
Submitted:
07 June 2024
Posted:
10 June 2024
You are already at the latest version
Abstract
Summary Genome assembly projects have grown exponentially due to breakthroughs in sequencing technologies and assembly algorithms. Evaluating the quality of genome assemblies is critical to ensure the reliability of downstream analysis and interpretation. To fulfil this task, we have developed the AssemblyQC pipeline that performs file-format validation, contaminant checking, contiguity measurement, gene- and repeat-space completeness quantification, telomere inspection, taxonomic assignment, synteny alignment, scaffold examination through Hi-C contact-map visualisation, and assessments of completeness, consensus quality and phasing through K-mer analysis. It produces a comprehensive HTML report with method descriptions, tables, and visualisations. Availability and Implementation The pipeline uses NextFlow for workflow orchestration and adheres to the best-practice established by the nf-core community. This pipeline offers a reproducible, scalable, and portable method to assess the quality of genome assemblies – the code is available online.GitHub: https://github.com/Plant-Food-Research-Open/assemblyqc Supplementary information Pipeline usage documentation, parameter descriptions and example outputs are available on GitHub: https://github.com/Plant-Food-Research-Open/assemblyqc/tree/main/docs. A preview report is also hosted online: https://plant-food-research-open.github.io/assemblyqc
Keywords:
Genome
; Quality assessment
; Nextflow
Introduction
During the last 20 years, reference genome assemblies have been generated for over 700 plant species (Sun, et al., 2022). As of May 2024, the Assembly Database hosted by the National Center for Biotechnology Information (NCBI) returned more than 2.3 million search results (NCBI, 2024). This exponential growth in genome assemblies has been realised by the continuous and substantive decrease in the cost of whole-genome sequencing (NHGRI, 2023), coupled with advancements of sequencing technologies and assembly algorithms (Agarwal, et al., 2020; Dida and Yi, 2021). This trend is expected to persist as the pursuit for high quality genomes remains a major goal (Rhie, et al., 2020). Moreover, the reduced costs allow an increasingly wider community of research labs to routinely assemble new genomes. Aligned with this, fast and standardised assembly quality assessment has become critical and indispensable.
There are three major aspects of assembly quality: contiguity, completeness and correctness (Wang and Wang, 2023). The Earth Biogenome Project tracks an updated list of quality metrics and their minimum values which should be met by genome assemblies (EBP, 2023). A plethora of bioinformatics tools are available, each focusing on one specific aspect. To comprehensively assess an assembly, the researcher usually needs to run multiple tools to cover various aspects. This is challenging not only because of the open-source nature of many tools, which often require manual installation and configuration of correct dependencies, but also because it can be tedious and time-consuming to run different tools separately. Reproducibility of the quality assessment results is an even bigger challenge as the problem is compounded by varied runtime requirements across platforms and frequent version changes. For example, BUSCO (Seppey, et al., 2019; Simao, et al., 2015), which has been widely adopted to estimate the gene-space completeness of an assembly had 11 updates within a year (January 2022 to June 2023).
To facilitate a streamlined application of the quality assessment tools in a reproducible manner, GenomeQC was recently released (Manchanda, et al., 2020) with publicly available source code and a free R/Shiny webapp. The Vertebrate Genomes Project (VGP) assembly pipeline provides wrappers to execute different tools, including running individual quality evaluation tools (Rhie, et al., 2020). Incorporating additional tools for a thorough quality assessment as compared to existing pipelines, we have developed AssemblyQC, which adopts the highly portable NextFlow workflow management system in combination with the community-curated nf-core framework (Di Tommaso, et al., 2017; Ewels, et al., 2020; Langer, et al., 2024). AssemblyQC is a unified, fully automated, reproducible tool that can be executed on local machines, high-performance computers, or on the cloud to evaluate the quality of genome and transcriptome assemblies. Quality metrices chosen for report are based on community standards. The pipeline is implemented using nf-core modules, which are reviewed and regularly updated by a large open-source community (Langer, et al., 2024).
Materials and Methods
Design
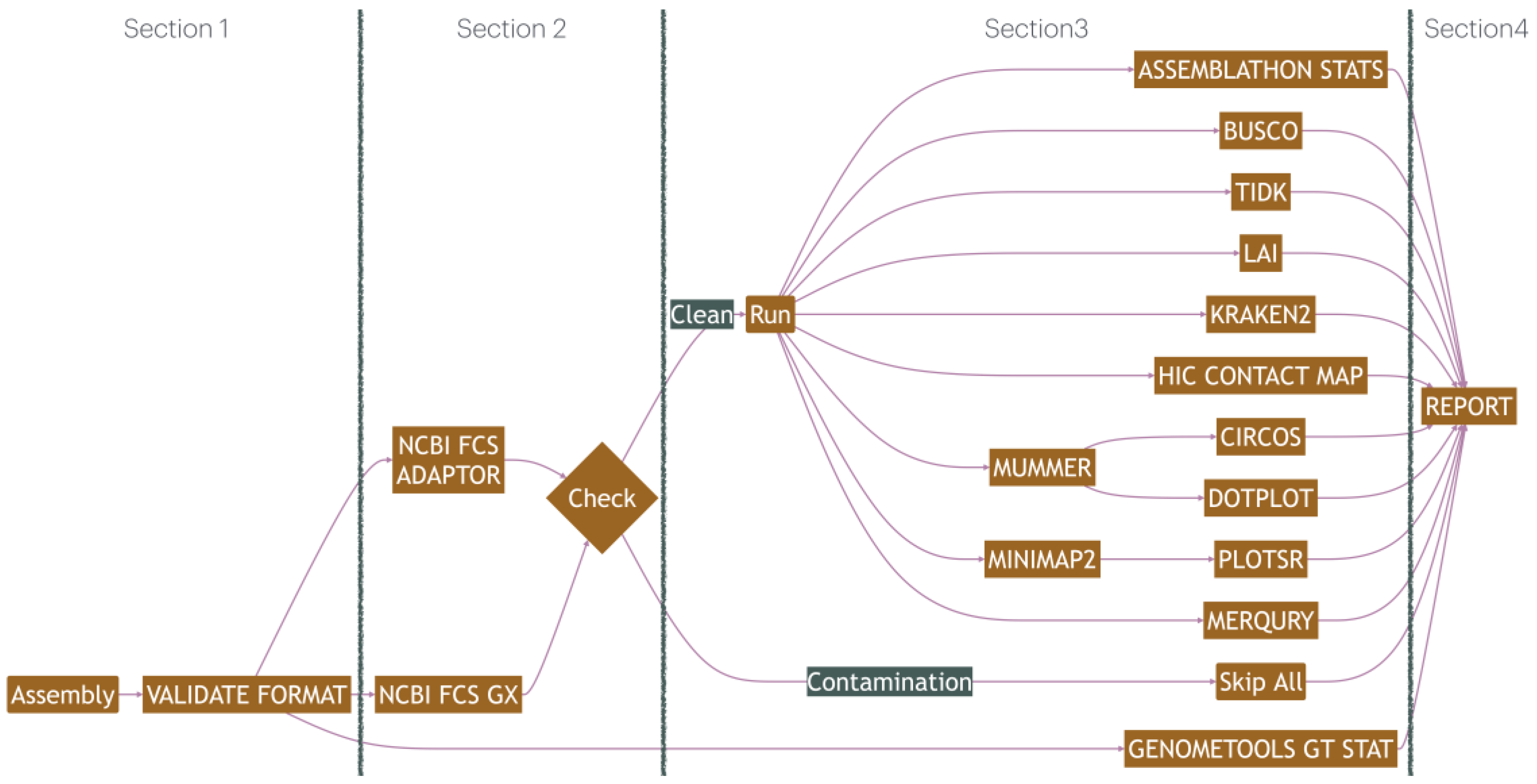
The pipeline is designed to evaluate multiple genome or transcriptome assemblies in parallel. The pipeline is divided into four major sections as shown in the flowchart in Figure 1. Within each section, data are processed in parallel where possible. In section 1, the pipeline checks the input FASTA and GFF3 annotation files using py_fasta_validator, SekQit rmdup and GenomeTools gt gff3validator (Edwards, 2019; Gremme, et al., 2013; Shen, et al., 2016), to ensure integrity of the input files, detect duplicate sequences and prevent failure of subsequent tools. In section 2, the pipeline uses NCBI’s Foreign Contamination Screen (FCS) and its database to detect contaminants such as adapters and sequences from foreign organisms (Astashyn, et al., 2023). Assemblies that successfully pass these checks move on to additional quality checks in section 3. The pipeline can be configured to skip quality checks for assemblies which contain contaminants.
Section 3 executes the remaining quality assessment in parallel for each assembly. For scaffold and contig-level contiguity statistics associated with FASTA sequences, assemblathon2-analysis is used (UCDAVIS-Bioinformatics, 2012). To break the assembly into contigs, the pipeline uses 100 ‘N’ bases as the default unknown gap size. This parameter can be changed and is quoted in the AssemblyQC final report atop the contig-level statistics. Statistics related to annotations in GFF3 files are computed with GenomeTools gt stat tool (Gremme, et al., 2013). The Benchmarking Universal Single-Copy Orthologs (BUSCO) tool is used for estimating the gene-space completeness of each assembly (Seppey, et al., 2019). The pipeline can be configured to evaluate each assembly against one or more BUSCO lineages in parallel. The quality of the repeat-space is evaluated with the Long Terminal Repeat (LTR) Assembly Index (LAI) (Ou, et al., 2018). The LTR_retriever workflow consisting of LTR_FINDER and LTRharvest for de novo detection of LTRs (Ellinghaus, et al., 2008; Ou and Jiang, 2018; Ou and Jiang, 2019). The LAI statistic is independent of the BUSCO statistics and can help improve overall assembly contiguity by isolating shortcomings in the repeat-space.
The pipeline can also assess chromosome completeness and correctness through telomere, Hi-C contact-frequency, and synteny visualisations. Telomere Identification toolKit (TIDK) is employed to estimate the presence of telomeres with a specified telomeric motif (Brown, 2023). To supplement assessment with this user-specified repeat motif, the toolkit also explores the most likely data-driven repeat sequence for each assembly from the assembly sequences. The results for TIDK are sorted in ascending order by sequence length using SeqKit (Shen, et al., 2016). Presence of the telomeric repeats throughout a sequence often indicates assembly errors, and in many cases, these can be corrected by rearranging fragments in the sequence.
Where Hi-C data are provided, a contact-frequency map is generated by the pipeline and added to the report for visualisation. FASTQC and FASTP are used to trim and quality check the Hi-C reads (Andrews, 2010; Chen, et al., 2018). The reads are then mapped to the assembly sequences using Burrow-Wheeler Aligner (BWA) (Li, 2013). The mapping quality is checked with hic_qc.py (Sullivan, 2022), and finally converted into hic format through a workflow consisting of samblaster, samtools and run-assembly-visualizer.sh (Danecek, et al., 2021; Dudchenko, et al., 2017; Faust and Hall, 2014). The interactive visualisation of the Hi-C map is added to the report using the Juicebox.js JavaScript library (Robinson, et al., 2018). Atypical Hi-C contact-frequency patterns such as gaps or non-diagonal contact concentrations can indicate gaps in the assembly or misassignment of contigs to scaffolds.
Two sub-workflows, the pair-wise mode, and the chromosome-level comparison, are implemented in the pipeline to generate synteny plots between input assemblies. The pair-wise workflow creates synteny plots between each combination of input assemblies. Firstly, cross-mapping is performed with MUMmer4 (Marcais, et al., 2018). The pipeline can be configured to include or exclude many-to-many mappings detected by MUMmer4. The mappings are then plotted with Circos (Krzywinski, et al., 2009). A dot-plot can also be created which is helpful in identifying inversions (Cabanettes and Klopp, 2018). This result makes it easy to visualise gaps and large structural variations (SVs) between assemblies and helps to consistently assign chromosomal numbers to those assemblies. Chromosome-level comparison sub-workflow maps corresponding chromosomes in all the input assemblies with Minimap2 (Li, 2018), and generates synteny plots with plotsr (Goel and Schneeberger, 2022; Li, 2018), allowing chromosome-by-chromosome visualisation of the key synteny blocks and large SVs from all input. This sub-workflow is helpful for creating a single summary synteny visualisation.
Kraken2 is used to assign taxonomic labels to each assembly sequence (Wood, et al., 2019) and an interactive report is produced with Krona (Ondov, et al., 2011). This can be useful in detecting cross-domain contamination in the assemblies (Cornet and Baurain, 2022).
Merqury is also included in the pipeline to facilitate k-mer analysis. The pipeline supports both mixed-haplotype and diploid assembly assessment with and without the availability of parental reads. In addition to K-mer completeness and consensus quality, Merqury is very useful in evaluating the extent of haplotype phasing (Rhie, et al., 2020).
In section 4 of the pipeline, outputs from various assessment tools are gathered, parsed, and converted into a HyperText Markup Language (HTML) report using Jinja templating language in Python. Other than file format validation, the remaining assessment tools are optional and can be bypassed via settings in the configuration file.
Implementation Details
The implementation of the pipeline follows the NextFlow good-practice developed by the nf-core community (Ewels, et al., 2020; Langer, et al., 2024). It is built with the nf-core pipeline template, and whenever feasible, utilising the nf-core modules and sub-workflows. The nf-test is used for unit testing, with each module executing a single script or tool. The modules do not rely on installation of dependencies in the system environment; instead, they are implemented using version-locked Bioconda Docker/Singularity/Apptainer containers (da Veiga Leprevost, et al., 2017; Gruning, et al., 2018; Kurtzer, et al., 2017; Merkel, 2014) from public repositories including https://quay.io and https://hub.docker.com. Pipeline code, documentation and exemplar reports are available on GitHub: https://plant-food-research-open.github.io/assemblyqc.
Conclusions
We have created a highly portable and reproducible pipeline for comprehensive assessment of genome and transcriptome assemblies. The pipeline evaluates contiguity, completeness, correctness, and contamination of assemblies in multiple ways with various well adopted bioinformatics tools. The quality assessment results are presented in a shareable HTML report. Furthermore, the actionable insights from the report such as locations of contaminants, locations and abundance of telomeric motifs inside chromosome sequences, presence of gaps or off diagonal contacts in the Hi-C map, and structural variations borne out by the synteny plots, can be used to guide iterative improvement of the assembly.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Pipeline usage documentation, parameter descriptions and example outputs are available on GitHub: https://github.com/Plant-Food-Research-Open/assemblyqc/tree/main/docs. A preview report is also hosted online: https://plant-food-research-open.github.io/assemblyqc.
Funding
This work was funded by the Traits for Life Indoors (TFLI) programme in the Horticulture Production Goes Urban research direction supported by Strategic Science Investment Funding (SSIF) received by The New Zealand Institute for Plant and Food Research Limited.
Data Availability Statement
The pipeline uses NextFlow for workflow orchestration and adheres to the best-practice established by the nf-core community. This pipeline offers a reproducible, scalable, and portable method to assess the quality of genome assemblies – the code is available online. GitHub: https://github.com/Plant-Food-Research-Open/assemblyqc.
Acknowledgments
The plant-food-research-open/assemblyqc uses code and infrastructure developed and maintained by the nf-core community. We are thankful to Mahesh Binzer-Panchal from National Bioinformatics Infrastructure Sweden, Uppsala University, Sweden for his help in porting pipeline modules to the nf-core template. We acknowledge the Ministry of Business, Innovation and Employment (MBIE) for providing the Strategic Science Investment Funding (SSIF).
References
- Agarwal, T., et al. Recent Advances in Gene and Genome Assembly: Challenges and Implications. In.; 2020.
- Andrews, S. FastQC: a quality control tool for high throughput sequence data. In.: Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom; 2010.
- Astashyn, A., et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. bioRxiv 2023:2023.2006.2002.543519. [CrossRef]
- Brown, M., González De la Rosa, P. M. and Mark, B. A Telomere Identification Toolkit. In.; 2023.
- Cabanettes, F. and Klopp, C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018;6:e4958. [CrossRef]
- Chen, S., et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34(17):i884-i890. [CrossRef]
- Cornet, L. and Baurain, D. Contamination detection in genomic data: more is not enough. Genome Biol 2022;23(1):60. [CrossRef]
- da Veiga Leprevost, F., et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics 2017;33(16):2580-2582. [CrossRef]
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10(2):giab008. [CrossRef]
- Di Tommaso, P., et al. Nextflow enables reproducible computational workflows. Nat Biotechnol 2017;35(4):316-319.
- Dida, F. and Yi, G. Empirical evaluation of methods for de novo genome assembly. PeerJ Comput Sci 2021;7:e636.
- Dudchenko, O., et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017;356(6333):92-95. [CrossRef]
- EBP. Report on Assembly Standards Version 5.0. In.: Earth Biogenome Project; 2023.
- Edwards, R. 2019. linsalrob/fasta_validator: Initial Release. Release v0.1. [CrossRef]
- Ellinghaus, D., Kurtz, S. and Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 2008;9(1):18. [CrossRef]
- Ewels, P.A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 2020;38(3):276-278. [CrossRef]
- Faust, G.G. and Hall, I.M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 2014;30(17):2503-2505. [CrossRef]
- Goel, M. and Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 2022;38(10):2922-2926. [CrossRef]
- Gremme, G., Steinbiss, S. and Kurtz, S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform 2013;10(3):645-656. [CrossRef]
- Gruning, B., et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods 2018;15(7):475-476. [CrossRef]
- Krzywinski, M., et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19(9):1639-1645. [CrossRef]
- Kurtzer, G.M., Sochat, V. and Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS One 2017;12(5):e0177459. [CrossRef]
- Langer, B.E., et al. Empowering bioinformatics communities with Nextflow and nf-core. bioRxiv 2024:2024.2005.2010.592912.
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 2013.
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34(18):3094-3100. [CrossRef]
- Manchanda, N., et al. GenomeQC: a quality assessment tool for genome assemblies and gene structure annotations. BMC Genomics 2020;21(1):193. [CrossRef]
- Marcais, G., et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol 2018;14(1):e1005944. [CrossRef]
- Merkel, D. Docker: lightweight linux containers for consistent development and deployment. Linux j 2014;239(2):2.
- NCBI. NCBI Assembly Database. In.; 2024.
- NHGRI. DNA Sequencing Costs: Data. In.; 2023.
- Ondov, B.D., Bergman, N.H. and Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 2011;12(1):385. [CrossRef]
- Ou, S., Chen, J. and Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 2018;46(21):e126. [CrossRef]
- Ou, S. and Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol 2018;176(2):1410-1422. [CrossRef]
- Ou, S. and Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob DNA 2019;10(1):48. [CrossRef]
- Rhie, A., et al. Towards complete and error-free genome assemblies of all vertebrate species. bioRxiv 2020:2020.2005.2022.110833. [CrossRef]
- Rhie, A., et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 2020;21(1):245. [CrossRef]
- Robinson, J.T., et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst 2018;6(2):256-258 e251. [CrossRef]
- Seppey, M., Manni, M. and Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 2019;1962:227-245. [CrossRef]
- Shen, W., et al. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 2016;11(10):e0163962. [CrossRef]
- Simao, F.A., et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31(19):3210-3212. [CrossRef]
- Sullivan, S. 2022. hic_qc. Release 6881c33. https://github.com/phasegenomics/hic_qc. (28 June 2023 date last accessed).
- Sun, Y., et al. Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci 2022;27(4):391-401. [CrossRef]
- UCDAVIS-Bioinformatics. assemblathon2-analysis. In.; 2012.
- Wang, P. and Wang, F. A proposed metric set for evaluation of genome assembly quality. Trends Genet 2023;39(3):175-186. [CrossRef]
- Wood, D.E., Lu, J. and Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol 2019;20(1):257. [CrossRef]
Figure 1.
Pipeline flowchart.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



