
Submitted:
07 June 2024
Posted:
11 June 2024
You are already at the latest version
Abstract
The delayed effects of radiation alone and combined with skin injury on the kidney are poorly understood. We aimed to unravel and compare the inflammatory, oxidative stress, and survival signaling pathways in the kidney cortex before mice manifested renal dysfunction after these two injuries. Mice were analyzed 30 days post irradiation (9.5 Gy for radiation alone, 9.0 Gy for radiation combined with a skin wound). Radiation alone did not significantly alter BUN, NGAL, or KIM-1 protein levels, indicating preserved kidney function. However, the combined injury had increased KIM-1 protein levels, indicating a nuanced effect on renal health. Radiation and combined injury activated distinct inflammatory pathways. Radiation increased STAT3-Y705 phosphorylation, while combined injury boosted STAT1-Y701 phosphorylation. Additionally, radiation increased mRNA abundance of IFNγR1, IFNγR2, heparanase and IL-10, while combined injury tended to increase heparanase mRNA levels and reduced IL-4 mRNA levels. Both injuries increased the abundance of HO-1 protein, indicating oxidative stress, but radiation alone also reduced MnSOD and catalase proteins. Both injuries promoted AKT1-S473 phosphorylation and diminished p53 protein levels, suggesting inhibition of apoptosis. In summary, despite the distinct activation of inflammatory and oxidative stress pathways, both radiation alone and combined injury activated protective mechanisms such as HO-1 and AKT1, offering insights into molecular events before manifesting renal dysfunction.
Keywords:
Inflammation
; oxidative stress
; apoptosis
; STAT1
; STAT3
; IL-10
; Heme Oxygenase-1
; AKT
; p38 and p53.
1. Introduction
Ionizing radiation injuries (RI), often in conjunction with other injuries namely combined injury (CI), have historically and contemporarily manifested in both warfare scenarios, such as Hiroshima and Nagasaki, and nuclear accidents like Chernobyl and Fukushima. The persistent relevance of these events underscores the need for a deep understanding of the biological impacts of such injuries. RI and CI manifest across a spectrum of symptoms classified temporally into acute, consequential, and late effects. Acute effects occur during or immediately after exposure, consequential effects evolve from ongoing damage after the initial exposure, and late effects, such as kidney injury, may not surface until months or years later [1,2,3,4,5,6,7].
The pathophysiology of radiation-induced kidney damage is complex and multi-faceted. Key mechanisms include direct DNA damage, inflammation, oxidative stress, microvascular damage, mitochondrial impairment, all of which culminate in renal tubular cell death [3,8,9,10,11,12]. There is no specific treatment for radiation-induced kidney injury or any types of acute kidney injury (AKI) [13,14].
Transcription factors like STAT1 and STAT3 are instrumental in regulating inflammatory mediators. They are activated by phosphorylation of certain amino acids, including tyrosine residuals. Protein tyrosine phosphatase SHP-1 modulates these pathways by dephosphorylating STAT proteins, thus acting as a critical regulatory checkpoint [15,16,17]. Ionizing radiation induces reactive oxygen species (ROS), which include superoxide anion and hydrogen peroxide. The body's defense against ROS involves several antioxidative enzymes, including heme oxygenase-1 (HO-1), superoxide dismutase (SOD), catalase, and peroxidase [18,19,20,21,22]. HO-1, in particular, is crucial for mitigating oxidative stress by breaking down heme, producing antioxidants like biliverdin and bilirubin, generating the protective gas carbon monoxide, and preventing iron from contributing to oxidative damage [23,24,25]. The interplay between inflammation and ROS can establish a self-reinforcing cycle, where immune cells augment ROS levels, which then escalates further inflammation, thereby exacerbating organ injury [20,26]. AKT, also known as protein kinase B (PKB), is a serine/threonine kinase and plays a protective role in organ injury by promoting cell survival, reducing apoptosis, enhancing cell proliferation, suppressing inflammation, and preserving mitochondrial function [27,28]. AKT often inhibits p53 in order to exert its pro- cell survival and anti-apoptotic cell death function [29,30,31].
Although the pathophysiology of RI and CI-induced kidney damage has become increasingly understood, the molecular pathways affected before any noticeable functional kidney damage remain largely unknown. Investigating these early alterations offers insight into the radiation's effects at the cellular and molecular levels, potentially elucidating why the kidney is resilient to immediate radiation-induced injury and identifying new therapeutic targets for intervention. In this study, we explored and contrasted the molecular pathways associated with inflammation, oxidative stress, and cell survival 30 days after mice were injured by radiation alone or in combination with a skin wound.
2. Results
Radiation alone or in combination with a skin wound has little immediate effect on renal function. As expected, RI did not significantly increase BUN levels (Figure 1a), serum NGAL levels (Figure 1b), or protein abundance of KIM-1 (Figure 1c), a marker for renal tubular damage, in the mouse renal cortex compared to the sham group 30 days after mice were exposed to 9.5 Gy of 60Co radiation. CI did not significantly affect BUN or NGAL levels (Figure 1d and e), but increased KIM-1 protein abundance (Figure 1f) in the renal cortex when compared to the wound group. Compared with the sham group, wound alone had no significant effects on BUN (sham 42.7±2.8 mg/dL vs. wound 35.2±3.0 mg/dL) or serum NGAL levels (sham 3.2±0.7 ng/mL vs. wound 2.1±0.7 ng/mL) (Figure 1a, b, d and e). These data suggest that neither RI nor CI reduce kidney function within 30 days after injury, but CI has a nuanced effect at the molecular level.
Radiation alone and combined injury upregulate inflammatory pathways, but they do so in distinct ways in the renal cortex. While RI had no significant effect on STAT1-Y701 phosphorylation (Figure 2a), it significantly increased STAT3-Y705 phosphorylation compared with the sham group (Figure 2b). In contrast, CI significantly increased STAT1-Y701 phosphorylation (Figure 2c), but it had no significant effects on STAT3-Y705 phosphorylation (Figure 2d). Wound alone significantly increased phosphorylation of both STAT1-Y701 and STAT3-Y705 compared with the sham group (Figure 2e and f). Having found that RI, CI and wound alone activated STAT3 and STAT1, we sought to determine whether the insults affected the upstream signaling pathway of STATs. None of the insults had a significant effect on SHP-1-S591 phosphorylation or protein abundance of SHP-1 (Figure 2g to i). However, RI decreased p38 phosphorylation (Figure 2j), whereas CI tended to increase p38 protein abundance (Figure 2k). Wound alone had no significant effects on phosphorylation of p38 or p38 protein abundance when compared with the sham group (Figure 2l). These data indicate that RI and CI activate inflammatory pathways through different mechanisms without SHP-1 involvement in either pathway.
Radiation alone and combined injury have different effects on pro- and anti-inflammatory gene mRNA levels in the renal cortex. We found that while RI and CI had no significant effects on the majority of pro-inflammatory gene mRNA levels that were examined, RI significantly up-regulated mRNA abundance of IFNγR1, IFNγR2, and heparanase compared with the sham group (Figure 3a), whereas CI only tended to increase mRNA levels of heparanase (Figure 3b). Likewise, RI and CI had different effects on mRNA levels of immune-regulatory genes. RI remarkably increased IL-10 mRNA levels by 287.2%, whereas CI had no significant effects on the IL-10 mRNA abundance, but reduced the IL-4 mRNA abundance by 61.5% compared with their respective controls (Figure 3c and d). Wound alone did not notably impact the mRNA levels of most pro-inflammatory and immune-regulatory genes. However, it led to a significant decrease in IFNγR1 mRNA levels when compared to the sham group, as shown in Figure 3e and f.
Radiation alone and combined injury upregulate HO-1. Both RI and CI increased the HO-1 protein abundance by 80.7% and 38.3%, respectively, in the renal cortex when compared to their respective control groups, indicating the presence of oxidative stress after injuries (Figure 4a and b). However, RI had an additional effect of reducing the protein abundance of MnSOD and catalase (Figure 4c and g), whereas CI did not exhibit this reduction (Figure 4d and h). To gain a comprehensive understanding of how these two injuries affected the anti-oxidative stress pathways, we conducted additional experiments to assess their impact on CuZnSOD, PRDX1, and ferritin heavy chain. Our findings indicate that neither RI nor CI had a significant effect on the levels of these proteins (Figure 4e, f, and i to l). When compared with the sham group, wound alone had no significant effects on these anti-oxidant protein levels except for reducing catalase protein abundance (Figure 4p).
Radiation alone and combined injury upregulate cell survival and downregulate cell apoptosis pathways. Both RI and CI led to a remarkable increase in the phosphorylation of AKT-S473 (149.0% and 208.8% respectively, Figure 5a and b), while CI reduced protein abundance of AKT1 and AKT2 compared with wound alone (Figure 5b). However, RI and CI had different effects on the phosphorylation of GSK-3β-S9, one of AKT's downstream targets. While RI increased the phosphorylation of GSK-3β-S9 and concurrently decreased the overall GSK-3β protein levels, CI did not significantly affect either parameter, highlighting the complexity of AKT signaling pathways (Figure 5c and d). Despite any changes in protein abundance of HSC-70 (heat shock cognate protein 70), a pro-survival molecule (Figure 5e and f), both injuries resulted in a decrease in protein abundance of p53 when compared with their respective controls (Figure 5g and h). Wounding by itself did not significantly alter the levels of any of these proteins in comparison to the sham group (Figure 5i to l).
3. Discussion
Inflammatory Pathways
While it is well known that inflammation plays a critical role in RI- and CI-induced organ injuries, specific studies of the effects of these injuries on STAT1 and STAT3 remain scarce. X-ray intraperitoneal irradiation induced structural and functional damage in the mouse kidney from 3 to 5 months after the injury. This radiation-induced nephropathy was associated with increased protein abundance of STAT1 and STAT3 in the kidney, coupled with elevated blood levels of multiple pro-inflammatory cytokines, such as TNF-α, IL-6, and IFN-γ [33]. CI (radiation+hemorrhage) activated STAT3 in the mouse ileum, a radiation-sensitive organ, on day 1 after the injury, whereas RI did not [34].
In the present study, we found that both STAT1 and STAT3 were activated prior to the manifestation of kidney dysfunction (Figure 2). However, they were activated distinctly. RI primarily increased STAT3-Y705 phosphorylation, while CI upregulated STAT1-Y701 phosphorylation in the mouse kidney cortex. In contrast to what was observed with X-ray intraperitoneal irradiation-induced nephropathy [33], both RI and CI either significantly reduced or tended to reduce protein abundance of STAT1 and STAT3 (Figure 2). Subsequently, these differential activations resulted in different expression patterns of inflammation genes in the kidney cortex. RI increased mRNA abundance of IFNγR1, IFNγR2, and heparanase, while CI primarily influenced heparanase (Figure 3). Heparanase exacerbates inflammation by promoting cell migration, angiogenesis, the release of inflammatory mediators, and immune cell activation [35,36,37]. Similarly, cytokines exhibited diverse responses, with RI significantly elevating IL-10 mRNA levels but CI reducing IL-4 mRNA abundance (Figure 3). Both IL-10 and IL-4 are well known for their immune regulatory effects [38,39,40].
It appears that RI induced a stronger immune response than CI, possibly influenced by the higher radiation dose used in RI (9.5 Gy compared to 9.0 Gy for CI) and differential activation of STAT3 and STAT1. STAT3 is known for its role in promoting inflammation, but it can also be involved in anti-inflammatory processes depending on the context. This dual role might allow STAT3 to more effectively initiate a strong but controlable immune response. In contrast, STAT1 is generally associated with promoting inflammation [41,42,43]. Indeed, the increase in IL-10 mRNA levels following RI may indicate a regulatory response to the RI-induced inflammation, potentially contributing to the delayed renal function damage. It is known that activation of STAT1 inhibits expression of IL-4 [44,45]. The reduction of IL-4 mRNA following CI points to a suppression of anti-inflammatory signaling, which could exacerbate inflammatory damage. Since STAT1 could have transient effects on cytokine expression [46,47], it remains to be seen whether CI might induce pro-inflammatory cytokines at earlier time points. Our findings indicate that different mechanisms govern the inflammation between RI and CI, highlighting the specificity of their effects, which could influence long-term outcomes.
The role of p38 in inflammation is complex and context-dependent. The activation of p38 is generally regarded as pro-inflammatory, although in certain situations, p38 activation can have anti-inflammatory effects [48,49,50]. Research indicates that p38 activation is linked to more severe radiation-induced injury. Male mice are more susceptible to such injuries than females, with males displaying increased p38 activation in the ileum following irradiation, a response not observed in females [51]. Similarly, CI with hemorrhage induced activation of p38, whereas RI alone did not, in the ileum [34]. Correspondingly, CI induced more pro-inflammatory cytokine expression and greater structural damage in the ileum than RI [34]. Wang et al. demonstrated a causal effect of p38 on radiation-induced residual bone marrow injury by showing that inhibiting p38 in irradiated mice with SB203580 boosted the frequency of bone marrow hematopoietic stem and progenitor cells [52]. We have found that RI reduced activation of p38, which was associated with a lack of expression of various pro-inflammatory cytokine observed in the ileum. This mechanism could add an explaination for the renal resilience to RI.
Oxidative Stress and Antioxidant Defense
Oxidative stress is one of the main pathophysiological mechanisms for radiation-induced nephropathy [53,54]. Increased HO-1 protein abundance was a shared feature of both RI and CI, indicating a response to oxidative stress (Figure 4). However, RI also decreased MnSOD and catalase protein levels (Figure 4). This suggests that RI induced stronger oxidative stress than CI, likely because RI involved a higher radiation dose than CI. Additionally, RI triggered a more potent and regulated immune response in the kidney cortex (Figure 3). It is well known that inflammation induces oxidative stress, whereas anti-inflammation does the opposite [55]. It is evident that IL-10 increases HO-1 expression in the heart [56]. Reflecting the dynamic immune interactions, RI increased HO-1 protein abundance by 80.7% compared to 38.3% in CI, attempting to mitigate the oxidative stress. These observations suggest that the mechanisms for RI and CI in generate oxidative stress are not entirely the same.
Cell Survival Pathways
Exposure to ionizing radiation can trigger apoptotic pathways in cells, leading to tissue damage in organs. Cells developed anti-apoptotic mechanisms such as activation of AKT and inhibition of p53 to inhibit apoptosis. AKT activation can promote cell survival and inhibit apoptosis by phosphorylating and inactivating pro-apoptotic proteins [27,30]. We previously demonstrated that in RI- and CI-induced-injuries in brain and ileum, activation of AKT was reduced associated with increased activation of caspase-3 [57]. Ghrelin mitigated the injuries associated with increased activation of AKT and decreased activation of caspase-3 in the tissues [57]. In the present study, we found that both RI and CI increased activation of AKT in the kidney (Figure 5), suggesting a self-defensive mechanism for the delay in radiation-induced damage to the kidney compared to the brain and ileum.
p53 induces apoptosis by activating the transcription of pro-apoptotic genes, while repressing anti-apoptotic genes. In normal conditions, Mdm2 (Mouse double minute 2) sequesters p53 in the cytosol by binding to p53, preventing its transcriptional activity [58,59]. RI- and CI-induced brain hemorrhage reduced association of Mdm2 with p53, suggesting that more free form p53 was available in the brain [57]. Ghrelin attenuated brain hemorrhage associated with increasing association of p53 with Mdm2 [57]. We report that RI and CI reduced p53 protein abundance, another mechanism for inhibition of p53, in the kidney cortex (Figure 5). It is important to point out that since inflammation, oxidative stress and apoptosis can intricately affect each other, HO-1 can inhibit inflammation and apoptosis, and activation of AKT and down regulation of p53 can inhibit inflammation and oxidative stress as well. Our research has suggested the complex interplay between inflammatory, oxidative and cell survival pathways in the kidney in early response to RI and CI.
Although wound alone did not significantly increase BUN and serum NGAL levels, it significantly increased phosphorylation of STAT1-Y701 and STAT3-Y705 (Figure 2), and decreased IFNγR1 mRNA (Figure 3) and catalase protein (Figure 4) abundance in the renal cortex. Nonetheless, the significance of these findings remains unknown.
Limitations of the Study
While our study provides valuable insights into the early molecular responses of the kidney to RI and CI, several limitations should be acknowledged. The study used two different doses of γ-irradiation (9.5 Gy for RI and 9.0 Gy for CI). While the reduced dose of radiation in CI was necessary to ensure sufficient survival of mice available for molecular analysis, this precluded a direct statistical comparison between the two groups using two-way analysis of variance. It would be beneficial to investigate a range of doses to better understand dose-dependent effects and to generalize the findings across various radiation exposure scenarios. Another limitation is that our study focused on early responses, specifically 30 days post-injury. This time point may be too late for some immune and oxidative responses and too early for others, as it typically takes months or years to observe kidney dysfunction following the injuries.
Summary
In summary, although neither RI nor CI compromised renal function in our study, we observed that CI elicited more sub-clinical alterations at a microscopic level in the kidney cortex, as evidenced by the increased abundance of KIM-1 protein than RI. It appears that inflammation and oxidative stress are among the earliest affected pathways, but RI and CI induced inflammation and oxidative stress distinctly. RI enhanced phosphorylation of STAT3-Y705 and increased mRNA levels of IFNγR1 and IFNγR2, heparanase, and IL-10. In contrast, CI predominantly increased phosphorylation of STAT1-Y701, tended to raise heparanase mRNA levels, and decreased IL-4 mRNA levels. While RI led to an increase in IL-10 mRNA levels and a decrease in p38 MAP kinase activation (suggesting a particular anti-inflammatory and stress response), CI exhibited a lack of these responses, potentially explaining the observed differences in subclinical kidney changes. RI reduced MnSOD and catalase proteins, whereas CI did not. However, despite the distinct activation patterns of inflammatory and oxidative stress pathways induced by both types of injury, our findings reveal a common activation of cellular defense mechanisms against oxidative stress, exemplified by increased HO-1 protein abundance, and survival mechanisms, such as increased AKT activation and reduced p53 protein abundance (Figure 6).
4. Methods and Materials
Animal and experimental design. All procedures were approved by the Institutional Animal Care and Use Committee of Uniformed Services University (MED-22-099). B6D2F1/J female mice (12 weeks old, approximately 20-26 g) purchased from Jackson Laboratory (Bar Harbor, ME) were maintained in a facility accredited by AAALAC in plastic microisolator cages with hardwood chip bedding and allowed to acclimate to their surroundings for at least 3 days prior to the study. Male mice were not used in this study because of potential problems associated with male mouse aggression, such as fight wounds which were not desirable during the experimental period. Previous injury studies [57] also used female mice for this reason. As such, we continued to conduct this study with female mice so that data collected could be compared with previous ones. These mice were provided with commercial rodent chow (Rodent Diet #8604, Harlan Teklad, Madison, WI) and acidified tap water (pH=2.5-2.8) ad libitum. Rooms holding animals were maintained at 22°C ± 2°C with 50% ± 20% relative humidity using at least 10-15 air changes/h of 100% conditioned fresh air with a 12-h 0600 (light) to 1800 (dark) full-spectrum lighting cycle. Mouse tails were tattooed for individual identification during acclimation. B6D2F1/J female mice were randomly divided into 4 groups: 1) sham (N=10); 2) wound (N=10); 3) Radiation (N=20) ; 4) Radiation+wound (N=20).The sham-irradiated animals were treated in the same manner as the irradiated animals but not exposed to the radiation source. The experiments were performed twice with a half number of mice in each experiment. However, some mice in both RI and CI groups did not survive to day 30, resulting in fewer than 20 mice remaining in these groups by the study's end. The mice were euthanized 30 days after exposure to irradiation by exsanguination under isoflurane, and cervical dislocations were performed as a secondary physical method.
Radiation. Animals were subjected to irradiation as previously described [57]. Briefly, mice in the RI group received 9.5 Gy of 60Co gamma photons, delivered at 0.4 Gy/min in the Uniformed Services University 60Co facility. For the CI group, mice were exposed to 9.0 Gy of 60Co, delivered at 0.4 Gy min-1 because other insults are known to increase radiation sensitivity and the use of 9.5 Gy in the CI group resulted in excessive lethality (data not shown). Sham mice and wounded mice were handled the same way, but remained in the staging room outside the 60Co facility. During irradiation, mice were kept awake in ventilated, acrylic plastic boxes with four compartments. The accuracy of the delivered doses was confirmed using an ionization chamber calibrated to measure the radiation dose to the midline soft tissue of the mice [57].
Wounding. Within 1 hr after irradiation, animals were anesthetized with isoflurane, and a wound was created 19±1.3 mm from the occipital bone and between the scapulae, using a punch [32]. Wounded and CI mice were injected i.p. with 0.5 ml saline containing 150 mg/kg acetaminophen (OFIRMEV injection, NDC 43825-102-01; Mallinckrodte Pharmaceuticals, Hazelwood, MO, USA) immediately after wounding to alleviate the pain. Sham mice and irradiated only mice received 0.5 ml saline i.p. The animal procedures are summarized in Figure 7.
Blood urea nitrogen (BUN) assay. Blood was collected via a cardiac puncture under isoflurane on day 30. BUN levels in serum were measured using the diacetyl monoxime method, as previously described [60,61]. Briefly, serum samples were diluted tenfold with deionized water. Proteins in the samples were precipitated by adding a 0.61 M trichloroacetic acid solution, and then separated by centrifuging for 10 minutes at room temperature, at 10,000 g. The resulting supernatants were mixed with a chromogen and incubated for 5 min at 80°C, and absorption measurements were taken with a microplate reader (Elx800, BioTek, Winooski, Vermont, United States) at 490 nm. The chromogen was created by freshly mixing Reagent A and Reagent B in a 2:1 ratio. Reagent A was prepared by dissolving 10 mg of ferric chloride in 10 ml of 1.48 M phosphoric acid, which was then mixed with 60 ml of distilled water and 30 ml of 5.52 M sulfuric acid. Reagent B consisted of 50 mg of diacetyl monoxime with 1 mg of thiosmicarbazide dissolved in 10 ml of distilled water [60,61].
Serum neutrophil gelatinase-associated lipocalin (NGAL) assay. Serum NGAL was detected using a kit from Abcam (ab199083) according to the manufacturer’s protocol.
Western blot analysis. The kidney cortex was homogenized in a lysis buffer consisting of 10 mM triethanolamine, 250 mM sucrose, pH 7.4 plus a protease inhibitor cocktail tablet (Roche, Catolog # 11784500), 2mM of NaF (MilliporeSigma, Catalog# S6776) and 2 mM of Na3VO4 (MilliporeSigma, Catalog# 450243) to inhibit phosphatases. The homogenates were centrifuged at 13000 rpm at 40C for 8 min [62]. The protein concentrations in supernatants were measured with the BCA assay (Thermo Scientific, Product# 23228 and 1859078). After assay, samples were dissolved in an SDS loading buffer for immediate use or frozen at -800C for future use. Equal quantities of protein samples (between 25-60 μg protein/lane) were loaded into 15 well or 17 well 4%–12% Bis-Tris gels (ThermoFisher, Catalog# NP0336BOX or NW04127BOX). After gel electrophoresis, the proteins were transferred onto nitrocellulose membranes (ThermoFisher, Catalog# LC2001). The membranes were soaked in blocking buffer (Odyssey, Part # 927-90001) at room temperature for 1 hour and the gels were stained with SimplyBluetm SafeStain (ThermoFisher, Catalog # LC6060) overnight to examine sample loadings. After soaking, the membranes were probed overnight with primary antibodies typically at 1:500 or 1:1000 dilution at 4°C. The membranes were then washed with PBS+0.1% Tween-20 and probed with an Alexa Fluor® secondary antibody for 1 hour at room temperature then examined with Odyssey, an infrared imaging scanner (Li-Cor). The data is presented without normalization, as stained gels were used to confirm equal sample loading. Reserving the first lane for protein markers, the maximum number of samples we could accommodate was 14 or 16. Protein samples were loaded into gels according to the randomly assigned numbers of mice, without any selection bias. If a significant difference in a protein abundance was observed in the first Western blot analysis, the experiment was repeated once with reproducible results presented. If there was no clear trend for a significant difference of protein levels in the first Western blot analysis, the experiment was stopped. The inconsistencies in sample numbers could arise from several factors. Occasionally, a bubble formed in the first well, or a distortion occurred in the last well, preventing their inclusion in the analysis. The data presented in the manuscript represents only a subset of the analyses we conducted. At times, we faced a shortage of samples for certain groups but proceeded with the Western blot analysis regardless. These results are included in the manuscript. The primary antibodies against proteins are listed as follows: Kidney injury molecule-1 (KIM-1, Invitrogen, PA520244), STAT1-Y701-P (Cell Signaling, 9167), STAT1 (Cell Signaling, 9176), STAT3-Y705-P (Cell Signaling, 9145S), STAT3 (Cell Signaling, 9139S), SHP-1-S591(ECM Biosciences, SP1531), SHP-1 (Santa Cruz, sc-287), p38-P (Cell Signaling, 9215S), p38 (Upstate, 05-454), MnSOD (Upstate, 06-984), Cu/ZnSOD (Upstate, 07-403), Peroxiredoxin 1 (PRDX) (Cell Signaling, 8732S), HO-1 (Cell Signaling, 43966S), p53 (Cell Signaling, 2524), AKT-S473 (Cell Signaling, 4060S), AKT (Cell Signaling, 2920S), GSK-3β-S9 (Cell Signaling, 9336), and GSK-3β (Cell Signaling, 9832).
qPCR. Quantitative PCR (qPCR) analysis of mRNA abundance were performed as described previously [60]. Briefly, total RNA was isolated with the TRIzol kit (ThermoFisher, Ref# 15596026) and measured with NanoDrop 8000 Spectrophotometer (ThermoFisher). cDNAs were synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Part# 4368814). mRNAs were measured using the Fast SYBR Green Master Mix (Applied Biosystems, Ref# 4385612) in a Cycler 480 (Roche) and normalized to a housekeeping gene, L32. Fold difference was calculated as previously described [60]. The primers used are listed in Table 1.
Statistical analysis. Data are expressed as means ± standard errors. All analyses were normalized to the average value of the respective control groups. Shapiro-Wilk normality test and Q-Q plot revealed normal distributions of our data. Statistical analyses were performed using a non-paired two-tailed t-test with Microsoft Excel. Two-way ANOVA could not be performed because two different doses of radiation were used in the RI and CI groups: 9.5 Gy and 9.0 Gy, respectively. A p value less than 0.05 was considered significant.
5. Conclusions
In conclusion, our findings have highlighted the complexity of the body's response to RI and CI, revealing both unique and shared pathways in inflammation, oxidative stress, and cellular survival mechanisms. These intriguing insights have laid the groundwork for understanding the mechanisms underlying delayed functional impairment in the kidney and open new targets such signaling molecules for future research into therapeutic strategies to protect renal function in the events of nuclear attacks and accidents, with a particular focus on the distinct and common molecular pathways activated by RI and CI.
Author Contributions
Investigation: G.D., H.W., B.P., G.C., J.G.K, and X.Z.; writing—original draft preparation: G.D. and X.Z.; writing—review and editing: G.D., B.P., G.C., J.G.K, and X.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the United States of America Department of Defense grants MED-83-12715 (to XZ), NIAID 5U19AI06777-18 SAPO: GI7070 (to JGK), and AFR-B2-12812 (to JGK).
Institutional Review Board Statement
The study was conducted according to the USUHS Institutional Animal Care and Use Committee (IACUC) under protocol MED-22-099 approved on October 28, 2022.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Acknowledgments
The authors thank Dr. Regina Day for her help in obtaining the approval of our animal protocol from the Institutional Animal Care and Use Committee of Uniformed Services University.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Cohen, E.P.; Hankey, K.G.; Farese, A.M.; Parker, G.A.; Jones, J.W.; Kane, M.A.; Bennett, A.; MacVittie, T.J. Radiation Nephropathy in a Nonhuman Primate Model of Partial-body Irradiation with Minimal Bone Marrow Sparing-Part 1: Acute and Chronic Kidney Injury and the Influence of Neupogen. Health Phys 2019, 116, 401–408. [Google Scholar] [CrossRef]
- Parker, G.A.; Cohen, E.P.; Li, N.; Takayama, K.; Farese, A.M.; MacVittie, T.J. Radiation nephropathy in a nonhuman primate model of partial-body irradiation with minimal bone marrow sparing—Part 2: histopathology, mediators, and mechanisms. Health physics 2019, 116, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Farese, A.M.; Booth, C.; Tudor, G.L.; Cui, W.; Cohen, E.P.; Parker, G.A.; Hankey, K.G.; MacVittie, T.J. The Natural History of Acute Radiation-induced H-ARS and Concomitant Multi-organ Injury in the Non-human Primate: The MCART Experience. Health Phys 2021, 121, 282–303. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.; Hegi-Johnson, F. Radiotherapy toxicity. Nature Reviews Disease Primers 2019, 5, 13. [Google Scholar] [CrossRef]
- Ahmad, A.; Shi, J.; Ansari, S.; Afaghani, J.; Molina, J.; Pollack, A.; Merscher, S.; Zeidan, Y.H.; Fornoni, A.; Marples, B. Noninvasive assessment of radiation-induced renal injury in mice. Int J Radiat Biol 2021, 97, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Ozasa, K.; Akiba, S.; Niwa, O.; Kodama, K.; Takamura, N.; Zaharieva, E.K.; Kimura, Y.; Wakeford, R. Long-term effects of radiation exposure on health. Lancet 2015, 386, 469–478. [Google Scholar] [CrossRef]
- Cohen, E.P.; Fish, B.L.; Moulder, J.E. Late-onset effects of radiation and chronic kidney disease. Lancet 2015, 386, 1737–1738. [Google Scholar] [CrossRef]
- Klaus, R.; Niyazi, M.; Lange-Sperandio, B. Radiation-induced kidney toxicity: molecular and cellular pathogenesis. Radiat Oncol 2021, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Kiang, J.G.; Blakely, W.F. Combined radiation injury and its impacts on radiation countermeasures and biodosimetry. Int J Radiat Biol 2023, 99, 1055–1065. [Google Scholar] [CrossRef]
- Mostaghimi, S.; Mehrvar, S.; Foomani, F.H.; Narayanan, J.; Fish, B.; Camara, A.K.S.; Medhora, M.; Ranji, M. Vascular regression in the kidney: changes in 3D vessel structure with time post-irradiation. Biomed Opt Express 2022, 13, 4338–4352. [Google Scholar] [CrossRef]
- Yamaga, S.; Aziz, M.; Murao, A.; Brenner, M.; Wang, P. DAMPs and radiation injury. Front Immunol 2024, 15, 1353990. [Google Scholar] [CrossRef] [PubMed]
- Molinar-Inglis, O.; DiCarlo, A.L.; Lapinskas, P.J.; Rios, C.I.; Satyamitra, M.M.; Silverman, T.A.; Winters, T.A.; Cassatt, D.R. Radiation-induced multi-organ injury. Int J Radiat Biol 2024, 100, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. Reducing Oxygen Demand to Alleviate Acute Kidney Injury. Front Biosci (Landmark Ed) 2023, 28, 62. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.A.; Kavanagh, B.D.; Paulino, A.C.; Das, S.K.; Miften, M.; Li, X.A.; Pan, C.; Ten Haken, R.K.; Schultheiss, T.E. Radiation-associated kidney injury. Int J Radiat Oncol Biol Phys 2010, 76, (3 Suppl), S108–15. [Google Scholar] [CrossRef]
- Trécul, A.; Morceau, F.; Dicato, M.; Diederich, M. Dietary compounds as potent inhibitors of the signal transducers and activators of transcription (STAT) 3 regulatory network. Genes Nutr 2012, 7, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.-C.; Teng, H.-W.; Shiau, C.-W.; Tai, W.-T.; Hung, M.-H.; Yang, S.-H.; Jiang, J.-K.; Chen, K.-F. Pharmacological targeting SHP-1-STAT3 signaling is a promising therapeutic approach for the treatment of colorectal cancer. Neoplasia 2015, 17, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; Huang, T.T.; Spitz, D.R.; Oberley, L.W.; Li, J.J. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol.Cell Biol. 2003, 23, 2362–2378. [Google Scholar] [CrossRef]
- Wei, J.; Wang, B.; Wang, H.; Meng, L.; Zhao, Q.; Li, X.; Xin, Y.; Jiang, X. Radiation-Induced Normal Tissue Damage: Oxidative Stress and Epigenetic Mechanisms. Oxid Med Cell Longev 2019, 2019, 3010342. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Miao, B.; Cao, H.; Tian, X.; Shen, L.; Yang, Z.; Yuan, F.; Ding, Y. Monkfish (Lophius litulon) Peptides Ameliorate High-Fat-Diet-Induced Nephrotoxicity by Reducing Oxidative Stress and Inflammation via Regulation of Intestinal Flora. Molecules 2023, 28, 245. [Google Scholar] [CrossRef] [PubMed]
- Nath, M.; Agarwal, A. New insights into the role of heme oxygenase-1 in acute kidney injury. Kidney Res Clin Pract 2020, 39, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, R.; Pu, Q.; Jiang, S.; Yu, F.; Yang, Z.; Han, T. Investigation of nephrotoxicity on mice exposed to polystyrene nanoplastics and the potential amelioration effects of DHA-enriched phosphatidylserine. Sci Total Environ 2023, 892, 164808. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, K.; Han, Z.; Chi, M.; Sai, X.; Zhu, P.; Ding, Z.; Song, L.; Liu, C. Immunomodulatory Effects of Heme Oxygenase-1 in Kidney Disease. Front Med (Lausanne) 2021, 8, 708453. [Google Scholar] [CrossRef] [PubMed]
- Gambini, J.; Stromsnes, K. Oxidative Stress and Inflammation: From Mechanisms to Therapeutic Approaches. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Covington, S.M.; Bauler, L.D.; Toledo-Pereyra, L.H. Akt: A Therapeutic Target in Hepatic Ischemia-Reperfusion Injury. J Invest Surg 2017, 30, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, R.; Yan, Y.; He, B.; Miao, C.; Fang, Y.; Wan, H.; Zhou, G. Exosomes-Mediated Signaling Pathway: A New Direction for Treatment of Organ Ischemia-Reperfusion Injury. Biomedicines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Kiang, J.G.; Smith, J.T.; Anderson, M.N.; Umali, M.V.; Ho, C.; Zhai, M.; Lin, B.; Jiang, S. A novel therapy, using Ghrelin with pegylated G-CSF, inhibits brain hemorrhage from ionizing radiation or combined radiation injury. Pharm Pharmacol Int J 2019, 7, 133–145. [Google Scholar]
- Abraham, A.G.; O'Neill, E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans 2014, 42, 798–803. [Google Scholar] [CrossRef]
- Liu, A.; Zhu, Y.; Chen, W.; Merlino, G.; Yu, Y. PTEN Dual Lipid- and Protein-Phosphatase Function in Tumor Progression. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Li, X.; Wang, L.; Lin, B.; Zhai, M.; Hull, L.; Zizzo, A.; Cui, W.; Kiang, J.G. Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Deng, L.; Wang, S.; Leng, X.; Yao, P.; Li, C.; Zheng, Y. Combining network pharmacology and in vitro and in vivo experiments to study the mechanism of Keluoxin in the treatment of radiation nephropathy†. J Radiat Res 2023, 64, 769–782. [Google Scholar] [CrossRef]
- Kiang, J.G.; Smith, J.T.; Anderson, M.N.; Elliott, T.B.; Gupta, P.; Balakathiresan, N.S.; Maheshwari, R.K.; Knollmann-Ritschel, B. Hemorrhage enhances cytokine, complement component 3, and caspase-3, and regulates microRNAs associated with intestinal damage after whole-body gamma-irradiation in combined injury. PLoS One 2017, 12, e0184393. [Google Scholar] [CrossRef]
- Abassi, Z.; Goligorsky, M.S. Heparanase in Acute Kidney Injury. Adv Exp Med Biol 2020, 1221, 685–702. [Google Scholar] [PubMed]
- Li, J.C.; Wang, L.J.; Feng, F.; Chen, T.T.; Shi, W.G.; Liu, L.P. Role of heparanase in sepsis-related acute kidney injury (Review). Exp Ther Med 2023, 26, 379. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Impact of heparanase on renal fibrosis. Journal of Translational Medicine 2015, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation 2017, 14, 117. [Google Scholar] [CrossRef]
- Kushwah, R.; Hu, J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci 2011, 1, 20. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Anderton, S.M. Cytokines in the induction and resolution of experimental autoimmune encephalomyelitis. Cytokine 2005, 32, 81–84. [Google Scholar] [CrossRef]
- Multhoff, G.; Radons, J. Radiation, inflammation, and immune responses in cancer. Front Oncol 2012, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Liongue, C.; Sobah, M.L.; Ward, A.C. Signal Transducer and Activator of Transcription Proteins at the Nexus of Immunodeficiency, Autoimmunity and Cancer. Biomedicines 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, C.; Leung, S.; Salvekar, A.; Mano, H.; Schindler, U. Repression of IL-4-induced gene expression by IFN-gamma requires Stat1 activation. J Immunol 1999, 162, 4053–4061. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.L.; Newcomb, D.C.; Parekh, V.V.; Van Kaer, L.; Collins, R.D.; Zhou, W.; Goleniewska, K.; Chi, M.H.; Mitchell, D.; Boyce, J.A.; Durbin, J.E.; Sturkie, C.; Peebles, R.S. Jr. , STAT1 negatively regulates lung basophil IL-4 expression induced by respiratory syncytial virus infection. J Immunol 2009, 183, 2016–2026. [Google Scholar] [CrossRef]
- Tolomeo, M.; Cavalli, A.; Cascio, A. STAT1 and Its Crucial Role in the Control of Viral Infections. International Journal of Molecular Sciences 2022, 23, 4095. [Google Scholar] [CrossRef]
- Menon, P.R.; Staab, J.; Gregus, A.; Wirths, O.; Meyer, T. An inhibitory effect on the nuclear accumulation of phospho-STAT1 by its unphosphorylated form. Cell Communication and Signaling 2022, 20, 42. [Google Scholar] [CrossRef]
- Schieven, G.L. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem 2005, 5, 921–928. [Google Scholar] [CrossRef]
- Sisakht, M.; Darabian, M.; Mahmoodzadeh, A.; Bazi, A.; Shafiee, S.M.; Mokarram, P.; Khoshdel, Z. The role of radiation induced oxidative stress as a regulator of radio-adaptive responses. Int J Radiat Biol 2020, 96, 561–576. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, H.; Guo, W.; Yu, L. Potential role of ghrelin in the regulation of inflammation. Faseb j 2022, 36, e22508. [Google Scholar] [CrossRef]
- Kiang, J.G.; Cannon, G.; Olson, M.G.; Smith, J.T.; Anderson, M.N.; Zhai, M.; Umali, M.V.; Ho, K.; Ho, C.; Cui, W.; Xiao, M. Female Mice are More Resistant to the Mixed-Field (67% Neutron + 33% Gamma) Radiation-Induced Injury in Bone Marrow and Small Intestine than Male Mice due to Sustained Increases in G-CSF and the Bcl-2/Bax Ratio and Lower miR-34a and MAPK Activation. Radiat Res 2022, 198, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Zhou, D. Inhibition of p38 MAPK attenuates ionizing radiation-induced hematopoietic cell senescence and residual bone marrow injury. Radiat Res 2011, 176, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Kato, T.; Isogai, T.; Gu, Y.; Yamashita, T. The Potential Effects of Taurine in Mitigation of Radiation Nephropathy. Adv Exp Med Biol 2019, 1155, 497–505. [Google Scholar] [PubMed]
- Geng, R.; Fang, J.; Kang, S.G.; Huang, K.; Tong, T. Chronic exposure to UVB induces nephritis and gut microbiota dysbiosis in mice based on the integration of renal transcriptome profiles and 16S rRNA sequencing data. Environ Pollut 2023, 333, 122035. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Wilkinson, F.L.; Sandhu, M.A.; Lightfoot, A.P. The Interplay of Oxidative Stress and Inflammation: Mechanistic Insights and Therapeutic Potential of Antioxidants. Oxidative Medicine and Cellular Longevity 2021, 2021, 9851914. [Google Scholar] [CrossRef]
- Gupta, R.; Liu, L.; Zhang, X.; Fan, X.; Krishnamurthy, P.; Verma, S.; Tongers, J.; Misener, S.; Ashcherkin, N.; Sun, H.; Tian, J.; Kishore, R. IL-10 provides cardioprotection in diabetic myocardial infarction via upregulation of Heme clearance pathways. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Kiang, J.G.; Smith, J.T.; Cannon, G.; Anderson, M.N.; Ho, C.; Zhai, M.; Cui, W.; Xiao, M. Ghrelin, a novel therapy, corrects cytokine and NF-κB-AKT-MAPK network and mitigates intestinal injury induced by combined radiation and skin-wound trauma. Cell Biosci 2020, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Thomasova, D.; Anders, H.J. Cell cycle control in the kidney. Nephrol Dial Transplant 2015, 30, 1622–1630. [Google Scholar] [CrossRef]
- Packialakshmi, B.; Stewart, I.J.; Burmeister, D.M.; Feng, Y.; McDaniel, D.P.; Chung, K.K.; Zhou, X. Tourniquet-induced lower limb ischemia/reperfusion reduces mitochondrial function by decreasing mitochondrial biogenesis in acute kidney injury in mice. Physiol Rep 2022, 10, e15181. [Google Scholar] [CrossRef]
- Rahmatullah, M.; Boyde, T.R.C. Improvements in the determination of urea using diacetyl monoxime; methods with and without deproteinisation. Clinica Chimica Acta 1980, 107, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Packialakshmi, B.; Xiao, Y.; Nurmukhambetova, S.; Lees, J.R. Progression of experimental autoimmune encephalomyelitis is associated with up-regulation of major sodium transporters in the mouse kidney cortex under a normal salt diet. Cell Immunol 2017, 317, 18–25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Radiation alone had no significant effect on renal function as determined by BUN levels (a), serum NGAL levels (b), and KIM-1 protein abundance (c) compared with the sham group in the female B6D2F1/J mouse renal cortex, 30 days post injury. Combined injury did not significantly affect BUN or serum NGAL levels (d and e), but significantly increased KIM-1 protein abundance (f) in the renal cortex compared with the wound group, 30 days post injury. Radiation alone injury was induced by 9.5 Gy of 60Co radiation, delivered at 0.4 Gy min-1. In the combined injury group, mice were first exposed to 9.0 Gy of 60Co, delivered at 0.4 Gy min-1, because the dose of 9.5 Gy induced excessive lethality. A wound was then created 19±1.3 mm from the occipital bone and between the scapulae, using a punch within 1 h after irradiation [32]. (** p<0.01, non-paired two-tailed t-test).
Figure 1.
Radiation alone had no significant effect on renal function as determined by BUN levels (a), serum NGAL levels (b), and KIM-1 protein abundance (c) compared with the sham group in the female B6D2F1/J mouse renal cortex, 30 days post injury. Combined injury did not significantly affect BUN or serum NGAL levels (d and e), but significantly increased KIM-1 protein abundance (f) in the renal cortex compared with the wound group, 30 days post injury. Radiation alone injury was induced by 9.5 Gy of 60Co radiation, delivered at 0.4 Gy min-1. In the combined injury group, mice were first exposed to 9.0 Gy of 60Co, delivered at 0.4 Gy min-1, because the dose of 9.5 Gy induced excessive lethality. A wound was then created 19±1.3 mm from the occipital bone and between the scapulae, using a punch within 1 h after irradiation [32]. (** p<0.01, non-paired two-tailed t-test).
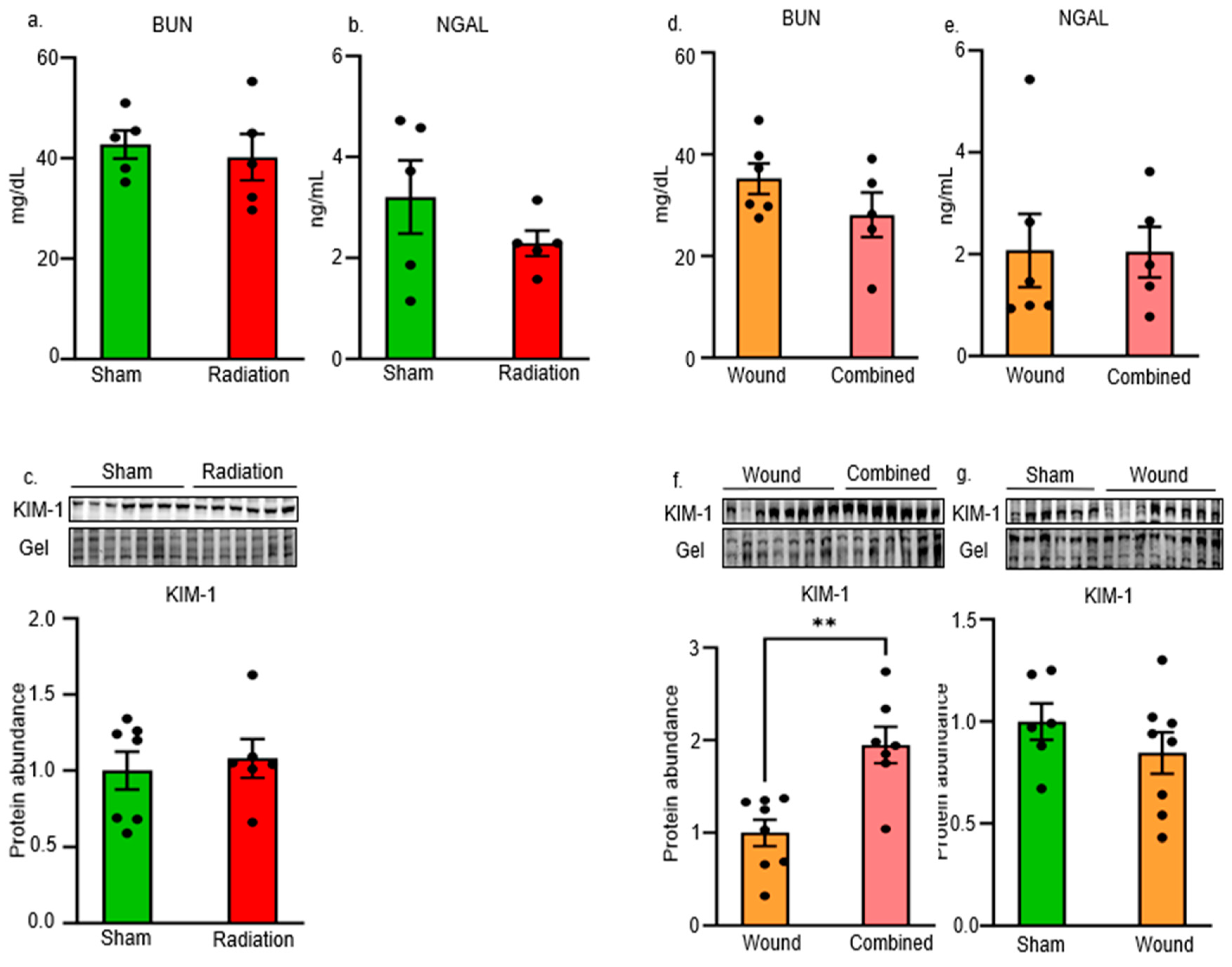
Figure 2.
Radiation alone had no significant effect on STAT1-Y701 phosphorylation (a), but it significantly increased STAT3-Y705 phosphorylation compared with the sham group (b). In contrast, combined injury significantly increased STAT1-Y701 phosphorylation (c), but it had no significant effect on STAT3-Y705 phosphorylation compared with the wound group (d). Wound alone significantly increased phosphorylation of both STAT1-Y701 and STAT3-Y705 compared with the sham group (e and f). Neither radiation alone, combined injury, nor wound alone had a significant effect on SHP-1-S591 phosphorylation or SHP-1 protein abundance (g to i). Radiation alone reduced p38 phosphorylation (j), whereas combined injury or wound alone did not (k and l). (* p<0.05, non-paired two-tailed t-test). .
Figure 2.
Radiation alone had no significant effect on STAT1-Y701 phosphorylation (a), but it significantly increased STAT3-Y705 phosphorylation compared with the sham group (b). In contrast, combined injury significantly increased STAT1-Y701 phosphorylation (c), but it had no significant effect on STAT3-Y705 phosphorylation compared with the wound group (d). Wound alone significantly increased phosphorylation of both STAT1-Y701 and STAT3-Y705 compared with the sham group (e and f). Neither radiation alone, combined injury, nor wound alone had a significant effect on SHP-1-S591 phosphorylation or SHP-1 protein abundance (g to i). Radiation alone reduced p38 phosphorylation (j), whereas combined injury or wound alone did not (k and l). (* p<0.05, non-paired two-tailed t-test). .
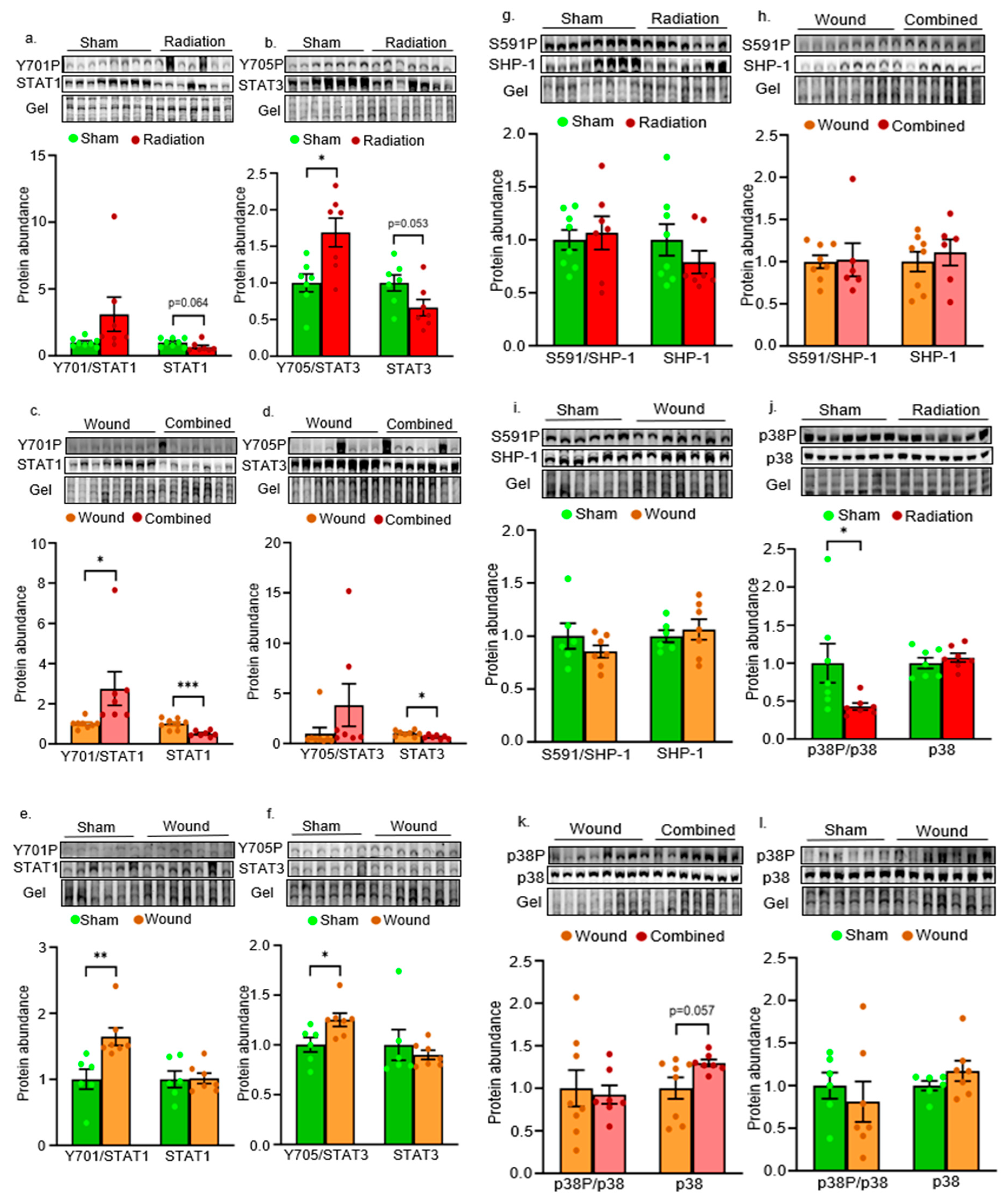
Figure 3.
Radiation alone upregulated pro- and anti-inflammatory gene mRNA levels such as IFNγR1, IFNγR2, Heparanase and IL-10 (a and c), whereas combined injury reduced IL-4 mRNA levels in the mouse renal cortex (d). Wound alone had no significant effects on a majority of pro-inflammatory and immune-regulatory gene mRNA levels, but significantly reduced IFNγR1 mRNA levels, when compared with the sham group (Figure 3e and f). (* p<0.05, ** p<0.01, non-paired two-tailed t-test). .
Figure 3.
Radiation alone upregulated pro- and anti-inflammatory gene mRNA levels such as IFNγR1, IFNγR2, Heparanase and IL-10 (a and c), whereas combined injury reduced IL-4 mRNA levels in the mouse renal cortex (d). Wound alone had no significant effects on a majority of pro-inflammatory and immune-regulatory gene mRNA levels, but significantly reduced IFNγR1 mRNA levels, when compared with the sham group (Figure 3e and f). (* p<0.05, ** p<0.01, non-paired two-tailed t-test). .
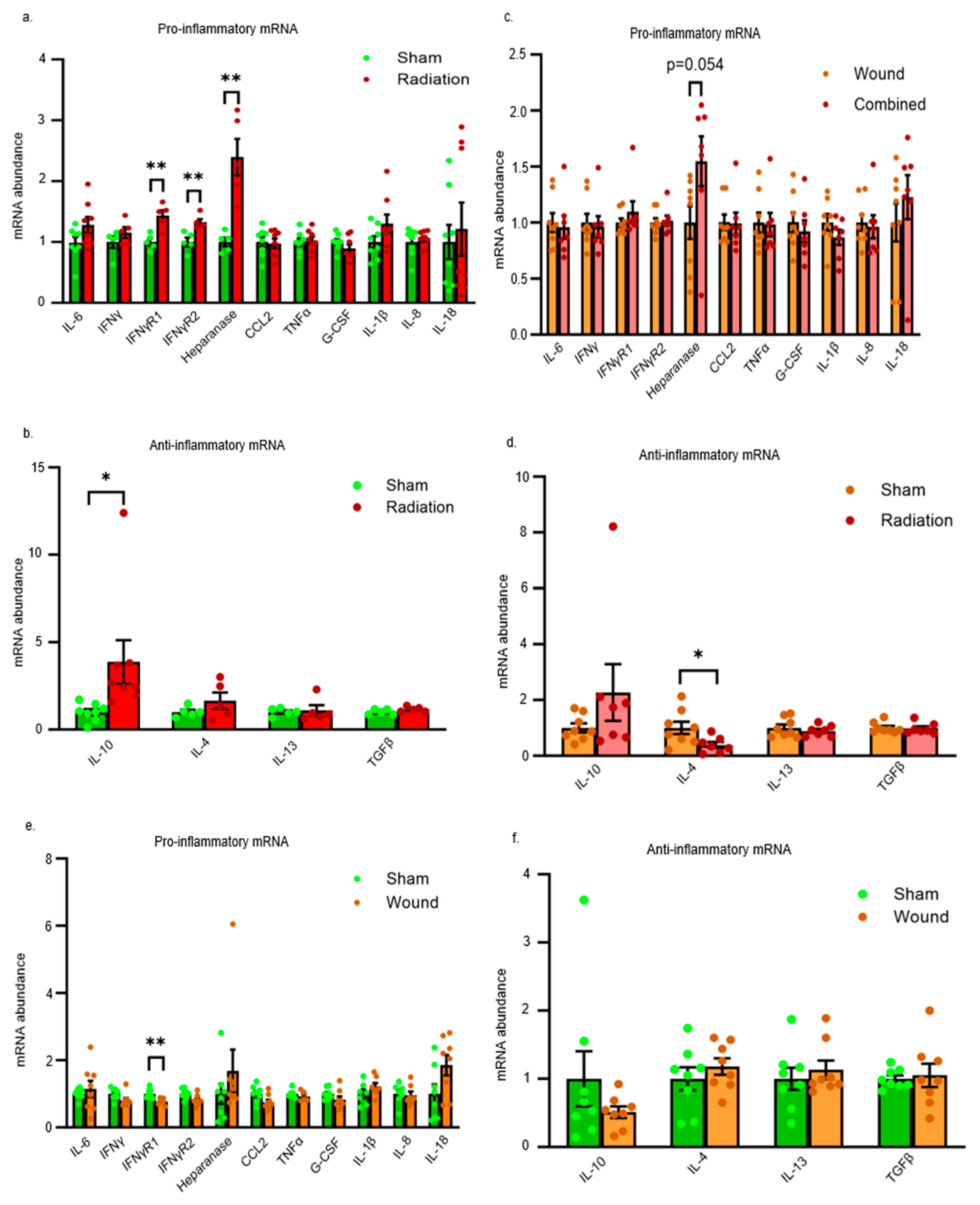
Figure 4.
Both radiation alone and combined injury induced oxidative stress as determined by increased protein abundance of HO-1 when compared with their respective control (a and b). Moreover, radiation alone also reduced protein abundance of MnSOD and catalase (c and g). Wound alone reduced catalase protein abundance when compared with the sham group (p). (* p<0.05, ** p<0.01, non-paired two-tailed t-test).
Figure 4.
Both radiation alone and combined injury induced oxidative stress as determined by increased protein abundance of HO-1 when compared with their respective control (a and b). Moreover, radiation alone also reduced protein abundance of MnSOD and catalase (c and g). Wound alone reduced catalase protein abundance when compared with the sham group (p). (* p<0.05, ** p<0.01, non-paired two-tailed t-test).
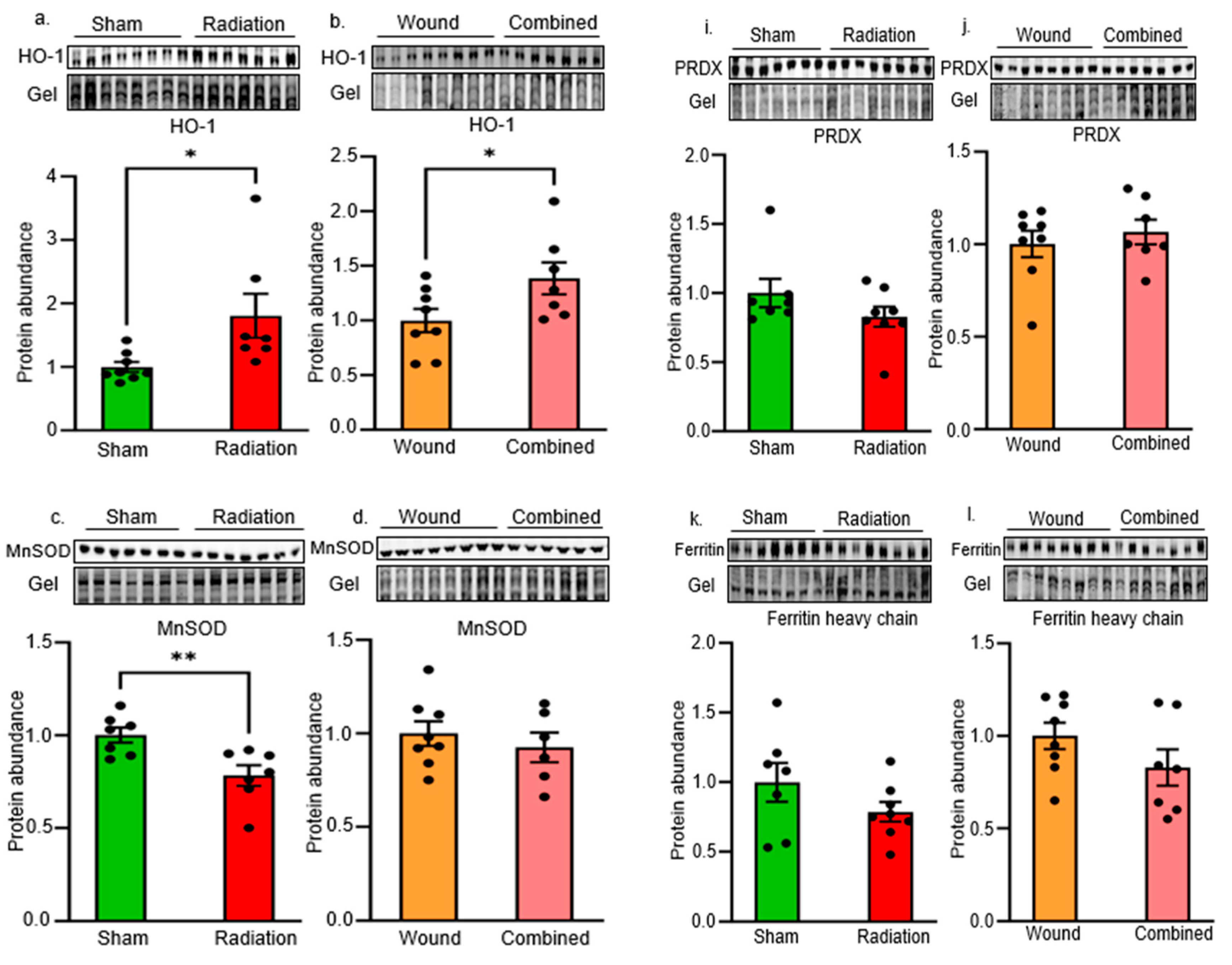
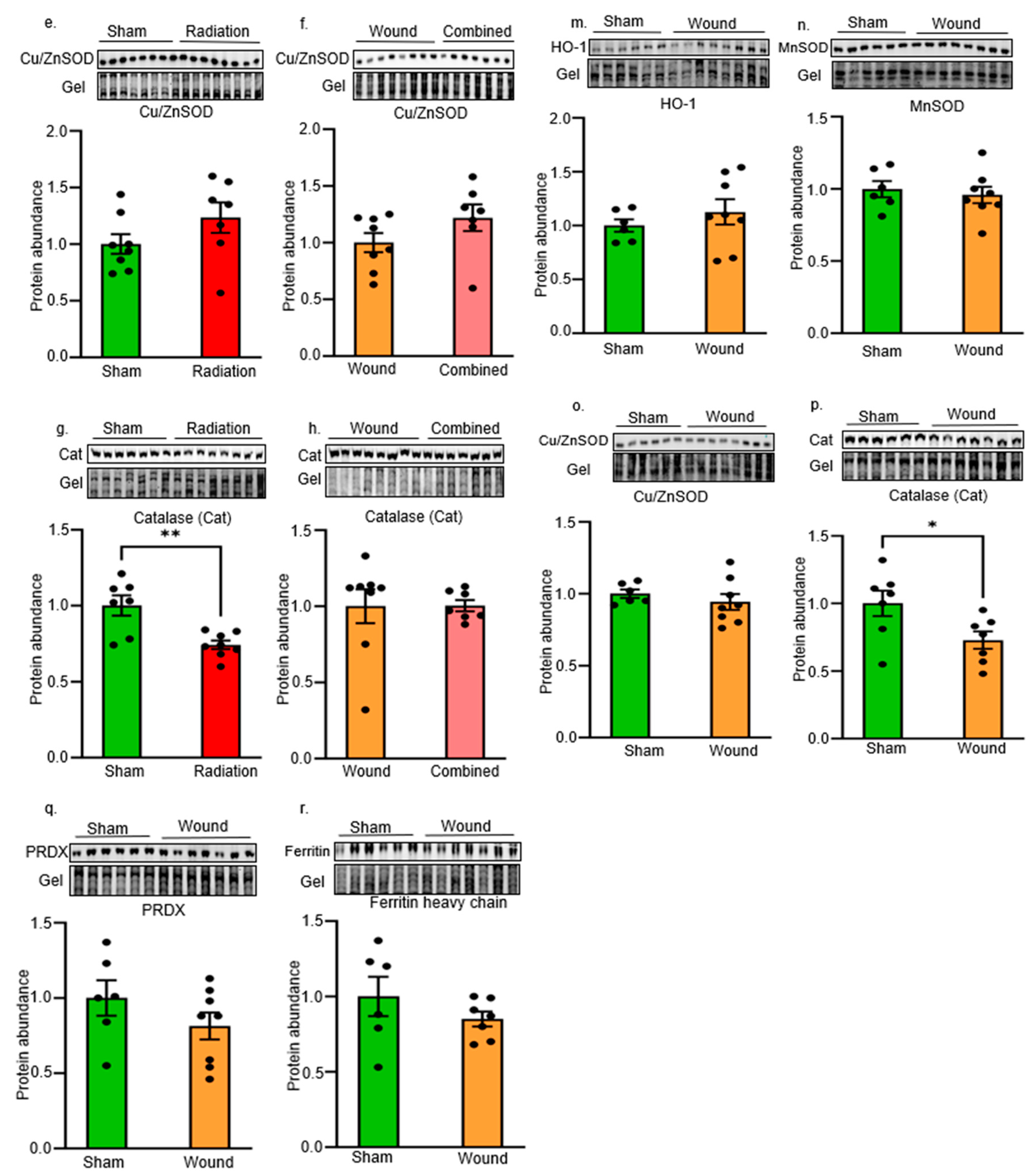
Figure 5.
Both radiation alone and combined injury increased AKT-S473 phosphorylation (a and b). In addition, radiation alone increased GSK-3β-S9 phosphorylation and reduced GSK-3β protein abundance (c). Both radiation alone and combined injury reduced p53 protein levels (g and h). Wound alone had no significant effect on any of these protein levels when compared with the sham group (i to l). (* p<0.05, *** p<0.005, non-paired two-tailed t-test).
Figure 5.
Both radiation alone and combined injury increased AKT-S473 phosphorylation (a and b). In addition, radiation alone increased GSK-3β-S9 phosphorylation and reduced GSK-3β protein abundance (c). Both radiation alone and combined injury reduced p53 protein levels (g and h). Wound alone had no significant effect on any of these protein levels when compared with the sham group (i to l). (* p<0.05, *** p<0.005, non-paired two-tailed t-test).
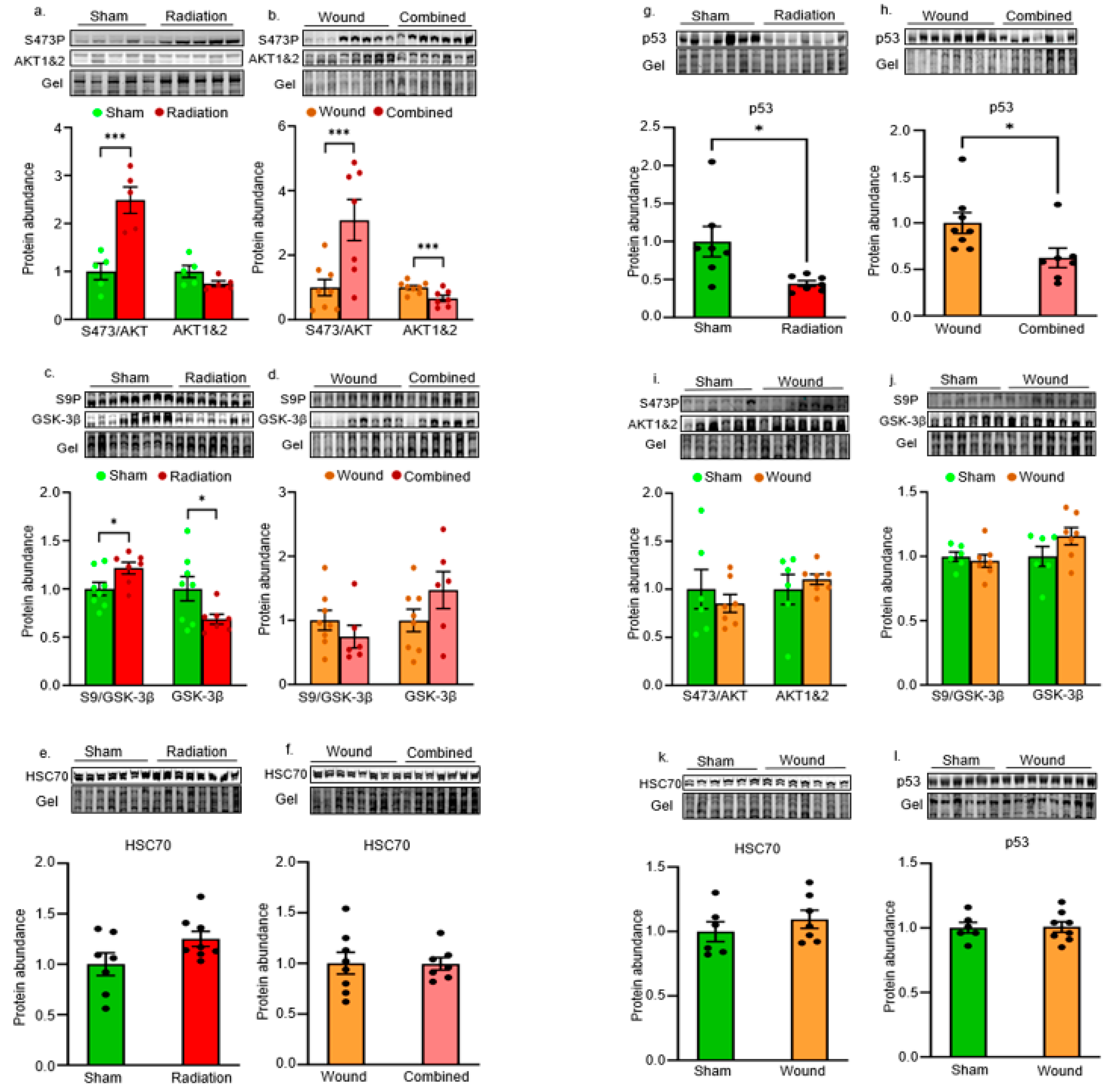
Figure 6.
The schema of molecular abnormalities preceding renal dysfunction following radiation and combined injuries.
Figure 6.
The schema of molecular abnormalities preceding renal dysfunction following radiation and combined injuries.
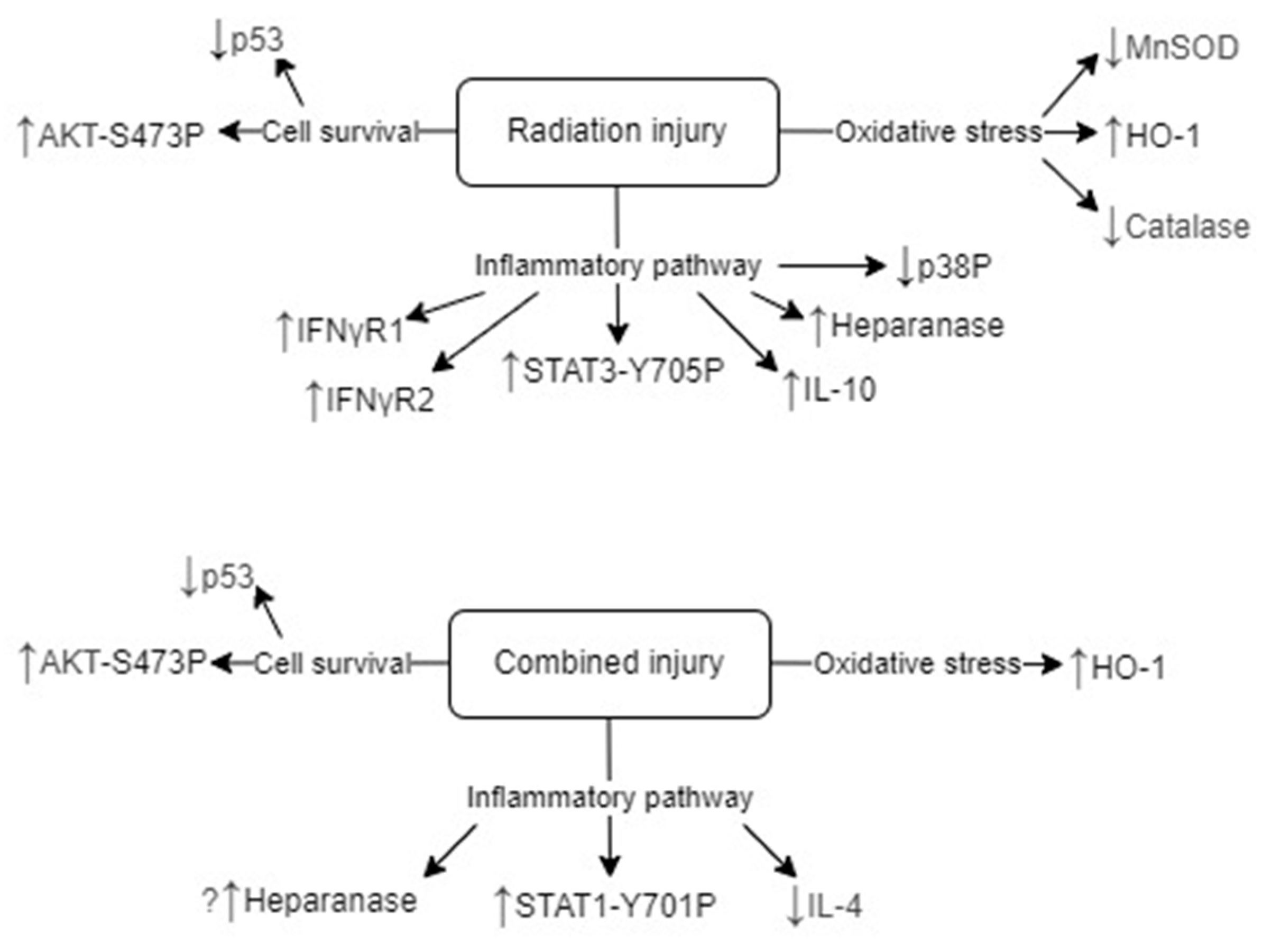
Figure 7.
The outline of animal procedures for the sham, radiation alone, wound alone, and combined injury groups.
Figure 7.
The outline of animal procedures for the sham, radiation alone, wound alone, and combined injury groups.
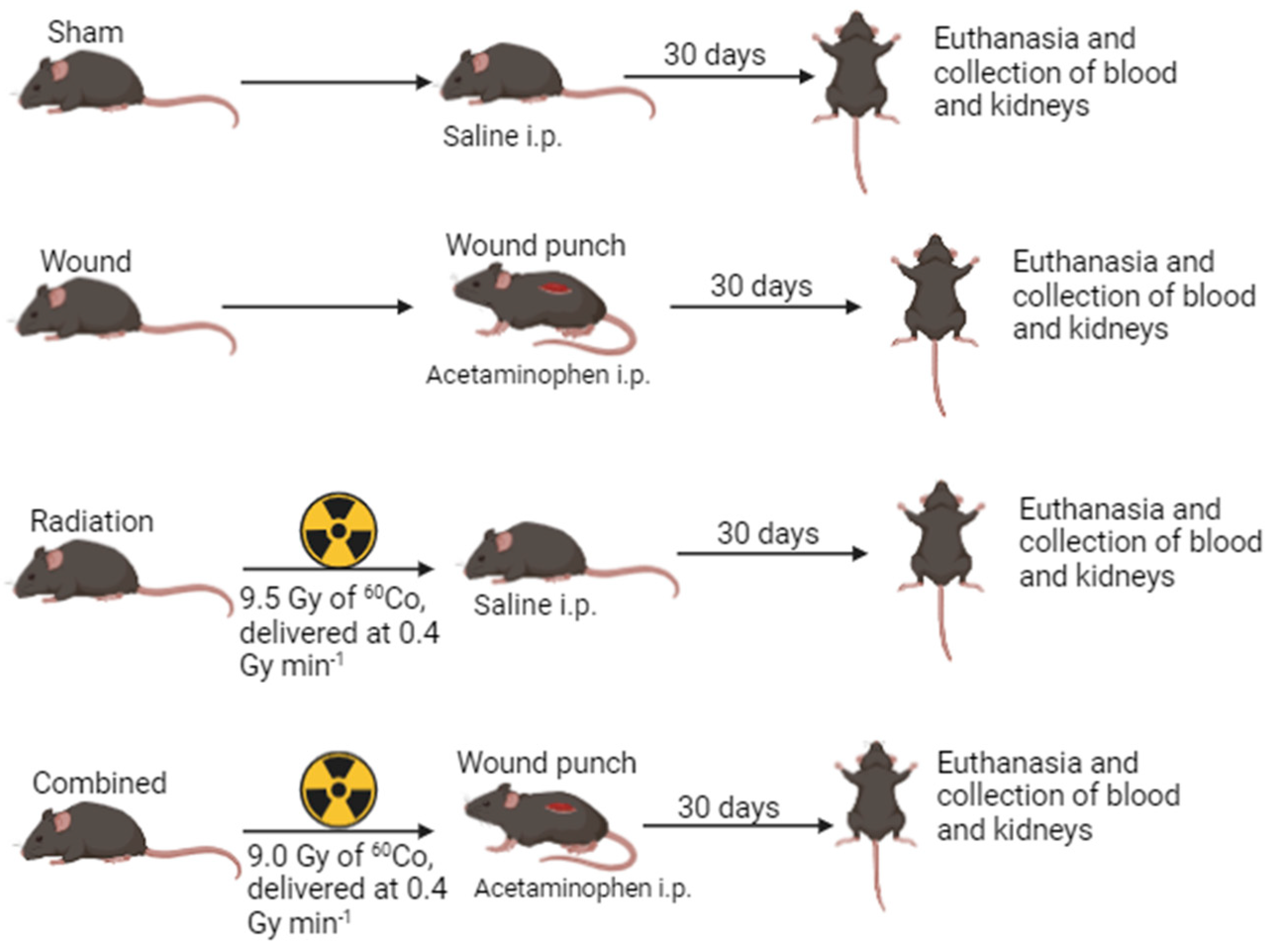

Table 1.
The list of primers.
| Gene names | Forward | Reverse |
|---|---|---|
| IL-6 | AGTGGCTAAGGACCAAGACC | ACCACAGTGAGGAATGTCCA |
| IFNγ | CTGCTGATGGGAGGAGATGT | CACATTCGAGTGCTGTCTGG |
| IFNγR1 | CGAAGCAGCAGAACAGGAAG | CGTCTTTGTGTCGGAGTTGG |
| IFNγR2 | GTTCCCTCGTCATACACTTCTC | CCTGTTCCTGTTGGGTTTCT |
| Heparanase | GCCTCGAGGGAAGACAGTTA | GCGATGCGTCCATTCAAGTA |
| CCL2 | AACTGCATCTGCCCTAAGGT | CTGTCACACTGGTCACTCCT |
| TNFα | AGCCCATATACCTGGGAGGA | TGGATGAACACCCATTCCCT |
| G-CSF | CAGCCTTCACTTCTGCCTTC | CTGGAAAGGGCTTTCTGCTC |
| IL-18 | CTTCTGCAACCTCCAGCATC | GTGAAGTCGGCCAAAGTTGT |
| IL-1β | ACTCATTGTGGCTGTGGAGA | AGCCTGTAGTGCAGTTGTCT |
| IL-8 | GCACCCAAACCGAAGTCATA | TCTGAACCAAGGGAGCTTCA |
| IL-10 | AGAGAAGCATGGCCCAGAAA | ACACCTTGGTCTTGGAGCTT |
| IL-4 | AACGAGGTCACAGGAGAAGG | CTGCAGCTCCATGAGAACAC |
| IL-13 | GGATTCCCTGACCAACATCTC | AGGGATGGTCTCTCCTCATT |
| TGFβ | CAGACATTCGGGAAGCAGTG | TTCCACATGTTGCTCCACAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



