
Submitted:
08 June 2024
Posted:
10 June 2024
You are already at the latest version
Abstract
Delta 9 tetrahydrocannabinol (Δ-9-THC or THC), the primary psychoactive constituent of marijuana, has been found to affect the human body in a variety of ways. According to the Substance Abuse and Mental Health Services Administration (SAMHSA) of the U.S. Department of Health and Human Services, the amount of THC in marijuana has steadily climbed over the past few decades, with today's marijuana having three times the concentration of THC compared to 25 years ago. Inhalation is a major route of exposure, allowing substances to enter the body via the respiratory tract. Therefore, it is imperative to understand the molecular underpinning of THC exposure in the lung epithelium, as lung cells receive significant amounts of THC during marijuana smoking. In this study, the transcriptional alterations caused by various concentrations of THC in non-cancerous human bronchial epithelial cell line (BEAS-2B) were studied. THC exposure (0, 800, 1000, 1200, and 1500 ng/mL) caused a significant dose-dependent decrease in cell viability after 24-hour exposure. Transcriptome analysis showed a distinct dose-dependent response. HIF-1 signaling, ferroptosis, AMPK signaling, and immunogenic pathways were activated by upregulated genes associated with dosing concentration of THC. Glutathione and fatty acid metabolic pathways were significantly altered by THC-dependent downregulated genes. and downregulated genes and their associated pathways in BEAS-2B cells. Ingenuity Pathway Analysis revealed several top canonical pathways altered by THC exposure, among which ferroptosis, NRF-2 mediated oxidative stress response, caveolar mediated endocytosis (loss of cell adhesion to the substrate), tumor microenvironment, HIF1alpha signaling, and the unfolded protein response pathways were the major pathways affected. The top genes that were significantly altered as a result of THC exposure were HMOX1, CDK7, HLA-C, and SLC39A4. Present results suggested that THC exposure can induce a variety of effects, including cell death, which may involve ferroptosis and the NRF-2 signaling pathway. Moreover, the Δ-9-THC-induced cell death was ameliorated by inhibiting the ferroptosis pathway. In contrast, the ferroptosis agonist exacerbated the cell death process, suggesting that Δ-9-THC utilizes the ferroptosis pathway to induce cell death in bronchial epithelial cells.
Keywords:
Delta-9 THC
; Bronchial epithelial cells
; Transcriptome
; in vitro
; Ferroptosis
1. Introduction
Marijuana, derived from Cannabis sativa, is one of the most used illicit drugs in the United States for both medicinal and recreational purposes (1). Although marijuana is one of the oldest multi-usage cultivated crops, its primary use has always been medicinal (2). This annual herb has gained a lot of attention recently as many states are legalizing and decriminalizing its use for recreational purposes. Due to the associated health benefits, extensive research is being conducted on the secondary components of marijuana, referred to as cannabinoids (3-5). Marijuana is composed of over 400 different chemical compounds with approximately 66 Phyto cannabinoids (6). These Phytocannabinoids are naturally occurring oxygen-containing aromatic hydrocarbon compounds found in cannabis sativa. The common cannabinoids that are the active ingredients of marijuana are Delta 9 tetrahydrocannabinol (Δ-9-THC) and cannabidiol (CBD). Cannabidiol is the non-psychoactive constituent of the plant and is known to act as an antagonist of the natural cannabinoid receptors in the body. Delta 9 tetrahydrocannabinol (Δ-9-THC), the primary psychoactive constituent, is a partial agonist of the cannabinoid receptors and mimics the endogenous cannabinoids of the body, producing a wide range of biological and behavioral responses (7).
The endogenous cannabinoid receptors are part of a larger unit called the endocannabinoid system, which is composed of cell surface G protein-coupled cannabinoid CB1 and CB2 receptors and are known to regulate cellular processes such as cell survival, cell death and cell proliferation (8, 9). The CB1 receptors are located throughout the body and can be found in the brain, liver, kidneys, ovaries, and the lungs (10-12). The lungs are a major route of exposure as inhalation allows substances to enter the body and interact with the components of the respiratory tract. Epidemiological studies have found an increase in the prevalence of symptoms indicative of a wide variety of respiratory diseases and conditions, such as cough, shortness of breath, acute bronchodilation, wheezing, and airway obstruction (13-19). Taken together, marijuana exposure to airway cells can have adverse effects. However, the underlying mechanism is not fully understood.
Human cells or cell lines exposed to THC resulted in cell death, inflammation and oxidative stress (19-21). Oxidative stress induced by marijuana exposure may also mediate the release of pro-inflammatory cytokines. In an epidemiological study, patients who had a cannabis dependence exhibited elevated levels of IL-6 and IL-8, indicating that exposure can induce inflammation in humans (19). Primary airway epithelial cells that were exposed to Δ-9-THC showed a decreased mitochondrial membrane potential as well as cell viability, causing mitochondrial membrane damage. These findings were consistent with neural studies, showing that neural cells exposed to Δ-9-THC induced cell death via DNA damage, caspase activation and cytochrome-c translocation in neural cells (22). Previous studies have also demonstrated that exposure to cannabinoids induced oxidative stress (20). Oxidative stress occurs as a result of overproduction of reactive oxygen species (ROS). The human body has many defense mechanisms to counteract oxidative stress. The antioxidant glutathione (GSH) and the protein senstrin2 (SENS2) are known to repress and detoxify ROS. Δ-9-THC has been found to increase hydrogen peroxide production, which can also lead to oxidative stress, whereas CBD has been found to decrease ROS by increasing GSH levels (20, 23). The literature clearly suggests that Δ-9-THC exposure can result in cell death. However, the mechanism behind the cell death is not clearly understood.
According to the Substance Abuse and Mental health Services Administration (SAMHSA, https://www.samhsa.gov/marijuana) of the U.S. Department of Health and Human Services (1), the amount of THC in marijuana has steadily increased over the past few decades with today’s marijuana having three times the concentration of THC compared to 25 years ago. Given the higher concentration of THC and increased use of marijuana for medicinal and recreational purposes, it is imperative to understand the molecular underpinning of THC exposure in the lung epithelium as lung cells receive significant amounts of THC during marijuana smoking before it goes into other parts of the body via circulation. The present study aims to determine transcriptional alterations induced by THC in normal human bronchial cells. We determined dose-responses of Δ-9-THC exposure using human relative concentrations on bronchial epithelial cells (BEAS-2B) in vitro and examined transcriptional responses of bronchial epithelial cells by RNA sequencing followed by gene ontology enrichment (GO) and pathway analyses. Results revealed a novel cell death pathway caused by THC exposure in bronchial epithelial cells.
2. Materials and Methods
2.1. Cell Culture, THC Exposure, and Sample Collection
Immortalized human bronchial epithelial cells (BEAS-2B), obtained from ATCC in Manassas, Virginia, were used in the present study. Cells were grown in LHC-9 (1x) media and maintained at 37° C in a humidified incubator with 5% CO2 and 95% air in vented T-75 flasks (Corning). Delta-9 THC was purchased from Sigma Aldrich (T4764, Missouri). When cells were 95-100% confluent, they were seeded in 24 well plates and exposed to 800, 1000, 1200, and 1500 ng/ml of Δ-9-THC and vehicle for 24 hours. The cells were harvested after the 24-hour exposure for further downstream analysis.
2.2. Cell Viability Analysis
To determine the cell viability, cells were exposed to a range of concentrations (0.06 ng/ml-2400 ng/ml). After exposure was completed, cells were evaluated for their viability using a 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) cell proliferation assay (Thermo Fisher). Briefly, the MTT assay measures the conversion of MTT (3-(4,5-Dimethylthiazol-2-Yl)-2,5-10 Diphenyltetrazolium Bromide) to an insoluble formazan. The MTT stock solution (2 mg/ml of PBS) was made, diluted to a 1:9 ratio with media, and cells were incubated in it for two hours. After incubation, the MTT solution was decanted, and cells were rinsed with 1X PBS. Then DMSO was added and scanned in a Biotek microplate reader at an absorbance of 540nm.
2.3. RNA/DNA Extraction and cDNA Synthesis
According to the user’s guide, total RNA was extracted from the cells using the ZR Duet kit (Zymo Research). Following extraction, cDNA was synthesized from 2ug of total RNA per sample using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Oligo(dT) and random primer mix were used following the guidelines provided by the user’s guide.
2.4. Library Prep and RNA Sequencing
The RNA-sequencing library was prepared with the NEBNext® Ultra II RNA Library Prep Kit for Illumina (E7775, NEB) according to the manufacturer’s instructions. RNA was then purified, fragmented, isolated and primed for double stranded cDNA synthesis. Sequence adaptors and indexes were then ligated to the end of the dscDNA sample. Polymerase chain reaction was then used to amplify the sample through 13 cycles. Quality control was then completed by cleaning up, assessing and quantifying the amplified cDNA on a Bioanalyzer. RNA sequencing was performed on the HiSeq X ten (Illumina) platform (150bp PE) at Novogene Corporation (Davis, California) at a target depth of 40 million reads per sample.
2.5. Pathway Analysis
Enrichment analysis of significant genes were performed using Gene Ontology and Ingenuity Pathway Analysis (IPA). IPA software was also used to reveal the pathways that were most altered as a result of exposure to different concentrations. Overlapping DEGs between each treatment group were exhibited by using Venn diagram produced by VENNY (http://bioinfogp.cnb.csic.es/tools/venny/index.html). Shiney GO (24), and Cytoscape (25) were used for making disease specific pathways. The TLSEA was used for lncRNA (26) set enrichment.
2.6. Gene Validation by Real-time Quantitative PCR (qPCR)
Real Time qPCR was conducted using PowerUp SYBR TM Green PCR Master Mix (Applied Biosystems) with a QuantStudio 3 (Applied Biosystems). Primers specific to Ferroptosis (HO1, FTH, FTL, CHAC1, GCLC and GSH), unfolded protein response (BIP, PERK, ELF2A and SREPB) and the NRF2 pathway (NRF2, SOD, CAT, GST and SQSTM1), oxidative stress (SENS2, IL8), DNA Methylation (DNMT and DNMT3a) and a cannabinoid receptor (CB1) were examined.
2.7. Role of Ferroptosis in Cell Viability Test
To characterize the role of ferroptosis in Δ-9-THC treated cells, we examined the effects of inhibiting or enhancing ferroptosis in BEAS-2B cells exposed to Δ-9-THC. Cells were cultured as described above. When cells were 95-100% confluent, they were seeded in 24 well plates and exposed to only 1200 ng/ml Δ-9-THC, vehicle, ferrostatin-1 (Fer-15µM), erastin (ERA .1 µM) and ERA + FER-1, for 24hrs days. The cells were harvested after the 24hr exposure. Delta-9 THC (T4764), ferrostatin-1 (SML0583), and erastin (E7781-1mg) were purchased from Sigma Aldrich (St. Louis, MO). FER-1 and ERA were both dissolved in dimethyl sulfoxide (DMSO) and FER-1 was treated 1 hr before Delta-9 THC exposure. 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) cell proliferation assay (Thermofisher) was used to evaluate cell viability (27).
Cells were collected and re-suspended in an ice-cold 50 mm potassium phosphate buffer. The cell suspension was sonicated (provide parameters and time), followed by centrifugation at 13,000 x g for 10 minutes at 4°C to remove cell debris. The supernatants were then collected, and protein concentrations were quantified using the Bio-Rad protein assay (Bradford, Hercules, CA) using bovine serum albumin (BSA) as the standard. Protein lysate from treated cells was used to measure total cellular glutathione (GSH) according to the procedures (28). Fluorescence intensity was measured at an excitation wavelength of 350 nm and an emission wavelength of 420 nm. The sample GSH content was calculated using a GSH (Sigma–Aldrich) standard curve and expressed as nanomoles of GSH per milligram of sample protein.
Intracellular reactive oxygen species were determined using an orange dye to quantify ROS. Cells were stained with the ROS orange working solution and incubated for 60 minutes at 37°C. After incubation, cells were scanned in a Biotek microplate reader at an absorbance of 540/570nm.
Lipid peroxides were detected as malondialdehyde (MDA) reacting with thiobarbituric acid (TBA) to form a 1:2 adduct (color complex, TBARS) measurable by spectrofluorometric analysis at 530nm. The concentrations of TBARS were calculated using MDA as a reference standard. The quantities of TBARS were expressed in terms of amount (pmol) per mg protein.
RNA was isolated from cells and reverse transcribed to cDNA, as described above. Realtime qPCR was conducted using PowerUp SYBR Green PCR Master Mix (Applied Biosystems) with a QuantStudio 3 (Applied Biosystems). Expression of ferroptosis-specific (HO-1, FTH, TRFC, SQSTM1) and ROS-scavenging proteins (NRF2, GCLC) was examined.
2.8. Statistical analysis
The viability and qPCR gene expression data were expressed as mean ±SEM. Differences between the two groups were analyzed using a T-test. Following One-way ANOVA. Dunnett’s post-hoc test was used to compare the three groups. Statistical significance was considered at p˂0.05 unless otherwise stated.
3. Results
3.1. Δ-9-THC Dose-Dependent Viability of BEAS-2B Cells
As illustrated in Figure 1, varying concentrations of Δ-9-THC (800, 1000, 1200, 1500, 1800 ng/ml, and 2400 ng/mL) were treated for a 24-hour period, compared with the control group, the cell viability was significantly decreased in a dose-dependent manner except for 800 ng/ml treatment group of Δ-9-THC. Cell viability was significantly declined in the treatment group of 1000, 1200, 1500, 1800, and 2400 ng/mL of Δ-9-THC. The 800, 1000, 1200, and 1500 concentrations were further selected for RNA sequencing.
3.2. Δ-9-THC Dose-Mediated Global Alterations in Gene Expression Profile, Biomarkers, Transcription Factors, and Canonical Pathways
To investigate the effect of marijuana exposure on molecular pathways of BEAS-2B cells, RNA-seq analysis was performed. Ingenuity Pathway Analysis ® (IPA ® v. 9.0) and R scripts were used to generate and determine the pathway of differentially expressed genes (DEGs) in the different Δ-9-THC concentrations and associated heatmaps. To illustrate the closeness of DEGs found in BEAS-2B mediated by the doses of Δ-9-THC, vehicle, and media, a dendrogram was plotted. A similar type of DEGs was found in BEAS-2B cells in the treatment group of 800 ng/mL of Δ-9-THC THC, media, and vehicles, while DEGs found in 1200 ng/mL and 1500 ng/mL of Δ-9-THC treatment group were more similar but different compared to the rest of the concentrations. (Figure 2A). The Venn diagram was used to demonstrate the degree of overlap of DEGs among the treatment groups; in BEAS-2B cells, the most significant number of overlapped DEGs was found in the treatment groups of 1200 ng/mL and 1500 ng/mL Δ-9-THC exposure (Figure 2B). The global alteration of gene expression profiles in BEAS-2B cells associated with the treatment groups of Δ-9-THC compared to the vehicle was illustrated in the heatmap (Figure 2C and Supplementary Figure S1). Not only in similar DEGs, but their expression patterns were similar between the vehicle and treatment group of 800 ng/mL of Δ-9-THC THC but different compared to other treatment groups.
Transcriptomic biomarkers were identified from RNA-Seq datasets using -2<Log2FC > 3 and FDR 0.05. MMP1 and IL1RL1 were significantly up-regulated biomarkers in BEAS-2B cells at Δ-9-THC doses of 800 ng/mL and 1000 ng/mL in comparison to vehicle control (Supplementary Table S1). However, HSPA1A, HSPA1B, HSPA6B, and HMOX were identified as biomarker in treatment group of 1500 ng/mL and 1200 ng/mL of Δ-9-THC (Supplementary Table S1). To determine the specific transcription factors associated with aberrant gene expression in BEAS-2B cells controlling the global gene expression among the various treatment group of Δ-9-THC, transcription factor networks were illustrated. In BEAS-2B cells, FOXP3, TPS3, NFKB1 and MYC, ESR1, STAT2, FOXP3 were associated with 800 ng/mL and 1000 ng/mL of Δ-9-THC respectively (Figure 2D,E). In addition, SP1, MTF2, E2F4, and ESR2, MYC, HIF1A, ATF2 were found in BEAS-2B cells to be associated with treatment groups of 1200 ng/mL and 1500 ng/mL of Δ-9-THC, respectively (Figure 2F,G). Ferroptosis signaling pathways and caveolar-mediated endocytosis were found to be commonly triggered in BEAS-2B cells across all treatment groups of Δ-9-THC. NRF2-mediated oxidative pathway, and LXR/RXR activation pathway were found in BEAS-2B cells due to 1000 ng/mL and 1200 ng/mL of Δ-9-THC treatment (Figure 2H).
3.3. Δ-9-THC Mediated Enrichment of Long Non-Coding RNA (lncRNA) in BEAS-2B Cells Associated in Activation of Lung Cancerous Pathways
Global gene expression study identified several types of long non-coding RNA (lncRNA) in the BEAS-2B cells that were associated with Δ-9-THC dosage concentration. In BEAS-2B cells, the percentage of lncRNA compared to protein-coding genes was 4.58%, 5.44%, 3.86%, and 9.70% at Δ-9-THC concentrations of 800 ng/mL, 1000 ng/mL, 1200 ng/mL, and 1500 ng/mL, respectively, compared to vehicle control (Figure 3A–D). To illustrate the distribution of lncRNAs in BEAS-2B cells, a Venn diagram was plotted (Figure 3E), and three common lncRNAs were identified in the BEAS-2B cells across all dosing concentrations of Δ-9-THC. Using the lncRNA pathway enrichment, treatment groups of 800 ng/mL, 1200 ng/mL, and 1500 ng/mL of Δ-9-THC in the BEAS-2B cells were linked to common non-small cell lung cancer (Figure 3F). In BEAS-2B cells, 1000 ng/mL of Δ-9-THC-driven lncRNA was found to be associated with malignant glioma development. This finding revealed that THC-driven lncRNA could activate lung carcinogenesis in humans in a dose-dependent manner.
3.4. HIF-1 Signaling, Ferroptosis, AMPK Signaling, and Immunogenic Pathways were Triggered by Δ-9-THC Dose-Dependent Upregulated Genes in BEAS-2B Cells
To find common and distinctively expressed upregulated genes in BEAS-2B cells across all treatment groups of Δ-9-THC compared to vehicle control, total upregulated genes from RNA sequencing were plotted in a Venn diagram. A total of fourteen genes were found to be commonly upregulated in BEAS-2B cells among all experimental dosing concentrations of Δ-9-THC (Figure 4A), and those genes were associated with activation of HIF1 signaling, multiple glucose metabolism, and metabolic pathways (Supplementary Figure S2A). Further, common 100 genes were significantly upregulated in the treatment groups of 1200 ng/mL and 1500 ng/mL of Δ-9-THC (Figure 4A). Compared to vehicle control, upregulated genes in BEAS-2B cells were accountable for activating the HIF-1 pathway at 800 ng/mL and 1000 ng/mL of Δ-9-THC (Figure 4B), while the ferroptosis pathway was enriched by upregulated genes found in BEAS-2B cells by exposure effects at 1000 ng/mL and 1500 ng/mL of Δ-9-THC (Figures 3E and 4C). AMPK signaling was found to be triggered by 1200 ng/mL of Δ-9-THC in BEAS-2B cells (Figure 3D). Furthermore, in 1500 ng/mL of the Δ-9-THC treatment group, most immunogenic pathways, such as NF-kappa B signaling, TNF signaling, NOD-like receptor signaling, cytokine-cytokine receptor interaction, and IL-17 signaling, were being activated by upregulated genes in BEAS-2B cells (Figure 3E). The results indicated that upregulated genes in BEAS-2B cells were linked to disease-specific molecular pathways, including stress, immunogenic, and carcinogenic.
3.5. Cysteine-Methionine Metabolism, Glutathione Metabolism, Amino Acid Metabolism, and Fatty Acid Metabolism were Significantly Enriched by THC-Mediated Downregulated Genes in Human Lung Cells
In total downregulated genes, the thirteen genes were commonly downregulated across all Δ-9-THC treatment groups compared to the vehicle control in BEAS-2B cells (Figure 4F). Carbon metabolism, alanine, aspartate and glutamate metabolism, glycine, serine, and threonine metabolism, and arginine biosynthesis were found to be enriched by commonly expressed downregulated genes (Supplementary Figure S2B) across the treatment groups. Downregulated genes found in BEAS-2B cells treated with 800 ng/mL of the Δ-9-THC were found to be associated with cysteine and methionine metabolism and glutathione metabolism (Figure 4G). In 1000 ng/mL of the Δ-9-THC treatment group, p53 signaling, pyruvate metabolism, and lysine degradation pathways were linked to downregulated genes found in BEAS-2B cells (Figure 4H). Furthermore, fat metabolism linked to fatty acid degradation, fatty acid biosynthesis, and PPAR signaling pathways were significantly perturbed by downregulated genes in the treatment group of 1200 ng/mL dose of Δ-9-THC (Figure 4I). In the highest concentration (1500 ng/mL), focal adhesion, metabolic pathways, lysosome, and O-glycan biosynthesis were significantly altered by downregulated genes in BEAS-2B cells (Figure 4J).
Functional Network Analysis and enrichment of mammalian phenotypes associated with DEGs driven by THC exposure
To investigate the molecular effects driven by DEGs in human lung epithelial cells due to Δ-9-THC, molecular function (MF), biological process (BP), and Cellular component (CC) were studied under Gene Ontology (GO). The biological pathways driven by DEGs in BEAS-2B cells, such as response to hypoxia (P value 2.408E-04), monosaccharide metabolic process (P value 1.004E-03) were significantly altered by 800 ng/mL of Δ-9-THC dosing concentration (Supplementary Figure S3A). In 1000 ng/mL of Δ-9-THC, Protein binding (MF, P value 1.98E-11), programmed cell death (BP, P value 1.455E-06), autophagy, organelle disassembly (BP, P value 9.64E-03), ubiquitin-dependent ERAD pathway (BP, P value 3.45E-03), and endoplasmic reticulum chaperon complex (CC, P value 2.96E-03) were significantly perturbed in BEAS-2B cells (Supplementary Figure S3B). 2-oxoglutarate-dependent dioxygenase activity (MF, P value 3.9E-10), cellular response to stress (BP, P value 3.22 E-03), autophagy (BP, P value 3.71E-02) were found to be altered by 1200 ng/mL of Δ-9-THC in BEAS-2B cells (Supplementary Figure S3C). In the highest concentration of Δ-9-THC (1500 ng/mL), protein folding chaperone (MF, P value 1.533E-05), response to unfolded protein (BP, P value 9.056E-18), cellular stress response (BP, P value 4.94E-13), inflammatory response (BP, P value 1.33E-03) were significantly enriched by DEGs. GO results indicated that Δ-9-THC dose-dependent disease-specific pathways were being triggered in lung epithelial cells (BEAS-2B). In lower concentrations of Δ-9-THC (800 ng/mL), genes such as ENO2, PGK1, and HK2 were significantly upregulated in activation of HIF-1 signaling (Figure 5A), glycogen biosynthesis and abnormal muscle fiber morphology as mammalian phenotype (Supplementary Figures S4 and S5). MAP1LC3B, FTH1, GCLC, GCLM, HMOX, SAT2, and FTL were associated with the activation of the ferroptosis pathway in BEAS-2B cells lung cells due to 1000 ng/mL concentration of THC (Figure 5B). Furthermore, a significant number of genes such as TPP1, GFPT1, HSPA5, SERP1, and HYOU1 were found to be associated with IRE1-mediated unfolded protein response, endoplasmic stress response, and impaired glucose tolerance pathways, decreased circulating insulin levels were associated with mammalian phenotype in BEAS-2B cells triggered by 1000 ng/mL of Δ-9-THC. (Supplementary Figures S6 and S7). In BEAS-2B cells, Impaired glucose tolerance (Figure 5C), endoplasmic reticulum to Golgi vesicle-mediated transport, SNARE interaction in vesicular transport, and impaired glucose tolerance were found to be enriched by a significant number of genes triggered by 1200 ng/mL of Δ-9-THC (Supplementary Figure S8). Abnormal circulating protein level and decreased cellular hemoglobin content were found to be linked with mammalian phenotype associated with 1200 ng/mL of Δ-9-THC (Supplementary Figure S9) in BEAS-2B cells. The high Δ-9-THC (1500 ng/mL) concentration were found to activate significant number of immunogenic genes such as CXCL2 and 3, IL6, MMP1, MAPK6 with activation of IL17 signaling pathway (Figure 5D) in BEAS-2B cells and common DEGs triggered in RNA seq enriched with oxidative stress pathway, response to endoplasmic reticulum stress (Supplementary Figure S10).
3.6. Cell Viability- Ferroptosis
To characterize the role of ferroptosis in Δ-9-THC treated cells, cells were treated with 1200 ng/mL, with and without the ferroptosis inhibitor Fer-1 or agonist ERA for 24 hr. As expected, Δ-9-THC alone decreased cell viability at 1200 ng/mL. Treatment with Fer-1 at 5 μM increased cell viability in the Δ-9-THC exposed cells but not control (without Δ-9-THC) levels (Figure 6A). In contrast, the ferroptosis agonist ERA caused a further reduction in cell viability in Δ-9-THC exposed cells but only at 0.1 μM (Figure 6B). At the higher concentrations (0.5 and 1 μM) there was no significant effect compared to the Δ-9-THC only control. Overall, these results suggest that the ferroptosis pathway is at least partially involved in the response of the BEAS-2B cells to Δ-9-THC exposure. Our data showed that BEAS-2B cells exposed to Δ-9-THC + Fer-1, ERA + Δ-9-THC, and Δ-9-THC + Fer-1+ERA did not cause a significant change in cellular GSH levels, as seen in Figure 6C. However, there was a slight decrease in cells co-treated with ERA + Δ-9-THC (Figure 6C). To determine if there was a significant increase in reactive oxygen species due to Δ-9-THC-induced cell death, cells were treated with Δ-9-THC or ferroptosis inhibitors and ferroptosis agonists. As shown in Figure 6D, ROS levels were not altered in any of the conditions compared to the vehicle control. The levels of MDA, an indicator of lipid peroxidation, were measured following a 24hr cotreatment of Δ-9-THC + ERA and a 1hr pretreatment of Fer-1 followed by a 24hr exposure to Δ-9-THC in bronchial epithelial cells. Δ-9-THC exposure did not significantly increase MDA levels compared (Figure 6E). Furthermore, there was not a significant difference in MDA levels in cells exposed to THC together with the Fer-1 inhibitor and the ERA agonist.
3.7. Aberrant Expression Of Ferroptosis, Autophagy, and ER Response Gene
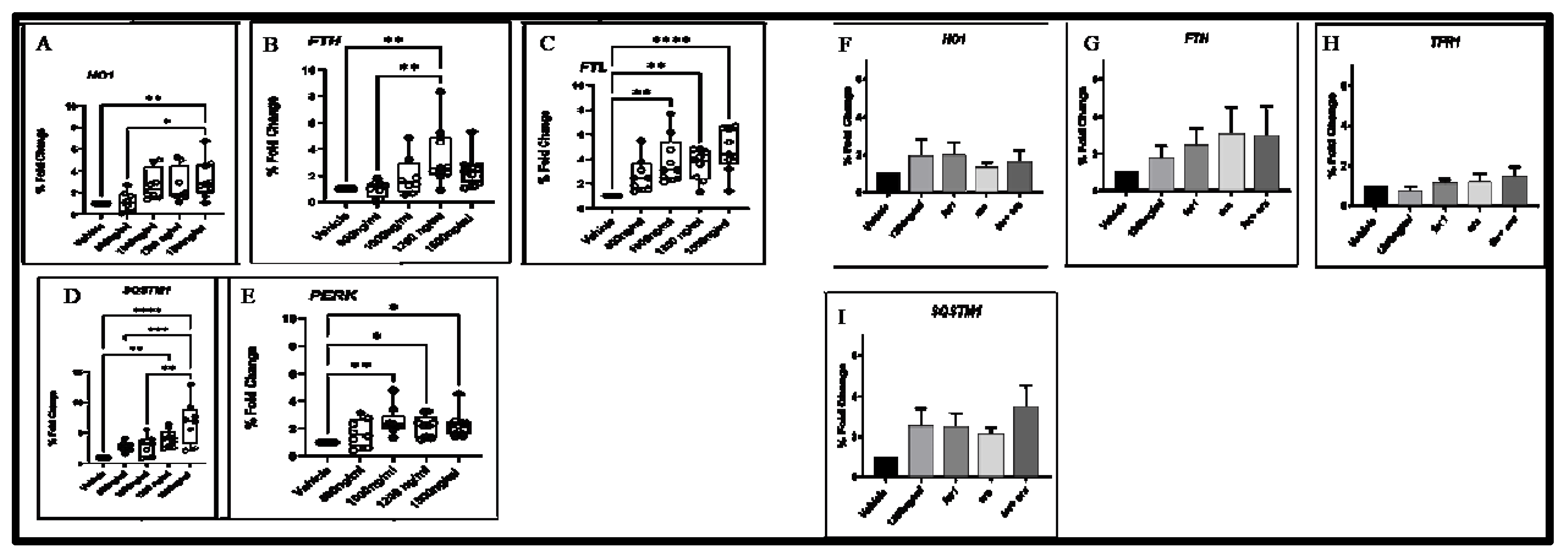
To validate the gene expression data, we selected several up-regulated targets identified by RNA seq data and examined their transcript levels by RT-qPCR. A significant up-regulation of the gene encoding essential ferroptosis enzyme, HO1 (Figure 7A), and the gene encoding iron storage proteins FTH and FTL (Figure 7B,C) were determined. Furthermore, a significant up-regulation of SQSTM1 (Figure 7D) and PERK (Figure 7E) in response to autophagy and ER stress, respectively, was also observed. Gene expression pattern in 1200 Δ-9-T group, fer-1, era and with the combination of era with fer1was examined in HO1, FTH, FTL, SQSTM and percentage of foldchange was insignificant, but differential expression pattern was observed in each treatment condition (Figure 7F–I).
4. Discussion
As the use of marijuana for medicinal and recreational purposes is increasing rapidly, it is imperative to understand the alterations in the lung epithelium caused by the constituents of marijuana. The present study investigated the impact of 24-hour exposure to Δ-9-THC on the transcriptional profiles of bronchial epithelial cells by RNA sequencing. The cell viability was confirmed by MTT assay, and Δ-9-THC exposure resulted in decreased cell viability in a dose-dependent manner. Bronchial cells exposed to marijuana smoke condensate showed a similar pattern of decreased cell viability (29). In human lung cancer cells, Δ-9-THC caused a dose-dependent decrease in mitochondrial membrane potential (30). This decrease in mitochondrial membrane potential is tied directly with cell death and a decreased production of ATP, which plays an essential role in apoptotic cell death (31).
Several top canonical pathways were altered by THC, among which ferroptosis, tumor microenvironmental, and unfolded protein response pathways were the major pathways. A gene ontology enrichment analysis showed that the genes significantly altered due to exposure were H01, CDK7, HLA-C, and SLC39A4. Finally, to validate the gene expression data, RT-qPCR was used to examine upregulated genes, H01, FTL, FTH, and PERK which are associated with ferroptosis and unfolded protein response pathway.
Literature suggests that Δ-9-THC exposure can lead to cell death in various cell types including airway cells. Δ-9-THC caused the initiation of apoptosis in oral cancer cells, cortical neurons as well as immune cells (32-35). In the present study, bronchial airway cells that are exposed to Δ-9-THC concentrations, less than the human relative concentration, resulted in cell death. Δ-9-THC induced upregulated genes were associated with immunogenic IL17 signaling pathway, TNF signaling pathway, and cytokine-cytokine receptor interaction that might trigger cell death mechanism. Increased levels of IL-17 (36) and TNFα (37) were positively correlated to serum C-reactive protein levels in chronic obstructive pulmonary disease (COPD) patients. The present study revealed for the first time a link between Δ-9-THC and the immunogenic pathway-driven COPD pathogenesis in human lung epithelial cells.
Ferroptosis is a cell death process that is iron-dependent and characterized by lipid peroxidation. This process is mediated by phospholipid peroxidation in association with free iron-mediated Fenton reactions (38-39). A correlation between Δ-9-THC exposure and cell death has been demonstrated; however, iron-dependent cell death has not been demonstrated yet. In a study with the same cells used in the present study, ferroptosis was found to be one of the major pathways affected by exposure to cigarette smoke condensate (40). Another study that exposed airway cells to cigarette smoke showed an up-regulation in HMOX1, as seen in our studies (35). HMOX1, a widely used biomarker for oxidative stress, plays a critical role in ferroptosis as it processes heme and releases Fe2+ (41). This is highlighted in a study that used EF24 on osteosarcoma cells and determined cell death, in which ferroptosis was mediated by HMOX1 (42). When HMOX1 was upregulated, it resulted in ferroptosis in the osteosarcoma cells and had the exact opposite effect when HMOX1 was knocked down, thus showing its involvement in ferroptosis. This corresponds with the up-regulation of HMOX1 in our study, thus showing that ferroptosis is occurring as a result of Δ-9-THC exposure.
Another key component of ferroptosis is ferritin, which is a major intracellular iron storage protein that is involved in intracellular iron homeostasis. Ferritin is a heteropolymer, which includes FTL1 (Ferritin light polypeptide 1) and FTH1 (Ferritin heavy polypeptide 1) that protects cells from iron-mediated damage (43). In the present study, we saw an increase in both FTH1 and FTL1 expression because of exposure to Δ-9-THC. However, studies by Hou et al. showed a decrease in FTH levels in the DHA-exposed leukemia cells as a result of the degradation of ferritin by autophagy, leading to an increase and accumulation of free iron (44). In contrast, as in our studies, FTL expression was increased in RAW264.7 cells that were exposed to LPS (45). In addition to being an iron storage, FTL is also known to be an antioxidant. The significant increase in FTL1 gene expression observed in the present study can be the result of oxidative stress, specifically lipid peroxidation that may be occurring because of Δ-9-THC exposure. It is reasonable to assume that if the level of lipid peroxidation increases, the FTL and FTH proteins may increase, as shown in our study, in a protective manner. Similar to our study, HeLa cells that were exposed to DOX had an increase in ferritin mitochondrial expression to reduce the cytotoxicity that occurred as a result (46).
It was previously reported that BEAS-2B airway cells, when exposed to Δ-9-THC, resulted in cytotoxicity, oxidative stress, and cell death (39, 47-48). To further evaluate the mechanism behind cell death and involvement of ferroptosis in Δ-9-THC -induced cytotoxicity, BEAS-2B cells were treated with FER-1 (5µM) to ameliorate Δ-9-THC induced effects. When airway cells were pretreated with Fer-1 for one hour and then immediately treated with Δ-9-THC for 24 hours, there was a significant increase in cell viability compared to treatment with Δ-9-THC alone. These observations supported previous studies in that Fer-1 prevents oxidative lipid damage and ferroptosis in auditory hair, neuronal, and airway cells (49-51). Ferroptosis is a form of non-apoptotic cell death primed by lipid peroxidation. Fer-1 is a potent ferroptosis inhibitor that prevents lipid peroxidation. Recent studies have shown that Fer-1 has ROS scavenging capabilities, particularly scavenging alkoxyl radicals (52). Our results indicate that Δ-9-THC could induce ferroptosis in airway cells, and Fer-1, a potent inhibitor of ferroptosis, can inhibit cell death associated with Δ-9-THC. These findings provide insight into the cell death pathway associated with Δ-9-THC -induced cytotoxicity and suggest that ferroptosis is involved.
Erastin (ERA) is an inducer of ferroptosis as it inhibits cystine import, thus depleting GSH, decreasing GPX4 expression. GPX4 is a key regulator of ferroptosis as it mediates lipid peroxidation. In this study, when airway cells were cotreated with Δ-9-THC and ERA for 24 HRs, there was a noticeable decrease in cell viability compared to exposure solely to Δ-9-THC. However, this decrease was not significant. Similar to our results, human pancreatic, neuroblastoma, and airway cells, when exposed to ERA, as the exposure, led to an increase in cell death and ferroptosis (53-54). The visible decrease in viability in BEAS-2B cells cotreated with ERA (0.1µM), and Δ-9-THC resulted in a significant increase in viability when pretreated with Fer-1, again supporting the notion that ferroptosis is the underlying mechanism related to Δ-9-THC induced cytotoxicity.
Literature suggests that Δ-9-THC exposure can lead to cell death in various cell types, including airway cells. Specifically, it causes the initiation of apoptosis in oral cancer cells, cortical neurons, and immune cells (39). Induction of apoptosis has been found in colon cancer cells, glioma cells, and prostate cancer cells that were exposed to cannabinoids (55-58). Cannabinoids-induced apoptosis seems to occur in a ceramide-dependent manner, activating caspase and resulting in cell death (59, 60). In contrast, lung cancer cells exposed to marijuana smoke and THC exhibited signs of induction of necrosis but inhibition of apoptosis. Specifically, marijuana smoke and THC exposure led to inhibition of caspase-3 and an increase in reactive oxygen species (56).
Deregulation of ferroptosis has been linked to Chronic obstructive pulmonary disease (COPD) in airway cells exposed to cigarette smoke extract (41). Despite ferroptosis, a number of other interesting findings in our study aligned with COPD pathogenesis, including aberrant regulation of glycogen biosynthesis, glycolysis, gluconeogenesis, impaired glucose tolerance as well as abnormal muscle fiber morphology. Although glycogen is being synthesized by the liver and skeletal muscle, chronic COPD patients experience muscle weakness and inadequate cellular energy supplements linked with disturbance in glucose metabolism (61, 62). Taken together, we speculate that Δ-9-THC exposure may lead to ferroptosis and abnormal glucose metabolism, which may play a role in COPD pathogenesis.
In the present study, we observed an opposing relationship between the NRF2 and osteoarthritis pathway. Osteoarthritis is characterized by the death of chondrocytes and the increase of oxidative stress and inflammation (63-64). These results were consistent with a study in IL-1β-stimulated human chondrocytes, which showed that the overexpression of NRF2 resulted in a decrease in oxidative stress and apoptosis markers, which are both upregulated in osteoarthritis (65). Comparably, a similar study showed IL-1β-stimulated human chondrocytes that were treated with eriodictyol, which has antioxidant and anti-inflammatory properties, inhibited the anti-inflammatory pathway, and activated the NRF2 signaling pathway (66). Thus elucidating the connection between the upregulation of the NRF2 pathway attenuating the inflammation and oxidative stress drive osteoarthritis pathway. We can assume that Δ-9-THC is working in the same manner, upregulating NRF2 expression, leading to a decrease in inflammatory expression in bronchial cells exposed to Δ-9-THC. NRF2 (nuclear factor E2-related factor 2) pathway is an inducer of cytoprotective enzymes such as HMOX1, GSH, FTH1, and FTL1, which are all major components in the ferroptosis pathway and are highlighted in our study. NRF2 is activated by oxidative stress, resulting in an increase in antioxidants that detoxify and eliminate reactive oxidants (67-68). Δ-9-THC has been shown to induce oxidative stress in different cell types, such as human placental, neuron, and glioma cells (20, 69-70). NRF2 plays a significant role in the mitigation of ferroptosis and lipid peroxidation (71). Specifically, NRF2 activates the antioxidant-responsive element-dependent genes that counteract and balance oxidative mediators to restore and maintain cellular redox homeostasis (72). NRF2 also mediates the Hepatic Fibrosis/ Hepatic Stellate Cell Activation Pathway, which is characterized by a net accumulation of extracellular matrix and scar resulting from chronic liver injury (73). This process is mediated by several novel pathways, such as oxidative stress, autophagy, and endoplasmic reticulum stress (74). NRF2 can also induce the unfolded protein response pathway, which was upregulated in our studies, via activation of PERK (75). In accordance with NRF2-mediated oxidative stress, downregulated genes induced by Δ-9-THC were found to be linked with glutathione metabolism. Excessive ROS accumulation and failure of proper scavenging mechanisms by glutathione could have triggered cell death (76). The results from this study suggest that Δ-9-THC induces the NRF2 pathway in lung epithelium. NRF2, which is typically upregulated as a response to stress, may be playing an essential role and as demonstrated from previous studies, NRF2 regulates many pathways in the body. This leaves us to believe that Δ-9-THC is dysregulating many pathways, as demonstrated by the results of this study, and in response to this, NRF2 is possibly becoming activated and responding accordingly.
Unfolded protein response is another pathway that was up-regulated because of exposure to Δ-9-THC. Unfolded protein response (UPR) is a cellular stress pathway that is associated with endoplasmic reticulum (ER) stress. When a cell undergoes UPR, an increase in the expression of PERK can be expected as it is one of the primary signal transducers of the UPR pathway. This was displayed in our studies, which showed an increase in PERK expression as a result of bronchial epithelial cells exposed to Δ-9-THC. The ferroptosis and unfolded protein response pathways are connected as half of the lipids in the cells are in the endoplasmic reticulum (56), and therefore, the ER may play a major role in the initiation of ferroptosis. PERK is a known mediator of ferroptosis as it has been shown to have a direct impact on NRF2, causing phosphorylation and increasing levels of HMOX1, thus activating ferroptosis (49). The unfolded protein response is also known to be activated and upregulated in the tumor microenvironment which is a pathway that was upregulated in our studies (49). The tumor microenvironment is the environment that surrounds the tumor and consists of surrounding blood vessels, fibroblasts, immune cells, signaling molecules, lymphatic cells, and the extracellular matrix (57-80). This environment has been shown to be modulated in response to cigarette smoke exposure, not only resulting in phenotypic changes and inducing autophagy but also contributing to aggressiveness in vitro as well as in vivo (81-82). Ferroptosis has also been shown to contribute to the tumor microenvironment by driving the polarization of macrophages in the tumor microenvironment (83). In addition to the tumor microenvironment, bronchial cells also activate the caveolar-mediated endocytosis pathway. The caveolar-mediated endocytosis pathway is responsible for internalizing, uptake, and regulating signaling organelles and bulk membranes (83). As a result of cigarette smoke exposure, airway cells have been shown to induce the caveolar-mediated endocytosis pathway, with the increase of this pathway being associated with lung infections in smokers (84). Similarly, overexpression of HIF1- alpha has been associated with lung cancer, which is correlated with cigarette smoke (85). Specifically, genes linked to the HIf1- alpha signaling pathway are shown to have a significant impact on lung carcinogenesis and genomic instability (86). Moreover, activation and overexpression of the HIf1-alpha signaling pathway have also been linked to COPD and decreased lung function (87). Relatedly, the LXR/RXR activation pathway also has a negative impact on lung function. The LXR/RXR activation pathway was slightly increased in most concentrations in our studies. This pathway regulates cholesterol homeostasis and lipid and carbohydrate metabolism (88-89). When cells are dying, there is typically an increase in lipid production in which the LXR/RXR pathway breaks down and removes this surplus of lipids (90). LXR/RXR activation pathway also can induce airway inflammation and airway hyperresponsiveness, as researchers who used an agonist against the LXR/RXR activation pathway were able to attenuate airway inflammation, asthma, and pulmonary emphysema in-vivo in murine and rat models (91-92). The glucocorticoid signaling pathway is another pathway that was deregulated because of Δ-9-THC exposure. Glucocorticoids, which are a class of corticosteroids, are typically prescribed to treat chronic inflammation and asthma in the respiratory tract (93-94). However, research shows that an increasing number of people do not respond to glucocorticoid administration to treat inflammation associated with asthma, and glucocorticoid inhibitors may decrease this associated inflammation, but studies are still ongoing (95). Studies are now trying to find new therapeutic strategies to treat different types of inflammation. A recent study combined glucocorticoid with ferroptosis-inducing agents in order to find a novel therapy for eosinophilic airway inflammation; the results showed the induction of ferroptosis-like cell death relieved the allergic airway inflammation in mice eosinophils (96). As a result of that study subsequent studies are looking into this pathway to cell death as a novel pathway in drug abuse.
In summary, the transcriptome analysis of bronchial epithelial cells exposed to Δ-9-THC revealed genes with distinctly altered patterns of expression, particularly those specific to ferroptosis, unfolded protein response, and tumor microenvironment pathways. The extent of the gene deregulation was dependent on the concentration of Δ-9-THC. To our knowledge and based on literature, the ferroptosis pathway is likely a novel pathway associated with Δ-9-THC -induced cytotoxicity in bronchial epithelial cells. The present findings also show that Fer-1, an inhibitor of ferroptosis, ameliorates airway cells from Δ-9-THC induced cell death, suggesting ferroptosis is one of the mechanisms behind cell death. Many pathways were highlighted in this study due to Δ-9-THC exposure, and many were seen across multiple concentrations (97). However, with the cell death phenotype in the Δ-9-THC exposed airway cells, the follow-up study will focus on the ferroptosis pathway and the mechanism behind Δ-9-THC-induced cell death.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
RKB, MSD, JZ designed the experiment; MSD performed cell culture and molecular assays; XW prepared RNAseq library; XW and SA performed bioinformatic analysis; MF prepared figures; SM performed post-bioinformatic data analysis; MD, SC, MF, and RKB wrote the manuscript, JZ critically reviewed the manuscript.
Acknowledgements
The authors declare no conflict of interest. They thank the Bhandari Research Group members for their assistance with the experiment. Undergraduate students, particularly Coryanna Jefferies, Jasmine Allen, and Marlee Vassall, assisted MSD with her cell culture experiments. The research project was supported by funds from the Graduate School of the University of North Carolina Greensboro to MSD.
References
- Bose, J.; Hedden, S.L.; Lipari, R.N.; Park-Lee, E. Key substance use and mental health indicators in the United States: Results from the 2017 National Survey on Drug Use and Health (HHS Publication No. SMA 2018: 17-5044, NSDUH Series H-52.
- Holland, J. The pot book: A complete guide to cannabis: Simon and Schuster; 2010.
- Lukhele, S.T.; Motadi, L.R. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement Altern Med 2016, 16, 335. [Google Scholar] [CrossRef]
- Mucke, M.; Weier, M.; Carter, C.; Copeland, J.; Degenhardt, L.; Cuhls, H.; Radbruch, L.; Hauser, W.; Conrad, R. Systematic review and meta-analysis of cannabinoids in palliative medicine. J Cachexia Sarcopenia Muscle 2018, 9, 220–234. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Magallanes, J.A.M.; Shau, H.; Tashkin, D.; Roth, M.D. Oxidative stress produced by marijuana smoke: An adverse effect enhanced by cannabinoids. Am J Respir Cell Mol Biol 1999, 20, 1286–1293. [Google Scholar] [CrossRef]
- Baron, E.P. Comprehensive review of medicinal marijuana, cannabinoids, and therapeutic implications in medicine and headache: What a long, strange trip it’s been. Headache: J Head and Face Pain. 2015, 55, 885–916. [Google Scholar] [CrossRef]
- Cooper, Z.D.; Haney, M. Actions of delta-9-tetrahydrocannabinol in cannabis: Relation to use, abuse, dependence. Int Rev Psychiatry 2009, 21, 104–112. [Google Scholar] [CrossRef]
- Goode, E. Marijuana: Routledge; NY, USA 2017.
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Bouaboula, M.; Rinaldi, M.; Carayon, P.; Carillon, C.; Delpech, B.; Shire, D.; Le Fur, G.; Casellas, P. Cannabinoid-receptor expression in human leukocytes. Eur J Biochem 1993, 214, 173–180. [Google Scholar] [CrossRef]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; LE Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J biochem 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Turcotte, C.; Blanchet, M.-R.; Laviolette, M.; Flamand, N. Impact of cannabis, cannabinoids, and endocannabinoids in the lungs. Front Pharmacol. 2016, 7, 317. [Google Scholar] [CrossRef]
- Aldington, S.; Williams, M.; Nowitz, M.; Weatherall, M.; Pritchard, A.; McNaughton, A.; Robinson, G.; Beasley, R. The effects of cannabis on pulmonary structure, function and symptoms. Thorax. 2007. 62, 1058–1063. [CrossRef]
- Hancox, R.J.; Shin, H.H.; Gray, A.R.; Poulton, R.; Sears, M.R. Effects of quitting cannabis on respiratory symptoms. Eur Respir J 2015, 46, 80–87. [Google Scholar] [CrossRef]
- Macleod, J.; Robertson, R.; Copeland, L.; McKenzie, J.; Elton, R.; Reid, P. Cannabis, tobacco smoking, and lung function: A cross-sectional observational study in a general practice population. Br J Gen Pract 2015, 65, e89–e95. [Google Scholar] [CrossRef]
- Mégarbane, B.; Chevillard, L. The large spectrum of pulmonary complications following illicit drug use: Features and mechanisms. Chem Biol Interact 2013, 206, 444–451. [Google Scholar] [CrossRef]
- Owen, K.P.; Sutter, M.E.; Albertson, T.E. Marijuana: Respiratory tract effects. Clin Rev Allergy Immunol 2014, 46, 65–81. [Google Scholar] [CrossRef]
- Tan, W.C.; Lo, C.; Jong, A.; Xing, L.; FitzGerald, M.J.; Vollmer, W.M.; Buist, S.A.; Sin, D.D. Marijuana and chronic obstructive lung disease: A population-based study. CMAJ 2009, 180, 814–820. [Google Scholar] [CrossRef]
- Bayazit, H.; Selek, S.; Karababa, I.F.; Cicek, E.; Aksoy, N. Evaluation of oxidant/antioxidant status and cytokine levels in patients with cannabis use disorder. Clin Psychopharmacol Neurosci 2017, 15, 237. [Google Scholar] [CrossRef]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. BioMed Res Int 2015, 2015, 323706. [Google Scholar] [CrossRef]
- Zamberletti, E.; Gabaglio, M.; Prini, P.; Rubino, T.; Parolaro, D. Cortical neuroinflammation contributes to long-term cognitive dysfunctions following adolescent delta-9-tetrahydrocannabinol treatment in female rats. European Neuropsychopharmacology 2015, 25, 2404–2415. [Google Scholar] [CrossRef]
- Campbell, V.A. Tetrahydrocannabinol-induced apoptosis of cultured cortical neurones is associated with cytochrome c release and caspase-3 activation. Neuropharmacol 2001, 40, 702–709. [Google Scholar] [CrossRef]
- Park, E.-J.; Park, Y.-J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol Lett 2019, 303, 55–66. [Google Scholar] [CrossRef]
- Lemecha, M.; Chalise, J.P.; Takamuku, Y.; Zhang, G.; Yamakawa, T.; Larson, G.; Itakura, K. Lcn2 mediates adipocyte-muscle-tumor communication and hypothermia in pancreatic cancer cachexia. Molecular Metabolism. 2022, 66, 101612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome research. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, J.; Li, Z.; Wang, Y.; Lin, H.; Wu, B. TLSEA: A tool for lncRNA set enrichment analysis based on multi-source heterogeneous information fusion. Frontiers in Genetics. 2023, 14, 1181391. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell proliferation and cytotoxicity assays. Current pharmaceutical biotechnology. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Xie, F.; Liu, K.; Liu, H.; Xie, J. Evaluation of whole cigarette smoke induced oxidative stress in A549 and BEAS–2B cells. Environmental toxicology and pharmacology. 2017, 54, 40–7. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res 2013, 14, 97. [Google Scholar] [CrossRef]
- Zou, Y.; Chen, X.; Liu, J.; Zhou, D.B.; Kuang, X.; Xiao, J.; Yu, Q.; Lu, X.; Li, W.; Xie, B.; et al. Serum IL-1β and IL-17 levels in patients with COPD: Associations with clinical parameters. International journal of chronic obstructive pulmonary disease. 2017 24, 1247–54.
- Lai, H.; Rogers, D.F. New pharmacotherapy for airway mucus hypersecretion in asthma and COPD: Targeting intracellular signaling pathways. Journal of aerosol medicine and pulmonary drug delivery. 2010, 23, 219–231. [Google Scholar] [CrossRef]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Campbell, V.A. Tetrahydrocannabinol-induced apoptosis of cultured cortical neurones is associated with cytochrome c release and caspase-3 activation. Neuropharmacol 2001, 40, 702–709. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Tashkin, D.P.; Roth, M.D. Marijuana smoke and Δ9-tetrahydrocannabinol promote necrotic cell death but inhibit Fas-mediated apoptosis. Toxicol Appl Pharmacol 2001, 174, 264–272. [Google Scholar] [CrossRef]
- Zeissler, I.; Weiner, L.; Vogel, Z. Δ9-tetrahydrocannabinol increases C6 glioma cell death produced by oxidative stress. Neurosci 2005, 134, 567–574. [Google Scholar] [CrossRef]
- Semlali, A.; Beji, S.; Ajala, I.; Rouabhia, M. Effects of tetrahydrocannabinols on human oral cancer cell proliferation, apoptosis, autophagy, oxidative stress, and DNA damage. Arch Oral Biol 2021, 105200. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Kouyoumjian, S.; Khoshaghideh, F.; Tashkin, D.P.; Roth, M.D. Δ9-Tetrahydrocannabinol disrupts mitochondrial function and cell energetics. Am J Physiol-Lung Cell Mol Physiol 2003, 284, L298–L306. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun 2019, 10, 1–14. [Google Scholar]
- Park, E.-J.; Park, Y.-J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol Lett 2019, 303, 55–66. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X. Ferroptosis as a target for protection against cardiomyopathy. Proc Nat Acad Sci USA. 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Lin, H.; Chen, X.; Zhang, C.; Yang, T.; Deng, Z.; Song, Y.; Huang, L.; Li, F.; Li, Q.; Lin, S. EF24 induces ferroptosis in osteosarcoma cells through HMOX1. Biomed Pharmacother. 2021, 136, 111202. [Google Scholar] [CrossRef]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol Lett 2018, 15, 5405–5411. [Google Scholar]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Rad Biol Med 2019, 131, 356–69. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, J.; Cai, L.; Wang, S.; Liu, C.; Zhang, Y.; You, L.; Fu, Y.; Shi, Z.; Yin, Z. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim Biophys Acta Mol Cell Res 2014, 1843, 2775–2783. [Google Scholar] [CrossRef]
- Cocco, E.; Porrini, V.; Derosas, M.; Nardi, V.; Biasiotto, G.; Maccarinelli, F.; Zanella, I. Protective effect of mitochondrial ferritin on cytosolic iron dysregulation induced by doxorubicin in HeLa cells. Mol Bio Rep 2013, 40, 6757–6764. [Google Scholar] [CrossRef]
- Sarafian, T.; Habib, N.; Mao, J.T.; Tsu, I.H.; Yamamoto, M.L.; Hsu, E.; Tashkin, D.P.; Roth, M.D. Gene expression changes in human small airway epithelial cells exposed to Δ9-tetrahydrocannabinol. Toxicol Lett 2005, 158, 95–107. [Google Scholar] [CrossRef]
- Aguiar, J.A.; Huff, R.D.; Tse, W.; Stämpfli, M.R.; McConkey, B.J.; Doxey, A.C.; Hirota, J.A. Transcriptomic and barrier responses of human airway epithelial cells exposed to cannabis smoke. Physiol Rep 2019, 7, e14249. [Google Scholar] [CrossRef]
- Hu, B.; Liu, Y.; Chen, X.; Zhao, J.; Han, J.; Dong, H.; Zheng, Q.; Nie, G. Ferrostatin-1 protects auditory hair cells from cisplatin-induced ototoxicity in vitro and in vivo. Biochem Biophys Res Commun 2020, 533, 1442–1448. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett 2020, 25, 1–14. [Google Scholar] [CrossRef]
- Miotto, G.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol 2020, 28, 101328. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D.; Constantin, D. Molecular mechanisms of muscle fatigue. International Journal of Molecular Sciences. 2021, 22, 11587. [Google Scholar] [CrossRef]
- Mallia, P.; Webber, J.; Gill, S.K.; Trujillo-Torralbo, M.B.; Calderazzo, M.A.; Finney, L.; Bakhsoliani, E.; Farne, H.; Singanayagam, A.; Footitt, J.; et al. Role of airway glucose in bacterial infections in patients with chronic obstructive pulmonary disease. Journal of Allergy and Clinical Immunology. 2018, 142, 815–823. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, D.-H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol Cancer Res 2018, 16, 1073–1076. [Google Scholar] [CrossRef]
- Wei, R.; Zhao, Y.; Wang, J.; Yang, X.; Li, S.; Wang, Y.; Yang, X.; Fei, J.; Hao, X.; Zhao, Y. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci 2021, 17, 2703. [Google Scholar] [CrossRef]
- Ramirez, M.U.; Hernandez, S.R.; Soto-Pantoja, D.R.; Cook, K.L. Endoplasmic reticulum stress pathway, the unfolded protein response, modulates immune function in the tumor microenvironment to impact tumor progression and therapeutic response. Int J Mol Sci 2020, 21, 169. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J Cell Sci 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Domingo-Vidal, M.; Whitaker-Menezes, D.; Martos-Rus, C.; Tassone, P.; Snyder, C.M.; Tuluc, M.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Cigarette smoke induces metabolic reprogramming of the tumor stroma in head and neck squamous cell carcinoma. Mol Cancer Res 2019, 17, 1893–1909. [Google Scholar] [CrossRef]
- Blanco, F.J.; Guitian, R.; Vázquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis: A possible pathway for osteoarthritis pathology. Arthritis Rheum 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Zahan, O.-M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med Pharm Reports. 2020, 93, 12. [Google Scholar] [CrossRef]
- Rigoglou, S.; Papavassiliou, A.G. The NF-κB signalling pathway in osteoarthritis. Int J Biochem Cell Biol 2013, 45, 2580–2584. [Google Scholar] [CrossRef]
- Khan, N.M.; Ahmad, I.; Haqqi, T.M. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic Biol Med 2018, 116, 159–71. [Google Scholar] [CrossRef]
- Ankita Bansal, M. Celeste Simon; Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Chen, Y.; Zhou, B.; Shan, X.; Yang, G. Eriodictyol inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes. Biomed Pharmacother 2018, 107, 1128–1134. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Goncharov, I.; Weiner, L.; Vogel, Z. Δ9-tetrahydrocannabinol increases C6 glioma cell death produced by oxidative stress. Neurosci 2005, 134, 567–574. [Google Scholar] [CrossRef]
- Zeissler, M.-L.; Eastwood, J.; McCorry, K.; Hanemann, C.O.; Zajicek, J.P.; Carroll, C.B. Delta-9-tetrahydrocannabinol protects against MPP+ toxicity in SH-SY5Y cells by restoring proteins involved in mitochondrial biogenesis. Oncotarget 2016, 7, 46603. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 2019, 23, 101107. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.; Chen, D.; Wruck, C.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017, 6, e371. [Google Scholar] [CrossRef]
- Lu, C.; Xu, W.; Zhang, F.; Shao, J.; Zheng, S. Nrf2 knockdown attenuates the ameliorative effects of ligustrazine on hepatic fibrosis by targeting hepatic stellate cell transdifferentiation. Toxicol 2016, 365, 35–47. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free Radic Biol Med 2015, 88, 417–426. [Google Scholar] [CrossRef]
- Hou, H.H.; Pan, H.J.; Liao, W.Y.; Lee, C.H.; Yu, C.J. Autophagy in fibroblasts induced by cigarette smoke extract promotes invasion in lung cancer cells. Int J Cancer 2020, 147, 2587–2596. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]
- Kiss, A.L.; Botos, E. Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J Cell Mol Med 2009, 13, 1228–1237. [Google Scholar] [CrossRef]
- Duffney, P.F.; Embong, A.K.; McGuire, C.C.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Cigarette smoke increases susceptibility to infection in lung epithelial cells by upregulating caveolin-dependent endocytosis. PLoS ONE 2020, 15, e0232102. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.; Harris, A. Relation of hypoxia inducible factor 1 α and 2 α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Q.; Song, Y.; Li, X.; Wang, Y.; Wan, S.; Zhang, Z.; Su, H. Polymorphisms of HIF1A gene are associated with prognosis of early stage non-small-cell lung cancer patients after surgery. Med Oncol 2014, 31, 1–9. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med 2018, 16, 4553–4561. [Google Scholar] [CrossRef]
- Ulven, S.M.; Dalen, K.T.; Gustafsson, J.-Å.; Nebb, H.I. LXR is crucial in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids 2005, 73, 59–63. [Google Scholar] [CrossRef]
- Cha, J.-Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis: The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 2007, 282, 743–751. [Google Scholar] [CrossRef]
- Larrede, S.; Quinn, C.M.; Jessup, W.; Frisdal, E.; Olivier, M.; Hsieh, V.; Kim, M.-J.; Van Eck, M.; Couvert, P.; Carrie, A. Stimulation of cholesterol efflux by LXR agonists in cholesterol-loaded human macrophages is ABCA1-dependent but ABCG1-independent. Arterioscler Thromb Vasc Biol 2009, 29, 1930–1936. [Google Scholar] [CrossRef]
- Morichika, D.; Miyahara, N.; Fujii, U.; Taniguchi, A.; Oda, N.; Senoo, S.; Kataoka, M.; Tanimoto, M.; Kakuta, H.; Kiura, K. A retinoid X receptor partial agonist attenuates pulmonary emphysema and airway inflammation. Respir Res 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Fujii, U.; Miyahara, N.; Taniguchi, A.; Oda, N.; Morichika, D.; Murakami, E.; Nakayama, H.; Waseda, K.; Kataoka, M.; Kakuta, H. Effect of a retinoid X receptor partial agonist on airway inflammation and hyperresponsiveness in a murine model of asthma. Respir Res 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb Exp Pharmacol 2017, 237, 171–196. [Google Scholar]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013, 34, 518–530. [Google Scholar] [CrossRef]
- Palumbo, M.L.; Prochnik, A.; Wald, M.R.; Genaro, A.M. Chronic stress and glucocorticoid receptor resistance in asthma. Clin Ther 2020, 42, 993–1006. [Google Scholar] [CrossRef]
- Enweasor, C.; Flayer, C.H.; Haczku, A. Ozone-Induced Oxidative Stress, Neutrophilic Airway Inflammation, and Glucocorticoid Resistance in Asthma. Front Immunol 2021, 12, 192. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, H.; Xuan, N.; Zhou, L.; Wu, Y.; Zhu, C.; Li, M.; Weng, Q.; Shen, J.; Zhang, H. Induction of ferroptosis-like cell death of eosinophils exerts synergistic effects with glucocorticoids in allergic airway inflammation. Thorax. 2020, 75, 918–927. [Google Scholar] [CrossRef]
- Vassall, M.; Chakraborty, S.; Feng, Y.; Faheem, M.; Wang, X.; Bhandari, R.K. Transcriptional alterations induced by delta-9 tetrahydrocannabinol in the brain and gonads of adult medaka. Journal of Xenobiotics. 2023, 13, 237–251. [Google Scholar] [CrossRef]
Figure 1.
BEAS-2B cells treated with Δ-9-THC for 24 hours. Data represents mean ± SEM. Statistical significance (*** p<0.001).
Figure 1.
BEAS-2B cells treated with Δ-9-THC for 24 hours. Data represents mean ± SEM. Statistical significance (*** p<0.001).
Figure 2.
Global transcriptional alteration in BEAS-2B cell by Δ-9-THC. (A) Dendrogram visualizing the hierarchical relationship between concentrations. (B) Venn diagram showing highly significant DEGs in all treatment groups and their overlap with/among each other. (C) A heatmap showing all DEGs in BEAS-2B cells in response to various concentrations of THC exposure for 24 hours. List of transcription factors associated with Δ-9-THC dosing concentration of (D) 800 ng/mL (E) 1000 ng/mL (F) 1200 ng/mL and (G) 1500 ng/mL. (H) Canonical pathways activated by Δ-9-THC in BEAS-2B cells.
Figure 2.
Global transcriptional alteration in BEAS-2B cell by Δ-9-THC. (A) Dendrogram visualizing the hierarchical relationship between concentrations. (B) Venn diagram showing highly significant DEGs in all treatment groups and their overlap with/among each other. (C) A heatmap showing all DEGs in BEAS-2B cells in response to various concentrations of THC exposure for 24 hours. List of transcription factors associated with Δ-9-THC dosing concentration of (D) 800 ng/mL (E) 1000 ng/mL (F) 1200 ng/mL and (G) 1500 ng/mL. (H) Canonical pathways activated by Δ-9-THC in BEAS-2B cells.
Figure 3.
Percentage of lncRNA found in the Δ-9-THC treatment group of (A) 800 ng/mL, (B) 1000 ng/mL, (C) 1200 ng/mL, and (D) 1500 ng/mL. (E) Venn diagram showing highly significant lncRNAs in all treatment groups and their overlap with/among each other. (F) Pathway enrichment by lncRNA in each treatment group.
Figure 3.
Percentage of lncRNA found in the Δ-9-THC treatment group of (A) 800 ng/mL, (B) 1000 ng/mL, (C) 1200 ng/mL, and (D) 1500 ng/mL. (E) Venn diagram showing highly significant lncRNAs in all treatment groups and their overlap with/among each other. (F) Pathway enrichment by lncRNA in each treatment group.
Figure 4.
(A) Venn diagram showing overlap of upregulated DEGs associated with Δ-9-THC. Upregulated DEGs driven pathways in the treatment group of (B) 800 ng/mL (C) 1000 ng/mL (D) 1200 ng/mL and (E) 1500 ng/mL. (F) Venn diagram showing overlap of downregulated DEGs associated with Δ-9-THC. Downregulated DEGs driven pathways in the treatment group of (G) 800 ng/mL (H) 1000 ng/mL (I) 1200 ng/mL and (J) 1500 ng/mL.
Figure 4.
(A) Venn diagram showing overlap of upregulated DEGs associated with Δ-9-THC. Upregulated DEGs driven pathways in the treatment group of (B) 800 ng/mL (C) 1000 ng/mL (D) 1200 ng/mL and (E) 1500 ng/mL. (F) Venn diagram showing overlap of downregulated DEGs associated with Δ-9-THC. Downregulated DEGs driven pathways in the treatment group of (G) 800 ng/mL (H) 1000 ng/mL (I) 1200 ng/mL and (J) 1500 ng/mL.

Figure 5.
Genes associated with (A) HIF-1 signaling pathway triggered in 800 ng/mL, (B) ferroptosis pathway mediated with 1200 ng/mL, (C) impaired glucose tolerance in 1500 ng/mL, (D) IL17 signaling pathways 1500 ng/mL of treatment group.
Figure 5.
Genes associated with (A) HIF-1 signaling pathway triggered in 800 ng/mL, (B) ferroptosis pathway mediated with 1200 ng/mL, (C) impaired glucose tolerance in 1500 ng/mL, (D) IL17 signaling pathways 1500 ng/mL of treatment group.
Figure 6.
The effect of Delta 9 THC 1200ng/ml and Fer-1 cotreatment (A), and Erastin and Delta 9 THC 1200ng/ml cotreatment on bronchial epithelial cells (B). Data represents ±SEM. Statistical significance *** p<0.001(n=3). The effect of Δ-9-THC, Fer-1(5µM), ERA(.1µM) + Δ-9-THC and Δ-9-THC + Fer-1+ERA on GSH content (C), ROS level (D) and lipid Peroxidation (E). Data represents ±SEM. Statistical significance *** p<0.001. (n=3).
Figure 6.
The effect of Delta 9 THC 1200ng/ml and Fer-1 cotreatment (A), and Erastin and Delta 9 THC 1200ng/ml cotreatment on bronchial epithelial cells (B). Data represents ±SEM. Statistical significance *** p<0.001(n=3). The effect of Δ-9-THC, Fer-1(5µM), ERA(.1µM) + Δ-9-THC and Δ-9-THC + Fer-1+ERA on GSH content (C), ROS level (D) and lipid Peroxidation (E). Data represents ±SEM. Statistical significance *** p<0.001. (n=3).
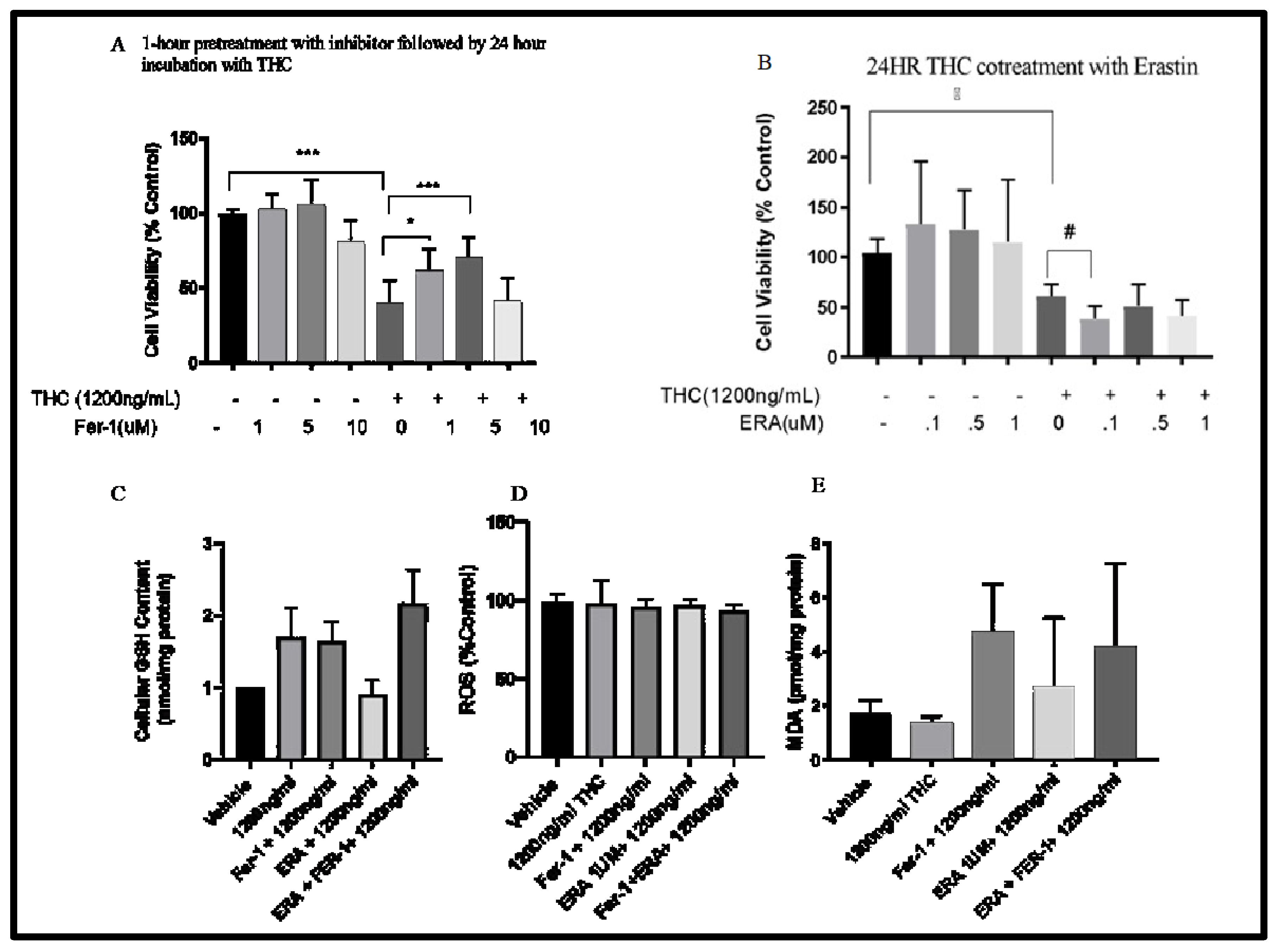
Figure 7.
Validation of the RNA-seq results by RT-qPCR. The relative expression levels of the DEGs between the bronchial cells exposed to different concentrations of Δ-9-THC. Asterisks indicate statistical significance (* p<0.05, ** p<0.01, *** p<0.001, p<0.0001).
Figure 7.
Validation of the RNA-seq results by RT-qPCR. The relative expression levels of the DEGs between the bronchial cells exposed to different concentrations of Δ-9-THC. Asterisks indicate statistical significance (* p<0.05, ** p<0.01, *** p<0.001, p<0.0001).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



