
Submitted:
07 June 2024
Posted:
07 June 2024
You are already at the latest version
Abstract
Background: In response to inflammation, the absorption of nutritional iron is restricted. Multiple inflammation-related signaling pathways can lead to ferroptosis, an iron-dependent mechanism of programmed cell death. Since the pathophysiological significance of the presence and uptake of iron in chronic inflammation is still unknown, we tested the effect of a low iron diet on the clinical course of arthritis in the mouse model of collagen-induced arthritis (CIA).
Methods: Six- to eight-week-old male DBA/1 mice were fed either a normal or a low iron diet starting three weeks before arthritis induction. From day four after the collagen-booster made on day 25, the development of arthritis was regularly monitored until the end of the experiment (day 34), using a standard clinical arthritis score. The complete blood count was determined as well as Fe2+, Fe3+, oxidized and reduced glutathione (GSH and GSSG) and malondialdehyde (MDA) in whole paw tissue by ELISA. Quantitative PCR was performed from whole paw tissue for glutathione peroxidase 4 and other key regulator genes of iron metabolism and ferroptosis. We used nonparametric tests to com-pare markers of iron metabolism and ferroptosis as well as nonlinear regression models for longitu-dinal data for signs of arthritis.
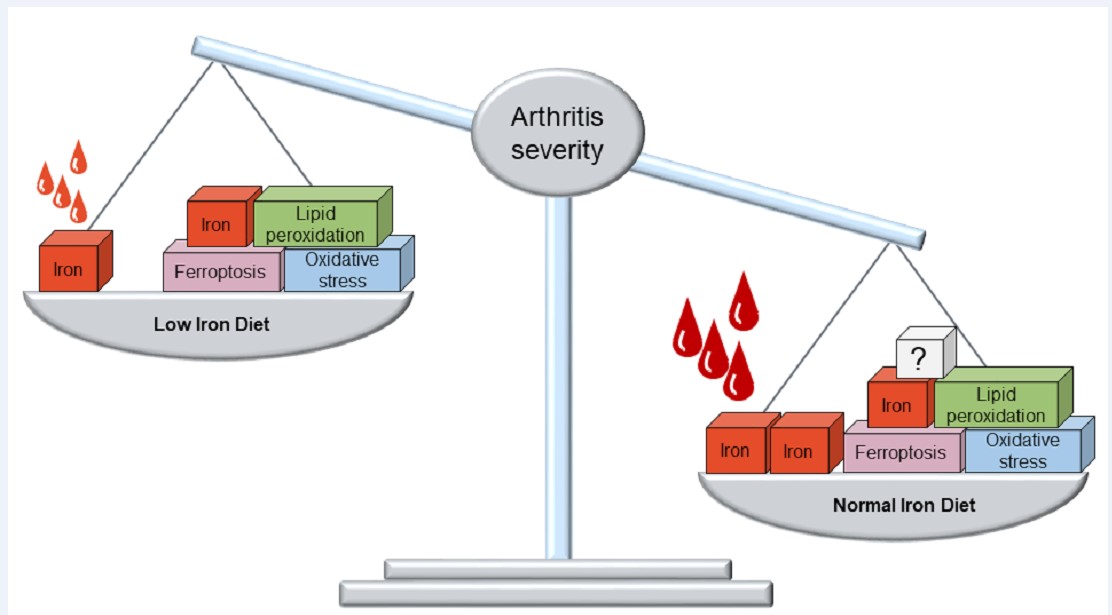
Results: Mice fed a low iron diet showed a significantly less severe course of arthritis compared to mice fed a normal iron diet (p
Keywords:
inflammation
; iron
; ferroptosis
; collagen-induced arthritis
; clinical arthritis score
1. Introduction
Synovial inflammation is one of the hallmarks of rheumatoid arthritis (RA) [1]. Abundant production of pro-inflammatory cytokines and chemokines triggers invasion of macrophages and lymphocytes into the joint, proliferation of synovial fibroblasts, extensive angiogenesis and is characterized by hypoxia and oxidative stress [2]. Since crucial for oxygen transport, iron homeostasis is tightly regulated [3]. Free Fe3+ from the blood is transported into the cell by transferrin (TF). When interacting with the transferrin receptor (TFR), receptor-bound iron is taken up into endosomes, whereFe3+ is reduced to Fe2+ before entering the cytoplasm. Intracellular Fe2+ is stored bound to ferritin, formed by ferritin light and heavy chain 1 (FTH1) and, to a lesser extent, as labile iron pool. In inflammation, intestinal iron availability and the amount of circulating iron is limited due to identical mechanisms on different cell types [3]. Ferroportin (FPN), the only known cellular iron exporter, is expressed in macrophages, duodenal enterocytes and hepatocytes. It is a target for the liver-expressed hormone hepcidin, which promotes the internalization and degradation of FPN and inhibits iron efflux [4,5]. Hepcidin transcription is activated by pro-inflammatory cytokines, most prominently interleukin (IL-) 6, via signal transducer and activator of transcription (STAT) 3 pathway [6,7]. An alternative mechanism for inhibiting FPN-mediated iron export is its transcriptional repression by pro-inflammatory cytokines [8]. In addition, in the hypoxic inflammatory state, synovial fibroblasts have been shown to produce stroma cell-derived factor 1 (SDF1) and vascular endothelial growth factor (VEGF), the latter leading to neovascularization [9]. As a consequence, synovial inflammation in RA is characterized by increased iron storage in the reticuloendothelial system [10] and limited intestinal iron absorption [11], often leading to a mixed picture of functional iron deficiency and true iron deficiency anemia. Since the impact of these hypoferric states on the underlying disease course remains unknown, it represents a challenge for the clinical practitioner in determining whether and how to treat them. In 2012, an iron-dependent form of non-apoptotic cell death has been discovered and termed ferroptosis [12]. Although the current understanding of ferroptosis is not comprehensive, similar features between ferroptosis and RA have been identified, providing a potential link between iron metabolism, oxidative stress and inflammation [13]. Ferroptosis is characterized by the intracellular accumulation of reactive oxygen species (ROS) arising from the reaction between redox-active iron and lipid peroxides [13], which are generated by the oxidation of membrane phospholipids containing polyunsaturated fatty acids [14]. The heavy chain subunit of SLC3A2 and the light chain subunit of SLC7A11 together form system Xc-, a functional cystine/glutamate antiporter. Intracellular cystine is reduced to cysteine and becomes part of reduced glutathione (GSH), which counteracts the accumulation of lipid peroxides until this pathway is exhausted. In addition, high amounts of ROS can be controlled by glutathione peroxidase 4 (GPX4). Thus, system Xc-, GSH and GPX4 are considered as “classic pathway” of ferroptosis regulation [15]. In addition, a probably context-dependent role of acyl-CoA synthetase long-chain family member 4 (ACSL4) has been described which is involved in the activation of polyunsaturated fatty acids and their incorporation into membrane phospholipids [16].
To study the clinical effect of limited iron absorption in immune-mediated inflammation and to investigate its association with ferroptosis, we further restricted nutritive iron availability in collagen-induced arthritis (CIA), a well-established mouse model of RA. We assessed the clinical arthritis course and measured total blood counts as well as Fe2+ and Fe3+ levels, the ratio of GSH and oxidized glutathione (GSSG) as readout of oxidative stress and the levels of malondialdehyde (MDA), indicating peroxidation of unsaturated lipids, in whole paws. We also measured key regulators of iron metabolism and ferroptosis in whole paws by qPCR. Notably, mice fed a low iron diet showed significantly lower red cell indices, indicating an additive dietary effect to iron restriction only by endogenous regulation in inflammation. Furthermore, mice fed a low iron diet experienced significantly less severe arthritis progression, indicating an anti-inflammatory defense mechanism, which could be utilized therapeutically. However, the anti-inflammatory properties of low iron diet take most likely take place independently of iron accumulation and ferroptosis in the inflamed tissues.
2. Methods
2.1. Mice
Six- to eight-week-old male DBA/1 mice were purchased from Envigo, Amsterdam, Netherlands. Upon arrival, mice were randomly assigned to groups of five per cage. Mice were housed under temperature- (22 ± 2°Celsius) and humidity- (40 to 60%) controlled conditions with unlimited access to fresh water and food in a 12/12-hour light/dark cycle. All experimental procedures have been approved by the cantonal veterinary office (license number BE83/2021).
2.2. Normal Iron Diet
The standard diet (article number: 343200PXV05, Granovit AG, Kaiseraugst, Switzerland) in the animal facility is referred to as normal iron diet, which contains 51 mg iron per kg.
2.3. Low Iron Diet
Low iron diet (catalogue number: D18756, Research Diets Inc., NJ, USA) contained 5 mg iron per kg, i.e. ten times less than the normal iron diet. Mice were placed on diet upon arrival four weeks before arthritis induction until the end of the experiment.
2.4. Arthritis Induction and Experimental Course
Arthritis was induced with bovine type II collagen (MD Biosciences, Zürich, Switzerland), provided as frozen solution in 0.05M glacial acetic acid, which was thawed overnight at 4°C before use. Followed by emulsification with complete Freund`s adjuvant (MD Biosciences, Zürich, Switzerland), on day 0 isoflurane-narcotized mice received an intra-cutaneous injection of 100µl emulsion (50µl per side) just above the tail base. On day 21, mice were boostered by intraperitoneal injection of 200µl of bovine type II collagen (MD Biosciences, Zürich, Switzerland) and sterile phosphate-buffered saline in a 1:1 mixture. On day 34, isoflurane-narcotized mice were sacrificed by a combination of cervical dislocation and intra-cardiac puncture (Figure 1A).
2.5. Clinical Arthritis Score
A standard clinical arthritis score [17] was assessed every day from day 21 onwards. GAS, SX or TA performed arthritis scoring of each paw separately in a blinded manner according to the following standardized system: 0: no clinical arthritis, 1: erythema and swelling confined to the digits, 2: erythema and swelling confined to the digits and pads, 3: erythema and swelling involving digits, pads and wrists/ankles; mice avoid using the affected paw. The scores from each paw were summed, resulting in a maximum achievable score of 12 per mouse. Divergent scores between two assessors were reassessed and discussed until an agreement was found.
2.6. Complete Blood Counts
A volume of 200µl blood was collected by intra-cardiac puncture of isoflurane-narcotized mice on day 34 in EDTA tubes (Sarstedt, Nümbrecht, Germany). Analysis was performed with ProCyte Dx (IDEXX Laboratories Inc., Maine, USA).
2.7. Tissue Homogenization
For determination of iron, GSH/GSSG and MDA levels, we mixed whole paws with 250µl extraction buffer. For real-time qPCR, 20% chloroform was added to each sample. Afterwards paws were homogenized for three minutes at 25 Hertz with a precooled TissueLyser II (Qiagen GmbH, Hilden, Germany).
2.8. Determination of Iron Levels
Paw iron levels were quantified by a colorimetric iron assay kit (Abcam, Cambridge, United Kingdom, catalogue number: ab83366), according to the manufacturer`s protocol. Absorbance was measured at 593nm with a BioTek microplate reader (Agilent Technologies, California, USA).
2.9. Determination of GSH/GSSG Levels
For quantification of GSH/GSSG levels, the GSH/GSSG Detection Assay Kit II (Abcam, Cambridge, United Kingdom, catalogue number: ab205811) was used, according to the manufacturer`s protocol. Absorbance was measured at Ex 490nm and Em 520nm with a BioTek microplate reader (Agilent Technologies, California, USA).
2.10. Determination of MDA Levels
For quantification of MDA levels, we used the Lipid Peroxidation (MDA) Assay Kit (Abcam, Cambridge, United Kingdom, catalogue number: ab118970). Optical density values were measured at 695nm with a BioTek microplate reader (Agilent Technologies, California, USA).
2.11. Real-Time qPCR
Total RNA was prepared with GENEzol™ reagent (Geneaid Biotech Ltd., New Taipei City, Taiwan). One ml of the reagent was added to 50mg of the homogenized paw tissue. Samples were shaken vigorously for 10 seconds and centrifuged at 4°C and 16,000 × g for 15 minutes to separate the phases. The aqueous phase was transferred and an equal volume of isopropanol was added. Samples were incubated at room temperature for ten minutes, followed by centrifugation at 4°C and 16,000 × g for ten minutes. Pellets of mRNA were washed twice in 70% ethanol, dried and re-suspended in nuclease-free water. The mRNA was reversely transcribed into cDNA with the High-Capacity RNA-to-cDNA™ Kit (Thermo Fisher Scientific, Massachusetts, USA). The mRNA levels were assessed by real-time qPCR, using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Massachusetts, USA) with the 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Massachusetts, USA). Ribosomal protein 29 (RPS29) was used as internal control. PCR amplification was performed over 40 cycles. All primers (synthesized by Microsynth AG, Balgach, Switzerland) are shown in Supplementary Table S1. Results are depicted using the 2(-Delta C(T)) method.
2.12. Statistical Analysis
Statistical analysis was performed with either Stata software (version 18) or GraphPadPrism software (version 10.1.2). Graphs were plotted with GraphPadPrism software. For statistical analysis of total blood count parameters, we performed an unpaired t-test. Nonlinear regression analysis was used for the longitudinal data of the clinical disease course. P-values <0.05 were regarded as statistically significant.
3. Results
3.1. Mice Fed a Low Iron Diet Develop Less Severe Arthritis
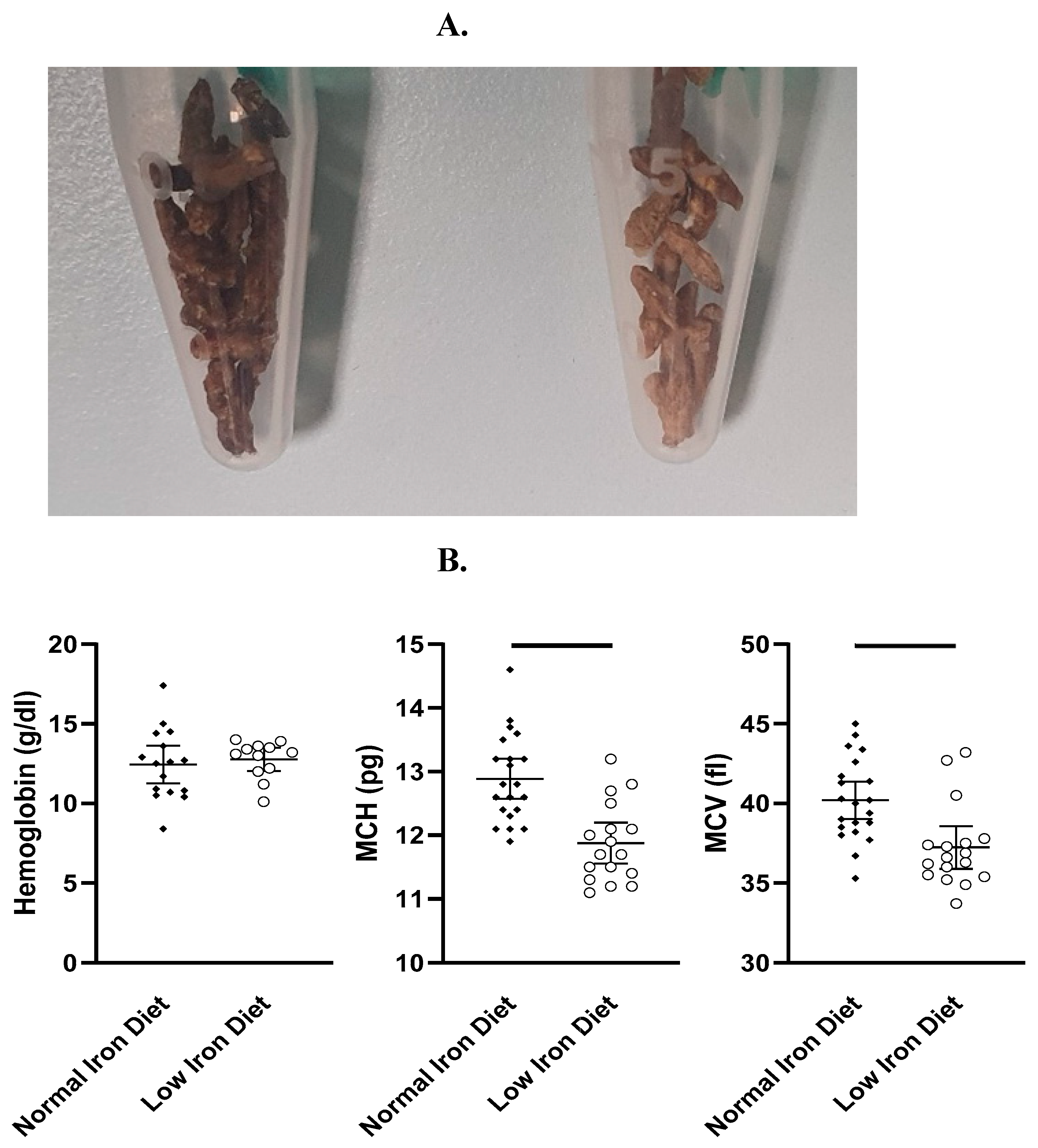
From days 25 to 34, we clinically assessed the severity of arthritis using the above described clinical score (Figure 1A). Of note, arthritis remained less severe in mice fed a low iron diet compared to mice fed a normal iron diet (Figure 1B), suggesting a protective mechanism. Measuring the ankle and paw diameters of the mice on day 34 revealed no differences between mice fed a normal iron or a low iron diet (Supplementary Table S2). In the feces, discoloration indicated low iron levels in mice fed a low iron diet (Figure 2A). On day 34, we measured hemoglobin levels (g/dl), mean corpuscular volumes (MCV, fl) and mean corpuscular hemoglobin levels (MCH, pg). As a low iron diet effect, we found reduced erythrocyte indices MCV and MCH. Interestingly, low iron diet had no effect on hemoglobin levels (Figure 2B). Furthermore, no diet related differences in weight gain or loss were found (Supplementary Table S3).
3.2. Articular Iron Accumulation and Ferroptosis Remain Unaffected by a Low Iron Diet
In order to assess the local iron distribution in relation to reduced arthritis severity in mice exposed to a low iron diet, we next set out to detect Fe2+ (and Fe3+) in total mouse paws. No differences were found between the normal iron diet group and the low iron diet group, nor were any differences associated with the degree of clinical arthritis score (Supplementary Table S4A). We then set out to assess key players of iron metabolism in total mouse paws by qPCR, namely FTH1, TFRC, FPN1, VEGF and SDF1 (Supplementary Table S5A). However, we could not identify differences in gene expression between normal iron diet and low iron diet groups. Next, in order to assess oxidative stress in synovial tissues, we measured levels of GSH and GSSG in total mouse paws (Supplementary Table S4B). In this comparison, we found no significant differences between normal iron diet groups and low iron diet groups; the GSH/GSSG ratio indicated a high oxidative state in the paws of both diet groups, with a maximum GSH/GSSG ratio of 1:5. Finally, MDA levels as read-out for unsaturated lipid peroxidation were measured in total mouse paws (Supplementary Table S4C). Again, we did not find differences in the expression between normal iron diet groups and low iron diet groups. Finally, we assessed gene expression of master regulators of ferroptosis by qPCR, namely GPX4, ACSL4, SLC7A11 and SLC3A2 (Supplementary Table S5B). However, we could not identify differences in gene expression between normal iron diet and low iron diet groups.
4. Discussion
By restricting the availability of dietary iron in the highly inflammatory state of the CIA mouse model, we observed a less severe course of arthritis compared to mice fed a normal iron diet. Lower erythrocyte indices in mice on low iron diet indicated a reduction of the available iron for hemoglobin synthesis, but without causing anemia. Importantly, we could demonstrate that a well-tolerated low iron diet had an additive and beneficial effect to the physiological regulation of iron uptake in response to inflammation.
Although the CIA mouse model has several differences from human disease [18], it is the standard animal model of arthritis for human RA, closely resembling several of its inflammatory features [19]. Against this background, our data have significant translational implications, as the clinical practitioner is often faced with the interpretation of combined functional and true hypoferric states in inflammatory diseases. While the impact of hypoferric states on the course of the disease remains completely unclear, treatments are nevertheless made to counter-regulate these states [20]. Essentially, the corresponding treatment strategies usually start with oral iron supplementation. However, importantly, oral supplementary strategies for functional iron deficiency remain more or less unsatisfactory, since iron absorption and export require FPN, which is degraded upon hepcidin production in response to inflammation. In addition, although parenteral iron supplementation appears to be safe in case of stimulated erythropoiesis, it was recently shown that at least the intravenous route of iron administration can even aggravate inflammation [21,22]. Accordingly, our data also argue against iron supplementation in inflammatory arthritis, but rather suggest a positive role of enteral iron restriction. As such, a hypoferric state in chronic inflammation should be rather understood as a biomarker of the body`s anti-inflammatory defense which should not be counter-regulated, but therapeutically supported. Of note, positive effects of iron chelator therapies in the management of chronic inflammatory disorders have been described [23]. However, in patients with inflammation-related anemia, fine-tuning of iron restriction could be quite relevant to not impair quality of life by inducing severe anemia, but to take advantage only of the anti-inflammatory effect. Interestingly, in our experiments low iron diet led to a reduction in MCV and MCH, but was not relevant for hemoglobin concentrations. Thus, beside deciphering the molecular mechanisms behind this observation, future clinical research will have to assess how such a best balance could look like and whether simple dietary iron restriction could be used as an adjunct to currently applied biological (b) or targeted synthetic (ts) disease-modifying anti-rheumatic drugs (DMARDs).
Due to common features of iron overload and lipid peroxidation, some evidence from other studies suggests the involvement of ferroptosis in the pathogenesis of RA. However, current findings are quite controversial and the question as to whether ferroptosis is protective or harmful in RA remains elusive [24]. In a lipopolysaccharide-induced synovitis model in one report an increase of iron content and TFRC as well as a decrease of GPX4, SLC7A11 and SLC3A2 has been described, leading to increased cell death [25]. In contrast, other authors have reported decreased ferroptosis associated with decreased ACSL4 levels and increased levels of FTH1, GPX4 and SLCA11 [26]. Further investigating this question, we hypothesized diet-related differences in iron accumulation in the mouse paws indicating different ferroptotic activity and providing a potential explanation for the different progression of arthritis severity. Somewhat unexpectedly, iron concentrations, oxidative stress and ferroptosis markers were not significantly affected by low iron diet, thereby either suggesting other mechanisms, which could even take place outside of the affected joints, or too subtle differences to be captured by our methods. In detail, since we did not find differences in Fe2+ or Fe3+ content between the paws of mice fed a low or a normal iron diet, this could suggest that ferroptosis may not be the key mechanism for the observed anti-inflammatory effect of a low iron diet. However, the effects of ferroptosis may be cell type-dependent, which might also explain the ongoing controversy about its beneficial or harmful effects, and perhaps needs to be deciphered on a single cell basis in the different tissues of the joint, whereas we can currently only provide global data for all cell types in the inflamed paws. In addition to our findings regarding the iron content, we could not find any diet-specific differences for FTH1, TFRC, FPN1, VEGF and SDF1 on gene expression level in the paws. While we were able to detect high oxidative stress levels in the paws of both diet groups based on GSH/GSSG ratio, there were no group-specific differences, also not for MDA, the read-out for lipid peroxidation. Finally, gene expression levels of the master regulators of ferroptosis GPX4, ACSL4, SLC7A11 and SLC3A2 were not significantly different between the two diet groups. Although tempting to speculate, all these findings together are strongly arguing against ferroptosis as a general key mechanism of arthritis.
In summary, we report here for the first time that non-hemoglobin-relevant oral iron restriction can positively influence the clinical course of arthritis and the induction of such a state could easily be harnessed therapeutically. The mechanism underlying our observation requires further investigation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Godehard Scholz, Tasneem Arsiwala, Monique Vogel, Martin Bachmann and Burkhard Möller; Data curation, Godehard Scholz, Sisi Xie, Tasneem Arsiwala and Burkhard Möller; Formal analysis, Godehard Scholz, Sisi Xie, Tasneem Arsiwala and Burkhard Möller; Funding acquisition, Martin Bachmann and Burkhard Möller; Investigation, Godehard Scholz, Sisi Xie and Tasneem Arsiwala; Methodology, Godehard Scholz, Tasneem Arsiwala, Monique Vogel, Martin Bachmann and Burkhard Möller; Project administration, Godehard Scholz and Monique Vogel; Resources, Martin Bachmann and Burkhard Möller; Software, Godehard Scholz, Sisi Xie, Tasneem Arsiwala and Burkhard Möller; Supervision, Godehard Scholz, Monique Vogel, Martin Bachmann and Burkhard Möller; Validation, Godehard Scholz, Sisi Xie, Tasneem Arsiwala, Monique Vogel, Martin Bachmann and Burkhard Möller; Visualization, Godehard Scholz and Burkhard Möller; Writing – original draft, Godehard Scholz and Burkhard Möller; Writing – review & editing, Godehard Scholz, Martin Bachmann and Burkhard Möller.
Funding
This research project was partly sponsored by CSL Vifor, Switzerland.
Acknowledgments
The authors sincerely thank Prof. Iain McInnes and his team, University of Glasgow, for the kind collaboration in the establishment of the mouse model. The authors also sincerely thank Dr. Judith Howard and her team, Vetsuisse Faculty, University of Bern, for the kind collaboration in the measurement of the full blood counts.
Conflicts of interest
With relation to this publication, all authors declare no conflicts of interest.
References
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA - J. Am. Med. Assoc. 2018.
- Ng, C.T.; Biniecka, M.; Kennedy, A.; McCormick, J.; FitzGerald, O.; Bresnihan, B.; Buggy, D.; Taylor, C.T.; O’Sullivan, J.; Fearon, U.; et al. Synovial Tissue Hypoxia and Inflammation in Vivo. Ann. Rheum. Dis. 2010, 69, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Scholz, G.A.; Leichtle, A.B.; Scherer, A.; Arndt, U.; Fiedler, M.; Aeberli, D.; Finckh, A.; Gabay, C.; Kyburz, D.; Villiger, P.M.; et al. The Links of Hepcidin and Erythropoietin in the Interplay of Inflammation and Iron Deficiency in a Large Observational Study of Rheumatoid Arthritis. Br. J. Haematol. 2019, 186, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of Hepcidin-Bound Ferroportin Reveals Iron Homeostatic Mechanisms. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.D.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science (80-. ). 2004. [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 Mediates Hypoferremia of Inflammation by Inducing the Synthesis of the Iron Regulatory Hormone Hepcidin. J. Clin. Invest. 2004. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Spasic, M.V.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 Mediates Hepatic Hepcidin Expression and Its Inflammatory Stimulation. Blood 2007. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczek, S.; Aigner, E.; Theurl, I.; Weiss, G. Cytokine-Mediated Regulation of Iron Transport in Human Monocytic Cells. Blood 2003. [Google Scholar] [CrossRef] [PubMed]
- Hitchon, C.; Wong, K.; Ma, G.; Reed, J.; Lyttle, D.; El-Gabalawy, H. Hypoxia-Induced Production of Stromal Cell-Derived Factor 1 (CXCL12) and Vascular Endothelial Growth Factor by Synovial Fibroblasts. Arthritis Rheum. 2002. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of Inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Iron Deficiency. Blood 2019.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016.
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014.
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-Induced Arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Hu, Z.; Cheng, J.; Zhang, R.; Yang, P.; Guo, H.; Nan, G.; Guo, N.; Gou, X. Applicability and Implementation of the Collagen-induced Arthritis Mouse Model, Including Protocols (Review). Exp. Ther. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mingyue, H.; Feng, Z.; Zongshun, D.; Ying, X.; Xiong, C.; Liang, L. Systematic Review of Robust Experimental Models of Rheumatoid Arthritis for Basic Research. Digit. Chinese Med. 2021.
- Lanser, L.; Fuchs, D.; Kurz, K.; Weiss, G. Physiology and Inflammation Driven Pathophysiology of Iron Homeostasis—Mechanistic Insights into Anemia of Inflammation and Its Treatment. Nutrients 2021.
- Ooi, M.; Hibbs, S.; Chen, F.E. The Safety of Modern Intravenous Iron Infusions in Patients with Rheumatoid Arthritis–a Review of the Literature. Hematol. (United Kingdom) 2020.
- de Souza, L.V.; Hoffmann, A.; Fischer, C.; Petzer, V.; Asshoff, M.; Theurl, I.; Tymoszuk, P.; Seifert, M.; Brigo, N.; Hilbe, R.; et al. Comparative Analysis of Oral and Intravenous Iron Therapy in Rat Models of Inflammatory Anemia and Iron Deficiency. Haematologica 2023. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Islam, S.; Jarosch, S.; Zhou, J.; Hoskin, D.; Greenshields, A.; Al-Banna, N.; Sharawy, N.; Sczcesniak, A.; Kelly, M.; et al. The Utility of Iron Chelators in the Management of Inflammatory Disorders. Mediators Inflamm. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yang, Q.; Xi, Y.; Xie, Z.; Shen, J.; Li, Z.; Li, Z.; Qin, D. Ferroptosis in Rheumatoid Arthritis: A Potential Therapeutic Strategy. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, R. Icariin Enhances Cell Survival in Lipopolysaccharide-induced Synoviocytes by Suppressing Ferroptosis via the Xc-/GPX4 Axis. Exp. Ther. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Li, M.; Yang, C.; Sun, S.; Zhang, W.; Zhao, L.; Xu, N.; Zhang, J.; Shen, Y.; Zhang, X.; et al. Glycine Increased Ferroptosis via SAM-Mediated GPX4 Promoter Methylation in Rheumatoid Arthritis. Rheumatology 2022, 61, 4521–4534. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A. Overview of the experimental course. B. Development of arthritis from day 25 to day 34, presented as clinical arthritis score in mice on normal iron diet or low iron diet. Shown are the mean and 95% confidence intervals of the clinical arthritis score from six independent experiments. Notably, arthritis remained less severe in mice fed a low iron diet in comparison to mice fed a normal iron diet. p<0.0001.
Figure 1.
A. Overview of the experimental course. B. Development of arthritis from day 25 to day 34, presented as clinical arthritis score in mice on normal iron diet or low iron diet. Shown are the mean and 95% confidence intervals of the clinical arthritis score from six independent experiments. Notably, arthritis remained less severe in mice fed a low iron diet in comparison to mice fed a normal iron diet. p<0.0001.
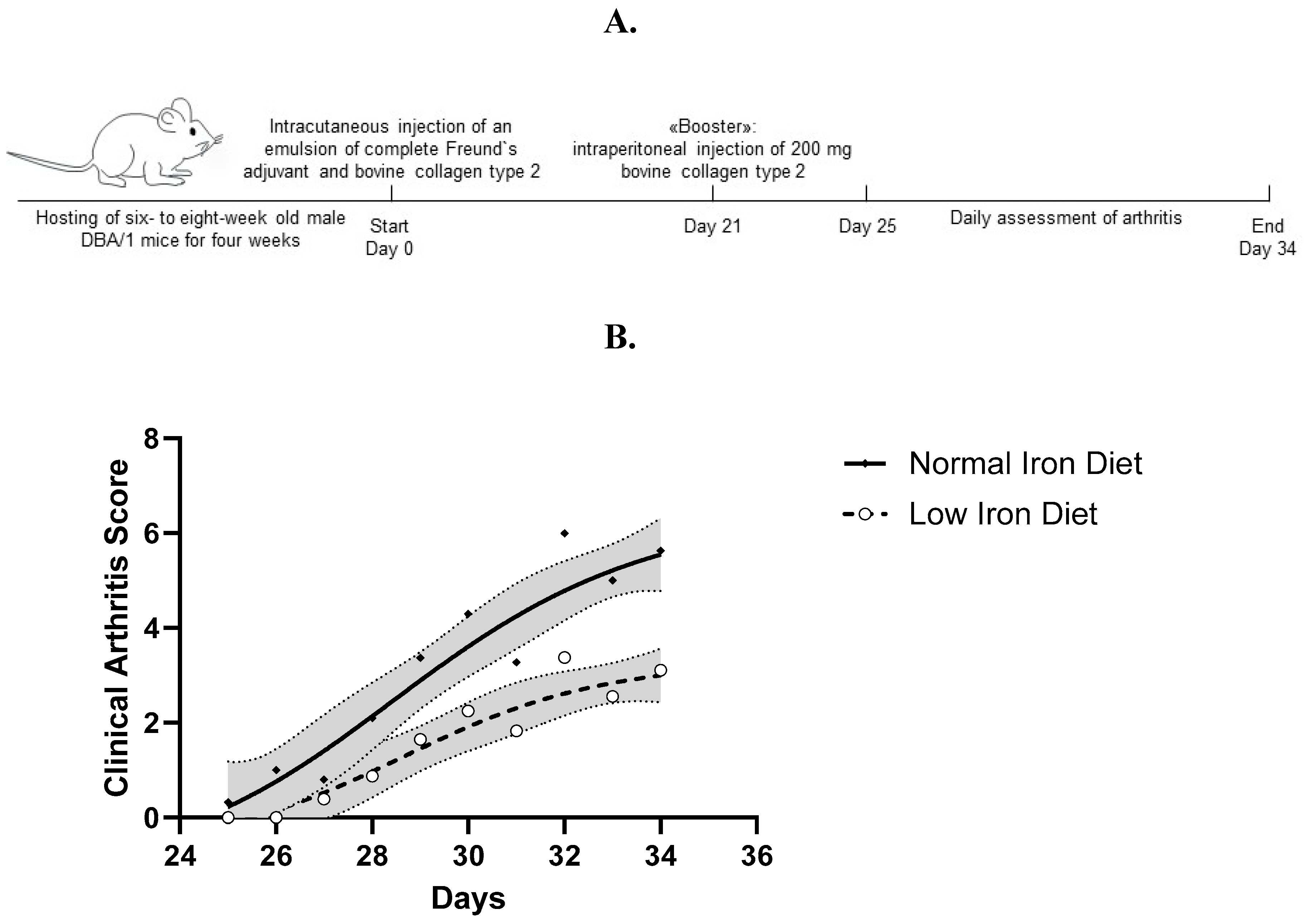
Figure 2.
A. Effect of a low iron diet due to discoloration of mouse feces. Left side: normal iron diet, right side: low iron diet. B. Hemoglobin levels (g/dl), mean corpuscular volume (MCV, fl) and mean corpuscular hemoglobin (MCH, pg) in mice fed a normal iron diet or a low iron diet. Of note, a reduction in MCV (p<0.001) and MCH (p<0.0001) were found in mice fed a low iron diet, although a low iron diet had no effect on hemoglobin.
Figure 2.
A. Effect of a low iron diet due to discoloration of mouse feces. Left side: normal iron diet, right side: low iron diet. B. Hemoglobin levels (g/dl), mean corpuscular volume (MCV, fl) and mean corpuscular hemoglobin (MCH, pg) in mice fed a normal iron diet or a low iron diet. Of note, a reduction in MCV (p<0.001) and MCH (p<0.0001) were found in mice fed a low iron diet, although a low iron diet had no effect on hemoglobin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



