
Submitted:
06 June 2024
Posted:
11 June 2024
You are already at the latest version
Abstract
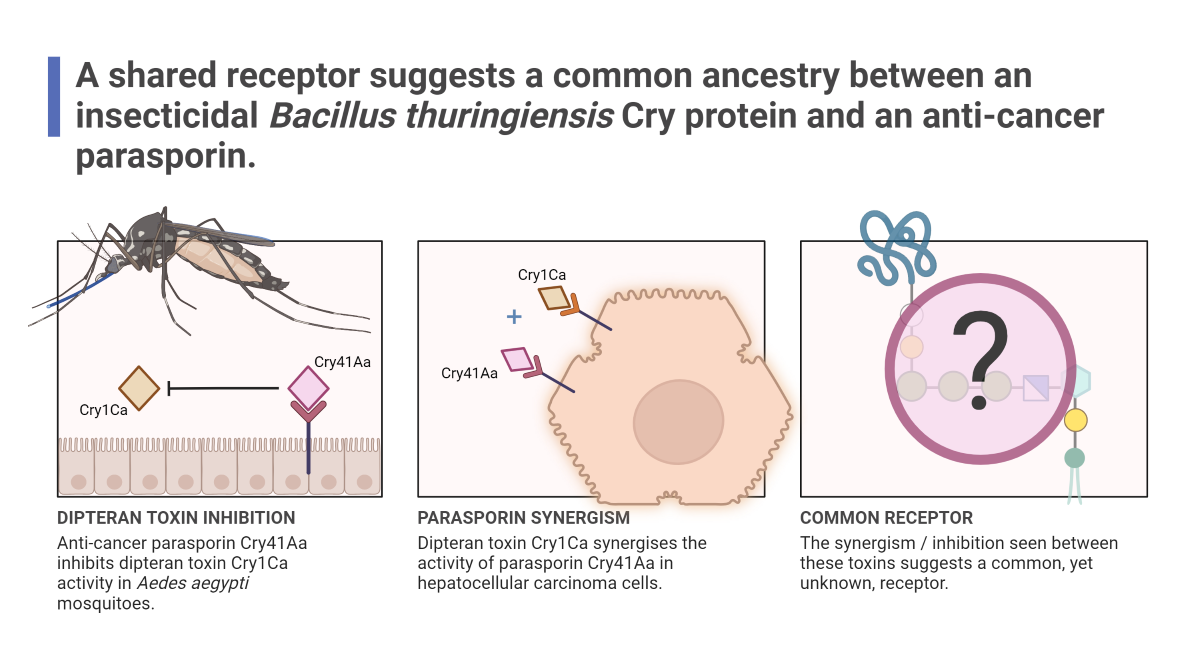
Cry toxins produced by the bacterium Bacillus thuringiensis are of significant agronomic value worldwide due to their potent and highly specific activity against various insect orders. However, some of these pore-forming toxins display specific activity against a range of human cancer cells whilst possessing no known insecticidal activity, Cry41Aa is one such toxin. Cry41Aa has similarities to its insecticidal counterparts in both its 3-domain toxic core structure and pore-forming abilities, but how it has evolved to target human cells is a mystery. This work shows that some insecticidal Cry toxins can enhance the toxicity of Cry41Aa against hepatocellular carcinoma cells, despite possessing no intrinsic toxicity themselves. This interesting crossover is not limited to human cancer cells, as Cry41Aa was found to inhibit some Aedes-active Cry toxins in mosquito larval assays. Here, we present findings that suggest that Cry41Aa shares a receptor with several insecticidal toxins indicating a stronger evolutionary relationship than their divergent activities might suggest.
Keywords:
Cry41Aa
; Cry1Ca
; Aedes aegypti
; HepG2
1. Introduction
Bacillus thuringiensis (Bt) is a gram-positive, spore-forming bacterium which produces crystalline inclusions during sporulation [1]. These crystalline bodies can house one or more proteins (such as Cry toxins) that possess considerable efficacy and specificity towards invertebrates, which has made these Bt Cry toxins valuable to agronomic interests, both as insecticidal agents and as genes expressed in transgenic crops [1]. Cry toxin insect specificity is derived from numerous factors, which include crystal dissolution via compatible pH conditions of the invertebrate midgut, proteolytic cleavage of the protoxin by means of a compatible gut protease, and receptor recognition and binding [1,2].
Cry toxins possess a three-domain toxic core structure which is highly conserved despite significant divergences in their sequence and target specificity [1]. They all require the same criteria for functionality, namely an environment in which to solubilise the crystal-encased protein, and proteolytic processing of the N-terminus required for activation [2]. Further still, many known receptors for these Cry toxins are shared across different orders of insects, such as the glycosyl-phosphatidylinositol (GPI) anchored aminopeptidases and alkaline phosphatases [2]. Though the Cry toxins are mostly known and commercialised for their efficacy against insects, a small subgroup of Cry toxins known as the parasporins possess activity against human cancer cells whilst having no known invertebrate targets. Parasporin-3, also known as Cry41Aa, is toxic towards some human cancer cells, which include hepatocellular carcinoma and myeloid leukaemia [3]. We also know that proteolytic activation is required for this activity [4]. Amongst all the specificity determinants between Cry proteins and their targets, receptor interaction is thought to be the most significant step in determining the cytotoxicity of a Cry protein [2]. Multiple receptors have been identified and their interactions with Cry toxins characterized including some for parasporins [5]. These include Beclin-1 for Parasporin 1 and GPI-anchor protein receptors for Parasporin 2 [6]. The mode of action of a Cry protein may not be limited to one type of receptor; studies have suggested that a toxin might interact with multiple receptors in each insect species and that interactions between these receptors might enhance the toxic effect [7]. In this work, we have explored the binding characteristics of the Cry41Aa protoxin against hepatocellular carcinoma cells and have demonstrated cross-over between Cry41Aa and some insecticidal Cry toxins, speaking to a closer evolutionary relationship.
2. Materials and Methods
Strains, plasmids, and constructs
Bacillus thuringiensis strain 4D7: An acrystalliferous strain of Bacillus thuringiensis subsp. kurstaki was used for the production of all Cry toxins within this work. This strain came from the Bacillus Genetic Stock Center (https://bgsc.org/).
Toxin expression plasmids: pHT315 is an E. coli / Bt shuttle vector containing an origin of replication for both E. coli and Bt, as well as resistance genes for ampicillin and erythromycin [8]. We have previously used this plasmid as the basis of an expression vector for Cry2Aa and Cry2Ab [9]. In this work pHTCry41Aa was created by replacing the ORF2 and cry2A genes from these constructs with cry41Aa and its ORF3 gene. For pHTCry41AaGFP the non-essential beta-trefoil domain at the C-terminus of Cry41Aa [3] was deleted and GFP was added after the 713th amino acid. To prevent cleavage of the GFP during activation a unique PreScission protease site (LEVLFQGP) was added after the 40th amino acid as previously described [4]. In pHTCry41AaHA an HA tag (YPYDVPDYA) was inserted in pHTCry41Aa after the 825th codon. To create the disabled (DIP) version of Cry41Aa an A126E mutation was introduced into pHTCry41AaHA. Cry1Ac, Cry1Ca, Cry1Da and Cry4Aa expression plasmids were created by replacing the cry2Aa gene from pHTCry2Aa with the alternative coding region. Finally, Cry11Aa was expressed from a construct in which the gene was cloned in pSVP27 [10].
Growth, solubilisation and activation of Cry toxins
Bacillus thuringiensis bacteria containing the Cry toxin genes used in these experiments, were inoculated to flasks containing 1L half concentration LB broth with the addition of 5 µg/ml erythromycin (chloramphenicol for Cry11Aa expression) and incubated for 3 days at 30 °C, 200 rpm. Several rounds of sonication and centrifugation were undertaken to fully lyse the cells before the spores and crystals were resuspended in ddH2O. All Cry toxins were solubilised by pelleting the crude cell lysate followed by resuspension in 50 mM Na2CO3 pH 10.5 containing 5 mM dithiothreitol (DTT) and incubation at 37 °C for one hour. Samples were centrifuged at 16,873 g for 2 minutes, and the supernatant was collected. For activation of Cry41AaGFP Pierce HRV 3C protease (1 Unit per 100 µg Thermo Fisher Scientific) was used in PreScission reaction buffer and left for 16 hours at 4 °C. The enzymatic reaction was stopped by addition of Complete mini EDTA-free proteinase inhibitor cocktail solution (Sigma-Aldrich). Cry41Aa and Cry41HADIP were activated by treating the solubilised samples with 1 mg/ml chymotrypsin at 37 °C for one hour. For Cry1Ca and Cry11Aa trypsin was used instead of chymotrypsin.
Cell culture
The adherent hepatocellular carcinoma cell line HepG2, purchased from ECACC, was routinely cultured in nunclon surface treated T-25, T-75, and T-175 flasks (Nunc) using a high glucose DMEM (4.5 g/l, Gibco Life Technologies) containing 1 % penicillin-streptomycin-L-glutamate (PSG; Gibco Life Technologies) and 10 % fetal bovine serum (FCS; Gibco Life Technologies) and incubated at 37 °C with 5 % CO2 humidified air.
Fluorescence microscopy using GFP-labelled Cry41Aa
HepG2 cells were placed on a sterile microscope cover glass at a density of 2.5 x 105 cells/ml in serum-free DMEM treated with PSG (1 %) for 16 h in a 6-well culture plate. Appropriate treatments were added to each well for one hour, the medium was aspirated, and each well washed with PBS. A 4 % formaldehyde solution (Sigma-Aldrich) was used to fix the samples. Coverslips were mounted on microscope glass slides using mounting media (plus DAPI), and the edges were sealed. The following parameters were set on the fluorescence microscope (Zeiss LSM880): a 64x objective (with immersion oil) was used to obtain a Z-stack scan of cells from bottom to top (cell surface) to obtain micrographs of the whole cell surface. A GFP filter was used to scan cells for bound GFP-tagged Cry41Aa on the surface of treated cells, whereas a blue filter was used to visualise DAPI staining.
Cellular competition assays
Cellular competition assays were carried out in 96-well culture plates (Nunc). Cells were seeded in a volume of 90 µl at a concentration of 2.5 x 105 cells/ml and allowed to adhere overnight whilst incubated at 37 °C with 5 % CO2 humidified air. 10 µl of either experimental or control samples were added to wells in triplicate for each condition. For the experimental wells, combinations of Cry41Aa at an EC50 dose of 2 µg/ml and 10 -180 fold amounts of competitor were added. Control wells included the application of 10 µl buffer only (50 mM Na2CO3 pH 10.5), Cry41Aa alone, competitor toxin at the highest concentration used, or Triton X-100 (0.1 %). DMEM was added to additional wells at a volume of 100 µl in triplicate to act as a background fluorescent control. Cells were then incubated for 24 hours.
Cell viability was measured the following day using the CellTiter-Blue cell viability assay kit (Promega). 20 µl of the CellTiter-Blue reagent alamarBlue was added to each well. The plate was incubated for 2 hours before reading using a GloMax – Multi Detection System from Promega. The fluorescence was measured using a green filter with an excitation wavelength of 525 nm and an emission wavelength range of 580-640 nm.
Cellular binding assays
Cells were seeded to 6-well culture plates (Nunc), 1 ml per well at a concentration of 1 x 106 cells/ml and incubated overnight to allow cells to adhere to the plates. Existing DMEM within the 6-well plates was aspirated off, and the toxin / DMEM solutions were added, with a control well receiving only 50 mM Na2CO3 pH 10.5 buffer. Plates were incubated in the presence of toxin for 1 hour. After incubation, DMEM and any unbound toxin were aspirated off, and each well was washed twice in PBS. 70 µl of resuspension buffer (20 mM Tris HCl pH 8 containing 1x phosphatase inhibitors and 1x protease inhibitors) was added to each well, and cells were transferred into the buffer using cell scrapers (Corning). 70 µl of whole cell lysate was collected from each well and placed in ice-cold Eppendorf tubes to be later analysed via western blotting. All binding experiments were repeated at least three times, and blots contained within this paper are representative of each experimental set.
Western blotting
Whole cell lysates (10 µl) were combined with 10 µl of 2x SDS-ME (SDS loading buffer + 5 % β-mercaptoethanol) and heated at 98 °C for 10 minutes. These samples were loaded onto a 7.5 % SDS PAGE gel at 15 µl per well and ran for 40 minutes at 200 V. The gel was removed and incubated in semi dry transfer buffer for 5 minutes while shaking. The protein samples were then transferred to a nitrocellulose membrane (Bio-Rad, 0.45 µm) using a Bio-Rad Trans-Blot Semi-Dry Transfer Cell system at 100 mA for 1 hour. The nitrocellulose membrane was then equilibrated in 20 ml of 1x PBS buffer for 5 minutes before incubation for 1 hour with 10 ml blocking buffer (1x PBS containing 0.02 % tween-20 and 5 % dry milk). The blocking buffer was poured off, and the membrane was incubated for a further hour in 5 ml of blocking buffer containing 5 µl of an HRP conjugated anti-HA antibody (Abcam). The membrane was washed in PBS-T 3 times with a 5-minute incubation with each wash. The last wash was poured off, and the membrane was incubated in an enhanced luminol-based chemiluminescent detection (ECL) buffer for 2 minutes (10 ml of 100 mM Tris HCl pH 8.5, 3 µl H2O2, 25 µl p-coumaric acid (14.7 mg/ml in DMSO), and 50 µl luminol (88.6 mg/ml in DMSO). The signal was then detected using a UVP ChemStudio imaging system with a 10-minute exposure time and displayed within the UVP Life Science VisionWorks software.
For detecting Cry1Ca the western blotting process remained the same, save for the use of an in house Cry1C specific primary antibody and a secondary anti-rabbit IgG HRP conjugated antibody (Cell Signalling Technology).
Aedes aegypti bioassays
Aedes aegypti eggs were originally provided by InfraVec2, adults were fed on defibrinated horse blood and a 10 % sucrose solution, larvae on goldfish flakes. The insects were kept at 27 °C, 65 % humidity. Larvae were selected at the 3rd instar stage of development and placed in 24-well plates, 5 larvae per well and 100 larvae total per condition. Cry1Ca was added at 25 µg/ml, and Cry41Aa was added as a competitor at various concentrations. Water-only and Cry41Aa-only conditions were used as controls. Water in the wells was topped up to a total volume of 3 ml. Larvae were incubated at 27 °C for 24 hours, after which the total alive and dead larvae for each condition were recorded.
3. Results
3.1. Fluorescence Microscopy Analysis of Binding of the GFP-Tagged Cry41Aa to the Surface of HepG2 Cells
Fluorescence microscopy was carried out to visualise binding of the Cry41Aa protein to its target cell. Results from the fluorescent micrographs (Figure 1) indicated that Cry41Aa in both its active and non-toxic protoxin form could bind to the surface of HepG2 cells. This suggested that regardless of proteolytic activation, interaction with the cell membrane still occurred, indicating that N-terminal cleavage has no significant effect on the binding of the protein to its targets.
3.2. Immunoblotting Confirms Cry41Aa’s Binding Capability
For many Bt Cry toxins N-terminal proteolytic cleavage has been shown to be a prerequisite for toxicity [9,11]. Though in vivo activation of Cry41Aa does not occur in the cellular environment, N-terminal cleavage using proteases such as chymotrypsin or proteinase K is nevertheless required for its activation and subsequent activity against cancer cells in vitro [4]. Following the fluorescent microscopy analysis, we compared the signals detected via immunoblotting from conditions where HepG2 cells were exposed to different concentrations of the Cry41AaHA protoxin, scraped, and the whole cell lysates subjected to immunoblotting. Figure 2A, lanes 2-6, show a dose-dependent increase in binding for the Cry41Aa protoxin. Lanes 1 and 7 show the HA-tagged protoxin-only and cell-only controls respectively, suggesting that this signal is specific to the Cry41AaHA protoxin. These results confirm that the protoxin can bind to HepG2 cells despite not undergoing N-terminal cleavage. Binding studies with the activated form of Cry41Aa were complicated by the fact that this form could lyse the target cells. To overcome this a mutant was created (Cry41AaHADIP) whose mutation (A126E) is thought to prevent pore-formation, rendering the mutant non-toxic. Figure 2B confirms that the activated form of Cry41Aa was also capable of binding in a dose-dependent manner. Compared to the protoxin however, binding of the activated toxin could be observed at a lower concentration of toxin.
A cellular competition assay utilising HepG2 cells was performed to clarify whether the binding shown in the immunoblotting experiment was specific (receptor-mediated) or nonspecific. For this assay, activated Cry41Aa toxin was competed against increasing amounts of the Cry41Aa protoxin, which possesses no toxicity towards HepG2 cells. As Figure 3 shows, the protoxin form of Cry41Aa has a dose-dependent inhibitory effect on the toxicity of the activated toxin. This indicated that not only is the protoxin form binding to the cells but that it is most likely binding to the same site as the activated form.
3.3. Cry41Aa and Insecticidal Cry Toxin Synergism
Following the inhibition seen in competition with the Cry41Aa protoxin, we investigated whether other Cry toxins could exert any effect on the toxicity of Cry41Aa. To test this, Cry41Aa was competed against increasing amounts of activated Cry1Ca, an insecticidal Cry toxin with efficacy against both lepidopteran and dipteran targets but possessing no toxicity towards HepG2 cells [3]. Unlike the inhibition seen with Cry41Aa and its protoxin form, synergism was observed between Cry41Aa and Cry1Ca, with the combination of the two toxins exerting increased toxicity compared to Cry41Aa alone. It was also observed that this increase in toxicity correlated with increasing amounts of Cry1Ca (Figure 4). This synergistic effect wasn't unique to just Cry1Ca, two other toxins, Cry11Aa and Cry4Aa, showed similar results, whereas Cry1Da, Cry1Ac, Cry2Aa, and Cry2Ab had no effect on Cry41Aa toxicity, (Table 1, Appendix Figure A1 and Figure A2). None of these insecticidal toxins had any direct effect on HepG2 themselves.
To determine if Cry1Ca was capable of binding to HepG2, cells were exposed to varying amounts of activated Cry1Ca before being scraped, and the whole cell lysates subjected to immunoblotting utilising an anti-Cry1C antibody. As Figure 5B shows, activated Cry1Ca bound HepG2 cells in a dose-dependent manner.
In contrast when the same experiment was carried out using the Cry1Ca protoxin no binding was observed at the concentrations used (Figure 5A). This result is consistent with previous findings that some insecticidal Cry toxins cannot efficiently bind to their intended targets without first undergoing N-terminal cleavage [11]. When the Cry1Ca protoxin was competed against Cry41Aa, it failed to produce the same synergistic effects as seen with the trypsin-activated Cry1Ca (Figure 6).
3.4. Cry41Aa Inhibits Cry1Ca Activity in Aedes aegypti Larvae
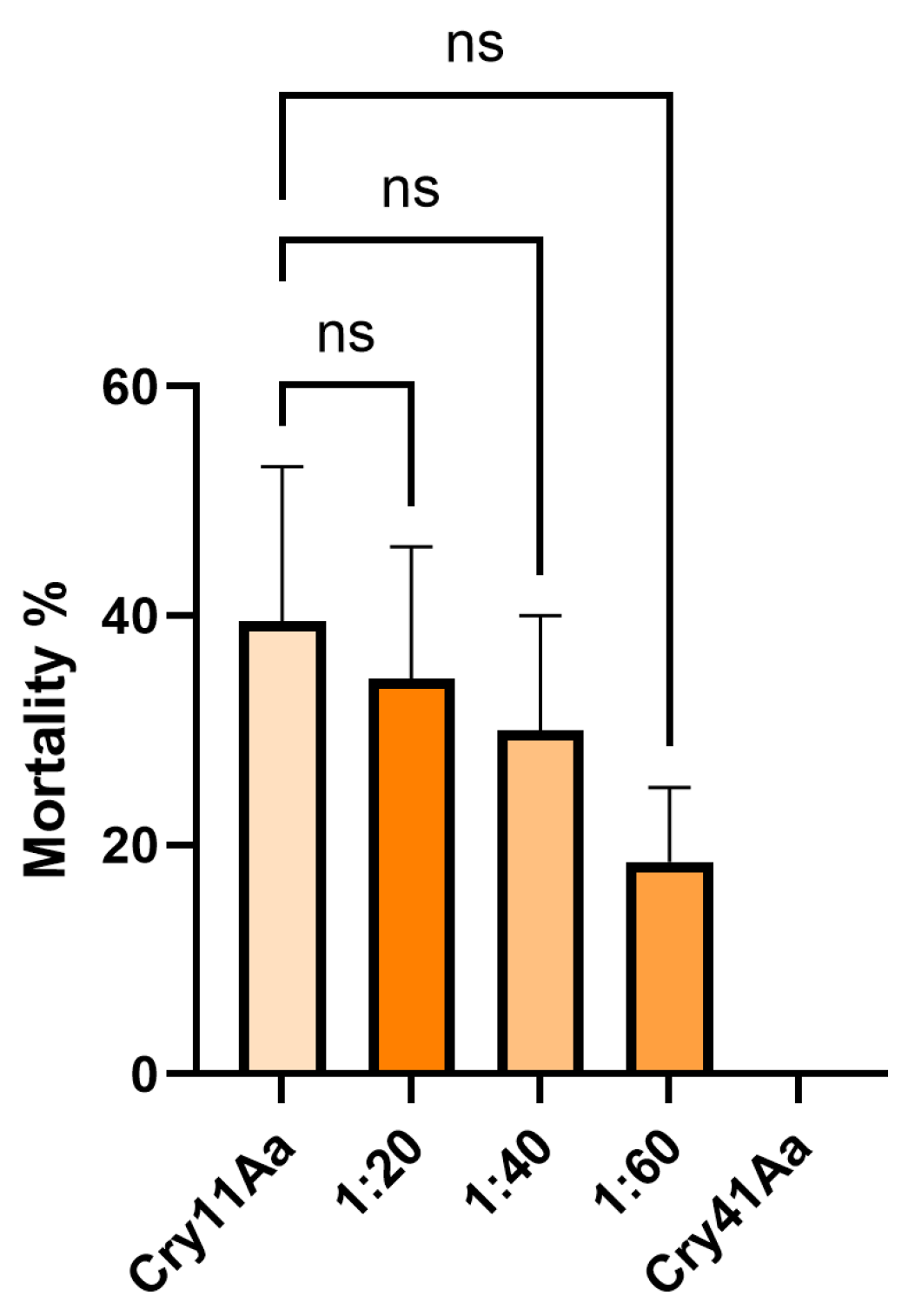
Due to Cry1Ca showing a synergistic effect against HepG2 cells with Cry41Aa, we investigated whether a similar effect might occur in a competition assay utilising an insect system. Aedes aegypti mosquitoes were chosen as they are known to be susceptible to Cry1Ca [12]. In these assays, Cry1Ca was administered at a dosage of 25 µg/ml along with increasing concentrations of Cry41Aa to 3rd instar Aedes aegypti larvae. Unlike the competition assays featuring HepG2 cells, the competitor inhibited rather than synergised. As Figure 7 shows, larval mortality dropped with increasing concentrations of Cry41Aa, indicating that this anti-cancer toxin can inhibit the effects of Cry1Ca through competition. This inhibition was not unique to Cry1Ca, as Cry41Aa was also capable of inhibiting another Aedes-active Cry toxin, Cry11Aa, in a similar manner (Appendix Figure A3).
4. Discussion
N-terminal activation is believed to be an important step in the mode of action of Cry toxins [11]. Cross-linking studies using disulfide bonds have shown that activation of Cry toxins through the cleavage of the N-terminal peptide segment results in a conformational change, leading to domain I moving away from domains II and III, insertion of the helical bundle into the membrane, and pore formation [13,14]. Bravo et al. 2007 postulated that this conformational change may expose a region within domain II responsible for toxin-receptor interaction [15]. Our work shows that N-terminal cleavage of Cry41Aa is not required for the toxin to bind to HepG2 cells. It's thought that Cry41Aa, like other Cry toxins, still requires proteolytic processing to insert and form a pore within the membrane, as evidenced by the fact the protoxin is not toxic towards cells [3]. Binding of the Cry41Aa protoxin was confirmed through both fluorescent microscopy and immunoblotting and it was found to compete with the activated toxin indicating shared binding sites. Though the Cry41Aa protoxin has been proven to be non-toxic towards HepG2 cells even at relatively high concentrations, lack of toxicity at any concentration cannot be ruled out [3]. Tabashnik et al. 2015 discussed a dual mode of action hypothesis following findings that Cry1Ab and Cry1Ac protoxins were more potent than the activated toxins in insects with resistance to these toxins, indicating that mechanisms of resistance can highlight alternative pathways for protoxins to exert toxicity [16].
This study also found that some insecticidal toxins could exert a synergistic effect with Cry41Aa against HepG2 cells despite these insecticidal toxins not possessing toxicity in their own right. Synergistic effects of Bt Cry toxins have been reported in studies since the 1980s [17] and can be the result of several different mechanisms in which combinations of two or more Cry toxins can exert a greater than expected influence [18,19]. The formation of hetero-oligomers between closely related Cry toxins has been shown to confer advantages in receptor binding and subsequent toxicity over monomers and homo-oligomers [20] and thus result in synergism. Toxins may even serve as membrane binding targets for other toxins, as Cyt1A was shown to serve as a functional, membrane bound receptor for Cry11Aa in Aedes aegypti [21].
Of the toxins tested within this study, several were found capable of exerting a synergistic effect with Cry41Aa. Interestingly all of those with this activity also have activity against Aedes aegypti although it is currently unclear if there is any significance to this observation. Among these, Cry1Ca was shown to bind HepG2 cells in its active, but not protoxin, form. Despite its ability to bind, exposure of HepG2 cells to activated Cry1Ca, or any of the other insecticidal Cry toxins used, failed to illicit any cell swelling, a characteristic early cellular response of HepG2 cells to Cry41Aa exposure [3]. This absence of cellular response suggests that, whilst possessing the ability to bind a putative receptor is certainly a prerequisite for Cry toxin specificity, other factors such as the orientation of binding or the ability to insert into the host membrane also affect toxicity. The binding and synergistic effect of Cry1Ca on Cry41Aa in HepG2 cells prompted the development of a two-receptor hypothesis. We hypothesise that two receptors exist for Cry41Aa and that one is less effective than the other. Cry41Aa can bind both targets, whilst Cry1Ca, Cry11Aa, and Cry4Aa Cry toxins can only bind to one, the hypothesised "less effective" target. In doing so, these Cry toxins facilitate a synergistic effect by blocking binding to the less effective site thereby allowing more of the Cry41Aa to bind to the "more effective" target. The observable result is a decrease in cell viability beyond that observed in controls featuring Cry41Aa alone where more of the toxin has bound to the less or non-effective receptor.
The synergism between the Cry toxins and Cry41Aa in HepG2 cells differs from the effect observed in Aedes aegypti larval competition studies. Competition studies have long been a means by which shared receptors and cross-resistance between different Cry toxins have been explored in insect systems [22]. We observed that Cry41Aa could inhibit the toxicity of Cry1Ca and Cry11Aa in larval assays, which suggests that Cry41Aa can bind the same receptor in the Aedes midgut. Though the putative receptor for Cry1Ca in Aedes aegypti is unknown, it has been elucidated that the receptor for Cry11Aa in Aedes is a glycosylphosphatidylinositol (GPI) anchored alkaline phosphatase [23]. Therefore, it's possible that an homologous alkaline phosphatase within mammalian cells may represent a shared binding site for Cry1Ca, Cry11Aa, Cry4Aa and Cry41Aa.
Though long considered to be a non-insecticidal and non-haemolytic Cry toxin, there exists the possibility that Cry41Aa does possess some form of insecticidal ability. Still, any insect species in which it may have an effect has yet to be found. Given Cry41Aa’s interesting crossover with the Aedes-active toxins detailed in this work, it could be argued that this parasporin may share a close evolutionary history with Cry toxins and that perhaps another factor, such as changes to the biochemistry of domain I, have imbued this protein with pore-forming capabilities in mammalian cells rather than in insects.
Author Contributions
Nicole Bryce-Sharron: data curation, formal analysis, methodology, investigation, visualisation, writing—original draft, and writing—review and editing. Mojtaba Nasiri: data curation, formal analysis, methodology, investigation, visualisation, and writing—original draft. Tom Powell: methodology, investigation, and visualization. Neil Crickmore: Conceptualisation, formal analysis, investigation, project administration, supervision, writing—original draft, and writing—review and editing. Michelle J. West: supervision.
Funding
Michelle J. West is funded by grant 20003 from Blood Cancer UK.
Data Availability Statement
All data included within article / supplementary materials. Raw data requests and further enquiries can be directed to the corresponding author.
Acknowledgments
We would like to thank Wided Souissi for support on a number of experiments contained within this work, and for providing the pHTCry41Aa-PreScission and pHTCry41AaHA constructs. We would also like to thank Juliette Bartlett, who cloned the genes for a number of the insecticidal Cry toxins featured in this work into the pHT315 vector.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
Figure A1.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry4Aa. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts (w/w) of activated Cry4Aa. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ***p=0.0006, ns=not significant.
Figure A1.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry4Aa. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts (w/w) of activated Cry4Aa. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ***p=0.0006, ns=not significant.
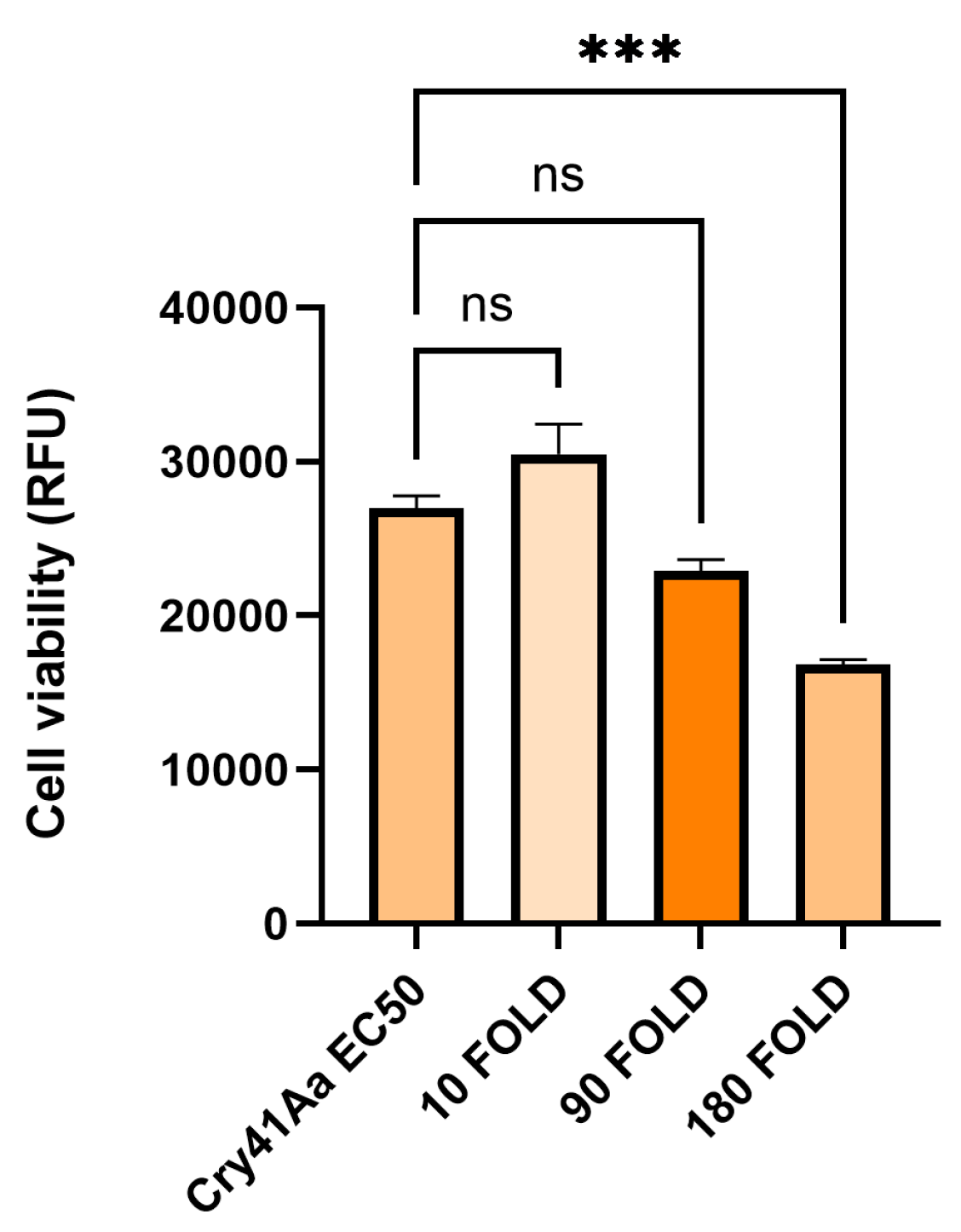
Figure A2.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry11Aa. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts (w/w) of activated Cry11Aa. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ****p=<0.0001.
Figure A2.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry11Aa. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts (w/w) of activated Cry11Aa. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ****p=<0.0001.
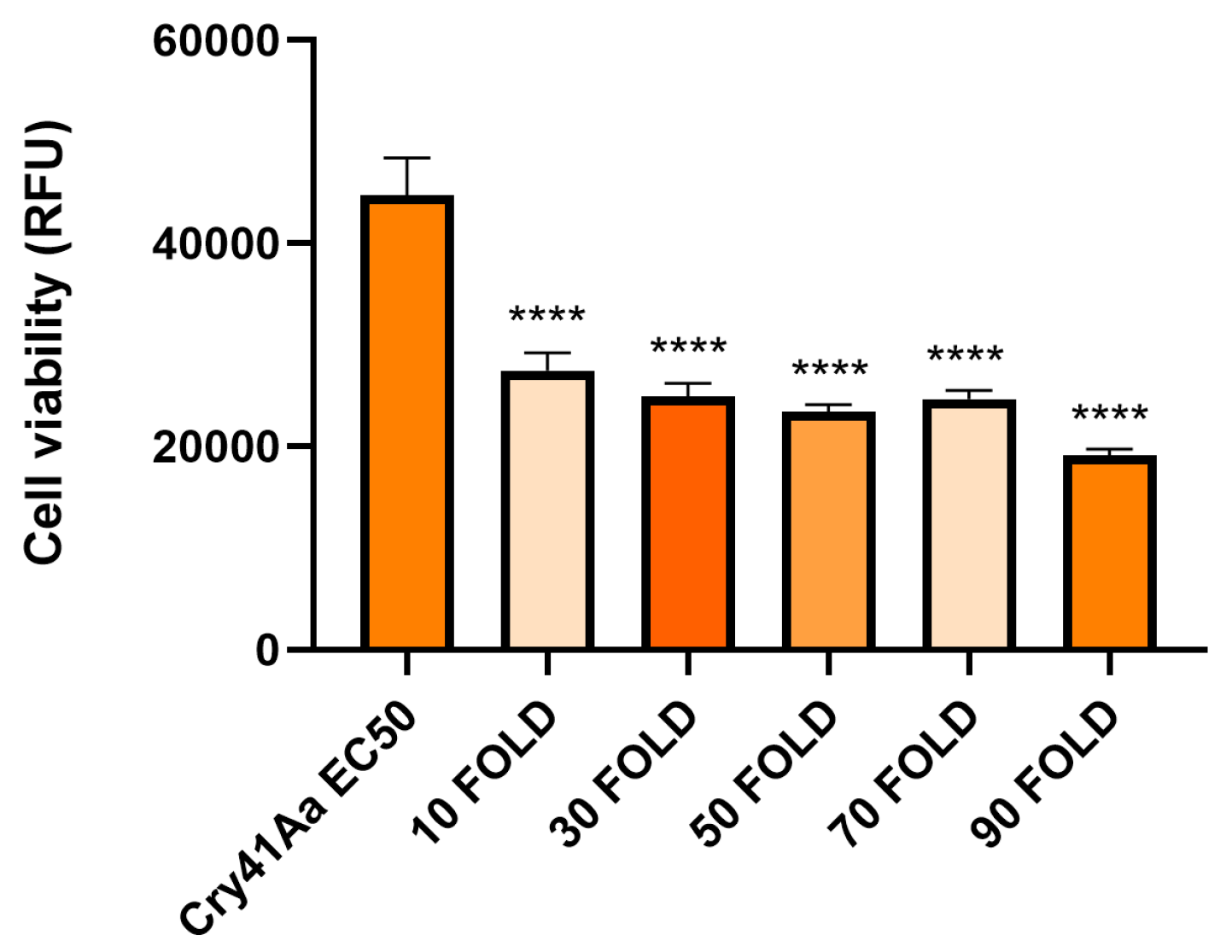
Figure A3.
Competition assay between Cry11Aa and excess Cry41Aa utilising 3rd instar Aedes aegypti larvae. Aedes-active Cry11Aa presented at 0.8 µg/ml competed with excess amounts (w/w) of Cry41Aa. Error bars show ±SEM. A one-way ANOVA was used to calculate significance. Ns=not significant.
Figure A3.
Competition assay between Cry11Aa and excess Cry41Aa utilising 3rd instar Aedes aegypti larvae. Aedes-active Cry11Aa presented at 0.8 µg/ml competed with excess amounts (w/w) of Cry41Aa. Error bars show ±SEM. A one-way ANOVA was used to calculate significance. Ns=not significant.
References
- R. de Maagd, A. Bravo, and N. Crickmore, “How Bacillus thuringiensis has evolved specific toxins to colonize the insect world,” Trends in Genetics, vol. 17, no. 4. pp. 193–199, Apr. 2001. [CrossRef]
- J. L. Jurat-Fuentes and N. Crickmore, “Specificity determinants for Cry insecticidal proteins: Insights from their mode of action,” Journal of Invertebrate Pathology, vol. 142, pp. 5–10, Jan. 2017. [CrossRef]
- V. Krishnan, B. Domanska, A. Elhigazi, F. Afolabi, M. J. West, and N. Crickmore, “The human cancer cell active toxin Cry41Aa from Bacillus thuringiensis acts like its insecticidal counterparts,” Biochemical Journal, vol. 474, no. 10, pp. 1591–1602, Apr. 2017. [CrossRef]
- W. Souissi, A. Kaloki, S. Etherington, B. Domanska, M. J. West, and N. Crickmore, “Differential proteolytic activation of the Bacillus thuringiensis Cry41Aa parasporin modulates its anticancer effect,” Biochemical Journal, vol. 476, no. 24, pp. 3805–3816, Dec. 2019. [CrossRef]
- R. Sato, “Utilization of Diverse Molecules as Receptors by Cry Toxin and the Promiscuous Nature of Receptor-Binding Sites Which Accounts for the Diversity,” Biomolecules, vol. 14, no. 4, pp. 425-425, Apr. 2024. [CrossRef]
- T. Akiba and S. Okumura, “Parasporins 1 and 2: Their structure and activity,” Journal of Invertebrate Pathology, vol. 142, pp. 44–49, Jan. 2017. [CrossRef]
- H. Endo, “Molecular and Kinetic Models for Pore Formation of Bacillus thuringiensis Cry Toxin,” Toxins, vol. 14, no. 7, p. 433, Jun. 2022. [CrossRef]
- O. Arantes and D. Lereclus, “Construction of cloning vectors for Bacillus thuringiensis,” Gene, vol. 108, no. 1, pp. 115–119, Dec. 1991. [CrossRef]
- F. A. Alzahrani and N. Crickmore, “N-terminal proteolysis determines the differential activity of Bacillus thuringiensis Cry2A toxins towards Aedes aegypti,” Journal of Invertebrate Pathology, vol. 204, pp. 108100–108100, Mar. 2024. [CrossRef]
- N. Crickmore and D. J. Ellar, “Involvement of a possible chaperonin in the efficient expression of a cloned CryIIA delta-endotoxin gene in Bacillus thuringiensis,” Molecular Microbiology, vol. 6, no. 11, pp. 1533–1537, Jun. 1992. [CrossRef]
- A. Bravo, J. Sánchez, T. Kouskoura, and N. Crickmore, “N-terminal Activation Is an Essential Early Step in the Mechanism of Action of the Bacillus thuringiensis Cry1Ac Insecticidal Toxin,” Journal of Biological Chemistry, vol. 277, no. 27, pp. 23985–23987, Jul. 2002 . [CrossRef]
- S. E. González-Villarreal, M. García-Montelongo, and J. E. Ibarra, “Insecticidal Activity of a Cry1Ca toxin of Bacillus thuringiensis Berliner (Firmicutes: Bacillaceae) and Its Synergism with the Cyt1Aa Toxin Against Aedes aegypti (Diptera: Culicidae),” Journal of Medical Entomology, vol. 57, no. 6, pp. 1852–1856, Jun. 2020. [CrossRef]
- J.-L. Schwartz et al., “Restriction of intramolecular movements within the CrylAa toxin molecule of Bacillus thuringiensis through disulfide bond engineering,” FEBS Letters, vol. 410, no. 2–3, pp. 397–402, Jun. 1997. [CrossRef]
- S. Pacheco, J. P. J. Quiliche, I. Gómez, J. Sánchez, M. Soberón, and A. Bravo, “Rearrangement of N-Terminal α-Helices of Bacillus thuringiensis Cry1Ab Toxin Essential for Oligomer Assembly and Toxicity,” Toxins, vol. 12, no. 10, p. 647, Oct. 2020. [CrossRef]
- A. Bravo, S. S. Gill, and M. Soberón, “Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control,” Toxicon, vol. 49, no. 4, pp. 423–435, Mar. 2007. [CrossRef]
- B. E. Tabashnik et al., “Dual mode of action of Bt proteins: protoxin efficacy against resistant insects,” Scientific Reports, vol. 5, no. 1, Oct. 2015. [CrossRef]
- D. Wu and F. N. Chang, “Synergism in mosquitocidal activity of 26 and 65 kDa proteins from Bacillus thuringiensis subsp. israelensis crystal,” FEBS Letters, vol. 190, no. 2, pp. 232–236, Oct. 1985. [CrossRef]
- R. Monnerat et al., “Synergistic activity of Bacillus thuringiensis toxins against Simulium spp. larvae,” Journal of Invertebrate Pathology, vol. 121, pp. 70–73, Sep. 2014. [CrossRef]
- N. Crickmore, E. J. Bone, J. A. Williams, and D. J. Ellar, “Contribution of the individual components of the δ-endotoxin crystal to the mosquitocidal activity of Bacillus thuringiensis subsp. israelensis,” FEMS Microbiology Letters, vol. 131, no. 3, pp. 249–254, Sep. 1995. [CrossRef]
- D. Pinos, N. Joya, S. Herrero, J. Ferré, and P. Hernández-Martínez, “Hetero-oligomerization of Bacillus thuringiensis Cry1A proteins enhance binding to the ABCC2 transporter of Spodoptera exigua,” Biochemical Journal, vol. 478, no. 13, pp. 2589–2600, Jul. 2021. [CrossRef]
- C. Perez et al., “Bacillus thuringiensis subsp. israelensis Cyt1Aa synergizes Cry11Aa toxin by functioning as a membrane-bound receptor,” Proceedings of the National Academy of Sciences, vol. 102, no. 51, pp. 18303–18308, Dec. 2005. [CrossRef]
- P. Hernández-Martínez et al., “Comparison of in vitro and in vivo binding site competition of Bacillus thuringiensis Cry1 proteins in two important maize pests,” Pest Management Science, vol. 78, no. 4, pp. 1457–1466, Jan. 2022. [CrossRef]
- Luisa E. Fernandez, Karlygash G. Aimanova, Sarjeet S. Gill, A. Bravo, and M. Soberón, “A GPI-anchored alkaline phosphatase is a functional midgut receptor of Cry11Aa toxin in Aedes aegypti larvae,” Biochemical Journal, vol. 394, no. 1, pp. 77–84, Jan. 2006. [CrossRef]
Figure 1.
Fluorescent micrographs of HepG2 cells treated with and without Cry41AaGFP. Micrographs show brightfield, GFP, DAPI, and merged (composite) images of each assay using appropriate treatments, z-Stack level for each image set to the corresponding surface level of cells.
Figure 1.
Fluorescent micrographs of HepG2 cells treated with and without Cry41AaGFP. Micrographs show brightfield, GFP, DAPI, and merged (composite) images of each assay using appropriate treatments, z-Stack level for each image set to the corresponding surface level of cells.
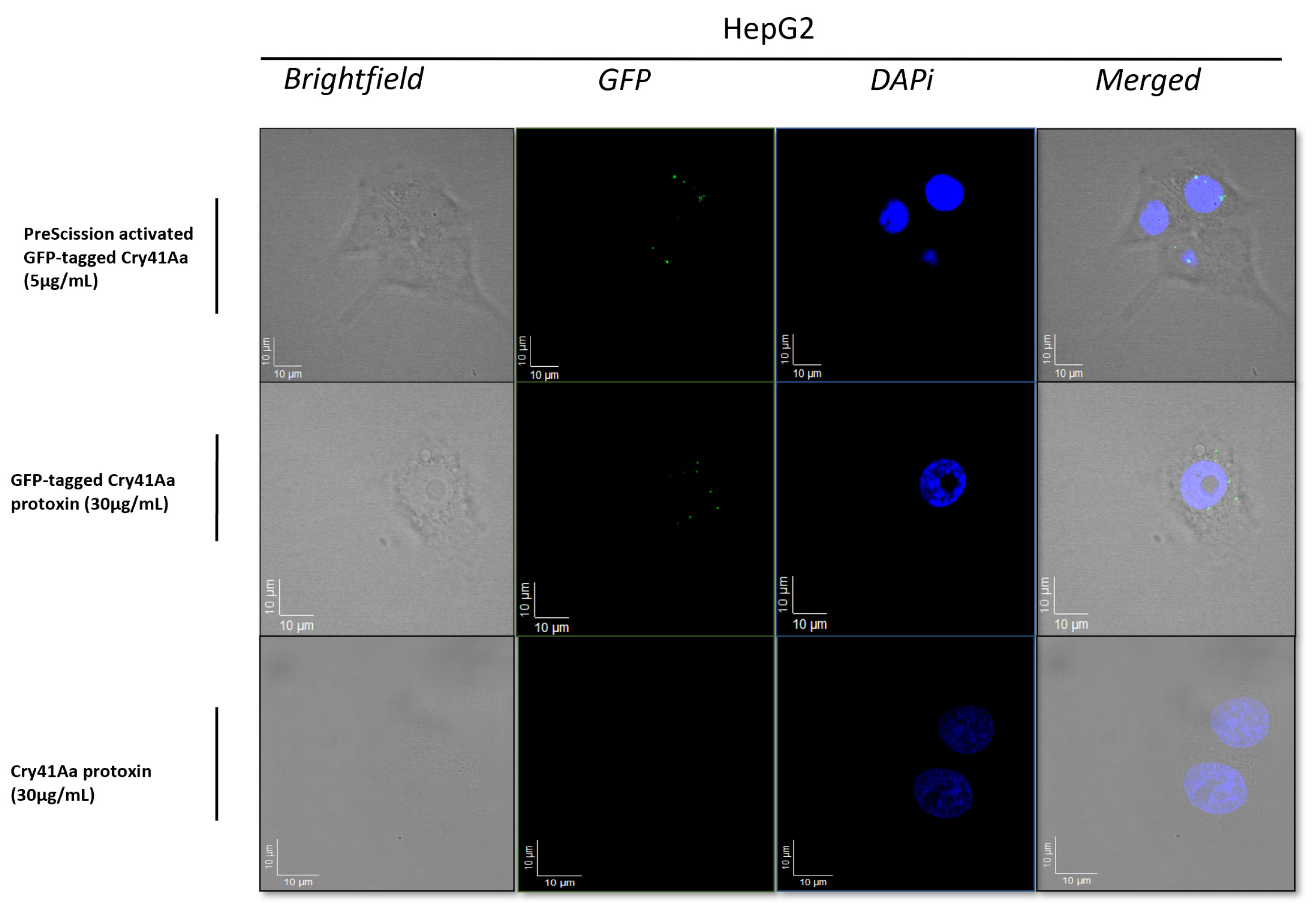
Figure 2.
Binding of Cry41AaHA protoxin and activated Cry41AaHADIP toxin to HepG2 cells. (A) lane 1, protoxin only; lanes 2-6 whole cell lysates of cells exposed to varying concentrations of Cry41AaHA protoxin (lane 2, 200 µg/ml; lane 3, 300 µg/ml; lane 4, 400 µg/ml; lane 5, 500 µg/ml; lane 6, 600 µg/ml); lane 7, cell only control. (B) lane 1, Cry41AaHADIP toxin only; lanes 2-6 show whole cell lysates of cells exposed to varying concentrations of activated Cry41AaHADIP (lane 2, 25 µg/ml; lane 3, 50 µg/ml; lane 4, 100 µg/ml; lane 5, 200 µg/ml; lane 6, 300 µg/ml); lane 7, cell only control.
Figure 2.
Binding of Cry41AaHA protoxin and activated Cry41AaHADIP toxin to HepG2 cells. (A) lane 1, protoxin only; lanes 2-6 whole cell lysates of cells exposed to varying concentrations of Cry41AaHA protoxin (lane 2, 200 µg/ml; lane 3, 300 µg/ml; lane 4, 400 µg/ml; lane 5, 500 µg/ml; lane 6, 600 µg/ml); lane 7, cell only control. (B) lane 1, Cry41AaHADIP toxin only; lanes 2-6 show whole cell lysates of cells exposed to varying concentrations of activated Cry41AaHADIP (lane 2, 25 µg/ml; lane 3, 50 µg/ml; lane 4, 100 µg/ml; lane 5, 200 µg/ml; lane 6, 300 µg/ml); lane 7, cell only control.

Figure 3.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess Cry41Aa protoxin. Activated Cry41Aa WT administered at an EC50 concentration of 2 µg/ml competed with fold excess amounts of Cry41Aa WT protoxin. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. **p=0.0016, ***p=0.0001, ****p=<0.0001, ns=not significant.
Figure 3.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess Cry41Aa protoxin. Activated Cry41Aa WT administered at an EC50 concentration of 2 µg/ml competed with fold excess amounts of Cry41Aa WT protoxin. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. **p=0.0016, ***p=0.0001, ****p=<0.0001, ns=not significant.
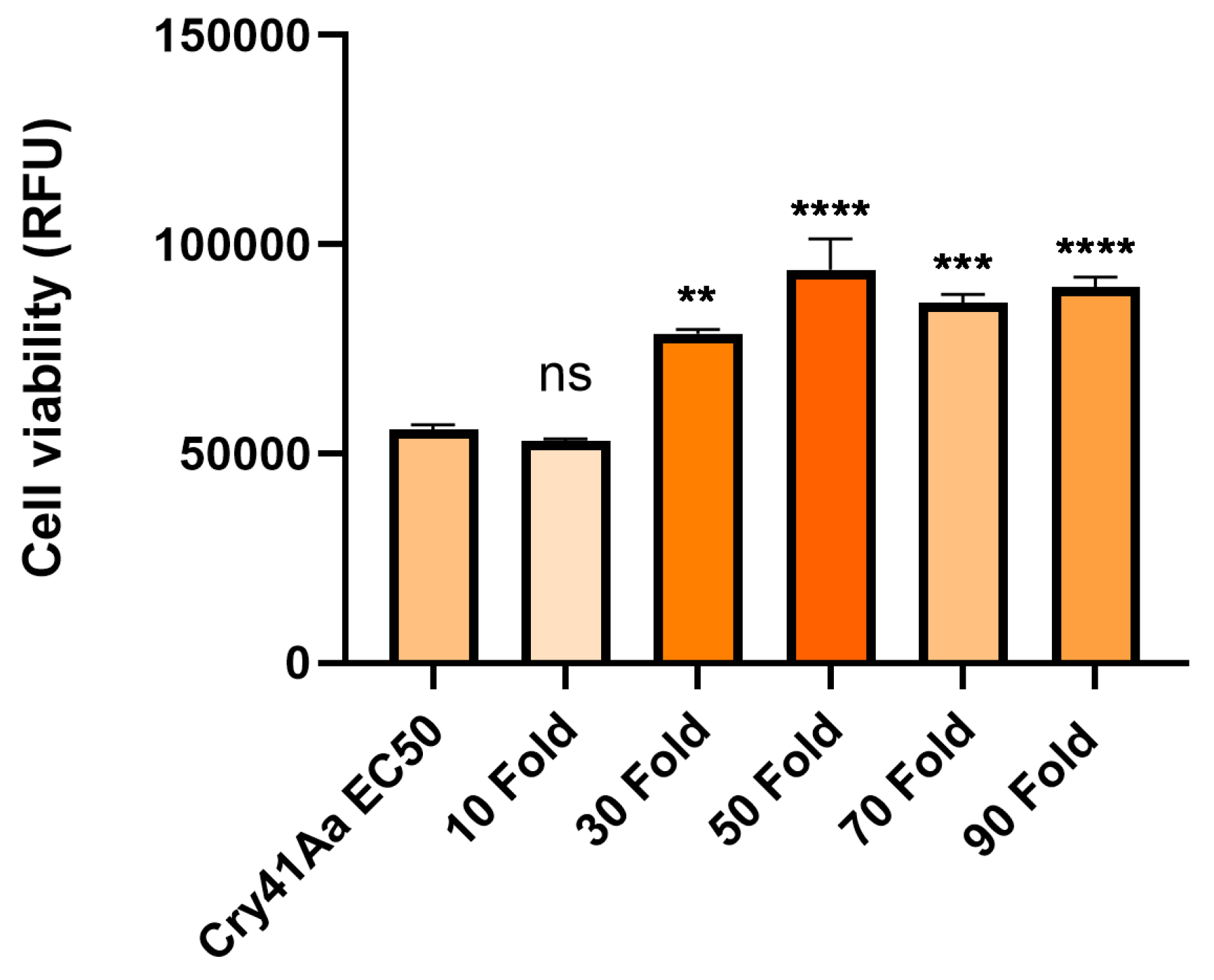
Figure 4.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry1Ca. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts of trypsin-activated Cry1Ca. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. *p=0.0166, **p= 0.0020, **p= 0.0018, ****p=<0.0001, ns=not significant.
Figure 4.
Cell viability of HepG2 cells against activated Cry41Aa competed with excess activated Cry1Ca. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts of trypsin-activated Cry1Ca. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. *p=0.0166, **p= 0.0020, **p= 0.0018, ****p=<0.0001, ns=not significant.
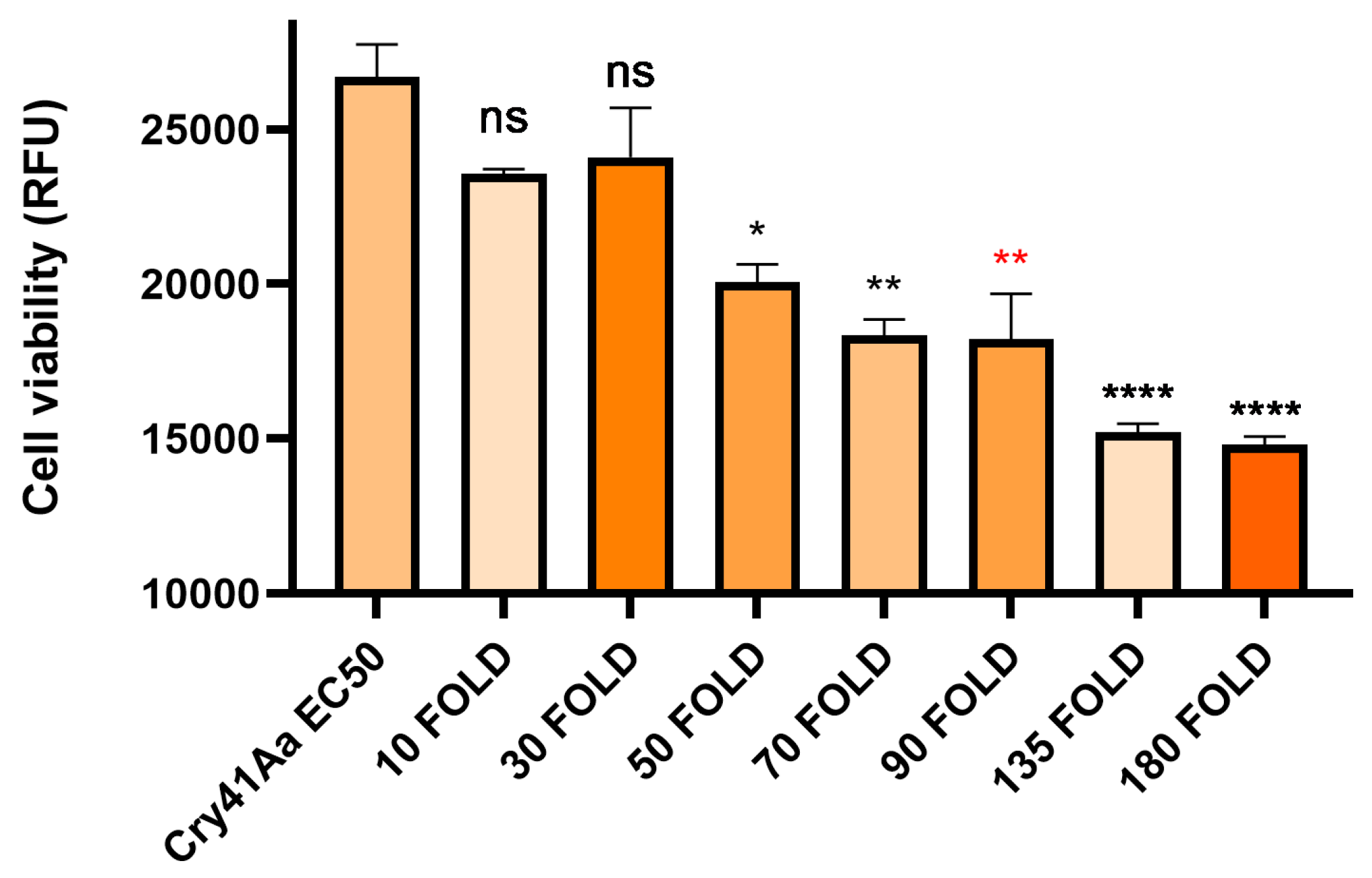
Figure 5.
Detection of bound Cry1Ca protoxin and active toxin to HepG2 cells. Lane 1, toxin-only; lanes 2-6 show whole cell lysates of cells exposed to varying concentrations of either the trypsin-activated Cry1Ca (A) or the Cry1Ca protoxin (B) lane 2, 200 µg/ml; lane 3, 300 µg/ml; lane 4, 400 µg/ml; lane 5, 500 µg/ml; lane 6, 600 µg/ml, lane 7, cell only control.
Figure 5.
Detection of bound Cry1Ca protoxin and active toxin to HepG2 cells. Lane 1, toxin-only; lanes 2-6 show whole cell lysates of cells exposed to varying concentrations of either the trypsin-activated Cry1Ca (A) or the Cry1Ca protoxin (B) lane 2, 200 µg/ml; lane 3, 300 µg/ml; lane 4, 400 µg/ml; lane 5, 500 µg/ml; lane 6, 600 µg/ml, lane 7, cell only control.

Figure 6.
Cell viability of HepG2 cells when treated with activated Cry41Aa and an excess of Cry1Ca protoxin. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts of Cry1Ca protoxin. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ns=not significant.
Figure 6.
Cell viability of HepG2 cells when treated with activated Cry41Aa and an excess of Cry1Ca protoxin. Activated Cry41Aa administered at an EC50 concentration of 2 µg/ml competed with excess amounts of Cry1Ca protoxin. Cell viability was assessed using the CellTiter-Blue assay. Cell viability was measured in relative fluorescence units (RFU). Error bars show ±SEM. A one-way ANOVA was used to calculate significance. ns=not significant.
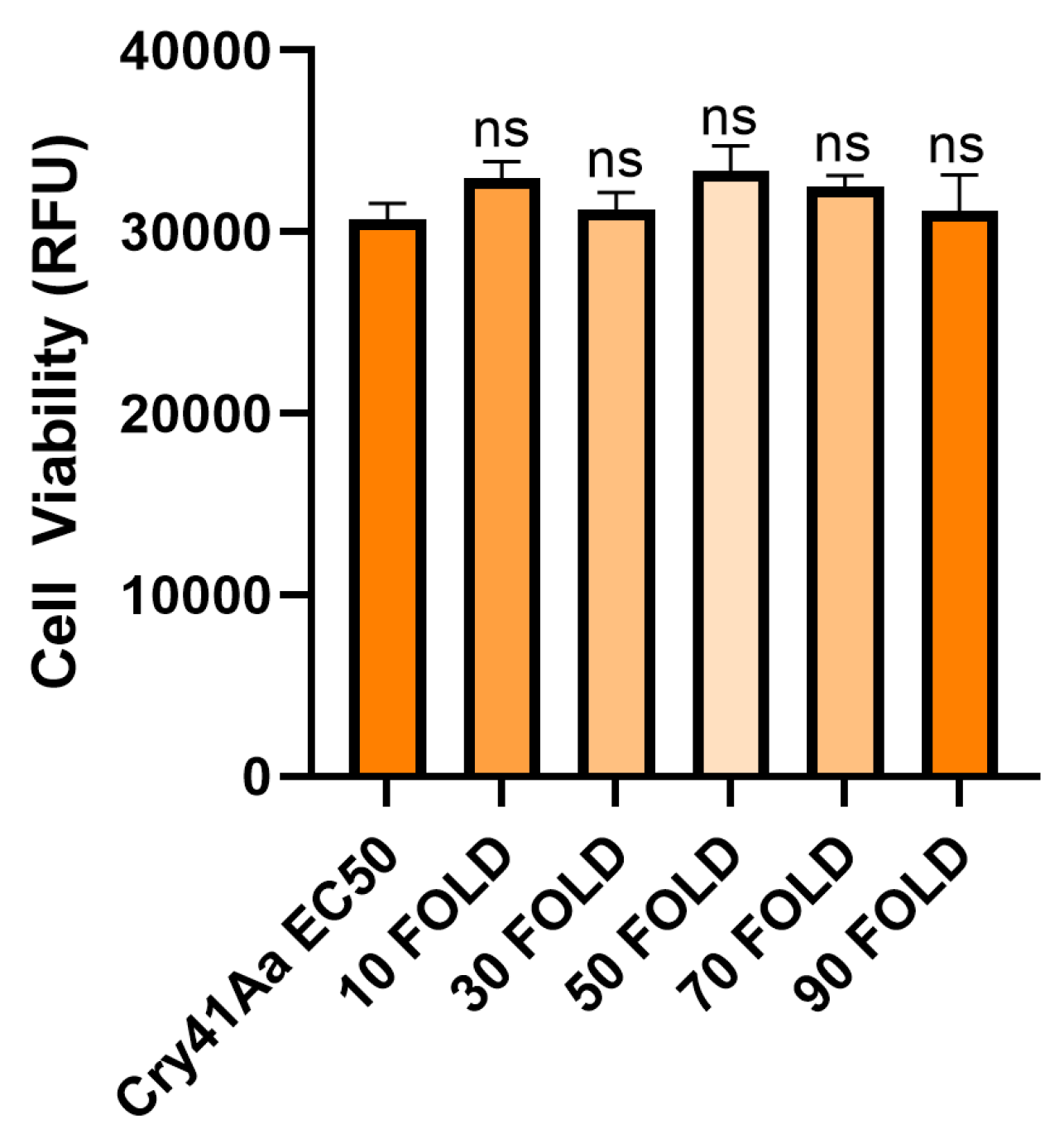
Figure 7.
Competition assay between Cry1Ca and excess Cry41Aa utilising 3rd instar Aedes aegypti larvae. Cry1Ca presented at 25 µg/ml competed with excess amounts (w/w) of Cry41Aa. Error bars show ±SEM. A one-way ANOVA was used to calculate significance. *p= 0.0130, ****p=<0.0001.
Figure 7.
Competition assay between Cry1Ca and excess Cry41Aa utilising 3rd instar Aedes aegypti larvae. Cry1Ca presented at 25 µg/ml competed with excess amounts (w/w) of Cry41Aa. Error bars show ±SEM. A one-way ANOVA was used to calculate significance. *p= 0.0130, ****p=<0.0001.

Table 1.
A list of insecticidal Cry toxins and their effects when presented with Cry41Aa against HepG2 cells.
Table 1.
A list of insecticidal Cry toxins and their effects when presented with Cry41Aa against HepG2 cells.
| Insecticidal Cry Toxin | Insecticidal Cry toxin (excess by weight) | Cry41Aa concentration | Effect when combined with Cry41Aa |
| Cry1Ca | 10-180 fold | 2 µg/ml | Synergism |
| Cry11Aa | 10-90 fold | 2 µg/ml | Synergism |
| Cry4Aa | 10-180 fold | 2 µg/ml | Synergism |
| Cry1Ac | 10-180 fold | 2 µg/ml | No effect |
| Cry1Da | 10-180 fold | 2 µg/ml | No effect |
| Cry2Aa | 10-90 fold | 2 µg/ml | No effect |
| Cry2Ab | 10-90 fold | 2 µg/ml | No effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



