
Submitted:
06 June 2024
Posted:
07 June 2024
You are already at the latest version
Abstract
A number of chemical modifications were carried out in order to synthesize new compounds based on the triterpenoid betulonic acid. Triazolation reactions were carried out for the synthesis of new 1,2,3-triazole derivatives based on betulonic acid. Chemical changes were made on the carbonyl group at the C-3 position of the initial molecule and on the carboxyl and hydroxyl groups at the C-28 position. Chromatographic methods were used to separate and purify the formed products. Spectroscopic (NMR-, mass-) methods were used to establish the structure of synthesized compounds. As a result of the chemical transformations carried out, a number of new derivatives of betulonic acid were obtained. All synthesized compounds were tested for antimicrobial and cytotoxic activity. As a result, compounds with pronounced antimicrobial activity were identified.
Keywords:
betulonic acid
; triterpenoid
; lupan
; natural product
; chemical transformation
; synthesis
; spectroscopic method
; cytotoxicity
; antimicrobial activity
1. Introduction
Terpenoids isolated from plant sources remain the starting material for various chemical transformations in terms of obtaining compounds with useful properties [1,2,3,4,5,6,7,8]. Terpenoid derivatives with cytotoxic, anti-inflammatory, analgesic, et al activity have been synthesized by chemical transformation [9,10,11,12,13,14,15,16]. The chemical transformation of triterpenoids makes it possible to solve various theoretical issues and search for promising ways of new biologically active compounds. Currently, it has been proven that triterpenoids and their derivatives are powerful agents of natural origin and can be used to create medicines.
The presence of functional groups such as hydroxyl, keto, carboxyl, double bonds in the triterpenoid molecule betulonic acid determines the possibilities of chemical modification of the molecule of this triterpenoid.
Our current aim was to carry out a number of chemical transformations based on betulonic acid and synthesize a number of new derivatives and study of the biological activity. In this regard, we have obtained new derivatives of betulonic acid.
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of Triazole Derivatives Based on Betulonic Acid
In order to synthesize new triazole derivatives of betulonic acid, triazolation reactions were carried out and a number of new derivatives were obtained. First, betulonic acid 2 was obtained by oxidizing betulin 1, which is found in natural sources.
Betulonic acid 2 was dissolved in toluene and the corresponding primary amine and 4-nitrophenylazide were added. The reaction was carried out with an interval of 100o C. The yield of the formed products was in the range of 60 - 68.3 %.
Scheme 1.
Reagent and conditions: a) Jones reagent, acetone, RT; b) Primary amine, 4-phenylnitroazide, toluene, 24 h, 100oC.
Scheme 1.
Reagent and conditions: a) Jones reagent, acetone, RT; b) Primary amine, 4-phenylnitroazide, toluene, 24 h, 100oC.
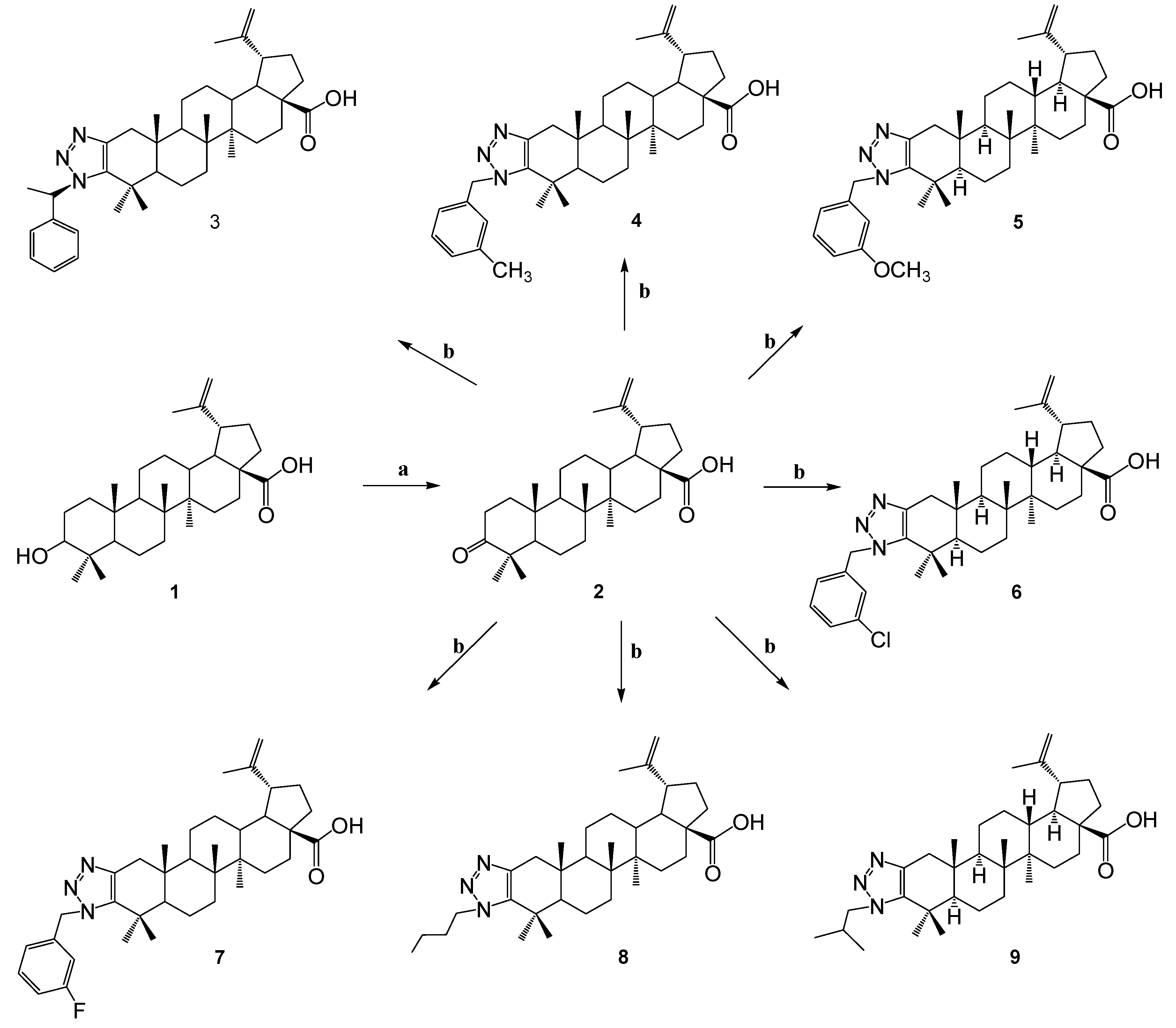
In the 1H NMR spectra of the synthesized betulonic acid triazole derivatives 3-7, the signals of the triazole residue were shown in the area δ 7.33-5.61 p.m. and in the spectrum of 8, 9 compounds, it is observed that additional signals are formed in comparison with the initial molecule, the signals are clearly shown in the area δ 2.91, 4.29 p.m. for 8, δ 2.92, 4.09 p.m. for 9. Loss of characteristic signal of the carbonyl group at the C-3 position of the initial molecule in the 13C NMR spectra of the resulting compounds, evidence of a triazolation reaction. The molecular ion peak [M+H]+ is present in the mass spectra of compounds.
2.1.2. Synthesis of Buthyl-Triazole Derivatives Based on Betulonic Acid
In order to continue obtaining new derivatives of betulonic acid, the triazolation reaction was carried out in the presence of ammonium acetate, as a result of which the compound 10 was obtained. Then the compound was dissolved in a methanol and brombutane and potassium tret-butoxide (PTB) were added to it. As a result, a mixture of two substances 11 and 12 was formed. In this case, it has been observed that a carboxyl group proton exchange situation occurs. The yield of the formed compounds was 46.4 and 20.3%, respectively.
By analyzing the spectral data and taking into account the molecular ionic peak in the mass spectra of the compounds, it was proved that molecules have the structures shown in Scheme 2. In 1H NMR spectrum of the compound 11, the signals of the protons of the exomethylene group at the C-20 position is presented in the area δ 4.76 and 4.63 p.m., for 12 in the area δ 4.75 and 4.61 p.m. The presence of butyl residue at C-28 is evidenced by the shift of the carbonyl group signal in 13C NMR spectrum to a strong field compared to molecule 10.
2.1.3. Synthesis of N-Derivatives of Betulonic Acid
To synthesize N-derivatives of betulonic acid, a number of triazole derivatives 3,5,7,8 were further reacted with 1,1'-carbonyldi-(1,2,4-triazole) in a THF. As a result colorless crystalline substances 13-16 were obtained.
Scheme 3.
Reagent and conditions: a) CDT, THF, 70oC.
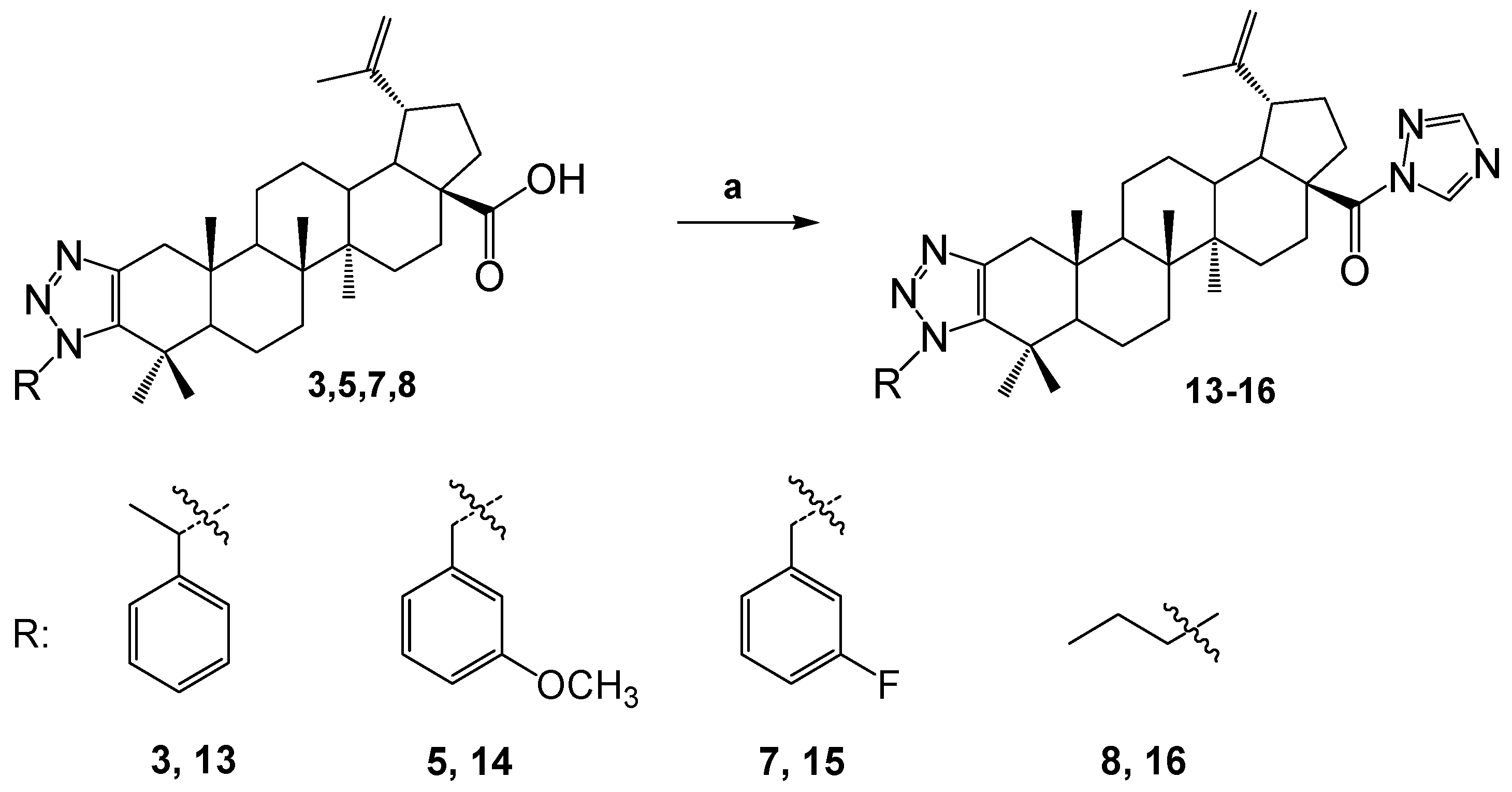
In 1NMR spectra of compounds, protons in the triazole ring at the C-28 position are observed as singlets at δ 8.93 and 7.99 p.m. for 13, δ 8.92 and 7.99 p.m. for 14, 15, and δ 8.93 and 8.00 p.m. for 16. In 13C NMR spectra, it is observed that due to the formation of a triazole ring in place C-28, the signal of the carbonyl group is shifted to a weak field, that is, it is manifested in regions δ 173.38, 173.39, 173.38, 173.39 p.m., respectively.
2.1.4. Synthesis of N-Derivatives of Phenylethyl-Triazole-Betulonic Acid
In plans to continue the synthesis of new compounds based on betulonic acid, the compound 3 reacted with diphenylphosphoryl azide (DPPA) in a toluene in the presence of triethylamine. The resulting carbamide derivative 17 is included in the further reaction. Then the corresponding amines were added. As a result, high-yield colorless crystalline substances 18-22 were obtained.
The structure of the synthesized compounds was determined by spectroscopic and high-resolution mass spectrometry methods.
Scheme 4.
Reagent and conditions: a) DPPA, toluene, Et3N; b) Amine, toluene, 110oC.
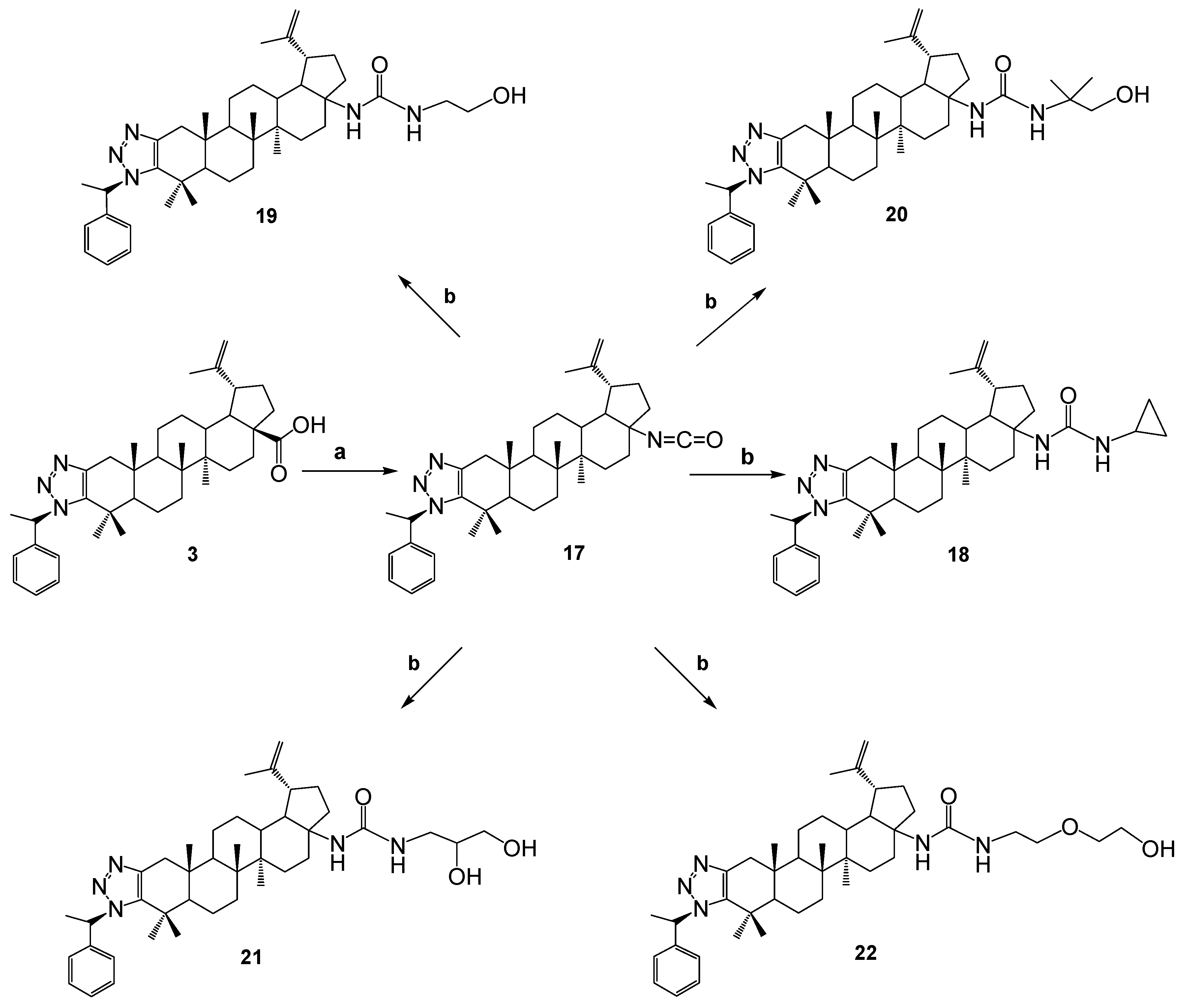
All synthesized compounds were tested for antimicrobial and cytotoxic activity.
2.2. Evaluation of the Biological Activity
2.2.1. Antimicrobial Activity
The antimicrobial activity of the samples was studied on recommended reference test microorganisms, facultative anaerobic gram-positive cocci Staphylococcus aureus ATCC 6538, aerobic gram-positive spore-forming Bacillus subtilis ATCC 6633, gram-negative bacillus facultative anaerobes Escherichia coli ATCC 25922 aerobic Pseudomonas aeruginosa ATCC 27853 and k yeast fungus Candida albicans ATCC 10231 by serial dilution with determination of the minimum inhibitory concentration (MIC). The test strains of microorganisms used in the study were obtained from the American collection of type cultures.
The results of the study of the antimicrobial activity of samples by serial dilution are shown in Table 1.
As a result of an antimicrobial study, it was found that the test samples exhibit antimicrobial activity against gram-positive test strains of Staphylococcus aureus, Bacillus subtilis, and gram-negative Escheriсhia coli, for which their minimum inhibitory concentration is between 12.5 and 50 µg/ml.
At the same time, of all the samples presented, the compound 3 and 18 showed simultaneously moderately pronounced anti-bacterial activity both against the gram-positive test strain Staphylococcus aureus ATCC 6538 and against the gram-negative test microorganism Escherichia coli, their minimally inhibitory concentrations were in relation to the test strains are 12.5 µg/ml.
The compound 6 showed pronounced antimicrobial activity against the gram-positive test strain Staphylococcus aureus ATCC 6538, and the compound 7 showed pronounced antimicrobial activity against the gram-negative test microorganism Escherichia coli ATCC 25922, their MIC = 6.3 µg/ml.
Samples 4, 7, 8, 13, 15, 18, 19, 21 showed gram-positive test strains of Staphylococcus aureus ATCC 65 showed moderate antibacterial activity, their MIC were 12.5 and 25 µg/ml.
For the Bacillus subtilis test strains ATCC 6633 and Escherichia coli ATCC 25922, the minimum inhibitory concentration of a number of tested compounds is in the range of 12.5-50 µg/ml.
At the same time, samples 3 and 18 showed antibacterial activity against the gram-negative test strain of Escherichia coli higher than the rest samples (MIC=12.5 µg/ml).
The compounds 2, 3, 5, 7, 19 and 20 showed antifungal action against the yeast-like fungus Candida albicans ATCC 10231, in concentrations of 25-50 µg/ml.
2.2.2. Cytotoxic Activity
These synthesized new compounds were subjected to the Artemia salina lethality test (Leach) in order to detect potential sources of new cytotoxic antitumor compounds. Larvicidal activity based on the percentage of larval mortality was evaluated after 24-hour exposure to treatments.
According to Meyer et al. [17], who classified the substances into toxic (LC50 value < 1000 mcg/ml) and nontoxic (LC50 value > 1000 mcg/ml), almost all tested compounds showed good cytotoxic activity of artemia compared to the reference compound.
The cytotoxicity of the compounds was evaluated in the survival test of larvae of Artemia salina (Leach) crustaceans in vitro cultivation conditions [17,18].
The results of testing the cytotoxic activity of samples against larvae of Artemia salina (Leach) crustaceans under in vitro cultivation conditions are shown in Table 2.
As a result of the study, it was found that the samples of the tested compounds were 2-4, 6-10, 13-15, 18-20, 22 exhibit cytotoxic activity against larvae of marine crustaceans Artemia salina (Leach).
These samples can be considered as potential candidates for an antitumor compound of plant origin. It has been found that these compounds can be sources of cytotoxic and antitumor compounds.
The pronounced antibacterial activity demonstrated in this study for some compounds indicates them as potential antimicrobial agents active against common microbial pathogens causing various socially significant infectious diseases.
3. Materials and Methods
3.1. General Chemistry Section
The 1H and 13C spectra of the compounds were measured on Bruker AMX 400 MHz instrument. In NMR spectra, tetramethylsilane and NMR solvent in some spectra were used as internal standards. The melting points were determined using the Reichert Thermovar. For column chromatography 70−230 mesh silica 60 (E.M. Merck) was used as the stationary phase. The dichlorometane, petroleum ether, ethylacetate were used as an eluent. The TLC (thin-layer chromatography) was checked on silica gel 0.20 mm 60 with a mixture of ethanol and sulfuric acid (10 ml of sulfuric acid and 90 ml of ethanol), and also used UV. Chemicals from commercial sources were used without further purification. Reaction dry solvents (toluene, THF, DMF, CH2Cl2) from commercial sources were used. UHPLC-ESI-Q-TOF-MS analyses of small molecules were performed using a Dionex Ultimate 3000 UHPLC coupled to a Zorbax RRHP Eclipse Plus C18 column (2.1x100mm, 1.8 µL) connected to a Bruker Impact II QTOF mass spectrometer. Mobile phases consisted of water (A) and acetonitrile (B), each supplemented with 0.1% formic acid. The following gradient was used at a flow rate of 0.200 mL/min: 0–2.5 min 5% B, 2.5–14 min 5-100% B, 14-19 min 100% B, 19-20.4 min 100-5% B, 20.4-25 min 5% B. The mass spectrometer was operated in positive ion mode with a scan range of 50-3000 m/z. Source conditions were: end plate offset at −500 V; capillary at −4500 V; nebulizer gas (N2) at 1.6 bar; dry gas (N2) at 8 L min−1; dry temperature at 180 °C. Ion transfer conditions were: ion funnel RF at 200 Vpp; multiple RF at 200 Vpp; quadrupole low mass at 55 m/z; collision energy at 5.0 eV; collision RF at 600 Vpp; ion cooler RF at 50–350 Vpp; transfer time at 121 μs; pre-pulse storage time at 1 μs. Calibration was performed with 1 mM sodium formate through a loop injection of 20 μL at the start of each run. Also, mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3uL/min and spectra were obtained in positive (or: negative) ionization mode with a resolution of 15000 (FWHM) using leucine enkephalin as lock mass.
3.2. Experimental Section
3-Oxo-lup-20(29)-en-28-oic Acid (Betulonic Acid, 2). Betulin (5.0 g, 11.3 mmol) was placed in a 0.5 l flask and filled with acetone (150 ml) and dissolved in an ultrasonic water bath. The freshly prepared Jones reagent (6.65 g Na2Cr2O7 and 6 ml H2SO4 in 50 ml water) was added drop by drop to the cooled (on an ice bath) solution. The color of the solution began to change. The reaction mixture was allowed to heat up to room temperature and continued to be stirred for 5 hours. The course of the reaction was checked by the TLC. Then MeOH (100 ml) was first added to the reaction mixture, and then water (50 ml). The resulting precipitate was filtered and washed with water (50 ml). The crude product was dried, then dissolved in Et2O (60 ml) and washed with water (30 ml), 7.5% hydrochloric acid (20 ml), water (20 ml), saturated aqueous solution NaHCO3 (20 ml) and water (20 ml). The ether layer was driven off on a rotary evaporator, and the remaining residue was purified by column chromatography (silica gel), a mixture of heptane and ethyl acetate (80:20) was used as an eluent. Compound 2 is collorless powder. The product yield is 1.55 g (30.2%).
1H NMR (400 MHz, CDCl3): δ 4.74 (d, J = 2.3 Hz, 1H), 4.66 – 4.58 (m, 1H), 3.01 (td, J = 10.7, 4.6 Hz, 1H), 2.56 – 2.35 (m, 2H), 2.33 – 2.17 (m, 2H), 2.07 – 1.83 (m, 2H), 1.70 (s, 4H), 1.64 (t, J = 11.4 Hz, 1H), 1.59 – 1.14 (m, 14H), 1.13 – 1.04 (m, 4H), 1.03 – 0.95 (m, 9H), 0.93 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 218.23, 181.72, 150.32, 109.79, 56.37, 54.94, 49.85, 49.19, 47.34, 46.89, 42.49, 40.64, 39.61, 38.51, 37.04, 36.92, 34.13, 33.60, 32.10, 30.55, 29.68, 26.64, 25.49, 21.37, 21.00, 19.63, 19.37, 15.96, 15.82, 14.63.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28 oic Acid (3). Betulonic acid (1.0 g, 1 equiv, 2.2 mmol), 4-nitrophenyl azide (360 mg, 1.3 equiv, 2.86 mmol), (S)- (‒)-α-methylbenzylamine (350 mg, 1.3 equiv, 2.86 mmol) and 4 Å molecular sieves (200 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (5 ml) and the reaction mixture was stirred at 100 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 3 is powdery with a yellowish tinge, m.p. 325-327 oC. Yield 790 mg (61 %).
1H NMR (400 MHz, CDCl3): δ 7.33 – 7.25 (m, 3H), 7.25 – 7.19 (m, 2H), 5.72 (q, J = 7.0 Hz, 1H), 4.77 (d, J = 2.3 Hz, 1H), 4.64 (m, 1H), 3.03 (td, J = 10.6, 4.5 Hz, 1H), 2.96 (d, J = 15.3 Hz, 1H), 2.32 – 2.20 (m, 2H), 2.15 (d, J = 15.3 Hz, 1H), 2.05 – 1.95 (m, 5H), 1.81-1.60 (m, 5H), 1.61 – 1.32 (m, 11H), 1.26 (s, 5H), 1.10 (s, 3H), 0.99 (s, 6H), 0.81 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.07, 150.22, 141.79, 141.05, 137.53, 128.64, 127.56, 126.24, 109.88, 59.28, 56.38, 54.78, 49.34, 49.21, 46.90, 42.45, 40.60, 38.91, 38.53, 38.32, 37.04, 33.80, 33.41, 32.08, 30.59, 29.80, 28.89, 25.50, 23.73, 21.36, 21.31, 19.42, 18.91, 16.25, 15.70, 14.65.
1′-(3-Methylbenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (4). Betulonic acid 2 (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv, 0.44 mmol), 3-methylbenzylamine (74.6 mg, 2.8 equiv, 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 4 is pale yellow powder, m.p. 261-264 oC. Yield 71.7 mg (68.3 %).
1H NMR (400 MHz, CDCl3): δ 7.17 (t, J = 7.6 Hz, 1H), 7.06 (d, J = 7.6 Hz, 1H), 6.85 (s, 1H), 6.81 (d, J = 7.7 Hz, 1H), 5.62 (m, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.64 (m, 1H), 4.12 (q, J = 7.2 Hz, 0H), 3.04 (td, J = 10.6, 4.5 Hz, 1H), 2.96 (d, J = 15.3 Hz, 1H), 2.25 (m, 6H), 1.99 (m, 2H), 1.77 (d, J = 12.8 Hz, 1H), 1.71 (s, 3H), 1.45 (m, 13H), 1.17 (s, 3H), 1.04 (s, 3H), 0.99 (d, J = 9.2 Hz, 6H), 0.90 (m, 1H), 0.78 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.38, 150.25, 141.84, 138.49, 138.00, 136.38, 128.59, 128.52, 127.03, 123.46, 109.86, 56.40, 54.59, 52.81, 49.29, 49.19, 46.90, 42.44, 40.56, 38.97, 38.51, 38.36, 37.04, 33.71, 33.35, 32.08, 30.58, 29.79, 28.77, 25.50, 21.41, 21.37, 21.31, 19.42, 18.91, 16.08, 15.70, 14.65.
HRMS (ESI+): m/z calculated for C38H54N3O2 [M+H]+: 584.42160, found: 584.4216.
1′-(3-Methoxybenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (5). Betulonic acid (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv, 0.44 mmol), 3-methoxybenzylamine (84.5 mg, 2.8 equiv, 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 18 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 5 is powdery with a yellowish tinge, m.p. 205-208oC. Yield 78 mg (60 %).
1H NMR (400 MHz, CDCl3): δ 7.21 (td, J = 7.9, 3.3 Hz, 1H), 6.83 – 6.75 (m, 1H), 6.65 – 6.54 (m, 3H), 5.62 (d, J = 3.8 Hz, 3H), 4.76 (d, J = 2.3 Hz, 1H), 4.67 – 4.63 (m, 1H), 3.74 (d, J = 1.6 Hz, 4H), 3.09 – 2.92 (m, 2H), 2.26 (ddd, J = 15.5, 9.7, 3.5 Hz, 2H), 2.21 – 2.14 (m, 1H), 2.06 – 1.82 (m, 3H), 1.81 – 1.75 (m, 2H), 1.71 (s, 3H), 1.68 (s, 0H), 1.61 – 1.36 (m, 10H), 1.35 – 1.18 (m, 4H), 1.17 (d, J = 2.6 Hz, 3H), 1.04 (s, 3H), 1.02 – 0.96 (m, 8H), 0.81 (s, 1H), 0.77 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 180.81, 159.97, 150.24, 141.89, 138.09, 138.04, 129.78, 118.65, 113.31, 111.96, 109.87, 56.36, 55.23, 54.79, 54.59, 52.74, 52.66, 49.30, 49.19, 46.88, 44.09, 42.45, 40.56, 40.36, 38.98, 38.48, 38.37, 37.03, 33.72, 33.35, 32.07, 30.57, 29.79, 28.73, 25.50, 21.38, 21.31, 19.42, 18.91, 16.08, 15.71, 15.19, 14.65.
HRMS (ESI+): m/z calculated for C38H54N3O3 [M+H]+: 600.4160, found: 600.4194.
1′-(3-Chlorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (6). Betulonic acid 2 (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv, 0.44 mmol), 3-chlorobenzylamine (87 mg, 2.8 equiv, 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (1:1) was used as an eluent. Compound 6 is pale yellow powder, m.p. 288-290 oC. Yield 65 mg (62 %).
1H NMR (400 MHz, CDCl3): δ 7.28 – 7.19 (m, 2H), 7.03 (d, J = 2.0 Hz, 1H), 6.89 (dt, J = 5.7, 2.1 Hz, 1H), 5.61 (d, J = 5.5 Hz, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.64 (t, J = 1.9 Hz, 1H), 3.04 (td, J = 10.6, 4.6 Hz, 1H), 2.96 (d, J = 15.4 Hz, 1H), 2.28 (ddd, J = 14.7, 9.0, 3.2 Hz, 2H), 2.23 – 2.15 (m, 1H), 2.00 (ddd, J = 17.8, 11.6, 5.5 Hz, 2H), 1.83 – 1.73 (m, 1H), 1.71 (s, 3H), 1.67 – 1.19 (m, 13H), 1.17 (s, 4H), 1.06 – 0.95 (m, 9H), 0.78 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.63, 150.23, 142.05, 130.06, 128.07, 126.53, 124.55, 109.87, 56.41, 54.51, 52.16, 49.27, 49.20, 46.91, 42.44, 40.55, 38.98, 38.53, 38.29, 37.03, 33.69, 33.31, 32.07, 30.58, 29.80, 28.83, 25.47, 21.41, 21.37, 19.42, 18.88, 16.09, 15.73, 14.64.
HRMS (ESI+): m/z calculated for C37H51ClN3O2 [M+H]+: 604.36698, found: 604.3661.
1’-(3-Fluorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (7). Betulonic acid (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv, 0.44 mmol), 3-fluorobenzylamine (77 mg, 2.8 equiv, 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 22 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 7 is powdery with a yellowish tinge, m.p. 302-305 oC. Yield 82 mg (64 %).
1H NMR (400 MHz, CDCl3): δ 7.27 (d, J = 5.5 Hz, 2H), 7.00 – 6.92 (m, 1H), 6.81 (ddd, J = 7.8, 1.8, 0.9 Hz, 1H), 6.71 (dt, J = 9.6, 2.2 Hz, 1H), 5.68 – 5.58 (m, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.67 – 4.62 (m, 1H), 3.03 (td, J = 10.6, 4.6 Hz, 1H), 2.96 (d, J = 15.4 Hz, 1H), 2.27 (td, J = 12.7, 11.3, 3.3 Hz, 2H), 2.23 – 2.15 (m, 1H), 2.07 – 1.93 (m, 2H), 1.77 (dd, J = 11.4, 4.1 Hz, 1H), 1.71 (s, 3H), 1.66 (t, J = 11.4 Hz, 1H), 1.62 – 1.33 (m, 9H), 1.33 – 1.18 (m, 3H), 1.17 (s, 3H), 1.04 (s, 3H), 0.99 (d, J = 8.9 Hz, 6H), 0.78 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.18, 150.22, 142.05, 138.10, 130.41, 130.32, 121.99, 113.63, 113.40, 109.88, 56.38, 54.53, 52.24, 49.28, 49.20, 46.91, 42.45, 40.56, 38.98, 38.53, 38.31, 37.02, 33.69, 33.32, 32.07, 30.58, 29.80, 28.78, 25.48, 21.37, 19.42, 18.89, 16.10, 15.73, 14.65.
HRMS (ESI+): m/z calculated for C37H51FN3O2 [M+H]+: 588.3960, found: 588.3966.
1′-Butyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (8). Betulonic acid (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (36 mg, 1 equiv, 0.22 mmol), n-butylamine (21 mg, 1.3 equiv, 0.286 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 8 is powdery with a yellowish tinge, m.p. 288-290 oC. Yield 74 mg (62.8 %).
1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.67 – 4.61 (m, 1H), 4.29 (td, J = 7.1, 1.6 Hz, 2H), 3.04 (td, J = 10.5, 4.5 Hz, 1H), 2.91 (d, J = 15.3 Hz, 1H), 2.34 – 2.20 (m, 2H), 2.13 (d, J = 15.4 Hz, 1H), 2.06 – 1.93 (m, 4H), 1.80 – 1.73 (m, 1H), 1.71 (s, 3H), 1.65 (d, J = 11.5 Hz, 1H), 1.62 – 1.33 (m, 11H), 1.30 (s, 3H), 1.26 (s, 4H), 1.18 (s, 3H), 1.04 – 0.93 (m, 10H), 0.93 – 0.80 (m, 1H), 0.77 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.21, 150.24, 141.03, 109.86, 56.39, 54.68, 49.37, 49.30, 49.21, 46.90, 42.46, 40.59, 38.98, 38.54, 38.27, 37.05, 33.66, 33.39, 32.85, 32.09, 30.60, 29.81, 29.71, 28.68, 25.50, 21.33, 20.18, 19.43, 18.98, 16.06, 15.72, 14.69, 13.70.
HRMS (ESI+): m/z calculated for C34H54N3O2 [M+H]+: 536.4211, found: 536.4214.
1′-Isobutyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid (9). Betulonic acid 2 (100 mg, 1 equiv, 0.22 mmol), 4-nitrophenyl azide (36 mg, 1 equiv, 0.22 mmol), isobutylamine (21 mg, 1.3 equiv, 0.286 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 ml) and the reaction mixture was stirred at 100 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 9 is pale yellow powder, m.p. 299-301 oC. Yield 67.8 mg (64.6 %).
1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.64 (t, J = 1.9 Hz, 1H), 4.16 – 4.03 (m, 2H), 3.04 (td, J = 10.6, 4.6 Hz, 1H), 2.92 (d, J = 15.3 Hz, 1H), 2.54 (dq, J = 13.8, 6.9 Hz, 1H), 2.35 – 2.19 (m, 2H), 2.14 (d, J = 15.3 Hz, 1H), 2.08 – 1.94 (m, 2H), 1.81 – 1.73 (m, 1H), 1.71 (s, 3H), 1.69 – 1.32 (m, 10H), 1.30 (s, 3H), 1.24 (d, J = 16.4 Hz, 3H), 1.17 (s, 3H), 1.15 – 1.03 (m, 1H), 1.03 – 0.93 (m, 13H), 0.93 – 0.83 (m, 1H), 0.77 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.59, 150.26, 140.94, 137.79, 109.83, 56.56, 56.42, 54.74, 49.30, 49.22, 46.91, 42.45, 40.58, 38.90, 38.55, 38.28, 37.05, 33.73, 33.38, 32.10, 30.61, 29.82, 29.38, 28.90, 25.50, 21.57, 21.36, 20.25, 20.21, 19.43, 19.00, 16.04, 15.74, 14.68.
HRMS (ESI+): m/z calculated for C34H54N3O2 [M+H]+: 536.42160, found: 536.4209.
1H′-Lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (10). Betulonic acid 2 (100 mg, 1 equiv, 0.22 mmol), ammonium acetate (84.8 mg, 5 equiv, 1.1 mmol) and 4-nitrophenyl azide (45.8 mg, 1.3 equiv, 0.28 mmol) were dissolved in dry DMF (1 mL). The reaction mixture was stirred at 80 °C for 24 hours. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 10 is pale yellow powder, m.p. 158 oC. Yield 48.3 mg (46 %). Spectroscopic data for compound 10 was consistent with previously reported data for this compound.
1H NMR (400 MHz, CDCl3): δ 4.77 (d, J = 2.5 Hz, 1H), 4.64 (d, J = 1.9 Hz, 1H), 3.05 (td, J = 10.7, 4.6 Hz, 1H), 2.90 (d, J = 15.5 Hz, 1H), 2.36 – 2.22 (m, 2H), 2.19 – 2.09 (m, 1H), 2.08 – 1.93 (m, 2H), 1.83 – 1.74 (m, 1H), 1.72 (s, 3H), 1.68 – 1.36 (m, 15H), 1.32 (s, 4H), 1.30 – 1.18 (m, 4H), 1.18 – 1.04 (m, 1H), 1.01 (d, J = 10.2 Hz, 6H), 0.97 – 0.80 (m, 6H), 0.77 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 181.38, 150.35, 149.92, 140.40, 109.80, 56.40, 53.42, 49.22, 49.09, 46.95, 42.51, 41.35, 40.75, 39.04, 38.51, 37.32, 37.07, 33.72, 33.38, 33.30, 32.16, 31.01, 30.62, 29.80, 29.06, 27.67, 25.51, 23.76, 22.63, 21.41, 20.45, 19.42, 19.16, 16.27, 15.69, 14.68.
Compound (11) and (12). Compound 10 (181 mg) was dissolved in dry methanol (1.5 ml) then added potassium tret-butoxide (PTB) (100.08 mg, 0.91 mmol). The reaction mixture was stirred ar room temperature for 3 hours. Arter a 1-bromobutane was slowly added the reaction mixture and stirred at 60oC for 12 hours. The solvent was distilled in a rotary evaporator. The crude reaction mixture was purified directly using column chromatography (silica gel), using a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 11 is colorless substance. Yield 102.9 mg (46.4 %).
1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.63 (t, J = 1.8 Hz, 1H), 4.32 (t, J = 7.3 Hz, 2H), 4.09 (qt, J = 10.8, 6.6 Hz, 2H), 3.04 (td, J = 10.8, 4.5 Hz, 1H), 2.83 (d, J = 15.4 Hz, 1H), 2.29 (td, J = 12.4, 11.7, 3.6 Hz, 2H), 2.07 (d, J = 15.4 Hz, 1H), 1.96 – 1.85 (m, 4H), 1.80 – 1.72 (m, 1H), 1.71 (s, 3H), 1.68 – 1.30 (m, 15H), 1.29 (d, J = 4.0 Hz, 4H), 1.21 (s, 2H), 1.17 (d, J = 5.7 Hz, 4H), 1.15 – 1.02 (m, 1H), 1.00 (s, 3H), 0.98 (d, J = 4.3 Hz, 4H), 0.96 – 0.81 (m, 11H), 0.79 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 176.22, 150.89, 150.54, 141.49, 109.63, 63.72, 56.58, 54.41, 53.50, 49.35, 49.17, 47.01, 42.46, 40.76, 38.90, 38.35, 37.62, 37.05, 33.50, 33.39, 32.14, 32.05, 31.08, 30.81, 30.68, 29.72, 25.61, 23.82, 21.44, 19.84, 19.44, 19.32, 19.21, 16.26, 15.63, 14.67, 13.72, 13.59.
HRMS (ESI+): m/z calculated for C38H62N3O2 [M+H]+: 592.48420, found: 592.4850.
Compound 12 is colorless substance. Yield 45 mg (20.3 %).
1H NMR (400 MHz, CDCl3) δ 4.75 (d, J = 2.4 Hz, 1H), 4.61 (m, 1H), 4.17 (m, 1H), 4.10 (m, 3H), 3.05 (td, J = 10.8, 4.4 Hz, 1H), 2.61 (d, J = 15.3 Hz, 1H), 2.37 – 2.24 (m, 2H), 2.05 – 1.98 (m, 1H), 1.97 – 1.85 (m, 2H), 1.84 – 1.73 (m, 3H), 1.70 (s, 3H), 1.68 – 1.37 (m, 13H), 1.33 (d, J = 10.5 Hz, 4H), 1.27 – 1.20 (m, 6H), 1.18 – 1.03 (m, 2H), 1.01 (s, 3H), 0.98 (d, J = 5.2 Hz, 3H), 0.95 (d, J = 6.8 Hz, 4H), 0.93 – 0.82 (m, 5H), 0.79 (s, 3H).
13C NMR (101 MHz, CDCl3) δ 176.17, 150.68, 150.15, 129.16, 109.61, 63.76, 56.52, 53.37, 49.34, 49.27, 47.36, 47.00, 42.49, 40.87, 39.24, 38.31, 37.03, 36.06, 33.65, 33.36, 32.16, 32.09, 30.81, 30.70, 30.59, 29.70, 25.58, 23.08, 21.59, 19.86, 19.38, 19.31, 19.00, 16.63, 15.63, 14.65, 13.72, 13.60.
HRMS (ESI+): m/z calculated for C38H62N3O2 [M+H]+: 592.48420, found: 592.4844.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (13). Compound 3 (100 mg, 1 equiv, 0.171 mmol) was dissolved in 2 ml of THF and 1,1′-carbonyldi-(1,2,4-triazole) (112.1 mg, 4 equiv, 0.684 mmol) was added. The reaction mixture was stirred at 70oC. The reaction was carried out for 5 hours. The solvent was distilled into a rotary evaporator. The remainder was chromatographed with silica gel in a column. When the column was eluted with a mixture of petroleum ether and ethyl acetate (5:3), compound 13 was isolated. Compound 13 is a colorless powder, m.p. 232-235oC. Yield 116 mg (107.4 %).
1H NMR (400 MHz, CDCl3): δ 8.93 (s, 1H), 7.99 (s, 1H), 7.34 – 7.26 (m, 3H), 7.24 – 7.18 (m, 3H), 5.72 (q, J = 7.0 Hz, 1H), 4.79 (d, J = 2.1 Hz, 1H), 4.68 (t, J = 1.8 Hz, 1H), 3.04 – 2.90 (m, 3H), 2.74 – 2.55 (m, 2H), 2.20 – 2.09 (m, 2H), 2.02 (d, J = 7.0 Hz, 3H), 1.90 – 1.75 (m, 3H), 1.73 (s, 3H), 1.72 – 1.57 (m, 2H), 1.57 – 1.30 (m, 6H), 1.30 – 1.13 (m, 9H), 1.09 (s, 3H), 0.99 (d, J = 10.7 Hz, 6H), 0.95 – 0.86 (m, 1H), 0.85 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 152.19, 145.19, 141.02, 128.64, 127.55, 126.23, 110.20, 59.28, 58.45, 54.84, 50.92, 49.54, 45.65, 42.33, 40.62, 38.94, 38.41, 37.19, 36.26, 33.81, 33.38, 31.47, 30.54, 29.97, 28.89, 25.53, 23.74, 21.52, 21.32, 19.40, 18.90, 16.29, 15.67, 14.65.
HRMS (ESI+): m/z calculated for C40H55N6O [M+H]+: 635.4432, found: 635.4393.
1′-(3-Methoxybenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (14). Compound 5 (93 mg, 1 equiv, 0.155 mmol) was dissolved in 2 ml of THF and 1,1′-carbonyldi-(1,2,4-triazole) (152.6 mg, 6 equiv, 0.93 mmol) was added. The reaction mixture was stirred at 70oC. The reaction was carried out for 24 hours. The solvent was distilled into a rotary evaporator. The remainder was chromatographed with silica gel in a column. When the column was eluted with a mixture of petroleum ether and ethyl acetate (4:1), compound 14 was isolated. Compound 14 is a colorless powder, m.p. 279-282 oC. Yield 52 mg (51.6 %).
1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H), 7.99 (s, 1H), 7.21 (t, J = 7.9 Hz, 1H), 6.82 – 6.77 (m, 1H), 6.63 – 6.58 (m, 1H), 6.56 (t, J = 2.1 Hz, 1H), 5.62 (s, 2H), 4.80 (d, J = 2.2 Hz, 1H), 4.71 – 4.64 (m, 1H), 3.74 (s, 3H), 3.05 – 2.90 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.60 (dd, J = 12.9, 7.3 Hz, 1H), 2.17 (s, 5H), 1.92 – 1.75 (m, 3H), 1.74 (s, 3H), 1.72 – 1.64 (m, 1H), 1.63 (d, J = 1.9 Hz, 2H), 1.60 – 1.34 (m, 5H), 1.35 – 1.19 (m, 2H), 1.18 (s, 3H), 1.06 (s, 3H), 1.03 (s, 3H), 0.97 (s, 4H), 0.80 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 173.39, 159.97, 152.21, 149.83, 145.18, 141.87, 138.10, 138.00, 129.78, 118.65, 113.27, 112.00, 110.20, 58.44, 55.23, 54.66, 52.74, 50.91, 49.49, 45.65, 42.33, 40.60, 39.01, 38.48, 37.18, 36.27, 33.74, 33.32, 31.47, 30.54, 29.96, 28.73, 25.53, 21.54, 21.35, 19.41, 18.91, 16.13, 15.65, 14.66.
HRMS (ESI+): m/z calculated for C40H55N6O2 [M+H]+: 651.4381, found: 651.4378.
1’-(3-Fluorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (15). Compound 7 (50 mg, 1 equiv, 0.085 mmol) was dissolved in 2 ml of THF and 1,1′-carbonyldi-(1,2,4-triazole) (55.7 mg, 4 equiv, 0.34 mmol) was added. The reaction mixture was stirred at 70oC. The reaction was carried out for 2.5 hours. The solvent was distilled into a rotary evaporator. The formed compound was washed three times with cold acetone. Compound 15 is a colorless powder, m.p. 307-310oC. Yield 60.7 mg (111.9 %).
1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H), 7.99 (s, 1H), 7.31 – 7.23 (m, 2H), 7.00 – 6.91 (m, 1H), 6.81 (dt, J = 7.8, 1.3 Hz, 1H), 6.72 (dt, J = 9.6, 2.1 Hz, 1H), 5.64 (d, J = 3.0 Hz, 2H), 4.80 (d, J = 2.2 Hz, 1H), 4.68 (t, J = 1.8 Hz, 1H), 3.75 (tdd, J = 5.8, 2.5, 1.6 Hz, 1H), 3.04 – 2.90 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.64 – 2.55 (m, 1H), 2.20 (d, J = 15.4 Hz, 1H), 1.91 – 1.75 (m, 4H), 1.74 (s, 3H), 1.70 – 1.34 (m, 8H), 1.25 (ddd, J = 25.0, 8.5, 3.2 Hz, 3H), 1.17 (s, 4H), 1.07 (s, 3H), 1.03 (s, 3H), 0.97 (s, 3H), 0.81 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 173.38, 164.29, 161.83, 152.21, 149.82, 145.18, 142.04, 138.07, 130.40, 130.32, 121.94, 114.94, 113.65, 110.21, 67.98, 58.44, 54.61, 52.25, 50.91, 49.49, 45.65, 42.34, 40.60, 39.01, 38.42, 37.18, 36.27, 33.72, 33.30, 31.46, 30.53, 29.96, 28.79, 25.52, 21.54, 21.41, 19.40, 18.90, 16.16, 15.65, 14.66.
HRMS (ESI+): m/z calculated for C39H52FN6O [M+H]+: 639.4181, found: 639.4180.
1′-Butyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (16). Compound 8 (50 mg, 1 equiv, 0.0934 mmol) was dissolved in 2 ml of THF and 1,1′-carbonyldi-(1,2,4-triazole) (61.2 mg, 4 equiv, 0.3736 mmol) was added. The reaction mixture was stirred at 70oC. The reaction was carried out for 2 hours. The solvent was distilled into a rotary evaporator. The formed compound was washed three times with cold acetone. Compound 16 is a colorless powder, m.p. 281-283oC. Yield 59 mg (107.7 %).
1H NMR (400 MHz, CDCl3): δ 8.93 (s, 1H), 8.00 (s, 1H), 7.27 (s, 1H), 4.79 (d, J = 2.2 Hz, 1H), 4.71 – 4.63 (m, 1H), 4.29 (td, J = 7.1, 1.4 Hz, 2H), 3.05 – 2.89 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.63 – 2.56 (m, 1H), 2.19 – 2.10 (m, 2H), 1.97 (pd, J = 7.1, 1.4 Hz, 2H), 1.89 – 1.75 (m, 3H), 1.71 (d, J = 17.7 Hz, 5H), 1.66 – 1.34 (m, 7H), 1.30 (s, 3H), 1.27 – 1.18 (m, 4H), 1.12 (td, J = 13.0, 4.3 Hz, 1H), 1.03 (s, 3H), 1.01 – 0.94 (m, 6H), 0.80 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 173.39, 152.21, 149.82, 145.19, 141.02, 137.26, 110.20, 58.45, 54.75, 50.92, 49.50, 49.36, 45.65, 42.34, 40.62, 39.01, 38.39, 37.19, 36.27, 33.68, 33.36, 32.86, 31.48, 30.54, 29.98, 28.68, 25.53, 21.52, 21.36, 20.18, 19.40, 18.97, 16.11, 15.67, 14.70, 13.70.
HRMS (ESI+): m/z calculated for C36H55N6O [M+H]+: 587.4432, found: 587.4438.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-isocyanate (17). Compound 3 (200 mg, 1 equiv., 0.34 mmol) was dissolved in toluene (6 ml) in an ultrasonic bath, then triethylamine (0.0464 ml, 1 equiv., 0.34 mmol) and diphenylphosphoryl azide (0.0772 ml, 1 equiv., 0.34 mmol) were added. The reaction was carried out at room temperature (21oC) for 6 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. Compound 17 colorless powder, m.p. 197-200 oC. Yield 120 mg (60.9 %).
1H NMR (400 MHz, CDCl3): δ 7.29 (m, 3H), 7.22 (m, 2H), 5.73 (q, J = 7.0 Hz, 1H), 4.77 (d, J = 2.1 Hz, 1H), 4.67 (m, 1H), 2.97 (d, J = 15.3 Hz, 1H), 2.56 (td, J = 10.9, 5.8 Hz, 1H), 2.13 (m, 2H), 2.02 (d, J = 7.0 Hz, 3H), 1.84 (m, 5H), 1.70 (d, J = 1.2 Hz, 3H), 1.52 (m, 8H), 1.29 (s, 3H), 1.20 (m, 2H), 1.11 (d, J = 1.8 Hz, 6H), 0.94 (s, 3H), 0.84 (m, 3H).
13C NMR (101 MHz, CDCl3): δ 148.71, 141.83, 140.99, 137.51, 128.64, 127.55, 126.24, 121.63, 110.63, 71.59, 59.27, 54.80, 49.32, 49.18, 48.10, 42.06, 40.66, 39.27, 39.17, 38.91, 38.42, 33.81, 33.57, 33.46, 29.29, 28.91, 27.81, 24.91, 23.74, 21.36, 21.34, 19.52, 18.89, 16.29, 15.69, 14.44.
HRMS (ESI+): m/z calculated for C38H53N4O [M+H]+: 581.4214, found: 581.4218.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative -1. (18). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 ml) in an ultrasonic bath, then triethylamine (0.024 ml, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 ml, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (20oC) for 7 hours. After cyclopropylamine (0.177 ml, 10 equiv., 1.7 mmol) in 0.5 ml of toluene was added to the reaction mixture. Then it was heated at 110°C for 4 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance 18 with a melting point of 200-203 oC was isolated. The yield was 65.9 mg (61 %).
1H NMR (400 MHz, CDCl3): δ 7.34 – 7.25 (m, 3H), 7.23 – 7.18 (m, 2H), 5.73 (q, J = 7.0 Hz, 1H), 4.95 (s, 1H), 4.75 (d, J = 2.1 Hz, 1H), 4.69 – 4.60 (m, 2H), 2.96 (d, J = 15.2 Hz, 1H), 2.67 (dt, J = 13.2, 3.3 Hz, 1H), 2.54 (dd, J = 12.5, 8.1 Hz, 1H), 2.44 (tq, J = 7.9, 4.6, 4.1 Hz, 2H), 2.16 (d, J = 15.2 Hz, 1H), 2.07 – 1.91 (m, 4H), 1.82 – 1.64 (m, 7H), 1.62 – 1.16 (m, 11H), 1.10 (d, J = 9.6 Hz, 6H), 0.99 (s, 3H), 0.95 – 0.75 (m, 6H), 0.69 – 0.56 (m, 2H).
13C NMR (101 MHz, CDCl3): δ 158.04, 149.40, 141.76, 140.94, 137.53, 128.66, 127.59, 126.23, 110.27, 63.72, 59.28, 54.74, 49.31, 49.23, 48.33, 42.07, 40.64, 38.89, 38.42, 38.37, 35.19, 33.83, 33.15, 29.86, 29.33, 28.89, 27.34, 25.11, 23.72, 22.70, 21.48, 21.35, 19.28, 18.90, 16.29, 15.58, 14.48, 7.82, 7.35.
HRMS (ESI+): m/z calculated for C41H60N5O [M+H]+: 638.4792, found:638.4750.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative -2. (19). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 ml) in an ultrasonic bath, then triethylamine (0.024 ml, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 ml, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (21oC) for 2 hours. After ethanolamine (0.1026 ml, 10 equiv., 1.7 mmol) in 0.5 ml of toluene was added to the reaction mixture. Then it was heated at 110°C for 7 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance 19 with a melting point of 202-204 oC was isolated. The yield was 126.2 mg (115.78 %).
1H NMR (400 MHz, CDCl3): δ 7.35 – 7.23 (m, 2H), 7.27 – 7.14 (m, 2H), 5.79 (s, 1H), 5.75 (q, J = 6.9 Hz, 1H), 5.18 (s, 1H), 4.69 (d, J = 2.3 Hz, 1H), 4.62 (t, J = 1.9 Hz, 1H), 4.12 (q, J = 7.2 Hz, 1H), 3.77 – 3.60 (m, 3H), 3.60 – 3.45 (m, 1H), 3.24 (s, 2H), 2.91 (d, J = 15.3 Hz, 1H), 2.68 – 2.60 (m, 1H), 2.55 (td, J = 11.0, 5.0 Hz, 1H), 2.46 (dd, J = 12.2, 8.1 Hz, 1H), 2.21 – 2.15 (m, 1H), 2.14 (d, J = 14.3 Hz, 1H), 2.08 (s, 1H), 2.03 (d, J = 12.8 Hz, 2H), 2.02 – 1.92 (m, 2H), 1.86 – 1.75 (m, 1H), 1.74 (s, 1H), 1.70 (s, 3H), 1.65 (d, J = 11.6 Hz, 1H), 1.62 – 1.54 (m, 1H), 1.57 – 1.47 (m, 4H), 1.47 – 1.35 (m, 1H), 1.38 – 1.28 (m, 1H), 1.28 (s, 4H), 1.26 (s, 9H), 1.30 – 1.19 (m, 1H), 1.13 (s, 3H), 1.19 – 1.03 (m, 2H), 1.02 (s, 3H), 0.96 (s, 3H), 0.96 – 0.85 (m, 2H), 0.89 – 0.81 (m, 2H), 0.79 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 174.49, 159.57, 149.75, 141.44, 140.91, 137.89, 128.73, 127.74, 126.14, 110.03, 64.03, 63.73, 59.28, 54.73, 49.53, 49.31, 47.47, 43.51, 42.07, 40.65, 38.86, 38.31, 37.51, 35.61, 33.85, 33.23, 31.93, 29.79, 29.71, 29.66, 29.37, 28.88, 27.43, 25.02, 23.58, 22.70, 21.49, 21.35, 19.23, 18.90, 16.35, 15.70, 14.41, 14.13.
HRMS (ESI+): m/z calculated for C40H60N5O2 [M+H]+: 642.4742, found: 642.470.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative -3. (20). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 ml) in an ultrasonic bath, then triethylamine (0.024 ml, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 ml, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (21oC) for 3 hours. After 2-amino-2-methyl-1-propanol (0.162 ml, 10 equiv., 1.7 mmol) in 0.5 ml of toluene was added to the reaction mixture. Then it was heated at 110°C for 3.5 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance 20 with a melting point of 176-178 oC was isolated. The yield was 95.9 mg (84.3 %).
1H NMR (400 MHz, CDCl3): δ 7.30 (dd, J = 8.2, 6.3 Hz, 3H), 7.23 – 7.16 (m, 2H), 6.45 (s, 1H), 5.75 (q, J = 7.0 Hz, 1H), 5.25 (s, 1H), 5.05 (s, 1H), 4.65 (d, J = 2.3 Hz, 1H), 4.60 (t, J = 1.9 Hz, 1H), 3.55 (s, 2H), 2.92 (d, J = 15.2 Hz, 1H), 2.67 (dt, J = 13.4, 3.4 Hz, 1H), 2.44 (ddd, J = 20.1, 14.0, 9.6 Hz, 2H), 2.17 (d, J = 15.2 Hz, 1H), 2.08 (s, 0H), 2.02 (d, J = 7.0 Hz, 3H), 1.99 – 1.90 (m, 1H), 1.75 (td, J = 14.7, 14.1, 8.5 Hz, 2H), 1.68 (s, 3H), 1.62 (d, J = 11.7 Hz, 1H), 1.62 – 1.40 (m, 5H), 1.39 – 1.24 (m, 7H), 1.23 (d, J = 1.9 Hz, 6H), 1.09 (d, J = 22.1 Hz, 8H), 0.96 (s, 3H), 0.93 – 0.78 (m, 7H).
13C NMR (101 MHz, CDCl3): δ 158.88, 149.68, 141.54, 140.91, 128.70, 127.69, 126.18, 109.99, 72.29, 63.89, 59.30, 54.78, 54.76, 49.51, 49.35, 47.37, 42.04, 40.62, 38.89, 38.36, 37.52, 35.61, 33.85, 33.24, 29.84, 29.55, 28.89, 27.40, 25.26, 25.22, 25.04, 23.60, 21.50, 21.35, 19.29, 18.89, 16.33, 15.74, 14.45.
HRMS (ESI+): m/z calculated for C42H64N5O2 [M+H]+: 670.5055, found: 670.5093.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid derivative -4. (21). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 ml) in an ultrasonic bath, then triethylamine (0.024 ml, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 ml, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (22oC) for 3 hours. After 3-amino-1,2-propanediol (0.1317 ml, 10 equiv., 1.7 mmol) was added to the reaction mixture. Then it was heated at 110°C for 2 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. Elution of a column with a mixture of ethyl acetate and methanol (10:1) isolated a colorless substance 21 with a melting point of 182-185 oC. The yield was 89.1 mg (78 %).
1H NMR (400 MHz, CDCl3): δ 7.35 – 7.23 (m, 3H), 7.18 (td, J = 7.3, 1.9 Hz, 2H), 5.95 (s, 1H), 5.76 (q, J = 7.3 Hz, 1H), 5.35 – 5.18 (m, 1H), 4.68 (s, 1H), 4.64 – 4.57 (m, 1H), 4.12 (q, J = 7.2 Hz, 1H), 3.82 – 3.70 (m, 1H), 3.70 – 3.58 (m, 1H), 3.59 – 3.39 (m, 2H), 3.26 (dt, J = 16.1, 5.3 Hz, 2H), 2.92 (dd, J = 20.5, 15.2 Hz, 1H), 2.69 – 2.59 (m, 1H), 2.54 (ddq, J = 16.3, 11.8, 6.9, 5.2 Hz, 1H), 2.42 (dd, J = 12.0, 8.1 Hz, 1H), 2.24 – 2.10 (m, 1H), 2.06 (d, J = 10.6 Hz, 1H), 2.01 (d, J = 6.9 Hz, 3H), 1.97 – 1.71 (m, 1H), 1.69 (s, 3H), 1.67 – 1.30 (m, 5H), 1.30 – 1.19 (m, 7H), 1.12 (d, J = 8.0 Hz, 4H), 1.03 – 0.89 (m, 6H), 0.89 – 0.80 (m, 1H), 0.78 (s, 2H).
13C NMR (101 MHz, CDCl3): δ 159.68, 149.68, 141.37, 140.94, 138.02, 128.76, 128.68, 127.79, 126.18, 126.08, 110.09, 71.98, 63.85, 63.83, 63.30, 59.27, 54.71, 49.45, 49.31, 47.39, 42.50, 42.06, 40.63, 38.86, 38.29, 37.50, 35.72, 33.86, 33.22, 30.84, 29.71, 28.85, 27.41, 25.62, 25.01, 23.63, 23.52, 21.52, 21.34, 19.22, 18.89, 16.40, 15.68, 14.43, 14.20.
HRMS (ESI+): m/z calculated for C41H62N5O3 [M+H]+: 672.4847, found: 672.4858.
1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative -5. (22). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 ml) in an ultrasonic bath, then triethylamine (0.024 ml, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 ml, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (22oC) for 3 hours. After 2-(2-aminoethoxy-ethanol (0.1705 ml, 10 equiv., 1.7 mmol) in 0.5 ml of toluene was added to the reaction mixture. Then it was heated at 110°C for 2 hours. At the end of the reaction, the solvent was distilled on a rotary evaporator. The remainder was chromatographed with silica gel in a column. Elution of a column with a mixture of ethyl acetate and methanol (20:1) isolated a colorless substance 22 with a melting point of 165-167oC. The yield was 76.2 mg (65 %).
1H NMR (400 MHz, CDCl3): δ 7.37 – 7.23 (m, 3H), 7.22 – 7.16 (m, 2H), 5.74 (q, J = 7.0 Hz, 1H), 5.49 (s, 1H), 4.76 – 4.67 (m, 2H), 4.62 (t, J = 1.9 Hz, 1H), 3.83 – 3.68 (m, 3H), 3.68 – 3.60 (m, 3H), 3.60 – 3.51 (m, 4H), 3.36 (d, J = 5.1 Hz, 2H), 2.93 (d, J = 15.2 Hz, 1H), 2.71 – 2.61 (m, 1H), 2.49 (ddt, J = 20.7, 12.4, 6.5 Hz, 2H), 2.16 (d, J = 15.2 Hz, 1H), 2.06 (d, J = 8.7 Hz, 0H), 2.01 (d, J = 7.0 Hz, 3H), 1.76 (d, J = 11.2 Hz, 2H), 1.70 (s, 3H), 1.68 – 1.28 (m, 7H), 1.27 (s, 7H), 1.19 – 1.02 (m, 7H), 0.96 (s, 3H), 0.90 – 0.82 (m, 5H), 0.80 (s, 3H).
13C NMR (101 MHz, CDCl3): δ 158.08, 149.69, 141.63, 140.96, 137.71, 128.69, 127.65, 126.19, 110.06, 72.36, 70.81, 63.60, 61.66, 61.14, 59.30, 54.74, 49.50, 49.27, 47.52, 42.07, 41.35, 40.63, 40.18, 38.87, 38.32, 37.63, 35.71, 33.83, 33.25, 29.81, 29.77, 28.88, 27.39, 25.03, 23.67, 22.63, 21.46, 21.35, 19.26, 18.91, 16.32, 15.71, 14.42.
HRMS (ESI+): m/z calculated for C42H64N5O3 [M+H]+: 686.5004, found: 686.4954.
3.3. In Vitro Biological Assays
3.3.1. Antimicrobial Activity
The antimicrobial activity of the samples was evaluated in relation to strains of Gram-positive bacteria Staphylococcus aureus, Bacillus subtilis, Gram-negative bacteria Escherichia coli, Pseudomonas aeruginosa and yeast fungus Candida albicans by serial dilution with determination of the minimum inhibitory concentration (MIC). The test strains of microorganisms used in the study were obtained from the American collection of type cultures. An antibacterial drug, ceftriaxone, benzylpenicillin sodium salt and an antifungal drug, nystatin, were used as comparison drugs.
MIC was determined by the method of serial dilution of ethanol solutions of the test samples in a nutrient broth. Suspensions of test strains at a concentration of 106 CFU/ml were used to carry out the method of serial dilutions. A suspension of test strains of microorganisms was prepared from daily cultures grown on mown agar at a temperature of 37 °С for 24 hours, for the yeast fungus Candida albicans 30 oC for 48 hours. The antimicrobial activity of the samples was studied at dilutions in the range of 1.56 - 50 µg /ml. 0.1 ml of microbial suspension at a concentration of 106 CFU/ml was added to each tube with a working dilution of each test sample. The procedure was repeated for all cultures tested. A suspension of microbes with a nutrient medium without a sample was placed in control tubes. The mixture was incubated in a thermostat for 24-48 hours, depending on the class of microorganism.
Then, visually determining the presence of turbidity in each of the tubes, the one that contained a transparent suspension and the lowest concentration of an antimicrobial agent was selected. This concentration corresponded to MIC. The results were averaged according to the data of three experiments.
3.3.2. Cytotoxic Activity
The cytotoxicity of the synthesized compounds against Artemia salina (Leach) was evaluated in accordance with the methodology proposed by Meyer et al. [17].
The cytotoxicity of the samples was evaluated in the survival test of larvae of Artemia salina (Leach) crustaceans.
The experiments were carried out on larvae of 2 days of age under in vitro cultivation conditions. The larvae were grown by immersing the eggs of Artemia salina (Leach) crustaceans in artificial seawater and incubated for 48 hours at a temperature of 37 0C. The sample was dissolved in 2 ml of ethanol, then 500 µl (3 parallels), 50 µl (3 parallels), 5 µl (3 parallels) were taken from this solution. After evaporation of ethanol, 5 ml of artificial seawater was added to each bottle.
Thus, if the initial weight of the sample was 2 mg, then the final concentrations of the sample were 100 µg/ml, 10 µg/ml and 1 µg/ml, respectively, of each concentration in 3 repetitions. 10 larvae of Artemia salina 2-day-old crustaceans were planted in each vial with the image using a Pasteur pipette. After that, all the vials were left at room temperature in the light for 24 hours.
After 24 hours, the surviving and dead larvae were counted. Then, using the obtained data on the upper and lower toxic limit, the half toxic dose of the sample was calculated. The control is DMSO in equal amounts.
The test was performed using ready–made samples, as well as a comparison drug - dactinomycin (actinomycin D), which has antitumor (cytotoxic) activity (Producer: Sigma Aldrich)
Lethal concentrations of these compounds, leading to 50% death of shrimp (LC50), and 95% confidence intervals were determined based on 24-hour calculations with probit analysis and obtaining an LC50 value with a 95% confidence interval [17].
4. Conclusions
Thus, for the synthesis of heterocyclic derivatives based on lupane-structured triterpenoid betulonic acid, 21 compounds were obtained, of which 18 compounds are new. The physico-chemical properties of the synthesized compounds have been proven by spectroscopic methods. As a result of the study of biological activity, it was found that most of the tested compounds exhibit antimicrobial and cytotoxic activity. Among those studied, compound 6 showed pronounced antimicrobial activity against the gram-positive Staphylococcus aureus test strain ATCC 6538, and compound 7 showed pronounced antimicrobial activity against the gram-negative Escherichia coli test strain ATCC 25922.
Supplementary Materials
The following supporting information can be downloaded at: The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR, HRMS spectra.
Author Contributions
Conceptualization, R.I.J. and W.D.; methodology, R.I.J. and W.D.; validation, R.I.J. and W.D.; investigation, R.I.J., A.Sh.A., R.B.S. and S.A.; resources, W.D., R.I.J., G.K.M.; writing—original draft preparation, R.I.J., Ye.M.S. and A.Sh.A.; writing—review and editing, R.I.J., W.D. and G.K.M.; visualization, R.I.J. and W.D.; supervision, R.I.J. and W.D.; project administration, R.I.J. and W.D.; funding acquisition, R.I.J., G.K.M., A.Sh.A. and R.B.S.. All authors have read and agreed to the published version of the manuscript.
Funding
The chemical part of the research was funded by the «Bolashak» International Program of the Republic of Kazakhstan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Authors thanks the staff for technical support with NMR and for technical assistance with mass spectrometry.
Conflicts of Interest
The authors declare no conflicts of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Xue, X.; Zhao, J.; Ma, Y.; Zhen Cao, Zh.; Guo, Z. Triterpenoids with Cytotoxicity from Abelia engleriana. Chemistry of Natural Compounds 2021, 57, 1163–1166. [Google Scholar] [CrossRef]
- Chen, H.-M.; Huang, Ch.-M.; Chen, L.-Ch.; Yang, Ch.-S.; Chang, T.-H.; Chen, Ch.-L.; Sung, P.-J.; Kuo, W.-L.; Cheng, M.-J.; Hsiao, J.-W.; Chen, J.-J. New Triterpenoid and Anti-Inflammatory Constituents of Eriobotrya deflexa f. deflexa. Chemistry of Natural Compounds 2022, 58, 496–500. [Google Scholar] [CrossRef]
- Ye, Ch.; Jin, M.; Sun, J.; Wang, J.; Li, S.; Zhou, W.; Li, G. ; A New Ursane-Type Triterpenoid from the Leaves of Rhododendron dauricum with Cytotoxic Activity. Chemistry of Natural Compounds 2021, 57, 327–330. [Google Scholar] [CrossRef]
- Salimova, E.V.; Magafurova, A.A.; Tretyakova, E.V.; Kukovinets, O.S.; Parfenova, L.V. Indole Derivatives of Fusidane Triterpenoids: Synthesis and the Antibacterial Activity. Chemistry of Heterocyclic Compounds 2020, 56, 800–804. [Google Scholar] [CrossRef]
- Ding, H.; Wang, Zh.; Chen, X.; Zhang, P.; Xu, F.; Xu, Y.; Manman, L.; Chen, Y. New Triterpenoid Saponin C-20 Epimers from the Alkaline-Degradation Products of Ginsenoside Re and Their Cytotoxic Activities. Chemistry of Natural Compounds 2018, 54, 490–495. [Google Scholar] [CrossRef]
- Hamid, A.A.; Aiyelaagbe, O.O.; Negi, A.S.; Luqman, S.; Kaneez, F.; Bukya, B.; Sathish, K.B. Triterpenoids from the Aerial Parts of Smilax kraussiana as Antitumor Agents. Chemistry of Natural Compounds 2017, 53, 1192–1195. [Google Scholar] [CrossRef]
- Olennikov, D.N.; Gornostai, T.G.; Penzina, T.A.; Borovskii, G.B. Lupane Triterpenoids and Sterols from Inonotus rheades Mycelium and their Anti-Glucosidase Activity. Chemistry of Natural Compounds 2017, 53, 988–990. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, L.; Zhou, F.; Ma, X.; Li, Zh.; Zhong, T.; Yonghong, Zh. A New Ursane Triterpenoid Possessing Cytotoxicity from the Fruits of Vitex trifolia var. simplicifolia. Chemistry of Natural Compounds 2016, 52, 660–663. [Google Scholar] [CrossRef]
- Gromova, M.A.; Kharitonov, Yu.V.; Rybalova, T.V.; Borisov, S.A.; Tolstikova, T.G.; Shults, E.E. Synthetic Transformations of Higher Terpenoids. 42*. Synthesis and Biological Activity of (Arylsulfonylureido)Diterpenoids with Various Substituents on the Aryl Fragment. Chemistry of Natural Compounds 2023, 59, 296–308. [Google Scholar] [CrossRef]
- Gromova, M.A.; Kharitonov, Yu.V.; Borisov, S.A.; Baev, D.S.; Tolstikova, T.G.; Shults, E.E. Synthetic Transformations of Higher Terpenoids. 39*. Synthesis and Analgesic Activity of Isopimaric Acid Derivatives. Chemistry of Natural Compounds 2021, 57, 474–481. [Google Scholar] [CrossRef]
- Kharitonov, Yu.V.; Shults, E.E.; Rybalova, T.V.; Pavlova, A.V.; Tolstikova, T.G. Synthetic Transformations of Higher Terpenoids. 40*. Synthesis and Assessment of Analgesic Activity of N-Containing Derivatives of Lambertianic Acid. Chemistry of Natural Compounds 2021, 57, 879–886. [Google Scholar] [CrossRef]
- Kharitonov, Yu.V.; Shults, E.E.; Shakirov, M.M.; Pokrovskii, M.A.; Pokrovskii, A.G.; Tolstikova, G.A. Synthetic Transformations of Higher Terpenoids. 31*. Synthesis of 1,2,3-triazolyl-containing furan labdanoids and studies of their cytotoxic activity. Russian Chemical Bulletin 2013, 62, 2046–2055. [Google Scholar] [CrossRef]
- Gromova, M.A.; Kharitonov, Yu.V.; Pokrovskii, M.A.; Bagryanskaya, I.Yu.; Pokrovskii, A.G.; Shults, E.E. Synthetic Transformations of Higher Terpenoids. 37*. Synthesis and Cytotoxicity of 4-(Oxazol-2-Yl)-18-Norisopimaranes. Chemistry of Natural Compounds 2019, 55, 52–59. [Google Scholar] [CrossRef]
- Spivak, A.Yu.; Shakurova, E.R.; Nedopekina, D.A.; Khalitova, R.R.; Khalilov, L.M.; Odinokov, V.N.; Bel’skii, Yu.P.; Ivanova, A.N.; Bel’skaya, N.V.; Danilets, M.G.; Ligacheva, A.A. Novel lupane triterpenoids containing allyl substituents in ring A: synthesis and in vitro study of antiinflammatory and cytotoxic properties. Russian Chemical Bulletin 2011, 60, 694–701. [Google Scholar] [CrossRef]
- Gur’eva, Y.A.; Zalevskaya, O.A.; Nikolaeva, N.S.; Aleksandrova, Yu.R.; Yandulova, E.Yu.; Neganova, M.E.; Slepukhin, P.A.; Kutchin, A.V. Chiral zinc complexes with terpene derivatives of ethylenediamine: synthesis and biological activity. Russian Chemical Bulletin 2022, 71, 2612–2620. [Google Scholar] [CrossRef]
- Yamansarov, E.Yu.; Saltykova, I.V.; Kovalev, S.V.; Petrov, R.A.; Shkil’; Seleznev, E.I.; Beloglazkina, E.K.; Majouga A.G. I.; Beloglazkina, E.K.; Majouga A.G. Synthesis and cytotoxicity of new alkyne derivatives of pentacyclic triterpenoids. Russian Chemical Bulletin, 2019, 68, 855–861. [Google Scholar] [CrossRef]
- Meyer, B.N., Ferrigni, N.R., Putnam, J.E., Jacobsen, L.B., Nicholsand, D.E., McLaughlin, J.L. Brine Shrimp: A Convenient General Bioassay for Active Plant Constituents. Planta Medica 1982, 45, 31–34. [Google Scholar]
- McLaughlin, L. Crown Gall Tumors on Potato Discs and Brine Shrimp Lethality: Two Simple Bioassays for Higher Plant Screening and Fractionation. Methods in Plant Boichemistry 1991, 6, 1–32. [Google Scholar]
- Genet, C.; Strehle, A.; Schmidt, C.; Boudjelal, G.; Lobstein, A.; Schoonjans, K.; Souchet, M.; Auwerx, J.; Saladin, R.; Wagner, A. ; Structure-Activity Relationship Study of Betulinic Acid, A Novel and Selective TGR5 Agonist, and Its Synthetic Derivatives: Potential Impact in Diabetes. J. Med. Chem. 2010, 53, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Samoshina, N.F.; Denisenko, M.V.; Denisenko, V.A.; Uvarova, N.I. Synthesis of glycosides of lupane-type triterpene acids. Chemistry of Natural Compounds 2003, 39, 575–582. [Google Scholar] [CrossRef]
- Barthel, A.; Stark, S.; Csuk, R. Oxidative transformations of betulinol. Tetrahedron 2008, 64, 9225–9229. [Google Scholar] [CrossRef]
Scheme 2.
Reagent and conditions: a) ammonium acetate, 4-nitrophenyl azide, DMF, 80 °C, 24 h; b) PTB, 1-bromobutane, methanol, 60 °C, 12 h.
Scheme 2.
Reagent and conditions: a) ammonium acetate, 4-nitrophenyl azide, DMF, 80 °C, 24 h; b) PTB, 1-bromobutane, methanol, 60 °C, 12 h.
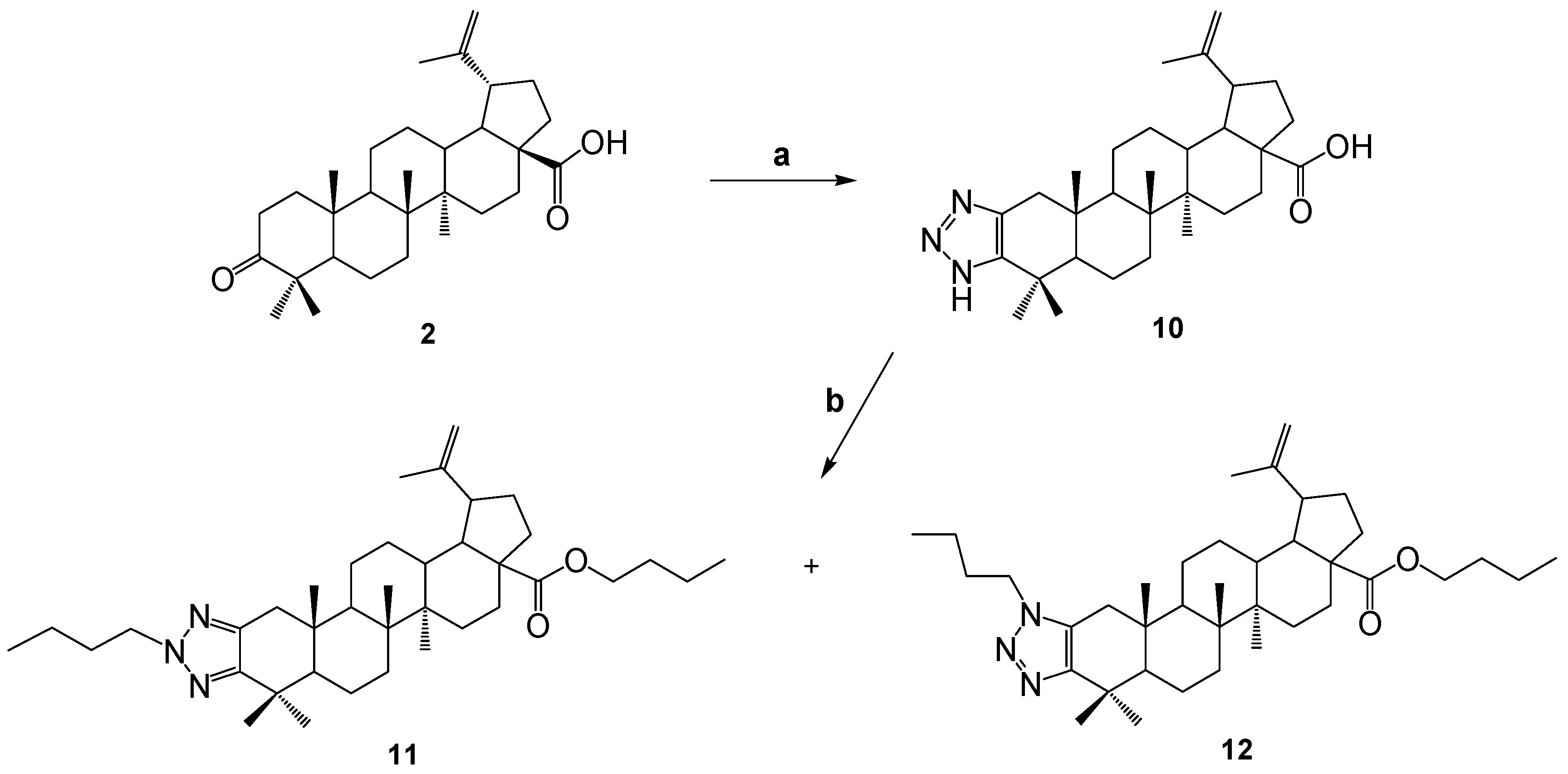

Table 1.
Antimicrobial activity of the compounds.
| Compounds |
Staphylococcus aureus АТСС 6538 |
Bacillus subtilis АТСС 6633 |
Escheriсhia coli АТСС 25922 |
Pseudomonas aeruginosa АТСС 27853 |
Сandida albicans АТСС 10231 |
|---|---|---|---|---|---|
| 3 | 25 | 50 | 12.5 | - | 50 |
| 8 | 25 | 50 | - | - | - |
| 7 | 25 | 25 | 6.3 | 50 | 25 |
| 5 | 50 | 25 | 25 | - | 50 |
| 13 | 25 | - | 50 | - | - |
| 16 | 50 | - | 50 | - | - |
| 15 | 12.5 | 50 | 25 | - | - |
| 14 | 50 | - | - | - | - |
| 17 | 50 | 50 | 50 | - | - |
| 18 | 25 | 50 | 12.5 | - | - |
| 19 | 25 | 25 | 50 | - | 25 |
| 20 | 50 | 25 | 25 | - | 50 |
| 21 | 25 | - | 50 | - | - |
| 22 | 50 | - | - | - | - |
| 6 | 6.3 | - | 25 | - | - |
| 9 | 50 | - | - | - | - |
| 4 | 25 | 50 | 50 | - | - |
| 10 | 50 | 50 | - | - | - |
| 2 | 50 | 25 | - | - | 50 |
| Ceftriaxone | 6.3 | 12.5 | 6.3 | 6.3 | - |
| Benzylpenicillin sodium salt | 12.5 | 25 | 25 | 50 | - |
| 16± 0,1 | 14± 0,1 | 14± 0,1 | - | - | |
| Nystatin | - | - | - | - | 12.5 |
Table 2.
Cytotoxic activity of the compounds.
| Compounds | LD50, µg/ml |
|---|---|
| 3 | 82.5 |
| 8 | 107.2 |
| 7 | 89.4 |
| 5 | - |
| 13 | 97.3 |
| 16 | 85.1 |
| 15 | 105.9 |
| 14 | 90.5 |
| 17 | - |
| 18 | 66.2 |
| 19 | 74.1 |
| 20 | 63.5 |
| 21 | - |
| 22 | 69.0 |
| 6 | 82.8 |
| 9 | 95.6 |
| 4 | 105.7 |
| 10 | 79.4 |
| 2 | 88.7 |
| Comparison drug: dactinomycin (actinomycin D) | 46.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



