Submitted:
07 June 2024
Posted:
11 June 2024
You are already at the latest version
Abstract
Inherited retinal diseases (IRDs) are extremely heterogeneous with at least 350 causative genes, complicating the process of genetic diagnosis. We analyzed samples of 252 index cases with IRDs using the Blueprint Genetics panel for “Retinal Dystrophy” that includes 351 genes. The cause of disease could be identified in 55% of cases. A clear difference was obtained between newly-recruited cases (74% solved) and cases that were previously analyzed by panels or whole exome sequencing (26% solved). As for the mode of inheritance, 75% of solved cases were autosomal recessive (AR), 10% were X-linked, 8% were autosomal dominant, and 7% were mitochondrial. Interestingly, in 12% of solved cases, structural variants (SVs) were identified as the cause of disease. The most commonly identified genes were ABCA4, EYS and USH2A, and the most common mutations were MAK-c.1297_1298ins353 and FAM161A- c.1355_1356del. In line with our previous IRD carrier analysis, we identified heterozygous AR mutations that are not the cause of disease in 36% of cases. The studied IRD panel was found to be efficient in gene identification. Some variants were mis-interpreted by the pipeline and therefore multiple analysis tools are recommended to obtain more accurate annotation of potential disease-causing variant.
Keywords:
Consanguinity
; gene panel
; Inherited retinal diseases
; retinal dystrophy
; targeted next generation sequencing
1. Introduction
Inherited retinal diseases (IRDs) encompass a broad spectrum of retinal phenotypes characterized by extensive clinical variability and profound genetic heterogeneity [1,2]. Among IRDs, retinitis pigmentosa (RP) stands out as one of the most prevalent and heterogeneous conditions in humans, caused by disease-causing mutations in over 60 genes with inheritance patterns spanning autosomal dominant (AD), autosomal recessive (AR), digenic, mitochondrial and X-linked (XL) modes [1].
The advent of substantial sequencing breakthroughs over the past decades as well as improved bioinformatics and functional analyses has heralded a remarkable advancement in mutation detection methods as well as mutation characterization [3]. These include next-generation sequencing (NGS), whole exome sequencing (WES), whole genome sequencing (WGS), functional genomic techniques (such as CRISPR-Cas9 gene editing, RNA sequencing, and functional assays), bioinformatics analysis, single-cell genomics, epigenomic profiling techniques (including ChIP-seq and DNA methylation profiling), and multi-omics integration. Each of these modern methodologies offers unique advantages in elucidating the genetic underpinnings of various diseases.
From providing a comprehensive view of disease mechanisms to facilitating the identification of novel mutations and understanding complex diseases with heterogeneous genetic backgrounds, these techniques are often employed synergistically to enhance diagnostics, tailor personalized treatments, and refine disease management strategies.
However, the selection of an appropriate method is fraught with challenges due to several inherent limitations. These include cost considerations, the complexity of data analysis, the potential for false positives and false negatives, limitations in functional validation, incomplete genomic coverage, ethnic diversity, ethical and privacy concerns surrounding genomic data, and the intricacies of clinical utility and interpretation.
In light of these challenges, we endeavor to navigate the landscape of available methodologies, striving to identify the most suitable approach that aligns with our goal of providing comprehensive genetic insights to many patients. Our aim is to unveil all pertinent mutations contributing to the heterogeneity of IRDs.
Here we used a single gene panel to screen the DNA of 252 unsolved cases with IRDs. The analysis revealed the identification of the cause of disease in 55% of cases, with variability in detection rates between sub-groups.
2. Materials and Methods
2.1. Patient Recruitment
Blood and saliva samples were collected from 252 individuals diagnosed with IRDs, representing a diverse mix of newly recruited patients and those who remained unsolved following previous screening tests. The sample collection process adhered to ethical standards, receiving explicit approval from the Hadassah Hospital Institutional Review Board. Informed consent, obtained in writing, ensured compliance with all relevant ethical regulations and safeguarded the rights and privacy of participants.
2.2. DNA Extraction
Genomic DNA extraction was carried out from blood samples using the Promega kit and Promega Maxwell (Madison, WI) DNA extraction device. DNA extraction from buccal swab samples was conducted at the Blueprint Genetics company.
2.3. Next-Generation Sequencing (NGS)
The Retinal Dystrophy Panel (Blueprint Genetics; Espoo, Finland; test code OP0801: https://blueprintgenetics.com/tests/panels/ophthalmology/retinal-dystrophy-panel/), including 351 IRD genes, was used to analyze DNA of 252 IRD cases. A clinical grade NGS sequencing assay was employed allowing comprehensive coverage across various genomic IRD genes. The analysis included detection of single nucleotide variants, insertions, deletions, indels, and copy number variants (CNVs- deletions or duplications spanning at least a single exon). Sequencing depth at the nucleotide level was 174x at the nucleotide level, ensuring unparalleled sensitivity and accuracy in variant detection.
3. Results
3.1. Patient Demographics
A total of 252 index cases, selected from a cohort of over 2000 families, were enrolled to this study. Of these, 230 DNA samples were extracted from blood, with 98 undergone testing with a negative output. Additionally, 22 fresh saliva samples from newly recruited patients were shipped for analysis. Our cohort exhibited ethnic diversity, with 66% Jews (including 26% Ashkenazi Jews, 12% North African Jews, and 10% Moroccan Jews), 31% Arab Muslims (including 3% Bedouins), 2% Arab Christians, and other ethnicities with less than 1% each.
3.2. Efficiency of the Gene Panel
The genetic cause of disease (pathogenic or likely pathogenic mutation/s) was identified in 138 out of the 252 analyzed samples, a yield rate of 55% (Figure 1A, Table S1). Consequently, 114 cases remained unsolved (Figure 1A, Table S1). Further analysis revealed varying rates of gene identification between two distinct groups: newly recruited samples exhibited a 74% success rate, while previously analyzed samples with negative results (the analysis included genotyping of founder mutations, IRD gene panels, and WES) showed a 24% success rate (Figure 1B).
3.3. Types of Solved Cases and Gene Frequency Analysis
3.4. Frequency of the Most Common Mutations
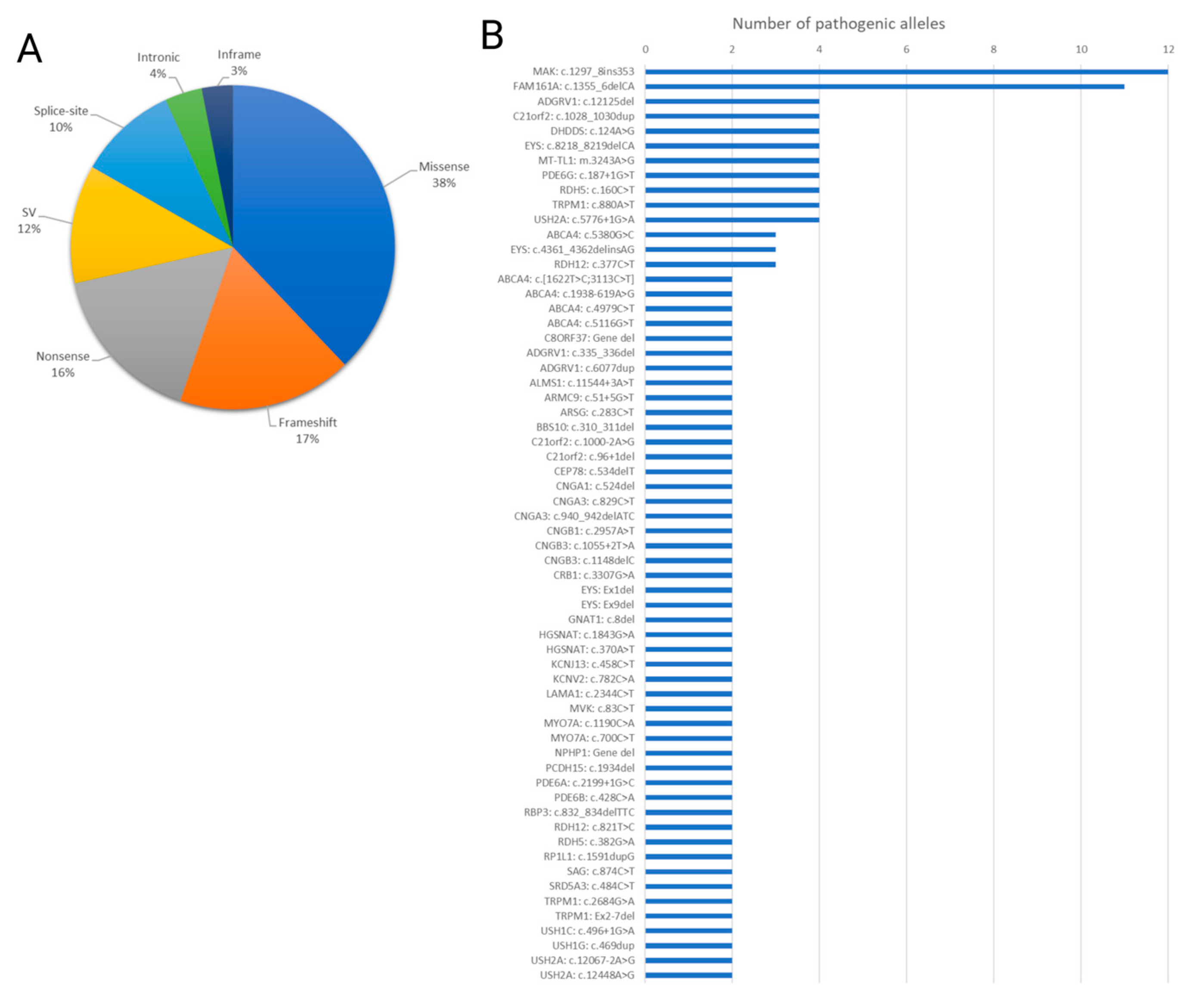
Missense mutations constituted the most common variant type (38%), followed by frameshift mutations (17%), nonsense mutations (16%), structural variants (SVs) (12%), splice site mutations (10%), intronic mutations (4%), and in-frame mutations (3%) (Figure 3A). Notably, the most recurrent mutations identified were c.1297_1298ins353 in MAK and c.1355_1356del in FAM161A (Figure 3B).
3.5. KIF11 and Non-Syndromic Chorioretinopathy
The KIF11 gene encodes a homo-tetrameric motor protein crucial for spindle polarity during mitosis. Variants in this ubiquitously expressed protein are typically de-novo and associated with various developmental syndromes, including impacts on retinal vasculature development. Considered rare, only one family in Israel is documented with a KIF11 mutation [4]. Surprisingly, among the 142 cases solved in our study, KIF11 emerged as the causal gene in two unrelated Ashkenazi Jewish patients.
Case MOL1984-1 was diagnosed at the age of 10 years with reduced visual acuity, mild myopia (-3 D), and photophobia. His Best-corrected visual acuity (BCVA) at the age of 16 was 0.6 / 0.5 in the right and left eye, respectively, and OCT showed a retinoschisis pattern. He has no family history of visual deficiencies. ERG at the age of 18 years revealed sub-normal responses, including cone flicker, mixed cone-rod b-wave and rod responses. Color vision was within normal range. Ophthalmic evaluation at the age of 19 years revealed atrophic chorioretinal patches next to the optic disc and the inferior arcade. The patient was therefore diagnosed with non-syndromic chorioretinopathy. Interestingly, gene panel analysis revealed a heterozygous frameshift mutation in KIF11 (Table S1).
Case MOL2101-1 was diagnosed with a congenital retinal disease, including chorioretinal scars and retinoschisis. Visual acuity deteriorated and at the age of 11 reached 0.4 in the right eye and hand motion in the left eye with mild myopia (-3.5 D). Multiple electroretinography (ERG) examinations revealed non-progressive sub-normal ERG responses, including cone flicker, mixed cone-rod a and b-wave and rod responses. The patient was therefore diagnosed with non-syndromic chorioretinopathy. Interestingly, gene panel analysis revealed a heterozygous de-novo nonsense mutation in KIF11 (Table S1).
3.6. An Elusive Coding Mutation
Further illustrating the complexities of genetic diagnostics, an intriguing and elusive nonsense pathogenic mutation in the EYS gene, was identified.
The index case, MOL2142-1, was diagnosed with ARRP due to extinguished ERG responses at the age of 55 years, fundus appearance that include typical signs of RP (bone-spicule pigmentation and attenuated blood vessels), and reduced BCVA of 0.25. Copy number variation (CNV) analysis of IRD genes, as part of the panel analysis, revealed a heterozygous deletion of a single EYS exon (#34), as well as two nucleotide changes (one synonymous and the other nonsynonymous), affecting neighboring nucleotides (c.4361C>A, p.S1454Y and c.4362C>G, p.S1454=). Interestingly, this combination of variants was identified in multiple cases with RP of Arab-Muslim origin, and confirmed to be in-cis. The combination of the two variant results in a nonsense mutation (c.4361_4362delinsAG, p.S1454*; Table S1). Such complex mutation can be oversighted in many NGS pipelines, highlighting the challenges of variant interpretation and the importance of comprehensive genetic panels.
3.7. Novel Genetic Discoveries in Israeli Patients: Unveiling Rare Variants and First Cases
Joubert syndrome, a recessive and genetically heterogeneous neurodevelopmental ciliopathy, is characterized by a distinctive brain malformation, often presenting with a complex phenotype that may include retinal dystrophy. In our study, we identified two cases, MOL2246-1 and MOL22151-1, marking the first documented instances in Israel of Joubert syndrome associated with mutations in the ARMC9 and LAMA1 genes, respectively. Notably, these genes are rare contributors to this syndrome; in the literature, only 19 cases of ARMC9 mutations have been reported across five different articles from Japanese [5,6], Chinese [7], Turkish [8], and Indian [9] populations, while LAMA1 mutations have been documented in just five cases from British [10] and Japanese [11] cohorts spanning two articles. This underscores the rarity and global diversity of genetic variants contributing to Joubert syndrome.
In addition to the aforementioned cases, our study unveiled several other rare variants, each representing the first documented instances in Israel. One such case is patient MOL1437-1, who exhibits a compound heterozygosity for missense mutations in the WDR19 gene, resulting in non-syndromic retinitis pigmentosa (RP). Another noteworthy finding is a homozygous nonsense mutation in the SAG gene in patient MOL2008-1, leading to Oguchi disease. Lastly, patient MOL2079-1 was identified as homozygous for a nonsense mutation in the SRD5A3 gene, which is associated with congenital disorder of glycosylation [12]. These five cases represent novel discoveries in the Israeli population, highlighting the significance of our study in expanding the understanding of rare genetic variants and their clinical implications.
4. Discussion
In the current study we used a single IRD gene panel to screen 252 IRD samples for mutations in 351 IRD-causing genes. Previous studies reported a yield of positive genetic results in 40-75% of analyzed cases and our study, with 55%, falls within this range, as expected [13,14,15,16,17]. Our cohort can be divided to newly-recruited cases who were not screened to any IRD-causing mutations and those who underwent genetic analysis (including genotyping founder mutations, IRD gene panels, and WES). The power of the current panel analysis can be appreciated by the much higher detection rate of 74% compare to only 26% of those previously analyzed. This high rate can partly be attributed to SV analysis performed on the NGS data. Improved recent NGS analysis pipelines include detection of both homozygous or heterozygous SVs, that assisted in the identification of such mutations on 10-15% of pathogenic alleles [18,19,20]. Switching to WGS is likely to increase this rate and improve overall detection rate of pathogenic alleles in IRDs.
Genetic analysis of newly diagnosed IRD cases depends on the phenotypic complexity and the available information on IRDs in the relevant population. The spectrum of IRD mutations in the Israeli population was studied comprehensively, mainly by a nationwide collaborative effort of the Israeli inherited retinal diseases consortium (IIRDC), leading to enriched information on disease prevalence of various IRD phenotypes [21], development of a NGS founder mutation gene panel [22], and identification of a large number of mutations in each sub-population [23,24,25]. This continuous effort allows in some cases to pinpoint the pathogenic mutation quickly, without the need of time-consuming and costly analyses. However, in the majority of cases, either due to the lack of known common founder mutations or challenging phenotyping, gene panels, such as the one used in the current study, can aid in the identification of the cause of disease in about 75% of cases.
Quick and reliable gene identification is important for a more efficient genetic counseling and disease prevention in the coming generations [26,27], participation in gene/mutation-specific clinical trials [1,28], or FDA-approved IRD therapies [29]. The analysis performed in this study, shows that gene panels can be highly efficient in identifying the cause of disease, and in some cases identifying also additional IRD-causing mutations that one out of three IRD patients carry by chance, as we previously reported [30]. Including the mitochondrial genome and performing SV analyses on the NGS data, further improves the yield of gene and mutation identification, and therefore such panels are highly recommended.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: identified gene and mutations in studied cases.
Author Contributions
Conceptualization, M.A.E. and D.S.; methodology, M.A.E., S.M., M.S., A.B, and S.K.; formal analysis, S.K. and E.B.; writing—original draft preparation, M.A.E. and D.S.; writing—review and editing, S.M., M.S., A.B, S.K., and E.B.; supervision, D.S. and E.B.; funding acquisition, D.S. and E.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Israel Science Foundation, grant number 1778/20.
Institutional Review Board Statement
Ethical approval was obtained from Hadassah University Medical Center Institutional Review Board (approval code 21-03.08.07, approved on 3 August 2007). The tenets of the Declaration of Helsinki were followed. Participants provided written informed consent after receiving an explanation about the study and its possible consequences and before donating blood samples.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data of this study are presented within the article and its Supplementary Materials. The authors are willing to share materials, data sets, and protocols used in the acquisition of data presented in this publication with other researchers upon request (contact Dror Sharon, E-mail: dror.sharon1@mail.huji.ac.il).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited Retinal Diseases: Linking Genes, Disease-Causing Variants, and Relevant Therapeutic Modalities. Prog Retin Eye Res 2021, 101029. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-Syndromic Retinitis Pigmentosa. Prog Retin Eye Res 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Britten-Jones, A.C.; Gocuk, S.A.; Goh, K.L.; Huq, A.; Edwards, T.L.; Ayton, L.N. The Diagnostic Yield of Next Generation Sequencing in Inherited Retinal Diseases: A Systematic Review and Meta-Analysis. Am J Ophthalmol 2023, 249, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Buchert, R.; Sturm, M.; Haack, T.B.; Schelling, Y.; Mahajnah, M.; Sharkia, R.; Azem, A.; Balousha, G.; Ghanem, Z.; et al. First-Line Exome Sequencing in Palestinian and Israeli Arabs with Neurological Disorders Is Efficient and Facilitates Disease Gene Discovery. European Journal of Human Genetics 2020, 28, 1034–1043. [Google Scholar] [CrossRef]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.H.H.; Phelps, I.G.; et al. Mutations in ARMC9, Which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am J Hum Genet 2017, 101, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, G.; Hu, W.; Mei, J.; Shen, Y.; Wang, M.; Tan, Y.; Yang, Y.; Lu, C.; Zhao, Y.; et al. Whole Exome Sequencing Identified Novel ARMC9 Variations in Two Cases With Joubert Syndrome. Front Genet 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Dengzhi Zhao 1, Y.C.K.Y.X.H.X.L.Y.Y.C.Z.H.X.S.L. Clinical Features and Genetic Analysis of Two Chinese Pedigrees Affected with Joubert Syndrome.
- Aksu Uzunhan, T.; Ertürk, B.; Aydın, K.; Ayaz, A.; Altunoğlu, U.; Yarar, M.H.; Gezdirici, A.; İçağasıoğlu, D.F.; Gökpınar İli, E.; Uyanık, B.; et al. Clinical and Genetic Spectrum from a Prototype of Ciliopathy: Joubert Syndrome. Clin Neurol Neurosurg 2023, 224. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Phadke, S.R.; Das Bhowmik, A.; Dalal, A. Whole Exome Sequencing Reveals a Mutation in ARMC9 as a Cause of Mental Retardation, Ptosis, and Polydactyly. Am J Med Genet A 2018, 176, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.; Olinger, E.; Wedderburn, S.; Ramakumaran, V.S.; Kini, U.; Clayton-Smith, J.; Ramsden, S.C.; Rice, S.J.; Barroso-Gil, M.; Wilson, I.; et al. Identification of LAMA1 Mutations Ends Diagnostic Odyssey and Has Prognostic Implications for Patients with Presumed Joubert Syndrome. Brain Commun 2021, 3. [Google Scholar] [CrossRef]
- Noguchi, Y.; Hinokuma, N.; Tominaga, M.; Miyamoto, S.; Nakashima, M. LAMA1 Variants Were Identified Joubert Syndrome Patient. Pediatrics International 2022, 64. [Google Scholar] [CrossRef]
- Jaeken, J.; Lefeber, D.J.; Matthijs, G. SRD5A3 Defective Congenital Disorder of Glycosylation: Clinical Utility Gene Card. European Journal of Human Genetics 2020, 28, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular Genetic Analysis Using Targeted NGS Analysis of 677 Individuals with Retinal Dystrophy. Sci Rep 2019, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Sun, V.; Tuan, H.F.; Keser, V.; Wang, K.; Ren, H.; Lopez, I.; Zaneveld, J.E.; Siddiqui, S.; et al. Comprehensive Molecular Diagnosis of 179 Leber Congenital Amaurosis and Juvenile Retinitis Pigmentosa Patients by Targeted next Generation Sequencing. J Med Genet 2013, 50, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-Based next Generation Sequencing as a Reliable and Efficient Technique to Detect Mutations in Unselected Patients with Retinal Dystrophies. European Journal of Human Genetics 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; González-Del Pozo, M.; Rodríguez-De La Ruá, E.; Dopazo, J.; Borrego, S.; Antinõlo, G. Improving the Management of Inherited Retinal Dystrophies by Targeted Sequencing of a Population-Specific Gene Panel. Sci Rep 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl Vis Sci Technol 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Hanany, M.; Khalailah, A.; Beryozkin, A.; Meyer, S.; Abu-diab, A.; Turky, F.A.; Mizrahi-meissonnier, L.; Lieberman, S.; Ben-yosef, T.; et al. Identification of Genomic Deletions Causing Inherited Retinal Degenerations by Coverage Analysis of Whole Exome Sequencing Data. J Med Genet 2016, 53, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-Number Variation Contributes 9% of Pathogenicity in the Inherited Retinal Degenerations. Genetics in Medicine 2020, 22, 1079–1087. [Google Scholar] [CrossRef]
- Van Schil, K.; Naessens, S.; Van de Sompele, S.; Carron, M.; Aslanidis, A.; Van Cauwenbergh, C.; Mayer, A.K.; Van Heetvelde, M.; Bauwens, M.; Verdin, H.; et al. Mapping the Genomic Landscape of Inherited Retinal Disease Genes Prioritizes Genes Prone to Coding and Noncoding Copy-Number Variations. Genetics in Medicine 2018, 20, 202–213. [Google Scholar] [CrossRef]
- Shalom, S.; Ben-Yosef, T.; Sher, I.; Zag, A.; Rotenstreich, Y.; Poleg, T.; Birk, O.S.; Gradstein, L.; Ehrenberg, M.; Deitch, I.; et al. Nationwide Prevalence of Inherited Retinal Diseases in the Israeli Population. JAMA Ophthalmol 2024. [CrossRef]
- Shalom, S.; Hanany, M.; Eilat, A.; Chowers, I.; Ben-Yosef, T.; Khateb, S.; Banin, E.; Sharon, D. Simultaneous Detection of Common Founder Mutations Using a Cost-Effective Deep Sequencing Panel. Genes (Basel) 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Sharon, D. The Genetics of Inherited Retinal Diseases in the Israeli and Palestinian Populations: A Lesson from Populations with High Rates of Consanguinity. In Essentials in Ophthalmology: Advances in Vision Research, Volume II- Genetic Eye Research in Asia and the Pacific; Prakash, G., Itawa, T., Eds.; Springer, 2019.
- Hanany, M.; Allon, G.; Kimchi, A.; Blumenfeld, A.; Newman, H.; Pras, E.; Wormser, O. ; S. Birk, O.; Gradstein, L.; Banin, E.; et al. Carrier Frequency Analysis of Mutations Causing Autosomal-Recessive-Inherited Retinal Diseases in the Israeli Population. European Journal of Human Genetics 2018, 26, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg- Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A Nation-wide Genetic Analysis of Inherited Retinal Diseases in Israel as Assessed by the Israeli Inherited Retinal Disease Consortium (IIRDC). Hum Mutat 2019, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Runhart, E.H.; Bauwens, M.; Corradi, Z.; De Baere, E.; Roosing, S.; Haer-Wigman, L.; Dhaenens, C.-M.; Vulto-van Silfhout, A.T.; Cremers, F.P.M. Personalized Genetic Counseling for Stargardt Disease: Offspring Risk Estimates Based on Variant Severity. The American Journal of Human Genetics 2022, 109, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Macarov, M.; Schneider, N.; Eilat, A.; Yahalom, C. Genetic Counseling Practice for Inherited Eye Diseases in an Israeli Medical Center during the COVID-19 Pandemic. J Genet Couns 2021, 30, 969–973. [Google Scholar] [CrossRef]
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Gene Therapy for Inherited Retinal Disorders: Update on Clinical Trials. Klin Monbl Augenheilkd 2021, 238, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Bacci, G.; Falsini, B.; Iarossi, G.; Melillo, P.; Mucciolo, D.P.; Murro, V.; Salvetti, A.P.; Sodi, A.; Staurenghi, G.; et al. Voretigene Neparvovec for Inherited Retinal Dystrophy Due to RPE65 Mutations: A Scoping Review of Eligibility and Treatment Challenges from Clinical Trials to Real Practice. Eye (Lond) 2024. [CrossRef]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide Carrier Frequency and Genetic Prevalence of Autosomal Recessive Inherited Retinal Diseases. Proceedings of the National Academy of Sciences 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
Figure 1.
Insights into the effectiveness of the gene panel in different diagnosis histories. (A) A pie chart representing the distribution of solved and unsolved cases among a total of 252 samples analyzed using the Blueprint retinal dystrophy gene panel. (B) A bar chart that compares the success rates in comparison to two distinct groups: newly recruited samples and previously analyzed samples with negative results.
Figure 1.
Insights into the effectiveness of the gene panel in different diagnosis histories. (A) A pie chart representing the distribution of solved and unsolved cases among a total of 252 samples analyzed using the Blueprint retinal dystrophy gene panel. (B) A bar chart that compares the success rates in comparison to two distinct groups: newly recruited samples and previously analyzed samples with negative results.
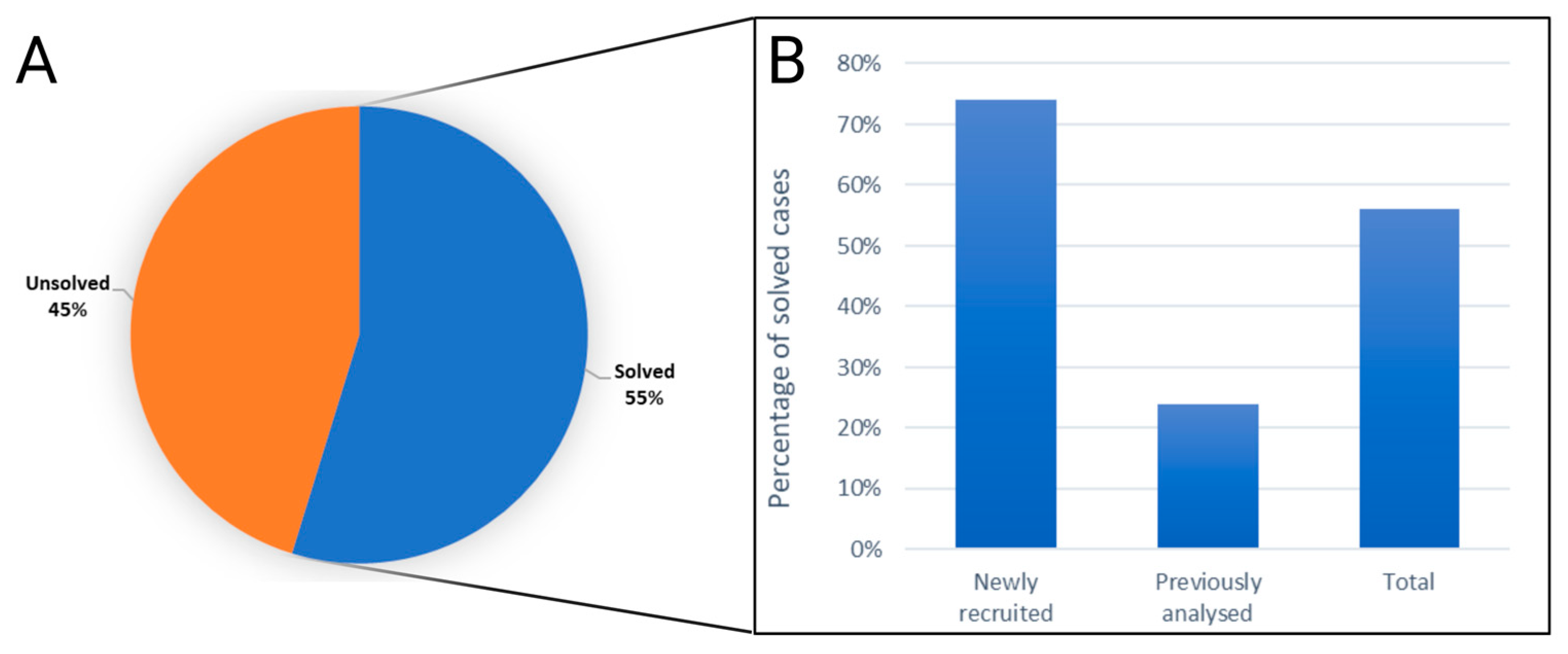
Figure 2.
Overview of genetic mechanisms in inherited retinal diseases. (A) Pie chart illustrating the distribution of solved cases based on the mode of inheritance, including autosomal recessive (AR), X-linked, autosomal dominant (AD), and mitochondrial genes. Each segment represents the percentage of cases solved under each mode of inheritance. (B) Bar chart presenting the frequency of the most implicated genes in the cohort of 138 solved cases. The X-axis displays the frequently identified genes, while the Y-axis represents the number of alleles in families for which the gene was identified as the cause of their phenotype.
Figure 2.
Overview of genetic mechanisms in inherited retinal diseases. (A) Pie chart illustrating the distribution of solved cases based on the mode of inheritance, including autosomal recessive (AR), X-linked, autosomal dominant (AD), and mitochondrial genes. Each segment represents the percentage of cases solved under each mode of inheritance. (B) Bar chart presenting the frequency of the most implicated genes in the cohort of 138 solved cases. The X-axis displays the frequently identified genes, while the Y-axis represents the number of alleles in families for which the gene was identified as the cause of their phenotype.
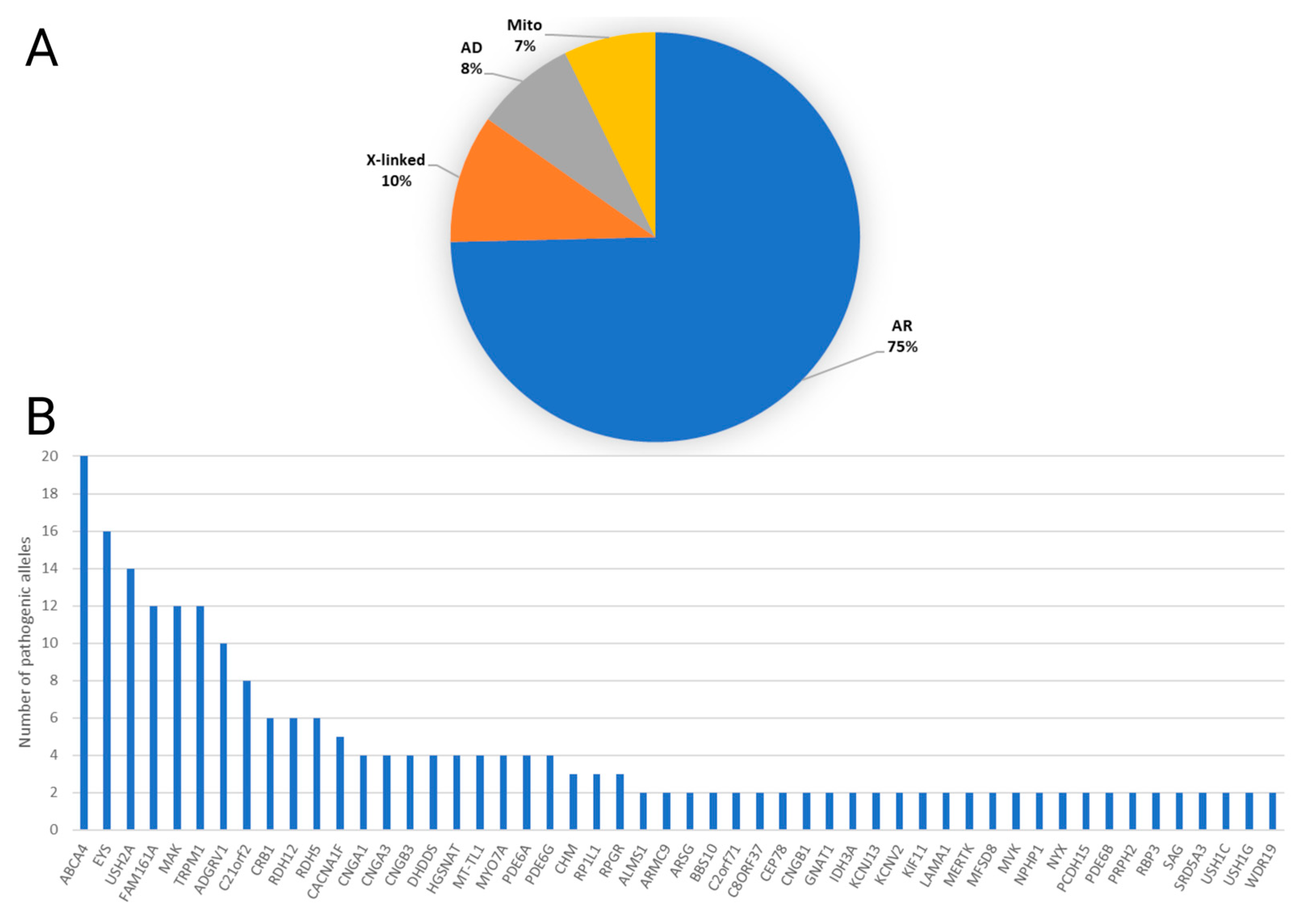
Figure 3.
Spectrum of genetic alterations contributing to inherited retinal diseases. (A) Pie chart visually representing the distribution of various mutation types identified in the solved cases, including missense, frameshift, nonsense, structural variants (SVs), splice site, intronic, and in-frame mutations. Each segment depicts the percentage of cases attributed to a specific mutation type. (A) Bar chart depicting the number of alleles for each of the most recurrent mutations detected among the indexed cases. The X-axis displays the identified disease-causing variants and the Y-axis represents the number of alleles in families for which the mutation was identified as the cause of their phenotype.
Figure 3.
Spectrum of genetic alterations contributing to inherited retinal diseases. (A) Pie chart visually representing the distribution of various mutation types identified in the solved cases, including missense, frameshift, nonsense, structural variants (SVs), splice site, intronic, and in-frame mutations. Each segment depicts the percentage of cases attributed to a specific mutation type. (A) Bar chart depicting the number of alleles for each of the most recurrent mutations detected among the indexed cases. The X-axis displays the identified disease-causing variants and the Y-axis represents the number of alleles in families for which the mutation was identified as the cause of their phenotype.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



