
Submitted:
06 June 2024
Posted:
07 June 2024
You are already at the latest version
Abstract
Human epidermal growth factor receptor 2 (HER2), a targetable transmembrane glycoprotein receptor of the epidermal growth factor receptor (EGFR) family, plays a crucial role in cell proliferation, survival, and differentiation. Aberrant HER2 signaling is implicated in various cancers, particularly breast and Gastric cancer, where HER2 overexpression or amplification correlates with aggressive tumor behavior and poor prognosis. Her2 activating mutations contribute to accelerated tumorigenesis and metastasis. This review provides an overview of HER2 biology, signaling pathways, mechanisms of dysregulation, diagnostic approaches, and therapeutic strategies targeting HER2 in cancer. Understanding the intricate details of HER2 regulation is essential for developing effective targeted therapies and improving patient outcomes.
Keywords:
HER2
; Cancer
; Metastasis
; Immune regulation
; ADC
; therapeutics
1. Introduction
Brief overview of HER2 and its significance in cancer biology
HER2 is a receptor tyrosine kinase, a surface protein of cells that helps regulate cell growth and division. When HER2 binds to specific growth factors, it activates signaling pathways that promote cell proliferation, survival, and migration. In some cases, HER2 is overexpressed with too many copies of the HER2 gene or too much HER2 protein on the surface of cancer cells. This overexpression can lead to uncontrolled cell growth and is associated with more aggressive forms of cancer[1].
Approximately 20-30% of breast cancers overexpress HER2. These tumors tend to be more aggressive and have a higher risk of recurrence compared to HER2-negative breast cancers. There are 5% HER2 activating mutations, which were shown negatively in the HER2 IHC clinical category with hyperactivated signaling.[2], which can lead to metastatic breast cancer[3]. The discovery of HER2 mutations led to targeted therapies to inhibit HER2 signaling. These therapies include drugs like neratinib, trastuzumab (Herceptin), pertuzumab (Perjeta), ado-trastuzumab emtansine (Kadcyla), and trastuzumab deruxtecan (T-DXd). These drugs can effectively block HER2 signaling, slowing or halting the growth of HER2-positive breast cancer cells.[4,5,6,7]. HER2 status is an essential prognostic and predictive marker in breast cancer. HER2-positive breast cancer tends to have a poorer prognosis compared to HER2-negative breast cancer.[8]However, targeted therapies have significantly improved outcomes for patients with HER2-positive breast cancer.[9,10]. Adaptive and acquired drug resistance have been challenges for breast cancer[11,12,13], especially HER2-mutated metastatic cancer[14,15,16]. Beyond breast cancer, HER2 alterations, including overexpression and activating mutations, have been identified in other cancer types, such as gastric and gastroesophageal cancer (4.4%-53.4%) [17,18] and non-small-cell lung cancer(4%) [19,20,21,22], endometrial(20%-40%)[23,24], and ovarian cancers[25]. Continue studies on HER2 and new therapies targeting HER2 signaling are developing in these cancers.
2. HER2-Regulated Signaling Pathways and Its Role in Cellular Function
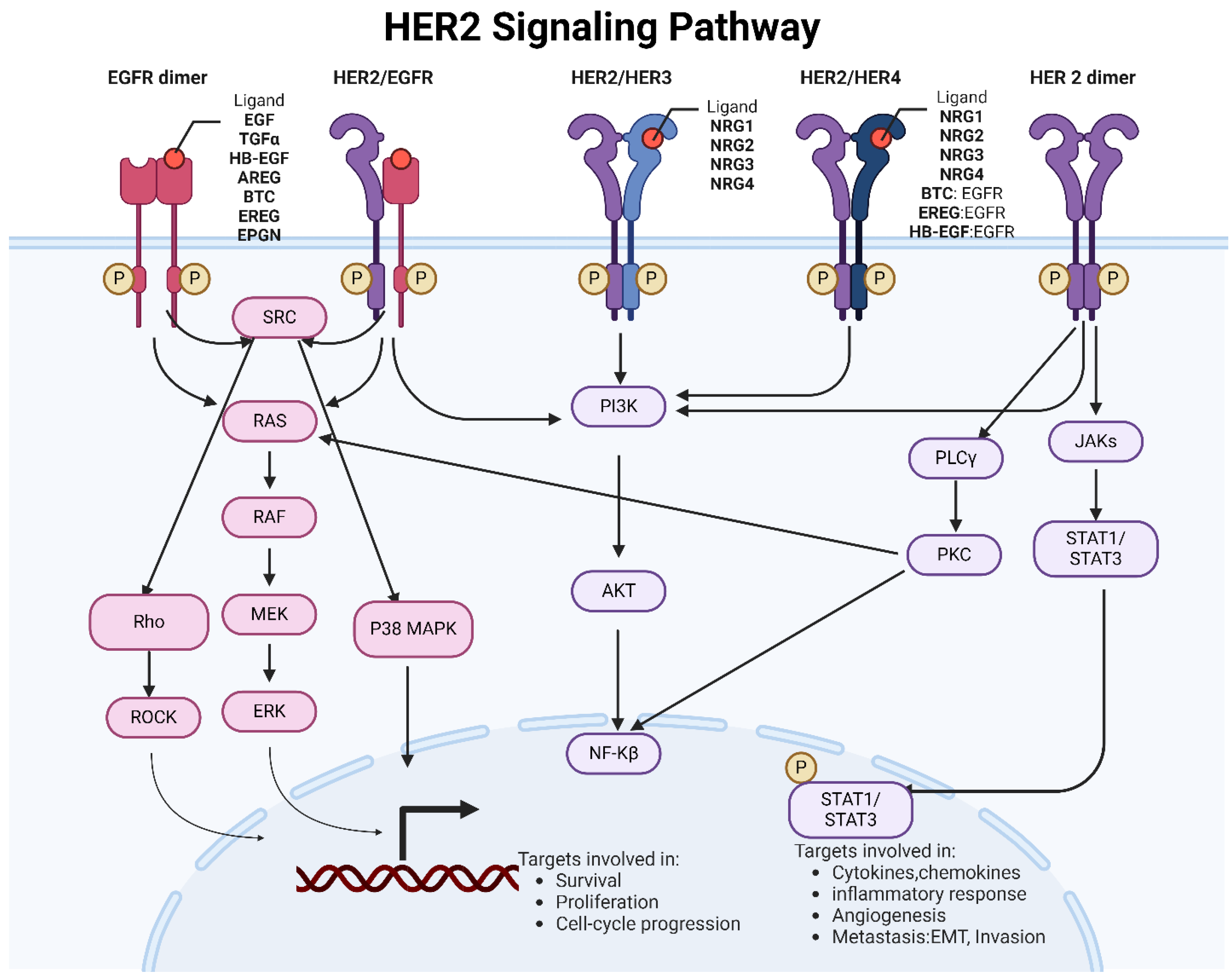
The HER2 signaling pathway is a critical regulatory system in cellular function, particularly in the context of cell growth, survival, and differentiation. Figure 1A shows a summary of the HER2 signaling pathway and its role in cellular function.
The HER2 receptor has no known ligand that directly binds to it. Instead, HER2 is the preferred partner for forming heterodimers (pairing) with other EGFR family members[26]. HER2 dimerization is a crucial aspect of its signaling mechanism and plays a significant role in the biology of various cancers. The formation of dimers—either homodimers or heterodimers—is a key step in the activation of HER2 signaling pathways[27]. The nature and consequences of HER2 dimerization can vary significantly across different cancer types. In breast cancer, HER2 frequently forms heterodimers with HER3[28]. The HER2-HER3 heterodimer is highly potent in activating downstream signaling pathways, such as PI3K/AKT and MAPK, promoting cell proliferation and survival. HER2 expression in gastric cancer can be more heterogeneous compared to breast cancer, with variable patterns of expression within and between tumors. HER2 in gastric cancer also dimerizes with HER3 and potentially with other EGFR family members, contributing to aggressive tumor behavior[29]. HER2 mutations, insertions in exon 20, can promote constitutive heterodimerization[30,31] and activation of HER2 without ligand binding in non-small cell lung cancer (NSCLC)[32].HER2 can dimerize with EGFR in colorectal cancer, influencing responsiveness to EGFR-targeted therapies like cetuximab[33]. HER2 is overexpressed in a subset of ovarian cancers, often forming dimers with other EGFR family members[34].
HER2 homodimers are generally less potent in signaling compared to heterodimers. However, HER2-HER3 heterodimers are particularly potent, activating robust downstream signaling pathways[1]. HER3, with its six binding sites for the p85 subunit of PI3K, plays a critical role in PI3K/AKT pathway activation when dimerized with HER2[35].
HER2 crosstalk with other signaling pathways through downstream targets. Akt activation not only promotes cell survival and growth but also cross-talks with other pathways. Akt can phosphorylate and activate mTOR (mammalian target of rapamycin), which regulates protein synthesis and cell growth[36]. Additionally, Akt can phosphorylate and inhibit TSC2 (tuberous sclerosis complex 2), activating the mTORC1 complex and subsequent phosphorylation of downstream targets involved in protein synthesis[37]. HER2 activation can also lead to the activation of Ras, a small GTPase protein. Ras activates Raf, which initiates a cascade leading to the activation of ERK (extracellular signal-regulated kinase)[38]. ERK activation can crosstalk with the PI3K/Akt/mTOR pathway by phosphorylating and activating mTORC1 or directly phosphorylating and inhibiting TSC2, leading to mTORC1 activation[39]. ERK can also crosstalk with the JNK (c-Jun N-terminal kinase) pathway, influencing cellular responses such as apoptosis and proliferation[40,41]. HER2 signaling can crosstalk with the Wnt/β-catenin pathway, crucial in regulating tumor progression[42]. Activation of HER2 signaling can lead to stabilization and nuclear translocation of β-catenin, a key mediator of Wnt signaling, promoting the transcription of Wnt target genes involved in cell proliferation and survival[43]. HER2 signaling can activate the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway, which regulates inflammation, immunity, and cell survival genes[44]. Activation of HER2 signaling can lead to phosphorylation and degradation of IκBα (inhibitor of NF-κB), resulting in the nuclear translocation of NF-κB and transcriptional activation of its target genes[45]. In hormone receptor-positive breast cancer, HER2 signaling can crosstalk with estrogen receptor (ER) signaling, influencing the response to endocrine therapy[46]. HER2 activation can lead to phosphorylation and activation of ER, enhancing its transcriptional activity and promoting cell proliferation[47].
Understanding the crosstalk between HER2 signaling and other pathways is critical for elucidating the complex mechanisms underlying cancer progression and treatment resistance. Targeting multiple signaling pathways simultaneously may be necessary to inhibit tumor growth and improve patient outcomes effectively.
2.1. Interaction with Other Members of the EGFR Family
HER2 is unique because it can form homodimers (pairs) without ligand binding. This ligand-independent activity contributes to its role as an oncogenic driver in cancer. HER2 can form heterodimers with HER1 (EGFR). This interaction is significant, especially in cancer biology, as HER2/HER1 heterodimers have been shown to enhance oncogenic signaling pathways more potently than HER1 homodimers or HER2 homodimers alone[48].
HER2 has a unique relationship with HER3. While HER3 has impaired kinase activity, it contains multiple docking sites for phosphotyrosine, making it an excellent partner for signaling amplification. HER2/HER3 heterodimers are particularly potent in activating downstream signaling pathways due to the strong recruitment and phosphorylation of HER3 by HER2[49].
HER2 can also form heterodimers with HER4. Like HER3, HER4 lacks strong kinase activity but possesses multiple tyrosine phosphorylation sites. HER2/HER4 heterodimers have been implicated in mammary gland development, where they play a role in lactation[50]. However, their significance in cancer biology is less clear than HER2/HER1 and HER2/HER3 heterodimers. The ligand-independent activity of HER2 contributes to its role as an oncogenic driver in various cancers.
Understanding the intricate interactions between HER2 and other EGFR family members is crucial for elucidating the complexities of cellular signaling, particularly in cancer biology. Targeted therapies that disrupt these interactions, such as monoclonal antibodies and small molecule inhibitors, have shown significant clinical efficacy in treating HER2-driven malignancies like HER2-positive breast cancer.
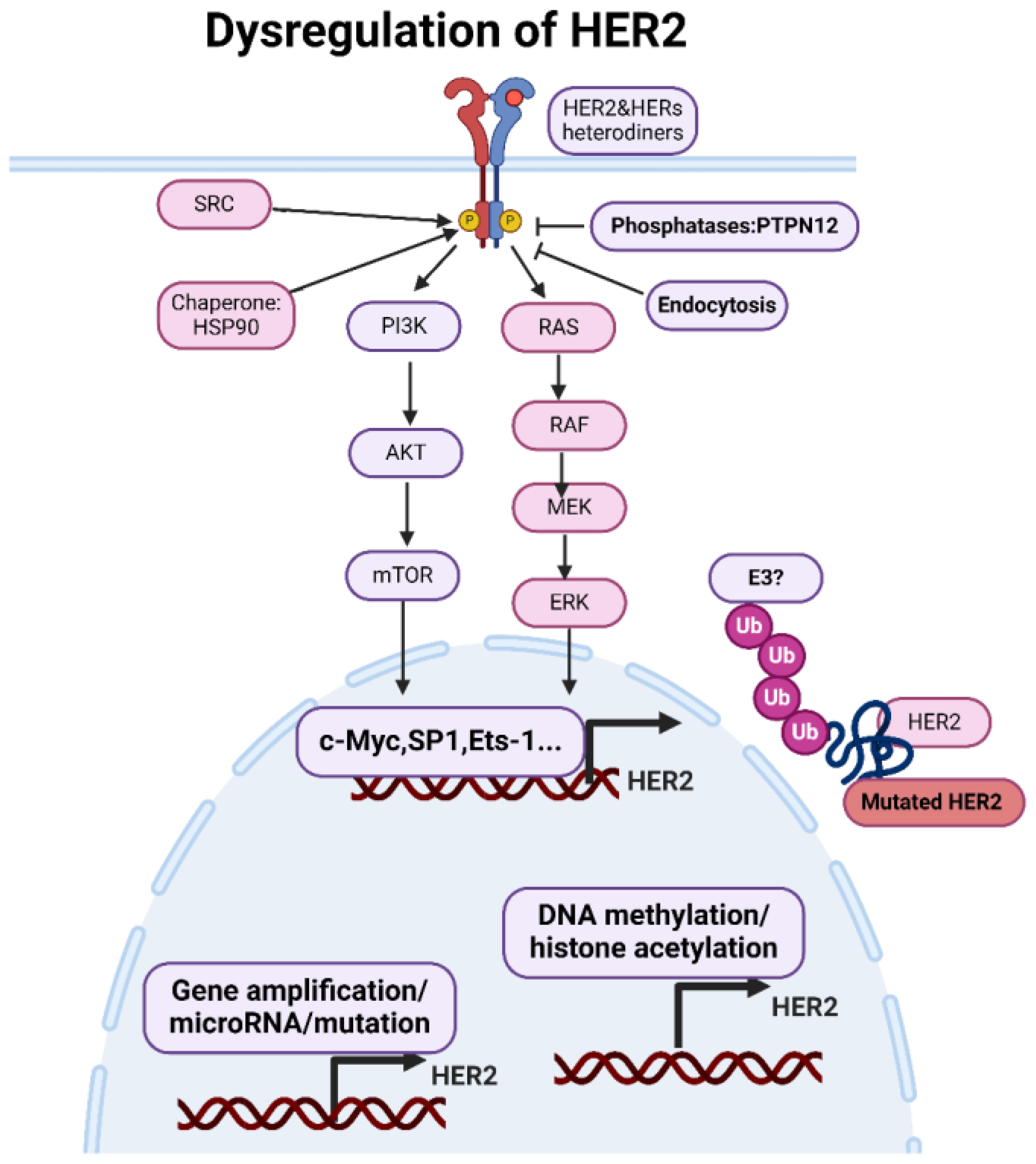
Figure 2.
Dysregulations of HER2. The expression of HER2 is primarily regulated at the level of gene transcription. Transcription factors. Epigenetic modifications can regulate HER2 gene expression by modulating chromatin structure and accessibility to transcriptional machinery. MicroRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression by binding to the 3' untranslated region (UTR) of target mRNAs and inhibiting translation or promoting mRNA degradation. The chaperone protein Hsp90 stabilizes HER2 and facilitates its maturation and activation. Conversely, phosphatases such as PTPN12 can dephosphorylate HER2, attenuating its signaling activity. Internalization of activated HER2 receptors via endocytosis serves as a mechanism for downregulating HER2 signaling. Endocytosed receptors can undergo lysosomal degradation, leading to the attenuation of HER2-mediated signaling pathways. Figure made using Biorender.
Figure 2.
Dysregulations of HER2. The expression of HER2 is primarily regulated at the level of gene transcription. Transcription factors. Epigenetic modifications can regulate HER2 gene expression by modulating chromatin structure and accessibility to transcriptional machinery. MicroRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression by binding to the 3' untranslated region (UTR) of target mRNAs and inhibiting translation or promoting mRNA degradation. The chaperone protein Hsp90 stabilizes HER2 and facilitates its maturation and activation. Conversely, phosphatases such as PTPN12 can dephosphorylate HER2, attenuating its signaling activity. Internalization of activated HER2 receptors via endocytosis serves as a mechanism for downregulating HER2 signaling. Endocytosed receptors can undergo lysosomal degradation, leading to the attenuation of HER2-mediated signaling pathways. Figure made using Biorender.
3. Dysregulations of HER2
3.1. Regulation of HER2 Expression and Activation and Mechanisms of Dysregulation
Like other receptors, the expression and activation of HER2 are tightly regulated by various mechanisms at both transcriptional and post-translational levels.
The expression of HER2 is primarily regulated at the level of gene transcription. Transcription factors, such as Sp1, AP-2, and Ets-1, can bind to specific regulatory elements in the HER2 gene promoter region, cite, thereby activating or repressing HER2 gene transcription. Factors influencing transcription factor activity, such as growth factors, hormones, and oncogenes, can also indirectly affect HER2 expression levels. Epigenetic modifications, such as DNA methylation and histone acetylation, can regulate HER2 gene expression by modulating chromatin structure and accessibility to transcriptional machinery. Aberrant epigenetic changes, such as hypomethylation of the HER2 promoter region, can increase HER2 expression in cancer cells. MicroRNAs (miRNAs) are small non-coding RNAs that can post-transcriptionally regulate gene expression by binding to the 3' untranslated region (UTR) of target mRNAs and inhibiting translation or promoting mRNA degradation. Several miRNAs have been identified as regulators of HER2 expression, with some miRNAs suppressing HER2 expression in cancer cells.
Several regulatory proteins modulate HER2 activation by either promoting or inhibiting dimerization and/or tyrosine kinase activity. For example, the chaperone protein Hsp90 stabilizes HER2 and facilitates its maturation and activation. Conversely, phosphatases such as PTPN12 can dephosphorylate HER2, attenuating its signaling activity. Internalization of activated HER2 receptors via endocytosis serves as a mechanism for downregulating HER2 signaling. Endocytosed receptors can undergo lysosomal degradation, leading to the attenuation of HER2-mediated signaling pathways.
HER2 dysregulation, characterized by overexpression or aberrant activation, can occur through various mechanisms, leading to increased HER2 signaling and contributing to cancer development and progression. One of the most common mechanisms of HER2 dysregulation is gene amplification, where multiple copies of the HER2 gene are present in cancer cells. Gene amplification resulting in overexpression of the HER2 receptor on the cell surface.HER2 gene amplification can occur through chromosomal abnormalities, such as gene amplification at the 17q12-q21 locus, where the HER2 gene is located. Transcription factors such as c-Myc, Sp1, and Ets-1 can bind to regulatory elements in the HER2 gene promoter region and promote HER2 gene transcription. Aberrant activation of signaling pathways, such as the Ras/Raf/MEK/ERK pathway or the PI3K/Akt pathway, can also upregulate HER2 transcription. Post-translational modifications, such as phosphorylation, ubiquitination, and glycosylation, can modulate HER2 protein stability, localization, and activity. Phosphorylation of HER2 tyrosine residues by kinases, such as Src or HER family members, can enhance HER2 signaling activity. Ubiquitination of HER2 by E3 ubiquitin ligases can target HER2 for proteasomal degradation, regulating HER2 protein levels. Aberrant interactions with other receptors or signaling molecules can enhance HER2 signaling activity and promote cancer progression.
Mutations in the HER2 gene can lead to alterations in the structure and function of the HER2 protein, contributing to cancer development and progression. While HER2 gene amplification and overexpression are more common in cancer, mutations in HER2 have also been identified in certain cancer types (Figure 4). Missense mutations result in substituting one amino acid for another in the HER2 protein sequence. These mutations can alter the protein's structure and function, affecting its signaling activity. Frameshift mutations result from inserting or deleting nucleotides in the HER2 gene, leading to a shift in the reading frame during translation, produce a truncated or altered HER2 protein. Nonsense mutations lead to the premature termination of protein synthesis, resulting in the production of a truncated and likely non-functional HER2 protein. Mutations in the HER2 gene are relatively rare compared to HER2 gene amplification and overexpression. HER2 mutations have been identified in various cancer types, including breast cancer, gastric cancer, lung cancer, and colorectal cancer.
HER2 mutations can result in constitutive activation of HER2 signaling pathways, leading to uncontrolled cell growth, proliferation, and survival. Some HER2 mutations may confer resistance to HER2-targeted therapies. Clinical trials are ongoing to evaluate the efficacy of HER2-targeted therapies in patients with HER2-mutant cancers, to improve outcomes for these patients.
Overall, while less common than HER2 gene amplification, mutations in the HER2 gene can significantly impact cancer biology and treatment outcomes. Understanding the functional consequences of HER2 mutations and their clinical implications is essential for developing personalized treatment strategies for patients with HER2-mutant cancers. Dysregulation of HER2 expression and activation, often observed in cancer, underscores the importance of targeted therapies aimed at inhibiting HER2 signaling in cancer treatment.
3.2. HER2 Crosstalk with Hormone Receptor Pathways
In breast cancer, HER2 signaling cross-talks with hormone receptor pathways, such as the estrogen receptor (ER) and the progesterone receptor (PR). This interaction is crucial in tumor development, progression, and therapeutic response.
HER2 activation can enhance the transcriptional activity of hormone receptors, such as ER and PR, by activating downstream signaling pathways. HER2-mediated PI3K/Akt/mTOR pathway activation can phosphorylate and activate ER, leading to increased ER transcriptional activity and estrogen-dependent cell proliferation. HER2 signaling can also modulate the expression and function of co-regulatory proteins that interact with hormone receptors, further enhancing hormone receptor activity.
Hormone receptors, particularly ER, can also regulate the expression of HER2 in breast cancer cells. Estrogen stimulation of ER-positive breast cancer cells can induce HER2 expression, leading to increased HER2 signaling activity. Conversely, inhibition of ER signaling through hormone therapy (e.g., tamoxifen) or estrogen deprivation can downregulate HER2 expression and attenuate HER2 signaling. Hormone receptor signaling can influence the response of HER2-positive breast cancer cells to HER2-targeted therapies, such as trastuzumab and lapatinib. Activation of ER signaling can promote resistance to HER2-targeted therapies by activating survival pathways and attenuating the cytotoxic effects of HER2 inhibition. Conversely, inhibition of ER signaling, either alone or in combination with HER2-targeted therapies, can enhance the efficacy of HER2-targeted treatments by sensitizing HER2-positive breast cancer cells to therapy.
The crosstalk between HER2 and hormone receptor pathways has important clinical implications for managing HER2-positive breast cancer. Combination therapies targeting both HER2 and hormone receptor pathways, such as dual HER2 blockade (e.g., trastuzumab plus pertuzumab) and endocrine therapy (e.g., aromatase inhibitors or selective estrogen receptor modulators), have been shown to improve outcomes in patients with HER2-positive, hormone receptor-positive breast cancer. Biomarkers such as hormone receptor status and HER2 expression are used to guide treatment decisions and predict response to targeted therapies in breast cancer patients. Understanding the complex crosstalk between HER2 and hormone receptor pathways is essential for developing optimal treatment strategies and improving outcomes for patients with HER2-positive breast cancer. Combination therapies targeting both HER2 and hormone receptor signaling pathways represent a promising approach for personalized treatment in this patient population.
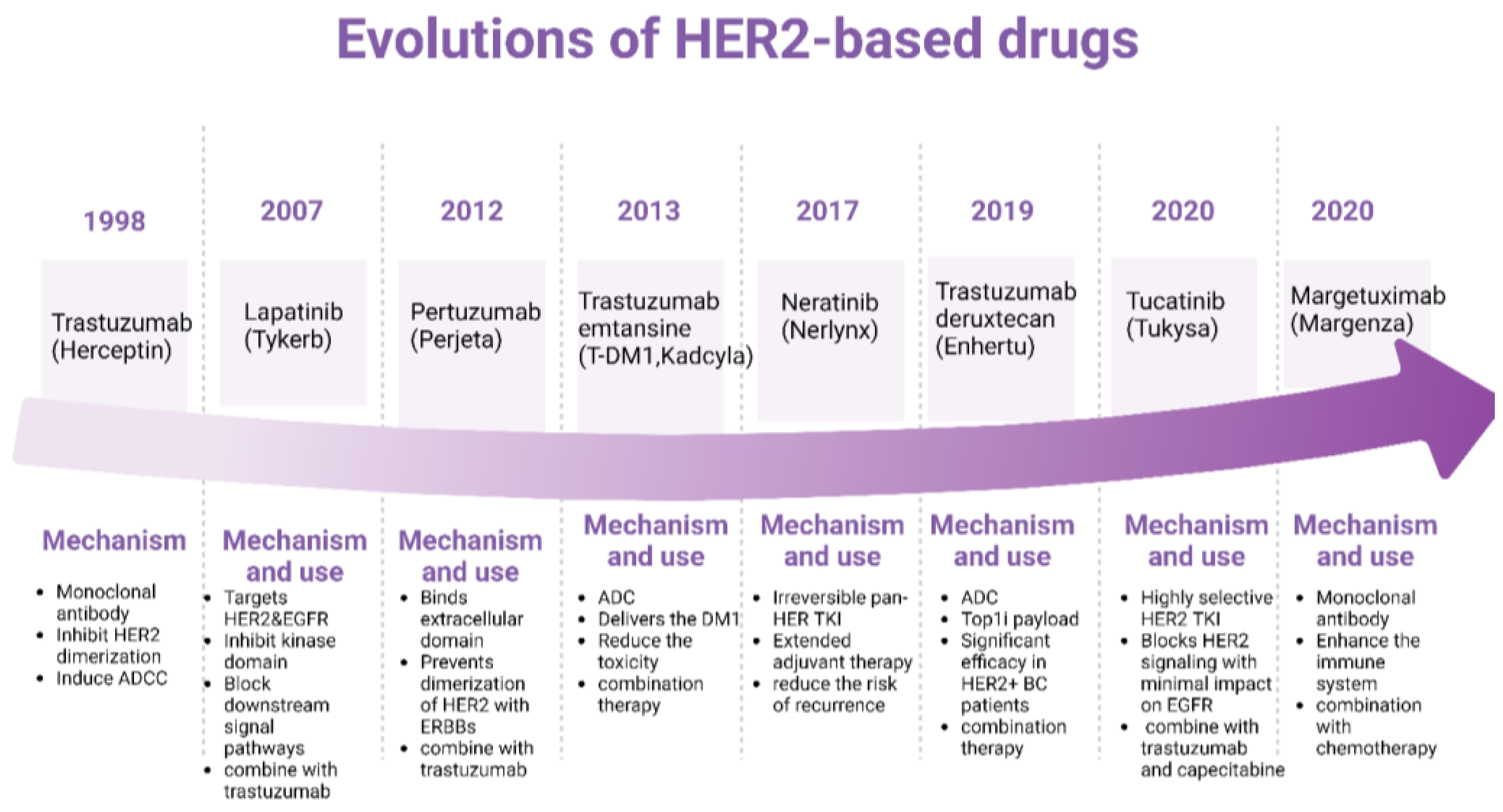
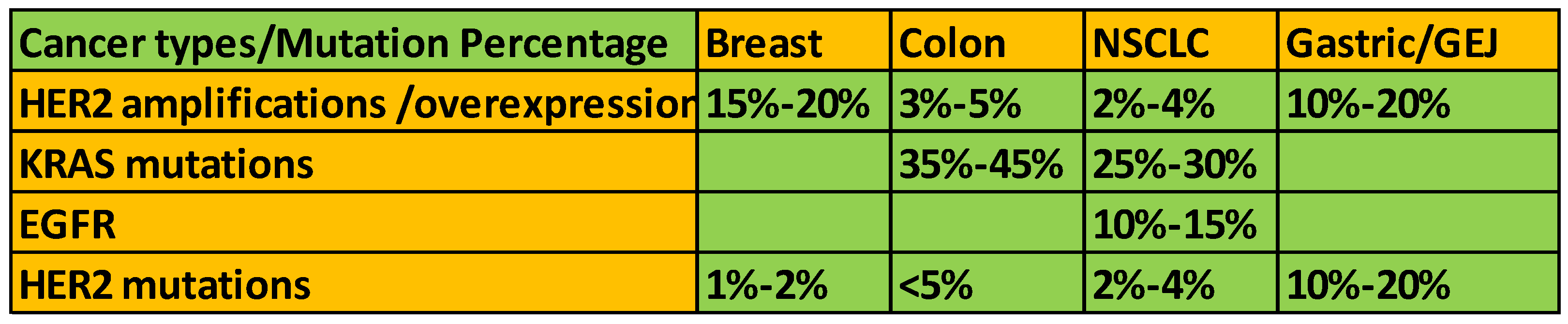
Figure 3.
Evolutions of HER2-based drugs and mechanism. The upper timeline shows the corresponding HER2-based drugs approved by the FDA. At the bottom are the corresponding mechanisms. Figure made using Biorender.
Figure 3.
Evolutions of HER2-based drugs and mechanism. The upper timeline shows the corresponding HER2-based drugs approved by the FDA. At the bottom are the corresponding mechanisms. Figure made using Biorender.
Figure 4.
HER2 alterations in different cancer types.
4. HER2-Based Therapeutic Strategies and Future Direction
4.1. Evolutions of HER2-Based Drugs
The evolution of HER2-targeted drugs has significantly transformed the treatment landscape for HER2-positive cancers, particularly breast cancer. The development of these drugs has been a milestone in precision medicine, providing more effective and personalized treatment options. Figure 3 is the overview of the key HER2-targeted drugs and their evolution. Trastuzumab is a monoclonal antibody that binds to the extracellular domain of the HER2 protein, inhibiting its dimerization and signaling. It also induces antibody-dependent cellular cytotoxicity (ADCC). It was the first HER2-targeted therapy and significantly improved outcomes for HER2-positive breast cancer patients [51]. Lapatinib is a small tyrosine kinase inhibitor (TKI) that targets HER2 and EGFR (ErbB1). It inhibits the intracellular tyrosine kinase domain, blocking downstream signaling pathways. Often used in combination with other therapies for patients with advanced or metastatic HER2-positive breast cancer who have progressed on trastuzumab[52]. Pertuzumab is a monoclonal antibody that binds to a different epitope on the HER2 extracellular domain than trastuzumab. It prevents dimerization of HER2 with other ErbB receptors. Typically used in combination with trastuzumab and chemotherapy, significantly enhancing the anti-tumor activity[53]. T-DM1 is an antibody-drug conjugate that combines trastuzumab with a cytotoxic agent (DM1). It delivers the cytotoxic drug directly to HER2-positive cells, reducing systemic toxicity. Provides targeted delivery of chemotherapy, improving efficacy and reducing side effects[54]. Neratinib is an irreversible pan-HER TKI that inhibits HER1, HER2, and HER4.Used as extended adjuvant therapy for early-stage HER2-positive breast cancer to reduce the risk of recurrence after initial trastuzumab therapy[55]. Tucatinib is a highly selective HER2 TKI that blocks HER2 signaling pathways with minimal impact on EGFR. Often combined with trastuzumab and capecitabine for treating advanced unresectable or metastatic HER2-positive breast cancer[56]. Enhertu is an antibody-drug conjugate that combines trastuzumab with a topoisomerase I inhibitor payload. It provides a potent cytotoxic effect upon internalization by HER2-positive cells[57]. Demonstrated significant efficacy in patients with advanced HER2-positive breast cancer, even those who have previously received multiple lines of HER2-targeted therapies. Margetuximab is a monoclonal antibody similar to trastuzumab but engineered to enhance the immune system's ability to kill cancer cells[58]. Used in combination with chemotherapy for patients with advanced HER2-positive breast cancer who have received prior anti-HER2 therapies.
Bispecific antibodies are currently being developed to target HER2 and another antigen, potentially increasing efficacy simultaneously. Immunotherapy strategies are being explored to induce a more robust immune response against HER2-positive cancer cells. Research continues on next-generation TKIs and ADCs with improved efficacy and safety profiles. The continuous evolution of HER2-targeted therapies highlights the importance of understanding the molecular mechanisms of cancer and developing treatments that can more precisely target cancer cells while sparing normal tissues.
4.2. HER2 Diagnostic Approaches
Diagnosing HER2-positive breast cancer involves several approaches to assess HER2 expression or gene amplification accurately. Immunohistochemistry, fluorescence in situ hybridization, and Chromogenic in situ hybridization are molecular techniques to detect HER2 gene amplification in tumor cells. Like FISH, CISH uses DNA probes that target the HER2 gene region, but the probes are labeled with chromogenic substances. Silver in situ hybridization (SISH) is a variation of CISH that uses silver staining to visualize HER2 gene amplification. SISH provides clear and crisp signals that can be easily interpreted by pathologists. Next-generation sequencing is a high-throughput molecular technique that can detect HER2 gene mutations and gene amplification and provide comprehensive genomic profiling of tumors. NGS may be used in research settings or in clinical practice for patients with advanced or metastatic breast cancer to guide targeted therapy decisions. Multiplexed assays, such as the PAM50 gene expression assay or the Oncotype DX Breast Recurrence Score, may provide additional information about HER2 status and tumor biology, aiding in treatment decision-making and prognosis assessment.
These diagnostic approaches are crucial in accurately identifying HER2-positive breast cancer patients who may benefit from HER2-targeted therapies such as neratinib, trastuzumab deruxtecan, pertuzumab, and ado-trastuzumab emtansine (T-DM1). With advances in molecular profiling and biomarker testing, personalized treatment approaches based on HER2 expression, hormone receptor status, and genomic alterations are increasingly utilized to tailor therapy to individual patients. Biomarker-driven clinical trials evaluate targeted therapies and combination regimens in specific subgroups of HER2-positive breast cancer patients, such as those with HER2 mutations or amplifications, hormone receptor positivity, or specific genomic alterations.
Overall, HER2 status plays a critical role in guiding treatment decisions and predicting response to therapy in breast cancer. Ongoing clinical trials continue to expand the treatment landscape for HER2-positive breast cancer, to further improve outcomes and quality of life for patients with this subtype of cancer.
5. The Role of HER2 in Metastasis
Clinical studies have shown that HER2 overexpression, amplification and mutations are associated with increased metastatic potential and poor prognosis in breast, gastric, and ovarian cancers. HER2-positive breast cancer is associated with a higher risk of metastasis.
HER2 activation leads to the activation of downstream signaling pathways which promote cell cycle progression and proliferation. ERK is activated, which phosphorylates and activates transcription factors that promote cell cycle progression, such as c-Myc and cyclin D1. HER2 activation of the PI3K/Akt/mTOR pathway leads to the phosphorylation and inactivation of pro-apoptotic proteins such as Bad[59], thereby preventing apoptosis. HER2 signaling can also activate the NF-κB pathway through AKT, which regulates cell survival and inflammation genes[60].
HER2 has been implicated in promoting metastasis. HER2 overexpression is associated with increased invasiveness and metastatic potential in cancer cells. HER2 signaling promotes epithelial-mesenchymal transition (EMT)[61], a process in which epithelial cells acquire mesenchymal characteristics, allowing them to migrate and invade surrounding tissues. HER2 signaling can upregulate the expression of matrix metalloproteinases (MMPs) and facilitate cancer cell invasion and metastasis[62]. These enzymes degrade the extracellular matrix (ECM) and facilitate tumor cell invasion through the surrounding tissue, thus promoting invasion and intravasation. Once cancer cells invade through the ECM, HER2 signaling can also promote intravasation, the process by which cancer cells enter into blood or lymphatic vessels, enabling them to disseminate to distant organs.
Additionally, HER2 signaling can promote angiogenesis, the formation of new blood vessels, which is essential for supplying nutrients and oxygen to metastatic tumors by regulating HIF1α [63]. The formation of new blood vessels, within the primary tumor microenvironment. Angiogenesis provides a blood supply to the growing tumor and facilitates nutrient and oxygen delivery. Increased angiogenesis in the primary tumor creates a favorable microenvironment for cancer cell intravasation and dissemination into the bloodstream, promoting metastasis. Once cancer cells disseminate through the bloodstream or lymphatic system, HER2 signaling can promote their survival and colonization at distant metastatic sites.HER2-driven PI3K/Akt/mTOR pathway, enhance cancer cell survival and migration in the metastatic microenvironment, establishing secondary tumors[3].
Clinical studies have shown that HER2 is associated with increased metastatic potential and poor prognosis in various cancers. Understanding the role of HER2 in metastasis is crucial for developing effective therapeutic strategies to target HER2-driven metastatic disease. HER2-targeted therapies have shown promise in treating HER2-positive metastatic cancers, highlighting the importance of HER2 as a therapeutic target in metastatic disease.
6. The Role of HER2 in Immune Regulation and Response
6.1. HER2 and Immune
HER2-positive cancer has implications for the immune response within the tumor microenvironment. HER2 is a tumor-associated antigen that can be recognized by the immune system. Overexpression of HER2 on the surface of cancer cells can lead to the presentation of HER2 peptides by major histocompatibility complex (MHC) molecules, eliciting an immune response[64]. HER2-targeted therapies, such as trastuzumab and pertuzumab, can enhance immune recognition of HER2-positive tumor cells by promoting antibody-dependent cellular cytotoxicity (ADCC) and immune-mediated tumor cell death[65].
HER2-positive breast tumors often exhibit increased immune infiltration compared to HER2-negative tumors. Tumor-infiltrating lymphocytes (TILs), particularly cytotoxic T cells, are commonly found in HER2-positive breast cancer.The presence of TILs in HER2-positive breast tumors has been associated with improved prognosis and response to HER2-targeted therapies, suggesting a role for the immune system in controlling HER2-positive breast cancer[66]. Expression of immune checkpoint molecules, such as programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), may modulate the immune response in HER2-positive breast cancer[67]. Preclinical and clinical studies have shown that HER2-positive breast tumors can express PD-L1, which may contribute to immune evasion and resistance to HER2-targeted therapies[68]. Combination therapies targeting HER2 and immune checkpoints are being investigated in clinical trials to overcome immune evasion and enhance antitumor immune responses in HER2-positive breast cancer.
Immunotherapy, particularly immune checkpoint inhibitors targeting PD-1/PD-L1 or CTLA-4, has shown limited efficacy as monotherapy in HER2-positive breast cancer. However, combination approaches combining HER2-targeted therapies with immune checkpoint inhibitors or other immunomodulatory agents are being explored in clinical trials to enhance antitumor immune responses and improve outcomes for patients with HER2-positive breast cancer[68].
Overall, HER2-positive breast cancer is associated with immune infiltration and interactions between HER2-targeted therapies and the immune system. Understanding the complex interplay between HER2 and the immune response is essential for developing effective immunotherapeutic strategies to improve outcomes for patients with HER2-positive breast cancer.
6.2. HER2 and TME
The tumor microenvironment (TME) plays a crucial role in the development, progression, and response to therapy in HER2-positive breast cancer. The TME of HER2-positive tumors is characterized by a complex interplay of various cell types, including cancer cells, stromal cells, immune cells, endothelial cells, and extracellular matrix components. HER2-positive tumors often exhibit increased immune infiltration, with higher tumor-infiltrating lymphocyte (TILs) levels than HER2-negative tumors.
HER2-positive tumors can attract immune cells to the TME through various mechanisms, including the secretion of chemokines and cytokines. Tumor-infiltrating immune cells, such as T, B, natural killer (NK), and dendritic cells, interact with HER2-positive cancer cells and influence tumor growth, invasion, and metastasis. Despite immune infiltration, HER2-positive tumors often exhibit immunosuppressive features within the TME, such as the expression of immune checkpoint molecules (e.g., PD-L1) and regulatory T cells (Tregs). These immunosuppressive mechanisms can impair antitumor immune responses and contribute to immune evasion and resistance to therapy in HER2-positive breast cancer. HER2 signaling can directly influence the composition and function of the TME in breast cancer. HER2 activation in cancer cells can produce cytokines, chemokines, and growth factors that modulate the TME. HER2-positive breast tumors may exhibit alterations in angiogenesis, extracellular matrix remodeling, and immune cell recruitment due to dysregulated HER2 signaling.
Understanding the interactions between HER2 and the TME is essential for developing effective therapeutic strategies for HER2-positive breast cancer. Combination therapies targeting HER2 and the TME components, such as immune checkpoint inhibitors, angiogenesis inhibitors, and stromal-targeting agents, are being investigated in preclinical and clinical studies to improve treatment outcomes in HER2-positive breast cancer. TME plays a critical role in shaping the behavior and response to therapy in HER2-positive breast cancer. Elucidating the complex interactions between HER2 signaling and the TME components is essential for developing novel therapeutic approaches to overcome treatment resistance and improve outcomes for patients with HER2-positive breast cancer. Modulating HER2 signaling may impact immune regulation within the TME and enhance antitumor immune responses.
6.3. HER2 and Immune Regulation
HER2 downstream, the JAK/STAT pathway, is Involved in immune response regulation and can influence inflammation. HER2-positive tumors often exhibit altered immune cell infiltration. HER2 signaling can attract immunosuppressive cells like regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), creating an immunosuppressive TME. HER2 signaling can upregulate cytokines such as IL-6 and chemokines like CCL2, CXCR4, promoting a pro-inflammatory and immunosuppressive environment conducive to tumor growth and metastasis[69]. HER2 signaling can induce the expression of PD-L1 on tumor cells, which binds to PD-1 on T cells, leading to T cell exhaustion and immune evasion. HER2-positive tumors can downregulate MHC class I molecules, impairing antigen presentation and reducing CTL-mediated killing of tumor cells[70]NK cells can target HER2-positive tumors through antibody-dependent cellular cytotoxicity (ADCC). Therapeutic antibodies like trastuzumab enhance ADCC by binding to HER2 on tumor cells and recruiting NK and macrophage cells to mediate tumor cell lysis[71]. HER2-targeted therapies can improve the presentation of HER2-derived peptides to T cells, boosting anti-tumor T-cell responses.
HER2 expression levels in breast cancer may serve as a potential biomarker for predicting response to immune checkpoint inhibitors and other immunotherapies. Tumors with high HER2 expression may be more immunogenic and responsive to immunotherapy approaches. Addressing the immunosuppressive mechanisms associated with HER2 signaling can help overcome resistance to HER2-targeted therapies and immunotherapies. The rationale is that HER2-targeted therapies can enhance antigen presentation and T-cell activation, making tumors more susceptible to checkpoint blockade[72]. The interplay between HER2 signaling and immune regulation is complex and has significant implications for developing and optimizing cancer therapies. HER2-targeted treatments inhibit tumor growth directly and modulate the immune response, offering potential synergistic benefits when combined with immunotherapies. Ongoing research into this relationship is crucial for advancing therapeutic strategies and improving outcomes for patients with HER2-positive cancers.
7. Conclusions and Future Perspectives
HER2 (human epidermal growth factor receptor 2) research continues to be a dynamic and rapidly evolving field, driven by the ongoing need to improve outcomes for patients with HER2-positive cancers. Development of next-generation ADCs aims to improve efficacy and reduce side effects compared to earlier ADCs like T-DM1 and T-DXd is needed. Bispecific Antibodies are the creation of antibodies that can simultaneously target HER2 and another tumor-specific antigen, enhancing specificity and reducing resistance, combining HER2-targeted therapies with immune checkpoint inhibitors (like PD-1/PD-L1 inhibitors) to enhance anti-tumor immune responses and combining HER2 inhibitors with drugs targeting other pathways (like PI3K/AKT/mTOR) to overcome resistance and improve efficacy. Precision Medicine and novel biomarker development for detection are needed. Utilizing genomic and proteomic profiling to tailor HER2-targeted treatments to individual patients based on specific molecular characteristics of their tumors. Identifying biomarkers that can predict which patients will benefit from HER2-targeted therapies or develop resistance will allow for more precise treatment adjustments.
Still, it is crucial to investigate the molecular mechanisms that lead to resistance to HER2-targeted therapies, such as activating mutations in the HER2 gene. Despite the substantial bulk, scRNA and spatial transcriptomics data on the acknowledgment of their heterogeneity, there remains a lack of timing resolution regarding the phenotypic state during cancer and metastasis development and progression. This analytical gap prevents us from obtaining unbiased spatiotemporal information on dynamic changes in gene expression, limiting our comprehension of the functional roles played by HER2 during drug resistance and metastasis. Addressing this gap will significantly advance our knowledge and pave the way for developing more targeted therapeutical interventions.
Identifying and targeting new molecules involved in resistance mechanisms to develop novel therapies to bypass or overcome resistance. Expanding HER2-targeted therapies to other cancers that express HER2, such as gastric, colorectal, and non-small cell lung cancers. Investigating the efficacy of HER2-targeted treatments in a tumor-agnostic manner, where HER2 overexpression or amplification determines treatment eligibility regardless of the cancer type. Using nanoparticles and other advanced drug delivery systems to improve the delivery and efficacy of HER2-targeted therapies, potentially reducing side effects and improving patient outcomes. Implementing adaptive clinical trial designs that allow for modifications based on interim results enhances clinical trial efficiency and success rate. Leveraging real-world data to complement clinical trials, providing insights into the long-term effectiveness and safety of HER2-targeted therapies in diverse patient populations.
Utilizing AI and machine learning to develop predictive models for patient response to HER2-targeted therapies, aiding in the decision-making process for personalized treatment plans, and employing advanced computational methods to analyze large datasets from clinical trials and real-world evidence, uncovering new insights and potential therapeutic targets. The future of HER2 research lies in an integrated approach that combines advanced therapeutic strategies, precision medicine, and cutting-edge technologies. By addressing the challenges of resistance, expanding the indications for HER2-targeted therapies, and leveraging innovations in drug delivery and clinical trial design, researchers aim to improve outcomes for patients with HER2-positive cancers significantly.
Funding
N/A.
Acknowledgments
N/A.
Conflicts of Interest
The author declares no conflict of interest.
References
- Iqbal, N. and N. Iqbal, Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol Biol Int, 2014. 2014: p. 852748. [CrossRef]
- Bose, R., et al., Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov, 2013. 3(2): p. 224-37. [CrossRef]
- Cheng, X., et al., Breast Cancer Mutations HER2V777L and PIK3CAH1047R Activate the p21-CDK4/6-Cyclin D1 Axis to Drive Tumorigenesis and Drug Resistance. Cancer Res, 2023. 83(17): p. 2839-2857. [CrossRef]
- Guo, L., et al., Neratinib for HER2-positive breast cancer with an overlooked option. Molecular Medicine, 2023. 29(1): p. 134. [CrossRef]
- Chan, A., Neratinib in HER-2-positive breast cancer: results to date and clinical usefulness. Ther Adv Med Oncol, 2016. 8(5): p. 339-50. [CrossRef]
- Cortes, J., et al., Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N Engl J Med, 2022. 386(12): p. 1143-1154. [CrossRef]
- Tolaney, S., New HER2-positive targeting agents in clinical practice. Curr Oncol Rep, 2014. 16(1): p. 359. [CrossRef]
- Abbasvandi, F., et al., Tumor characteristics and survival rate of HER2-low breast cancer patients: a retrospective cohort study. Sci Rep, 2023. 13(1): p. 16719. [CrossRef]
- Bose, R. and C.X. Ma, Breast Cancer, HER2 Mutations, and Overcoming Drug Resistance. N Engl J Med, 2021. 385(13): p. 1241-1243. [CrossRef]
- Ma, C.X., et al., The Phase II MutHER Study of Neratinib Alone and in Combination with Fulvestrant in HER2-Mutated, Non-amplified Metastatic Breast Cancer. Clin Cancer Res, 2022. 28(7): p. 1258-1267. [CrossRef]
- Li, Z., et al., Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell, 2018. 34(6): p. 893-905.e8. [CrossRef]
- Li, Q., et al., INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov, 2022. 12(2): p. 356-371. [CrossRef]
- Rimawi, M.F., C. De Angelis, and R. Schiff, Resistance to Anti-HER2 Therapies in Breast Cancer. American Society of Clinical Oncology Educational Book, 2015(35): p. e157-e164. [CrossRef]
- Jackson, C., et al., Clinical Significance of HER-2 Splice Variants in Breast Cancer Progression and Drug Resistance. International Journal of Cell Biology, 2013. 2013: p. 973584. [CrossRef]
- Xu, X., et al., HER2 Reactivation through Acquisition of the HER2 L755S Mutation as a Mechanism of Acquired Resistance to HER2-targeted Therapy in HER2(+) Breast Cancer. Clin Cancer Res, 2017. 23(17): p. 5123-5134. [CrossRef]
- Marín, A., et al., Acquired Secondary HER2 Mutations Enhance HER2/MAPK Signaling and Promote Resistance to HER2 Kinase Inhibition in Breast Cancer. Cancer Res, 2023. 83(18): p. 3145-3158. [CrossRef]
- Abrahao-Machado, L.F. and C. Scapulatempo-Neto, HER2 testing in gastric cancer: An update. World J Gastroenterol, 2016. 22(19): p. 4619-25. [CrossRef]
- Pous, A., et al., HER2-Positive Gastric Cancer: The Role of Immunotherapy and Novel Therapeutic Strategies. International Journal of Molecular Sciences, 2023. 24(14): p. 11403. [CrossRef]
- Riudavets, M., et al., Targeting HER2 in non-small-cell lung cancer (NSCLC): a glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open, 2021. 6(5): p. 100260. [CrossRef]
- Mazières, J., et al., Lung Cancer That Harbors an HER2 Mutation: Epidemiologic Characteristics and Therapeutic Perspectives. Journal of Clinical Oncology, 2013. 31(16): p. 1997-2003. [CrossRef]
- Garrido-Castro, A.C. and E. Felip, HER2 driven non-small cell lung cancer (NSCLC): potential therapeutic approaches. Translational Lung Cancer Research, 2013. 2(2): p. 122-127. [CrossRef]
- Mazières, J., et al., Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Annals of Oncology, 2016. 27(2): p. 281-286. [CrossRef]
- Balestra, A., D. Larsimont, and J.C. Noël, HER2 Amplification in p53-Mutated Endometrial Carcinomas. Cancers (Basel), 2023. 15(5). [CrossRef]
- Diver, E.J., et al., The Therapeutic Challenge of Targeting HER2 in Endometrial Cancer. Oncologist, 2015. 20(9): p. 1058-68. [CrossRef]
- Nasioudis, D., et al., Molecular landscape of ERBB2/HER2 gene amplification among patients with gynecologic malignancies; clinical implications and future directions. Gynecologic Oncology, 2024. 180: p. 1-5. [CrossRef]
- Trenker, R., et al., Structural dynamics of the active HER4 and HER2/HER4 complexes is finely tuned by different growth factors and glycosylation. 2023, Cold Spring Harbor Laboratory. [CrossRef]
- Bai, X., et al., Structure and dynamics of the EGFR/HER2 heterodimer. Cell Discovery, 2023. 9(1): p. 18. [CrossRef]
- Lee-Hoeflich, S.T., et al., A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res, 2008. 68(14): p. 5878-87. [CrossRef]
- Tang, D., et al., Assessment and prognostic analysis of EGFR, HER2, and HER3 protein expression in surgically resected gastric adenocarcinomas. OncoTargets and therapy, 2014. 8: p. 7 - 14. [CrossRef]
- Tsutsumi, H., et al., Mutant forms of EGFR promote HER2 trafficking through efficient formation of HER2-EGFR heterodimers. Lung Cancer, 2023. 175: p. 101-111. [CrossRef]
- Hirata, A., et al., HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res, 2005. 65(10): p. 4253-60. [CrossRef]
- Zhao, J. and Y. Xia, Targeting HER2 Alterations in Non–Small-Cell Lung Cancer: A Comprehensive Review. JCO Precision Oncology, 2020(4): p. 411-425. [CrossRef]
- Martin, V., et al., HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. British Journal of Cancer, 2013. 108(3): p. 668-675. [CrossRef]
- Chung, Y.W., et al., Overexpression of HER2/HER3 and clinical feature of ovarian cancer. J Gynecol Oncol, 2019. 30(5): p. e75. [CrossRef]
- Diwanji, D., et al., Structures of the HER2-HER3-NRG1β complex reveal a dynamic dimer interface. Nature, 2021. 600(7888): p. 339-343. [CrossRef]
- Dan, H.C., et al., Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKKα). J Biol Chem, 2014. 289(36): p. 25227-40. [CrossRef]
- Huang, J. and B.D. Manning, A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans, 2009. 37(Pt 1): p. 217-22. [CrossRef]
- Matkar, S., C. An, and X. Hua, Kinase inhibitors of HER2/AKT pathway induce ERK phosphorylation via a FOXO-dependent feedback loop. Am J Cancer Res, 2017. 7(7): p. 1476-1485.
- Mendoza, M.C., E.E. Er, and J. Blenis, The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci, 2011. 36(6): p. 320-8. [CrossRef]
- Pandey, G., A.T. Kuykendall, and G.W. Reuther, JAK2 inhibitor persistence in MPN: uncovering a central role of ERK activation. Blood Cancer J, 2022. 12(1): p. 13. [CrossRef]
- Jin, H., N.J. Lanning, and C. Carter-Su, JAK2, But Not Src Family Kinases, Is Required for STAT, ERK, and Akt Signaling in Response to Growth Hormone in Preadipocytes and Hepatoma Cells. Molecular Endocrinology, 2008. 22(8): p. 1825-1841. [CrossRef]
- Schade, B., et al., β-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer Res, 2013. 73(14): p. 4474-87. [CrossRef]
- Piedra, J., et al., Regulation of β-Catenin Structure and Activity by Tyrosine Phosphorylation*. Journal of Biological Chemistry, 2001. 276(23): p. 20436-20443. [CrossRef]
- Merkhofer, E.C., P. Cogswell, and A.S. Baldwin, Her2 activates NF-κB and induces invasion through the canonical pathway involving IKKα. Oncogene, 2010. 29(8): p. 1238-1248. [CrossRef]
- Pianetti, S., et al., Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene, 2001. 20(11): p. 1287-99. [CrossRef]
- Pegram, M., C. Jackisch, and S.R.D. Johnston, Estrogen/HER2 receptor crosstalk in breast cancer: combination therapies to improve outcomes for patients with hormone receptor-positive/HER2-positive breast cancer. npj Breast Cancer, 2023. 9(1): p. 45. [CrossRef]
- Shou, J., et al., Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2–Positive Breast Cancer. JNCI: Journal of the National Cancer Institute, 2004. 96(12): p. 926-935. [CrossRef]
- Lemmon, M.A., J. Schlessinger, and K.M. Ferguson, The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol, 2014. 6(4): p. a020768. [CrossRef]
- Burgess, A.W., EGFR family: Structure physiology signalling and therapeutic targets†. Growth Factors, 2008. 26(5): p. 263-274. [CrossRef]
- Jones, F.E., et al., ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. J Cell Biol, 1999. 147(1): p. 77-88. [CrossRef]
- Drebin, J.A., et al., Monoclonal antibodies identify a cell-surface antigen associated with an activated cellular oncogene. Nature, 1984. 312(5994): p. 545-548. [CrossRef]
- Tevaarwerk, A.J. and J.M. Kolesar, Lapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin Ther, 2009. 31 Pt 2: p. 2332-48. [CrossRef]
- Ishii, K., N. Morii, and H. Yamashiro, Pertuzumab in the treatment of HER2-positive breast cancer: an evidence-based review of its safety, efficacy, and place in therapy. Core Evid, 2019. 14: p. 51-70. [CrossRef]
- Minckwitz, G.v., et al., Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. New England Journal of Medicine, 2019. 380(7): p. 617-628. [CrossRef]
- Santamaria, S., et al., Imaging of Endocytic Trafficking and Extracellular Vesicles Released Under Neratinib Treatment in ERBB2(+) Breast Cancer Cells. J Histochem Cytochem, 2021. 69(7): p. 461-473. [CrossRef]
- Martin, M. and S. López-Tarruella, Emerging Therapeutic Options for HER2-Positive Breast Cancer. American Society of Clinical Oncology Educational Book, 2016(36): p. e64-e70. [CrossRef]
- D'Arienzo, A., et al., Toxicity profile of antibody-drug conjugates in breast cancer: practical considerations. EClinicalMedicine, 2023. 62: p. 102113. [CrossRef]
- Gradishar, W.J., et al., Margetuximab in HER2-positive metastatic breast cancer. Future Oncol, 2023. 19(16): p. 1099-1112. [CrossRef]
- Liu, Y., et al., Rapamycin induces Bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther, 2012. 11(1): p. 45-56. [CrossRef]
- Merkhofer, E.C., P. Cogswell, and A.S. Baldwin, Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene, 2010. 29(8): p. 1238-48. [CrossRef]
- Ingthorsson, S., et al., HER2 induced EMT and tumorigenicity in breast epithelial progenitor cells is inhibited by coexpression of EGFR. Oncogene, 2016. 35(32): p. 4244-4255. [CrossRef]
- Jafari, E., et al., Study of the Relationship between MMP-2 and MMP-9 and Her2/neu Overexpression in Gastric Cancer: Clinico- Pathological Correlations. Asian Pac J Cancer Prev, 2021. 22(3): p. 811-817. [CrossRef]
- Laughner, E., et al., HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol, 2001. 21(12): p. 3995-4004. [CrossRef]
- Sotiriadou, R., et al., Peptide HER2(776–788) represents a naturally processed broad MHC class II-restricted T cell epitope. British Journal of Cancer, 2001. 85(10): p. 1527-1534. [CrossRef]
- Bianchini, G. and L. Gianni, The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol, 2014. 15(2): p. e58-68. [CrossRef]
- Luque, M., et al., Tumor-Infiltrating Lymphocytes and Immune Response in HER2-Positive Breast Cancer. Cancers (Basel), 2022. 14(24). [CrossRef]
- Padmanabhan, R., et al., Crosstalk between HER2 and PD-1/PD-L1 in Breast Cancer: From Clinical Applications to Mathematical Models. Cancers (Basel), 2020. 12(3). [CrossRef]
- Krasniqi, E., et al., Immunotherapy in HER2-positive breast cancer: state of the art and future perspectives. Journal of Hematology & Oncology, 2019. 12(1): p. 111. [CrossRef]
- Li, Y.M., et al., Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell, 2004. 6(5): p. 459-69. [CrossRef]
- Inoue, M., et al., Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology, 2012. 1(7): p. 1104-1110. [CrossRef]
- Shi, Y., et al., Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with Fcγ receptors on macrophages. J Immunol, 2015. 194(9): p. 4379-86. [CrossRef]
- Song, P.N., et al., CD4 T-cell immune stimulation of HER2 + breast cancer cells alters response to trastuzumab in vitro. Cancer Cell Int, 2020. 20(1): p. 544. [CrossRef]
Figure 1.
Schematic representation of HER2 signaling regulation HER2 receptor does not have a known ligand that directly binds to it, unlike other members of the EGFR family. Instead, HER2 is the preferred partner for forming heterodimers (pairing) with other EGFR family members, such as HER1 (EGFR), HER3, and HER4. When a ligand (such as epidermal growth factor, EGF, particularly neuregulin) binds to other EGFR family members, it induces conformational changes in the receptor, leading to dimerization with HER2. This dimerization activates the intrinsic tyrosine kinase activity of HER2, resulting in the autophosphorylation of tyrosine residues within the receptor. Phosphorylated tyrosine residues on HER2 are docking sites for various intracellular signaling proteins, initiating downstream signaling cascades. These signaling pathways include the Ras/Raf/MEK/ERK pathway, the PI3K/Akt pathway, JAK-STAT and the PLCγ/PKC pathway. Upon activation, HER2 can phosphorylate and activate Ras, a small GTPase protein. Activated Ras then initiates a signaling cascade involving Raf, MEK (MAPK/ERK kinase), and ERK (extracellular signal-regulated kinase). ERK activation leads to the phosphorylation of various downstream targets involved in cell proliferation, survival, and differentiation. HER2 activation leads to phosphatidylinositol 3-kinase (PI3K) recruitment and activation. Activated PI3K generates phosphatidylinositol-3,4,5-triphosphate (PIP3), which recruits Akt (protein kinase B) to the cell membrane. Akt is then phosphorylated and activated, leading to the phosphorylation and inhibition of various downstream targets involved in cell survival, protein synthesis, and metabolism. Akt also phosphorylates and inhibits the pro-apoptotic protein Bad, promoting cell survival. Akt activation also leads to the activation of the mammalian target of rapamycin (mTOR), which regulates protein synthesis and cell growth. HER2 activation can lead to phospholipase C gamma (PLCγ) activation through direct phosphorylation or indirect activation via Ras. Activated PLCγ hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces calcium release from intracellular stores, leading to calcium signaling. DAG activates protein kinase C (PKC), which phosphorylates and regulates various downstream targets involved in cell growth, proliferation, and differentiation. HER2 signaling can also crosstalk with other signaling pathways, such as the JAK/STAT pathway, Wnt/β-catenin pathway, and NF-κB pathway, further modulating cellular responses. These pathways contribute to regulating gene expression, cell cycle progression, cell motility, and other cellular functions. Figure made using Biorender.
Figure 1.
Schematic representation of HER2 signaling regulation HER2 receptor does not have a known ligand that directly binds to it, unlike other members of the EGFR family. Instead, HER2 is the preferred partner for forming heterodimers (pairing) with other EGFR family members, such as HER1 (EGFR), HER3, and HER4. When a ligand (such as epidermal growth factor, EGF, particularly neuregulin) binds to other EGFR family members, it induces conformational changes in the receptor, leading to dimerization with HER2. This dimerization activates the intrinsic tyrosine kinase activity of HER2, resulting in the autophosphorylation of tyrosine residues within the receptor. Phosphorylated tyrosine residues on HER2 are docking sites for various intracellular signaling proteins, initiating downstream signaling cascades. These signaling pathways include the Ras/Raf/MEK/ERK pathway, the PI3K/Akt pathway, JAK-STAT and the PLCγ/PKC pathway. Upon activation, HER2 can phosphorylate and activate Ras, a small GTPase protein. Activated Ras then initiates a signaling cascade involving Raf, MEK (MAPK/ERK kinase), and ERK (extracellular signal-regulated kinase). ERK activation leads to the phosphorylation of various downstream targets involved in cell proliferation, survival, and differentiation. HER2 activation leads to phosphatidylinositol 3-kinase (PI3K) recruitment and activation. Activated PI3K generates phosphatidylinositol-3,4,5-triphosphate (PIP3), which recruits Akt (protein kinase B) to the cell membrane. Akt is then phosphorylated and activated, leading to the phosphorylation and inhibition of various downstream targets involved in cell survival, protein synthesis, and metabolism. Akt also phosphorylates and inhibits the pro-apoptotic protein Bad, promoting cell survival. Akt activation also leads to the activation of the mammalian target of rapamycin (mTOR), which regulates protein synthesis and cell growth. HER2 activation can lead to phospholipase C gamma (PLCγ) activation through direct phosphorylation or indirect activation via Ras. Activated PLCγ hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces calcium release from intracellular stores, leading to calcium signaling. DAG activates protein kinase C (PKC), which phosphorylates and regulates various downstream targets involved in cell growth, proliferation, and differentiation. HER2 signaling can also crosstalk with other signaling pathways, such as the JAK/STAT pathway, Wnt/β-catenin pathway, and NF-κB pathway, further modulating cellular responses. These pathways contribute to regulating gene expression, cell cycle progression, cell motility, and other cellular functions. Figure made using Biorender.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



