O-glycosylated proteins, S-glycosylation involving protein cysteines is considered a rare post-translational modification (PTM). The S-linked glycans identified to date are mono-saccharides or short oligo-saccharides made of Glc, Gal, and GlcNAc residues. The distinctive mono-ADP-S-ribosyl unit attached to proteins can also be categorized as S-glycans [8].
The experimental results of MS/MS-sequencing of glycopeptides show that S-glycosidic bonds in CID-type MS fragmentation are more stable than O-glycosidic linkages. This feature can help to determine the location of S-glycosylated cysteines in many proteins [9,10,11,12]. For example, by employing a routine proteomic workflow with final LC-ESI tandem mass spectrometry, researchers have detected S-glycosylated catalytic cysteines transiently occurring in glycosyl-enzyme intermediates. Based on these findings, a retaining mechanism for catalysis of two mutant glycosyltransferases has been proposed [13]. Nonetheless, detecting low-level S-linked glycosylation among ubiquitous O-, and N-glycosylated proteins remains challenging and requires new tailored analytical methods [14].
Our previous work described a chemically modified lysozyme (P-00698) randomly S-glucosylated at cysteine positions available in the protein molecule. The locations of the S-modified sites were detected directly using routine mass spectrometric methods, and indirectly after specific S-tagging of the S-glucosylated positions. As a result, a large library of CID/HCD fragmentation spectra was created for the model S-glucosylated and S-tagged peptides. Here, we present the results of a manual analysis of complex MS/MS spectra of selected S-glucosylated peptides from the previously created library. In the CID spectra, signals showing 162 Da neutral loss from the precursor ions dominate, accompanied by series of y/b-fragment ions retaining an intact S-glucosyl moiety. In addition, numerous deciphered signals provide spectral evidence of glucosyl transfer from cysteine onto Lys/Arg side chains and the N-terminals of the fragmenting peptides. Progressing the S2192→N glucosyl transfer leads to the formation of N-glucosylated derivatives and N-fructosylated derivatives [15]. We propose hypothetical mechanisms for these gas phase transformations.
2. Results
The present study builds on our previous work [14], in which CID/HCD fragmentation spectra of S-glucopeptides were identified using the Byonic sequence search engine. The created CID/HCD spectral library contained 90 unique sequences of S-glucopeptides. In the present study, a set of spectra from the CID/HCD spectral library was selected for meticulous manual analysis. The selected spectra included precursor ions of charge state z=+2 or z=+3, with one S-glucosylated cysteine position. The omitted spectra possessed long sequences bearing more modifications and a precursor ion charge state z>3.
Initial manual analysis was focused on identifying m/z signals corresponding to glucosyl oxocarbenium ion (Glc+, 163 Da) transfer from the Cys residue onto side chains of other amino acid residues.
2.1. General Characteristics of S-Glucopeptides MS-Fragmentation
The CID spectra showed a characteristic strong signal representing 1,2-anhydroglucose neutral loss (162 NL) from the +2 charged precursors (Figures S1–S5). Occasionally, glucosyl loss (Glc+, 163 Da) occurred from the +3-charged precursor ion, leading to a reduction of its charge state, as illustrated in Figure S2. Other signals of neutral losses from precursor ions, denoted as [M+zH+]/z, show the loss of hexose (180 NL), pentose (150 NL), tetrose (120 NL), and triose (90 NL). The CID spectra of S-glucosylated peptides signals show the loss of one (18 NL), two (36 NL), or three (54 NL) water molecules.
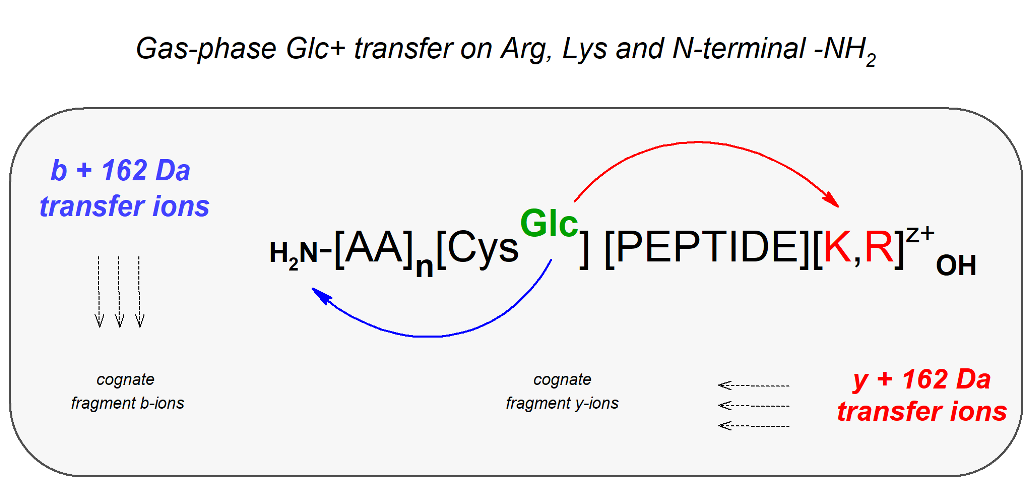
The automatically annotated long series of b/y fragment ions with retained C[S-Glc]-modification were typical for most of the CID spectra, and prove the moderate stability of the modification. Manual searching revealed the presence of b/y fragment ions with no Cys in their sequences but bearing a mass gain of 162 Da. This was linked to Glc+ ion transfer from cysteine to other amino acid residues within fragmenting peptides. In particular, [b+162]+/++ fragment ions corresponding to short N-terminal sequences and [y+162]+/++ comprising Lys or Arg on C-termini were detected. Unexpectedly, fragment ions denoted as [y/b+162+162-losses]+/++ were also revealed. This implies the transfer of two Glc+ ions onto fragments with no Cys residue in their sequences.
In the HCD spectra, [M+zH+-162]/z ions were weak or absent. A long series of y-type fragment ions from +2 and +3 charged precursors was observed, as can be seen in Figure 4; Figure 5 in Section 2.3.1.
2.2. Literature-Based Characteristics of Peptidyl N-Glucosylated Arg and Lys MS-Fragmentation
Although not previously described in the literature, Glc+ ion migration onto Lys primary amine groups or peptide N-termini in a gas phase seemed plausible and worth further exploration. Yet, a similar glucosyl transfer onto protonated arginine residues raised questions about the mechanism of transformation. The assumed formation of N-glucosylated arginine with subsequent Amadori rearrangement would lead to the cognate isobaric product (i.e., 1-amino-1-deoxy-D-fructoside), which should be distinguishable in the CID spectra [16]. Therefore, we conducted a thorough literature survey on the use of mass spectrometry in protein glycation studies. In particular, we focused on published data referring to the diagnostic fragmentation features of peptidyl N-glucosides, N-fructosides, and selected N-glycopeptides. The results are summarized in Table 1.
Our review of the literature data enabled us to establish a general map of N-glucosylated peptidyl arginine CID/HCD fragmentation, which is presented in Scheme 1. This map is applicable also to the CID fragmentation of N-glucosylated peptidyl lysine—i.e., if the pictured guanidinium group is replaced by the primary amino group. Then, 204 Da, 54 Da, and 24 Da diagnostic ions are not expected. The distinguished paths A, B, and C illustrate the main fragmentation routes, termed as follows:
- Path A: peptidyl N-glucoside fragmentation (Schiff base fragmentation path)
- Path B: peptidyl N-fructoside fragmentation (Amadori product fragmentation path)
- Path C: peptidyl N-fructoside dehydration (Amadori product dehydration path)
Figure 1.
CID fragmentation pathways of N-glucosylated peptides containing Arg or Lys.
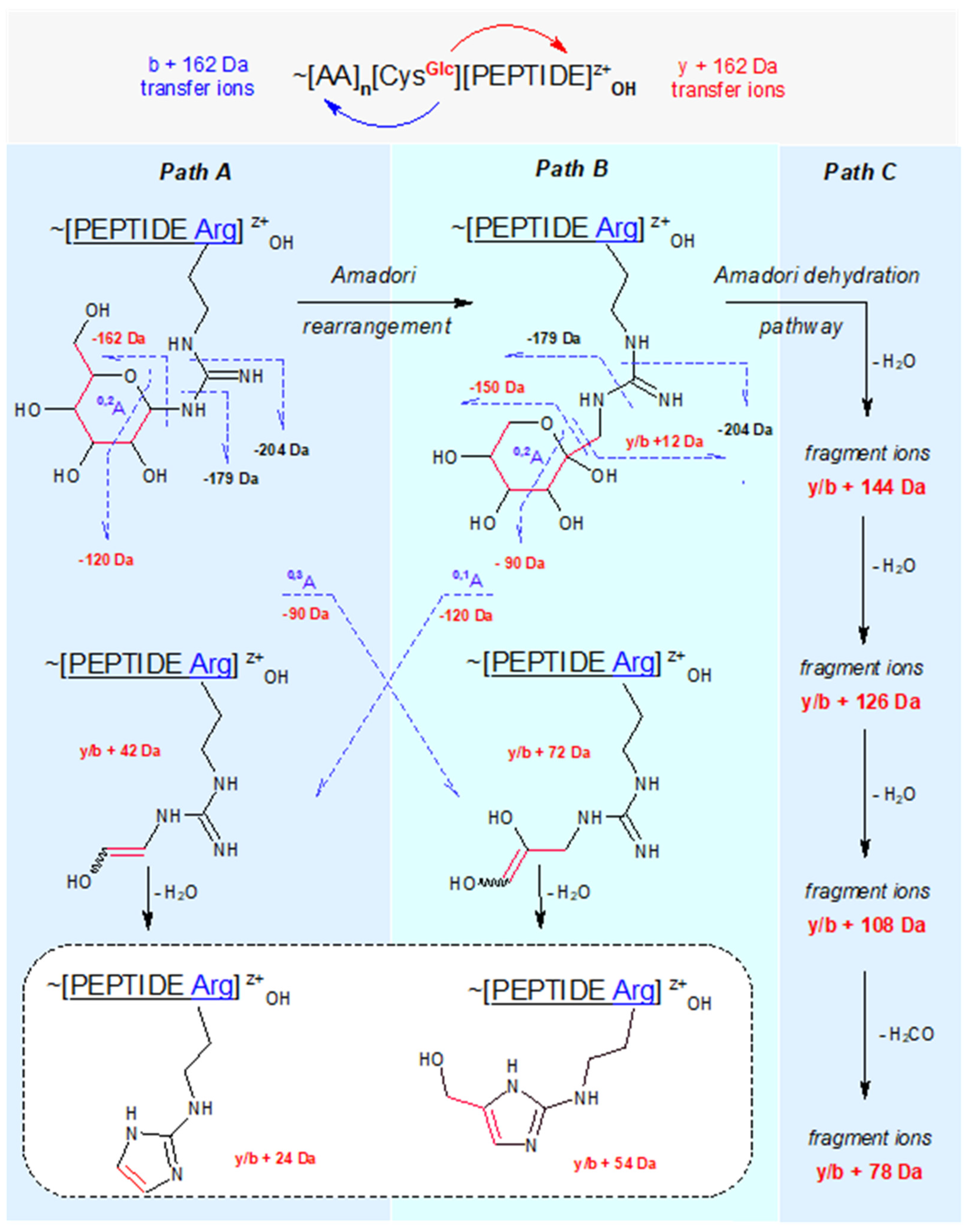
N-glucosides of primary amines are cyclic hemi-aminals (in the open form known as Schiff bases). Under physiological conditions, they hydrolyze easily or undergo Amadori rearrangement catalyzed by protic acids into isobaric N-fructosides (i.e., 1-amino -1-deoxy -D-fructosides) [16,17]. To the best of our knowledge, the gas phase Amadori rearrangement of N-glucosylated peptides (under CID/HCD fragmentation conditions) has not been described previously. However, it is worth highlighting previous studies that describe the CID fragmentation patterns of N-glucosylated asparagine in peptides [18] and discuss 0,2A n cross-ring cleavage as a general diagnostic tool for glycan assignment in glycoconjugate mixtures [19].
The detected signs of glycosyl (Glc+) transfer onto protonated arginine residue (ArgH+) led us to review the literature on the application of mass spectrometry in studies of protein arginine glycosylation [20]. It is known that arginine residue in vivo can be N-glucosylated, N-GlcNAcylated, N-rhamnosylated [21], and N-mono-ADP-ribosylated due to enzyme-catalyzed processes [22]. These modifications are prone to glycan and carbodiimide (42 Da, CN2H2) loss during CID. Therefore, it seemed that the 204 NL signal (162 Da + carbodiimide = 204 Da) might be a useful diagnostic marker for detecting N-glucosylated arginine in the CID spectra of our S-glucosylated peptides.
Under physiological conditions, the primary amino groups within proteins may follow non-enzymatic condensation with aldehydes, typically reducing sugars such as glucose. This reaction proceeds through an acyclic imine (Schiff base), followed by Amadori rearrangement to form so-called glycation products (e.g., N-fructosylated derivatives) [23,24]. Protein glycation can alter biomolecule natural functionalities and ultimately lead to abnormal biological processes. This important topic has been discussed in a large number of studies. In many of those studies, mass spectrometry was instrumental in detecting glycation structures [25,26,27,28,29].
2.3. Detailed Analysis of MS-Fragmentation Spectra for S-Glucopeptides
Fragmentation spectra of the following tryptic S-glucosylated peptides were subjected to detailed manual analysis:
- C[+162.]KGTDVQAWIR
- C[+162]ELAAAMK
- WWC[+162]NDGR
- SLGNWVC[+162]AAK
- RC[+162]ELAAAMK
These sequences have C-terminal lysine or arginine and single S-glucosylated cysteine located at the N-termini or within the peptide chain. As the exemplary sequence, C[+162.]KGTDVQAWIR was selected due to the accessibility of CID and HCD spectra for both +2 and +3 charged precursors. The results of the manual analysis are presented in Section 2.3, corresponding to the logic of Scheme 1. The centrally positioned peptide sequence guides the reader over the data arranged in vertical and horizontal lines. The b- and y- ions were automatically annotated. Their total abundance was calculated separately for each ion type and considered as 100% or the value of 1. Manually deciphered fragment ions arranged in columns represent entities of the same cleavage position but differing by mass loss or gain. Appropriately marked, diagnostic fragment ions derived from the decomposition of b-ions are placed in the upper rows. They indicate gas phase events on the precursor N-terminus. Changes in the precursor C-terminus are manifested by y-type diagnostic ions, analogically ordered in the lower rows. Their total abundance was expressed as the relative abundance versus the total abundance calculated for y ions.
2.3.1. CID-Fragmentation Analysis of C[+162.]KGTDVQAWIR Sequence
Fragment ions revealed in the CID spectrum of the +2 charged precursor (Figure 2) are presented in Table 2 and Table 3. The long series of signals corresponding to this particular type of diagnostic ions provides evidence of molecular transformation processes ongoing in the gas phase, which are explained in an additional sub-table (Table 2a).
Figure 2.
CID MS/MS spectrum of doubly charged C[+162.]KGTDVQAWIR).
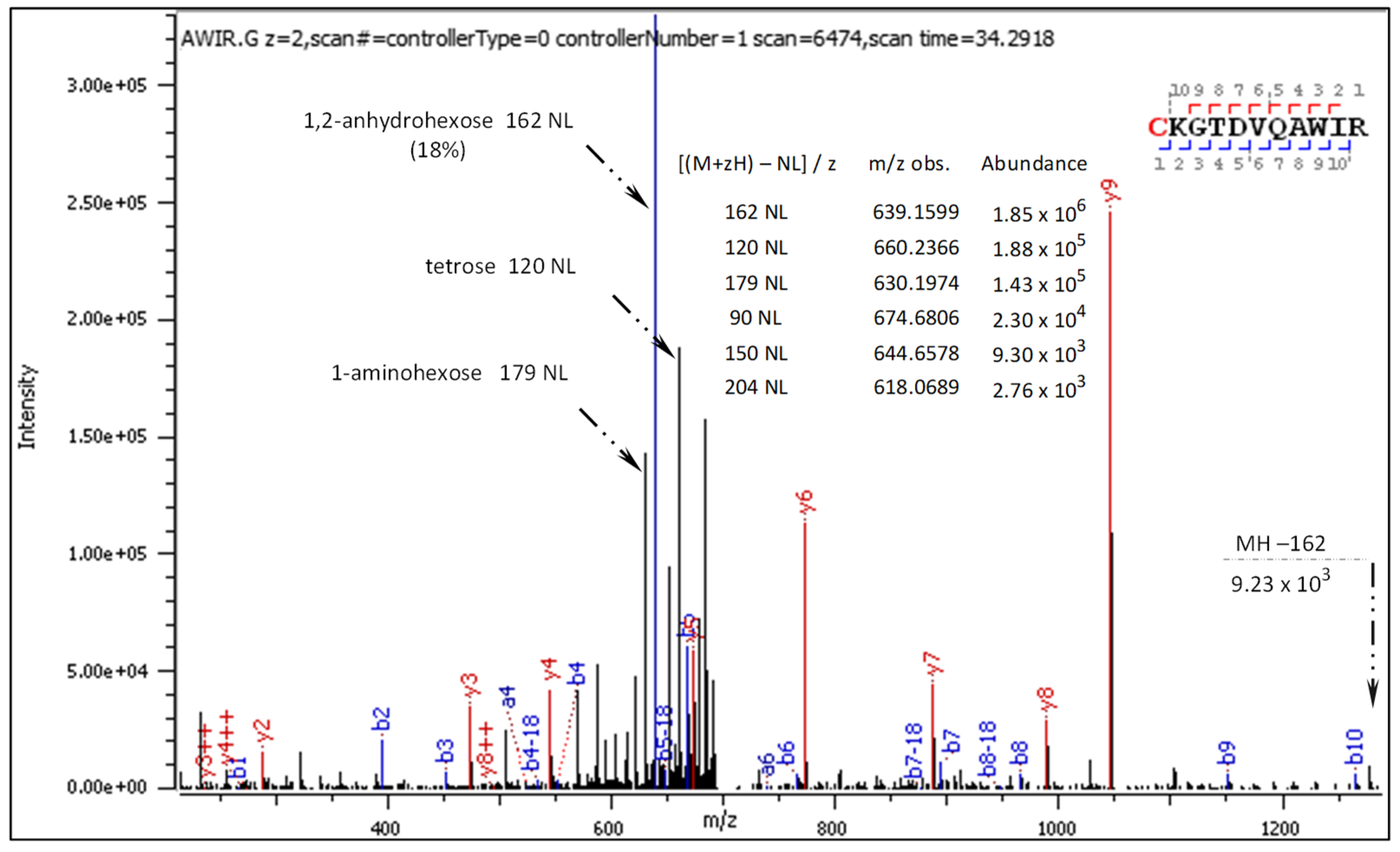

Table 2.
The set of fragment ions from the +2 charged precursor, as evidence of fragmentations following paths A, B, and C.
Table 2.
The set of fragment ions from the +2 charged precursor, as evidence of fragmentations following paths A, B, and C.
| [b-3H2O]+ | b1 | b2 | b3 | b4 | b5 | b6 | b7 | b8 | b9 | b10 | 0.20 | Path C | ||
| [b-2H2O]+ | b2 | b3 | b4 | b5 | b6 | b7 | b8 | b9 | b10 | 0.82 | ||||
| [b-H2O]+ | b2 | b3 | b10 | 0.03 | ||||||||||
| [b-150]+ | b3 | b4 | b5 | b6 | b8 | b9 | 0.004 | Path B | ||||||
| [b-90]+ | b2 | b3 | b4 | b5 | b6 | b9 | b10 | 0.22 | ||||||
| [b-120]+ | b2 | b3 | b5 | b6 | b7 | b9 | b10 | 0.24 | Path A | |||||
| [b]+ | b1 | b2 | b3 | b4 | b5 | b6 | b7 | b8 | b9 | b10 | # |
1.00 Total abundance (reference) |
||
|
Total abundance (reference) 1.00 |
CGlc | K | G | T | D | V | Q | A | W | I | R | |||
| # | y9 | y8 | y7 | y6 | y5 | y4 | y3 | y2 | [y]+ | |||||
| Path A | 0.02 | y9 | y7 | y5 | y3 | [y+162]+ | ||||||||
| 0.10 | y8 | y7 | y4 | y3 | y1 | [y+42]+ | ||||||||
| 0.04 | y7 | y6 | y4 | y3 | y2 | [y+24]+ | ||||||||
| Path B | 0.03 | y9 | y6 | y4 | y3 | y1 | [y+72]+ | |||||||
| 0.02 | y8 | y7 | y5 | y3 | y2 | y1 | [y+12]+ | |||||||
| 0.01 | y9 | y3 | y2 | y1 | [y+54]+ | |||||||||
| Path C | 0.03 | y7 | y4 | y3 | [y+144]+ | |||||||||
| 0.04 | y5 | y4 | y3 | y2 | y1 | [y+126]+ | ||||||||
| 0.03 | y10 | y9 | y7 | y4 | y3 | y2 | [y+108]+ | |||||||
| 0.01 | y4 | y1 | [y+78]+ | |||||||||||
Table 2.
a. Gas phase transformations of C[+162.]KGTDVQAWIR precursor of charge state +2.
| Transformations | Diagnostic ion(s) | Transformation path |
|---|---|---|
| N-glucosylation of Arg | [y+162]+ | A |
| N-glucosylation of Arg | [y+42; y+24]+ | A |
| N-fructosylation of Arg | [y+72; y+54; y+12]+ | B |
| N-fructosylation of Arg | [y+144; y+126; y+108; y+78]+ | C |
| S→N (Glc+) migration and tetrose loss | [b-120]+ | A |
| S→N (Glc+) migration and tiose loss | [b-90]+ | B |
| S→N (Glc+) migration and tiose loss | [b-150]+ | B |
Table 3.
The set of doubly glycosylated fragment ions and nascent diagnostic ions from the +2 charged precursor, as evidence of fragmentations following paths A, B, and C.
Table 3.
The set of doubly glycosylated fragment ions and nascent diagnostic ions from the +2 charged precursor, as evidence of fragmentations following paths A, B, and C.
| [b+108]+ | b1 | b3 | b4 | b5 | b9 | 4.37 | Path C | ||||||
| [b+126]+ | b1 | b3 | b6 | b7 | b8 | b9 | 0.28 | ||||||
| [b+144]+ | b1 | b3 | b5 | 0.48 | |||||||||
| [b+12]+ | b3 | b4 | b5 | b6 | b8 | b9 | 0.13 | Path B | |||||
| [b+72]+ | b2 | b3 | b4 | b5 | b6 | b9 | b10 | 0.33 | |||||
| [b+42]+ | b2 | b5 | b6 | b7 | b9 | b10 | 0.68 | Path A | |||||
| [b+162]+ | b2 | b3 | b8 |
1.00 Total abundance |
|||||||||
| CGlc | K | G | T | D | V | Q | A | W | I | R | |||
As can be seen in Table 3, the fragment ions b2+162, b3+162, and b8+162 carry an additional hexosyl residue, most likely attached either to the lysine residue or to the cysteine N-terminus, apparently due to an intermolecular glucosyl transfer. The diagnostic ions b1+144, b1+126, and b1+108 likely indicate an S→N glucosyl shift followed by Amadori rearrangement to N-fructosylated cysteine.
The general characteristics of the CID fragmentation patterns for the S-glucosylated peptides were presented in the Results sub-section 2.1. Figure 2 displays multiple neutral loss signals from the +2 charged precursor with dominating 162 NL. The relatively strong 179 NL peak (1.45 x 105) corresponds to 1-aminoglucose loss from N-glucosylated arginine. The less abundant 204 NL (2.76 x 103) indicates the combined loss of 1,2-anhydroglucose and carbodiimide.
Diagnostic fragment ions from the CID spectrum of the +3 charged precursor are collected in Table 4 and Table 5 and commented on in a sub-table (Table 4a).
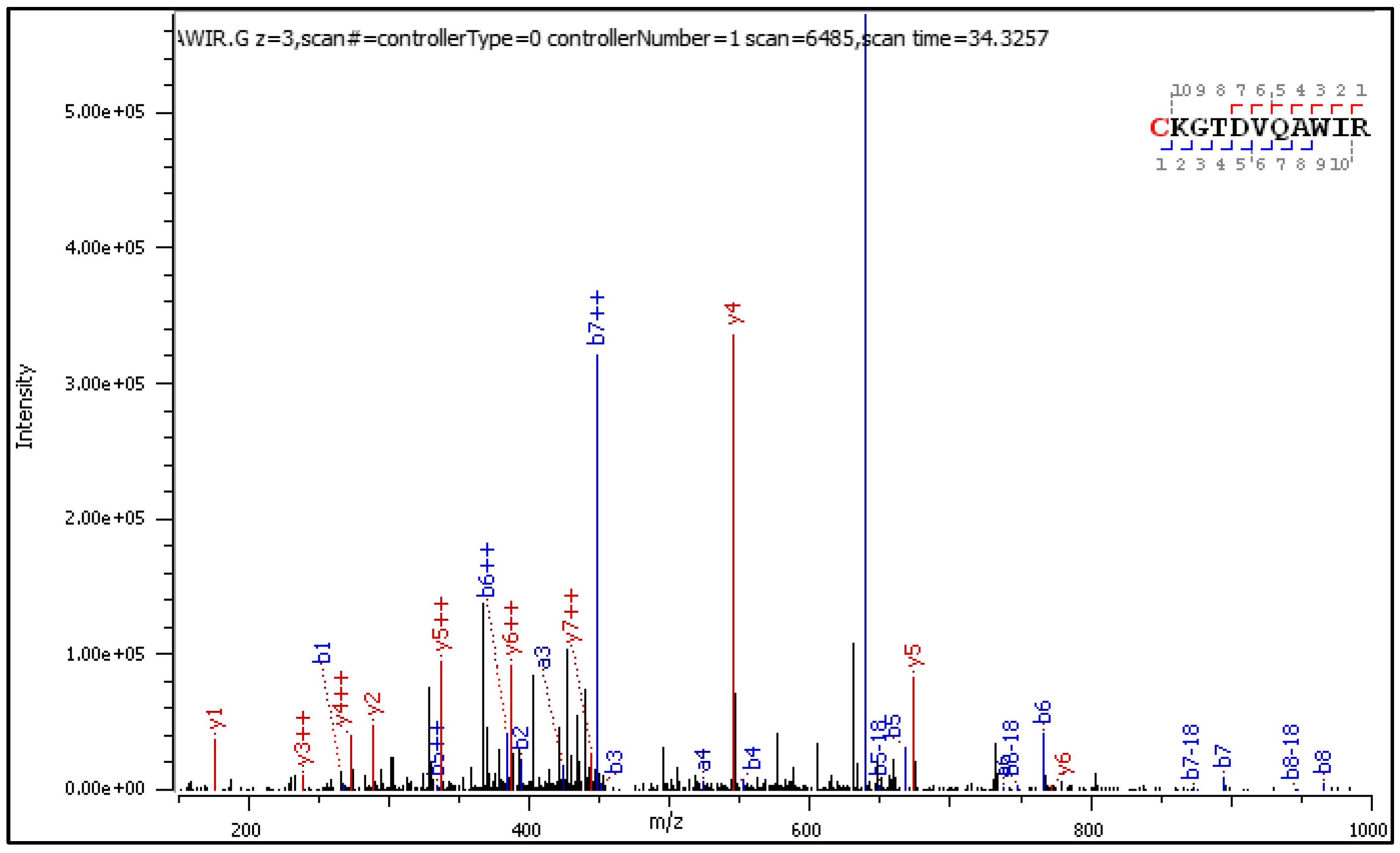
CID fragmentation of the +2 and +3 charged C[+162.]KGTDVQAWIR delivers similar sets of diagnostic fragment ions, experiencing a mass loss or gain according to paths A, B, and C from Scheme 1. Yet, in the CID spectrum of the +3 precursor shown in Figure 3, singly charged diagnostic ions are accompanied by longer series of the same but doubly charged diagnostic ions with higher abundance.
Figure 3.
CID MS/MS spectrum of triply charged C[+162.]KGTDVQAWIR).
2.3.2. HCD-Fragmentation Analysis of the C[+162.]KGTDVQAWIR Sequence
A few ions confirming glucosyl transfer and subsequent Amadori rearrangement populate the HCD spectra of the +2 and +3 charged precursors. The fragment ions containing a hydroxymethyl-imidazole ring [y+54]+/++ are slightly more abundant than the others. Manual analysis of selected peptides with N-terminal lysine and S-glucosylated cysteine located within the sequence confirmed all observations and conclusions presented in the previous sections. These results are presented in the Supplementary Materials (Tables S5 and S6, Figures S6 and S7).
Figure 4.
HCD MS/MS spectrum of doubly charged C[+162.]KGTDVQAWIR).
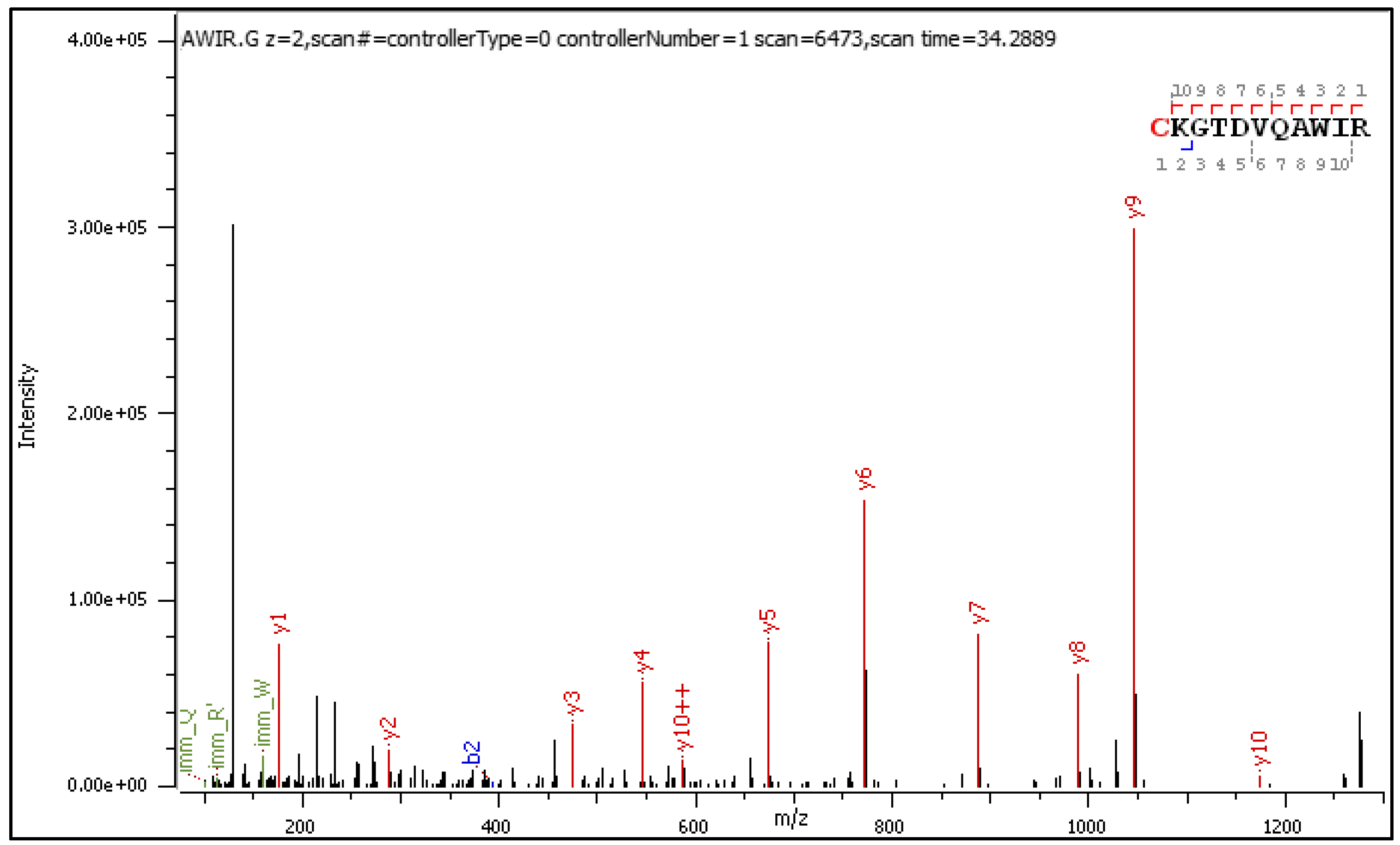
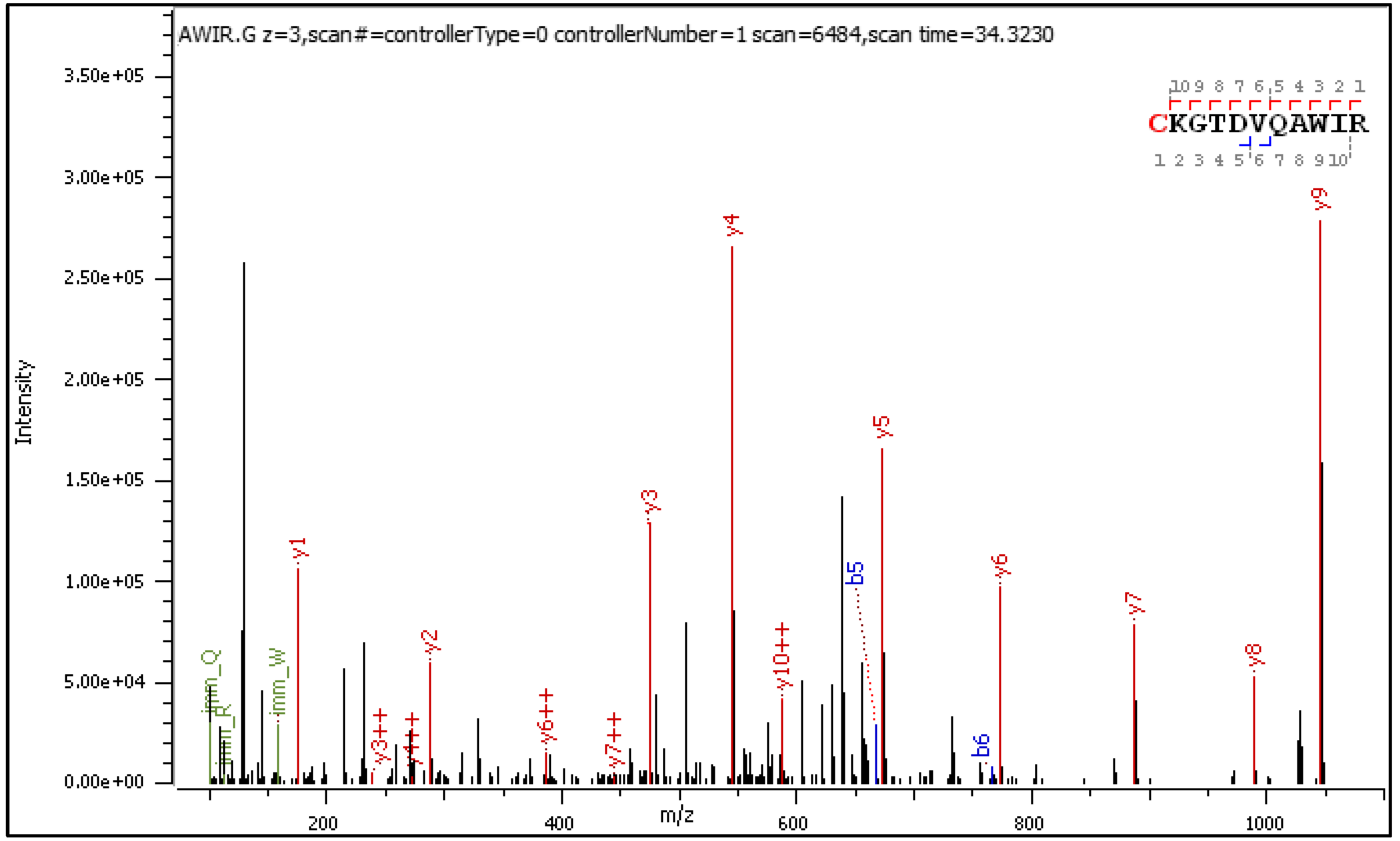
Figure 5.
HCD MS/MS spectrum of triply charged C[+162.]KGTDVQAWIR).
3. Discussion
The CID spectra of S-glucosylated peptides reveal the presence of automatically assigned principal fragment ions along with numerous unidentified m/z signals, including high-intensity peaks. Manual examination of the spectra enabled us to assign the fragment ions to many previously undefined m/z signals. Their presence sheds light on the side transformations concurrent with the MS-sequencing process.
We identified fragments [y/b+162]+/++, recognized as linked to the putative migration of glucocarbenium ion (Glc+, i.e.,163 Da) from the Cys to Lys and Arg residues. We further identified the signal of a general notation [y/b+162-losses]+/++, corresponding to initially formed N-glucosides which can further undergo Amadori rearrangement into isobaric N-fructosides (Figure 1). We detected doubly glycosylated fragment ions, labeled as [y/b+162+162-losses]+/++, indicating ongoing Glc+ transfers between ion fragments.
The mechanisms of these molecular transformations in a gas phase demand explication, in the context of analogous phenomena reported in the literature. The postulated transfer of the glucocarbenium ion Glc+ onto the guanidinium moiety of arginine (ArgH+) seems puzzling, for it would require difficult deprotonation of (ArgH+), enabling N-glucosylation of the uncharged arginine side chain.
3.1. Reported Spectral Evidence of Gas Phase Glycosyl Transfer
The following examples are drawn from mass spectrometric studies on unique gly-cans derived from various glycoconjugates. Currently, glycan rearrangement is observed via the migration of small monosaccharides to other intra-glycan positions, using CID analysis. In such cases, the glycan molecule plays a double function as the glycosyl donor and acceptor. Intra-molecular xylose migration has been observed using tandem mass spectrometry of N-linked glycans [30]. In studies on the Lewis X (Lex) and blood group antigen H-2, the CID-induced migration of fucose assisted by mobile proton led to the formation of a new O-glycosidic linkage [31]. Another intriguing outcome relates to CID-MS/MS analysis of a synthetic neo-glycolipid. As a result of an intramolecular mechanism of O-to-C glycosyl transfer, a C-glycosylated cholesterol derivative formed [32].
3.2. Glucosyl Transfer Evidence in CID/HCD Spectra of S-Glucopeptides
Tryptic peptides must cover a long analytical pathway, from protein digestion through chromatographic separation and complex ESI-MS/MS spectra acquisition, to be-come sequenced [33]. In the dry gas phase, charged peptides are flexible and continuously shape their conformations via intramolecular hydrogen bonds, salt bridges, hydrophobic interactions, and collisional interactions. The flexibility of peptides can be determined by measuring their end-to-end collision frequency [34,35,36,37,38,39,40,41].
In the present study, we selected CID/HCD spectra of S-glucosylated peptides containing the C[S-Glc] modification as a glycosyl ion [Glc+] source and diverse reactive groups located in their side chains. We assumed that under CID conditions the S-glycosidic bonds undergo “trashless” activation due to multiple collisions of the peptide with neutral gas molecules, as well as self-collisional interactions. As a result, a reactive transient ion pair, [Glc+/anh-Glc+][: S-Cys][Peptide]z+ OH, composed of Glc-oxocarbenium ion/protonated 1,2-anhydro-glucose [42] and peptidyl thiolate, may appear. This contact ion pair would be held together by electrostatic attractions. In most cases, collisional activation leads to neutral loss of 1,2-anhydro-glucose (162 Da) from the precursor ion. The 163 m/z signal of Glc+ may also appear in the MS/MS spectra.
The existing transient ion pair can collide with its conformationally accessible side chains, bearing nucleophilic/electrophilic functional groups—e.g., those located at the peptide C- or N-terminus. In anhydrous gaseous environments, the cysteine thiolate ion, [: S-Cys][Peptide]z+ OH, exhibits variable nucleophilicity and basicity, which depend on the peptide sequence and conformation. Therefore, we postulate that in favored spatial arrangements self-collisional peptide interactions may result in glucosyl transfer from Cys to side chains of Lys and Arg, or N-terminal amine groups. Such a collisional state brings together the critical structural elements of the glycopeptide—i.e., the glucosyl donor group, acceptor side chains, and groups involved in proton transfer (-NH2, -SH, -COOH). Thus, in certain respects such a collisional state resembles the catalytic center environment of some glycosyltransferases.
Generally, the stereochemistry of collisions and the energies of the colliding partners may control the mechanisms of glucosyl transfer to the Arg guanidinium end. Two routes, denoted Route N and Route B, are postulated. In Route N, the peptidyl thiolate acts as a catalytic nucleophile (N), whereas following Route B it behaves as a catalytic base (B). Figure 6 illustrates these two possible mechanisms of arginine N-glycosylation of peptides in a gas phase, with route B also applying for N-glycosylation of the protonated lysine and the peptide N-terminus.
Figure 6.
Hypothetical mechanisms of gas phase glucosyl transfer from Cys residue to the guanidinium group of Arg residue.
Figure 6.
Hypothetical mechanisms of gas phase glucosyl transfer from Cys residue to the guanidinium group of Arg residue.
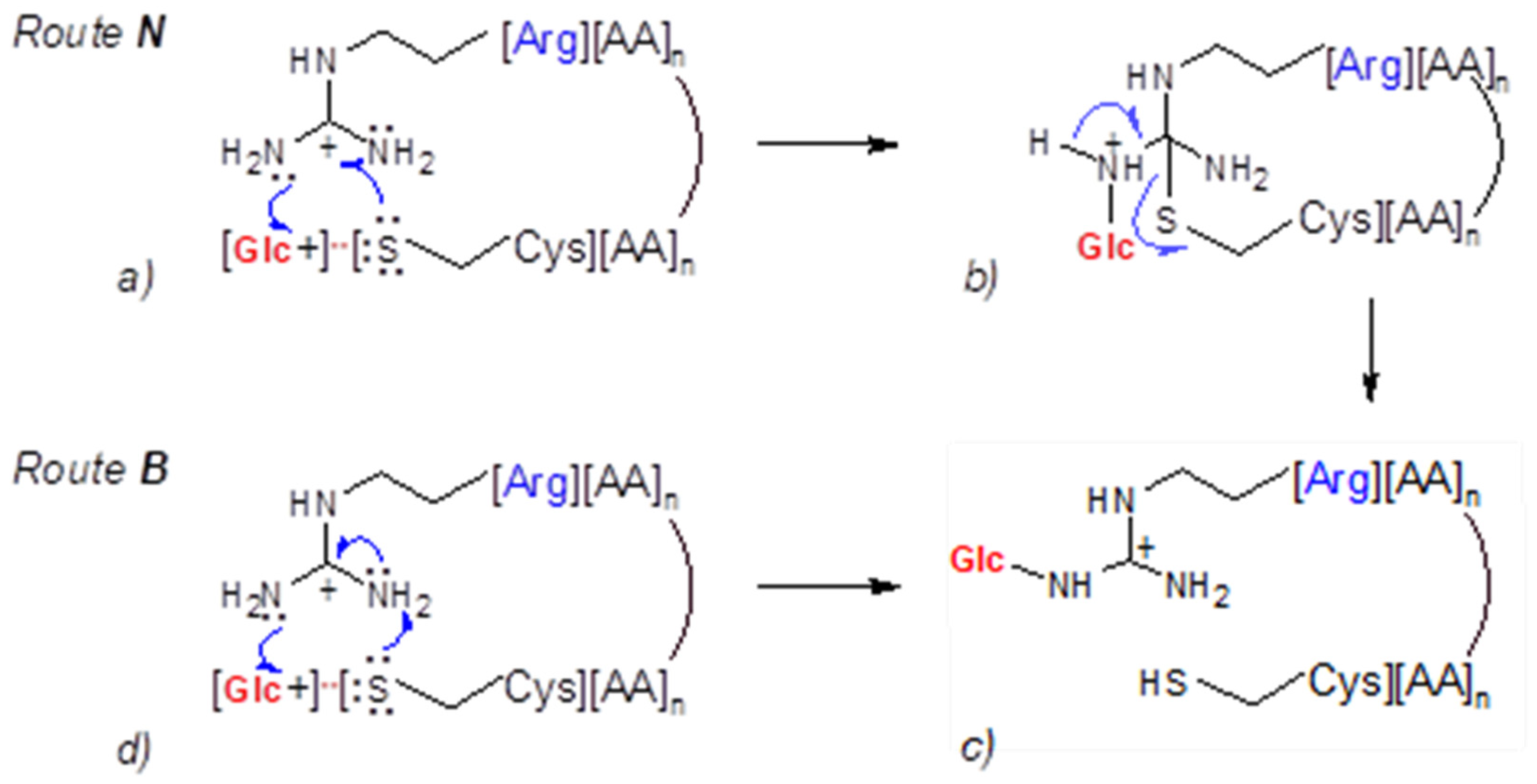
3.2.1. Route N: Cysteine Thiolate Acts as a Catalytic Nucleophile
We postulate that the [Glc+/anh-Glc+][: S-Cys][Peptide]z+ OH ion pair can collide with a guanidinium group of the C-terminal arginine (ArgH+). Steric interactions and Columb attraction/repulsion forces affect the collisional transient structure and impact the S→N glucosyl transfer process.
The guanidinium function of arginine is a planar resonance-stabilized structure comprising a central carbon atom of sp2 hybridization, which is linked to three nitrogen atoms by single C-N bonds [43]. The approaching thiolate anion neutralizes the positive charge of guanidinium carbon and changes the orbital hybridization of both carbon and nitrogen atoms. The resulting uncharged transient thioether cyclopeptide has tetrahedral carbon connected to three sp3-hybridized amine groups. This sterically crowded and electron-rich structural element traps the proximate Glc+ ion to form a C1-N+ glycosidic linkage with an accessible primary amine group. The positive charge of the created transitional structure is located on a single nitrogen atom and is not resonance-stabilized. Finally, the reaction of a thioether ring opening liberates the side chain’s cysteine thiol and restores the energetically favorable planar guanidinium N-glucosylated function.
In summary, the described mechanism of glucosyl transfer appeare feasible through the sp2 to sp3 hybridization changes in the carbon and nitrogen atoms of the arginine guanidinium function. Such a rehybridization can be described as “structural pyramidalization”, which enhances the nucleophilicity of nitrogen atoms.
The concept of enzymatic electrophilic functionalization of the arginine guanidinium group, omitting its deprotonation, was developed in studies on arginine kinases by Falcioni et al. [44]. These authors proposed a sophisticated mechanism of Arg phosphorylation, involving a process they described as “polarization–pyramidalization” of nitrogen in the arginine side chain. They also suggested that this phenomenon is exploited by many classes of enzymes mediating the post-translational modification of arginine, including N-glycosylation.[30]
The presented series of transformations proceeding in a gas phase and resulting in arginine residue N-glucosylation appears analogous to the arginine deiminase mode of action. Arginine deiminase (EC 3.5.3.6) uses the catalytic Cys406 as an essential nucleophile to form an intermediate covalent adduct with the guanidinium carbon of the substrate, followed by ammonia elimination [45]. Other less adequate examples exist of liquid phase reactions employing protonated guanidine derivatives and thiolates to perform particular target-directed transformations [46]. It has also been reported that the arginine guanidinium ion reacts with a carbanion derived from the condensation of glutaraldehyde with the lysine ε-amine group [47].
3.2.2. Route B: Cysteine Thiolate Behaves as a Catalytic Base
In the alternative route B, the cysteine thiolate in the [Glc+/anh-Glc+][: S-Cys][Peptide]z+ OH ion pair deprotonates a conformationally accessible guanidinium moiety of protonated ArgH+ (Figure 6, panel d). The resulting (unprotonated) guanidino group, while retaining its planar geometry, acquires nucleophilic properties. Next, it attacks the proximate Glc+ ion to form a charged N-glucosylated Arg+ (Figure 6, panel c). In this variant of the arginine N-glycosylation mechanism, the basicity of the thiolate in a gas phase dictates its role as an initial proton acceptor triggering the subsequent transformation steps.
Some experimental facts support this hypothetical sequence of rearrangements. In a liquid phase, the Arg of pKa 13.8 [48] can be N-functionalized using a stronger base—e.g., the Barton base of pKa 15.3 [49,50]. Analogously, with NCS reagent in a gas phase, N-acylation of the peptidyl arginine residue was observed exclusively if it existed in an un-protonated form [51,52]. It has also been reported that the pKa parameter, calculated for the cysteine residues present in some human kinases, ranges in scope from 7 to even 24 units [53,54]. This indicates enormous differences in the reactivity of the cysteine thiols dictated by the protein cavity characteristics. Thus, in favorable surroundings cysteine can be much more basic than arginine. Studies on the Rhodobacter sphaeroides mitochondrial proton pump provide evidence of the role of Cys-139 as an initial proton acceptor acting in the altered form of Cytochrome c oxidase (CcO) [55]. The cited scientific data demonstrate the experimental conditions in which the [:S-Cys]Peptide thiolate behaves either as a strong nucleophile or as a super-basic proton acceptor.
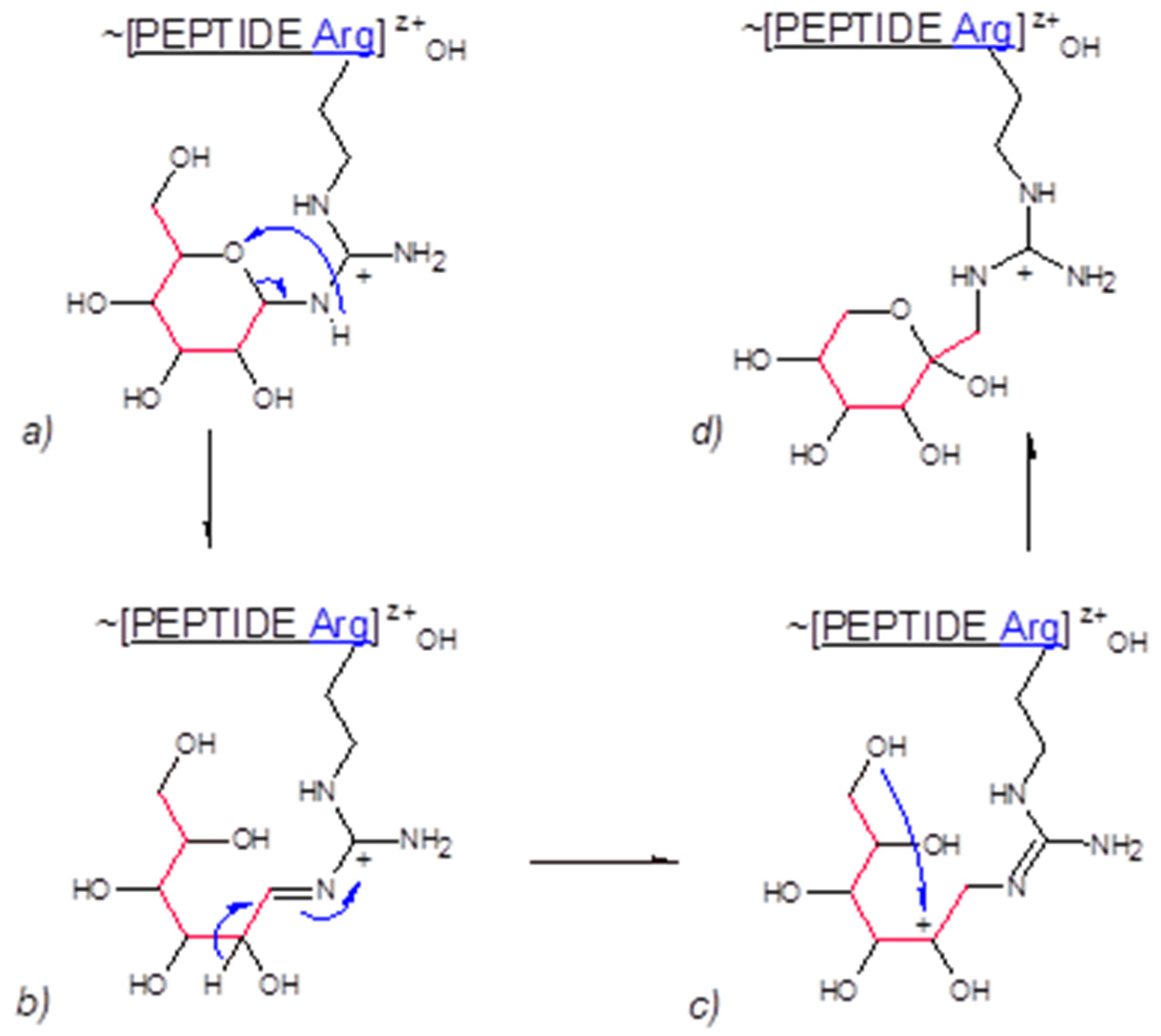
3.3. Gas Phase Amadori Rearrangement
We demonstrated in the results section that Lys and Arg N-glucosylated gas phase transfer products experience Amadori rearrangement to N-fructosylated structures. To the best of our knowledge, such events have not been observed previously. The hypothetical mechanism of these transformations is depicted in Figure 7. The process is initiated by the collisional splitting of the C(5)O-C(1) bond in the glucose hemiaminal (Figure 7, panel a), leading to the formation of the guanidinium Schiff base (Figure 7, panel b). Hydride 1,2-migration from C(2) to C(1) results in a new cationic species (Figure 7, panel c). Subsequent ring closure involving the C(6)-OH and C(2)+ cationic center produces N-fructosylated guanidinium residue of Arg (Figure 7, panel d). The postulated mechanism seems compatible with the low proton mobility environment of a gas phase, although 1,2-hydride shift mechanisms have also been proposed to explain enzymatic glucose to fructose interconversion [56].
The 1,2-hydride shift explains the fragmentation pattern of the S-adenosyl-L-methionine observed during CID analysis [57]. Analogously, the same phenomenon is proposed for elucidating differences in CID fragmentation of L-leucine and L-isoleucine [58].
Figure 7.
Hypothetical mechanism of Amadori rearrangement in gas phase.
4. Materials and Methods
The fragmentation spectra used in this study were derived from a library reported in a previous study. The list of CID and HCD spectral libraries containing S-glucosylated peptides is accessible in [14] (Table 1), and their sequences can be viewed in the Supplementary Materials at https://link.springer.com/article/10.1007/s00726-022-03208-7ใ
4.1. MS/MS Spectra Acquisition
All CID and HCD fragmentation spectra were generated using an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bergen) spectrometer working in the regime of data-dependent acquisition. The normalized collision energy was set to 30%. Dynamic exclusion was disabled. Raw instrument data (.RAW) were processed using the Byonic search engine (Protein Metrics Inc, v.0–25) against the Uni-Protaccession_p00698.decoys.fasta. Protein FDR was set to 1% FDR. All searches used a 6-ppm precursor and 20-ppm fragment ion tolerance for HCD, with 0.500 Da tolerance for CID fragmentation. All searches considered tryptic peptides with a maximum of two missed cleavages.
4.2. Manual Curation of Diagnostic Fragment Ions
The fragmentation pathways shown in Scheme 1 display ions and their lost or gained neutral fragments, the sizes of which were determined using a monoisotopic mass calculator accessible at https://www.sisweb.com/referenc/tools/exactmass.htm. The results are presented in Table 6.
Theoretical values of y/b fragment ions for a particular peptide sequence were determined using an MS/MS fragmentation calculator provided by the University of Washington’s Proteomics Resource available online at https://proteomicsresource.washington.edu/cgi-bin/fragment.cgi. The obtained data were used to calculate theoretical values for the target diagnostic fragment ions listed in Table 1, Section 2.2. Next, a manual search of selected MS/MS spectra was carried out to identify the m/z signals corresponding to particular diagnostic ions. The Excel sheets containing exemplary calculations and outcomes of manual spectra searches are also included in the Supplementary Materials (Supp Excel files).
5. Conclusions
The CID/HCD-fragmentation of S-glucopeptides is accompanied by a transfer of glucosyl ion Glc+ from cysteine to C-terminal Lys, Arg, and N-terminal amine groups. The moderate stability of the S-glycosidic bond under CID conditions enables the S-glucosylated precursor ions and their nascent fragment ions to play the double role of donor/acceptor in inter- and intramolecular glycosyl transfer processes.
We postulate that collisional activation of the S-glucosidic linkage may generate species carrying a reactive ion pair structure. The peptidyl thiolate component of the ion pair can display capabilities of both a super-active base and a strong nucleophile.
Such a duality can explain two alternative pathways of glycosyl transfer onto protonated arginine residue. While behaving as a nucleophile, the peptidyl thiolate attacks the guanidinium carbon, neutralizes its positive charge, and drives changes in orbital hybridization—a process we termed “pyramidalization”. The resulting transient electron-rich tetrahedral structure captures a nearby Glc+ cation and is stabilized by peptidyl thiol elimination. Ultimately, a charged planar N-glucosylated arginine residue is formed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: CID MS/MS spectrum of doubly charged C[+162]KGTDVQAWIR_(PID 1603); Figure S1a: CID MS/MS spectrum of doubly charged C[+162]KGTDVQAWIR_(PID 1603); 610-680 m/z range; Figure S2: CID MS/MS spectrum of triply charged C[+162]KGTDVQAWIR_ (PID 1606); Figure S2a: CID MS/MS spectrum of triply charged C[+162]KGTDVQAWIR_ (PID 1606); 405-455 m/z range; Figure S3: CID MS/MS spectrum of doubly charged C[+162]ELAAAMK_ (PID 222); Figure S3a: CID MS/MS spectrum of doubly charged C[+162]ELAAAMK_ (PID 222); 371-471 m/z range; Figure S4: CID MS/MS spectrum of doubly charged RC[+162]ELAAAMK_(PID 745); Figure S4a: CID MS/MS spectrum of doubly charged RC[+162]ELAAAMK_ (PID 745); 465-565 m/z range; Figure S5: CID MS/MS spectrum of doubly charged WWC[+162.]NDGR_ (PID 1189); Figure S5a: CID MS/MS spectrum of doubly charged WWC[+162.]NDGR_ (PID 1189); 425-525 m/z range; Figure S6: HCD MS/MS spectrum of doubly charged C[+162.]KGTDVQAWIR_ (PID 18335); Figure S7: HCD MS/MS spectrum of triply charged C[+162.]KGTDVQAWIR_ (PID 18338); Table S1: The set of fragment ions from the +2 charged precursor C[+162]ELAAAMK_ (PID 222), as evidence of CID fragmentations following paths A, B, and C; Table S1a: The set of doubly glycosylated fragment ions and nascent diagnostic ions from the +2 charged precursor C[+162]ELAAAMK_ (PID 222), as evidence of CID fragmentations following paths A, B, and C; Table S2: The set of fragment ions from the +2 charged precursor RC[+162]ELAAAMK_ (PID 754), as evidence of CID fragmentations following paths A, B, and C; Table S3: The set of fragment ions from the +2 charged precursor WWC[+162.]NDGR_ (PID 1189), as evidence of CID fragmentations following paths A, B, and C; Table S4: The set of fragment ions from the +2 charged precursor SLGNWVC[+162]AAK_ (PID 1684), as evidence of CID fragmentations following paths A, B, and C; Table S5: The set of fragment ions from the +2 charged precursor C[+162.]KGTDVQAWIR_(PID 18335), as evidence of HCD fragmentations following paths A, B, and C; Table S5a: The set of doubly glycosylated fragment ions and nascent diagnostic ions from the +2 charged precursor C[+162.]KGTDVQAWIR_(PID 18335), as evidence of HCD fragmentations following paths A, B, and C. No diagnostic signals were detected; Table S6: The set of fragment ions from the +3 charged precursor C[+162.]KGTDVQAWIR _ (PID 18338), as evidence of HCD fragmentations following paths A, B, and C; Supp Excel files.
Funding
This research was funded by the POLISH MINISTRY of SCIENCE and HIGHER EDUCATION, grant number 3195/B/P01/2007/33.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Bagdonaite, I.; Malakar, S.A.; Polasky, D.A.; Riley, N.M.; Schjoldager, K.; Vakhrushev, S.Y.; Halim, A.; Aoki-Kinoshita, K.F.; Nesvizhskii, A.I.; Bertozzi, C.R.; et al. Glycoproteomics. Nature Reviews Methods Primers 2022, 2. [Google Scholar] [CrossRef]
- Grabarics, M.; Lettow, M.; Kirschbaum, C.; Greis, K.; Manz, C.; Pagel, K. Mass Spectrometry-Based Techniques to Elucidate the Sugar Code. Chem Rev 2022, 122, 7840–7908. [Google Scholar] [CrossRef] [PubMed]
- Illiano, A.; Pinto, G.; Melchiorre, C.; Carpentieri, A.; Faraco, V.; Amoresano, A. Protein Glycosylation Investigated by Mass Spectrometry: An Overview. Cells 2020, 9, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Krasnova, L.; Wong, C.H. Oligosaccharide Synthesis and Translational Innovation. J Am Chem Soc 2019, 141, 3735–3754. [Google Scholar] [CrossRef] [PubMed]
- Doelman, W.; van Kasteren, S.I. Synthesis of Glycopeptides and Glycopeptide Conjugates. Org Biomol Chem 2022, 20, 6487–6507. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.G.; Demchenko, A.V. Intramolecular Glycosylation. Beilstein Journal of Organic Chemistry 2017, 13, 2028–2048. [Google Scholar] [CrossRef] [PubMed]
- Escopy, S.; Demchenko, A.V. Transition-Metal-Mediated Glycosylation with Thioglycosides. Chemistry – A European Journal 2022, 28. [Google Scholar] [CrossRef] [PubMed]
- Buch-Larsen, S.C.; Hendriks, I.A.; Lodge, J.M.; Rykær, M.; Furtwängler, B.; Shishkova, E.; Westphall, M.S.; Coon, J.J.; Nielsen, M.L. Mapping Physiological ADP-Ribosylation Using Activated Ion Electron Transfer Dissociation. Cell Rep 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.C.; Burlingame, A.L.; Medzihradszky, K.F. Cysteine S-Linked N-Acetylglucosamine (S-GlcNAcylation), a New Post-Translational Modification in Mammals. Molecular and Cellular Proteomics 2016, 15, 3405–3411. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Wu, R. Global and Site-Specific Analysis Revealing Unexpected and Extensive Protein S-GlcNAcylation in Human Cells. Anal Chem 2017, 89, 3656–3663. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Ahlawat, S.; Rao, A. Biochemical Characterization of an Inverting S/O-HexNAc-Transferase and Evidence of S-Linked Glycosylation in Actinobacteria. Glycobiology 2022, 32, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.Q.; Zhang, X.; Zou, Y.; Shi, R.; Cao, J.G.; Xu, S.Y.; Deng, L.F.; Zhou, L.; Gong, Y.; Shu, X.; et al. Nonenzymatic Stereoselective S-Glycosylation of Polypeptides and Proteins. J Am Chem Soc 2021, 143, 11919–11926. [Google Scholar] [CrossRef] [PubMed]
- Soya, N.; Fang, Y.; Palcic, M.M.; Klassen, J.S. Trapping and Characterization of Covalent Intermediates of Mutant Retaining Glycosyltransferases. Glycobiology 2011, 21, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Buchowiecka, A.K. Protein Cysteine S-Glycosylation: Oxidative Hydrolysis of Protein S-Glycosidic Bonds in Aqueous Alka-line Environments. Amino Acids 2023, 55, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Moises, J.E.; Regl, C.; Hinterholzer, A.; Huber, C.G.; Schubert, M. Unambiguous Identification of Glucose-Induced Glycation in MAbs and Other Proteins by NMR Spectroscopy. Pharm Res 2023, 40, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Mossine, V.V.; Yaylayan, V. Identification of MS/MS Diagnostic Ions to Distinguish Schiff Bases of Nα- or Nε-Mono-Glycated and Nα,Nε-Di-Glycated Lysines from Their Amadori Isomers. European Food Research and Technology 2022, 248, 2753–2763. [Google Scholar] [CrossRef]
- Xing, H.; Yaylayan, V. Insight into Isomeric Diversity of Glycated Amino Acids in Maillard Reaction Mixtures. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Giangrande, C.; Auberger, N.; Rentier, C.; Papini, A.M.; Mallet, J.M.; Lavielle, S.; Vinh, J. Multi-Stage Mass Spectrometry Analysis of Sugar-Conjugated β-Turn Structures to Be Used as Probes in Autoimmune Diseases. J Am Soc Mass Spectrom 2016, 27, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Vakhrushev, S.Y.; Zamfir, A.; Peter-Katalinić, J. 0,2A n Cross-Ring Cleavage as a General Diagnostic Tool for Glycan Assignment in Glycoconjugate Mixtures. J Am Soc Mass Spectrom 2004, 15, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Luo, J.; Li, S. Bacteria-Catalyzed Arginine Glycosylation in Pathogens and Host. Front Cell Infect Microbiol 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Corcilius, L.; Sharp, P.P.; Rajkovic, A.; Ibba, M.; Parker, B.L.; Payne, R.J. Synthesis of Rhamnosylated Arginine Glycopeptides and Determination of the Glycosidic Linkage in Bacterial Elongation Factor P. Chem Sci 2017, 8, 2296–2302. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, P.M.; Nowak, K.; Panse, C.; Leutert, M.; Grossmann, J.; Schlapbach, R.; Hottiger, M.O. Gas-Phase Fragmentation of ADP-Ribosylated Peptides: Arginine-Specific Side-Chain Losses and Their Implication in Database Searches. J Am Soc Mass Spectrom 2021, 32, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Moises, J.E.; Regl, C.; Hinterholzer, A.; Huber, C.G.; Schubert, M. Unambiguous Identification of Glucose-Induced Glycation in MAbs and Other Proteins by NMR Spectroscopy. Pharm Res 2023, 40, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.-R.; Zhao, L.-J.; Sun, Q.; Xie, H.-J.; Lei, Q.-F.; Fang, W.-J. Explore the Reaction Mechanism of the Maillard Reaction: A Density Functional Theory Study. J Mol Model 2015, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Korwar, A.M.; Vannuruswamy, G.; Jagadeeshaprasad, M.G.; Jayaramaiah, R.H.; Bhat, S.; Regin, B.S.; Ramaswamy, S.; Giri, A.P.; Mohan, V.; Balasubramanyam, M.; et al. Development of Diagnostic Fragment Ion Library for Glycated Peptides of Human Serum Albumin: Targeted Quantification in Prediabetic, Diabetic, and Microalbuminuria Plasma by Parallel Reaction Monitoring, SWATH, and MSE. Molecular and Cellular Proteomics 2015, 14, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.T.; Hemmler, D.; Diederich, P.; Rychlik, M.; Marshall, J.W.; Schmitt-Kopplin, P. Open Search of Peptide Glycation Products from Tandem Mass Spectra. Anal Chem 2022, 94, 5953–5961. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.; Schmidt, R.; Vikhnina, M.; Grishina, T.; Frolov, A. Maillard Proteomics: Opening New Pages. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Hemmler, D.; Schmitt-Kopplin, P. Discovery of Glycation Products: Unraveling the Unknown Glycation Space Using a Mass Spectral Library from In Vitro Model Systems. Anal Chem 2024, 96, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Keilhauer, E.C.; Geyer, P.E.; Mann, M. HCD Fragmentation of Glycated Peptides. J Proteome Res 2016, 15, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Hecht, E.S.; Loziuk, P.L.; Muddiman, D.C. Xylose Migration During Tandem Mass Spectrometry of N-Linked Glycans. J Am Soc Mass Spectrom 2017, 28, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Lettow, M.; Mucha, E.; Manz, C.; Thomas, D.A.; Marianski, M.; Meijer, G.; von Helden, G.; Pagel, K. The Role of the Mobile Proton in Fucose Migration. Anal Bioanal Chem 2019, 411, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Banoub, J.; Boullanger, P.; Lafont, D.; Cohen, A.; El Aneed, A.; Rowlands, E. In Situ Formation of C-Glycosides during Electrospray Ionization Tandem Mass Spectrometry of a Series of Synthetic Amphiphilic Cholesteryl Polyethoxy Neoglycolipids Containing N-Acetyl-D-Glucosamine. J Am Soc Mass Spectrom 2005, 16, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.A.; Fort, K.L.; Kim, D.; Servage, K.A.; Pierson, N.A.; Clemmer, D.E.; Russell, D.H. From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance p Reveals the Origin of Peptide Conformations. J Am Chem Soc 2013, 135, 19147–19153. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Nau, W.M. A Conformational Flexibility Scale for Amino Acids in Peptides. Angewandte Chemie - International Edition 2003, 42, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bordas-Nagy, J. Peptide Conformation in Gas Phase Probed by Collision-Induced Dissociation and Its Correlation to Conformation in Condensed Phases. J Am Soc Mass Spectrom 2006, 17, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Meroueh, O.; Hase, W.L. Collisional Activation of Small Peptides. Journal of Physical Chemistry A 1999, 103, 3981–3990. [Google Scholar] [CrossRef]
- Brintaki, A.N.; Lai-Yuen, S.K. EBGF: An Enhanced Geometric Hierarchical Representation for Protein Modeling and Rapid Self-Collision Detection. Comput Aided Des Appl 2009, 6, 625–638. [Google Scholar] [CrossRef]
- Foreman, D.J.; Mcluckey, S.A. Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry. Anal Chem 2020, 92, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Prentice, B.M.; Mc Luckey, S.A. Gas-Phase Ion/Ion Reactions of Peptides and Proteins: Acid/Base, Redox, and Covalent Chemistries. Chemical Communications 2013, 49, 947–965. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hudgins, R.R.; Nau, W.M. Primary and Secondary Structure Dependence of Peptide Flexibility Assessed by Fluorescence-Based Measurement of End-to-End Collision Rates. J Am Chem Soc 2004, 126, 16665–16675. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.G.; Komives, E.A. Charge State Coalescence during Electrospray Ionization Improves Peptide Identification by Tandem Mass Spectrometry. J Am Soc Mass Spectrom 2012, 23, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Franconetti, A.; Ardá, A.; Asensio, J.L.; Blériot, Y.; Thibaudeau, S.; Jiménez-Barbero, J. Glycosyl Oxocarbenium Ions: Structure, Conformation, Reactivity, and Interactions. Acc Chem Res 2021, 54, 2552–2564. [Google Scholar] [CrossRef]
- Moriarty, N.W.; Liebschner, D.; Tronrud, D.E.; Adams, P.D. Arginine Off-Kilter: Guanidinium Is Not as Planar as Restraints Denote. Acta Crystallogr D Struct Biol 2020, 76, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Falcioni, F.; Molt, R.W.; Jin, Y.; Waltho, J.P.; Hay, S.; Richards, N.G.J.; Blackburn, G.M. Arginine Kinase Activates Arginine for Phosphorylation by Pyramidalization and Polarization. ACS Catalysis 2024, 6650–6658. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Galkin, A.; Herzberg, O.; Dunaway-Mariano, D. Arginine Deiminase Uses an Active-Site Cysteine in Nucleophilic Catalysis of L-Arginine Hydrolysis. J Am Chem Soc 2004, 126, 5374–5375. [Google Scholar] [CrossRef] [PubMed]
- Novichkov, A.I.; Hanopolskyi, A.I.; Miao, X.; Shimon, L.J.W.; Diskin-Posner, Y.; Semenov, S.N. Autocatalytic and Oscillatory Reaction Networks That Form Guanidines and Products of Their Cyclization. Nat Commun 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Mauguen, Y.; Prangé, T. Revisiting Glutaraldehyde Cross-Linking: The Case of the Arg-Lys Intermolecular Doublet. Acta Crystallogr Sect F Struct Biol Cryst Commun 2010, 66, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Fitch, C.A.; Platzer, G.; Okon, M.; Garcia-Moreno, B.E.; McIntosh, L.P. Arginine: Its PKa Value Revisited. Protein Science 2015, 24, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Grundler, V.; Gademann, K. Direct Arginine Modification in Native Peptides and Application to Chemical Probe Development. ACS Med Chem Lett 2014, 5, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of Novel Uncharged Organic Superbases: Merging Basicity and Functionality. Acc Chem Res 2021, 54, 3108–3123. [Google Scholar] [CrossRef] [PubMed]
- McGee, W.M.; Mentinova, M.; McLuckey, S.A. Gas-Phase Conjugation to Arginine Residues in Polypeptide Ions via N-Hydroxysuccinimide Ester-Based Reagent Ions. J Am Chem Soc 2012, 134, 11412–11414. [Google Scholar] [CrossRef] [PubMed]
- Foreman, D.J.; Mcluckey, S.A. Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry. Anal Chem 2020, 92, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Awoonor-Williams, E.; Rowley, C.N. How Reactive Are Druggable Cysteines in Protein Kinases? J Chem Inf Model 2018, 58, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhan, S.; Che, Y.; Shen, J. Reactivities of the Front Pocket N-Terminal Cap Cysteines in Human Kinases. J Med Chem 2022, 65, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Varanasi, L.; Hosler, J. Alternative Initial Proton Acceptors for the D Pathway of Rhodobacter Sphaeroides Cytochrome c Oxidase. Biochemistry 2011, 50, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, S.; Saravanamurugan, S.; Riisager, A. Glucose Isomerization by Enzymes and Chemo-Catalysts: Status and Current Advances. ACS Catal 2017, 7, 3010–3029. [Google Scholar] [CrossRef]
- Barnett, Z.M.E.; Feketeová, L.; O’Hair, R.A.J. The Major Product Ion of S-Adenosyl-L-Methionine Arises from a Neighboring Group Reaction. Rapid Communications in Mass Spectrometry 2010, 24, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Arthur, C.J.; Gates, P.J. A Computational and Experimental Study of the Fragmentation of L-Leucine, l-Isoleucine and l-Allo-Isoleucine under Collision-Induced Dissociation Tandem Mass Spectrometry. Analyst 2020, 145, 6632–6638. [Google Scholar] [CrossRef] [PubMed]
Table 1.
The set of diagnostic ions for investigating glycosyl transfer effects.
| Mark | Ion type (notation) | Moiety lost or gained |
|---|---|---|
| 162 NL | [M+zH+-162]/z | 1,2-anhydroglucose loss |
| NL ions | [M+zH+-180;-150;-120; -90]/z | hexose, pentose, tetrose, triose loss, respectively |
| 179 NL | [M+zH+-179]/z | aminoglucose/aminofructose loss |
| 204 NL | [M+zH+-204/z | 1,2-anhydroglucose loss + carbodiimide loss |
| NL ions | [M+zH+-nH2O]/z, n=1, 2, 3 | water loss,-18 Da, -36 Da, -54 Da |
| 162 NG | y+/b+ [+162] | glucosyl gain |
| 144 NG etc. |
y+/b+ [+144; +126; +108] | from 162 NG ions loss of 1, 2, and 3 water molecules, respectively |
| 78 NG etc. |
y+/b+ [+78; +72; +42] | from 162 NG ions loss of (3H2O+ CH2O), triose, tetrose, respectively |
Table 4.
The set of fragment ions from the +3 charged precursor, as evidence of fragmentations following paths A, B, and C.
Table 4.
The set of fragment ions from the +3 charged precursor, as evidence of fragmentations following paths A, B, and C.
| [b-3H2O]++ | b2 | b3 | b4 | b5 | b6 | b7 | b10 | 0.15 | Path C | |||||
| [b-2H2O]++ | b4 | b6 | b7 | b10 | 0.10 | |||||||||
| [b-H2O]++ | b1 | b3 | b4 | b5 | b6 | b7 | b9 | 0.26 | ||||||
| [b-150]++ | b6 | b7 | b9 | b10 | 0.02 | Path B | ||||||||
| [b-90]++ | b2 | b3 | b5 | b6 | b7 | b9 | b10 | 0.29 | ||||||
| [b-120]++ | b4 | b5 | b6 | b7 | b10 | 0.14 | Path A | |||||||
| [b]++ | b5 | b6 | b7 | 1.00 | ||||||||||
| [b-3H2O]+ | b1 | b2 | b3 | b4 | b5 | b6 | b8 | 0.16 | Path C | |||||
| [b-2H2O]+ | b1 | b2 | b4 | b5 | b6 | 0.70 | ||||||||
| [b-H2O]+ | b1 | b2 | b5 | b6 | b7 | b8 | 0.06 | |||||||
| [b-150]+ | b2 | b3 | b4 | b5 | b6 | b7 | 0.81 | Path B | ||||||
| [b-90]+ | b1 | b2 | b3 | b5 | b6 | b7 | b8 | 0.13 | ||||||
| [b-120]+ | b1 | b2 | b3 | b4 | b5 | b6 | b7 | b8 | 0.41 | Path A | ||||
| [b]+ | b1 | b2 | b3 | b4 | b5 | b6 | b7 | b8 | # |
1.00 Total abundance |
||||
|
Total abundance 1.00 |
CGlc | K | G | T | D | V | Q | A | W | I | R | |||
| # | y7 | y6 | y5 | y4 | y2 | y1 | [y]+ | |||||||
| Path A | 0.02 | y3 | y2 | [y+162]+ | ||||||||||
| 0.06 | y8 | y5 | y4 | y3 | y2 | y1 | [y+42]+ | |||||||
| 0.03 | y8 | y6 | y5 | y3 | y2 | y1 | [y+24]+ | |||||||
| Paty B | 0.14 | y4 | y3 | [y+72]+ | ||||||||||
| 0.07 | y7 | y6 | y3 | y2 | y1 | [y+12]+ | ||||||||
| 0.02 | y5 | y1 | [y+54]+ | |||||||||||
| Path C | 0.02 | y3 | y2 | [y+144]+ | ||||||||||
| 0.04 | y3 | y2 | y1 | [y+126]+ | ||||||||||
| 0.03 | y3 | y2 | y1 | [y+108]+ | ||||||||||
| 0.01 | y3 | y1 | [y+78]+ | |||||||||||
| 1.00 | y10 | y7 | y6 | y5 | y4 | y3 | y2 | [y]++ | ||||||
| Path A | 0.20 | y10 | y9 | y8 | y7 | y5 | y4 | y2 | y1 | [y+162]++ | ||||
| 0.17 | y6 | y5 | y4 | y3 | y2 | [y+42] ++ | ||||||||
| 0.12 | y9 | y8 | y6 | y5 | y4 | y3 | [y+24]++ | |||||||
| Paty B | 0.10 | y9 | y6 | y5 | y4 | y3 | y1 | [y+72]++ | ||||||
| 0.19 | y10 | y9 | y8 | y7 | y6 | y5 | [y+12]++ | |||||||
| 0.08 | y9 | y6 | y4 | [y+54]++ | ||||||||||
| Path C | 0.11 | y8 | y7 | y4 | y3 | y2 | y1 | [y+144]++ | ||||||
| 0.17 | y10 | y9 | y8 | y7 | y5 | y3 | [y+126]++ | |||||||
| 0.04 | y10 | y9 | y7 | y4 | y2 | y1 | [y+108]++ | |||||||
| 0.14 | y9 | y8 | y6 | y5 | y4 | y2 | y1 | [y+78]++ | ||||||
Table 4.
a. Gas phase transformations of the C[+162.]KGTDVQAWIR precursor of charge state +3.
| Transformations | Diagnostic ion(s) | Transformation path |
|---|---|---|
| N-glucosylation of Arg | [y+162]+/++ | A |
| N-glucosylation of Arg | [y+42; y+24]+/++ | A |
| N-fructosylation of Arg | [y+72; y+54; y+12]+/++ | B |
| N-fructosylation of Arg | y+144; y+126; y+108; y+78 | C |
| S→N (Glc+) migration and tetrose loss | [b-120]+/++ | A |
| S→N (Glc+) migration and tiose loss | [b-90]+/++ | B |
| S→N (Glc+) migration and tiose loss | {b-150]+/++ | B |
Table 5.
The set of doubly glycosylated and nascent fragment ions from the +3 charged precursor, as evidence of fragmentations following paths A, B, and C.
Table 5.
The set of doubly glycosylated and nascent fragment ions from the +3 charged precursor, as evidence of fragmentations following paths A, B, and C.
| [b+108]++ | b1 | b4 | b5 | b6 | b7 | b8 | b9 | b10 | 13.23 | Path C | |||
| [b+126]++ | b2 | b3 | b4 | b5 | b6 | b7 | b8 | b10 | 7.82 | ||||
| [b+144]++ | b2 | b3 | b4 | b5 | b7 | b8 | b9 | b10 | 2.41 | ||||
| [b+12]++ | b2 | b3 | b4 | b5 | b8 | b9 | 2.45 | Path B | |||||
| [b+72]++ | b1 | b2 | b5 | b6 | b8 | b9 | b10 | 2.67 | |||||
| [b+42]++ | b4 | b9 | 0.33 | Path A | |||||||||
| [b+162]++ | b1 | b2 | b3 | b6 | b8 | b9 | 1.00 | ||||||
| [b+108]+ | b1 | b2 | b3 | b4 | b5 | 1.27 | Path C | ||||||
| [b+126]+ | b1 | b3 | 1.64 | ||||||||||
| [b+144]+ | b1 | b2 | b3 | b5 | 0.57 | ||||||||
| [b+12]+ | b1 | b2 | b3 | b4 | b6 | 1.15 | Path B | ||||||
| [b+72]+ | b1 | b4 | b7 | 0.70 | |||||||||
| [b+42]+ | b1 | b2 | b6 | 0.89 | Path A | ||||||||
| [b+162]+ | b1 | b2 | b3 | b4 | b5 |
1.00 Total abundance (reference) |
|||||||
| CGlc | K | G | T | D | V | Q | A | W | I | R | |||
Table 6.
List of mass losses and gains for precursors and fragment ions.
| Entry | Notation | Monoisotopic mass [Da] | Chemical formula | Chemical formula calculation |
|---|---|---|---|---|
| 1 | 204 | 204.074623 | C7H12O5N2 | 1,2-anhydro-Glc + carbodiimide |
| 2 | 180 | 180.063390 | C6H12O6 | Glc |
| 3 | 179 | 179.079374 | C6H13O5N | 1-amino-Glc |
| 4 | 162 | 162.052824 | C6H10O5 | 1,2-anhydro-Glc |
| 5 | 150 | 150.052824 | C5H10O5 | pentose |
| 6 | 144 | 144.042260 | C6H8O4 | 162 – H2O |
| 7 | 126 | 126.031695 | C6H6O3 | 162 – 2H2O |
| 8 | 120 | 120.042260 | C4H8O4 | tetrose |
| 9 | 108 | 108.021130 | C6H4O2 | 162 - 3H2O |
| 10 | 90 | 90.031695 | C3H6O3 | triose |
| 11 | 78 | 78.010565 | C5H2O | 162 - 3H2O – CH2O |
| 12 | 72 | 72.021130 | C3H4O2 | 162 - triose |
| 13 | 54 | 54.031695 | H6O3 | 3H2O |
| 14 | 54 | 54.010565 | C3H2O | 162 – triose – H2O |
| 15 | 42 | 42.021798 | CH2N2 | carbodiimide |
| 16 | 42 | 42.010565 | C2H2O | 162- tetrose |
| 17 | 36 | 36.021130 | H4O2 | 2H2O |
| 18 | 24 | 24.000000 | C2 | 162 - tetrose – H2O |
| 19 | 18 | 18.010565 | H2O | H2O |
| 20 | 12 | 12.00000 | C | C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



