
Submitted:
13 June 2024
Posted:
14 June 2024
You are already at the latest version
Abstract
This study aims to characterize the molecular profile of hepatitis B virus (HBV) among socially vulnerable immigrants residing in Brazil to investigate the introduction of uncommon HBV strains into the country. Serum samples from 102 immigrants with positive serology for HBV core antibody (anti-HBc) were tested for the presence of HBV DNA by PCR assays. Among these, 24 were also positive for the HBV surface antigen (HBsAg). The full or partial genome was sequenced to determine genotype by phylogenetic analysis. Participants were from Haiti (79.4%), Guinea Bissau (11.8%), Venezuela (7.8%), and Colombia (1%). Of the 21 HBV DNA-positive samples, subgenotypes A1 (52.4%), A5 (28.6%), E (9.5%), F2 (4.8%), and F3 (4.8%) were identified. Among the 78 HBsAg-negative participants, four were positive for HBV DNA, resulting in an occult HBV infection rate of 5.1%. Phylogenetic analysis suggested that most strains were likely introduced to Brazil by migration. Importantly, 80% of A5 sequences had the A1762T/G1764A double mutation, linked to an increased risk of hepatocellular carcinoma development. In conclusion, this study is the first report of HBV subgenotype A5 in Brazil, shedding new light on the diversity of HBV strains circulating in the country. Understanding the genetic diversity of HBV in immigrant communities can lead to better prevention and control strategies, benefiting both immigrants and the broader society.
Keywords:
HBV
; molecular epidemiology
; genotypes
; phylogenetic analysis
; immigration
1. Introduction
Hepatitis B is a highly prevalent viral infection in humans and a serious global public health problem. Hepatitis B virus (HBV) has infected one-third of the global population, with an estimated 296 million chronic infections [1]. In 2019, the World Health Organization estimated that hepatitis B was linked to approximately 820,000 deaths, primarily associated with cirrhosis and hepatocellular carcinoma (HCC). Despite the availability of a prophylactic vaccine since the 1980s, around 1.5 million new infections are reported annually [1]. The spread of chronic HBV infection varies greatly by geographical location. In regions with a high prevalence of chronic carriers, HBV infection is frequently transmitted at birth or during early childhood, while in areas with a low prevalence, it is typically acquired during adulthood through percutaneous or sexual transmission [2]. HBV infection acquired in adulthood results in chronic hepatitis in fewer than 5% of cases, whereas infection in infancy and early childhood causes chronicity in about 95% of cases. HBV surface antigen (HBsAg) is the established serological marker for the diagnosis of acute or chronic HBV infection. However, the presence of HBV DNA in the blood and/or liver of individuals who test negative for HBsAg is indicative of a unique form of chronic viral infection known as occult HBV infection (OBI). Low HBV replication rate due to the host's immune response may contribute to OBI in a majority of cases. Mutations altering the antigenicity of HBsAg, known as immune escape mutations, can also result in occult infection, impeding detection by commercial assays [3].
HBV is the prototype member of the family Hepadnaviridae, containing a partially double-stranded relaxed circular DNA genome approximately 3,200 nucleotides (nt) in length [4,5]. This family is characterized by a distinctive viral replication cycle including the activity of an error-prone reverse transcriptase, resulting in the formation of significant variability across HBV isolates [6,7]. HBV has been classified into nine genotypes (A-I), based on a greater than 7.5% genomic sequence divergence, with a potential genotype (J) recovered from a single individual. In addition, due to the large intra-genotypic diversity, genotypes A-D, F, and I have been further divided into subgenotypes based on 4% to 7.5% nucleotide divergence in the complete genome [8]. Frequencies of clinically relevant mutations (i.e., immune escape-, drug resistance-, and HCC-associated mutations) vary significantly between HBV genotypes [9], while differences in transmission routes, disease progression, response to antiviral therapies, and clinical outcome have also been demonstrated [10,11,12].
Recent findings of HBV DNA in archeological remains demonstrate that humans have been infected with HBV for millennia [13]. Given that HBV lacks environmental or animal reservoirs, its spread is closely associated with human dispersion. The global distribution of HBV genotypes and, occasionally, subgenotypes displays a significant geographic association, which most likely reflects historical human migration patterns [14,15,16]. Genotype A is frequently found in Africa (subgenotypes A1, A3 and A4), Caribbean (A1 and A5), Europe and North America (A2), and South America (A1 and A2), while genotypes B and C are found essentially in Asia. Genotype E is limited to West and Central Africa, and genotypes F and H are found in Amerindian populations. Genotypes D and G have a ubiquitous geographic distribution, while the more recent genotypes I and J were described in China, India, Laos and Vietnam (I), and Japan (J) [17,18].
The 2016 World Health Assembly endorsed the Global Health Sector Strategy including a goal to eliminate viral hepatitis as a public health threat by 2030, reducing new infections by 90% and mortality by 65% [19]. However, in order to meet these objectives, it is critical to reach socially vulnerable subpopulations at risk of acquiring or transmitting HBV infection, such as immigrants and refugees, a population that is growing globally. The molecular characteristics of HBV infection in immigrants reflect those of their home countries. The flow of immigrants may contribute to changes in the frequencies of HBV (sub)genotypes and the introduction of uncommon strains in host countries [20,21].
Brazil is the largest country in the Southern Hemisphere and traditionally a destination country for different migratory flows that have occurred throughout history. Despite not receiving significant immigration numbers today, Brazil is experiencing a gradual growth in migratory movements. In 2021, over 150,000 immigrants entered the country, most of them coming from geographical areas with intermediate or high HBV endemicity and living in conditions of poverty and social vulnerability [22]. In Brazil, HBV genotypes A, D, and F are mainly found countrywide, reflecting the contribution of the African (A1), European (A2 and D), and Amerindian (F) people in the formation of the Brazilian population [23,24,25]. Data on the molecular characterization of HBV in immigrant populations in Brazil is extremely scarce. Therefore, this study investigated the HBV (sub)genotypes, and the occurrence of OBI and clinically relevant viral mutations among socially vulnerable immigrants from Latin America and Africa residing in Brazil, to determine the potential introduction of uncommon HBV strains into the country.
2. Results
A total of 102 immigrants residing in Goiás, Central-West Brazil, with positive serology for anti-HBc were enrolled in this study. From these, 24 (23.5%) were also positive for HBsAg. Participants self-declared the following countries of origin: Haiti (n=81; 79.4%), Guinea Bissau (n=12; 11.8%), Venezuela (n=8; 7.8%) and Colombia (n=1; 1%). Most of them were male (68.6%) and aged 31–50 years (56.9%). HBV DNA was detected in 21/102 (20.6%) individuals (Table 1). Among the 78 HBsAg-negative subjects, four were positive for HBV DNA, resulting in an OBI rate of 5.1%.
Phylogenetic analysis of the 21 samples positive for HBV DNA demonstrated the presence of subgenotypes A1 (n=11; 52.4%), A5 (n=6; 28.6%), E (n=2; 9.5%), F2 (n=1; 4.8%), and F3 (n =1; 4.8%). In addition to amplifying the S and pre-C/C regions, complete genome amplification was specifically performed for samples classified as genotypes A5, E, F2, and F3, which are rare in Brazil. Phylogenetic trees representing the complete genome (n=7), S (n=21), and X/pre-C/C (n=12) sequences obtained in this study are shown in Figure 1. Sample classification into genotypes and subgenotypes remained unchanged in all three phylogenetic analyses (Figure 1).
Individuals infected with HBV subgenotype A1 were immigrants from Haiti (n=10) and Guinea-Bissau (n=1), while those infected with subgenotype A5 were exclusively from Haiti. The two cases of genotype E infection were from Haiti and Guinea-Bissau, respectively. Subgenotypes F2 and F3 were both found in Venezuelan immigrants. The four cases of OBI corresponded to genotypes A1 (n=3) and A5 (n=1) and were detected in immigrants from Guinea-Bissau (A1) and Haiti (A1 and A5) (Table 2).
Given that subgenotypes A1 and F2 are commonly found in Brazil, additional phylogenetic analyses were conducted using 270 S gene sequences (A1) and 36 full-length sequences (F2) to gain potential insights into whether the individuals may have brought these strains to Brazil or became infected after their arrival. Among the 10 sequences of subgenotype A1 obtained from the Haitian participants, eight (80%) grouped into clusters composed almost exclusively of sequences from Haiti, whereas the remaining two clustered with Brazilian (ID 188) and African (ID 313) sequences, respectively. The single A1 sequence obtained from the immigrant from Guinea-Bissau (ID 67) grouped in the cluster of sequences from Haiti (Figure 2). The subgenotype F2 complete genome retrieved from a Venezuelan participant (ID 358) grouped with Venezuelan sequences, more specifically into the F2a clade (Figure 3).
Regarding the presence of mutations of clinical relevance, none of the sequences showed immune escape- or drug resistance-associated mutations. Nonetheless, mutations linked to HCC were identified, including the A1762T/G1764A mutation in 58.3% (7/12) of pre-C/C sequences and the G1896A stop-codon mutation in 16.7% (2/12) of these sequences. Remarkably, 4 out 5 (80%) subgenotype A5 sequences were positive for A1762T/G1764A. Additionally, 12-nucleotide insertions in the S region were found in two sequences (genotypes A1 and E), while a 21-nucleotide deletion was detected in the pre-S2 region of the F2 subgenotype sequence (Table 2).
3. Discussion
To our knowledge, this is the first study on the molecular characterization of HBV in Latin American and African immigrants residing in Brazil. Furthermore, this study demonstrates for the first time the circulation of the rare HBV subgenotype A5 in the country. This subgenotype exhibits a distinct distribution pattern, including West and Central African countries, and Haiti, a country on the island of Hispaniola in the Greater Antilles archipelago of the Caribbean Sea. HBV A5 strains are thought to have spread during the slave trade from African nations to Haiti, where >90% of the population descended from African slaves [26]. The main causes of the recent migratory flows from Haiti have been economic hardship, exacerbated by natural disasters such as the earthquake in 2010 and Hurricane Matthew in 2016 [27]. The migration crisis in Haiti is particularly relevant to Brazil's migration policies. The present study confirms the predominance of this nationality, with 79.4% of participants being Haitians. HBV (sub)genotypes A1, A5 and E were found in 58.8%, 35.3% and 5.9% of them. These findings are consistent with those of Andernach and colleagues (2009) [26], who reported that among Haitian pregnant women surveyed, A1, A5, and E were found in 43%, 19.6%, and 6.1%, respectively. The introduction of HBV A5 strains from Haiti to Brazil is plausible, as this subgenotype had not previously been detected in the country. However, since subgenotype A1 is the most prevalent subgenotype in Brazil [23], an expanded phylogenetic analysis with 270 sequences of this subgenotype from different locations worldwide was conducted to explore the potential origin of these strains. Interestingly, 80% of A1 sequences obtained from Haitian participants clustered within Haitian sequences, which may suggest an introduction of these A1 strains from Haiti to Brazil.
The discovery of two HBV genotype E strains among immigrants from Haiti and Guinea-Bissau is also of epidemiological relevance, considering that this genotype is extremely rare in Brazil, with a few reports of detection mainly in individuals with connections to the African continent [28,29]. Differing from Brazil and Haiti, HBV genotype E is primarily found in Guinea-Bissau, a country on the western coast of Africa [26,30]. Despite being the most prevalent genotype in West Africa, genotype E has been understudied [31]. Notably, the low prevalence of this genotype in the African descendant populations in the Americas suggests that genotype E was not circulating during the slave trade period (16th to 19th centuries). Furthermore, the low genetic diversity, phylogeographic analyzes, and evidence of genotype E in ancient HBV samples point to a relatively recent reintroduction of this genotype into the human population [26,31].
HBV genotype F is assumed to represent the original genotype of the aboriginal populations of the Americas, since it has been detected in high frequencies in various South and Central American countries [32], as well as in native Alaskan communities [33]. Six subgenotypes have been described for HBV genotype F (F1–F6), and different clades have been proposed for subgenotype F1 (F1a, b, c, and d) and F2 (F2a and b) [34]. Unlike other Latin American countries, Brazil has a low prevalence of genotype F, even in the northern region, which includes the Amazon basin, where the indigenous Amerindian population is predominant [24]. Despite its low prevalence, genotype F strains are widespread throughout the country, with F2a being the predominant F subgenotype and the original native HBV of the Brazilian population [35]. In this study, subgenotypes F2a and F3 were identified in immigrants from Venezuela, where these subgenotypes are common [32]. The complete F2 genome sequence grouped within the F2a Venezuelan monophyletic cluster, suggesting that the individual might have acquired the infection in their home country. Similarly, the F3 strain was likely introduced to Brazil by the Venezuelan immigrant, as reports of subgenotype F3 in the country are scarce [36].
Mutations in the basal core promoter region (A1762T/G1764A) and a stop-codon mutation in the pre-C region (G1896A), which decrease/abolish HBeAg expression while enhancing viral replication, as well as deletions in the pre-S region, are associated with an elevated risk of HCC [37]. Importantly, A1762T/G1764A was present at much higher frequencies in A1 and A5 subgenotype sequences (75% and 80%, respectively) obtained in this study, compared to previous observations (26.4% and 31.3%, respectively) [9]. These results draw attention to the introduction of HBV strains with greater oncogenic potential in Brazil.
OBI raises concerns among researchers worldwide, as little is known about the clinical impact of this infection and its role in the transmission of the virus. Among the immigrant population studied, OBI was detected in three individuals infected with subgenotype A1 and in one infected with A5, yielding an OBI prevalence of 5.1% (n=4/78) in this group. This prevalence is higher than that found in a previous study conducted among Brazilian blood donors, which reported a prevalence of 0.6% (n=6/976) [38]. Numerous studies have shown that OBI rates vary substantially across different populations, due to a combination of factors such as the endemicity of HBV infection, the sensitivity of HBV DNA assays, and the biological sample analyzed [3]. Previous studies conducted in Central Brazil have shown OBI rates of 0% (n=0/522) in men who have sex with men [39], 3.8% (n=19/505) in treatment-naïve HIV-infected patients [40], and 12.7% (n=19/149) in injecting drug users [41].
This study has some limitations that should be acknowledged. First, the analysis was conducted on a limited number of sequences, potentially constraining the depth of insight into the epidemiological context of this population. Second, owing to limits in resources and time, this study exclusively enrolled individuals who tested positive for anti-HBc. The incorporation of seronegative individuals could offer a broader understanding of the prevalence of OBI within this population. Third, conducting Bayesian phylogeographic analyses would enable the reconstruction of the spatial and temporal diversification of the rare genotypes A5 and E in Brazil.
In conclusion, this study underscores the importance of investigating HBV (sub)genotypes and mutations in immigrant populations. Our results suggest the potential involvement of Haitian immigrants in the introduction of the rare HBV subgenotype A5 into Brazil. The introduction of uncommon (sub)genotypes and variants carrying clinically relevant mutations presents a potential challenge to countries of destination. However, it is important to approach this issue with sensitivity and respect for the experiences and perspectives of immigrant communities. A better understanding of the genetic diversity of HBV in these populations could guide focused epidemiological studies and preventive measures, potentially identifying specific viral strains and transmission patterns, thereby benefiting both immigrants and the broader population. Furthermore, this research can contribute to the global effort to combat HBV, as sharing research findings across regions can lead to more targeted and efficient interventions.
4. Materials and Methods
4.1. Study population
This cross-sectional study was conducted among socially vulnerable immigrants and refugees residing in Goiás, Central-West Brazil. Between July 2019 and January 2020, participants were recruited for a study investigating the seroprevalence of hepatitis B and C infections and associated factors [42]. Of the original 365 participants from the previous study, 102 with positive tests for the HBV core antibody (anti-HBc), a serological marker of exposure to HBV, were included in the present investigation. Informed written consent was obtained at the time of sampling from all participants. The Ethics Committee of Federal University of Goiás approved the study protocol (3.243.845 CAAE 06871019.7.0000.5083).
4.2. Viral DNA extraction and PCR amplification
Viral nucleic acids were extracted from 0.2 mL serum using a High Pure Viral Nucleic Acid kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions. Serum samples were tested for the presence of HBV DNA by qualitative polymerase chain reaction (PCR) assays. Negative controls were added in both extraction and PCR procedures. HBV partial S region (581 base pairs, bp) was amplified by semi-nested PCR. The first round of amplification was performed with 2 µL of DNA, 1U of Platinum Taq DNA Polymerase (Invitrogen, Waltham, MA), and sense PS1 (5’-CCATATTCTTGGGAACAAGA-3’, nts 2826–2845) and antisense SR (5’-CGAACCACTGAACAAATGGC-3’, nts 704–685) oligonucleotide primers. Second round PCR was performed with 2 µL of the first round product, 1U of Platinum Taq DNA Polymerase (Invitrogen), sense S1 (5’-CTTCTCGAGGACTGGGGACC-3, nts 124–143) and antisense SR primers. PCR conditions were as follows: 94˚C for 2 min, followed by 35 cycles of 94˚C for 30 sec, 52˚C (first step) or 55˚C (nested PCR) for 30 sec and 72˚C for 1 min 30 sec, and a final extension step at 72˚C for 7 min.
HBV partial X and pre-C/C regions (428 bp) were amplified in a nested PCR assay carried out with 4 µL of DNA, 1U of Platinum Taq DNA Polymerase (Invitrogen), and sense X1 (5’-ACCTCCTTTCCATGGCTGCT-3’, nts 1363–1382) and antisense C2 (5’-CTAACATTGAGATTCCCGAGATTGAGA-3’, nts 2458–2432) oligonucleotide primers. Second round PCR was performed with 4 µL of the first round product, 1U of Platinum Taq DNA Polymerase (Invitrogen), sense X4 (5’-AAGGTCTTACATAAGAGGAC-3, nts 1644–1663) and antisense C3 (5’-TTGCCTGAGTGCAGTATGGT-3’, nts 2071–2052) primers. PCR conditions for both rounds were 94°C for 2 min, 35 cycles at 94°C for 30 sec, 52°C for 30 sec, and 72°C for 1 min 30 sec, followed by a final elongation at 72°C for 7 min.
Almost the entire HBV genome (3,164 bp) was amplified by nested PCR. The first round was attempted with a protocol modified from Günther and colleagues (1995) [43] using 2 μL of DNA, primers P1-mod (5’-TTTTTCACCTCTGCCTAATCA-3’, nts 1821–1841) and P2-mod (5’-AAAAAGTTGCATGGTGCTGG-3’, nts 1825–1806), and the following PCR profile: denaturation at 94˚C for 4 min, followed by 10 cycles at 94˚C for 40 sec, 55˚C for 1 min, and 72˚C for 3 min; 10 cycles of 94˚C for 40 sec, 60˚C for 1 min, and 72˚C for 5 min; 10 cycles of 94˚C for 40 sec, 62˚C for 1 min, and 72˚C for 7 min; 10 cycles of 94˚C for 40 sec, 62˚C for 1 min, and 72˚C for 9 min; and a final extension step at 72˚C for 10 min. Second round PCR was performed with 5 µL of the first round product, and sense P1-nested (5’-ATGTCCTACTGTTCAAGCCTCC-3’, nts 1852-1873) and antisense P2-nested (5’-ATTTATGCCTACAGCCTCCT-3’, nts 1794-1775) primers. PCR conditions were as follows: 94°C for 4 min, 30 cycles at 94℃ for 30 sec, 50℃ for 30 sec, and 72℃ for 4 min, followed by a final elongation at 72°C for 10 min. Both PCR assays were performed using 1U of Platinum Taq DNA polymerase and supplied reagents (Invitrogen) in accordance with product instructions.
4.3. Nucleotide sequencing
Samples with a positive result in the PCR assays were subsequently subjected to nucleotide sequencing. PCR products were purified using the QIAquick PCR Purification kit (QIAGEN, Hilden, Germany). HBV sequences were determined via direct sequencing using a BigDye Terminator Kit v3.1 (Applied Biosystems, Waltham, MA), and sequencing reactions analyzed on an ABI3730xl automated sequencer (Applied Biosystems). Nucleotide sequences obtained during this study were deposited in the GenBank database under accession numbers PP093835-PP093848, PP101105-PP101109, and PP116453-PP116459.
4.4. HBV genotyping and mutational analysis
Multiple sequence alignments of the amplified genomic regions were performed using the MUSCLE program implemented in MEGA 11 software, with HBV reference sequences for genotypes A to J [44], and subsequently subjected to Maximum Likelihood phylogenetic analysis for genotyping. The phylogenetic trees were inferred with MEGA 11 software under the best nucleotide substitution model selected from Maximum Likelihood fits of 24 different nucleotide substitution models. The visualization of the phylogenetic trees was performed using the MEGA 11 and FigTree v1.4.4 software programs (http://tree.bio.ed.ac.uk/software/figtree/). For mutational analysis, the presence of 21 clinically relevant positions according to previous reports [9] were examined, including 10 amino acid sites within HBsAg (120, 126, 129, 130, 133, 141, 142, 143, 144, and 145) related to immune escape, three nucleotide positions within the core (C) promoter gene (1753, 1762, and 1764), the pre-C stop-codon mutation G1896A, and pre-S deletions related to HCC development, and seven amino acid positions within the reverse transcriptase (RT) domain of HBV polymerase (180, 181, 184, 202, 204, 236, and 250) related to antiviral resistance.
Author Contributions
Conceptualization, T.B.S., K.A.C. and N.M.A.; methodology, T.B.S., and T.L.S.M.; validation, T.B.S. and N.M.A; formal analysis, T.B.S. and N.M.A.; investigation, T.B.S., K.A.C. and N.M.A.; resources, M.S.C., S.A.T., K.A.C. and N.M.A.; data curation, T.B.S. and N.M.A.; writing—original draft preparation, T.B.S. and N.M.A.; writing—review and editing, T.B.S., T.L.S.M., M.S.C., S.A.T., K.A.C. and N.M.A.; supervision, N.M.A.; funding acquisition, K.A.C. and N.M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), grant number E-26/210.450/2019, and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grant number 309649/2022-6.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Federal University of Goiás (study protocol 3.243.845 CAAE 06871019.7.0000.5083).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Nucleotide sequences obtained during this study were deposited in the GenBank database under accession numbers PP093835-PP093848, PP101105-PP101109, and PP116453-PP116459.
Acknowledgments
The authors thank the members of NECAIH (Nucleus of Studies in Epidemiology and Care for Transmissible Infections in Human Health Disorders) for their assistance in carrying out the study, and the DNA Sequencing Platform (RPT01A-VPPCB/FIOCRUZ) of the Oswaldo Cruz Foundation for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- WHO World Health Organization. Fact sheets. Hepatitis B. https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (08 November 2023).
- Edmunds, W.J.; Medley, G.F.; Nokes, D.J.; O'Callaghan, C.J.; Whittle, H.C.; Hall, A.J. , Epidemiological patterns of hepatitis B virus (HBV) in highly endemic areas. Epidemiol Infect 1996, 117, 313–325. [Google Scholar] [CrossRef]
- Raimondo, G.; Locarnini, S.; Pollicino, T.; Levrero, M.; Zoulim, F.; Lok, A.S. , Update of the statements on biology and clinical impact of occult hepatitis B virus infection. J Hepatol 2019, 71, 397–408. [Google Scholar] [CrossRef]
- Karayiannis, P. , Hepatitis B virus: virology, molecular biology, life cycle and intrahepatic spread. Hepatol Int 2017, 11, 500–508. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. , Hepatitis B virus biology and life cycle. Antiviral Res 2020, 182, 104925. [Google Scholar] [CrossRef]
- Osiowy, C.; Giles, E.; Tanaka, Y.; Mizokami, M.; Minuk, G.Y. , Molecular evolution of hepatitis B virus over 25 years. J Virol 2006, 80, 10307–10314. [Google Scholar] [CrossRef]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. , The evolution and clinical impact of hepatitis B virus genome diversity. Nat Rev Gastroenterol Hepatol 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Araujo, N.M.; Teles, S.A.; Spitz, N. , Comprehensive Analysis of Clinically Significant Hepatitis B Virus Mutations in Relation to Genotype, Subgenotype and Geographic Region. Front Microbiol 2020, 11, 616023. [Google Scholar] [CrossRef]
- Araujo, N.M.; Waizbort, R.; Kay, A. , Hepatitis B virus infection from an evolutionary point of view: how viral, host, and environmental factors shape genotypes and subgenotypes. Infect Genet Evol 2011, 11, 1199–1207. [Google Scholar] [CrossRef]
- Chen, J.; Li, L.; Yin, Q.; Shen, T. , A review of epidemiology and clinical relevance of Hepatitis B virus genotypes and subgenotypes. Clin Res Hepatol Gastroenterol 2023, 47, 102180. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. , The clinical implications of hepatitis B virus genotype: Recent advances. J Gastroenterol Hepatol 2011, 26 Suppl 1, 123–130. [Google Scholar] [CrossRef]
- Kocher, A.; Papac, L.; Barquera, R.; Key, F.M.; Spyrou, M.A.; Hubler, R.; Rohrlach, A.B.; Aron, F.; Stahl, R.; Wissgott, A.; van Bommel, F.; Pfefferkorn, M.; Mittnik, A.; Villalba-Mouco, V.; Neumann, G.U.; Rivollat, M.; van de Loosdrecht, M.S.; Majander, K.; Tukhbatova, R.I.; Musralina, L.; Ghalichi, A.; Penske, S.; Sabin, S.; Michel, M.; Gretzinger, J.; Nelson, E.A.; Ferraz, T.; Nagele, K.; Parker, C.; Keller, M.; Guevara, E.K.; Feldman, M.; Eisenmann, S.; Skourtanioti, E.; Giffin, K.; Gnecchi-Ruscone, G.A.; Friederich, S.; Schimmenti, V.; Khartanovich, V.; Karapetian, M.K.; Chaplygin, M.S.; Kufterin, V.V.; Khokhlov, A.A.; Chizhevsky, A.A.; Stashenkov, D.A.; Kochkina, A.F.; Tejedor-Rodriguez, C.; de Lagran, I.G.; Arcusa-Magallon, H.; Garrido-Pena, R.; Royo-Guillen, J.I.; Novacek, J.; Rottier, S.; Kacki, S.; Saintot, S.; Kaverzneva, E.; Belinskiy, A.B.; Veleminsky, P.; Limbursky, P.; Kostka, M.; Loe, L.; Popescu, E.; Clarke, R.; Lyons, A.; Mortimer, R.; Sajantila, A.; de Armas, Y.C.; Hernandez Godoy, S.T.; Hernandez-Zaragoza, D.I.; Pearson, J.; Binder, D.; Lefranc, P.; Kantorovich, A.R.; Maslov, V.E.; Lai, L.; Zoledziewska, M.; Beckett, J.F.; Langova, M.; Danielisova, A.; Ingman, T.; Atienzar, G.G.; de Miguel Ibanez, M.P.; Romero, A.; Sperduti, A.; Beckett, S.; Salter, S.J.; Zilivinskaya, E.D.; Vasil'ev, D.V.; von Heyking, K.; Burger, R.L.; Salazar, L.C.; Amkreutz, L.; Navruzbekov, M.; Rosenstock, E.; Alonso-Fernandez, C.; Slavchev, V.; Kalmykov, A.A.; Atabiev, B.C.; Batieva, E.; Calmet, M.A.; Llamas, B.; Schultz, M.; Krauss, R.; Jimenez-Echevarria, J.; Francken, M.; Shnaider, S.; de Knijff, P.; Altena, E.; Van de Vijver, K.; Fehren-Schmitz, L.; Tung, T.A.; Losch, S.; Dobrovolskaya, M.; Makarov, N.; Read, C.; Van Twest, M.; Sagona, C.; Ramsl, P.C.; Akar, M.; Yener, K.A.; Ballestero, E.C.; Cucca, F.; Mazzarello, V.; Utrilla, P.; Rademaker, K.; Fernandez-Dominguez, E.; Baird, D.; Semal, P.; Marquez-Morfin, L.; Roksandic, M.; Steiner, H.; Salazar-Garcia, D.C.; Shishlina, N.; Erdal, Y.S.; Hallgren, F.; Boyadzhiev, Y.; Boyadzhiev, K.; Kussner, M.; Sayer, D.; Onkamo, P.; Skeates, R.; Rojo-Guerra, M.; Buzhilova, A.; Khussainova, E.; Djansugurova, L.B.; Beisenov, A.Z.; Samashev, Z.; Massy, K.; Mannino, M.; Moiseyev, V.; Mannermaa, K.; Balanovsky, O.; Deguilloux, M.F.; Reinhold, S.; Hansen, S.; Kitov, E.P.; Dobes, M.; Ernee, M.; Meller, H.; Alt, K.W.; Prufer, K.; Warinner, C.; Schiffels, S.; Stockhammer, P.W.; Bos, K.; Posth, C.; Herbig, A.; Haak, W.; Krause, J.; Kuhnert, D. , Ten millennia of hepatitis B virus evolution. Science 2021, 374, 182–188. [Google Scholar] [CrossRef]
- Paraskevis, D.; Angelis, K.; Magiorkinis, G.; Kostaki, E.; Ho, S.Y.; Hatzakis, A. , Dating the origin of hepatitis B virus reveals higher substitution rate and adaptation on the branch leading to F/H genotypes. Mol Phylogenet Evol 2015, 93, 44–54. [Google Scholar] [CrossRef]
- Spitz, N.; Mello, F.C.A.; Moreira, A.S.; Gusatti, C.S.; Martins, R.M.B.; Gomes, S.A.; Bello, G.; Araujo, N.M. , Reconstruction of the spatial and temporal dynamics of hepatitis B virus genotype D in the Americas. PLoS One 2019, 14, e0220342. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Sorrentino, C.; Lo Presti, A.; Tanzi, E.; Ciccozzi, M.; Galli, M. , Enigmatic origin of hepatitis B virus: an ancient travelling companion or a recent encounter? World J Gastroenterol 2014, 20, 7622–7634. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Xu, M.; Li, X.; Zhang, Z. , Distribution of hepatitis B virus genotypes and subgenotypes: A meta-analysis. Medicine (Baltimore) 2021, 100, e27941. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. , The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes (Basel) 2018, 9. [Google Scholar] [CrossRef]
- WHO World Health Organization. Global hepatitis report. https://www.who.int/publications/i/item/9789241565455 (09 November 2023).
- Coppola, N.; Alessio, L.; Pisaturo, M.; Macera, M.; Sagnelli, C.; Zampino, R.; Sagnelli, E. , Hepatitis B virus infection in immigrant populations. World J Hepatol 2015, 7, 2955–2961. [Google Scholar] [CrossRef]
- Razavi-Shearer, D.; Gamkrelidze, I.; Pan, C.Q.; Razavi-Shearer, K.; Blach, S.; Estes, C.; Mooneyhan, E.; Razavi, H. , The impact of immigration on hepatitis B burden in the United States: a modelling study. Lancet Reg Health Am 2023, 22, 100516. [Google Scholar] [CrossRef]
- OBMigra Ministério da Justiça e Segurança Pública, Observatório das Migrações Internacionais (OBMigra) - Relatório Anual 2022 https://portaldeimigracao.mj.gov.br/pt/dados/relatorios-a (14 November 2023).
- Lago, B.V.; do Espirito-Santo, M.P.; Costa, V.D.; Marques, V.A.; Villar, L.M.; Lewis-Ximenez, L.L.; Lampe, E.; Mello, F.C.A. , Genetic Diversity of the Hepatitis B Virus Subgenotypes in Brazil. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Lampe, E.; Mello, F.C.A.; do Espirito-Santo, M.P.; Oliveira, C.M.C.; Bertolini, D.A.; Goncales, N.S.L.; Moreira, R.C.; Fernandes, C.A.S.; Nascimento, H.C.L.; Grotto, R.M.T.; Pardini, M.; On Behalf Of The Brazilian Hepatitis, B.R.G. , Nationwide overview of the distribution of hepatitis B virus genotypes in Brazil: a 1000-sample multicentre study. J Gen Virol 2017, 98, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.M.; Simon, D.; Lunge, V.R. , Hepatitis B virus genotypes in Brazil: Introduction and dissemination. Infect Genet Evol 2021, 93, 104936. [Google Scholar] [CrossRef] [PubMed]
- Andernach, I.E.; Nolte, C.; Pape, J.W.; Muller, C.P. , Slave trade and hepatitis B virus genotypes and subgenotypes in Haiti and Africa. Emerg Infect Dis 2009, 15, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Lenders, S. Bolivians, Haitians and Venezuelans – three cases of immigration in Brazil (Bolivianos, haitianos e venezuelanos – três casos de imigração no Brasil). https://br.boell.org/pt-br/2019/04/15/bolivianos-haitianos-e-venezuelanos-tres-casos-de-imigracao-no-brasil#_ftn1 (23 November 2023.
- Motta-Castro, A.R.; Martins, R.M.; Araujo, N.M.; Niel, C.; Facholi, G.B.; Lago, B.V.; Mello, F.C.; Gomes, S.A. , Molecular epidemiology of hepatitis B virus in an isolated Afro-Brazilian community. Arch Virol 2008, 153, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Sitnik, R.; Sette, H., Jr.; Santana, R.A.; Menezes, L.C.; Graca, C.H.; Dastoli, G.T.; Silbert, S.; Pinho, J.R. , Hepatitis B virus genotype E detected in Brazil in an African patient who is a frequent traveler. Braz J Med Biol Res 2007, 40, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Honge, B.L.; Jespersen, S.; Medina, C.; Te Dda, S.; da Silva, Z.J.; Lewin, S.; Ostergaard, L.; Erikstrup, C.; Wejse, C.; Laursen, A.L.; Krarup, H.; Bissau, H.I.V. c. s. g. , Hepatitis B and Delta virus are prevalent but often subclinical co-infections among HIV infected patients in Guinea-Bissau, West Africa: a cross-sectional study. PLoS One 2014, 9, e99971. [Google Scholar] [CrossRef] [PubMed]
- Ingasia, L.A.O.; Wose Kinge, C.; Kramvis, A. , Genotype E: The neglected genotype of hepatitis B virus. World J Hepatol 2021, 13, 1875–1891. [Google Scholar] [CrossRef]
- Pujol, F.; Jaspe, R.C.; Loureiro, C.L.; Chemin, I. , Hepatitis B virus American genotypes: Pathogenic variants ? Clin Res Hepatol Gastroenterol 2020, 44, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Kowalec, K.; Minuk, G.Y.; Borresen, M.L.; Koch, A.; McMahon, B.J.; Simons, B.; Osiowy, C. , Genetic diversity of hepatitis B virus genotypes B6, D and F among circumpolar indigenous individuals. J Viral Hepat 2013, 20, 122–130. [Google Scholar] [CrossRef]
- Mojsiejczuk, L.N.; Torres, C.; Pisano, M.B.; Re, V.; Campos, R.H.; Flichman, D.M. , New pieces on genetic diversity and evolutionary history of hepatitis B virus: Characterization of the novel subgenotype F6. Infect Genet Evol 2017, 47, 140–142. [Google Scholar] [CrossRef]
- Mello, F.C.; Araujo, O.C.; Lago, B.V.; Motta-Castro, A.R.; Moraes, M.T.; Gomes, S.A.; Bello, G.; Araujo, N.M. , Phylogeography and evolutionary history of hepatitis B virus genotype F in Brazil. Virol J 2013, 10, 236. [Google Scholar] [CrossRef]
- Sousa, D.D.; Silva, C.R.S.; Lima Junior, W.P.; Barros, J.A.; Nascimento, I.; Souza, V.C.; Naveca, F.G.; Granja, F. , Phylogenetic analysis and genotype distribution of Hepatitis B Virus (HBV) in Roraima, Brazil. Rev Inst Med Trop Sao Paulo 2018, 60, e35. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. , Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst 2009, 101, 1066–1082. [Google Scholar] [CrossRef]
- Nishiya, A.S.; Levi, J.E.; de Almeida-Neto, C.; Witkin, S.S.; Ferreira, S.C.; Bassit, L.; Sabino, E.C.; Di-Lorenzo-Oliveira, C.; Salles, N.A.; Coutinho, A.S.; Bellesa, M.A.; Rocha, V.; Mendrone-Jr, A. , Occult and active hepatitis B virus detection in donated blood in Sao Paulo, Brazil. Transfusion 2021, 61, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.P.; Matos, M.A.; Silva, A.M.; Lopes, C.L.; Teles, S.A.; Matos, M.A.; Spitz, N.; Araujo, N.M.; Mota, R.M.; Kerr, L.R.; Martins, R.M. , Prevalence, Risk Behaviors, and Virological Characteristics of Hepatitis B Virus Infection in a Group of Men Who Have Sex with Men in Brazil: Results from a Respondent-Driven Sampling Survey. PLoS One 2016, 11, e0160916. [Google Scholar] [CrossRef]
- Oliveira, M.P.; Lemes, P.S.; Matos, M.A.; Del-Rios, N.H.; Carneiro, M.A.; Silva, A.M.; Lopes, C.L.; Teles, S.A.; Aires, R.S.; Lago, B.V.; Araujo, N.M.; Martins, R.M. , Overt and occult hepatitis B virus infection among treatment-naive HIV-infected patients in Brazil. J Med Virol 2016, 88, 1222–1229. [Google Scholar] [CrossRef]
- Matos, M.A.; Ferreira, R.C.; Rodrigues, F.P.; Marinho, T.A.; Lopes, C.L.; Novais, A.C.; Motta-Castro, A.R.; Teles, S.A.; Souto, F.J.; Martins, R.M. , Occult hepatitis B virus infection among injecting drug users in the Central-West Region of Brazil. Mem Inst Oswaldo Cruz 2013, 108, 386–389. [Google Scholar] [CrossRef]
- Martins, T.L.S.; Silva, G.; Silva, C.A.; Gomes, D.O.; Diniz, E.S.B.V.; Carneiro, M.; Pacheco, L.R.; Araujo, N.M.; Zanchetta, M.S.; Teles, S.A.; Caetano, K.A.A. , Hepatitis B and C in Immigrants and Refugees in Central Brazil: Prevalence, Associated Factors, and Immunization. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Gunther, S.; Li, B.C.; Miska, S.; Kruger, D.H.; Meisel, H.; Will, H. , A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol 1995, 69, 5437–5444. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Revill, P.A.; Littlejohn, M.; Matthews, P.C.; Ansari, M.A. , Analysis of genomic-length HBV sequences to determine genotype and subgenotype reference sequences. J Gen Virol 2020, 101, 271–283. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic analysis of HBV sequences inferred by using the Maximum Likelihood method. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). HBV reference sequences are indicated by their accession numbers followed by their genotype/subgenotype. The sequences generated in this study are identified with a black dot. Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. (a) Analysis of the full-length sequences. (b) Analysis of partial S gene region (581 bp). (c) Analysis of partial X and pre-C/C gene regions (428 pb).
Figure 1.
Phylogenetic analysis of HBV sequences inferred by using the Maximum Likelihood method. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). HBV reference sequences are indicated by their accession numbers followed by their genotype/subgenotype. The sequences generated in this study are identified with a black dot. Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. (a) Analysis of the full-length sequences. (b) Analysis of partial S gene region (581 bp). (c) Analysis of partial X and pre-C/C gene regions (428 pb).
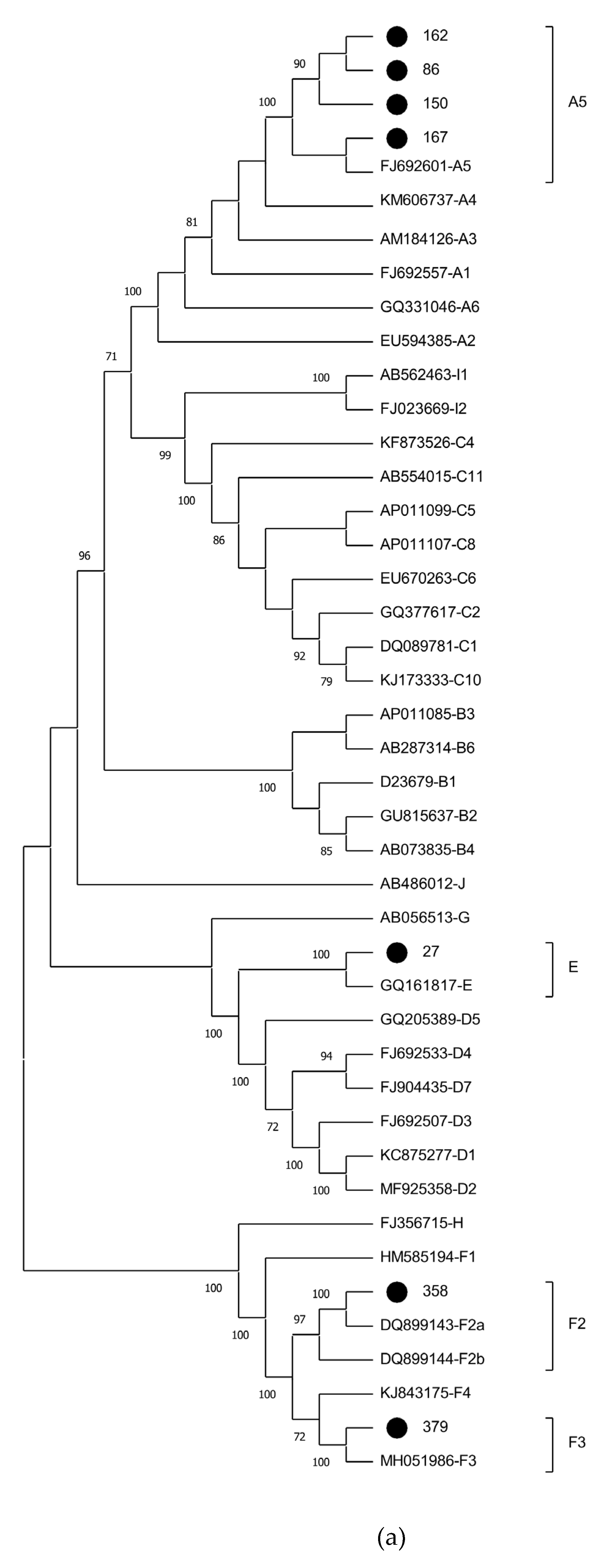
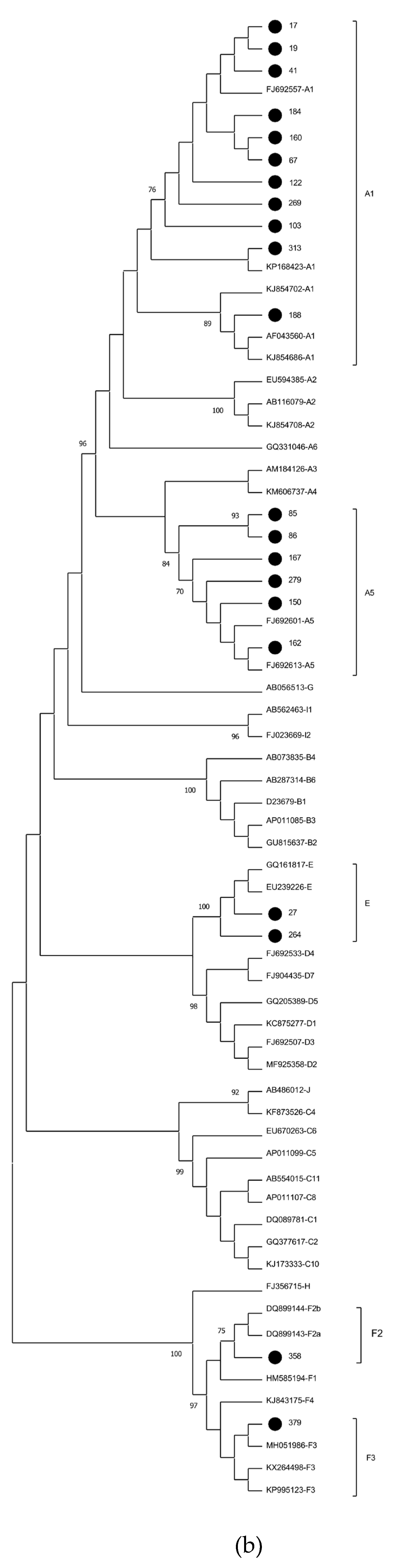
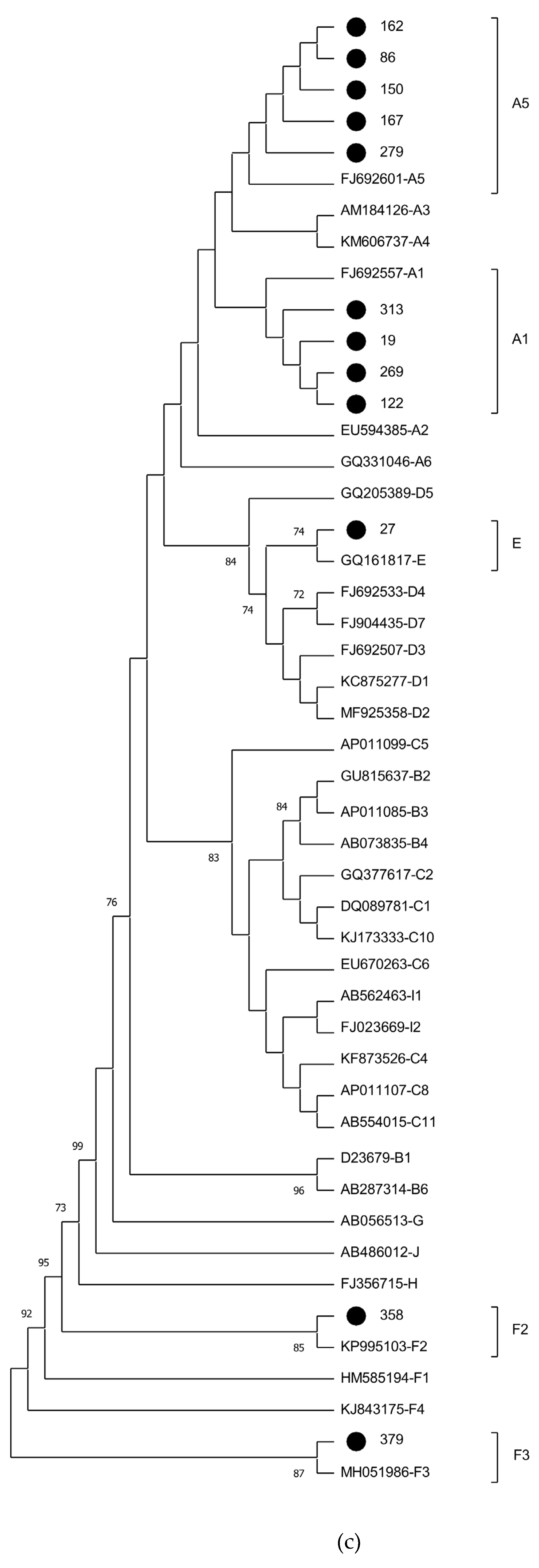
Figure 2.
Phylogenetic analysis of HBV partial S gene region (581 bp) inferred by using the Maximum Likelihood method. Phylogenetic tree of 270 subgenotype A1 sequences (11 sequences recovered in this study and identified with a black dot, and 259 worldwide sequences identified by their accession number and country of origin, obtained from a dataset previously published [9] and subsequently updated). The sequence with GenBank accession number KJ843175 (subgenotype F4) was used as an outgroup. The sequence clusters from Brazil and Haiti are highlighted in green and pink, respectively. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. .
Figure 2.
Phylogenetic analysis of HBV partial S gene region (581 bp) inferred by using the Maximum Likelihood method. Phylogenetic tree of 270 subgenotype A1 sequences (11 sequences recovered in this study and identified with a black dot, and 259 worldwide sequences identified by their accession number and country of origin, obtained from a dataset previously published [9] and subsequently updated). The sequence with GenBank accession number KJ843175 (subgenotype F4) was used as an outgroup. The sequence clusters from Brazil and Haiti are highlighted in green and pink, respectively. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. .
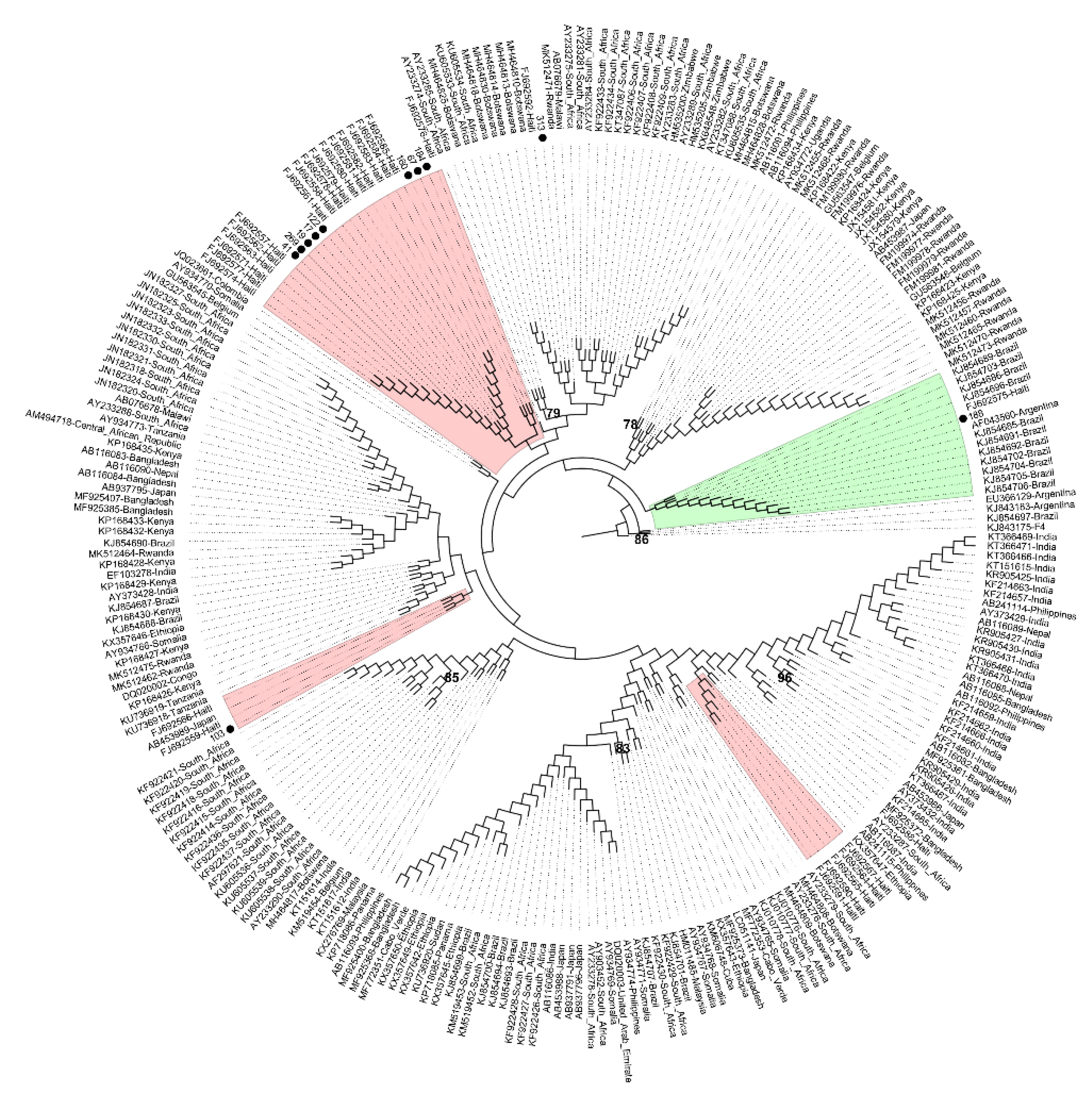
Figure 3.
Phylogenetic analysis of HBV complete genome sequences inferred by using the Maximum Likelihood method. Phylogenetic tree of 36 subgenotype F2 sequences (one sequence recovered in this study and identified with a black dot, and 35 worldwide sequences identified by their accession number and country of origin, obtained from a dataset previously published [9] and subsequently updated). The sequence with GenBank accession number FJ692557 (subgenotype A1) was used as an outgroup. The sequence clusters from Brazil and Venezuela are highlighted in green and yellow, respectively. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. .
Figure 3.
Phylogenetic analysis of HBV complete genome sequences inferred by using the Maximum Likelihood method. Phylogenetic tree of 36 subgenotype F2 sequences (one sequence recovered in this study and identified with a black dot, and 35 worldwide sequences identified by their accession number and country of origin, obtained from a dataset previously published [9] and subsequently updated). The sequence with GenBank accession number FJ692557 (subgenotype A1) was used as an outgroup. The sequence clusters from Brazil and Venezuela are highlighted in green and yellow, respectively. Values at internal nodes indicate percentage of 1000 bootstrap replicates that support the group (values below 70 were hidden). Phylogenetic tree displayed without scale due to logarithmic scaling option ('Toggle scaling of the tree') in MEGA 11 software. .
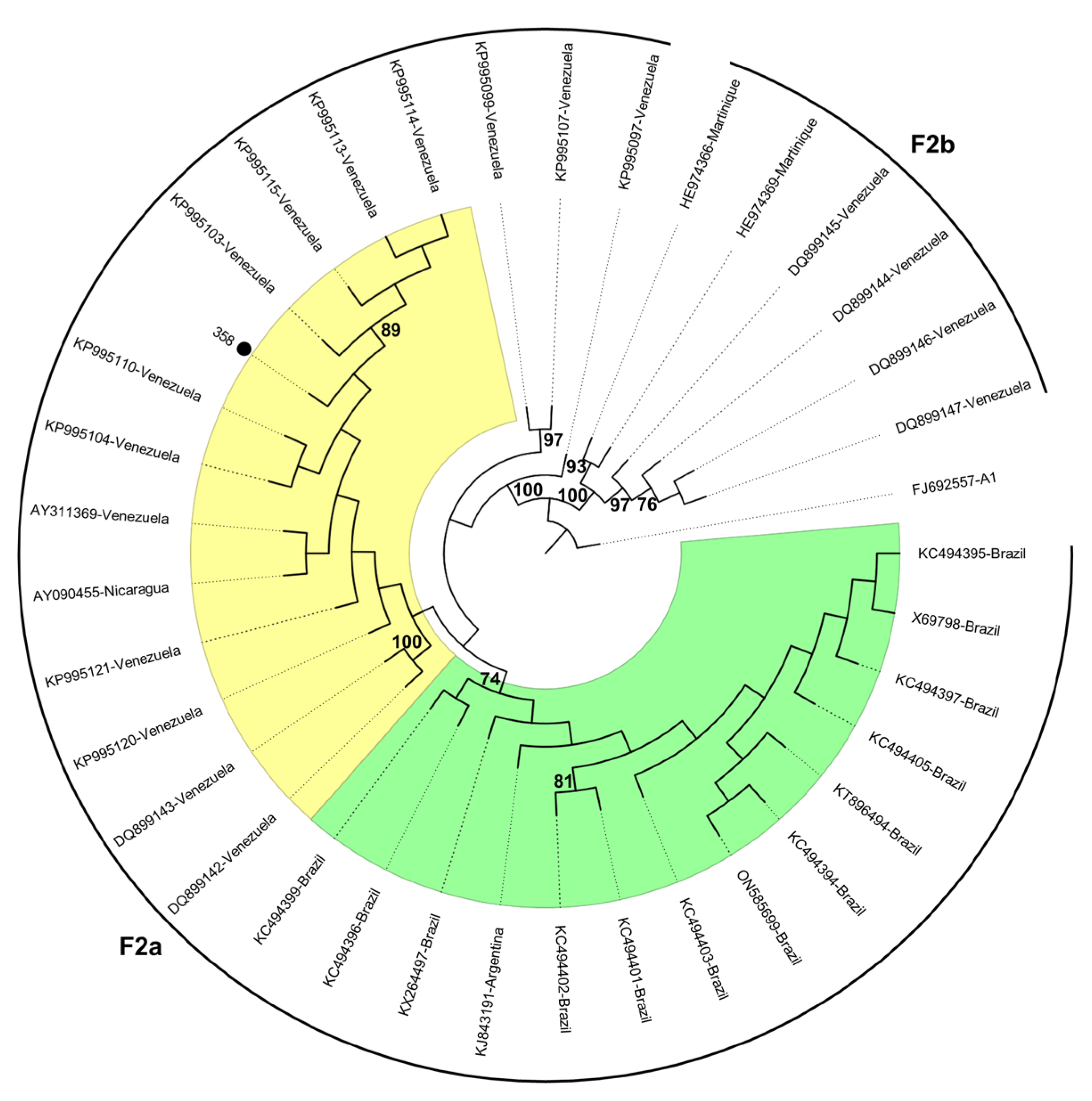

Table 1.
Sociodemographic and virological characteristics of 102 anti-HBc positive immigrants residing in Brazil.
Table 1.
Sociodemographic and virological characteristics of 102 anti-HBc positive immigrants residing in Brazil.
| Variables | n = 102 | (%) |
| Sex | ||
| Male | 70 | 68.6 |
| Female | 32 | 31.4 |
| Age | ||
| 2 – 11 | 0 | 0 |
| 12 – 17 | 0 | 0 |
| 18 – 30 | 38 | 37.3 |
| 31 – 50 | 58 | 56.9 |
| ≥51 | 6 | 5.9 |
| Country of Origin | ||
| Colombia | 1 | 1 |
| Guinea-Bissau | 12 | 11.8 |
| Haiti | 81 | 79.4 |
| Venezuela | 8 | 7.8 |
| Continent of Origin | ||
| Africa | 12 | 11.8 |
| Caribbean | 81 | 79.4 |
| South America | 9 | 8.8 |
| HBsAg | ||
| Positive | 24 | 23.5 |
| Negative | 78 | 76.5 |
| HBV DNA | ||
| Positive | 21 | 20.6 |
| Negative | 81 | 79.4 |
Table 2.
This is a table. Tables should be placed in the main text near to the first time they are cited.
Table 2.
This is a table. Tables should be placed in the main text near to the first time they are cited.
| ID | Country of origin | HBV serology | Amplified genomic region | Mutation sites | Subgenotype | ||||
|---|---|---|---|---|---|---|---|---|---|
| HBsAg | Anti-HBc | HBsAgb | pre-C/Cc | RTd | Other | ||||
| 17 | Haiti | Pos | Pos | S | None | ND | Nonee | A1 | |
| 19 | Haiti | Nega | Pos | S, pre-C/C | None | A1762T/G1764A | Nonee | A1 | |
| 41 | Haiti | Pos | Pos | S | None | ND | Nonee | A1 | |
| 67 | Guine Bissau | Nega | Pos | S | None | ND | Nonee | A1 | |
| 103 | Haiti | Pos | Pos | S | None | ND | Nonee | 12-nt insertion at S region | A1 |
| 122 | Haiti | Pos | Pos | S, pre-C/C | None | None | Nonee | A1 | |
| 160 | Haiti | Pos | Pos | S | None | ND | Nonee | A1 | |
| 184 | Haiti | Pos | Pos | S | None | ND | Nonee | A1 | |
| 188 | Haiti | Nega | Pos | S | None | ND | Nonee | A1 | |
| 269 | Haiti | Pos | Pos | S, pre-C/C | None | A1762T/G1764A | Nonee | A1 | |
| 313 | Haiti | Pos | Pos | S, pre-C/C | None | A1762T/G1764A | Nonee | A1 | |
| 85 | Haiti | Nega | Pos | S | None | ND | Nonee | A5 | |
| 86 | Haiti | Pos | Pos | complete genome | None | A1762T/G1764A, G1896A | None | A5 | |
| 150 | Haiti | Pos | Pos | complete genome | None | A1762T/G1764A | None | A5 | |
| 162 | Haiti | Pos | Pos | complete genome | None | A1762T/G1764A | None | A5 | |
| 167 | Haiti | Pos | Pos | complete genome | None | A1762T/G1764A | None | A5 | |
| 279 | Haiti | Pos | Pos | S, pre-C/C | None | None | Nonee | A5 | |
| 27 | Haiti | Pos | Pos | complete genome | None | G1896A | None | E | |
| 264 | Guine Bissau | Pos | Pos | S | None | ND | Nonee | 12-nt insertion at S region | E |
| 358 | Venezuela | Pos | Pos | complete genome | None | None | None | 21-nt deletion at pre-S2 region | F2 |
| 379 | Venezuela | Pos | Pos | complete genome | None | None | None | F3 | |
aOccult HBV infection (OBI).; bHBsAg amino acid mutations related to immune escape: P120L/Q/S/T, I/T126A/N/S, Q129H/N/R, G130N/R, M133I/L/T, K141E/I, P142L/S, S/T143L, D144A/E/G, G145R.; cPre-C/C nucleotide mutations related to HCC development: T1753V, A1762T/G1764A, G1896A.; dReverse transcriptase (RT) nucleotide mutations related to antiviral resistance: L180M, A181T/V, T184G/S, S202G/I, M204I/V, N236T, M250V.; eOnly RT nucleotide mutations L180M, A181T/V, T184G/S were analyzed. ND, not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



