
Submitted:
13 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
Pumpo is a Simmental breed and an essential livestock resource in the nucleus genetic cattle of Peru. This study provides a draft genome sequence of a top bull using a de novo assembly approach on the Illumina Novaseq X platform, yielding 208 GB of raw sequencing data with 150 bp paired-end reads. The final genome assembly resulted in a size of 2.06 Gb with an N50 contig length of 108 Mb and a completeness of 95.7% according to BUSCO analysis. A total of 973,925 simple sequence repeats (SSRs) were identified, with a predominance of mononucleotide repeats. The genome showed low heterozygosity (0.568%) and moderate repeatability (11.5%), aligning with other Bos taurus genomes. Reference-guided scaffolding improved the assembly quality significantly, producing an N50 scaffold value of 108 Mb. The SSR analysis of the Pumpo genome identified 973,925 SSRs with a frequency of 2,808 SSRs per kilobase, predominantly mononucleotide repeats, and 85,453 found in compound formations. Obtaining knowledge of the genome of a breeding Simmental bull is essential to optimize breeding programs and improve productivity.
Keywords:
Draft-Genome
; Pumpo
; BUSCO
; Genome Assembly
; SSR Analysis
1. Summary
Recent initiatives have brought to light the significance of exploring genetic variation in bovine populations through the lens of pangenomics [1]. The journey began with the landmark sequencing of the Bos taurus genome in 2004, originating from a Hereford individual, laying the foundation for subsequent advancements. Notably, the latest update to this genome, ARS-UCD1.2, showcases the use of continuous long-reading sequencing technology from Pacific Biosciences, underscoring the evolving methodologies driving genomic research forward [2]. In 2021, Heaton et al. [3] presented a reference genome for Fleckvieh-type Simmental cattle, however, they indicated that having an individual reference genome is insufficient to completely capture the range of genetic diversity seen within a species. Additionally, the spread of several Simmental genotypes with origins worldwide makes it challenging to establish the breed’s global standard, although there is no risk of this breed’s gene pool becoming smaller, according to reports [4].
Fleckvieh-Simmental breed, because of its high growth rate, milk yield, milk fat and protein ratio, docility, and adaptability is preferred worldwide in pure and crossbreeding [4]. In Peru, the first individuals arrived in 1970, due to an agreement of the Peru-Germany International Technical Cooperation, additionally, to their previously mentioned characteristics, it has an excellent aptitude for grazing, which is why their breeding has been growing steadily in different parts of the country [5]. In 2020, the National Institute of Agrarian Innovation (INIA for its acronym in Spanish) of the Ministry of Agrarian Development and Irrigation (MIDAGRI) of the Peruvian Government established 10 genetic nuclei across various geographical departments, resulting in the production of 4000 embryos and 710,000 high-quality semen straws of breeds such as Simmental [6] and as per the 2022 National Agricultural Survey, approximately 238,125 cattle farmers in Peru were using high-quality purebred or improved breeds in their herds, mostly because Peruvian cattle population is mainly conformed by creole cattle (64.03%) [7].
“Pumpo”, is a Fleckvieh-Simmental sire located at the Central Genetic Nucleus of INIA, with more than 300 offspring, its semen straws and embryos are highly requested for animal breeders across the country because of their high genetic value (C. Quilcate, pers. comm, May 2024) , also the influence of production traits on fertility can fluctuate widely across herds, breeds, and individuals, but dual-purpose breeds like the Simmental exhibits less susceptibility to herd-level factors compared to dairy breeds such as Holstein-Friesians or Brown Swiss [8,9], facilitating the production of high-quality semen.
We present a draft genome sequence and SSR data mining from a Fleckvieh-Simmental sire (B. taurus), using two algorithms improved through reference-guided scaffolding and MISA for SSR Mining. Assembly and SSR data analysis of a cattle genetic material donor provides valuable information, which is useful for both scientific research and livestock population management. The inception of such endeavors marks a pivotal moment in understanding the intricate genetic landscape of cattle, with implications ranging from breeding programs to agricultural sustainability.
2. Data Description
The total number of raw pair reads was 888,585,052 sequences, with an average length of 150 bp, a GC content of 41.87%, and a total sequencing data output of 208 GB. After the trimming, we retained 86.17% of sequencing data, and more than 765,713,810 million high-quality sequence reads with approximately 182.9 GB of total sequencing data were generated. In addition, the average quality per read was 38.
2.1. Genome Size and K-mer Analysis
The analysis revealed low heterozygosity (0.568%), moderate repeatability (11.5%), and a genome size estimate of 2.06 Gb (Figure 1), which aligns closely with other Bos taurus (ARS-LIC_NZ_Jersey: 2.64 Gb, ARS-LIC_NZ_Holstein-Friesian_1: 2.66 Gb, ARS-UCD1.3: 2.71 Gb, Btau_5.0.1: 2.72 Gb, and Brown Swiss: 2.66 Gb). Specifically, the genome repeat length ranged between 237.8 and 238.2 million base pairs, and the single genome length spanned between 1.82 and 1.82 billion base pairs. The model fit was high, between 96.34% and 97.89% (Table 1), indicating robust and accurate genome assembly.
2.2. Assembly De Novo, Reference-Assisted Scaffolding and Validation
The scaffolding approach significantly improved the quality of the genome assembly. The N50 value increased from 8,350 bp in contigs to 108,009,562 bp in scaffolds, demonstrating a substantial improvement in assembly continuity. The MaSuRCA assembly was enhanced through reference-guided scaffolding, achieving a total length of 2.74 Gb. This assembly included 14,156 contigs (≥1000 bp) with a GC content of 41.87%. The longest contig reached 163,170,176 bp. BUSCO (Benchmarking Universal Single-Copy Orthologs) analysis for the structured genome assembly reveals a total of 8,831 complete BUSCOs, indicating a high level of completeness. Among these, 8,688 are complete and single-copy BUSCOs. Additionally, there are 143 complete and duplicate BUSCOs, indicating potential regions of genome duplication. The analysis also identified 194 fragmented BUSCOs and 201 missing BUSCOs.
Comparing the initial contig assembly to the scaffold assembly highlights substantial improvements in the genomic assembly. In the initial contig assembly, there were 4,025 complete BUSCOs (43.6%), including 3,921 complete and single-copy BUSCOs (42.5%) and 104 complete and duplicated BUSCOs (1.1%). Additionally, the contig assembly had 2,058 fragmented BUSCOs (22.3%) and 3,143 missing BUSCOs (34.1%). In contrast, the scaffold assembly showed significant enhancement, with 8,831 complete BUSCOs (95.7%), including 8,688 complete and single-copy BUSCOs (94.2%) and 143 complete and duplicated BUSCOs (1.5%). The scaffold assembly also exhibited fewer fragmented and missing BUSCOs, with 194 (2.1%) and 201 (2.2%), respectively.

Table 2.
Statistics of the completeness of the de novo assembly of the Pumpo cattle genome.
| Statistic | Contigs | Scaffolds |
|---|---|---|
| N50 | 8,350 | 108,009,562 |
| N90 | 2,324 | 52,732,619 |
| L50 | 86,076 | 11 |
| L90 | 307,212 | 26 |
| Largest contig | 108,709 | 163,170,176 |
| Total length | 2,582,951,014 | 2,735,224,841 |
| GC (%) | 41.9 | 41.87 |
| # contigs (≥1000 bp) | 419,259 | 14,156 |
| # contigs (≥5000 bp) | 172,406 | 1,754 |
| # contigs (≥10,000 bp) | 63,792 | 623 |
| # contigs (≥25,000 bp) | 7,999 | 161 |
| # contigs (≥50,000 bp) | 432 | 68 |
| # N’s per 100 kbp | 0 | 4820.63 |
Table 3.
Summary of the BUSCO approach in the Pumpo assembly (contigs and scaffolds).
| Terms | Contigs | Scaffold |
|---|---|---|
| Complete BUSCOs | 4,025 (43.6%) | 8,831 (95.7%) |
| Complete and single-copy BUSCOs | 3,921 (42.5%) | 8,688 (94.2%) |
| Complete and duplicated BUSCOs | 104 (1.1%) | 143 (1.5%) |
| Fragmented BUSCOs | 2,058 (22.3%) | 194 (2.1%) |
| Missing BUSCOs | 3,143 (34.1%) | 201 (2.2%) |
2.3. SSR Data Mining
Simple sequence repeat (SSR) analysis revealed a heterogeneous distribution based on repeat unit size (Figure 2). Most SSRs were mononucleotide in size, with a total of 578,874 sequences, making it the most abundant. After this, the dinucleotide SSRs represented 250,768 sequences. Trinucleotide SSRs were less frequent, with 118,909 sequences. There was a significant reduction in the abundance of tetranucleotide and pentanucleotide unit sizes, with 11,348 and 13,704 sequences, respectively. Finally, hexanucleotide SSRs were the least common, with only 322 sequences. The summary of SSR in the Pumpo genome revealed a total of 973,925 identified SSRs, with a frequency of 2,808 SSRs per kilobase. Additionally, 85,453 SSRs were found in compound formations (Table 4).
3. Methods
3.1. Sample Collection and DNA Extraction
A healthy Simmental–Fleckvieh bull (Peruvian National Register Number 135, born in 2016) from the Central Genetic Nucleus of INIA, located at the Donoso Agricultural Experiment Station (EEA Donoso in Spanish) in Huaral, Lima (128 masl; 11°31′18″ S and 77°14′06″ W), was the focus of this study. This government facility houses a genetic nucleus herd. The blood sample was collected from the bull’s tail using vacutainers with EDTA as an anticoagulant and immediately transported to the laboratory for DNA extraction. Pumpo, the bull, has produced 30,382 semen straws from May 2021 to the present, as recorded in data logs. This research complied with Peruvian National Law No. 30407: “Animal Protection and Welfare”. Following the manufacturer’s protocols, genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Fitchburg, WI, USA). The extracted DNA’s integrity was verified via agarose gel electrophoresis, and its concentration was measured using a Qubit 2.0 Fluorometer (ThermoFisher Scientific, Waltham, MA, USA).
3.2. DNA Sequencing and Estimation of Genome Size
A 300 bp insert size library was constructed for paired-end DNA sequencing on the Illumina Novaseq X platform by GENEWIZ (South Plainfield, NJ, USA), yielding approximately 2x150 bp reads. To enhance the draft genome assembly, a reference-scaffolding methodology was utilized, and raw reads were assessed for quality using FastQC v.0.11.9 software. Quality control measures, including trimming (Phred Q > 25) and adapter removal, were executed with Trimmomatic v.0.36 [10] and TrimGalore [11], respectively. For the genomic survey, Jellyfish v.2.0 [12] was employed. GenomeScope v.1.0.0 [13] was used to estimate genomic parameters such as genome size, repeat content, and heterozygosity rate, utilizing the output from Jellyfish. The k-mer frequency distribution analysis revealed a common single-peak pattern, with k-depth calculations performed accordingly.
3.3. De Novo Assembly and Validation
For the de novo assembly, two distinct algorithms were employed: SOAPdenovo2 v.2.04 [14] and MaSuRCA v.4.0.6 [15]. These assemblers were chosen due to their varied methodological approaches, which allowed for a comparative analysis to identify the most effective algorithm for this genome. SOAPdenovo2 is known for its rapid and efficient assembly of short reads into contigs, whereas MaSuRCA utilizes a hybrid strategy to produce longer contigs and scaffolds, particularly in complex genomic regions. Subsequently, QUAST v.5.2.0 [16] was used to generate assembly statistics. MaSuRCA produced superior assembly metrics and was further improved using Samba Scaffolder v.1.0 [17] for scaffolding and gap-filling. Scaffolding based on the reference genome utilized the B. taurus genome (GenBank: GCA_002263795.4). Additional QUAST analysis was performed on the resulting scaffold output.
Assembly validation was conducted using two distinct methods: (i) errors in the assembly were identified by remapping filtered paired-end Illumina reads with Bowtie2 v.2.4.2 [18] and SamTools v.1.7 [19], and (ii) genome assembly completeness and gene space were evaluated using the BUSCO [20] strategy with a mammalian-specific profile consisting of 4,104 single-copy genes expected in mammals. Mitochondrial DNA sequences were removed following BLAST v.2.2.26 [21] searches against mitochondrial sequence databases. Contaminated scaffolds and vectors were ultimately discarded before submission to the NCBI database.
3.4. SSR-Mining
For the extraction and identification of SSR, MISA (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa) was used. Monomer (one nucleotide, n > 12), dimer (two nucleotides, n > 6), trimer (three nucleotides, n > 4) , tetramer (four nucleotides, n > 3), pentamer (five nucleotides, n > 3) and hexamer (six nucleotides, n > 3) motifs were searched in the sequences.
Author Contributions
Conceptualization, C.I.A. and W.A.; methodology, D.R. R.E., D.F., Y.R. and W.A.; software, R.E.; validation, W.A. D.C., Y.R., and D.R.; formal analysis, C.Q. and J.M.; investigation, R.E., D.F., Y.R., C.I.A., H.V. and W.A.; resources, J.L.M.; data curation, R.E., D.F. and C.I.A.; writing—original draft preparation, R.E., D.F., Y.R., C.I.A. and C.Q.; writing—review and editing, R.E., D.F., Y.R., W.A. and C.I.A.; visualization, D.V., H.V. and C.Q.; supervision, D.C. and C.I.A.; project administration, C.Q. and D.C.; funding acquisition, J.L.M., H.V., C.I.A. and W.A.
Funding
This research was funded by the following research project: “Mejoramiento de la disponibilidad de material genético de ganado bovino con alto valor a nivel nacional 7 departamentos” of the Ministry of Agrarian Development and Irrigation (MIDAGRI) of the Peruvian Government, with grant number CUI 2432072.
Institutional Review Board Statement
The study was conducted according to Peruvian National Law No. 30407: “Animal Protection and Welfare”.
Data Availability Statement
The associated Bioproject, Biosample, and Sequence Read Archive (SRA) numbers are PRJNA1097623, SAMN40874274, and SRR29269954, respectively.
Acknowledgments
We acknowledge the ‘PROMEG NACIONAL’ team for supporting the logistic activities. C.I.A. thanks Vicecerrectorado de Investigación of UNTRM.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bickhart, D.M.; The Bovine Pan-Genome Consortium. Nick Bickhart’s. Available online: https://njdbickhart.github.io/ (accessed on 5 June 2024).
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Heaton, M.P.; Smith, T.P.L.; Bickhart, D.M.; Vander Ley, B.L.; Kuehn, L.A.; Oppenheimer, J.; Shafer, W.R.; Schuetze, F.T.; Stroud, B.; McClure, J.C.; et al. A Reference Genome Assembly of Simmental Cattle, Bos taurus taurus. J. Hered. 2021, 112, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Koç, A. A review on Simmental Raising: 1. Simmental raising in the World and in Turkey, Adü Ziraat Derg., 2016, 2, 97–102. [CrossRef]
- AgroPerú. Avanza la crianza de vacunos Fleckvieh Simmental. Available online: https://www.agroperu.pe/avanza-la-crianza-de-vacunos-fleckvieh-simmental/ (accessed on 5 June 2024).
- 2020. Available online: https://www.inia.gob.pe/2020-nota-089/ (accessed on 5 June 2024).
- Instituto Nacional de Estadística e Informática IV Censo Nacional Agropecuario 2012. Available online: http://censos.inei.gob.pe/Cenagro/redatam/# (accessed on 5 June 2024).
- Toledo-Alvarado, H.; Cecchinato, A.; Bittante, G. Fertility traits of Holstein, Brown Swiss, Simmental, and Alpine Grey cows are differently affected by herd productivity and milk yield of individual cows. J. Dairy Sci. 2017, 100, 8220–8231. [Google Scholar] [CrossRef] [PubMed]
- Windig, J.J.; Calus, M.P.L.; Veerkamp, R.F. Influence of herd environment on health and fertility and their relationship with milk production. J. Dairy Sci. 2005, 88, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 2047–217X-1-18. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Salzberg, S.L. The SAMBA Tool Uses Long Reads to Improve the Contiguity of Genome Assemblies. PLoS Comput. Biol. 2022, 18, e1009860. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of k-mers in the draft genome of Pumpo cattle.
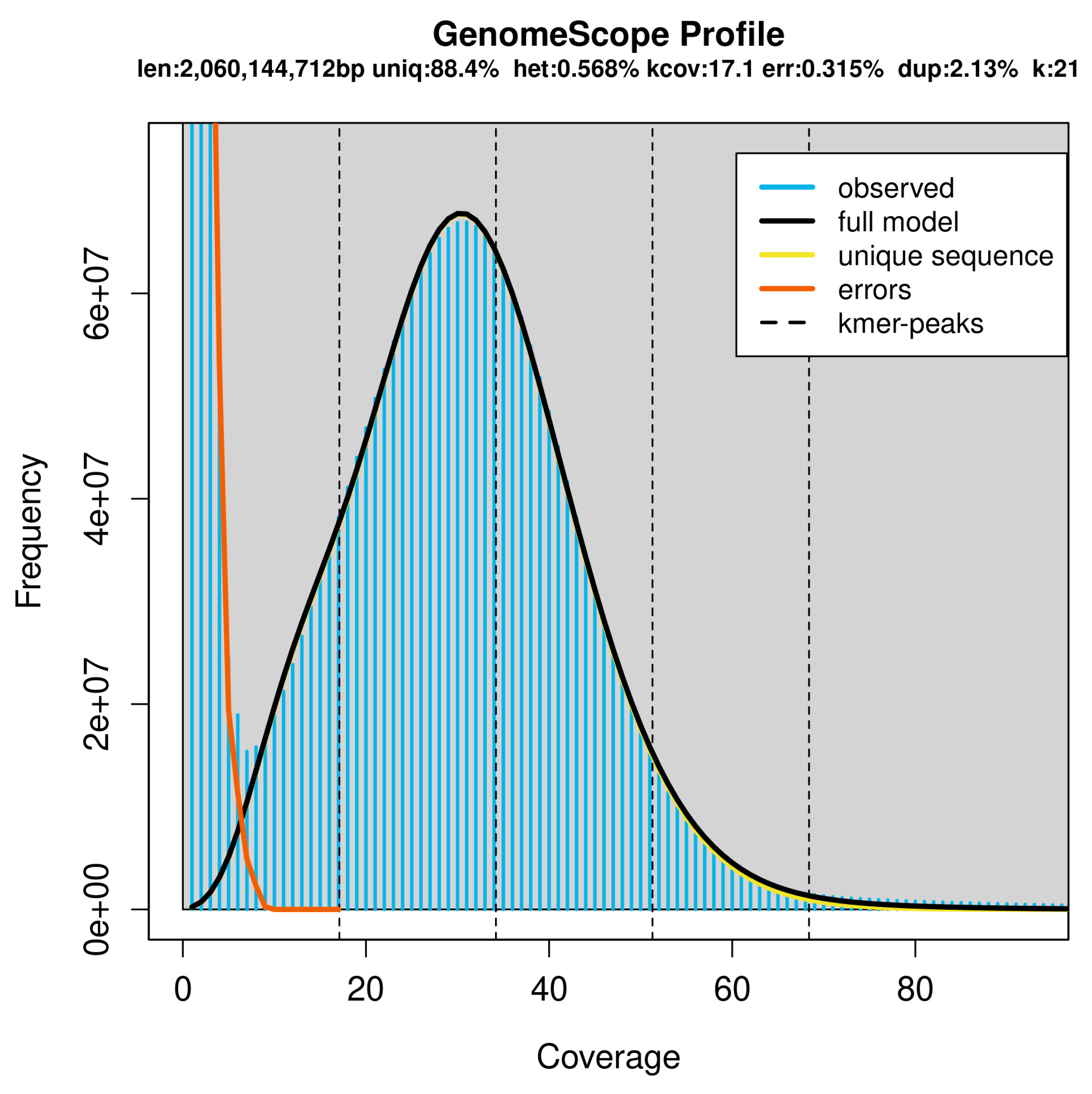
Figure 2.
Distribution of SSR in the draft genome of Pumpo.
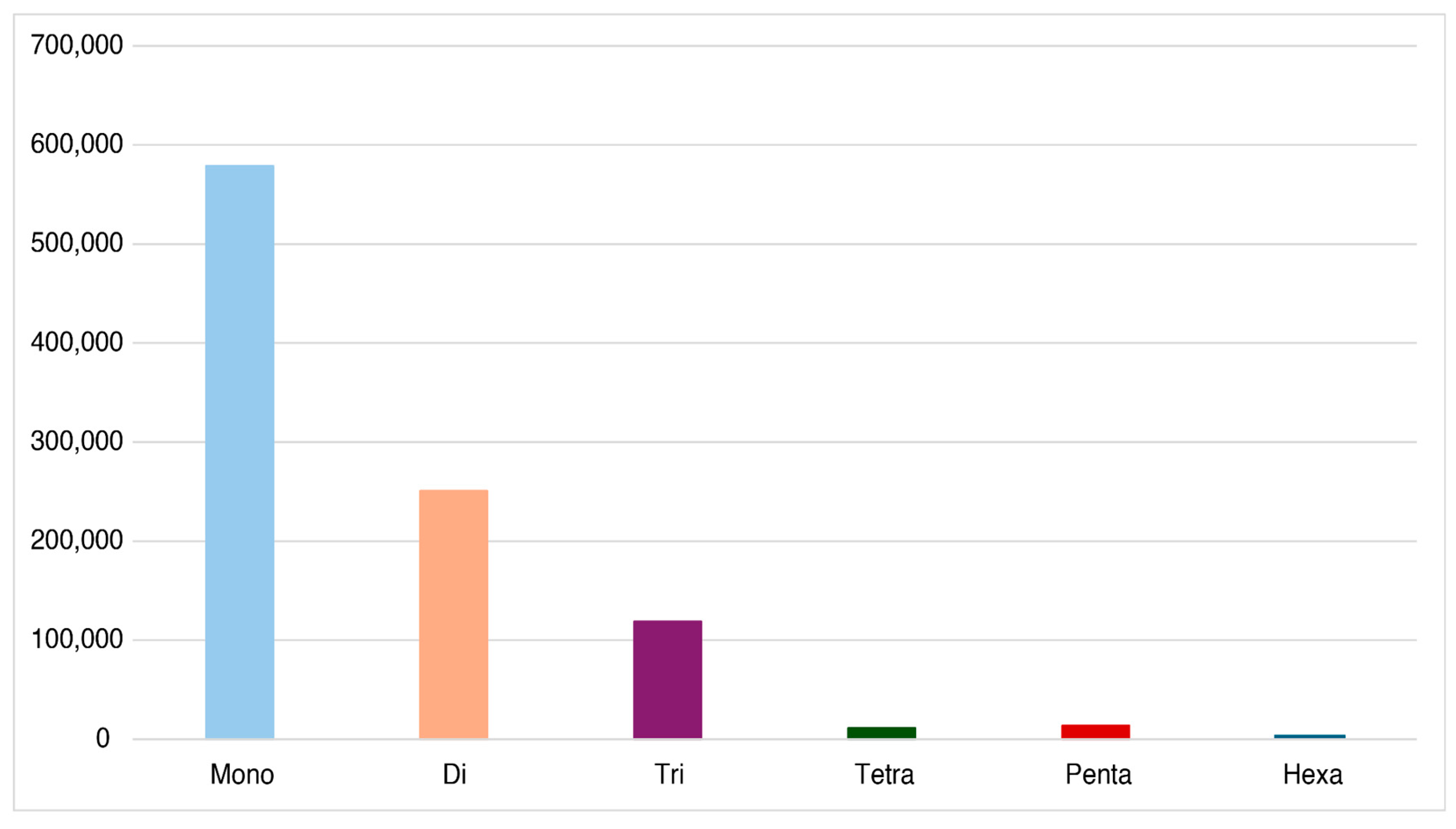
Table 1.
Summary of GenomeScope analysis.
| Property | Min. | Max. |
|---|---|---|
| Heterozygosity | 0.56% | 0.57% |
| Genome haploid length | 2,056,357,349 bp | 2,060,144,712 bp |
| Genome repeat length | 237,799,733 bp | 238,237,708 bp |
| Genome unique length | 1,818,557,616 bp | 1,821,907,004 bp |
| Model fit | 96.34% | 97.89% |
| Read error rate | 0.32% | 0.32% |
Table 4.
Summary of the SSR in the Pumpo genome.
| Type | Pumpo |
|---|---|
| Total number of identified SSRs | 973,925 |
| Frequency (SSR/Kb) | 2,808 |
| Number of SSRs present in compound formation | 85,453 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



