
Submitted:
11 June 2024
Posted:
11 June 2024
You are already at the latest version
Abstract
Poor therapeutic outcomes have been attributed to inter-individual and interethnic variability in cytochrome P450 (CYP450)-dependent metabolism and altered drug retention associated with P-glycoprotein (P-gp). An individualized pharmacotherapeutic approach would benefit the genetically diverse South African population. The study aim was to develop a validated, targeted, analytical method to quantify seven probe drugs and their metabolites in dried blood spots (DBS) using the Mitra™ volumetric absorptive micro-sampling device for blood collection. An Agilent liquid chromatography (LC) system coupled to a Sciex 4000 QTRAP tandem mass spectrometer (MS) was used for method optimization and validation. Targeted LC-MS/MS methods were validated according to ICH guidelines. The validated LC-MS/MS method met the required bioanalytical standards for specificity, sensitivity, linearity, accuracy, precision, carry-over, and stability. This study successfully validated the use of DBS, collected with the Mitra™ microsampling device, to measure expected probe drug and metabolite concentrations using the “Geneva phenotyping cocktail” for the purpose of simultaneous phenotyping of in vivo CYP450 metabolic activity of the CYP1A2, -2B6, -2C9, -2C19, -2D6, and -3A4 enzymes, and P-gp transport activity. However, statistical analysis using Bland-Altman plots showed that not all analytes exhibited linear distribution pharmacokinetics between DBS and plasma, which influenced the predicted vs real plasma concentrations based on measurements in a DBS matrix. These findings were attributed to the blood-to-plasma concentration ratio, the physicochemical properties and stability of the analytes, and the extraction efficiency of the Mitra™ sampler. More research is required before DBS sampling can be substituted for conventional plasma sampling
Keywords:
Phenotyping
; Personalised medicine
; LCMSMS method validation
; Dried blood spot sampling
; CYP450 metabolism
; P-gp transport
1. Introduction
Individual pharmacokinetic variability in pharmacotherapy remains a problem in clinical practice contributing to adverse drug reactions, drug-drug interactions and therapeutic failure. [1,2] Inter-individual and interethnic variability in cytochrome P450 (CYP450)-dependent metabolism[3] and altered drug absorption via differentially expressed transport channels such as P-glycoprotein (P-gp)[4], contribute to poor therapeutic response. An individualised approach in therapeutic management would be beneficial in the South African population considering the country’s large genetic diversity[5]. There is a need for personalised pharmacotherapy to tailor drug treatment to individual patients, taking varied inter-individual response to drug therapy into account to ensure effective and safe treatment. Single time point, non-invasive capillary sampling, combined with a low dose probe drug cocktail (to simultaneously quantify in vivo drug and metabolite concentrations), would enhance the feasibility and cost-effectiveness of routine phenotyping in clinical practice and guide personalised prescribing for individual patients.[6,7,8] A recent development in dried blood spot (DBS) sampling is the Mitra™ device, using Volumetric Absorptive Micro Sampling (VAMS™) technology to collect an accurate volume (10-30 µL) of whole blood onto a hydrophilic polymeric tip as an alternative to plasma sampling.[9] Whole volume extraction from the polymeric tip further aims to overcome issues with sample homogeneity and haematocrit.[9] Small volume blood sampling however presents bioanalytical challenges in terms of the reproducibility and sensitivity of the quantitative method and the agreement between quantitative measurements from a DBS versus plasma sampling. Agreement between the two measurements will determine whether the DBS method using the Mitra™ device can replace plasma sampling in our South African clinical setting. Furthermore, the physicochemical diversity of structurally related aromatic probe drugs contained in a single drug cocktail, require optimised analytical procedures for simultaneous quantification.
The purpose of this study was to use the validated “Geneva phenotyping drug cocktail”[7] to develop a validated, targeted, analytical LC-MS/MS method to quantify the seven probe drugs and six respective metabolites in dried blood spots using the Mitra™ volumetric absorptive micro-sampling device for blood collection. The aim was to assess inter-method agreement of measured probe drug and metabolite concentrations between the low sample volume dried blood spot, and the conventional plasma sampling as recommended by the International Consortium Microsampling Working Group.[10]
The study was conducted in accordance with the Declaration of Helsinki and ethics approval was obtained from the University of Pretoria’s Faculty of Health Sciences Research Ethics Committee (ref. no. 209/2016) prior to conducting the study. Written informed consent was obtained from volunteers and confidentiality was maintained throughout.
2. Materials and Methods
2.1. Analytical Methods
2.1.1. Chemicals and Reagents
Reference standards caffeine (batch # BCBR6677V), bupropion as hydrochloric salt (batch # 063M4707V), flurbiprofen (batch # SLBD4598V), hydroxy-omeprazole (batch # BCBS0382V), dextromethorphan (batch # SLBQ0513V) and dextrorphan (batch # 065K3257) were purchased from Sigma Aldrich (Pty) Ltd. (Johannesburg, South Africa), paraxanthine (batch # FN11121501), hydroxy-bupropion (batch # FN0213150Z), omeprazole (batch # FN02201501) and α-hydroxy-midazolam (batch # FN02041502) from Cerilliant (Pty) Ltd. (Texas, USA) supplied by Sigma-Aldrich (Cambridge, UK) and 4-hydroxy-flurbiprofen (batch # CRC-0151-048-F) and fexofenadine (batch # S-FF-516) from Clearsynth (Pty) Ltd. (Mumbai, India). Midazolam (batch # F1058F03, Roche) was obtained as Dormicum™ 15 mg.3 mL-1 ampoules from a local hospital pharmacy. Internal standards (IS) imipramine (batch # 107K0697) for positive mode was supplied by Sigma Aldrich (Pty) Ltd. (Johannesburg, South Africa) and probenecid as European Pharmacopoeia standard by Cayman Chemicals (Ann Harbour, MI, USA).
All solvents used during sample preparation and chromatography were HPLC grade. Acetonitrile (Romil® purity >99.9%), methanol (Romil® purity >99.9%) and Romil® HPLC-water were purchased from Microsep (Pty) Ltd. (Johannesburg, South-Africa). Analytical grade formic acid (purity ≥98%), ammonium formate (batch # MKCF2569), were obtained from Sigma Aldrich (Pty) Ltd. (Johannesburg, South Africa). In-house double deionised pyrogen-free water (>18 MΩ and < 5 ppm TOC), used during sample preparation, was produced using an ELGA Genetics water purification unit (ELGA, Wycombe, UK) housed in the Department of Pharmacology.
2.1.2. Preparation of Stock and Working Standard Solutions
Standard stock solutions of all seven probe drugs, six CYP450 metabolites and IS at concentrations of 1 mg.mL-1 were prepared in methanol. Working standard solutions were diluting from stock solutions, in methanol (v/v), on each day of the validation and stored at -20ºC. The working standard concentrations were 20 µg.mL-1 and 2 µg.mL-1 for caffeine and flurbiprofen, 10 µg.mL-1 and 1 µg.mL-1 for paraxanthine, bupropion, hydroxy-bupropion, hydroxyflurbiprofen, omeprazole, hydroxy-omeprazole, dextromethorphan, dextrorphan and fexofenadine, 4 µg.mL-1 and 0.4 µg.mL-1 for midazolam and hydroxy-midazolam, taking into account the expected in vivo concentrations in human whole blood and plasma after administration of phenotyping doses.
2.1.3. Instrumentation
The LC-MS/MS system consisted of an Agilent 1100/1200 combined series HPLC system (Agilent Technologies, Palo Alto, CA, USA) coupled to an ABSciex 4000 triple quadrupole mass spectrometer, equipped with a Turbo-V® electrospray ionisation (ESI) source (Sciex, Concord, Canada). Analyst™ Software, version 1.5.2 (Sciex, Concord, Canada), was used to operate the system and manage the optimisation, data acquisition and perform the data analysis.
2.1.4. Mass Spectrometric Conditions
Optimised MS fragmentation source conditions were determined by infusing each analyte at a concentration of 500 ng.mL-1 directly into the ESI source of the Sciex 4000 QTrap mass spectrometer at a constant flow rate of 10 or 20 µL.min-1 using a Harvard syringe pump. The Turbo V™ (ESI) ionisation source parameters were optimised with a curtain gas pressure of 23 units, nebuliser gas (GS1) pressure of 35 units and auxiliary gas (GS2) pressure of 30 units. The ESI capillary potential was operated at -4500 V for negative mode and +5000 V for positive mode acquisitions respectively. The source temperature was 450ºC with an entrance potential ± 10 V. Final targeted MRM transitions and optimised conditions are given in Table 1.
2.1.5. Chromatographic Conditions
Separation was achieved on a Kinetex™ Biphenyl column (100 mm × 2.1 mm, 2.6 µm) with optimised binary gradient elution at a flow rate of 300 μL.min-1 and mobile phase composition A: H2O 0.1% formic acid and B: 80:20 MeOH: ACN 0.1% formic acid. The analyte mixture was dissolved in 10 mM NH4COOH 50:50 methanol: deionised water and prepared just prior to sample injection and made up freshly on each day of the validation. The sample injection volume for all injections was set at 10 µL. The total run time of the final acquisition methods was 9 minutes and 13 minutes for negative and positive mode respectively. Typical chromatograms with chromatographic methods and elution times of all analytes are depicted in Figure 1 A and B.
2.1.6. Human Plasma and Whole Blood Collection and Storage
Caffeine-free human blank whole blood was collected by individual venepuncture from healthy volunteers into four purple 4 mL ethylene-diamine-tetra acetic acid (EDTA) vacutainers for each volunteer. The whole blood was stored immediately at 4°C and used within four hours after collection to prepare blank and spiked dried blood spot samples for method validation. Blank plasma was obtained from separate EDTA tubes, collected from the same volunteers, by centrifugation (1500 xg for 15 minutes) at room temperature. Aliquoted human plasma samples were stored at -80°C, and were thawed at room temperature and vortex mixed before sample preparation and extraction.
2.1.7. Preparation of Calibration Standards for Validation in Plasma and Dried Blood Spots
Appropriate volumes from working standard analyte mixtures, containing all probe drugs and metabolites, were added to 2 mL Eppendorf tubes to prepare the calibration range for each analyte to cover the expected in vivo concentrations as follows : Caffeine and flurbiprofen 0.06 µg.mL-1 – 9.0 µg.mL-1; Paraxanthine, bupropion, hydroxy-bupropion, hydroxy-flurbiprofen, omeprazole, hydroxy-omeprazole, dextromethorphan, dextrorphan and fexofenadine 0.03 µg.mL-1 – 4.5 µg.mL-1; Midazolam and hydroxymedazolam 0.01 µg.mL-1 – 1.8 µg.mL-1. (Supplementary Table S1) The solvent was evaporated under vacuum at 30°C and reconstituted with 100 µL of thawed pooled plasma or whole blood followed by vortex mixing. To further prepare the whole blood samples, Mitra™ tips were used to volumetrically soak up exactly 20 µL of blank or spiked whole blood at an angle of 45°, taking care not to submerge the tip into the samples. The sample filled Mitra™ tips were allowed to air dry at room temperature for approximately 2 hours and then stored in closed containers at room temperature until extraction. For assessment of matrix effects samples were spiked post extraction at three separate concentration levels (Supplementary Table S2).
2.1.8. Extraction from Plasma and DBS Mitra Tips
The plasma extraction was performed by a 3-step protein precipitation method. A 100 µL of acetonitrile was added to plasma samples, followed by sonication for 15 minutes and 5 minutes vortex mixing. The sonication and vortex mixing steps were repeated twice more after further additions of 100 µL acetonitrile each. After protein precipitation the samples were centrifuged at 14,000 xg for 10 minutes to remove all precipitated plasma proteins. A 100 µL aliquot of the supernatant was transferred, with an Eppendorf pipette, to glass tapered autosampler vial inserts in 2 mL amber glass LC vials and diluted with 100 µL of a 10 mM ammonium formate aqueous solution that contained the IS for positive and negative ESI mode acquisition respectively, just prior to the LC-MS/MS analysis. The same procedure was followed for blank and spiked solvent samples.
The spiked and blank sample filled Mitra™ tips were inserted into clean 2 mL Eppendorf vials and analytes extracted with an organic 1:1 mixture of methanol and acetonitrile according to the method by Ye and Gao[11] reported to allow better extraction of hydrophobic analytes with a more consistent recovery across a wide haematocrit range. The extraction was carried out in two steps by adding 100 µL of a 1:1 methanol: acetonitrile mixture to the Eppendorf vial containing the Mitra™ tip followed by ultrasonication for 30 minutes and vortex mixing at maximum rpm for another 15 minutes. The sonication and vortex mixing steps were repeated once more after addition of another 100 µL of the 1:1 methanol: acetonitrile mixture. The final extracts were combined and a 100 µL aliquot of the supernatant transferred into tapered glass autosampler vial inserts in 2 mL amber glass autosampler vials and diluted with 100 µL of aqueous 10 mM ammonium formate solution containing IS just prior to the LC-MS/MS analysis.
2.1.9. LCMSMS Method Validation
The analytical method was validated according to ICH guidelines[12] for matrix effects, recovery, linearity, limits of quantitation and detection, carry-over, inter and intraday precision and accuracy and analyte stability in plasma and DBS matrixes. Relative matrix effects were evaluated from three batches on different days at three different concentration levels. The signal response of each individual analyte in post extracted matrix was compared to pure solvent spiked at the same concentration level. Recovery was assessed at three different concentration levels on three separate days, injected in triplicate and the mean analyte responses calculated for all analytes measured in DBS and plasma compared to the mean analyte responses obtained in solvent. The average recovery over three days was assessed as the percentage ratio of the experimentally determined calibration slopes in both matrixes and their corresponding solvents, spiked before extraction, and the precision calculated (% CV). The linearity of the method, over the expected concentration ranges, was determined using the quantitation function in Analyst® 1.5.2 software. Post-extracted spiked plasma and DBS samples were injected in triplicate for LC-MS/MS analysis and repeated on three separate days. Matrix matched calibration curves were plotted as the ratio of each analyte peak area versus the internal standard peak area as a function of the analyte concentration using a weighted linear regression function (of 1/x) to account for the heteroscedasticity of the data in the lower concentration range. The LOD was estimated at three times the average signal to baseline noise ratio and LOQ at 10 times the average signal to baseline noise ratio at the analyte retention time measured when injecting a blank matrix extracted sample. Carry-over was assessed by injecting two blank samples after the highest concentration of standard in matrix or solvent and determining the average analyte peak areas of each analyte present in the second blank sample. The intraday and interday accuracy and precision was assessed from triplicate injections on the same day and three separate days respectively over the entire calibration range. Analyte stability was assessed under different storage and sample preparation conditions, spiked into methanol alone, 1:1 methanol: water containing 0.1% formic acid (pH ±2.6), 1:1 methanol: water containing 0.01% acetic acid (pH ± 4) and 1:1 methanol: water containing 0.025% ammonium formate (pH ± 8). Short term analyte stability was investigated at room temperature over a 6-hour run time, at -20°C for one month and for three freeze thaw cycles.
2.2. Pilot Pharmacokinetic Study in a Healthy Volunteer
As an initial proof of concept, a “home-made” phenotyping cocktail was given to a healthy volunteer followed by an in vivo pharmacokinetic study with sparse plasma and DBS sampling using the Mitra™ device for whole blood collection. Simultaneous venous (using EDTA vacutainers) and capillary sampling was done at baseline and at 1 hour, 2.5 hours, 3 hours and 3.5 hours after oral administration. Plasma was obtained by centrifugation (1500 xg for 15 minutes) of venous blood at room temperature within one hour after collection. The same sample storage, preparation and extraction procedures as described above were followed before LC-MS/MS analysis using the validated method.
2.3. Statistical Analysis of Agreement between DBS Collected with the Mitra™ Device and Plasma Sampling
Variability in the degree of affinity for erythrocyte and plasma protein binding sites and factors affecting the affinity was estimated in vitro from whole blood, collected from a representative study population as recommended by the Microsampling Working Group[10] Blood samples collected from healthy caffeine-shy volunteers were pooled and used in all experiments to eliminate, as best possible, any potential haematocrit bias in the dried blood spots. Whole blood calibration curves, from low volume dried blood spots, were used to predict the analyte concentration in plasma, using the linear regression equation, at the same expected (spiked) concentration levels. The percentage difference between the predicted values and the true average observed values were calculated with Excel and plotted against the true average values observed for the spiked plasma samples, using GraphPad prism version 8.0.2 statistical software for Windows (GraphPad Software, San Diego, California USA, www.graphpad.com). Variation within 20% of the true average observed values indicated agreement between the two sampling methods and possibly linear pharmacokinetics between the analyte concentrations obtained from the Mitra™ sampling device and plasma. A variation of more than 20% would indicate that a non-linear correlation exists between the concentration of the analyte in plasma and DBS.[10]
3. Results and Discussion
3.1. LCMSMS Method Validation
3.1.1. Evaluation of Matrix Effects
Ionisation suppression or enhancement were analyte specific with relative matrix effects between 90 and 110% for most analytes in post extracted plasma and DBS matrixes. This was with the exception of bupropion and hydroxy-bupropion where the ionisation suppression was more than 10% in both plasma and dried blood spots with precision between 14.01 and 23.66%, and the latter variance observed at the lowest concentration level in the DBS matrix. (Supplementary Table S3) Flurbiprofen and hydroxyflurbiprofen also showed ionisation suppression with matrix effects suppressing the peak areas to 64.32 and 71.12% respectively at the lowest spiked concentration, confirming findings by Matuszewski et al.[13] Spiked matrix-matched calibration curves, prepared from pooled population samples, were used to compensate for problems associated with matrix effects during method development and assessment of inter-method agreement of sampling methods.
3.1.2. Evaluation of Recovery from Plasma and Dried Blood Spots
The absolute recovery of all analytes (Table 2) consistently ranged from 97.35 to 113.73% in plasma with the coefficient of variation (CV) ≤ 14.16%. while mean recoveries from DBS across the three days were more inconsistent (CV 26.38% for caffeine; 30.11% for paraxanthine; 17.90% for dextromethorphan; 18.68% for dextrorphan; and 18.60% for midazolam). The positive recovery bias seen for caffeine and paraxanthine was consistent with prior literature.[14]
3.1.3. Linearity, LOD, LLOQ
Method linearity was established with analyte specific concentration ranges that fell within the expected in vivo concentrations for each analyte following the administered phenotyping cocktail dosages. The coefficient of determination (r2) for the calibration curves and the estimated LOD and LLOQ are shown in Table 2. The coefficient of determination exceeded 0.9936 for human plasma and 0.9929 for DBS.
3.1.4. Carry-Over
The carry-over did not exceed 20% of the lower limit of quantification when comparing the % area of a blank injection to that of the LLOQ after the highest concentration sample injection (Supplementary Figure S1).
3.1.4. Intra- and Inter-Day Precision and Accuracy
The intra-day accuracy and precision for each analyte were within the acceptable bias of 15% above and 20% below the LLOQ for plasma and DBS (Supplementary Tables S4 and S5). Inter-day variability calculated from separate working standard solutions over three non-consecutive days were acceptable: Plasma matrix accuracy between 91.08 – 105.99% for all analytes with precision less than 12.07%; and between 92.55 – 105.48% in DBS with precision less than 13.03% at higher concentration levels (Table 3).
3.1.5. Analyte Stability
As expected, the pH of the solution in the sample vial had an influence on the analyte signal intensities corresponding to the degree of ionisation, however this consistently showed a % CV between 0.6 – 6.9% for all analytes except omeprazole and its metabolite. Short term analyte stability at room temperature in the sample vial showed an average reduction of 86.3% (at pH ± 4) and 95.5% (at pH ±2.6) in the peak area of omeprazole over a period of 6 hours. A similar decrease in analyte signal was observed for hydroxy-omeprazole, with 59.9% (at pH ± 4) to 96.0% (at pH ±2.6). Both omeprazole and hydroxy-omeprazole remained stable at higher pH conditions, corresponding to physiological pH, in the sample vial. When the analyte mixtures were stored at -20°C for one month, the variation in the measured analyte peak areas was less than 15% for all analytes when stored in methanol. The analyte peak area of bupropion, when kept at room temperature for one month, decreased by between 19.8 and 28.3% and confirmed the findings of Bosilkovska et al.[15] Stability after 3 freeze-thaw cycles are depicted in Table 4. Results indicate that most analytes were stable at higher concentration levels in both matrixes, with the accuracy between 85% – 115%. Flurbiprofen showed a positive bias slightly higher than 15% in plasma at all QC levels and at higher concentrations in DBS, with a variance greater than 20% at the lowest spiked QC concentration. Analyte stability might be increased by extraction from the Mitra™ sampler shortly after sample collection and storage of extracted samples at -80°C before further analysis.[16]
3.2. Pilot Pharmacokinetic Study in a Healthy Volunteer
The validated LC-MS/MS method was adequate for phenotypic profiling and in combination with optimised analytical extraction protocols, was able to determine inter-individual variability to infer metabolic activity or drug transport activity, from a small-volume, biological DBS sample, after administration of a low dose “home-made” phenotyping cocktail. Extracted ion chromatograms at different PK sampling times are shown in Figure 2.
3.3. Assessment of Inter-Method Agreement between Dried Blood Spots and Plasma Sampling
The agreement between predicted and measured plasma and dried blood spot concentrations consistently fell within a 20% range for lipophilic analytes over three separate days, indicating fair agreement between the two sampling methods. These lipophilic analytes have log P values between 2.48 and 3.49 and are expected to be highly protein bound. Conversely, the DBS extraction conditions resulted in altered amounts of caffeine, paraxanthine, dextrorphan and bupropion, flurbiprofen, omeprazole and their hydroxylated metabolites liberated from the Mitra™ sampler. This indicated that blood cell distribution kinetics are regulated not only by the blood-to-plasma concentration ratio, but also by the physicochemical properties of the analytes, extraction temperature, total analyte concentration, analyte stability and time dependent equilibrium between different blood compartments. These factors most likely confounded the extent to which the extraction procedure liberated bound drug from either plasma proteins or erythrocytes for these analytes. The plasma concentrations of caffeine and its metabolite paraxanthine (Supplementary Figure S2), hydroxy-flurbiprofen metabolite, omeprazole and hydroxy-omeprazole (Supplementary Figure S3) indicated agreement between the two sampling methods within the acceptable 20% variation range at higher extraction temperature and sonication time, however a DBS calibration curve underestimated the plasma concentration for flurbiprofen at the same conditions and bupropion was unstable at high extraction temperature, which was consistent with literature.[17]
his study successfully validated the use of DBS, collected with the volumetrically controlled absorptive microsampling device Mitra™, to measure expected probe drug and metabolite concentrations using the “Geneva phenotyping cocktail”. The validated method met all the required standards accepted in bioanalytical chemistry for specificity, sensitivity, linearity, accuracy, precision, carry-over and stability. There are, however, a number of important factors that require careful consideration before agreement between in vivo plasma and DBS sampling with the Mitra™ sampler can be assessed. Analyte blood-to plasma ratios are governed by blood cell distribution kinetics, which in turn could be influenced by analyte concentrations, the time it takes to reach the erythrocyte-to-plasma equilibrium and temperature.[18] Acidic analytes are not expected to have a high affinity for protein components on or in erythrocytes, whereas the basic pharmaceuticals would be in their ionised state at physiological pH with a greater affinity for acidic phospholipids, carbonic anhydrase and intracellular cyclophilin.[19] Variability in the degree of affinity for erythrocyte and plasma protein binding sites and factors affecting the affinity should be estimated in vitro from whole blood, collected from a representative study population. This is relevant to any study aimed at using DBS to assess in vivo drug exposure, especially with respect to reliability and agreement between DBS and plasma concentrations. Another important consideration is the extent to which the extraction procedure liberates bound drug from either plasma proteins or erythrocytes. Different matrices and extraction protocols may result in a varied amount of drug being liberated from plasma proteins potentially influencing the measured concentration. It was evident from the initial in vitro assessment of agreement that blood cell distribution kinetics are regulated not only by the blood-to-plasma concentration ratio, but also by the physicochemical properties of the analytes, extraction temperature, total analyte concentration, analyte stability and time-dependent equilibrium between different blood compartments. Previous studies have confirmed the importance of optimising the extraction procedure, conditions and solvents.[11] The proposed relationship between analyte pKa and Log P and blood cell distribution with their hypothesised influence on predicted vs real plasma concentrations based on measurements in DBS matrix, is illustrated in Figure 3. In the case of more acidic compounds like flurbiprofen and hydroxy-flurbiprofen, that are not expected to have high affinity for erythrocytes, whole blood would show a lower concentration due to the reduced amount of plasma in the total volume collected and analysed, compared to the harvested plasma where erythrocytes are absent. When recovery from the DBS was optimised, the DBS calibration curve still underestimated the plasma concentration of flurbiprofen. Basic pharmaceuticals are however in their ionised state at physiological pH, which influences their binding affinity for erythrocytes. The measured concentration in plasma would thus be lower than in whole blood since whole blood would have analytes bound to the erythrocytes. A calibration curve developed from DBS would in theory over-estimate the concentration of the drug in plasma, which was observed for dextromethorphan and dextrorphan in this study. In addition to the differences in erythrocyte affinity seen for acidic versus basic pharmaceuticals, the lipophilicity of the analytes also contributed to the blood cell distribution kinetics in vitro. Fexofenadine, midazolam and hydroxy-midazolam are hydrophobic and have higher plasma protein binding than the more hydrophilic analytes caffeine and paraxanthine. Hydrophilic and polar analytes are expected to show poor plasma protein binding and usually do not enter erythrocytes. While caffeine does enter erythrocytes, it does not bind to the cell constituents implying highly correlated concentrations of the drug measured in both whole blood and plasma. This could explain why the harsher DBS extraction methods did not influence predicted vs measured plasma concentration in the case of fexofenadine, midazolam and hydroxy-midazolam since the high proportion of these drugs were bound to plasma proteins and were liberated under the optimised plasma extraction method. Conclusive data on the extent to which analytes bind to protein components in or on erythrocytes or to plasma proteins in an in vitro environment are lacking and might influence quantitative in-vitro-in-vivo correlation. Caution should therefore be exercised when predicting plasma concentration using DBS calibration curves, as these are highly dependent on the extraction protocol, where the resultant recovery of different analytes using different conditions may be affected by the matrix.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
The National Research Foundation (NRF) of South Africa and the Department of Pharmacology of the University of Pretoria for funding the mass spectrometer instrumentation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Johansson, I. and M. Ingelman-Sundberg, Genetic polymorphism and toxicology—with emphasis on cytochrome p450. Toxicological sciences, 2010. 120(1): p. 1-13. [CrossRef]
- Patel, T.K. and P.B. Patel, Mortality among patients due to adverse drug reactions that lead to hospitalization: a meta-analysis. European journal of clinical pharmacology, 2018. 74(6): p. 819-832. [CrossRef]
- Samer, C.F., et al., Applications of CYP450 testing in the clinical setting. Molecular diagnosis & therapy, 2013. 17(3): p. 165-184.
- Giacomini, K.M., et al., Membrane transporters in drug development. Nature reviews Drug discovery, 2010. 9(3): p. 215-236. [CrossRef]
- Rajman, I., et al., African genetic diversity: implications for cytochrome P450-mediated drug metabolism and drug development. EBioMedicine, 2017. 17: p. 67-74.
- Boer, T., et al., Application of dried blood spot sampling combined with LC-MS/MS for genotyping and phenotyping of CYP450 enzymes in healthy volunteers. Biomedical Chromatography, 2011. 25(10): p. 1112-1123.
- Bosilkovska, M., et al., Geneva cocktail for Cytochrome P450 and P-Glycoprotein activity assessment using dried blood spots. Clinical Pharmacology & Therapeutics, 2014. 96(3): p. 349-359.
- Donzelli, M., et al., The Basel cocktail for simultaneous phenotyping of human cytochrome P450 isoforms in plasma, saliva and dried blood spots. Clinical pharmacokinetics, 2014. 53(3): p. 271-282.
- Denniff, P. and N. Spooner, Volumetric absorptive microsampling: a dried sample collection technique for quantitative bioanalysis. Analytical chemistry, 2014. 86(16): p. 8489-8495.
- Evans, C., et al., Implementing dried blood spot sampling for clinical pharmacokinetic determinations: considerations from the IQ Consortium Microsampling Working Group. 2015, Springer.
- Ye, Z. and H. Gao, Evaluation of sample extraction methods for minimizing hematocrit effect on whole blood analysis with volumetric absorptive microsampling. Bioanalysis, 2017. 9(4): p. 349-357.
- Guideline, I.H.T. Validation of analytical procedures: text and methodology Q2 (R1). in International conference on harmonization, Geneva, Switzerland. 2005.
- Matuszewski, B., Standard line slopes as a measure of a relative matrix effect in quantitative HPLC–MS bioanalysis. Journal of Chromatography B, 2006. 830(2): p. 293-300. [CrossRef]
- De Kesel, P.M. E. Lambert, and C.P. Stove, Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Analytica chimica acta, 2015. 881: p. 65-73.
- Bosilkovska, M., et al., Simultaneous LC-MS/MS quantification of P-glycoprotein and cytochrome P450 probe substrates and their metabolites in DBS and plasma. Bioanalysis, 2014. 6(2): p. 151-164.
- Kok, M.G.and M. Fillet, Volumetric absorptive microsampling: current advances and applications. Journal of pharmaceutical and biomedical analysis, 2017. [CrossRef]
- O’byrne, P.M., et al., The aqueous stability of bupropion. Journal of pharmaceutical and biomedical analysis, 2010. 53(3): p. 376-381.
- Rowland, M. and G.T. Emmons, Use of dried blood spots in drug development: pharmacokinetic considerations. The AAPS journal, 2010. 12(3): p. 290-293.
- Emmons, G. and M. Rowland, Pharmacokinetic considerations as to when to use dried blood spot sampling. Bioanalysis, 2010. 2(11): p. 1791-6.
Figure 1.
A. Chromatogram obtained with scheduled MRM acquisition method for analytes in negative ESI mode. Separation was achieved on a Kinetex™ Biphenyl column (100 mm × 2.1 mm, 2.6 µm) with a flow rate of 300 μL.min-1 and mobile phase composition A: H2O 0.1% formic acid and B: 80:20 MeOH: ACN 0.1% formic acid with 10 mM ammonium formate in the sample vial. The elution order, with average retention times and % CV was: 1 – hydroxyflurbiprofen, 5.94 min. (0.21%); 2 – internal standard probenecid, 6.23 min. (0.15%) monitoring two MRM transitions; 3 – flurbiprofen, 6.45 min. (0.15%).
Figure 1.
A. Chromatogram obtained with scheduled MRM acquisition method for analytes in negative ESI mode. Separation was achieved on a Kinetex™ Biphenyl column (100 mm × 2.1 mm, 2.6 µm) with a flow rate of 300 μL.min-1 and mobile phase composition A: H2O 0.1% formic acid and B: 80:20 MeOH: ACN 0.1% formic acid with 10 mM ammonium formate in the sample vial. The elution order, with average retention times and % CV was: 1 – hydroxyflurbiprofen, 5.94 min. (0.21%); 2 – internal standard probenecid, 6.23 min. (0.15%) monitoring two MRM transitions; 3 – flurbiprofen, 6.45 min. (0.15%).
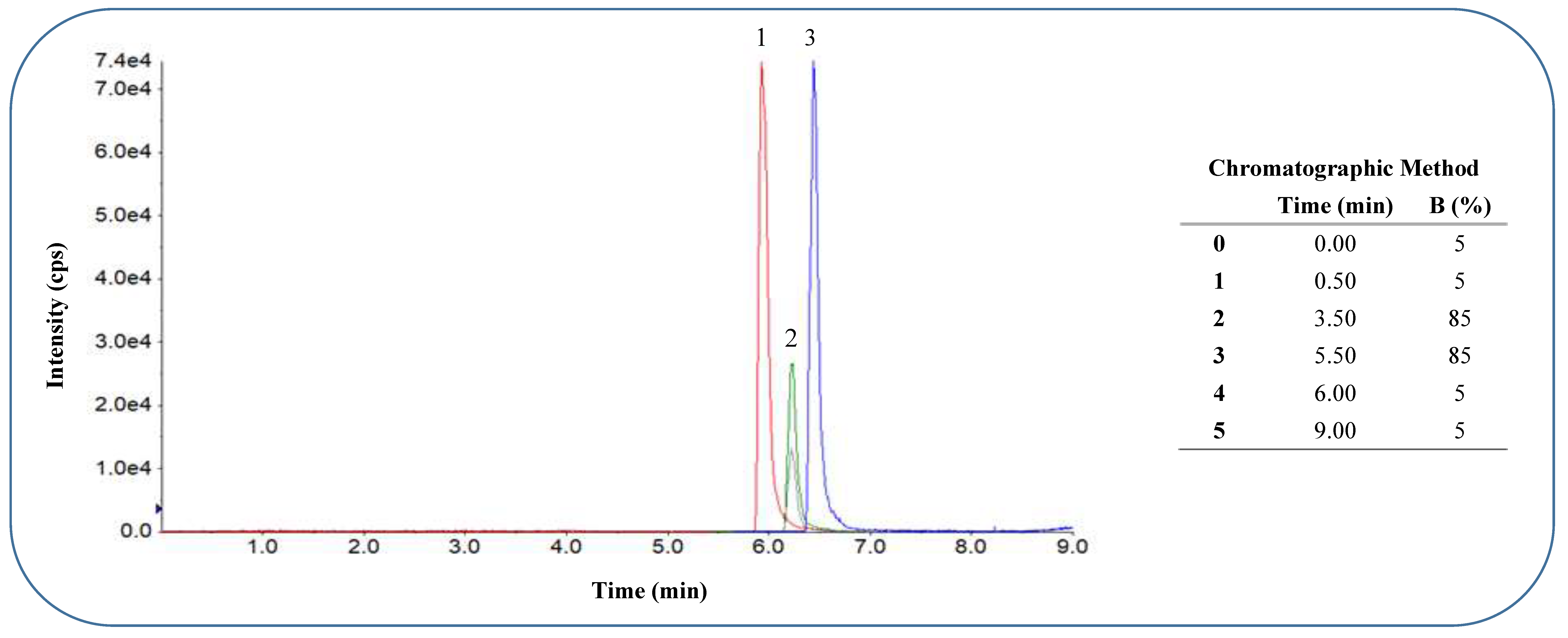
Figure 1.
B: Chromatogram obtained with scheduled MRM acquisition method for analytes in positive ESI mode at optimised elution gradient. Separation was achieved on a Kinetex™ Biphenyl column (100 mm × 2.1 mm, 2.6 µm) with a flow rate of 300 μL.min-1 and mobile phase composition A: H2O 0.1% formic acid and B: 80:20 MeOH: ACN 0.1% formic acid with 10 mM ammonium formate in the sample vial. The elution order with average retention times and % CV was: 1 – paraxanthine 3.83 min. (0.45%), 2 – caffeine 4.99 min. (0.35%), 3 – hydroxy-bupropion 5.79 min. (0.25%), 4 – dextrorphan 5.93 min. (0.36%), 5 – bupropion 7.16 min. (0.30%), 6 – hydroxy-omeprazole 7.32 min. (0.21%), 7 – omeprazole 8.30 min. (0.18%), 8 – dextromethorphan (two MRM transitions) 8.88 min. (0.19%), 9 – midazolam 9.04 min. (0.19%), 10 – hydroxymidazolam 9.18 min. (0.15%), 11 – IS imipramine 10.18 min. (0.24%), 12 – fexofenadine 11.13 min. (0.29%).
Figure 1.
B: Chromatogram obtained with scheduled MRM acquisition method for analytes in positive ESI mode at optimised elution gradient. Separation was achieved on a Kinetex™ Biphenyl column (100 mm × 2.1 mm, 2.6 µm) with a flow rate of 300 μL.min-1 and mobile phase composition A: H2O 0.1% formic acid and B: 80:20 MeOH: ACN 0.1% formic acid with 10 mM ammonium formate in the sample vial. The elution order with average retention times and % CV was: 1 – paraxanthine 3.83 min. (0.45%), 2 – caffeine 4.99 min. (0.35%), 3 – hydroxy-bupropion 5.79 min. (0.25%), 4 – dextrorphan 5.93 min. (0.36%), 5 – bupropion 7.16 min. (0.30%), 6 – hydroxy-omeprazole 7.32 min. (0.21%), 7 – omeprazole 8.30 min. (0.18%), 8 – dextromethorphan (two MRM transitions) 8.88 min. (0.19%), 9 – midazolam 9.04 min. (0.19%), 10 – hydroxymidazolam 9.18 min. (0.15%), 11 – IS imipramine 10.18 min. (0.24%), 12 – fexofenadine 11.13 min. (0.29%).
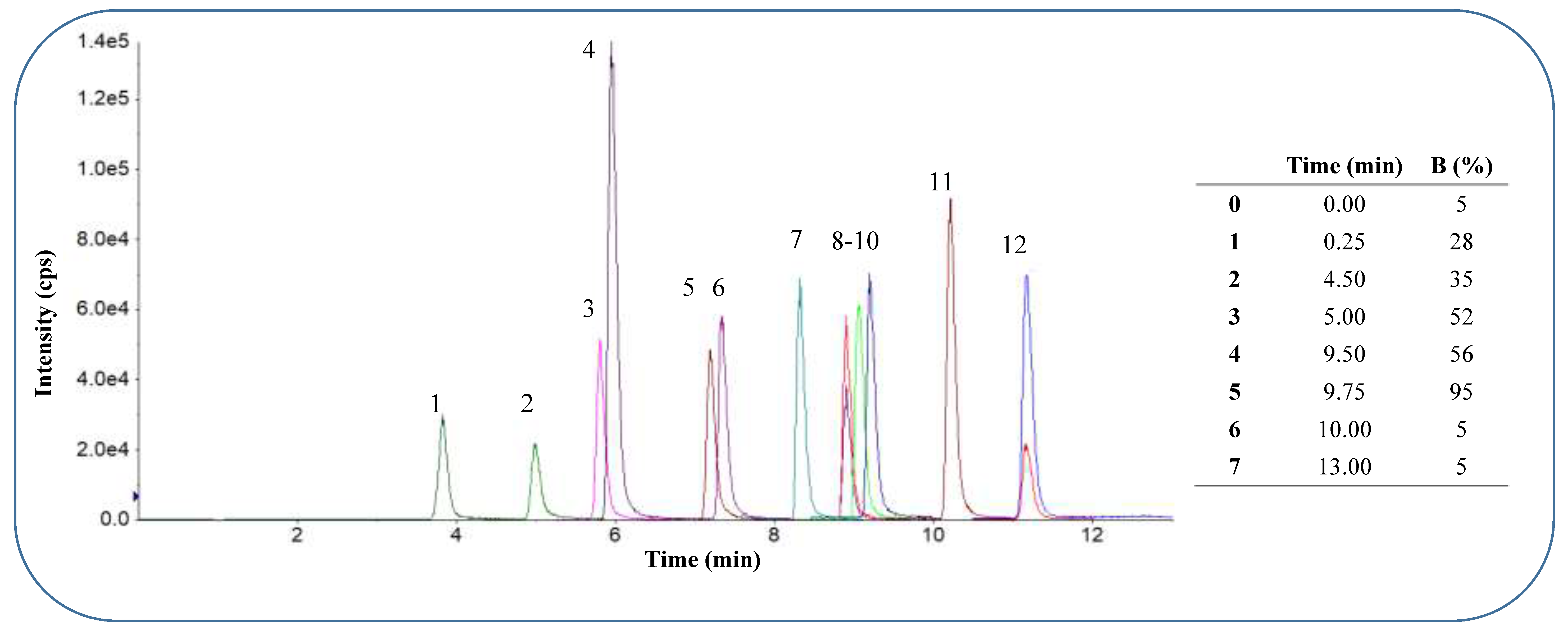
Figure 2.
Chromatograms obtained with a scheduled MRM acquisition method for analytes in positive ESI mode after oral administration of “home-made” phenotyping cocktail with plasma sampling (A) and DBS sampling (B) using the Mitra microsampling device. A1 and B1 – paraxanthine and caffeine 2.5 hours post dose; A2 and B2 – hydroxy-bupropion and bupropion 3.5 hours post dose; A3 and B3 – dextrorphan, hydroxy-omeprazole, omeprazole, dextromethorphan, hydroxy-midazolam, midazolam and fexofenadine 3 hours post dose.4. Discussion.
Figure 2.
Chromatograms obtained with a scheduled MRM acquisition method for analytes in positive ESI mode after oral administration of “home-made” phenotyping cocktail with plasma sampling (A) and DBS sampling (B) using the Mitra microsampling device. A1 and B1 – paraxanthine and caffeine 2.5 hours post dose; A2 and B2 – hydroxy-bupropion and bupropion 3.5 hours post dose; A3 and B3 – dextrorphan, hydroxy-omeprazole, omeprazole, dextromethorphan, hydroxy-midazolam, midazolam and fexofenadine 3 hours post dose.4. Discussion.
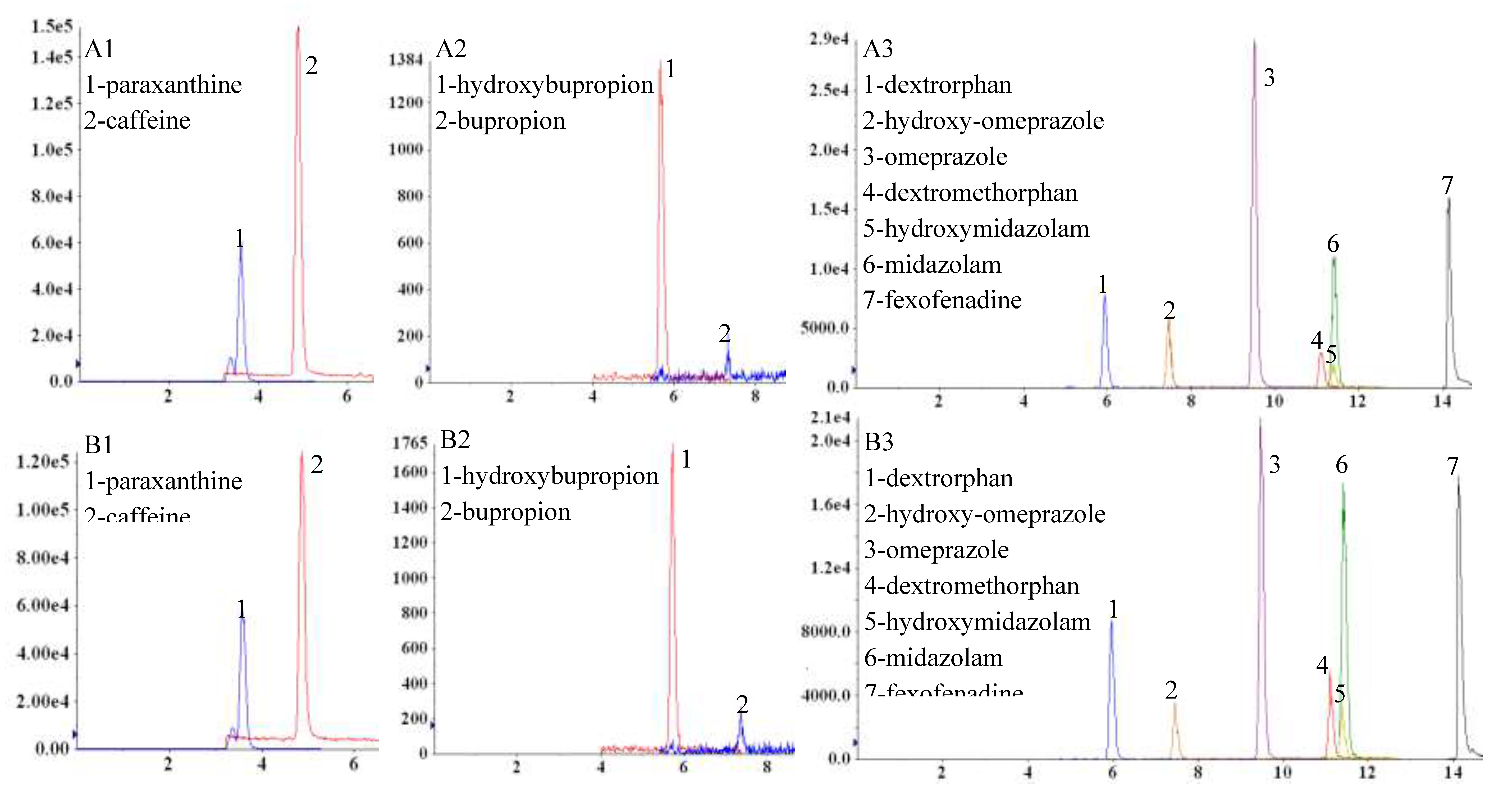
Figure 3.
Proposed relationship between analyte pKa and Log P and blood cell distribution with their hypothesised influence on predicted vs real plasma concentrations based on measurements in DBS matrix (Illustration created by the authors).
Figure 3.
Proposed relationship between analyte pKa and Log P and blood cell distribution with their hypothesised influence on predicted vs real plasma concentrations based on measurements in DBS matrix (Illustration created by the authors).
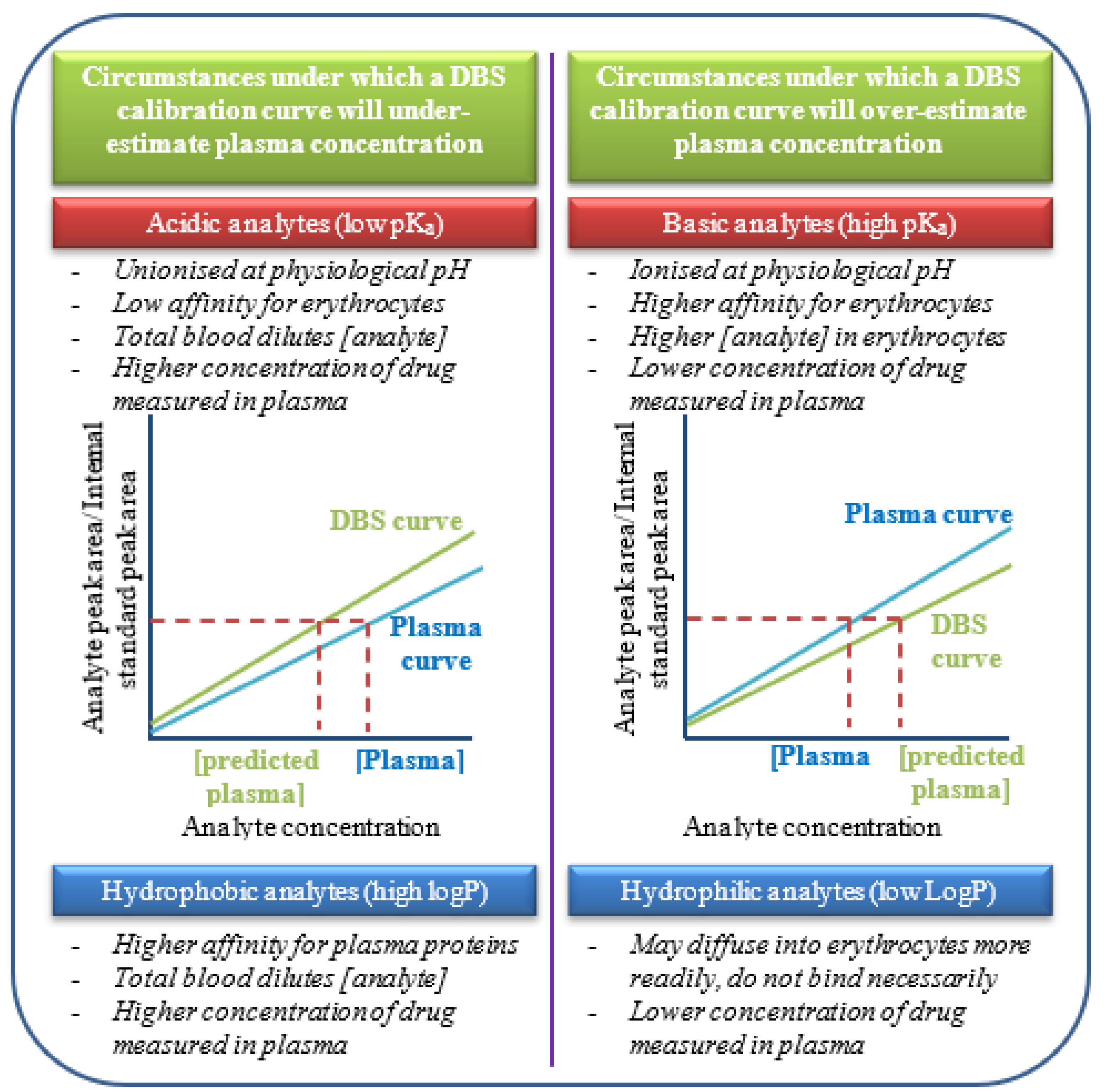

Table 1.
Optimised mass spectrometric fragmentation parameters.
| Negative ionisation mode [M – H] - | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | CYP probe | Q1 (m/z) | Q3 (m/z) | DP (V) | CE (V) | CXP (V) | |||||
| Flurbiprofen | CYP2C9 | 243.20 | 199.30 | -12 | -14 | -12 | |||||
| Hydroxyflurbiprofen | 259.20 | 215.20 | -40 | -11 | -4 | ||||||
| Internal Standard | |||||||||||
| Probenecid | 284.30 | 240.30 | -50 | -24 | -10 | ||||||
| Positive ionisation mode [M+H] + | |||||||||||
| Fexofenadine | P-gp | 502.7 | 466.6; 484.7 | 100 | 38 | 12 | |||||
| Caffeine | CYP1A2 | 195.3 | 138.2 | 20 | 25 | 6 | |||||
| Paraxanthine | 181.1 | 124.2 | 70 | 27 | 5 | ||||||
| Bupropion | CYP2B6 | 240.4 | 131.3 | 20 | 50 | 11 | |||||
| Hydroxybupropion | 256.4 | 103.1 | 50 | 52 | 3 | ||||||
| Omeprazole | CYP2C19 | 346.3 | 198.1 | 25 | 30 | 10 | |||||
| Hydroxyomeprazole | 362.1 | 214.4 | 50 | 15 | 10 | ||||||
| Dextromethorphan | CYP2D6 | 272.4 | 147.4; 171.5 | 90 | 50 | 10 | |||||
| Dextrorphan | 258.4 | 157.2 | 80 | 45 | 4 | ||||||
| Midazolam | CYP3A4 | 326.3 | 291.4 | 80 | 35 | 14 | |||||
| Hydroxymidazolam | 342.2 | 324.1 | 89 | 29 | 18 | ||||||
| Internal Standard | |||||||||||
| Imipramine | 281.5 | 86.1 | 50 | 50 | 10 | ||||||
P-gp = permeability glycoprotein; CYP = Cytochrome P450; m/z – mass to charge ratio; DP = declustering potential in volts; CE = collision energy in volts; collision cell exit potential in volts.
Table 2.
Linearity (coefficient of determination r2), LOD, LLOQ and relative recovery of all analytes in DBS and plasma.
Table 2.
Linearity (coefficient of determination r2), LOD, LLOQ and relative recovery of all analytes in DBS and plasma.
| Analyte | Human plasma | Dried Blood Spots (Mitra™) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Linearity (r2) | LOD (ng.mL-1) | LLOQ (ng.mL-1) |
Recovery (%) (CV%) | Linearity (r2) | LOD (ng.mL-1) |
LLOQ (ng.mL-1) |
Recovery (%) (CV%) | |||
| FEX | 0.9997 | 0.86 | 2.86 | 106.99 | (6.76) | 0.9994 | 1.02 | 3.41 | 100.16 | (13.88) |
| CAF | 0.9958 | 9.53 | 31.78 | 97.35 | (11.46) | 0.9971 | 8.65 | 28.85 | 118.23 | (26.38) |
| PAR | 0.9973 | 1.76 | 5.86 | 102.23 | (11.44) | 0.9929 | 1.45 | 4.84 | 116.30 | (30.11) |
| BUP | 0.9990 | 0.62 | 2.07 | 98.86 | (7.20) | 0.9983 | 0.94 | 3.13 | 32.01 | (0.15) |
| OH-BUP | 0.9997 | 0.68 | 2.26 | 108.18 | (2.74) | 0.9995 | 0.70 | 2.34 | 107.64 | (15.41) |
| FLB | 0.9936 | 10.42 | 34.72 | 99.43 | (4.21) | 0.9981 | 3.69 | 12.30 | 107.98 | (15.09) |
| OH-FLB | 0.9939 | 1.75 | 5.82 | 100.52 | (5.12) | 0.9983 | 2.30 | 7.65 | 93.25 | (8.77) |
| OPZ | 0.9968 | 0.22 | 0.72 | 113.73 | (12.46) | 0.9986 | 0.49 | 1.63 | 89.88 | (10.42) |
| OH-FLB | 0.9985 | 0.50 | 1.67 | 108.24 | (8.11) | 0.9979 | 0.82 | 2.73 | 94.29 | (4.28) |
| DEX | 0.9958 | 0.67 | 2.23 | 110.60 | (10.74) | 0.9997 | 0.74 | 2.46 | 94.23 | (17.90) |
| DTP | 0.9999 | 0.29 | 0.96 | 109.28 | (14.16) | 0.9996 | 0.34 | 1.14 | 97.34 | (18.68) |
| MDZ | 0.9976 | 0.27 | 0.90 | 112.68 | (13.21) | 0.9987 | 0.18 | 0.61 | 107.46 | (18.60) |
| OH-MDZ | 0.9978 | 0.71 | 2.38 | 102.87 | (11.37) | 0.9988 | 0.47 | 1.58 | 106.93 | (13.27) |
Table 3.
Inter-day accuracy (%) and precision (%) in human plasma and DBS (n=9), using the Mitra™ platform, spiked at 5 different concentrations on three separate days from separately made up working standard solutions.
Table 3.
Inter-day accuracy (%) and precision (%) in human plasma and DBS (n=9), using the Mitra™ platform, spiked at 5 different concentrations on three separate days from separately made up working standard solutions.
| Analyte | Spiked conc. (ng.mL-1) | Inter-day plasma (n=9) | Spiked conc. (ng.mL-1) | Inter-day DBS (n=9) (Mitra™) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | |||||||
| FEX | 75.00 | 73.08 | ± | 2.24 | 97.45 | 3.07 | 75.00 | 70.45 | ± | 3.95 | 93.93 | 5.61 |
| 150.00 | 153.58 | ± | 5.88 | 102.39 | 3.83 | 150.00 | 149.20 | ± | 2.63 | 99.47 | 1.76 | |
| 750.00 | 785.30 | ± | 5.76 | 104.71 | 0.73 | 750.00 | 739.89 | ± | 96.38 | 98.65 | 13.03 | |
| 1500.00 | 1522.38 | ± | 17.35 | 101.49 | 1.14 | 1500.00 | 1556.97 | ± | 25.56 | 103.80 | 1.64 | |
| 4500.00 | 4446.33 | ± | 25.58 | 98.81 | 0.58 | 4500.00 | 4404.97 | ± | 50.71 | 97.89 | 1.15 | |
| CAF | 150.00 | 150.67 | ± | 11.70 | 100.45 | 7.77 | 150.00 | 142.91 | ± | 7.73 | 95.27 | 5.41 |
| 300.00 | 299.66 | ± | 8.11 | 99.89 | 2.71 | 300.00 | 295.94 | ± | 12.07 | 98.65 | 4.08 | |
| 1500.00 | 1548.55 | ± | 44.42 | 103.24 | 2.87 | 1500.00 | 1451.72 | ± | 156.33 | 96.78 | 10.77 | |
| 3000.00 | 2980.31 | ± | 112.20 | 99.34 | 3.76 | 3000.00 | 3012.33 | ± | 53.93 | 100.41 | 1.79 | |
| 9000.00 | 8884.12 | ± | 137.15 | 98.71 | 1.54 | 9000.00 | 8931.49 | ± | 65.62 | 99.24 | 0.73 | |
| PAR | 75.00 | 75.36 | ± | 5.12 | 100.48 | 6.79 | 75.00 | 73.93 | ± | 3.42 | 98.58 | 4.63 |
| 150.00 | 144.48 | ± | 5.57 | 96.32 | 3.86 | 150.00 | 147.19 | ± | 6.86 | 98.12 | 4.66 | |
| 750.00 | 784.73 | ± | 33.99 | 104.63 | 4.33 | 750.00 | 774.79 | ± | 47.41 | 103.31 | 6.12 | |
| 1500.00 | 1517.20 | ± | 32.91 | 101.15 | 2.17 | 1500.00 | 1535.04 | ± | 87.08 | 102.34 | 5.67 | |
| 4500.00 | 4436.14 | ± | 69.15 | 98.58 | 1.56 | 4500.00 | 4428.43 | ± | 136.32 | 98.41 | 3.08 | |
| BUP | 75.00 | 79.49 | ± | 1.72 | 105.99 | 2.17 | 75.00 | 79.11 | ± | 3.64 | 105.48 | 4.60 |
| 150.00 | 137.87 | ± | 5.26 | 91.91 | 3.82 | 750.00 | 148.92 | ± | 2.57 | 99.28 | 1.72 | |
| 750.00 | 793.77 | ± | 36.09 | 105.84 | 4.55 | 1500.00 | 746.10 | ± | 18.83 | 99.48 | 2.52 | |
| 1500.00 | 1542.74 | ± | 34.11 | 102.85 | 2.21 | 3000.00 | 1468.00 | ± | 8.00 | 97.87 | 0.54 | |
| 4500.00 | 4405.34 | ± | 93.61 | 97.90 | 2.12 | 4500.00 | 4515.67 | ± | 154.22 | 100.35 | 3.42 | |
| Analyte | Spiked conc. (ng.mL-1) | Inter-day plasma (n=9) | Spiked conc. (ng.mL-1) | Inter-day DBS (n=9) | ||||||||
| Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | |||||||
| OH-BUP | 30.00 | 30.29 | ± | 3.66 | 100.96 | 12.07 | 75.00 | 72.59 | ± | 1.70 | 96.79 | 2.34 |
| 150.00 | 142.18 | ± | 7.68 | 94.79 | 5.40 | 150.00 | 145.60 | ± | 7.25 | 97.07 | 4.98 | |
| 750.00 | 766.23 | ± | 21.70 | 102.16 | 2.83 | 750.00 | 761.55 | ± | 33.97 | 101.54 | 4.46 | |
| 1500.00 | 1511.25 | ± | 43.93 | 100.75 | 2.91 | 1500.00 | 1514.68 | ± | 9.77 | 100.98 | 0.65 | |
| 4500.00 | 4459.10 | ± | 110.87 | 99.09 | 2.49 | 4500.00 | 4404.56 | ± | 109.85 | 97.88 | 2.49 | |
| FLB | 150.00 | 142.43 | ± | 6.32 | 94.95 | 4.44 | 150.00 | 148.60 | ± | 6.60 | 99.06 | 4.44 |
| 300.00 | 303.85 | ± | 8.42 | 101.28 | 2.77 | 300.00 | 307.07 | ± | 16.19 | 102.36 | 5.27 | |
| 1500.00 | 1544.51 | ± | 17.87 | 102.97 | 1.16 | 1500.00 | 1461.66 | ± | 90.79 | 97.44 | 6.21 | |
| 3000.00 | 3057.69 | ± | 151.34 | 101.92 | 4.95 | 3000.00 | 3044.17 | ± | 158.49 | 101.47 | 5.21 | |
| 9000.00 | 8936.24 | ± | 128.58 | 99.29 | 1.44 | 9000.00 | 8942.83 | ± | 529.40 | 99.36 | 5.92 | |
| OH-FLB | 75.00 | 71.56 | ± | 2.18 | 95.41 | 3.05 | 75.00 | 75.65 | ± | 2.79 | 100.87 | 3.68 |
| 150.00 | 149.78 | ± | 3.18 | 99.85 | 2.12 | 150.00 | 150.20 | ± | 6.35 | 100.13 | 4.23 | |
| 750.00 | 783.89 | ± | 20.70 | 104.52 | 2.64 | 750.00 | 734.04 | ± | 23.47 | 97.87 | 3.20 | |
| 1500.00 | 1520.17 | ± | 54.33 | 101.34 | 3.57 | 1500.00 | 1512.70 | ± | 57.85 | 100.85 | 3.82 | |
| 4500.00 | 4458.25 | ± | 47.52 | 99.07 | 1.07 | 4500.00 | 4474.10 | ± | 91.90 | 99.42 | 2.05 | |
| OPZ | 30.00 | 27.72 | ± | 1.29 | 92.39 | 4.67 | 75.00 | 71.33 | ± | 4.33 | 95.11 | 6.07 |
| 150.00 | 155.65 | ± | 10.34 | 103.77 | 6.65 | 150.00 | 151.96 | ± | 5.89 | 101.31 | 3.87 | |
| 750.00 | 785.52 | ± | 6.70 | 104.74 | 0.85 | 750.00 | 768.41 | ± | 31.38 | 102.46 | 4.08 | |
| 1500.00 | 1564.87 | ± | 26.32 | 104.32 | 1.68 | 1500.00 | 1522.43 | ± | 12.65 | 101.50 | 0.83 | |
| 4500.00 | 4423.55 | ± | 22.13 | 98.30 | 0.50 | 3000.00 | 4449.65 | ± | 23.97 | 98.88 | 0.54 | |
| OH-OPZ | 75.00 | 74.59 | ± | 2.88 | 99.46 | 3.86 | 30.00 | 70.46 | ± | 1.99 | 93.94 | 2.82 |
| 150.00 | 161.45 | ± | 14.09 | 107.63 | 8.73 | 75.00 | 149.75 | ± | 0.74 | 99.83 | 0.50 | |
| 750.00 | 784.63 | ± | 57.01 | 104.62 | 7.27 | 750.00 | 785.00 | ± | 71.40 | 104.67 | 9.10 | |
| 1500.00 | 1505.75 | ± | 13.97 | 100.38 | 0.93 | 1500.00 | 1503.30 | ± | 24.74 | 100.22 | 1.65 | |
| 4500.00 | 4098.65 | ± | 353.24 | 91.08 | 8.62 | 3000.00 | 4463.32 | ± | 18.64 | 99.18 | 0.42 | |
| Analyte | Spiked conc. (ng.mL-1) | Inter-day plasma (n=9) | Spiked conc. (ng.mL-1) | Inter-day DBS (n=9) | ||||||||
| Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | |||||||
| DEX | 75.00 | 72.71 | ± | 4.60 | 96.94 | 6.32 | 75.00 | 73.64 | ± | 1.55 | 98.19 | 2.10 |
| 150.00 | 147.26 | ± | 6.04 | 98.17 | 4.10 | 150.00 | 151.34 | ± | 2.26 | 100.89 | 1.50 | |
| 750.00 | 791.79 | ± | 44.43 | 105.57 | 5.61 | 750.00 | 743.62 | ± | 19.24 | 99.15 | 2.59 | |
| 1500.00 | 1554.21 | ± | 73.08 | 103.61 | 4.70 | 1500.00 | 1506.87 | ± | 21.87 | 100.46 | 1.45 | |
| 4500.00 | 4361.56 | ± | 181.78 | 96.92 | 4.17 | 4500.00 | 4477.94 | ± | 20.06 | 99.51 | 0.45 | |
| DTP | 75.00 | 71.69 | ± | 1.66 | 95.59 | 2.31 | 75.00 | 73.45 | ± | 2.82 | 97.94 | 3.84 |
| 150.00 | 151.68 | ± | 3.26 | 101.12 | 2.15 | 150.00 | 151.35 | ± | 2.75 | 100.90 | 1.82 | |
| 750.00 | 787.06 | ± | 31.41 | 104.94 | 3.99 | 750.00 | 740.02 | ± | 17.83 | 98.67 | 2.41 | |
| 1500.00 | 1548.95 | ± | 77.14 | 103.26 | 4.98 | 1500.00 | 1484.57 | ± | 14.08 | 98.97 | 0.95 | |
| 4500.00 | 4403.00 | ± | 118.14 | 97.84 | 2.68 | 4500.00 | 4508.65 | ± | 11.75 | 100.19 | 0.26 | |
| MDZ | 30.00 | 29.42 | ± | 2.01 | 98.07 | 6.82 | 30.00 | 27.77 | ± | 0.86 | 92.55 | 3.09 |
| 60.00 | 59.57 | ± | 3.30 | 99.28 | 5.54 | 60.00 | 60.37 | ± | 1.91 | 100.62 | 3.17 | |
| 300.00 | 306.27 | ± | 5.23 | 102.09 | 1.71 | 300.00 | 307.86 | ± | 21.64 | 102.62 | 7.03 | |
| 600.00 | 599.51 | ± | 2.43 | 99.92 | 0.41 | 600.00 | 615.30 | ± | 13.62 | 102.55 | 2.21 | |
| 1800.00 | 1795.05 | ± | 7.09 | 99.72 | 0.40 | 1800.00 | 1785.79 | ± | 19.80 | 99.21 | 1.11 | |
| OH-MDZ | 30.00 | 28.87 | ± | 0.31 | 96.25 | 1.09 | 30.00 | 28.59 | ± | 2.67 | 95.31 | 9.33 |
| 60.00 | 59.40 | ± | 0.60 | 99.00 | 1.01 | 60.00 | 60.01 | ± | 1.49 | 100.02 | 2.49 | |
| 300.00 | 310.92 | ± | 1.91 | 103.64 | 0.61 | 300.00 | 302.20 | ± | 14.90 | 100.73 | 4.93 | |
| 600.00 | 609.72 | ± | 13.92 | 101.62 | 2.28 | 600.00 | 609.52 | ± | 21.61 | 101.59 | 3.55 | |
| 1800.00 | 1782.55 | ± | 12.59 | 99.03 | 0.71 | 1800.00 | 1778.79 | ± | 34.44 | 98.82 | 1.94 | |
FEX – fexofenadine; CAF – caffeine; PAR – paraxanthine; BUP – bupropion; OH-BUP – hydroxy-bupropion; FLB – flurbiprofen; OH-FLB – hydroxyflurbiprofen; OPZ – omeprazole; OH-OPZ – hydroxy-omeprazole; DEX – dextromethorphan; DTP – dextrorphan; MDZ – midazolam; OH-MDZ – hydroxymidazolam.
Table 4.
Accuracy and precision of spiked QC samples in plasma and DBS after three freeze-thaw cycles at -80 ° C.
Table 4.
Accuracy and precision of spiked QC samples in plasma and DBS after three freeze-thaw cycles at -80 ° C.
| Human plasma | Dried Blood Spots ( Mitra™ ) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spiked conc. (ng.mL-1) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | Spiked conc. (ng.mL-1) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | |||||
| Fexofenadine | 150.00 | 142.17 | ± | 15.74 | 94.53 | 11.07 | 150.00 | 136.97 | ± | 1.86 | 91.31 | 1.36 |
| 600.00 | 587.21 | ± | 86.94 | 97.87 | 14.81 | 450.00 | 410.73 | ± | 29.24 | 91.27 | 7.12 | |
| 3000.00 | 2953.25 | ± | 349.62 | 98.44 | 11.84 | 3000.00 | 2529.27 | ± | 95.76 | 84.31 | 3.79 | |
| Caffeine | 150.00 | 105.25 | ± | 17.76 | 70.16 | 16.87 | 150.00 | 131.88 | ± | 2.28 | 87.92 | 1.73 |
| 600.00 | 566.56 | ± | 57.78 | 94.43 | 10.20 | 600.00 | 515.05 | ± | 9.32 | 85.84 | 1.81 | |
| 3000.00 | 2770.61 | ± | 378.45 | 92.35 | 13.66 | 3000.00 | 2642.36 | ± | 53.60 | 88.08 | 2.03 | |
| Paraxanthine | 75.00 | 58.02 | ± | 7.03 | 77.36 | 12.12 | 75.00 | 63.54 | ± | 4.04 | 84.73 | 6.36 |
| 300.00 | 285.58 | ± | 36.22 | 95.19 | 12.68 | 300.00 | 280.23 | ± | 17.40 | 93.41 | 6.21 | |
| 1500.00 | 1439.21 | ± | 184.95 | 95.95 | 12.85 | 1500.00 | 1389.55 | ± | 72.03 | 92.64 | 5.18 | |
| Bupropion | 150.00 | 117.58 | ± | 26.11 | 78.39 | 22.20 | 150.00 | 138.67 | ± | 10.00 | 92.44 | 7.21 |
| 750.00 | 671.12 | ± | 51.89 | 89.48 | 7.73 | 450.00 | 462.01 | ± | 36.69 | 102.67 | 7.94 | |
| 3000.00 | 3270.21 | ± | 217.32 | 109.01 | 6.65 | 3000.00 | 2822.43 | ± | 220.29 | 94.08 | 7.81 | |
| Hydroxybupropion | 75.00 | 66.98 | ± | 3.96 | 89.31 | 5.91 | 75.00 | 71.09 | ± | 2.51 | 94.78 | 3.54 |
| 300.00 | 320.10 | ± | 8.88 | 106.70 | 2.77 | 300.00 | 278.04 | ± | 21.46 | 92.68 | 7.72 | |
| 1500.00 | 1635.18 | ± | 12.05 | 109.01 | 0.74 | 1500.00 | 1576.19 | ± | 31.09 | 105.08 | 1.97 | |
| Flurbiprofen | 150.00 | 169.06 | ± | 18.58 | 112.71 | 10.99 | 150.00 | 193.96 | ± | 2.54 | 129.30 | 1.31 |
| 600.00 | 711.69 | ± | 54.04 | 118.61 | 7.59 | 600.00 | 650.30 | ± | 82.69 | 108.38 | 12.72 | |
| 3000.00 | 3497.46 | ± | 115.23 | 116.58 | 3.29 | 3000.00 | 3283.55 | ± | 493.34 | 109.45 | 15.02 | |
| Hydroxyflurbiprofen | 75.00 | 65.43 | ± | 0.90 | 87.24 | 1.38 | 150.00 | 122.48 | ± | 4.42 | 81.65 | 3.61 |
| 300.00 | 284.31 | ± | 16.94 | 94.77 | 5.96 | 300.00 | 299.39 | ± | 14.40 | 99.80 | 4.81 | |
| 1500.00 | 1511.98 | ± | 88.64 | 100.80 | 5.86 | 1500.00 | 1481.57 | ± | 6.22 | 98.77 | 0.42 | |
Table 4.
(continued): Accuracy and precision of spiked QC samples in plasma and DBS after three freeze-thaw cycles at -80 ° C.
Table 4.
(continued): Accuracy and precision of spiked QC samples in plasma and DBS after three freeze-thaw cycles at -80 ° C.
| Analyte | Human plasma | Dried Blood Spots (Mitra™ ) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spiked conc. (ng.mL-1) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | Spiked conc. (ng.mL-1) | Found (mean ± SD in ng.mL-1) | Accuracy (%) | Precision (%) | ||||||
| Omeprazole | 150.00 | 160.38 | ± | 19.83 | 106.92 | 12.37 | 75.00 | 54.21 | ± | 2.61 | 72.29 | 4.81 | |
| 300.00 | 251.81 | ± | 26.33 | 83.94 | 10.46 | 300.00 | 281.02 | ± | 5.71 | 93.67 | 2.03 | ||
| 1500.00 | 1399.75 | ± | 237.21 | 93.32 | 16.95 | 1500.00 | 1338.81 | ± | 55.19 | 89.25 | 4.12 | ||
| Hydroxy-omeprazole | 75.00 | 68.39 | ± | 1.65 | 91.19 | 2.41 | 75.00 | 71.15 | ± | 3.37 | 94.87 | 4.74 | |
| 300.00 | 313.34 | ± | 26.25 | 104.45 | 8.38 | 300.00 | 294.24 | ± | 12.17 | 98.08 | 4.13 | ||
| 1500.00 | 1457.62 | ± | 76.06 | 97.17 | 5.22 | 1500.00 | 1474.07 | ± | 39.84 | 98.27 | 2.70 | ||
| Dextromethorphan | 75.00 | 77.41 | ± | 6.88 | 103.21 | 8.89 | 75.00 | 75.36 | ± | 4.96 | 100.48 | 6.58 | |
| 300.00 | 333.85 | ± | 16.94 | 111.28 | 5.07 | 300.00 | 274.74 | ± | 10.84 | 91.58 | 3.95 | ||
| 1500.00 | 1629.79 | ± | 97.39 | 108.65 | 5.98 | 1500.00 | 1543.52 | ± | 50.39 | 102.90 | 3.26 | ||
| Dextrorphan | 150.00 | 150.04 | ± | 4.32 | 100.03 | 2.88 | 150.00 | 151.49 | ± | 5.92 | 100.99 | 3.91 | |
| 750.00 | 790.96 | ± | 48.57 | 105.46 | 6.14 | 750.00 | 659.63 | ± | 17.17 | 87.95 | 2.60 | ||
| 3000.00 | 3127.85 | ± | 104.21 | 104.26 | 3.33 | 3000.00 | 3131.03 | ± | 141.39 | 104.37 | 4.52 | ||
| Midazolam | 30.00 | 26.14 | ± | 3.14 | 87.14 | 12.01 | 30.00 | 24.78 | ± | 1.97 | 82.62 | 7.93 | |
| 120.00 | 137.37 | ± | 16.05 | 114.48 | 11.69 | 120.00 | 115.10 | ± | 2.26 | 95.91 | 1.96 | ||
| 600.00 | 650.37 | ± | 55.62 | 108.40 | 8.55 | 600.00 | 559.28 | ± | 36.62 | 93.21 | 6.55 | ||
| Hydroxy-midazolam | 30.00 | 34.19 | ± | 5.72 | 113.97 | 16.73 | 60.00 | 49.65 | ± | 1.37 | 82.74 | 2.75 | |
| 180.00 | 205.49 | ± | 12.36 | 114.16 | 6.01 | 180.00 | 164.88 | ± | 8.48 | 91.60 | 5.15 | ||
| 1200.00 | 1090.39 | ± | 129.74 | 90.87 | 11.90 | 1200.00 | 979.89 | ± | 31.29 | 81.66 | 3.19 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



