
Submitted:
05 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
Glycogen storage disease type III (GSDIII) is a hereditary glycogenosis caused by deficiency of the glycogen debranching enzyme (GDE), an enzyme, encoded by Agl, enabling glycogen degradation by catalyzing alpha-1,4-oligosaccharide side chain transfer and alpha-1,6-glucose cleavage. GDE deficiency causes accumulation of phosphorylase-limited dextrin, leading to liver disorder followed by fatal myopathy. We tested here the capacity of the new autophagosomal activator GHF-201 to alleviate disease burden by clearing pathogenic glycogen surcharge in the GSDIII mouse model Agl-/-. We used open field, grip strength and rotarod tests for evaluating GHF-201’s effects on locomotion, biochemistry panel to quantify hematological biomarkers, indirect calorimetry to quantify in vivo metabolism, transmission electron microscopy to quantify glycogen in muscle, and fibroblast image analysis to determine cellular features affected by GHF-201. GHF-201 was able to improve all locomotion parameters and partially reversed hypoglycemia, hyperlipidemia and liver and muscle malfunction in Agl-/- mice. Treated mice burnt carbohydrates more efficiently and showed significant improvement of aberrant ultrastructural muscle features. In GSDIII patient fibroblasts, GHF-201 restored mitochondrial membrane polarization and corrected lysosomal swelling. In conclusion, GHF-201 is a viable candidate for treating GSDIII as it recovered a wide range of its pathologies in vivo, in vitro, and ex vivo.
Keywords:
glycogen
; glycogen storage disease type III
; pharmacotherapy
1. Introduction
Glycogen is a branched polysaccharide with a molecular weight of nine to ten million Da. The average glycogen molecule contains about 55,000 glucose residues linked by α-1,4 (92%) and α-1,6 (8%) glycosidic bonds [1]. The synthesis of glycogen is primed by glycogenin [2] (even though aberrant forms of glycogen are also formed when glycogenin is suppressed [3]), and catalyzed by glycogen synthase (GYS) and glycogen branching enzyme (GBE): GYS links glucose residues to each other by α-1,4 glycosidic bonds to form linear chains. GBE, in turn, attaches a short linear stretch of α-1,4-linked glucose units from the outer non-reducing end of a growing glycogen chain into an α-1,6 position of a pre-existing chain thus generating a branch. Repetitive cycles of elongation and branching generate a large (110–290 nm in liver) [4] spherical molecule with a hydrophilic surface and increased number of reactive termini, facilitating glycogen synthesis and degradation [5,6]. Glycogen degradation, on the other hand, takes place in the cytosol and in lysosomes. In the cytosol, glycogen is degraded by glycogen phosphorylase (GP) and glycogen debranching enzyme (GDE). GP phosphorylates outer glucose residues and releases each one as glucose-1-hosphate. GP can degrade an outer chain only until 4 residues from the branch point. Then, GDE is required to complete glycogenolysis. GDE catalyzes two reactions: transferring a three-glucosyl glucan to the non-reducing end of another linearstrand (α-1,4-glucanotransferase reaction) and hydrolyzing the α-1-6-glycosidic bond remaining on the branch point, releasing glucose (amylo-α-1,6-glucosidase reaction, n.b., glucose, rather than glucose-1-phosphate, is released). In the lysosomes, glycogen degradation is catalyzed by α-glucosidase.
Glycogen is stored primarily in liver and muscle, where it respectively serves as a reservoir for regulating blood glucose levels and as a readily mobilizable energy reserve. Therefore, aberrations in glycogen metabolism, or glycogen storage disorders (GSDs) are primarily hepatic or muscular disorders, with two notable exceptions, APBD and LD, involving specifically the central and peripheral nervous systems. The most common disorder of glycogen metabolism is observed in diabetes, in which abnormal amount of insulin or abnormal insulin response result in accumulation or depletion of liver glycogen. However, GSDs are normally associated with hereditary deficiencies in enzymes of glycogen and glucose metabolism causing glycogen accumulation (except for GSD type 0, which is a GYS deficiency and is therefore associated with glycogen insufficiency).
In this work we focused on GSD type III (GSDIII, Cori disease), the third most prevalent muscle GSD (incidence of 1:100,000) after GSD types V and II [7,8,9], which is caused by GDE deficiency leading to accumulation of glycogen due to its reduced degradation. Phosphorylase-limit dextrin (PLD), the type of glycogen which accumulates in GSDIII, is structurally abnormal, containing shorter outer branches [10]. While phosphorylase-limit dextrin is sensitive to diastase digestion and does not form polyglucosan bodies [10], it is still associated with large vacuoles and can cause histological damage to myofibrils [11,12]. GSDIII usually starts as a liver disorder characterized by hepatomegaly, hypoglycemia, hyperlipidemia and hyperketonemia. These result from the limited ability to breakdown glycogen to glucose, leading to excessive use of lipid oxidation as an alternative energy source with ensuing attenuation of lipid uptake by adipocytes and increase in ketone bodies as byproducts of fatty acid oxidation (FAO). Later on, myopathy pursues [13], with occasional cardiomyopathy [14], liver cirrhosis and hepatocellular adenoma [15].
GSDIII implicates vacuolar myopathy and myofibrillar damage [12,16], also observed in GSDII [17], which shares with GSDIII muscle glycogen over accumulation (albeit lysosomal in GSDII and mostly cytoplasmic in GSDIII). We therefore tested here the GSDIII curative capacity of GHF-201, a novel safe compound capable of reversing glycogen accumulation and enhancing autophagy [18] (doi.org/10.1101/2023.02.20.529109). We show that GHF-201 was capable of improving motility and grip strength in a Agl-/- mouse model of GSDIII, where GDE, encoded by the murine Agl gene, was knocked out. At the same time, GHF-201 did not modify the deficient motor learning capacities of Agl-/- mice associated with a neurological deficiency not implicated in GSDIII. We further demonstrate that GHF-201 corrected hypoglycemia, hyperlipidemia and liver and muscle damage, as well as carbohydrate over fat fuel preference. Combined with reduction of glycogen levels and muscle atrophy at the tissue level, and improvement of lysosomal and mitochondrial phenotypes at the cell level (in GSDIII patients’ fibroblasts), our results strongly support that GHF-201 has a potential therapeutic capacity for treating GSDIII, either as a standalone, or as part of a combination therapy.
2. Materials and Methods
2.1. In Vivo Studies
In vivo studies were conducted on Agl-/- mice [7] and congenic C57Bl6J mice as wild type control. n=6–10 mice/arm were used. 50 µL of 250 mg/kg GHF-201 in 10% DMSO were intramuscularly injected to the gastrocnemius in alternate flanks twice a week. 10% DMSO vehicle was used as a vehicle control. Mice were treated from the age of 3 months to the age of 8 months. Animals’ weight was monitored biweekly. A weight decrease of >10% between sequential weightings, or >20% from treatment initiation led to removal of the animal from the study. We tested monthly moving distance and cumulative sojourn in the center of an open field, front paw grip strength using a grip strength meter (Columbus Instruments, Columbus, Oh, USA) , and rotarod performance (latency to fall from a rotating cylinder). The rotating cylinder (UGO Basile S.R.L., Gemonio, Italy) accelerated from 5 to 40 RPM in 99 sec. Mice were tested for 3 consecutive trials with a rest period of approximately 20 min between trials.
Blood biochemistry (enzyme activities), triglycerides and glucose were determined at the Hadassah central laboratory division. Indirect calorimetry and multi-parameter metabolic assessment (metabolic cages) were performed as in [18].
2.2. Ultrastructural Electron Microscopy Studies
Gastrocnemius muscle samples were fixed, embedded, sectioned and analyzed by transmission electron microscope as described in [7].
2.3. High Content Analysis of GSDIII Patient Fibroblasts
Primary Skin fibroblasts from 3 Patients were procured from Coriell Institute (New Jersey, USA) and expanded in DMEM supplemented with 1% Sodium Pyruvate, 1% Penicillin-Streptomycin-Amphotericin B, 10% heat inactivated fetal calf serum (Biological industries, Israel), and 1% 100x non-essential amino acids solution (NEAA) at 37 °C with 5% CO2. For live imaging, GSDIII and healthy control (HC) skin fibroblasts were seeded at 1,400 cells per well and cultured at 37 °C and 5% CO2 with or without GHF-201 (50 µM) for 24 h in specialized microscopy-grade 96-well plates (Cellvis P96-1.5H-N). Cells were then washed in HBSS, supplemented for 20 min with 1.6mM Hoechst 33342, 0.1 mM LysoTracker Deep Red, 0.05 mM TMRE and 0.4 mM calcein-AM Green (Thermo-Fisher Scientific), and washed again with HBSS. Subsequently, images were acquired using an Operetta G1 system at 20X magnification under environmental control (37 °C and 5% CO2) conditions. All assay parameters (including the acquisition exposure times, objective, and the analysis parameters) were kept constant for all assay repetitions. Images were analyzed by the Harmony image analysis software. Downstream analysis included removal of wells with outlier values, feature selection by backward feature elimination and normalization to HC levels prior to statistical analysis [19]. Multivariate analysis was performed in Python.
2.4. Statistical Analysis
Sample sizes were determined based on the effect sizes of key parameters measured (e.g., open field moving distance), as detailed in [20]. These sample sizes were sufficient to provide the acceptable power of at least 80%. Blinding in in vivo studies was obtained by encoding the animals in the different treatment groups so that the experimenters were blinded to treatment allocation. To obtain randomization, litters from mice born a few days apart were pooled and pups of similar weight were divided into vehicle and GHF-201-treated arms. Specific statistical tests are mentioned in the relevant figure legends. The TEE ANCOVA analysis was provided by the NIDDK Mouse Metabolic Phenotyping Centers (MMPC, www.mmpc.org) using their Energy Expenditure Analysis page (http://www.mmpc.org/shared/regression.aspx) and supported by grants DK076169 and DK115255.
3. Results
3.1. Animal Weight
GSDIII male mice presented with a higher weight as compared to WT mice. According to [11] GSDIII mice were lighter than WT mice, while in [21] no significant difference between the weights of GSDIII and WT mice was observed, together suggesting that such a difference might be batch-dependent. GHF-201 treatment did not significantly modify weight in GSDIII mice (Figure 1).
3.2. Locomotor Studies
We show that GHF-201 improved open field performance at a relatively advanced stage of the disease (last time point when mice were approximately 8 months old). This improvement was demonstrated as increases in both distance traveled (Figure 2A) and sojourn in the center of the open field (Figure 2B), which indicates a more exploratory behavior associated with decreased anxiety. Sojourn in the center was corrected by GHF-201 to WT value at the age of 8 months. Interestingly, while GHF-201 significantly increased grip strength (Figure 2C) and improved open field parameters, it did not increase the latency to fall from a rotating rod in the rotarod test, which was decreased in Agl-/- mice (Figure 2D). Wild type mice manifested training and motor learning capacity respectively demonstrated by increased latency between runs in each test and increased overall latency over time. Agl-/- mice, on the other hand, treated or not, did not show any training or learning capacities. Indeed rotarod measures motor coordination, which is considered a neurological function of the nigrostriatal dopamine system [22,23] not implicated in GSDIII. Motor activity measured by the open field test, on the other hand, is influenced by muscle strength, which was modified by GHF-201 (Figure 2C). There are several works (e.g., [24]), which demonstrate independence of rotarod and open field results.
Notably, all behavioral tests had to be discontinued following the 4th test post treatment initiation. The reason for that was that after the 4th test the weight of all mice was decreased by >10% from the last reading suggesting that GHF-201, increases muscle strength-associated mobility, but not survival, in GSDIII mice.
3.3. Blood Metabolic Panel
We tested whether GHF-201 is able to correct the hypoglycemia, hypertriglyceridemia and liver and muscle damage as measured by blood examinations [11,25,26]. Such effects are expected from an agent, such as GHF-201, capable of inducing the catabolism of liver glycogen with an ensuing rise in blood glucose and reduction in compensatory lipid flux to the liver. Our blood metabolic panel results show that GHF-201 is able to correct hypoglycemia and hyperlipidemia and partially restore muscle (creatine kinase) and liver (alanine transferase) damage, all of which characteristically accrue with time in the Agl-/- GSDIII mouse model (Figure 3).
3.4. In Vivo Metabolic Profile
The effect of GHF-201 on various in vivo metabolic parameters was determined by indirect calorimetry. Fuel preference at the whole animal level is determined by the respiratory exchange ratio (RER, the ratio of CO2 produced to O2 consumed). Lower RER indicates higher fat burn, while higher RER indicates higher carbohydrate burn. As our results (Figure 4A) show, GHF-201 significantly increased RER in treated v untreated Agl-/- mice in light, dark and overall. Total energy expenditure (TEE) is also significantly increased by GHF-201 overall (i.e., in light and dark conditions combined). In the light GHF-201 increases TEE to the extent that it is indiscriminable from WT (correction effect). GHF-201 correction of TEE to WT levels can also be revelaed by ANCOVA, which also accounts for the dependence of TEE on body weight: The difference between WT and Agl-/- in light and overall conditions became insignificant when Agl-/- mice were treated with GHF-201. Importantly, similar to TEE correction to WT level by GHF-201, a significant difference in TEE between vehicle and GHF-201 is also revealed by ANCOVA only under the light condition. Our observations that GHF-201 also increased fat oxidation during light, and, especially, carbohydrate burning, which was compromised in Agl-/- mice, strongly suggest that GHF-201 can increase glycogen catabolism. This is a therapeutic advantage as the accumulated, poorly digested, and malconstructed glycogen in GSDIII is considered a pathogenic factor. Stimulation of wheel running and correction of ambulatory activity in the light (Figure 4B) are in line with open field results (Figure 2A), also acquired in the light, and with the GHF-201-mediated stimulation of carbohydrate catabolism. Toghether these results suggest that GHF-201 can improve metabolic efficacy, compromised by the diseased state, at the whole animal level. Similar to our observation in the GSDIV mouse model [18], GHF-201 also increased food and water intake, even though not significantly (Figure 2C). This increase was mainy observed in the dark, where activity and food consumption are increasedin mice. A GHF-201-mediated increase in food and water intake with a parallel increase in fuel burning (Figure 2A) constitute an improvement in metabolic efficiency.
3.5. Muscle Glycogen Quantification
Blinded quantification of transmission EM sections of gastrocnemius muscle sections showed that in muscle sections derived from GSDIII mice, higher % of the area was occupied by dense glycogen granules as compared to muscle sections derived from wild type mice (Figure 5A). Myofibrils in GSDIII muscle sections were also more dissociated than wild type myofibrils due to the increased glycogen storage (Figure 5B). Both increase in glycogen granule area and myofibril dissociation were significantly corrected (even though not to wild type levels) by the GHF-201 treatment.
3.6. Glycogen Levels, Lysosomal and Mitochondrial Features of GSDIII Patient-Derived Skin Fibroblasts
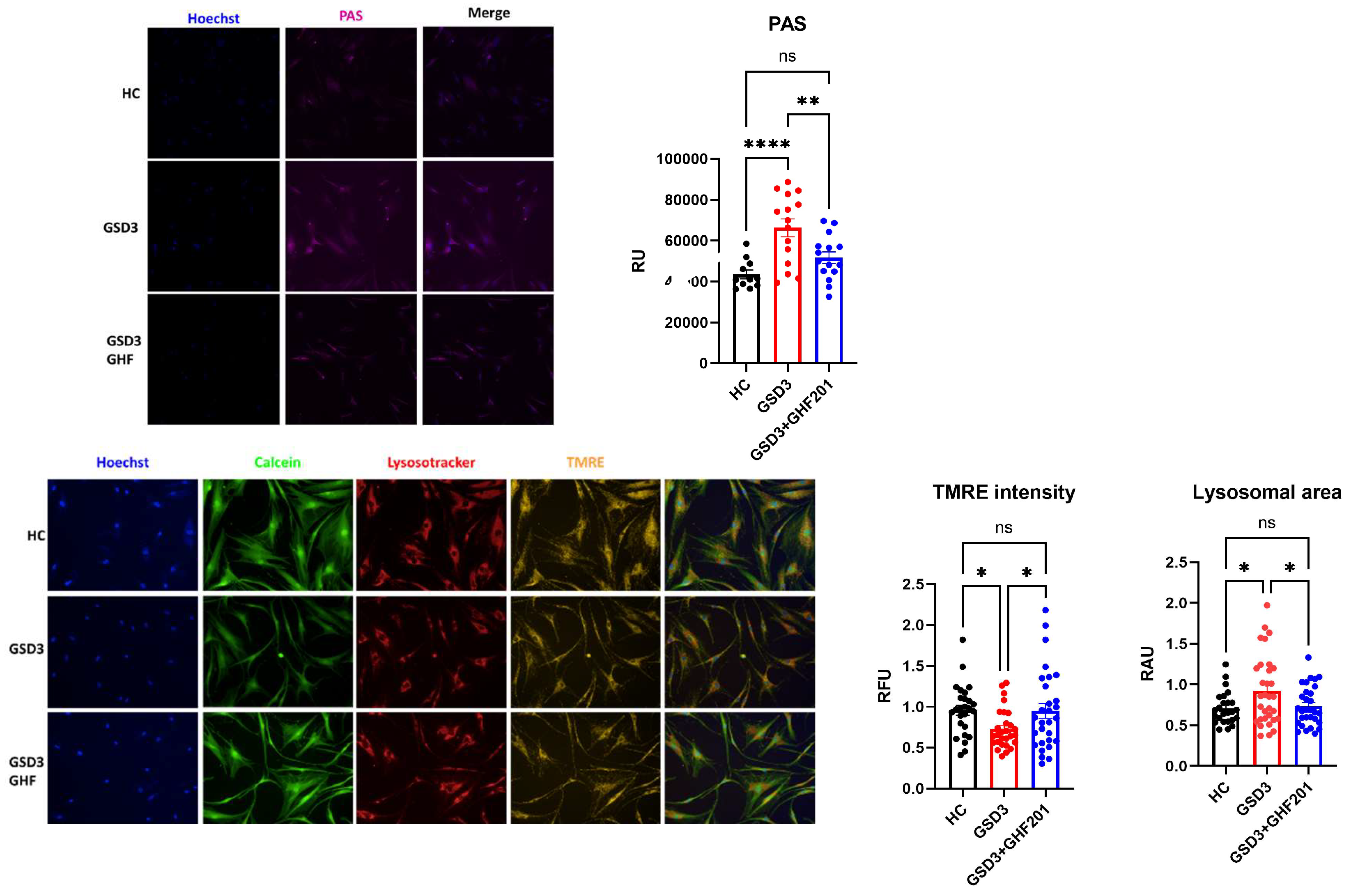
Consistent with the results in gastrocnemius muscle, in skin fibroblasts derived from GSDIII patients, GHF-201 also reduced glycogen levels and rendered them similar (not statistically different) to those of HC fibroblasts, as determined by periodic acid-Schiff’s reagent (PAS) staining (Figure 6A). This result suggests that the mechanism of action by which GHF-201 alleviated GSDIII symptoms in mice is autolysosomal degradation of glycogen, which is known to be mediated by GHF-201 in GSDIV [18] and GSDIa (doi.org/10.1101/2023.02.20.529109).
To confirm GHF-201 mechanism of action at a cellular level, we decided to test its effects on lysosomal and mitochondrial features in fibroblasts derived fron GSDIII patients. Since GHF-201 is an autophagic and catabolic activator, we anticipated that its significant effect at a cellular level will be at the lysosomal-mitochondrial axis, as shown, for instance, in GSDII [27]. Our results (Figure 6B) show that under starvation, which induces autophagic flux, GHF-201 reduced lysosomal area bringing it close to the area observed in HC fibroblasts. Reduced lysosomal area was also observed in other systems in healthy as compared to lysosomal impaired cells [28] and is presumably associated with increased autophagic flux and lysosomal function, which can clear overladen lysosomes from their excess content and thus reduce their size, which is increased by the lysosomal swelling this excess content causes. Mitochondrial depolarization was observed in GSDIII patient fiblroblasts as compared to HC fibroblasts. This mitochondrial depolarization is probably associated with the limited glycogen degradation and carbohydrate fuel production characteristic of GSDIII (Figure 4A). In accordance with an increase in mitochondrial fueling by enhanced sutophagic catabolism, GHF-201 treatment repolarized mitochondrial membrane potential, bringing it closer to that of HC fibroblasts (Figure 6C).
4. Discussion
The long-term aim of this study was to test the capacity of GHF-201 to alleviate disease symptoms in the Agl-/- mouse model of GSDIII. GHF-201 specifically binds the lysosomal membrane protein LAMP1 [18]. GHF-201 was further able to re-acidify the aberrant alkaline lysosomal pH. In adult GSDIV (APBD) models, GHF-201 ameliorated the diseased state by enhancing lysosomal glycogen catabolism and autophagy.
GSDIII starts with liver involvement followed by storage of glycogen in skeletal muscle provoking a myopathy. The disease is caused by cytoplasmic glycogen debranching enzyme (GDE) deficiency leading to accumulation of glycogen due to its reduced degradation. Glycogen accumulation in muscle is more extensive in GSDIII, as compared to the other myopathy implicating GSDs GSDII, GSDV, GSDIX, GSDX, and GSDXV [9]. In particular, human muscle biopsies from GSDIII patients show a typical and constant vacuolar myopathy, characterized by multiple and variably sized vacuoles filled with PAS-positive material [7]. Phosphorylase-limit dextrin (PLD), the type of glycogen which accumulates in GSDIII, is structurally abnormal, containing shorter outer branches [10]. While PLD is sensitive to diastase digestion and does not form polyglucosan bodies [10], it is still associated with large vacuoles and can cause histological damage to myofibrils [11,16]. In fact, as also shown by our results (Figure 5), disruption of myofibrillar structure by glycogen vacuoles is more pronounced in GSDIII than in other GSDs with prominent muscular involvement [9].
GSDIII usually starts as a liver disorder and later on myopathy pursues [13], with occasional cardiomyopathy [14], liver cirrhosis and hepatocellular adenoma [15]. Interestingly, possibly due to higher overall glycogen accumulation, GSDIII manifests with excess glycogen in both cytoplasm and lysosomes, as opposed to GSDII, where excess glycogen is primarily found in lysosomes due to the aberrant function of GAA [29]. The main pathogenic factor in GSDIII, as revealed by ultrastructural analysis of muscle specimens from patients, is autophagosome accumulation. Concordantly, Laforet et al., using electron microscopy, showed the presence of large non-membrane bound sarcoplasmic deposits of normally structured glycogen as well as smaller rounded sac structures lined by a continuous double membrane containing only glycogen, corresponding to autophagosomes. A consistent SQSTM1/p62 decrease and beclin-1 increase in human muscle biopsies suggested autophagic dysregulation [7]. The latter can impede the degradation of dysfunctional organelles and proteins, and, notably, also of glycogen, in a specific pathway mediated by the starch binding domain-containing protein 1 (Stbd1), which tags glycogen for its autophagy, interacting with autophagy machinery and glycogen related proteins, including GDE, in other domains [30]. Interestingly, among these glycogen related proteins, GDE has the highest number of Atg8 binding motifs [30], also suggesting an important role of autophagy in GSDIII, which might take place if GDE deficiency impairs the interaction with the autophagy machinery. Moreover, dysfunctional autophagy plays a pivotal role in disrupting muscle homeostasis and causing structural damage to sarcomeres [31]. The tight link between autophagic impairment and GSDIII pathology can thus explain the beneficial effects of GHF-201, a general autophagic activator [18], in improving motor function (Figure 2) and skeletal muscle health (Figure 5) in a GSDIII mouse model.
Our blood biochemistry panel demonstrated that GHF-201 treatment led to improved liver and muscle function and partially corrected blood hypoglycemia and hypertriglyceridemia (Figure 3), also observed in GSDIII patients [32]. Both hypoglycemia and hyperlipidemia are expected outcomes of glycogen surcharge since more glucose is shunted from blood to intracellular glycogen and since high glycogen levels can inhibit the energy sensor AMPK [33,34,35], leading to activation of acetyl CoA carboxylase, the rate limiting enzyme of fatty acid synthesis, which is phospho-inhibited by AMPK. Reduction of glycogen levels by GHF-201 (Figure 5) expectedly mitigated this hyperlipidemia. The blood lipid profile can also reflect fuel utilization. As glycogen mobilization is compromised in GSDIII and enhanced by GHF-201, we expected that GSDIII mice use lipid instead of carbohydrate fuel as a compensatory reaction. However, as suggested by the hypertriglyceridemia observed in untreated GSDIII mice (Figure 3), the mice are not capable of this metabolic compensation. Notably, as revealed by both indirect calorimetry (Figure 4) and the blood biochemistry panel (Figure 3), GHF-201 treatment enabled this metabolic compensation by boosting fat burn (Figure 4), which is possibly reflected in the reduction in blood triglyceride level (Figure 3). A similar effect of GHF-201 was observed in the GSDIV modeling Gbeys/ys mice [18]. Interestingly, GHF-201-mediated improvement of fat oxidation (Figure 4A), and other in vivo metabolic effects (TEE, (Figure 4A), ambulatory activity (Figure 4B), wheel running (Figure 2B)) were more pronounced in the light than in the dark. Since mice are nocturnal animals, in which food is mostly consumed in the dark (see also Figure 4C) and digested in the light, it can be conjectured that GHF-201, affecting fuel digestion, or autophagic substrate catabolism, exerts a larger effect in the light.
In conclusion, we show here that GHF-201, a LAMP-1 targeting small molecule acting as a general activator of autophagic flux, can partially correct aberrant metabolic, motor and myopathologic phenotypes in the Agl-/- mouse model of GDE deficiency, or GSDII, a disease implicating a particularly high glycogen burden, causing liver and myofibrillar injury.
Author Contributions
Conceptualization, OK, MW, and EM; Methodology, KM, SS, SB, US, MM, SB, and HG.; Writing—original draft preparation, OK.; Writing—review and editing, MW, EM; Supervision, JT, HR, MW, EM, OK; funding acquisition, MW, EM, OK. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research was funded by Association Francaise contre les Myopathies, grant number 23116.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of THE HEBREW UNIVERSITY OF JERUSALEM (protocol code 16403 and date of approval 30 March 2022).
Acknowledgments
The authors wish to thank Prof. Eran Perlson from Tel Aviv University for his help in collection and preparation of the muscle samples and Prof. Giuseppe Ronzitti from Genethon, France, for providing the mice used in this study.
Conflicts of Interest
OK and MW are advisors to the company Golden Heart Flower (GHF) to which GHF-201 is sublicensed.
References
- B. ILLINGWORTH, J. LARNER, and G. T. CORI, "Structure of glycogens and amylopectins. I. Enzymatic determination of chain length," (in eng), J Biol Chem, vol. 199, no. 2, pp. 631-40, Dec 1952.
- C. Smythe and P. Cohen, "The discovery of glycogenin and the priming mechanism for glycogen biogenesis," (in eng), Eur J Biochem, vol. 200, no. 3, pp. 625-31, Sep 1991. [CrossRef]
- G. Testoni et al., "Lack of Glycogenin Causes Glycogen Accumulation and Muscle Function Impairment," (in eng), Cell Metab, vol. 26, no. 1, pp. 256-266.e4, Jul 2017. [CrossRef]
- M. M. Adeva-Andany, M. Gonzalez-Lucan, C. Donapetry-Garcia, C. Fernandez-Fernandez, and E. Ameneiros-Rodriguez, "Glycogen metabolism in humans," BBA Clin, vol. 5, pp. 85-100, Jun 2016. [CrossRef]
- C. Prats, T. E. Graham, and J. Shearer, "The dynamic life of the glycogen granule," (in eng), J Biol Chem, vol. 293, no. 19, pp. 7089-7098, May 11 2018. [CrossRef]
- D. S. Froese et al., "Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design," Hum Mol Genet, vol. 24, no. 20, pp. 5667-76, Oct 15 2015. [CrossRef]
- P. Laforêt et al., "Deep morphological analysis of muscle biopsies from type III glycogenesis (GSDIII), debranching enzyme deficiency, revealed stereotyped vacuolar myopathy and autophagy impairment," (in eng), Acta Neuropathol Commun, vol. 7, no. 1, p. 167, Oct 2019. [CrossRef]
- Schreuder, A. Rossi, S. Grünert, and T. Derks Glycogen Storage Disease type III (GeneReviews ® [Internet]). Seattle (WA) University of Washington, Seattle, 2010 [updated 2022 Jan 6].
- W. B. Hannah, T. G. J. Derks, M. L. Drumm, S. C. Grünert, P. S. Kishnani, and J. Vissing, "Glycogen storage diseases," (in eng), Nat Rev Dis Primers, vol. 9, no. 1, p. 46, Sep 07 2023. [CrossRef]
- P. S. Kishnani et al., "Glycogen storage disease type III diagnosis and management guidelines," (in eng), Genet Med, vol. 12, no. 7, pp. 446-63, Jul 2010. [CrossRef]
- P. Vidal et al., "Rescue of GSDIII Phenotype with Gene Transfer Requires Liver- and Muscle-Targeted GDE Expression," (in eng), Mol Ther, vol. 26, no. 3, pp. 890-901, 03 2018. [CrossRef]
- S. DiMauro, T. S. M. d. a. m. In:, and N. Y. Myology. mcGraw-Hill, pp 1535–1558.
- V. Decostre et al., "Long term longitudinal study of muscle function in patients with glycogen storage disease type IIIa," (in eng), Mol Genet Metab, vol. 122, no. 3, pp. 108-116, 11 2017. [CrossRef]
- V. Valayannopoulos et al., "Successful treatment of severe cardiomyopathy in glycogen storage disease type III With D,L-3-hydroxybutyrate, ketogenic and high-protein diet," (in eng), Pediatr Res, vol. 70, no. 6, pp. 638-41, Dec 2011. [CrossRef]
- E. Demo et al., "Glycogen storage disease type III-hepatocellular carcinoma a long-term complication?," (in eng), J Hepatol, vol. 46, no. 3, pp. 492-8, Mar 2007. [CrossRef]
- S. DiMauro et al., "Debrancher deficiency: neuromuscular disorder in 5 adults," (in eng), Ann Neurol, vol. 5, no. 5, pp. 422-36, May 1979. [CrossRef]
- S. S. Peng, W. L. Hwu, N. C. Lee, F. J. Tsai, W. H. Tsai, and Y. H. Chien, "Slow, progressive myopathy in neonatally treated patients with infantile-onset Pompe disease: a muscle magnetic resonance imaging study," (in eng), Orphanet J Rare Dis, vol. 11, no. 1, p. 63, 05 2016. [CrossRef]
- Kakhlon et al., "Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism," (in eng), EMBO Mol Med, p. e14554, Sep 06 2021. [CrossRef]
- J. C. Caicedo et al., "Data-analysis strategies for image-based cell profiling," (in eng), Nat Methods, vol. 14, no. 9, pp. 849-863, Aug 31 2017. [CrossRef]
- J. Charan and N. D. Kantharia, "How to calculate sample size in animal studies?," (in eng), J Pharmacol Pharmacother, vol. 4, no. 4, pp. 303-6, Oct 2013. [CrossRef]
- K. M. Liu, J. Y. Wu, and Y. T. Chen, "Mouse model of glycogen storage disease type III," (in eng), Mol Genet Metab, vol. 111, no. 4, pp. 467-76, Apr 2014. [CrossRef]
- S. Kamel et al., "Experimental Evidence for Diiodohydroxyquinoline-Induced Neurotoxicity: Characterization of Age and Gender as Predisposing Factors," (in eng), Pharmaceuticals (Basel), vol. 15, no. 2, Feb 19 2022. [CrossRef]
- M. Lundblad, E. Vaudano, and M. A. Cenci, "Cellular and behavioural effects of the adenosine A2a receptor antagonist KW-6002 in a rat model of l-DOPA-induced dyskinesia," (in eng), J Neurochem, vol. 84, no. 6, pp. 1398-410, Mar 2003. [CrossRef]
- M. P. Hassan et al., "Photobiomodulation therapy improved functional recovery and overexpression of interleukins-10 after contusion spinal cord injury in rats," (in eng), J Chem Neuroanat, vol. 117, p. 102010, 11 2021. [CrossRef]
- S. Lucchiari et al., "Hepatic and neuromuscular forms of glycogenosis type III: nine mutations in AGL," (in eng), Hum Mutat, vol. 27, no. 6, pp. 600-1, Jun 2006. [CrossRef]
- S. Lucchiari, D. Santoro, S. Pagliarani, and G. P. Comi, "Clinical, biochemical and genetic features of glycogen debranching enzyme deficiency," (in eng), Acta Myol, vol. 26, no. 1, pp. 72-4, Jul 2007.
- J. A. Lim, L. Li, O. Kakhlon, R. Myerowitz, and N. Raben, "Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease," (in eng), Autophagy, vol. 11, no. 2, pp. 385-402, 2015. [CrossRef]
- M. E. G. de Araujo, G. Liebscher, M. W. Hess, and L. A. Huber, "Lysosomal size matters," (in eng), Traffic, vol. 21, no. 1, pp. 60-75, 01 2020. [CrossRef]
- C. Nascimbeni, M. Fanin, E. Masiero, C. Angelini, and M. Sandri, "The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII)," (in eng), Cell Death Differ, vol. 19, no. 10, pp. 1698-708, Oct 2012. [CrossRef]
- P. Koutsifeli et al., "Glycogen-autophagy: Molecular machinery and cellular mechanisms of glycophagy," (in eng), J Biol Chem, vol. 298, no. 7, p. 102093, Jul 2022. [CrossRef]
- M. Margeta, "Autophagy Defects in Skeletal Myopathies," (in eng), Annu Rev Pathol, vol. 15, pp. 261-285, Jan 24 2020. [CrossRef]
- M. LaBarbera, G. Milechman, and F. Dulbecco, "Premature coronary artery disease in a patient with glycogen storage disease III," (in eng), J Invasive Cardiol, vol. 22, no. 8, pp. E156-8, Aug 2010.
- J. I. Mobbs et al., "Determinants of oligosaccharide specificity of the carbohydrate-binding modules of AMP-activated protein kinase," (in eng), Biochem J, vol. 468, no. 2, pp. 245-57, Jun 01 2015. [CrossRef]
- X. Li et al., "Structural basis of AMPK regulation by adenine nucleotides and glycogen," (in eng), Cell Res, vol. 25, no. 1, pp. 50-66, Jan 2015. [CrossRef]
- Koay et al., "AMPK beta subunits display isoform specific affinities for carbohydrates," (in eng), FEBS Lett, vol. 584, no. 15, pp. 3499-503, Aug 04 2010. [CrossRef]
Figure 1.
Weight over time in WT mice, Agl-/- (KO) mice injected intramuscularly biweekly with 10% DMSO solvent control, and KO mice injected in the same way with 250 mg/kg GHF-201.
Figure 1.
Weight over time in WT mice, Agl-/- (KO) mice injected intramuscularly biweekly with 10% DMSO solvent control, and KO mice injected in the same way with 250 mg/kg GHF-201.
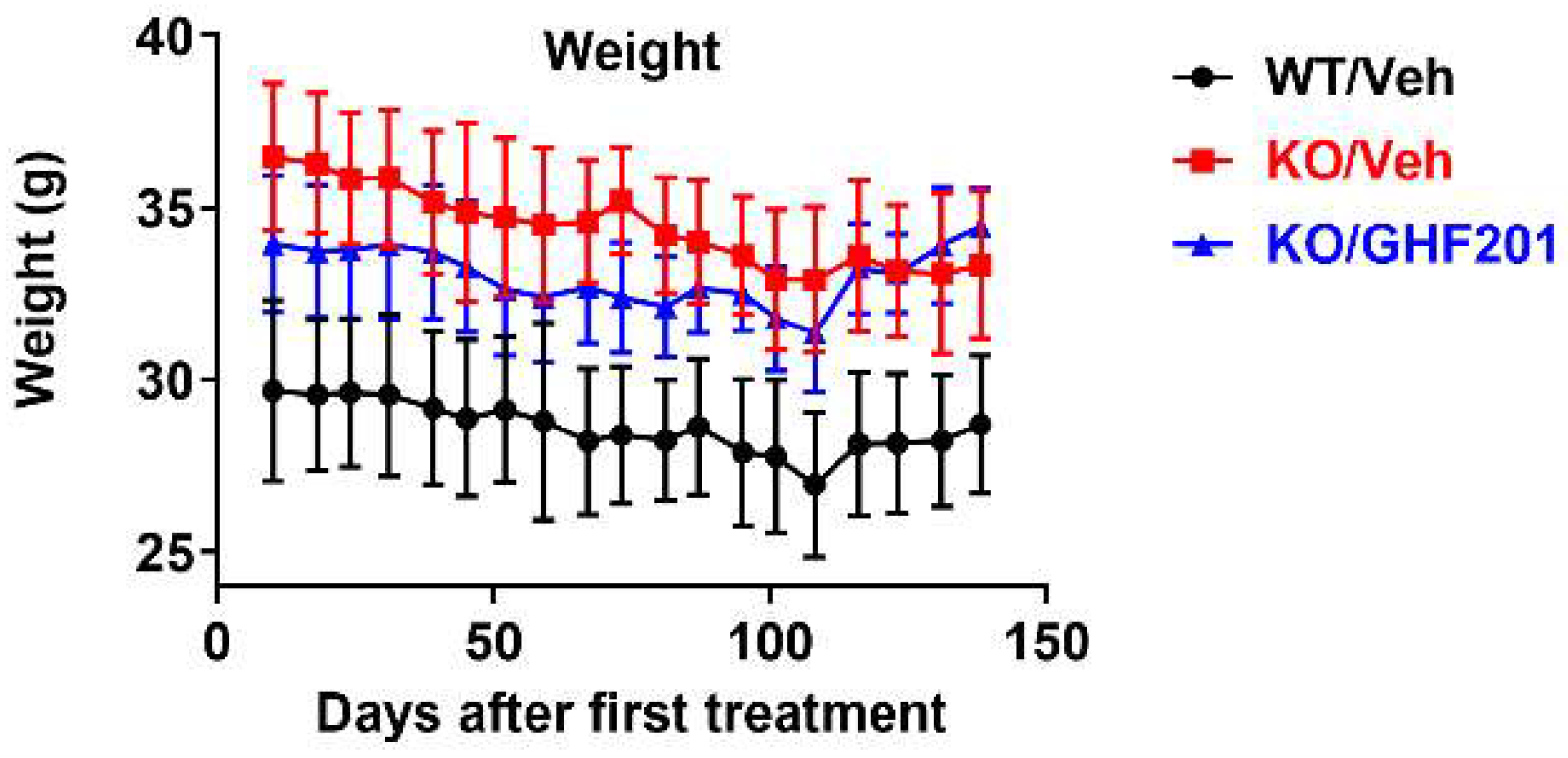
Figure 2.
Movement distance (A), sojourn in the center of open field arena (inset, representative open field sojourn heatmaps) (B), grip strength (C) and latency to fall from a rotating rod (D) were measured in n=8 Agl-/- mice treated with 10% DMSO vehicle (KO/Veh), n=6 wild type mice treated with vehicle (WT/Veh), and n=10 Agl-/- mice treated with GHF-201 (KO/GHF-201). In (D), each rotarod session included three consecutive runs (1-3), separated by 10 min pauses. Two-way ANOVA with repeated measures shows that only in (C) GHF-201 treatment values were higher than vehicle-treated values in KO mice throughout the period (p<0.05). *, KO/GH201 is significantly different from KO/Veh; *#, KO/GHF-201 is significantly different from both KO/Veh and WT/Veh; *^, KO/GHF-201 is significantly different only from KO/Veh and not from WT/Veh (correction effect). Difference significance determined by multiple t-tests with Sidak post-hoc correction. All error bars represent s.e.m.
Figure 2.
Movement distance (A), sojourn in the center of open field arena (inset, representative open field sojourn heatmaps) (B), grip strength (C) and latency to fall from a rotating rod (D) were measured in n=8 Agl-/- mice treated with 10% DMSO vehicle (KO/Veh), n=6 wild type mice treated with vehicle (WT/Veh), and n=10 Agl-/- mice treated with GHF-201 (KO/GHF-201). In (D), each rotarod session included three consecutive runs (1-3), separated by 10 min pauses. Two-way ANOVA with repeated measures shows that only in (C) GHF-201 treatment values were higher than vehicle-treated values in KO mice throughout the period (p<0.05). *, KO/GH201 is significantly different from KO/Veh; *#, KO/GHF-201 is significantly different from both KO/Veh and WT/Veh; *^, KO/GHF-201 is significantly different only from KO/Veh and not from WT/Veh (correction effect). Difference significance determined by multiple t-tests with Sidak post-hoc correction. All error bars represent s.e.m.
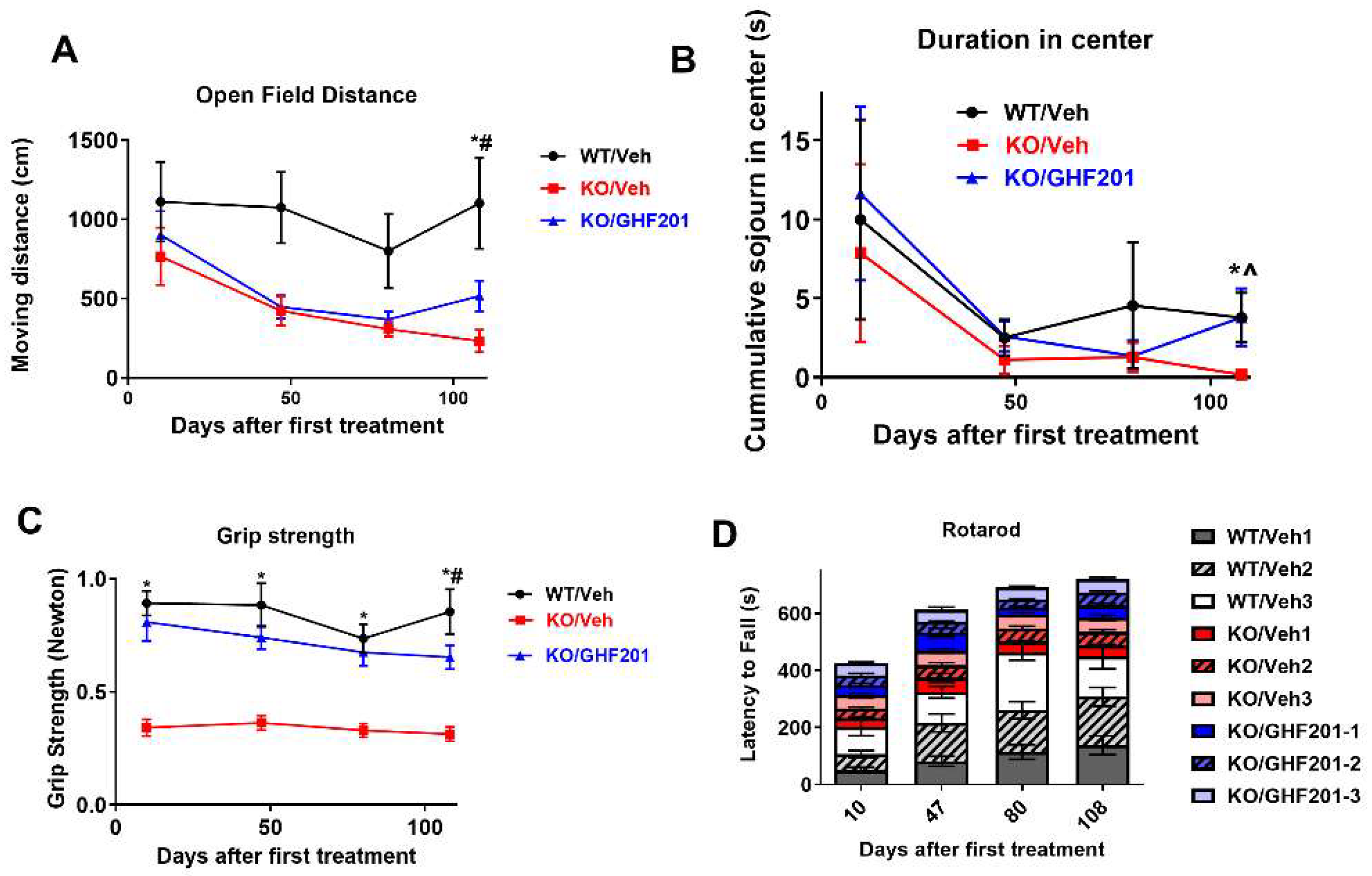
Figure 3.
Blood metabolic panel based on n=8 Agl-/- mice treated with 10% DMSO vehicle (KO/Veh), n=6 wild type mice treated with vehicle (WT/Veh), and n=10 Agl-/- mice treated with GHF-201, as indicated. At all time points, GHF-201 significantly reduced alanine transferase and creatine phosphokinase, demonstrating partial restoration of liver and muscle damages respectively. Additionally, blood glucose was increased and blood triglycerides were decreased by GHF-201 in treated Agl-/- mice. *, significant difference v KO/Veh determined by two-tailed t-tests.
Figure 3.
Blood metabolic panel based on n=8 Agl-/- mice treated with 10% DMSO vehicle (KO/Veh), n=6 wild type mice treated with vehicle (WT/Veh), and n=10 Agl-/- mice treated with GHF-201, as indicated. At all time points, GHF-201 significantly reduced alanine transferase and creatine phosphokinase, demonstrating partial restoration of liver and muscle damages respectively. Additionally, blood glucose was increased and blood triglycerides were decreased by GHF-201 in treated Agl-/- mice. *, significant difference v KO/Veh determined by two-tailed t-tests.
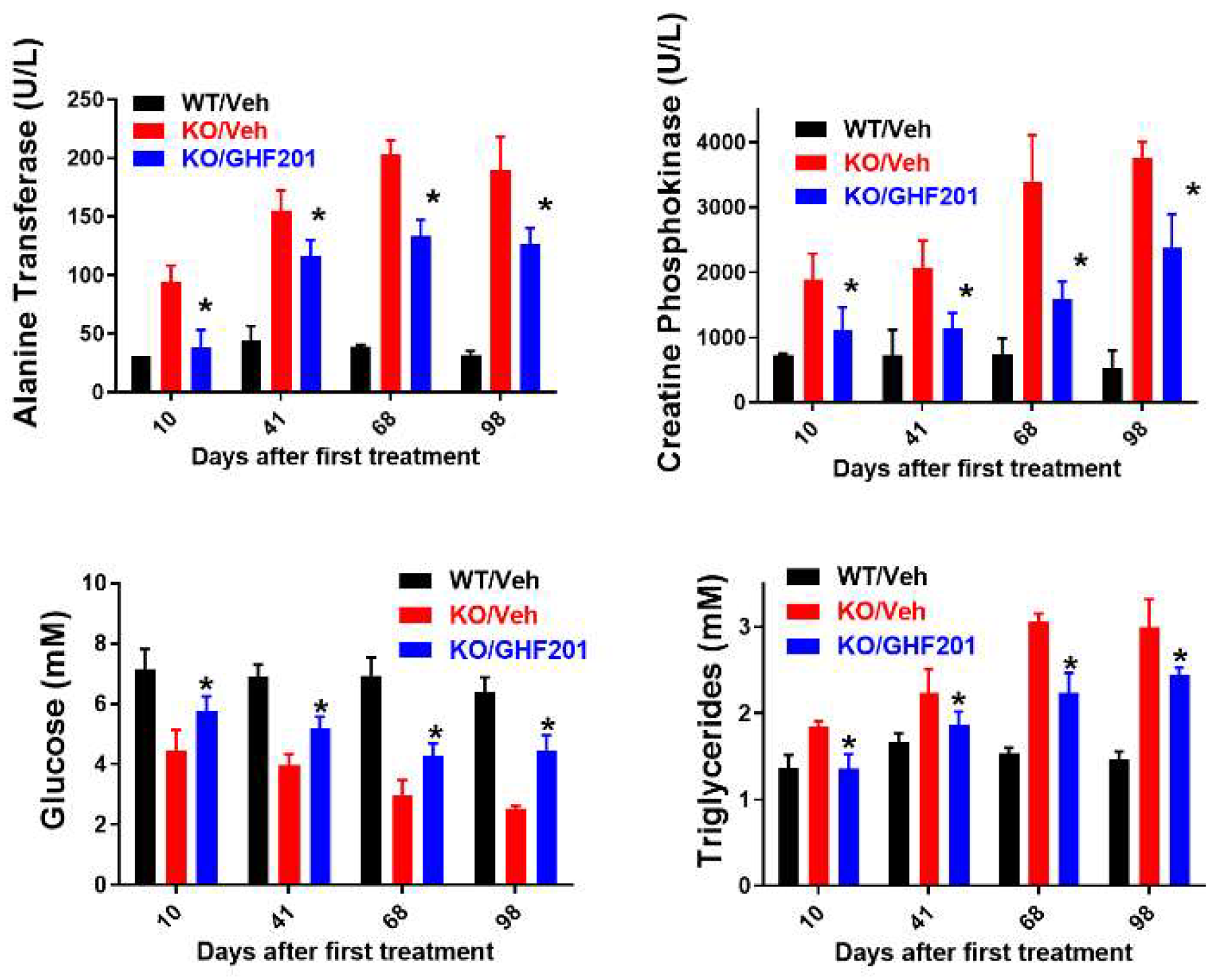
Figure 4.
(next page). Indirect calorimetry and metabolic cage analysis. Following a 48 h habituation period, mice (n=4 from each study arm indicated) were monitored over a 24-hr period. Data are mean±SEM from eight-month-old mice. (A) Vehicle-treated Agl-/- mice demonstrate lower respiratory exchange ratio (RER), total energy expenditure (TEE), and carbohydrate and fat oxidation. These parameters were increased or corrected by GHF-201 where indicated. ANCOVA results suggest that weight-dependent TEE is reduced in Agl-/- compared to WT mice in both light conditions and overall, and that GHF-201 corrects this TEE reduction in Agl-/- mice only in the light. (B) Ambulatory activity and wheel running were also increased or corrected by GHF-201 as indicated. (C) Food and water intake (total cumulative values are shown in the bar graphs) were increased, but not significantly, mainly in the dark period by GHF-201. *#, Significant difference between KO/GHF-201 and both KO/Veh and WT/Veh. *^, Significant difference between KO/GHF-201 and KO/Veh, but not between KO/GHF-201 and WT/Veh.
Figure 4.
(next page). Indirect calorimetry and metabolic cage analysis. Following a 48 h habituation period, mice (n=4 from each study arm indicated) were monitored over a 24-hr period. Data are mean±SEM from eight-month-old mice. (A) Vehicle-treated Agl-/- mice demonstrate lower respiratory exchange ratio (RER), total energy expenditure (TEE), and carbohydrate and fat oxidation. These parameters were increased or corrected by GHF-201 where indicated. ANCOVA results suggest that weight-dependent TEE is reduced in Agl-/- compared to WT mice in both light conditions and overall, and that GHF-201 corrects this TEE reduction in Agl-/- mice only in the light. (B) Ambulatory activity and wheel running were also increased or corrected by GHF-201 as indicated. (C) Food and water intake (total cumulative values are shown in the bar graphs) were increased, but not significantly, mainly in the dark period by GHF-201. *#, Significant difference between KO/GHF-201 and both KO/Veh and WT/Veh. *^, Significant difference between KO/GHF-201 and KO/Veh, but not between KO/GHF-201 and WT/Veh.
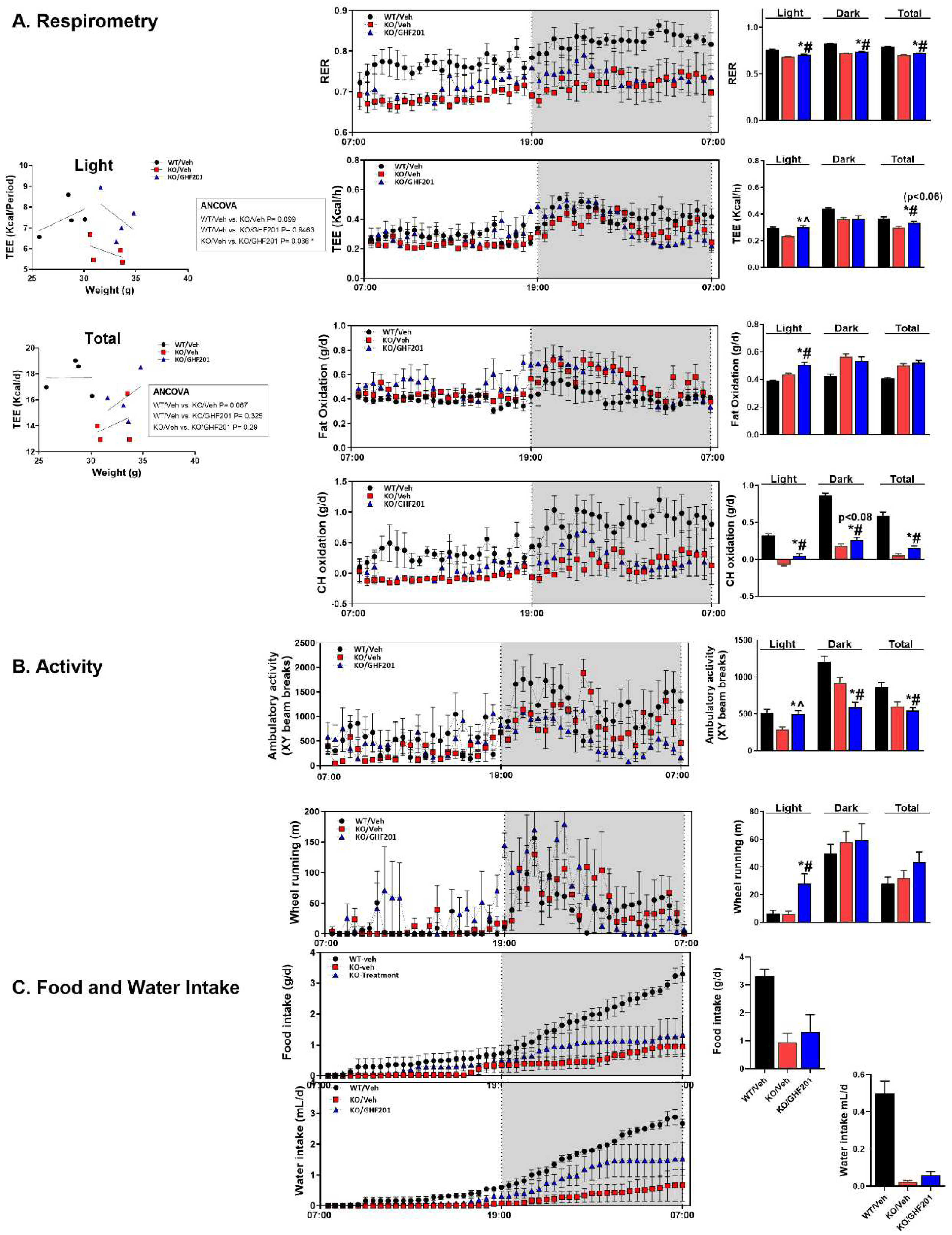
Figure 5.
(A) Transmission electron microscopy images of longitudinal sections of gastrocnemius muscle collected from 8-month-old animals treated as indicated. Note granular glycogen material (arrow) in sections from untreated Agl-/- mouse. Also note variable width of sarcomeres in the Agl-/- mouse sample. These ultrastructural phenotypes were partially corrected in the GHF-201-treated Agl-/- mouse. (B) Quantification of area % of glycogen based on analysis of TEM images.
Figure 5.
(A) Transmission electron microscopy images of longitudinal sections of gastrocnemius muscle collected from 8-month-old animals treated as indicated. Note granular glycogen material (arrow) in sections from untreated Agl-/- mouse. Also note variable width of sarcomeres in the Agl-/- mouse sample. These ultrastructural phenotypes were partially corrected in the GHF-201-treated Agl-/- mouse. (B) Quantification of area % of glycogen based on analysis of TEM images.
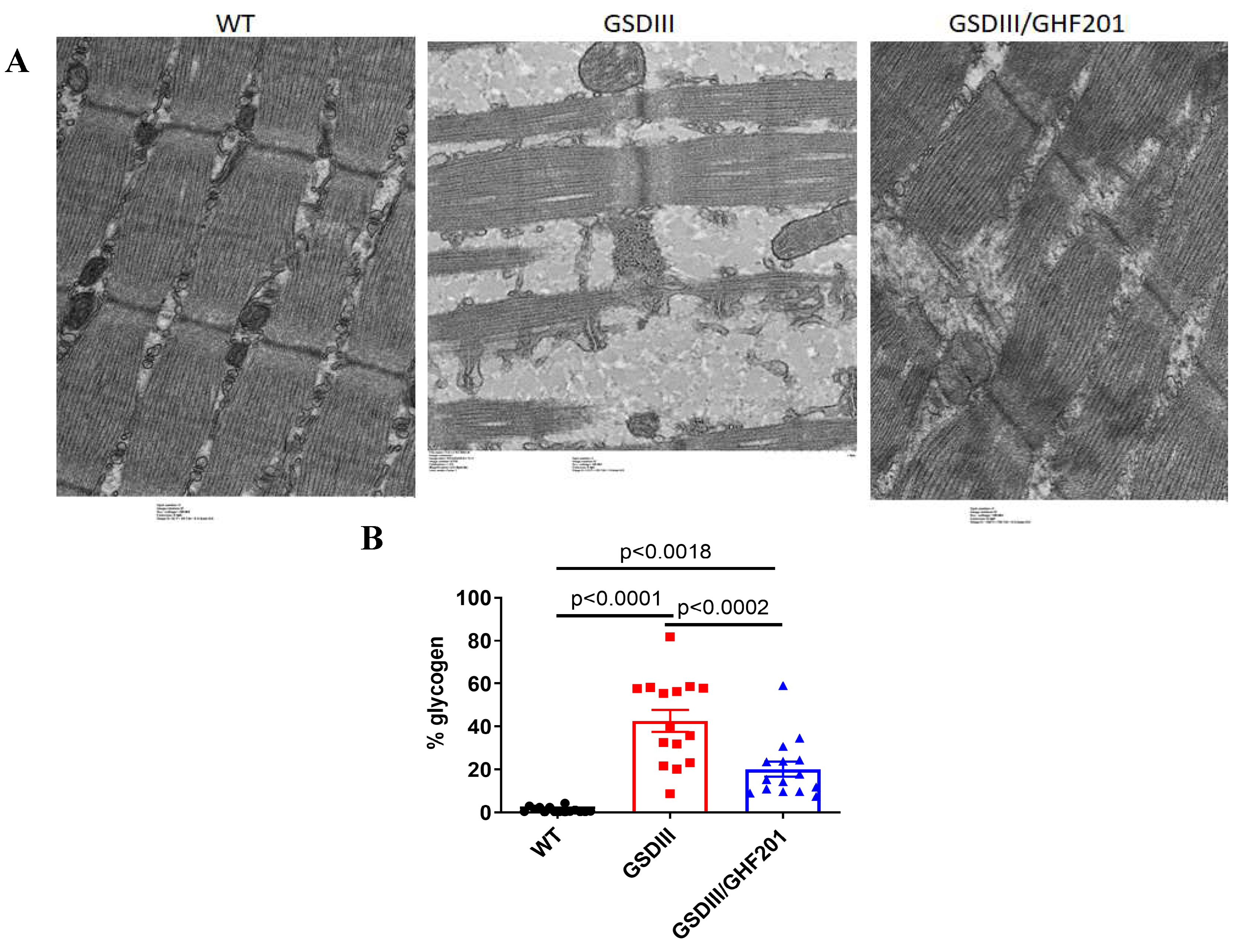
Figure 6.
Skin fibroblasts from 3 GSD3 and 3 HC individuals were cultured in 96 well plates for 48 h in starvation medium without FBS and glucose. Subsequently cells were live-stained with (A) PAS reagent to quantify intracellular glycogen, or (B) a mix of fluorescent dyes which included Hoechst, Calcein-AM, TMRE, and Lysortracker to respectively stain nuclei (blue), cytoplasm (green), respiring mitochondria (yellow) and lysosomes (red). Multiple images from the live-stained cells were automatically obtained by an Operetta G1 image analyzer under environmental controlled conditions. Shown are representative images and their quantification in relative units.
Figure 6.
Skin fibroblasts from 3 GSD3 and 3 HC individuals were cultured in 96 well plates for 48 h in starvation medium without FBS and glucose. Subsequently cells were live-stained with (A) PAS reagent to quantify intracellular glycogen, or (B) a mix of fluorescent dyes which included Hoechst, Calcein-AM, TMRE, and Lysortracker to respectively stain nuclei (blue), cytoplasm (green), respiring mitochondria (yellow) and lysosomes (red). Multiple images from the live-stained cells were automatically obtained by an Operetta G1 image analyzer under environmental controlled conditions. Shown are representative images and their quantification in relative units.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



