
Submitted:
04 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
The need for solvating and encapsulating hydro-sensitive molecules drives noticeable trends in the applications of cyclodextrins in pharmaceutical industry, food, polymer, materials, and agricultural science. Among them, β-cyclodextrin is one of the most used for the entrapment of phenolic acid compounds to mask the bitterness of wheat bran. In this regard, there is still a need for a good predictive model that assesses β-cyclodextrin bitterness masking capabilities for various phenolic compounds. This study uses a dataset of 20 phenolic acids docked to the β-cyclodextrin cavity to generate three different binding constants. The data from docking study were combined with topological, topographical and quantum-chemical features from the ligands in a machine learning-based structure-activity relationship study. Three different models for each binding constant were computed using a combination of Genetic Algorithm (GA) and Multiple Linear Regression (MLR) approaches. The developed ML/QSAR models showed a very good performance, with high predictive ability and correlation coefficients of 0.969 and 0.984, for training and test sets, respectively. The models revealed several factors responsible for a binding with cyclodextrin, showing positive contributions toward the binding affinity values, including such features as presence of six-membered rings in the molecule, branching, electronegativity values, and polar surface area.
Keywords:
flavors
; machine learning
; β-cyclodextrin
; binding affinity
; QSAR
1. Introduction
In higher organisms, the taste buds detect flavors in the oral cavity. Herein, wheat and wheat bran products give off the sensation of bitterness.[1] The bitter flavor is a taste modality, and the perception of bitterness in wheat products could suggest the presence of phenolic compounds.[2] Experimental studies conclude that phenolic compounds can bind to the oral cavity's taste genes (i.e., TAS2R16). The consequence of this phenomenon is the resultant bitter sensation that individuals perceive.[1] The phenolic acids are produce plants in response to oxidative stress and they are comprise of phenyl, hydroxyl, and carboxylic acid moieties.[3] They are classified into two subgroups: Hydroxybenzoic and hydroxycinnamic acids[4] which give them a variety of characteristics mainly attributed to their chemical activity and applications. [5], [6] Besides their bio-based origin, most phenolic acids are safe for consumption in various food products. Most phenolic acids display hydrophobic characteristics, but some exhibit a spectrum of solubilities depending on their chemical structure [7]. For example, Gallic and P-coumaric acids are phenolic acids soluble in water. [8], [9] They are also notorious for their capacity to form hydrogen bonds with other molecules that can influence their overall activity. However, this interaction can also affect the subsequent molecule's characteristics. They can undergo reactions like oxidation, esterification, and conjugation, modifying their structure, chemical activity, biological properties, and stability. [10] The phenyl ring in the chemical composition of phenolic compounds plays a fundamental role in their antioxidative, anticancer, and antibacterial properties. They are considered very beneficial to human health. [11] The presence of phenolic acids in food could influence the perception of food; hence they are often extracted or broken down before consumptive processes. [12] In the case of the fermentation process it involves the breakdown of molecules like phenols, polyphenols, and alkaloids, while solvent extraction involves using solvents like methanol, ethanol, and acetone to extract phenols from a sample of interest. Both methods mentioned are demanding and could raise health concerns due to the availability of solvent residuals or remnants. However, an alternative like cyclodextrin entrapment of phenolic compounds could reduce these drawbacks. [13] In previous studies, this process has shown several benefits, including improved kinetics, bioavailability, stability, and solubility of biomolecules. In summary, these oligosaccharides can form inclusion complexes with several compounds with distinctive hydrophobic characteristics. [14]
Cyclodextrins are cyclic oligosaccharides that are non-toxic derivatives of starch compounds. Complexing molecules with cyclodextrin is a process that has proven effective in applications like propagating polymers, improving solubility, and providing bioavailable environments for insoluble compounds. Additionally, entrapping ligands within cyclodextrins is a common practice for influencing the perception of flavonoids. [15]- [17] The three most commonly used forms of cyclodextrin compounds are: α-, β-, and γ- cyclodextrins. They are differentiated based on their different sizes; the oligosaccharide with the smaller ring is α-, the β- is mid-sized, and the larger is γ-cyclodextrin. They are derived from bio-based enzymatic activity and are safe for consumption, as FDA-funded studies declared. Their sizes are relative to their application and ability to encapsulate the molecules within their cavities. [18] It is important to remark that among all three cyclodextrins the β-cyclodextrin variant have showed promising characteristics related to cavity dimensions, non-toxicity and other aspects like the ability to manage specific reactions, forming complexes, solubilizing and stabilizing molecules. [19] Besides β-cyclodextrin is statistically the most occurring cyclodextrin, and it's very affordable and easy to use. [20]- [26]
In other hand, computational approaches have gained wide application in pharmaceutical industry, food industry and materials science. Thus, quantum-chemical and machine learning(ML)-based Quantitative Structure-Activity Relationship (ML/QSAR) methods are routinely applied to investigate property of various biologically-active compound and materials. [27]- [33] Moreover, nowadays the combination of two or more methods are getting more popular and beneficial in compounds/materials design and explanation/prediction of the underlying interactions of the chemical systems. [34]- [36] For example, in regard to cyclodextrins and their interactions with other compounds, a research conducted by Mirrahimi et al. combined molecular docking and Quantitative Structure-Property Relationship (QSPR) models with the aim to predict the stability constant attained when a guest molecule is within a β-cyclodextrin, and also to understand the underlying interactions between the guest ligands and the host β-cyclodextrin molecule. [37] In other work by Antonio et al. [38], authors attempted to understand the interaction mechanisms of stabilizing OBPs in β-cyclodextrins for bitter masking sensations. This study employed molecular dynamics to observe the polar and hydrophobic interactions of different OBPs in β-cyclodextrin. Furthermore, Tuba et al. combined experimental and computational studies to unveil the main interactions related with the phenolic acids – β-CD bindings. [39] For this purpose the authors used a computational process that involved molecular docking of phenolic acids into the molecular cavity of β-cyclodextrin, followed by the development of a QSPR model. Additionally, their study was on the notion that complexing phenolic acids with β-cyclodextrin would enable the masking of bitterness by the taste receptors. Although this study the combination of experimental and computational methodologies in β-cyclodextrin the process in only done for the encapsulation of three phenolic acid compounds. [39] Therefore, and following this idea we carried out a more broad study using 20 phenolic acid compounds of interest that were docked to β-cyclodextrin to generate the more reliable ligan-host complexes and obtain different binding constant values, to assess the stability of complexes. Afterward, these response values were connected to ML/QSPR models using topological, topographical, semi-empirical and quantum-chemical descriptors from the ligands to identify the factors that are related the most with the activities under consideration.
2. Materials and Methods
Dataset Preparation
The chemical structure of the 20 known single phenolic acids were drawn using the ChemSketch [23] and Marvin Sketch version 22.13.0. [40] In the first step the ligands β-cyclodextrin complexes were built by using Avogadro [41] and HyperChem software. [42] After that the structures were optimized using the Bio-CHARMM force field which is equivalent to CHARMM27 for the case of the biomolecules. [43] The final structures were put into Auto Dock Vina [44] to perform the molecular docking calculations.
The optimized ligand binding conformations were obtained, as well as all the necessary data for correlating phenolic acids' activity (binding affinities) to their physiochemical properties (descriptors). Additionally, AutoDock Vina served as the source of the Binding Score values, and related scholarly articles served as sources for activity data (binding affinities). [39], [45]- [51] In Table 1 are depicted the name of the ligands used in this study together with some properties.
After importing upright and inverted structures into the software Avogadro, [41] the necessary corrections and file conversion were done. Explicit hydrogens were added to investigated structures deducted during the docking process on AutoDock Vina. Afterward, the structures were imported directly into PyMol to visualize the interactions between the ligand and β-cyclodextrin and ensure a net charge of zero on the complex. Each neutralized complex is then imported to HyperChem for preliminary optimization using the semi-empirical PM3 forcefield.
Binding Constant Calculations
The binding affinity was calculated by employing the PM6 semi-empirical QM energy model in MOPAC software. [52] MOPAC is a computational chemistry-based software used to calculate quantum and semi-empirical related properties of molecules. It contains a set of semi-empirical methods, like AM1, PM3, PM6, and MNDO. The listed methods can be used to calculate electronic properties, spectral properties, reaction mechanisms, transition states, reactivity, and solubility of molecules. As stated, the PM6 model was selected as the best functional for its reputation in terms of accuracy in calculating molecular and quantum-chemical properties. Additionally, the computational accuracy of PM6 is comparable to experimental conditions and findings, with some exceptions. [53] A log units of binding affinity were used as the chemical activity or response variable in the second model and log binding energy in the case of the third model. JMOL software was used to visualize the output files from MOPAC calculations. [54] JMOL is a chemistry-based visualization software that can perform computational chemistry analysis. It pictures molecular orbitals and electron densities, visualization of crystal structures, and the 3D rendering of molecular structures.
Docking Procedures
The Auto Dock Vina software was selected for the molecular docking and scoring calculations. The software is a user-friendly chemistry-based program for protein-ligand docking studies. It performs a grid-based docking, flexible ligand-based docking, and multiple docking-ligands screening procedures. [44] The output analysis of the docking study was carried out with PyMol software [55] to visualize and investigate the intermolecular interactions of docked molecules. PyMol is a chemistry-based software used for molecular visualization and analysis. It provides 3D rendering of structures, the capacity to manipulate molecular structures, measure distances, angles, and torsional angles, and the ability to visualize molecular surfaces, interactions, electrostatic potentials, and molecular orbitals. As discussed earlier, the most convenient docking method was employed: the rigid receptor and flexible ligand approach. Afterward, Auto Dock Vina was used to create optimized versions of the ligand conformation within the cavity of β-cyclodextrin and outputs different binding affinities of each conformation. From this point, the two best ligand conformations were selected with the highest binding affinity scores. Each conformation chosen has either an upright or inverted conformation to the β-cyclodextrin. To be considered upright, most of the body of the ligand must be facing the position of the β-cyclodextrin, which has two hydroxyl groups extending from the ends of each glucose subunit. To be considered inverted, most of the body of the ligand must face the b-cyclodextrin position with one hydroxyl extending from the ends of each glucose subunit. The upright and inverted complexes were collected for each ligand to see which has the better binding affinity and more substantial molecular interactions.
Molecular Descriptors Generation
In the next step the Dragon 6 software was used to generate geometrical, topological, and constitutional descriptors. [56] Dragon 6 is a software used for calculating different types of molecular descriptors, varying from zero to 3-dimensionality (0D to 3D). The Dragon 6 algorithms are used to encode the chemical structures and capture essential aspects of the topological and structural information of the molecules. However, due to limitations in the software, the inclusion complexes were excluded from the descriptors generation and only ligands were considered in the generation of the molecular descriptors. In a posterior filtering out, all the invariant and close-to-constant descriptors were discarded keeping only 1200 descriptors, which were either 2D or 3D descriptors of the investigated phenolic acids.
Herein, we collected HyperChem descriptors to combine them with other descriptors obtained from the abovementioned Dragon software for further GA-MLR analysis. After optimization in HyperChem, the molecular structure of the complex was saved in the appropriate file format (.zmt), then used for semi-empirical calculations in the software MOPAC after editing. In MOPAC, the semi-empirical PM6 forcefield was used for the analyses. The PM6 method provides a desired level of accuracy in calculating the selected quantum-chemical descriptors for further use in developing QSAR models. MOPAC calculations mainly provided such properties as heat of formation (HF) and molecular orbital energies, like HOMO/LUMO energies. The orbitals (HOMO and LUMO) were evaluated in JMOL and further documented.
Variable Selection and QSAR Modeling
After generating the descriptors, the set of 20 phenolic acid compounds was split in a ratio of 4:1 (80%-20%), as training and test sets, respectively. This was done by sorting the binding constants for each study in ascending order by binding activity, and hence assigning the fifth compound to the test set. In result, 16 compounds were included in the training set and 4 compounds in test set.
The QSARINS software was applied to perform the feature selection and model selection by MLR and GA procedures with the aim to optimize the selection of descriptors for developing predictive models. Additionally, predictive models contain the combination of descriptors that best predict the property studied and calculate the applicability domain.
The feature selection in QSARINS aims to find the best combination of descriptors which leads to highly performing and accurate ML models. In this study implement the preliminary feature selection based on GA-MLR with the QSARINS 2.2.4 package. [57]- [61] In this scenario, the selection process is started with 500 random models and executed 3000 iterations of evolution. Additionally, a mutation rate of 0.35 was assigned for the GA feature selection process.
A genetic algorithm (GA) is a class of algorithms acquired from evolutionary algorithms and employed to solve the Knapsack problem of sorting through a series of weighted variables. In simpler terms, it uses natural selection to generate approximate solutions for complex problems. This process starts with a combination of possible solutions, for instance, various descriptors provided. QSAR assigns each descriptor a genome translated or encoded to the solutions generated. It creates a generation that includes all sets of possible combinations. Beginning from generation 0, contingent on randomness to develop a viable solution. A fitness function is applied, which evaluates how good each solution is. Solutions with higher fitness scores are then used as parents to generate new solutions based on the mutation rate parameter. After applying a crossover function on these parent solutions, GA creates solutions for the next generation, and this cycle is repeated depending on the number of generations requested. A considerable degree of randomness drives the selection and crossover processes. Top solutions with top scores per generation are chosen and moved to the next combinatorial generation. Then, a mutation is applied to change the randomness of a combination based on a probability function. The process explained above loops until the number of generations provided has been satisfied or until it attains a solution to the sorting problem.
As discussed above, the ML/QSAR approach aims to generate a mathematical relationship between an activity (chemical, biological, or physical) and a set of descriptors (molecular properties) that are best associated with this activity. Once a model is obtained and validated, this relationship/model is used to predict the activity of other molecules within the applicability domain, which is based on the set of molecules used to generate model. The quality of models should be assessed to get robust models and hence reliable predictions gathered from them, where the different external and internal validation procedures play a fundamental role at time to check for the robustness and stability of the QSAR models. The cross-validation technique “leave-one-out” was used in the internal validation process of the QSAR models obtained from the GA-MLR feature selection. This procedure consists in removing one molecule at each time from the training set and re-running the selected model against the individual molecules LOO). Later, the observed response values, and the predicted response values calculated by the models are used to obtain the correlation coefficients, R [2] (Eq. 1) and the root mean square errors RMSE (Eq. 2), as statistical parameters to prove the performance of each model. This process is done in the training, cross-validation data, and the external set (R [2]training, QLOO, R [2]test,).
where: yjobs – the experimental (observed) value of the property for the ith/ jth compound; yjpred – predicted value for ith/ jth compound; y, ŷ – the mean experimental value of the property in the training and validation set, respectively; n, k – the number of compounds in the training and validation set, respectively. These and other statistical parameters were calculated for the training, test and cross-validation series. Besides, all descriptors were normalized by using MATLAB software [62] implementations and the Python matplotlib library was used to generate detailed plots of our models for analysis. [63]
Applicability Domain
Additionally, to ensure the reliability of predictions our predictions, we employed a leverage approach in calculating the chemical applicability domain (AD). [64], [65] The Williams plot is the most common statistical tool in visual applicability domains. In this study, the Williams plot encompasses standard cross-validated residuals (RES) of the data set, plotted against the leverage values (Hat Diagonal, h) of the data set. The Residuals vs. leverage plot efficiently captures response outliers (Std. residuals) and structurally influential compounds (Leverage) within the data set.
Three sigma plots are graphical interpretations employed to ascertain outliers within the data set studied. We have used three sigma plots to detect molecules with response variables that are significantly different from the rest of the data set. The Y-axis (Std. residuals) shows the difference between the predicted and actual values of the chemical activities. At the same time, Std residuals axis shows the standard deviation of the residuals multiplied by three (3σ). Data points that fall outside the 3-sigma range are considered as outliers. In fact, these outliers could result from data entry errors, experimental errors, or other factors which concurrently impact the accuracy and reliability of QSAR models. The h* index and the 3-sigma plot identify data points that carry much weight to themselves and could skew the data distribution. Hence, the h* index aims to identify the data points that influence the QSAR model the most.
Descriptor Significance Plot
The significance plot is drawn directly from the QSAR model. As observed, the coefficient of each descriptor in the QSAR model represents the magnitude or influence of that descriptor on the model. Since these models are linear by the MLR methodology, it is much easier to represent their magnitudes as bar plots. The coefficient sign could be positive or negative, and the magnitude determines the influence of a descriptor on a QSAR model. The significance plot makes it easier to differentiate the impact of each descriptor in the model.
3. Results and Discussion
In this work, we have investigated interaction of a set of 20 phenolic compounds with β-CD by estimation of three different binding constants of β-CD–ligand complexes. A molecular protein-ligand docking approach was used, as well as ML/QSAR approach. As result, three ML/QSAR models were developed to describe and predict binding score affinity (Model 1), binding affinity (Model 2) and binding energy (Model 3), Table 2.
Binding Score/Affinity (Model 1)
The best model for the prediction of the binding affinity was obtained with three variables and is shown below in Eq. (3):
Log BSA= 0.078·nR06 + 0.353·ATS4m - 0.294·BELe3 +0.494
The statistical parameters of the model are depicted in Table 2 and the descriptors related to each model are shown in Table 3.
Figure 1A represents the influence of each descriptor towards the log of binding affinity. From this graph is observed that nR06 and ATS4m descriptors have a positive contribution to the activity and the other remaining descriptor, BELe3, have a negative impact to the activity, with ATS4m showing the highest contribution among all the variables and nR06 the lowest one. It is important to highlight that in the Model 1 (Eq.3) the correlation coefficient between the observed and predicted values - R [2] is 0.969 for the training set and R [2] = 0.984 for the test set, as can be seen from Figure 1B.
The Williams plot in Figure 1C addresses the applicability domain regarding the three-sigma deviation and the leverage threshold h*. As it can be seen from the figure the majority of the molecules fall within the (-3σ; +3σ) domain and inside the h critical value, with only one compound that falls outside the h*. It implies that a leverage value exceeds the threshold value (hi>h*). This result is mainly due to structural differences of this single molecule in our test set, which draws weight to itself, and at the same time balances the model. It reflects the high leverage of this molecule but doesn't diminish the validity of this model.
After confirming the validity and reliability of the Model 1, in the next step a mechanistic interpretation of the model was also assessed by doing a detailed analysis of the three descriptors involved in the model (see Figure 2A-C). For example, the nR06 descriptor has a small positive influence on the binding affinity in the model, the descriptor quantifies the number of 6-membered rings in a molecule and belonging to the class of fragment descriptors. In general, this molecular descriptor has a positive influence because of the π interactions that could stabilize the ligand within the β-CD pocket. [66] Besides, as it can be observed from Figure 2A, all the structures with zero value for this descriptor, no-rings structures have most of the lower values for the binding score affinity, below 0.5 approximately, and in the opposite way for the ligands with one ring that have a log binding score affinity above 0.5. Also, it should be noticed the case of eucalyptol (ligand 4) with three rings has the highest value in all the data but with a value around 0.6, and same for the case of ligands 3 and 17, with log binding score affinity not higher as it was expected. In these three cases this could happen because the model is multi-featured and the interactions with the other variables should always be considered.
The ATS4m descriptor is a 2D autocorrelation class descriptor, Broto-Moreau autocorrelation of lag 4 (log function) weighted by mass, and is calculated from molecular graph by summing the products of atom weights of the terminal atoms of all the paths of the considered path length (the lag). Therefore, there are two main aspects involved in this descriptor definition, the first is related to the frequency of the path. For the menthol and heptanol ligands there are counted 17 and 5 paths, respectively, which is connected to some extent to the branching. From this analysis it could be noticed that increase in branching positively impact the log binding score affinity as it can be seen from Figure 2B, where a linear positive correlation is observed for most of the cases. This finding supports the idea that with higher branching more hydrogen interactions can occur between the ligands and the β-CD.
The third descriptor, BELe3 is a Burden eigenvalue descriptor, and as it can be seen from Figure 2C there is no linear tendency within the values in the range 0.8 to 1.2, and only ligand 15 is staying apart with an extreme value about 1.6 and with a binding affinity in the intermediate range.
Although the functionality of BEle3 descriptor is ambiguous, there are some distinctive observations within the clusters. Molecules with a phenyl group or six-membered rings cluster together and tend to be higher on the binding affinity scale. Another observation is that hydroxyl branches off the phenyl are favored over methoxy branches. Additionally, shorter stem (Hydroxybenzoic) molecules are favored over the longer stem (Hydroxycinnamic) molecules. Gallic acid is of the hydroxybenzoic class and has three hydroxyl group branches, a short stem leading to carboxylic acid, and no methoxy groups. It has the highest binding affinity so far. In Figure S1A (Electrostatic Potential graph), Gallic acid shows more saturation of the electronegative charge than Vanillic acid (Figure S1B). This observation could be associated with the presence or the number of hydroxyl groups and related to their electronic orbitals. As seen in Figure S2A & S2B, both molecules are readily reactive in a complex with BCD and retain their HOMO-LUMO orbitals. In Figure S1C, has Maltol with a six-membered ring, and Figure S2C shows that it retains the HOMO-LUMO orbitals in a complex, which means it is readily reactive. On the contrary, Figure S1D shows that Menthol has more saturated positive and neutral charges. It has one hydroxyl group and Figure S2D shows it doesn’t retain its HOMO-LUMO orbital in a complex with BCD.
Binding Affinity (Model 2)
Second ML/QSAR model is related to the binding affinity (BA, Table 1 and Table 2), a model with 3-variables was selected as the best one with the EEig03r and H0e descriptors contributing positively to the binding constant values and the S3K with a negative impact in the activity (see Eq. 4). It is important to remark that the H0e molecular descriptor has the highest impact towards the response variable of our model as is depicted in Figure 3A. The statistical parameters of our Model 2 are shown in Table 2 that includes among others RMSE, MAE and CCC, and all of them showed very good performance values, such as R [2] = 0.859 for the training set and R [2] = 0.956 for the test set. In Figure 3B is plotted the actual vs predicted values of the log binding affinity where a good correlation is observed for both training and test sets.
Log BA= -0.116·S3K + 0.127·EEig03r + 0.145·H0e + 1.064
Also, an applicability domain analysis of the Model 2 was performed by the Williams plot, as shown in Figure 3C. As it can be observed from this plot, all the ligands for both training and test sets fall within the applicability domain range, which means inside the leverage critical threshold (h*) with values lower that h* = 0.75, and inside the (-3σ; +3σ) standard deviation.
A deeper analysis on the influence of each descriptor and mechanistic explanation was done for the three descriptors. The first descriptor S3K is the 3-path Kier alpha-modified shape index. It is a topological index that is related to the centrality of branching and encodes the number of paths with length k=3 in an H-depleted molecular graph for this descriptor. As can be seen from Figure 4A, the ligands with aromatics rings have the lower descriptor values and aliphatic chains have the higher values. This is interesting because although gallic acid (ligand 6) have the higher number of 3-length paths have the one of the lower values in the descriptor, that is because this descriptor also considers the different shape contribution of heteroatoms and hybridization states, with the last one having the lower values for the aromatic rings and the higher values for the aliphatic chains. Other factors, such as the point of origination of a branch or the groups of a branch, could also influence the binding affinity. For example, Reinskje et al. showed that the binding affinity of p-alkylbenzamidinium chloride to serin proteinase trypsin27 could decrease with an increase in branching from the C1 carbon of the phenyl ring [67].
The second descriptor in the model EEig03r quantifies the topological complexity of a molecule based on the eigenvalues of the edge adjacency matrix of the molecule. This descriptor is mainly related to molecular branching. As can be seen in Figure 4B the ligands that possess high values of EEig03r have a high degree of branching and high binding affinity values. In contrast, molecules with low values of EEig03r are more likely linear and have a less branched structure and lower binding affinity values, showing a clear positive trend related to the activity. This could be related to the concept that branching could enhance interactions with β-CD. For example, is important to remark that there are similarities in the molecular structures of syringic (ligand 18) and trans ferulic acid (ligand 19). Syringic acid has a binding affinity of -76 kJ/mol, and its structure shows several branches from the phenyl ring. In the case of trans ferulic acid the phenyl ring and three groups branching out, could be the factors that are related with a high binding affinity value and hence a strong interaction with β-CD.
The third descriptor in the model H0e is defined as an H autocorrelation of lag 0/weighted by Sanderson electronegativity and classified inside the GETAWAY descriptor class. The term H is related to the leverage matrix, since this descriptor is calculated from the leverage matrix obtained by the centered atomic coordinates, the other terms lag 0 refers to the topological distance with 0 only considering the atom itself and the last term ‘e’ is the weighting scheme, the electronegativity in the Sanderson scale for this descriptor. As can be observed from Figure 4C there is a clear and positive correlation between the molecular descriptor and the binding affinity with gallic acid (ligand 6) having the higher value of the descriptor as it has a high number of oxygens in the structure 5 in total, therefore increase the total electronegativity of the molecule and hence the binding affinity value. The opposite case is shown for 1-heptanol (ligand 8) with only one oxygen in the structure and hence a lower value in the descriptor and in the binding affinity. The increase in the total electronegativity in the molecules is also related somehow with the polarity and then that molecule will be more likely to participate in hydrogen bonding and other intermolecular interactions. Therefore, the H0e descriptor could be related to the influence of the distribution of electronegative groups across the molecular topology and consequently, those ligands with phenolic, carbonyl, hydroxyl, and carboxylic group in the molecular structure, should display higher capacity to form hydrogen bonds and stronger interactions within the β-CD cavity.
Binding Energy
The third model (Model 3) is developed for binding energy (BE) as a response variable. The model has 3 molecular descriptors that include PSA with a positive impact in the activity, GATS8e and Mor10u with a negative contribution to the binding energy, as can be observed from Eq. 5 and Figure 5A. The quality of the model was validated with the test set and using the statistical parameters RMSE, MAE, R [2] and others commonly used to prove the performance of the models. The R [2] values for training and test were 0.779 and 0.663, respectively with adequate values for the other parameters for both training and test sets, Table 3. Besides, the actual vs predicted values of the log binding energy are shown in Figure 5B for training and test sets.
Log BE= -0.214·GATS8e - 0.351·Mor10u + 0.248·PSA + 1.798
The Williams plot shown in Figure 5C validates the model with the implementation of three-sigma residuals and the leverage threshold h*. This figure shows that both training and test sets fall within the three-sigma standard deviations and all the ligands falls inside the h*, which implies that no leverage value is more significant than the leverage threshold value (hi<h*).
A set of important descriptors are included in the Model 3. Thus, the PSA or TPSA descriptor is related to the Topological Polar Surface Area. This molecular descriptor is calculated based on the summation of tabulated surface contributions of polar fragments that includes the atoms involved and the bonding pattern (single, double, triple bonds). These polar fragments are those containing oxygen and nitrogen, and some ‘less polar’ with phosphorus and sulfur. [68] In this way it explains the surface area of a molecule that is accessible to polar solvents. As can be seen from Figure 6A there is a positive trend between the binding affinity and the molecular descriptor, with ligand 18 (syringic acid) having the highest molecular descriptor value in correspondence with the high number of oxygens in the structure, five in total for this compound summing up a high contribution to the total polar surface area calculations as described above and a high binding energy value. In the other case we have ligand 17 (styrene) with no oxygen atoms in the structure and hence a low TPSA value. This tendency towards the binding energy could be to the effect that an increase in the number of oxygens in functional groups could enhance the occurrence of hydrogen bounds and hence stabilize the ligand- β-CD interactions. In Figure 6B,C, could be observed the density plot for the remaining two descriptors in the model and as can be seen there is not a linear observable tendency with all the values in the range and only ligand 10 for both descriptors and ligand 3 for Mor10u descriptor show extreme values.
Although the functionality of the GATS8e descriptor is uncertain in Figure 6B, there are some observations in the clustering pattern. Cluster one molecules were mostly Hydroxycinnamic acids like Sinapic, P-coumaric, and Trans ferulic acid. The common structural feature is that they possess three carbon stems, the C6-C3 side chain. The second cluster in the density graph contains Maltol, Gallic acid, menthol, and an outlier, Neral. The most common structural feature in the second cluster is the absence of a long side chain or stem. The third cluster contains Vanillic and Syringic acid, which possess similar structural properties to the previous cluster. They are mostly Hydroxybenzoic acids. It seems GATS8e segregated the structural difference between hydroxybenzoic acids and hydroxycinnamic acids. It scored the cluster containing mostly hydroxycinnamic acids higher than clusters containing hydroxybenzoic acids. From the significance graph, it is evident that the GATS8e has an inverse relationship with binding affinity. The presence of the C6-C3 side group could impact binding affinity adversely. It could affect the charge density, reducing hydroxycinnamic acids' interactive strength during BCD entrapment.
In Figure 6C, the Mor10u descriptor has a distinctive clustering pattern. Molecules with the phenyl group are distributed away to zero. In other cases, molecules with non-aromatic cyclic groups score higher and are the furthest away from zero. Conversely, linear molecules have scores closer to zero. It seems the Mor10u descriptor separates the molecules based on intermolecular distances and 3D conformational analysis in scoring molecules. Molecules like P-coumaric acid (14) and eucalyptol (4) have aromatic groups. Molecules like D-Limonene (3), Menthol (12), and Hydroxymethylfurfural (9) have cyclic groups. Molecules like Pinellic acid, Isoamyl-acetate (10), and Neral (13) are relatively linear. Additionally, with Mor10u, the molecules closer to zero have lower binding affinity values. The higher affinity molecules have phenyl groups or aromatic rings as a common attribute. They are also clustered toward the upper half of the density graph. Most linear structured molecules have lower binding affinity and have descriptor scores close to zero. However, Pinellic acid is linear but a member of the higher binding affinity cluster. This observation argues that the size/molecular volume could also be a determining factor.
In this study, the predictive R [2]train values for each ML/QSAR model (see Table) have the following order - Model 1 > Model 2 > Model 3, and show the proportion of variance within the binding affinity that is captured by the models. In this sense, the Model 1 suggests that it explains a larger portion of the variance within the response variable compared to Model 2 and Model 3. For test set the same tendency is observed for the R [2] and the other statistical parameters (RMSE, MAE, CCC) that were used to prove the performance of our models.
4. Conclusions
In this study we explored possible molecular mechanisms involved in masking bitterness in wheat bran-associated ligands with β-cyclodextrin. The applied computational approaches involve the combination of molecular docking and machine learning to unveil the mechanisms. Three ML/QSAR models were obtained with very good performances in both training and test sets, where model with the binding score affinity (BSA) having the highest values of the correlation coefficient R [2]train = 0.969 and R [2]test = 0.984. The model has as a main descriptor the number of 6-membered rings (nR06), which shows positive influence towards the binding constant, and promotes the stabilization through polar-π interactions. Another important descriptor, the 2D descriptor ATS4m is related mainly to path frequency shows a positive tendency towards the binding constant, and this descriptor is connected to the molecule branching and hence increase the interactions between the ligand and the β-CD in the complex by different modes. Besides, other descriptors weighted by electronegativity values and polar surface area (PSA) were able to show positive influence for the binding constants in Models 2 and 3. This aligns with the outcomes that hydrophobic and/or lipophilic interactions drive most β-CD−Ligand interactions, highlighting the importance of solvation in a polar environment like water and also the relation with hydrogen bonding interactions, and the instantaneous occurrences of dipole–dipoles. Other features that promote or hinder binding are the atomic mass of ligands and, understandably, the size of the ligand. Most of the ligands in this study fit within the size range and get entrapped in the β-CD molecular pocket. These findings could be very helpful to design new complex systems with better binding constants values and hence with improved bitterness masking properties.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, B.R. and S.S.; methodology, B.R.; software, K.I., M.N., A.D., G.C-M.; validation, K.I., M.N., A.D., G.C-M and B.R.; formal analysis, K.I., M.N., J.K., A.D., G.C-M.; investigation, K.I., M.N., J.K., A.D., G.C-M.; resources, B.R. and S.S.; data curation, K.I., M.N., A.D. and G.C-M; writing—original draft preparation, K.I., M.N., J.K., G.C-M; writing—review and editing, K.I., A.D., G.C-M., S.S. and B.R.; visualization, K.I., A.D. and G.C-M.; supervision, S.S. and B.R.; project administration, B.R.; funding acquisition, B.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Foundation under grant number NSF CHE-1800476, as well as by NSF MRI award OAC-2019077 and NSF ND EPSCoR award #IIA-1355466 and by the State of North Dakota.
Data Availability Statement
The main data are available in Supplementary Information file and the detailed information is available upon the request from authors.
Acknowledgments
Authors acknowledge the support from the National Science Foundation under grant number NSF CHE-1800476. This work is also supported in part by the NSF MRI award OAC-2019077, as well as ND EPSCoR award #IIA-1355466 and by the State of North Dakota. The authors thank Prof. Paola Gramatica for generously providing a free license for the QSARINS software. Supercomputing support from CCAST HPC System at NDSU is acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Khan, A. S.; Murtaza, B.; Hichami, A.; Khan, N. A. A cross-talk between fat and bitter taste modalities. Biochimie 2019, 159, 3-8. [CrossRef]
- Ardoin, R.; Smith, B.; Lea, J.; Boue, S.; Smolensky, D.; Santana, A. L.; Peterson, J. Consumer perceptions and antioxidant profiling of acidified cold-brewed sorghum bran beverages. Journal of Food Science 2023, 88 (6), 2301-2312, Article. Scopus. [CrossRef]
- Vuolo, M. M.; Lima, V. S.; Maróstica Junior, M. R. Chapter 2 - Phenolic Compounds: Structure, Classification, and Antioxidant Power. In Bioactive Compounds, Campos, M. R. S. Ed.; Woodhead Publishing, 2019; pp 33-50.
- Taofiq, O.; González-Paramás, A. M.; Barreiro, M. F.; Ferreira, I. C. F. R. Hydroxycinnamic Acids and Their Derivatives: Cosmeceutical Significance, Challenges and Future Perspectives, a Review. Molecules (Basel, Switzerland) 2017, 22 (2), 281. PubMed. [CrossRef]
- Heleno, S. A.; Martins, A.; Queiroz, M. J. R. P.; Ferreira, I. C. F. R. Bioactivity of phenolic acids: Metabolites versus parent compounds: A review. Food Chemistry 2015, 173, 501-513. [CrossRef]
- Luna-Guevara, M. L.; Luna-Guevara, J. J.; Hernández-Carranza, P.; Ruíz-Espinosa, H.; Ochoa-Velasco, C. E. Chapter 3 - Phenolic Compounds: A Good Choice Against Chronic Degenerative Diseases. In Studies in Natural Products Chemistry, Atta ur, R. Ed.; Vol. 59; Elsevier, 2018; pp 79-108.
- Rosa, L. A.; Moreno-Escamilla, J. O.; Rodrigo-Gracia, J.; Haard, N. F. Postharvest Physiology and Biochemistry of Fruits and Vegetables, Phenolic Compounds, Chapter 12. UK: Woodhead Publishing 2019, 253-271.
- Combes, J.; Clavijo Rivera, E.; Clément, T.; Fojcik, C.; Athès, V.; Moussa, M.; Allais, F. Solvent selection strategy for an ISPR (In Situ/In stream product recovery) process: The case of microbial production of p-coumaric acid coupled with a liquid-liquid extraction. Separation and Purification Technology 2021, 259, 118170. [CrossRef]
- Furia, E.; Beneduci, A.; Malacaria, L.; Fazio, A.; La Torre, C.; Plastina, P. Modeling the Solubility of Phenolic Acids in Aqueous Media at 37 °C. In Molecules, 2021; Vol. 26.
- Rodrigues, J. F.; Soares, C.; Moreira, M. M.; Ramalhosa, M. J.; Duarte, N. F.; Delerue-Matos, C.; Grosso, C. Moringa oleifera Lam. Commercial Beverages: A Multifaceted Investigation of Consumer Perceptions, Sensory Analysis, and Bioactive Properties. Foods 2023, 12 (11), Article. Scopus. [CrossRef]
- Zhang, S.; Shan, X.; Niu, L.; Chen, L.; Wang, J.; Zhou, Q.; Yuan, H.; Li, J.; Wu, T. The Integration of Metabolomics, Electronic Tongue, and Chromatic Difference Reveals the Correlations between the Critical Compounds and Flavor Characteristics of Two Grades of High-Quality Dianhong Congou Black Tea. Metabolites 2023, 13 (7), Article. Scopus. [CrossRef]
- Issaoui, M.; Delgado, A. M.; Caruso, G.; Micali, M.; Barbera, M.; Atrous, H.; Ouslati, A.; Chammem, N. Phenols, Flavors, and the Mediterranean Diet. J AOAC Int 2020, 103 (4), 915-924. [CrossRef]
- Kim, J. S. Synthesis and Characterization of Phenolic Acid/Hydroxypropyl-β-Cyclodextrin Inclusion Complexes. Prev Nutr Food Sci 2020, 25 (4), 440-448. [CrossRef]
- Cid-Samamed, A.; Rakmai, J.; Mejuto, J. C.; Simal-Gandara, J.; Astray, G. Cyclodextrins inclusion complex: Preparation methods, analytical techniques and food industry applications. Food Chemistry 2022, 384, 132467. [CrossRef]
- Gramage-Doria, R.; Armspach, D.; Matt, D. Metallated cavitands (calixarenes, resorcinarenes, cyclodextrins) with internal coordination sites. Coordination Chemistry Reviews 2013, 257 (3), 776-816. [CrossRef]
- Faisal, Z.; Fliszár-Nyúl, E.; Dellafiora, L.; Galaverna, G.; Dall’Asta, C.; Lemli, B.; Kunsági-Máté, S.; Szente, L.; Poór, M. Cyclodextrins Can Entrap Zearalenone-14-Glucoside: Interaction of the Masked Mycotoxin with Cyclodextrins and Cyclodextrin Bead Polymer. In Biomolecules, 2019; Vol. 9.
- Mathivet, T.; Méliet, C.; Castanet, Y.; Mortreux, A.; Caron, L.; Tilloy, S.; Monflier, E. Rhodium catalyzed hydroformylation of water insoluble olefins in the presence of chemically modified β-cyclodextrins: evidence for ligand-cyclodextrin interactions and effect of various parameters on the activity and the aldehydes selectivity. Journal of Molecular Catalysis A: Chemical 2001, 176 (1), 105-116. [CrossRef]
- Sandilya, A. A.; Natarajan, U.; Priya, M. H. Molecular View into the Cyclodextrin Cavity: Structure and Hydration. ACS Omega 2020, 5 (40), 25655-25667. [CrossRef]
- da Rocha Neto, A. C.; de Oliveira da Rocha, A. B.; Maraschin, M.; Di Piero, R. M.; Almenar, E. Factors affecting the entrapment efficiency of β-cyclodextrins and their effects on the formation of inclusion complexes containing essential oils. Food Hydrocolloids 2018, 77, 509-523. [CrossRef]
- Chodankar, D.; Vora, A.; Kanhed, A. β-cyclodextrin and its derivatives: application in wastewater treatment. Environmental Science and Pollution Research 2022, 29 (2), 1585-1604. [CrossRef]
- Tajbakhsh, M.; Naimi-Jamal, M. R. Copper-doped functionalized β-cyclodextrin as an efficient green nanocatalyst for synthesis of 1,2,3-triazoles in water. Scientific Reports 2022, 12 (1), 4948. [CrossRef]
- Hedges, A. Chapter 22 - Cyclodextrins: Properties and Applications. In Starch (Third Edition), BeMiller, J., Whistler, R. Eds.; Academic Press, 2009; pp 833-851.
- Crini, G.; Fourmentin, S.; Fenyvesi, É.; Torri, G.; Fourmentin, M.; Morin-Crini, N. Cyclodextrins, from molecules to applications. Environmental Chemistry Letters 2018, 16 (4), 1361-1375. [CrossRef]
- Braga, S. S.; Barbosa, J. S.; Santos, N. E.; El-Saleh, F.; Paz, F. A. A. Cyclodextrins in Antiviral Therapeutics and Vaccines. In Pharmaceutics, 2021; Vol. 13.
- Jiayue, L.; Tian, B. Selective modifications at the different positions of cyclodextrins: a review of strategies. Turkish Journal of Chemistry 2020, 44, 261-278. [CrossRef]
- Stella, V. J.; He, Q. Cyclodextrins. Toxicologic Pathology 2008, 36 (1), 30-42. (acccessed 2023/05/20). [CrossRef]
- Toropov, A.; Toropova, A.; Rasulev, B.; Benfenati, E.; Gini, G.; Leszczynska, D.; Leszczynski, J. CORAL: Binary Classifications (Active/Inactive) for Liver-Related Adverse Effects of Drugs. Current Drug Safety 2012, 7, 257-261. [CrossRef]
- Karuth, A.; Alesadi, A.; Xia, W.; Rasulev, B. Predicting Glass Transition of Amorphous Polymers by Application of Cheminformatics and Molecular Dynamics Simulations. Polymer 2021, 218, 123495. [CrossRef]
- Chen, M.; Jabeen, F.; Rasulev, B.; Ossowski, M.; Boudjouk, P. A computational structure-property relationship study of glass transition temperatures for a diverse set of polymers. Journal of Polymer Science Part B: Polymer Physics 2018, 56. [CrossRef]
- Rasulev, B.; Jabeen, F.; Stafslien, S.; Chisholm, B.; Bahr, J.; Ossowski, M.; Boudjouk, P. Polymer Coating Materials and Their Fouling Release Activity: A Cheminformatics Approach to Predict Properties. ACS applied materials & interfaces 2016, 9. [CrossRef]
- Toropova, A.; Toropov, A.; Rasulev, B.; Benfenati, E.; Gini, G.; Leszczynska, D.; J, L. QSAR models for ACE-inhibitor activity of tri-peptides based on representation of the molecular structure by graph of atomic orbitals and SMILES. Structural Chemistry 2012, 23, 1873-1878. [CrossRef]
- Rasulev, B.; Kusic, H.; Leszczynska, D.; Leszczynski, J.; Koprivanac, N. QSAR modeling of acute toxicity on mammals caused by aromatic compounds: The case study using oral LD50 for rats. Journal of Environmental Monitoring 2010, 12, 1037-1044. [CrossRef]
- Gooch, A.; Sizochenko, N.; Rasulev, B.; Gorb, L.; Leszczynski, J. In vivo toxicity of nitroaromatics: A comprehensive quantitative structure–activity relationship study. Environmental Toxicology and Chemistry 2017, 36 (8), 2227-2233. https://doi.org/10.1002/etc.3761 (acccessed 2022/06/06). [CrossRef]
- Golmohammadi, M.; Aryanpour, M. Analysis and evaluation of machine learning applications in materials design and discovery. Materials Today Communications 2023, 35, Review. Scopus. [CrossRef]
- Ji, H.; Pu, D.; Yan, W.; Zhang, Q.; Zuo, M.; Zhang, Y. Recent advances and application of machine learning in food flavor prediction and regulation. Trends in Food Science and Technology 2023, 138, 738-751, Review. Scopus. [CrossRef]
- Kou, X.; Shi, P.; Gao, C.; Ma, P.; Xing, H.; Ke, Q.; Zhang, D. Data-Driven Elucidation of Flavor Chemistry. Journal of Agricultural and Food Chemistry 2023, 71 (18), 6789-6802, Review. Scopus. [CrossRef]
- Mirrahimi, F.; Salahinejad, M.; Ghasemi, J. B. QSPR approaches to elucidate the stability constants between β-cyclodextrin and some organic compounds: Docking based 3D conformer. Journal of Molecular Liquids 2016, 219, 1036-1043. [CrossRef]
- Rescifina, A.; Chiacchio, U.; Iannazzo, D.; Piperno, A.; Romeo, G. β-Cyclodextrin and Caffeine Complexes with Natural Polyphenols from Olive and Olive Oils: NMR, Thermodynamic, and Molecular Modeling Studies. Journal of Agricultural and Food Chemistry 2010, 58 (22), 11876-11882. [CrossRef]
- Simsek, T.; Rasulev, B.; Mayer, C.; Simsek, S. Preparation and Characterization of Inclusion Complexes of β-Cyclodextrin and Phenolics from Wheat Bran by Combination of Experimental and Computational Techniques. In Molecules, 2020; Vol. 25.
- MarvinView; www.chemaxon.com: 2016. (accessed.
- Hanwell, M. D.; Curtis, D. E.; Lonie, D. C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics 2012, 4 (1), 17. [CrossRef]
- HyperChem(TM) Professional 8.0; 2019. (accessed.
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010, 31 (4), 671-690. [doi]. [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling 2021, 61 (8), 3891-3898. [CrossRef]
- Górnas, P.; Neunert, G.; Baczyński, K.; Polewski, K. Beta-cyclodextrin complexes with chlorogenic and caffeic acids from coffee brew: Spectroscopic, thermodynamic and molecular modelling study. Food Chemistry 2009, 114 (1), 190-196. [CrossRef]
- Santos, C.; Buera, P.; Mazzobre, M. Phase solubility studies and stability of cholesterol/β-cyclodextrin inclusion complexes. Journal of the science of food and agriculture 2011, 91, 2551-2557. [CrossRef]
- Pinho, E.; Soares, G.; Henriques, M. Cyclodextrin modulation of gallic acid in vitro antibacterial activity. Journal of Inclusion Phenomena and Macrocyclic Chemistry 2015, 81 (1), 205-214. [CrossRef]
- Karathanos, V.; Mourtzinos, I.; Yannakopoulou, K.; Andrikopoulos, N. Study of the solubility, antioxidant activity and structure of inclusion complex of vanillin with β-cyclodextrin. Food Chemistry 2007, 101, 652-658. [CrossRef]
- Narayanasamy, R.; Thammanna, M.; J. Photophysics of Caffeic, Ferulic and Sinapic Acids with α- and β-Cyclodextrins: Spectral and Molecular Modeling Studies. International Letters of Chemistry, Physics and Astronomy 2017, 72, 37-51. [CrossRef]
- Liu, B.; Zeng, J.; Chen, C.; Liu, Y.; Ma, H.; Mo, H.; Liang, G. Interaction of cinnamic acid derivatives with β-cyclodextrin in water: Experimental and molecular modeling studies. Food Chemistry 2016, 194, 1156-1163. [CrossRef]
- Lukasiewicz, M.; Jakubowski, P. Determination of Complexation Parameters for β-Cyclodextrin and Randomly Methylated β-Cyclodextrin Inclusion Complexes of p-Cumaric Acid Using Reversed-Phase Liquid Chromatography; 2014. [CrossRef]
- Stewart, J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. Journal of molecular modeling 2012, 19. [CrossRef]
- Stewart, J. J. P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. Journal of Molecular Modeling 2007, 13 (12), 1173-1213. [CrossRef]
- Hanson, R. M. Jmol SMILES and Jmol SMARTS: specifications and applications. Journal of Cheminformatics 2016, 8 (1), 50. [CrossRef]
- The PyMOL Molecular Graphics System; 2010. (accessed.
- Toddeschini; R.; Consonni, V.; Mauri, A.; Pavan, M. Dragon Software for the Calculation of Molecular Descriptors, Version 6 for Windows; Talete SRL: Milan, Italy. 2014.
- Cassani, S.; Kovarich, S.; Papa, E.; Roy, P. P.; van der Wal, L.; Gramatica, P. Daphnia and fish toxicity of (benzo)triazoles: Validated QSAR models, and interspecies quantitative activity-activity modelling. Journal of Hazardous Materials 2013, 258-259, 50-60, Article. Scopus. [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. Journal of Computational Chemistry 2013, 34, 2121 - 2132.
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. Journal of Computational Chemistry 2014, 35, 1036 - 1044.
- Katoch, S.; Chauhan, S. S.; Kumar, V. A review on genetic algorithm: past, present, and future. Multimedia tools and applications 2021, 80 (5), 8091-8126. PubMed. [CrossRef]
- Najafi, A.; Ardakani, S. S.; Marjani, M. Quantitative Structure-Activity Relationship Analysis of the Anticonvulsant Activity of Some Benzylacetamides Based on Genetic Algorithm-Based Multiple Linear Regression. Tropical Journal of Pharmaceutical Research 2011, 10, 483-490.
- MathWorks, I. MATLAB : the language of technical computing : computation, visualization, programming : installation guide for UNIX version 5; Natwick : Math Works Inc., 1996., 1996.
- Hunter, J. D. Matplotlib: A 2D graphics environment. Computing in Science and Engineering 2007, 9 (3), 99-104, Article. Scopus. [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Molecular Informatics 2010, 29 (6-7), 476-488, Review. Scopus. [CrossRef]
- Dieguez-Santana, K.; Pham-The, H.; Villegas-Aguilar, P. J.; Le-Thi-Thu, H.; Castillo-Garit, J. A.; Casañola-Martin, G. M. Prediction of acute toxicity of phenol derivatives using multiple linear regression approach for Tetrahymena pyriformis contaminant identification in a median-size database. Chemosphere 2016, 165, 434-441. [CrossRef]
- dos Passos Menezes, P.; Dória, G. A. A.; de Souza Araújo, A. A.; Sousa, B. M. H.; Quintans-Júnior, L. J.; Lima, R. N.; Alves, P. B.; Carvalho, F. M. S.; Bezerra, D. P.; Mendonça-Júnior, F. J. B.; et al. Docking and physico-chemical properties of α- and β-cyclodextrin complex containing isopulegol: a comparative study. Journal of Inclusion Phenomena and Macrocyclic Chemistry 2016, 85 (3), 341-354. [CrossRef]
- Talhout, R.; Villa, A.; Mark, A. E.; Engberts, J. B. F. N. Understanding Binding Affinity: A Combined Isothermal Titration Calorimetry/Molecular Dynamics Study of the Binding of a Series of Hydrophobically Modified Benzamidinium Chloride Inhibitors to Trypsin. Journal of the American Chemical Society 2003, 125 (35), 10570-10579. [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 2000, 43 (20), 3714-3717. [CrossRef]
Figure 1.
The performance of the Model 1 according to Eq.3. A. The magnitude of influence of different descriptors of 3-variable model on the binding score according to Eq.3; B. The correlation graph between the observed and predicted values of log binding score; C. Williams’s plot of standardized residual versus leverage of binding score. Training set (blue dots), test set (orange dots).
Figure 1.
The performance of the Model 1 according to Eq.3. A. The magnitude of influence of different descriptors of 3-variable model on the binding score according to Eq.3; B. The correlation graph between the observed and predicted values of log binding score; C. Williams’s plot of standardized residual versus leverage of binding score. Training set (blue dots), test set (orange dots).
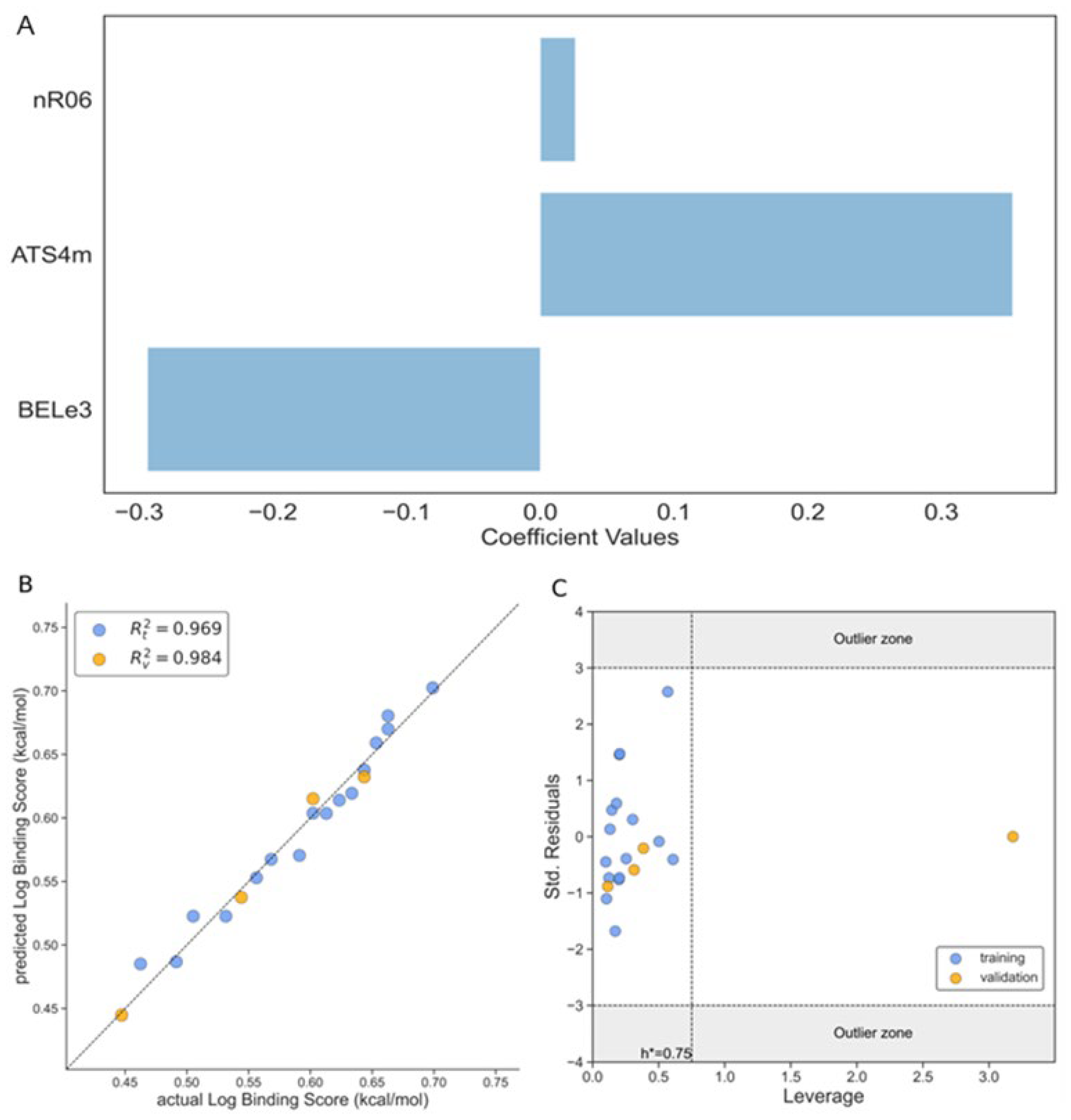
Figure 2.
Density plot for the 3-variable model on log binding score affinity for molecular descriptor influence in the property. A. Density plot for nR06 descriptor; B. Density plot for ATS4m descriptor; C. Density plot for BELe3 descriptor.
Figure 2.
Density plot for the 3-variable model on log binding score affinity for molecular descriptor influence in the property. A. Density plot for nR06 descriptor; B. Density plot for ATS4m descriptor; C. Density plot for BELe3 descriptor.
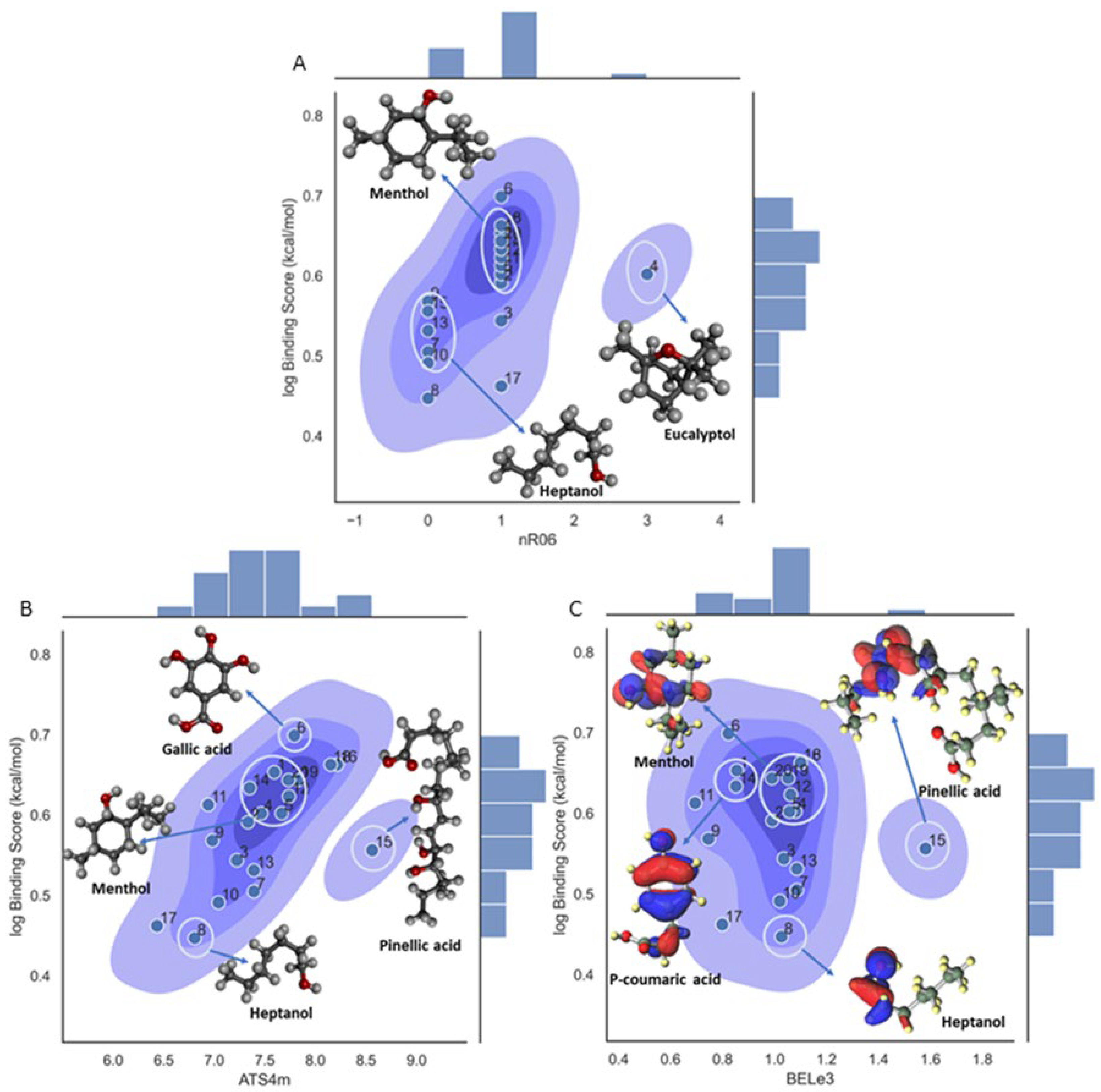
Figure 3.
The performance of the Model 2 according to Eq.4. A. The magnitude of influence of different descriptors of 3-variable model on the log binding affinity according to Eq 4; B. The correlation plot between the observed and predicted values of log binding affinity; C. Williams’s plot of standardized residual versus leverage of log binding affinity. Training set (blue dots), test set (orange dots).
Figure 3.
The performance of the Model 2 according to Eq.4. A. The magnitude of influence of different descriptors of 3-variable model on the log binding affinity according to Eq 4; B. The correlation plot between the observed and predicted values of log binding affinity; C. Williams’s plot of standardized residual versus leverage of log binding affinity. Training set (blue dots), test set (orange dots).
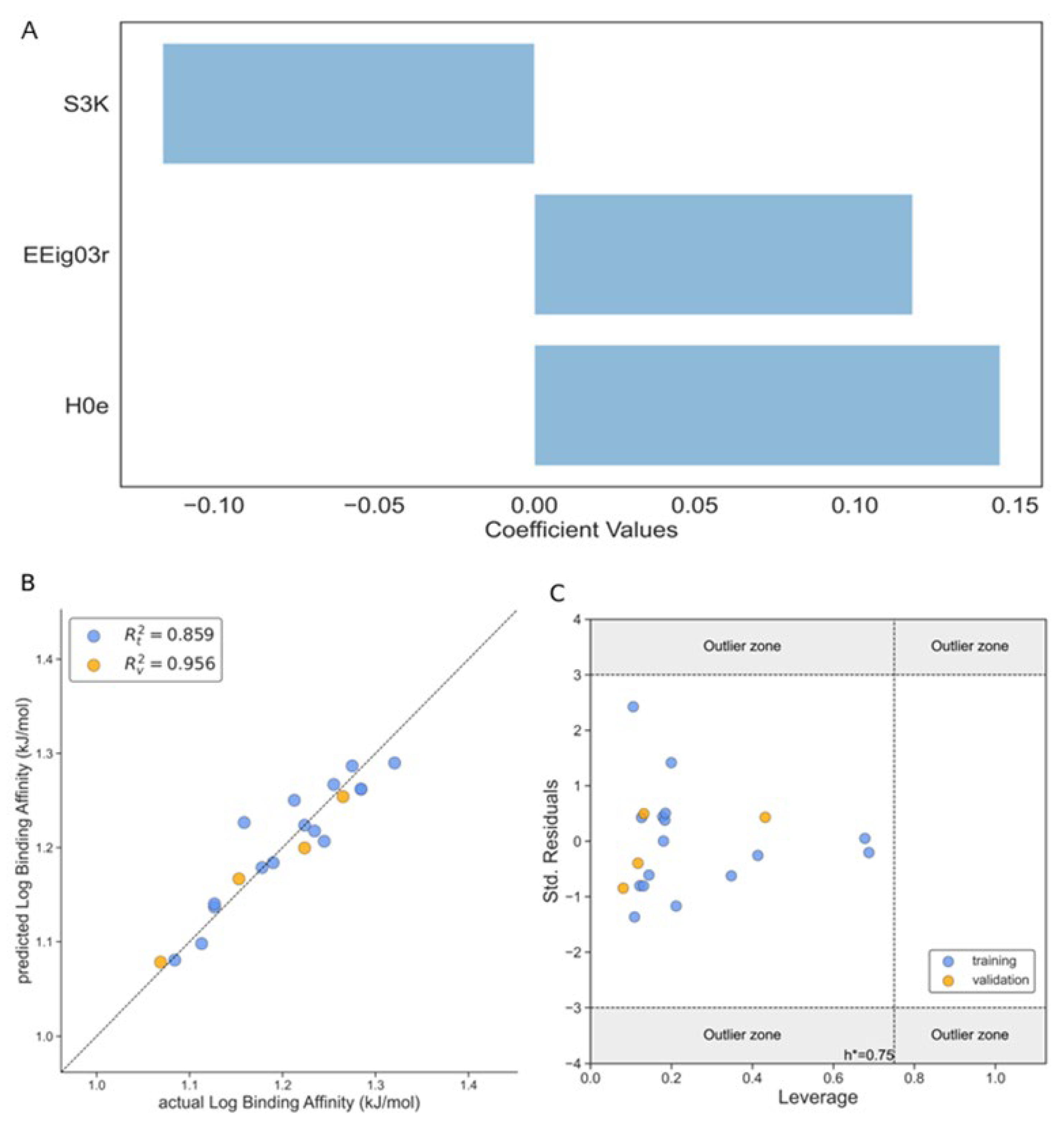
Figure 4.
Density plot for the 3-variable model on log binding affinity for molecular descriptor influence in the property. A. Density plot for S3K descriptor; B. Density plot for EEig03r descriptor; C. Density plot for H0e descriptor.
Figure 4.
Density plot for the 3-variable model on log binding affinity for molecular descriptor influence in the property. A. Density plot for S3K descriptor; B. Density plot for EEig03r descriptor; C. Density plot for H0e descriptor.
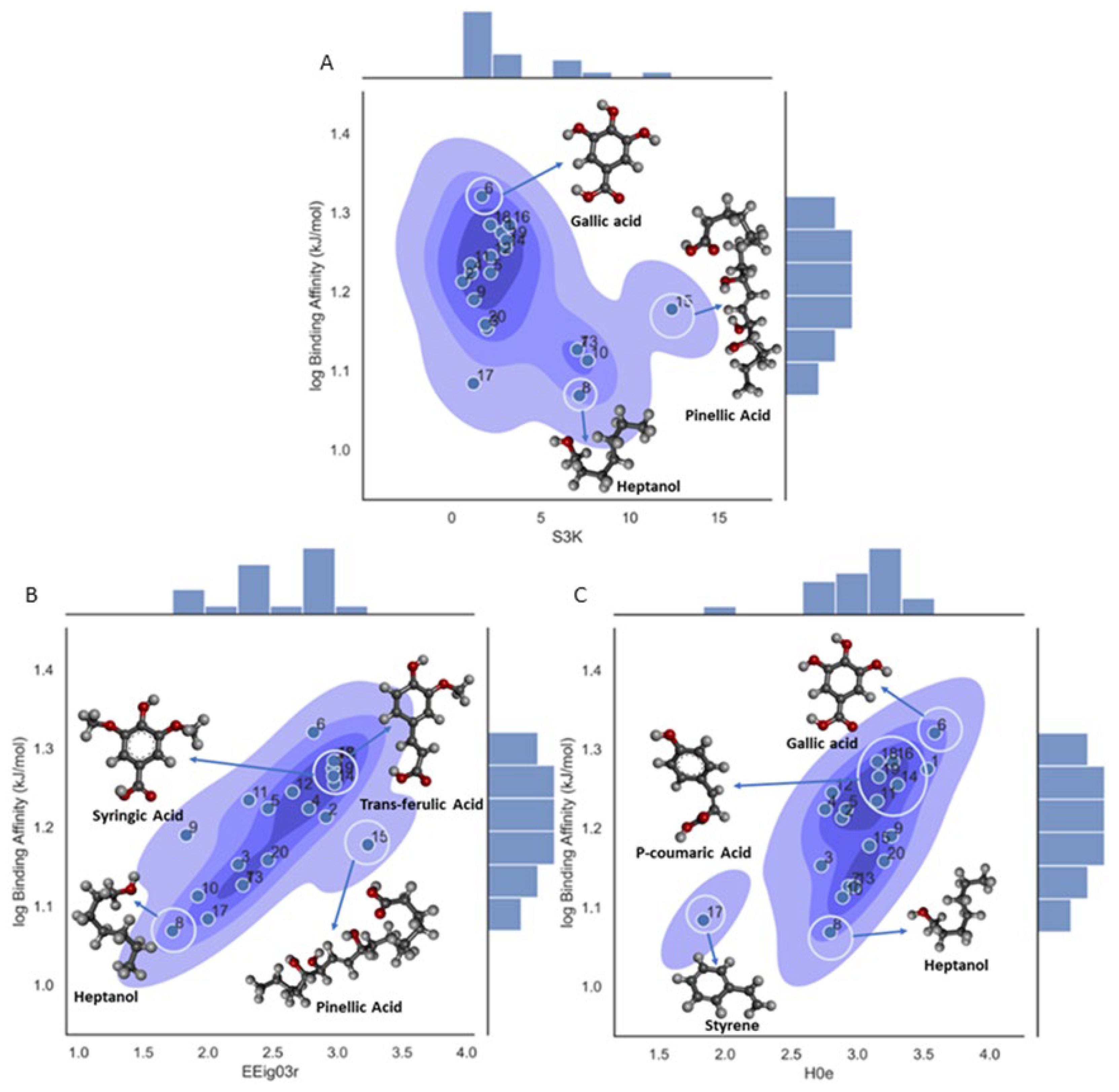
Figure 5.
The performance of the Model 3 according to Eq.5. A. The magnitude of influence of different descriptors of 3-variable model on the log binding energy according to Eq. 5; B. The correlation plot between the observed and predicted values of log binding energy; C. Williams’s plot of standardized residual versus leverage of log binding energy. Training set (blue dots), test set (orange dots).
Figure 5.
The performance of the Model 3 according to Eq.5. A. The magnitude of influence of different descriptors of 3-variable model on the log binding energy according to Eq. 5; B. The correlation plot between the observed and predicted values of log binding energy; C. Williams’s plot of standardized residual versus leverage of log binding energy. Training set (blue dots), test set (orange dots).
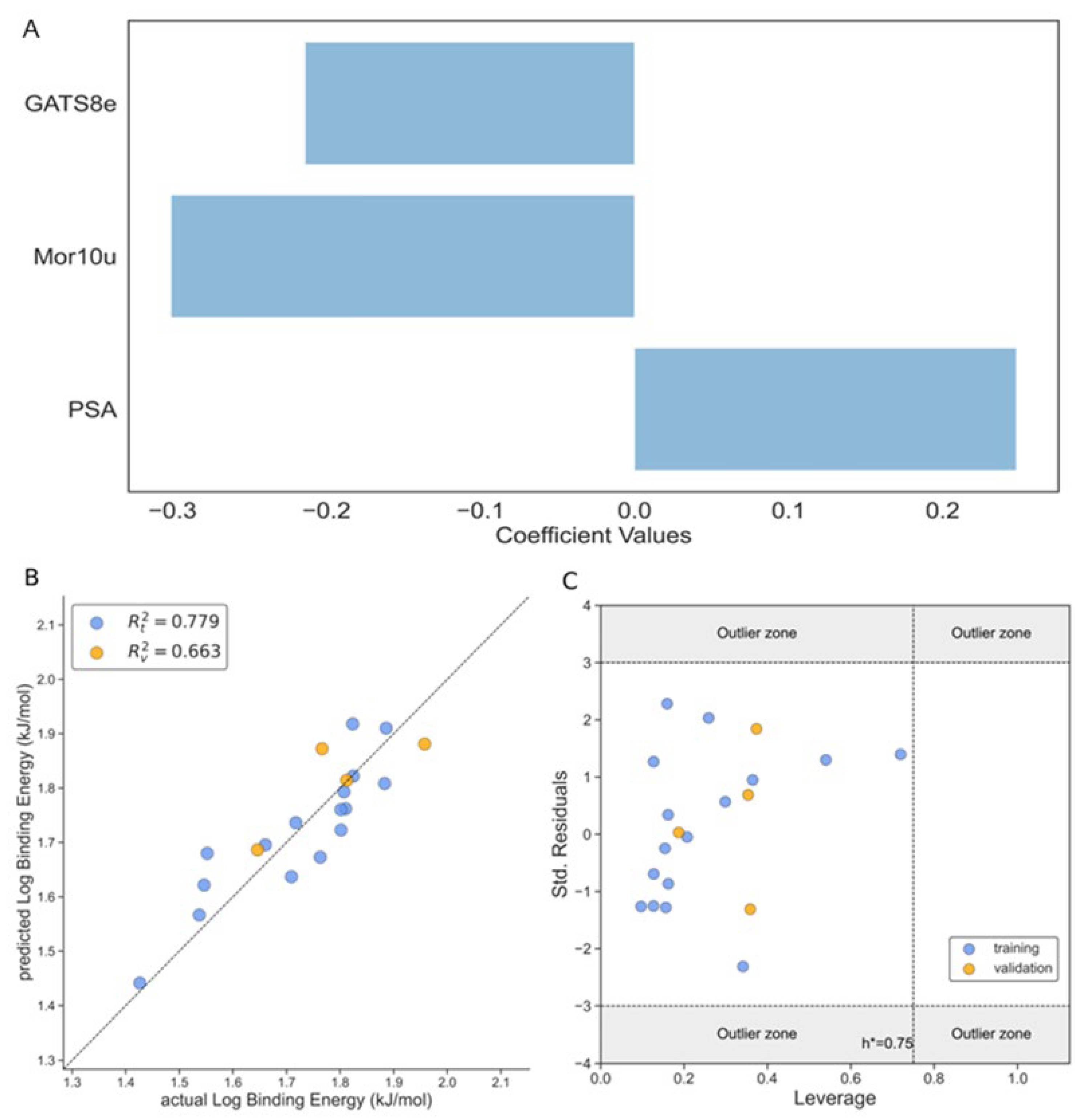
Figure 6.
Density plot for the 3-variable model on log binding affinity for molecular descriptor influence in the property. A. Density plot for PSA descriptor; B. Density plot for GATS8e descriptor; C. Density plot for Mor 10u descriptor.
Figure 6.
Density plot for the 3-variable model on log binding affinity for molecular descriptor influence in the property. A. Density plot for PSA descriptor; B. Density plot for GATS8e descriptor; C. Density plot for Mor 10u descriptor.
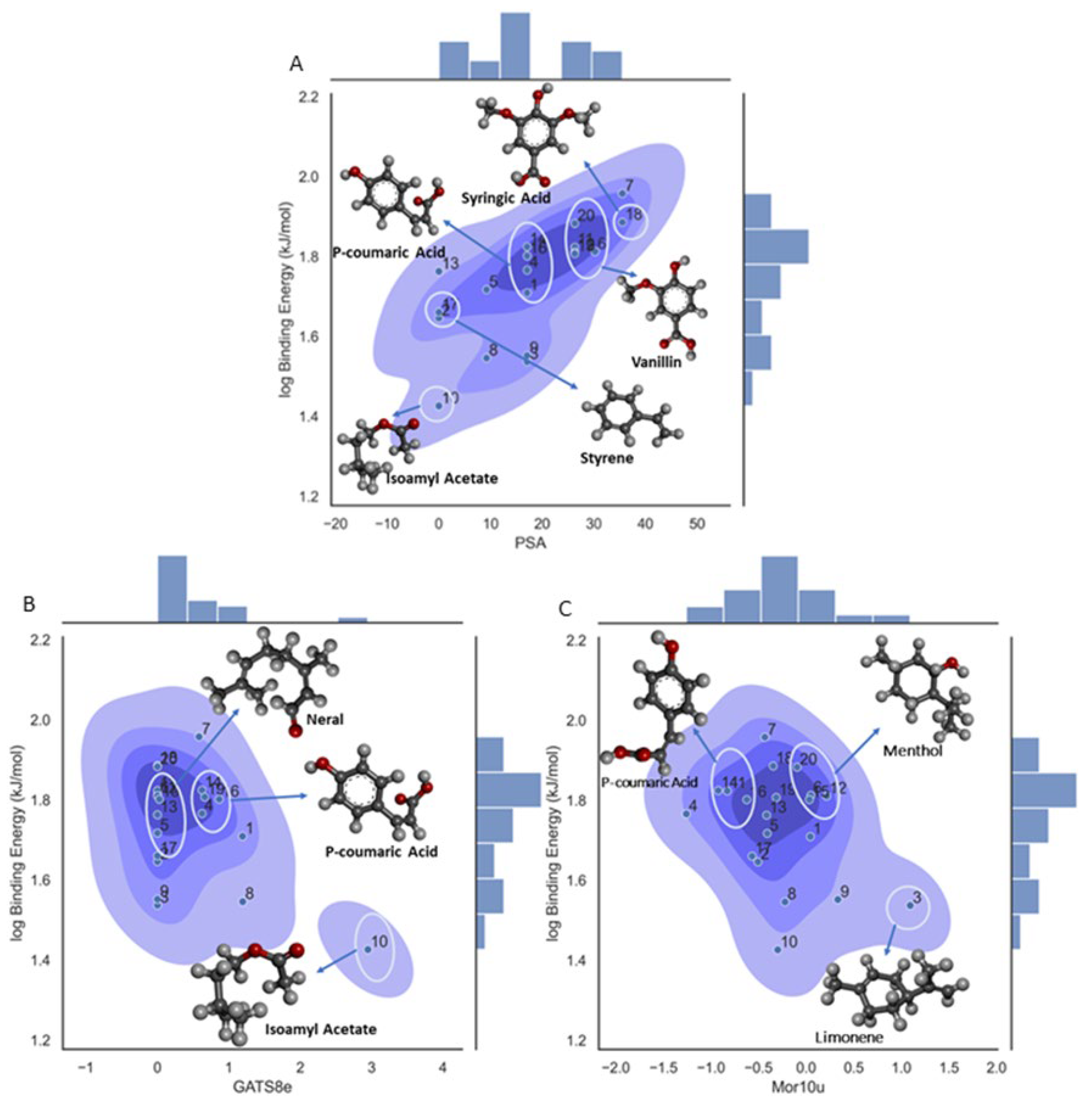

Table 1.
Phenolic acids and the quantum, semi-empirical and binding constant properties.
| ID | β -CD-Ligand Complex |
HOMO (eV) |
LUMO (eV) |
Gap (eV) |
Binding Score (kJ/mol) |
Binding Affinity (kJ/mol) |
|---|---|---|---|---|---|---|
| 1 | Caffeic acid-in | -9.1890 | -1.0514 | -8.1376 | -4.5 | -51.1831 |
| 2 | Camphor-in | -10.1147 | 0.0704 | -10.1851 | -3.9 | -34.4419 |
| 3 | D-Limonene-in | -9.3119 | 0.0092 | -9.3211 | -3.5 | -44.2580 |
| 4 | Eucalyptol-in | -9.9785 | 0.1605 | -10.139 | -4.0 | -52.1555 |
| 5 | Eugenol-up | -8.9658 | -0.1518 | -8.8140 | -4.0 | -35.1691 |
| 6 | Gallic acid-up | -9.5087 | -1.2760 | -8.2327 | -5.0 | -35.6459 |
| 7 | Geranial-up | -9.5651 | -0.2514 | -9.3137 | -3.2 | -58.3673 |
| 8 | Heptanol-up | -10.2024 | -0.1426 | -10.0598 | -2.8 | -26.6948 |
| 9 | Hydroxy Methyl Furfural-up | -9.2240 | -0.4026 | -8.8214 | -3.7 | -64.9081 |
| 10 | Isoamyl acetate-up | -10.1978 | -0.3113 | -9.8865 | -3.1 | -66.5904 |
| 11 | Maltol-up | -9.8792 | -1.3000 | -8.5792 | -4.1 | -64.6218 |
| 12 | Menthol-in | -10.1733 | -0.0167 | -10.1566 | -4.2 | -57.9313 |
| 13 | Neral-up | -9.7932 | -0.4443 | -9.3489 | -3.4 | -66.7781 |
| 14 | P-Coumaric acid-in | -9.3920 | -0.3048 | -9.0872 | -4.3 | -63.3347 |
| 15 | Pinellic acid-in | -9.8072 | 0.0819 | -9.8891 | -3.6 | -63.3198 |
| 16 | Sinapic acid-up | -9.1111 | -1.1979 | -7.9132 | -4.6 | -90.6849 |
| 17 | Styrene-up | -9.4684 | -0.4950 | -8.9734 | -2.9 | -45.7564 |
| 18 | Syringic acid-up | -9.2231 | -0.9025 | -8.3206 | -4.6 | -76.8800 |
| 19 | Trans Ferulic acid-up | -9.0647 | -1.4466 | -7.6181 | -4.4 | -64.1563 |
| 20 | Vanillic acid-in | -9.1477 | -0.9277 | -8.2200 | -4.4 | -76.3647 |
Table 2.
Statistical parameters’ values for binding constant MLR models.
| Parameters | Log BSA | Log BA | Log BE |
|---|---|---|---|
| Model # Number of variables |
1 (Eq.3) 3 |
2(Eq.4) 3 |
3 (Eq.5) 3 |
| R [2] (training set) | 0.969 | 0.859 | 0.779 |
| RMSE (training set) | 0.0116 | 0.0256 | 0.0631 |
| MAE (training set) | 0.0095 | 0.0192 | 0.0527 |
| CCC (training set) | 0.985 | 0.924 | 0.876 |
| F | 126.902 | 24.349 | 14.117 |
| R [2] (cross-validation) | 0.925 | 0.805 | 0.634 |
| RMSE (cross-validation) | 0.0182 | 0.0302 | 0.0812 |
| MAE (cross-validation) | 0.0135 | 0.0236 | 0.0698 |
| CCC (cross-validation) | 0.961 | 0.897 | 0.790 |
| R [2] (external test) | 0.984 | 0.956 | 0.663 |
| RMSE (external test) | 0.0093 | 0.0156 | 0.0685 |
| MAE (external test) | 0.0082 | 0.0146 | 0.0563 |
Table 3.
List of descriptors included in the QSAR models.
| Descriptor | Description | Class |
|---|---|---|
| Model 1 – Binding Score Affinity | ||
| nR06 | Number of 6-membered rings | Ring Descriptors |
| ATS4m | Broto-Moreau autocorrelation of lag 4 (log function) weighted by mass | 2D Autocorrelations |
| BEle3 | Lowest eigenvalue n. 3 of Burden matrix / weighted by atomic Sanderson electronegativities | BCUT Descriptors |
| Model 2 – Binding Affinity | ||
| S3K | 3-path Kier alpha-modified shape index | Topological Indices |
| EEig03r | Eigenvalues | Edge adjacency indices |
| H0e | H autocorrelation of lag 0 / weighted by Sanderson electronegativity | GETAWAY Descriptors |
| Model 3 – Binding Energy | ||
| Descriptor | Description | Class |
| GATS8e | Geary autocorrelation of lag 8 weighted by mass | 2D Autocorrelations |
| Mor10u | Signal 10 / unweighted | 3D-MoRSE Descriptors |
| TPSA | Topological polar surface area using N,O polar contributions | Molecular properties |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



