
Submitted:
15 June 2024
Posted:
17 June 2024
You are already at the latest version
Abstract
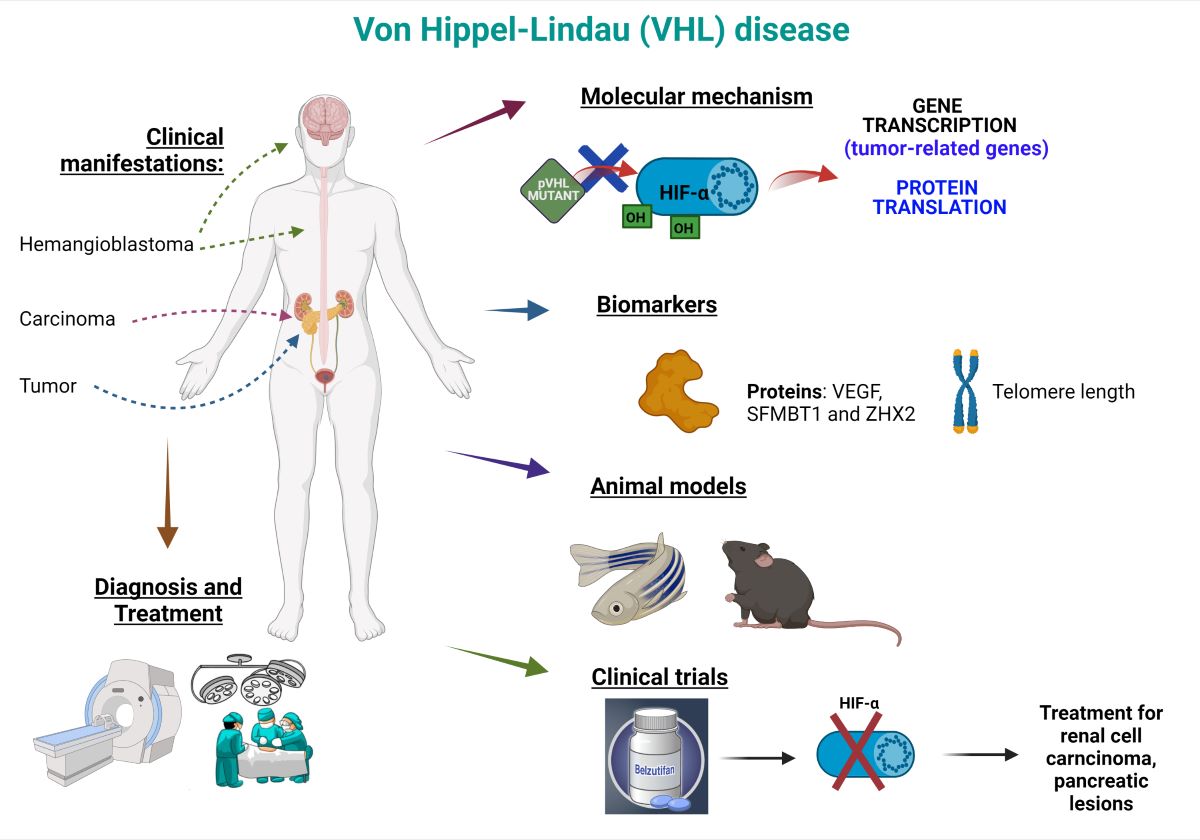
Von Hippel-Lindau disease (VHL) is a multiple organ neoplastic syndrome with autosomal dominant transmission, complete penetrance, and variable expression caused by mutations in the VHL gene. Although VHL disease is hereditary in many cases, new mutations cause up to 20 % of the incidence. Mutations in VHL may cause the development of cysts and tumors in many organs, including brain and spinal cord hemangioblastoma, renal cell carcinoma (RCC), retinal heman-gioblastoma (RH), pheochromocytoma, epididymal and broad ligament cystadenomas, endo-lymphatic sac tumor, pancreatic neuroendocrine tumors, and renal and pancreatic cysts. VHL is a syndrome associated with functional inactivation of the Von Hippel–Lindau protein (pVHL). pVHL is a tumor suppressor mainly known for its role as a regulator of hypoxia-inducible factor (HIF) activity. The prevalence of VHL disease is between 1 in 39 000 and 1 in 91 000 individuals in different regional populations. Here, we review the etiology, epidemiology, pathophysiology, genetics, clinical manifestations, diagnosis, and current treatments, as well as the molecular aspects of this disease. We also discuss the animal models used to study this disease, VHL biomarkers, and current VHL clinical trials for VHL according to NIH.
Keywords:
VHL
; pVHL
; tumor suppressor
; HIF
; treatments
; biomarkers
; clinical trials
1. Introduction
Von Hippel-Lindau disease (VHL) is a rare genetic disorder with autosomal dominant transmission resulting from deletions or mutations in the VHL gene. VHL disease affects 1 in 36,000 people (10,000 cases in the US and 200,000 cases worldwide). 20% of the cases are de novo. The age of onset is 26 years, and 97% of individuals with a VHL gene mutation have symptoms at 65 years of age. The VHL disease affects males and females and all ethnic groups equally. Patients with VHL disease may experience tumors and/or cysts in up to ten parts of the body, including the brain, spine, eyes, kidneys, pancreas, adrenal glands, inner ears, reproductive tract, liver, and lung. Most of their tumors are benign, but still can cause severe damage to the affected body structure. The manifestation of VHL in each patient can vary; thus, it is essential to check regularly for possible VHL manifestations throughout a person's lifetime. In general, the most common treatment is surgical, and it is considered that with good monitoring and early detection, it is possible to treat the disease and reduce its harmful consequences.
VHL can also cause malignant tumors; therefore, it is considered to be part of a group of familial cancer risk factors, which are transmitted genetically.
The current diagnosis is mainly made through DNA testing to verify VHL gene mutations.
The most common symptom of VHL is hemangioblastomas. These are benign tumors that occur in the brain, spinal cord, and retina. Hemangioblastomas are benign.
The VHL gene product (pVHL) acts as a key regulator of cellular hypoxia. pVHL through the HIF (hypoxia-inducible factor) complex is indirectly responsible for increased levels of growth factors, including vascular endothelial factor (VEGF), platelet-derived growth factor (PDGF), and transforming growth factor alpha (TGF-β). In the case of a nonfunctioning gene such as in VHL disease, regulation of the HIF complex does not occur. These results in increased levels of various growth factors allowing for enhanced blood vessel growth (angiogenesis) and tumor formation.
Due to multiple organ involvement, the symptoms and manifestations of VHL overlap with a wide range of diseases. These include the following: Kidney: sporadic kidney cancer, Birt-Hogg-Dubé (BHD) syndrome, hereditary leiomyomatosis and renal cell carcinoma (HLRCC), tuberous sclerosis complex, succinate dehydrogenase subunit (SDH); Adrenal or pheochromocytoma: succinate dehydrogenase subunit (SDH), multiple endocrine neoplasia 2 syndrome, types A and B (MEN2A and MEN2B); Inner ear: Meniere's disease; Pancreas: pancreatic cancer; Retina: retinal hemangioblastomas are unique to VHL. The presence of a retinal hemangioblastoma leads to a clinical diagnosis of VHL; Brain or spine: hemangioblastomas in the brain or spine are different from other forms of brain or spine tumors and their diagnosis is considered a criterion for a VHL DNA test.
2. Von Hippel-Lindau (VHL) Disease: Highlights
The VHL disease is a multiorgan neoplastic syndrome with autosomal dominant transmission, complete penetrance, and variable expression caused by mutations in the VHL gene. Although VHL disease is hereditary in many cases, new mutations cause up to 20% of the cases [1]. Pathogenic variants in the VHL gene predispose individuals to tumors and cysts in many organ systems. These include brain and spinal cord hemangioblastoma, renal cell carcinoma (RCC), retinal hemangioblastoma (RH), pheochromocytoma, epididymal and broad ligament cystadenomas, endolymphatic sac tumor, pancreatic neuroendocrine tumors, and renal and pancreatic cysts [2,3].
2.1. History
Observations relating to VHL disease first appeared in the 19th century. For example, in 1879, Panas and Rémy illustrated a retinal hemangioblastoma for the first time. Subsequently, von Hippel contributed to the description of the disease through clinical data obtained from a patient with multiple retinal lesions that appeared over several years. From all her observations, von Hippel concluded that the primary retinal lesion was a hemangioblastoma. Later, in 1926, Lindau published a monograph. In this document, he brought together into one coherent entity the retinal, cerebral, and visceral components of this disease. Lindau designated "central nervous system angiomatosis" as this disease entity because he believed that visceral concomitants did not manifest by symptoms. Finally, ‘Lindau’s disease’ was defined as an association of cerebellar hemangioblastoma with one or more lesions: retinal hemangioblastoma (the ‘von Hippel tumor”), spinal cord hemangioblastoma, pancreatic cysts, renal and epididymal abnormalities, and the existence of at least one other family member with the disease [4]. This term changed to the ‘von Hippel Lindau (VHL)’ disease in the 1970s. In 1988, a report explained the linkage of the VHL gene to chromosome 3 [5], and, in 1993, a research group identified the VHL tumor suppressor gene [6].
2.2. Etiology
Von Hippel-Lindau syndrome (VHL) is a syndrome associated with functional inactivation of the von Hippel–Lindau protein (pVHL) [7]. The von Hippel–Lindau protein (pVHL) is a tumor suppressor mainly known for its role as a regulator of hypoxia-inducible factor (HIF) activity [8]. The homonymous VHL gene codifies the pVHL protein, localizes on chromosome 3p25, and is expressed in fetal and adult tissues [9]. On the other hand, VHL-related tumor development follows Knudson’s ‘two-hit model’ of tumorigenesis [10]: Patients with VHL are born with a germline mutation in one copy of their VHL gene in all cells (first hit or event). Somatic mutation in the other copy of the VHL gene (second hit or event) initiates tumor development in a particular cell [11,12].
2.3. Epidemiology
The disease reports penetrance of > 80% by age 60 and an approaching 100% by age 75. The prevalence of VHL disease is between 1 in 39 000 and 1 in 91 000 individuals in different regional populations [13,14]. Most are >20 years old and are prone to selection bias due to the inclusion of clinically affected patients with VHL disease diagnosed before genetic testing was available. For example, VHL disease had a birth incidence of 1 in 36,000 live births in Eastern England and prevalences of 1 in 39,000 individuals in South-West Germany, and 1 in 53 000 individuals in eastern England [15,16]. However, in Denmark, in an unselected cohort of all known Danish carriers of a disease-causing variant is estimated a VHL prevalence of 1 in 46 900 individuals and an incidence of 1 in 27 300 live births [17].
2.4. Pathophysiology
Von Hippel-Lindau syndrome (VHL) is a rare hereditary cancer characterized by the development of benign or malignant tumors in specific topographic locations. In this sense, central nervous system hemangioblastoma and clear cell renal cell carcinoma (RCC) are the most frequently tumors [3].
Tumorigenesis in VHL syndrome is linked to the loss of function of the VHL tumor suppressor protein in cell differentiation [18] where hypoxia-inducible factors (HIF1 and HIF2) are activated and accumulate in the cell [19,20]. The consequences of this up-regulation include transcriptional activation of genes containing hypoxia-responsive elements [21,22]. However, it remains unclear why the loss of VHL function and subsequent HIF activation lead to tumorigenesis. An explanation suggests that the ‘second hit’ would cause loss of pivotal VHL function during organ development leading to maldeveloped structures that represent prerequisites for tumor formation [23].
3. Genetics
3.1. VHL Gene and Protein
In 1988, Seizinger and colleagues mapped the VHL gene. The findings showed that the VHL gene is located on the short arm of chromosome 3 in the 3p 25-25 region (Figure 1). Moreover, the VHL gene has 14,500 base pairs of genomic DNA, and 852 nucleotides with three exons that can generate two mRNAs through a process that causes the loss of exon 2. This gene is conserved in rodents, flies, and worms [24,25].
The VHL gene consists of three exons with the capacity to generate two mRNA transcripts. The mRNA1 transcript is composed of exons 1, 2, and 3, and the mRNA2 transcript is composed of exons 1 and 3 (Figure 2). The mRNA1 transcript encodes two pVHL proteins. When translation occurs, the complete protein from 1-213 amino acids (aa) with a molecular mass of 24 ~ 30 kDa, also known as pVHL30, and a small peptide from 54 to 213 aa is translated through an alternative start codon at codon 54 with a molecular mass of 18 ~ 19 kDa known as pVHL19. Both proteins (pVHL30 and pVHL19) are localized in the cytoplasm and cell nucleus, respectively [26,27].
The structure of the pVHL protein has two domains: the β domain that ranges from 63 to 155 aa and the α domain that includes amino acids 156 to 193, as shown in Figure 3. On the other hand, the protein pVHL30 contains an acidic domain located in amino acids 1-54, absent from the pVHL19 protein. The function of pVHL30 is present in several events. For example, protein degradation via the proteasome is the most studied, and in addition to both isoforms, it appears to maintain tumor suppression activity [28].
3.2. Inheritance
The VHL disease is autosomal dominant, that is, it is transmitted from parents to children. However, the disease can occur sporadically. The mutation is inherited when one parent has the VHL gen altered (mutated), so each child has a 50% chance of inheriting it. Consistent with the dominant inheritance pattern, it is enough to have an altered gene to develop the disease. Thus, an affected parent (who therefore has a mutated gene and a normal one) could pass on the mutated gene to his offspring, who in turn will develop the disease despite having the other normal gene (see figure 4) [29]. VHL disease also is autosomal, which means that men and women can suffer from it equally (Figure 4). People with parents, brothers, or sisters with VHL have a 50% risk of disease. If the person has an uncle, cousin, or grandparent with VHL, they are also at risk for developing VHL. Moreover, it has been reported the presence of the disease in individuals without family background, therefore it is necessary a differential diagnosis in each individual [30].
3.3. Mutations
In VHL gene mutation carriers the development of the disease begins with the loss or inactivation of the wild-type allele. Cytogenetic abnormalities, mutations, or hypermethylation that cause inactivation of the remaining ‘healthy’ allele (Figure 5). Currently, it is unknown why the second event occurs in some tissues and not in others [31]. On the other hand, some studies show that patients develop retinal hemangioblastoma with complete deletion of the VHL gene less frequently than those with a single amino acid substitution in the VHL gene. In addition, there were no differences between patients with a truncated variant and those with a single amino acid substitution [32,33].
Germline VHL mutations cause different protein defects that result in the clinical heterogeneity of VHL disease. For example, type 1 VHL disease (without pheochromocytoma) is associated with mutations that cause the complete unraveling of the protein structure (missense mutations in the hydrophobic core of the VHL protein, protein-truncating mutations, and partial gene deletions). On the other hand, VHL disease type 2 (with phaeochromocytoma) is associated with missense mutations at pVHL protein binding sites, causing local defects [34]. Table 1 shows several examples of the relationship between the type of VHLdisease and some mutations.
Furthermore, Nordstrom-O’Brien reports a mutation spectrum of 43.2% in exon 1, 17% in exon 2, and 39.8% in exon 3 [48]. Mutations are heterogeneous and are widely distributed throughout the coding sequence, especially in exons 1 and 3. Missense mutations are the most common type of mutation (61%), followed by frameshift (15.7%), nonsense (13.2%), in-frame insertions / deletions (6.6%) and splicing mutations (3.5%) [73]. Finally, wild-type allele inactivation may arise due to allelic loss, hypermethylation, or point mutations [74,75].
pVHL has two domains: domain alpha and domain beta. Most pathogenic missense mutations are found in two regions of the VHL protein that interact with the elongin C-binding site. In line with this, several tumor mutations locate in alpha-domain, in the amino acids that contact with elongin C (residues 158–184 of the α domain) [76]. The rest of the mutations are in the beta-domain (residue 65–117 of the β domain) [65,77]. Reports indicate that mutations in Ser111 and Trp117 of pVHL block HIF binding [78]. Most recurrent mutations are the result of de novo mutations in mutation-susceptible regions, known as hot spots. The most common hot spots are delPhe76, Asn78Ser, His, Thr, Pro86Leu, Arg 161 Stop, Cys 162Tyr, Arg167Gln, Trp, and Leu78Pro [37]. Fourteen mutations (5.5 %) locate at Ser65, nine mutations (3.5 %) at Trp117, (3.1 %) at Phe76, (2.8 % each) at Asn78, Ser80, Leu135, and Arg161, (2.4 %) at His115, and five mutations (2 % each) at Gly114 and Leu184 [79]. Common germline mutations in VHL are delPhe76, Asn78Ser, Argl61Stop, Arg167Gln, Argl67Trp, and Leu178Pro [27,37]. Moreover, it has been elucidated the effect of VHL Arg200Trp mutation on HIF-1 interaction and ubiquitination [80]. In line with this, specific missense mutations (Arg167Trp and Arg167Gln) are associated with a high risk (62%) of phaeochromocytoma [34]. Furthermore, amino acid substitution (Glu70Lys) at the HIF-α binding site increments specific risk of developing CNS hemangioblastoma [65], while missense mutations (Leu118Pro or Arg167Trp) are associated with renal cancer [81] (Figure 6). Finally, the Try98His and Asn (Asn78Ser) associate with a low risk of RCC (type 2A phenotype).
4. Manifestations, Diagnosis, and Treatment of VHL Disease
Symptoms of VHL disease vary among patients and depend on the size and location of the tumors. On the other hand, diagnosis can be made based on specific clinical criteria (signs, symptoms, and imaging), or when molecular genetic testing reveals a change in the VHL gene. Finally, treatment depends on the location and size of the tumors and usually involves surgical removal of the tumors. Radiation therapy may be used in some cases (Table 2).
5. Molecular Basis of VHL Disease
The best known pVHL function is the regulation of hypoxia-inducible factor-alpha (HIF-α) protein levels through degradation under normoxic conditions [99]. The interaction between pVHL and HIF-α requires the hydroxylation dependent on prolyl-4 hydroxylase domain enzymes (PHD1, -2 and -3) of at least one of two specific proline residues of HIF-α [100]. Subsequently, HIF-α is ubiquitylated and degraded via the proteasome. Under hypoxic conditions or in the presence of mutant pVHL [43,101], the pVHL complex cannot recognize HIF-α, then HIF-α accumulates in the cytoplasm. In the cytoplasm, HIF-α forms a heterodimer with HIF-β. Subsequently, the heterodimer translocates to the nucleus with the transcriptional coactivator p300, where it binds to response elements (HRE) [102] and promotes the transcription of many genes involved in angiogenesis, glucose metabolism, cell survival and tumor progression [103] (Figure 7).
However, the pathogenesis of clear-cell type renal cell carcinomas (ccRCC) implies mTOR complex 1 (mTORC1). For example, reports show that mTORC1 activates in 60% to 85% of ccRCCs [104].
mTOR is a serine/threonine protein kinase. mTOR nucleates two different complexes, mTORC1 and mTORC2 [105]. mTORC1 is composed of mTOR, the regulatory associated protein of mTOR (RAPTOR), and the protein mammalian lethal with sec-teen protein 8 [106]. Some mTORC1 substrates are S6 kinase 1 (S6K1) and the eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1). Finally, phosphorylation of S6K1 and 4E-BP1 by mTORC1 stimulates protein translation [107].
In response to hypoxia, the regulation of mTORC1 involves the protein regulated in development and the DNA damage response 1 (REDD1). In this condition, HIF binds to a response element in the REDD1 promoter for her induction and therefore negatively regulates mTORC1 [108] (Figure 7a right). Paradoxically, mTORC1 is broadly activated in ccRCCs, pVHL inactivated, HIF activated, and upregulated REDD1. These findings suggest that other mechanisms favor tumors to escape growth suppressor signals resulting from pVHL loss and up-regulation of REDD1 [109] (Figure 7b left).
Another HIF-dependent mechanism that activates mTORC1 is the down-regulation of the mTOR inhibitor, the DEP domain-containing mTOR-interacting protein (DEPTOR). DEPTOR is significantly down-regulated in pVHL-deficient ccRCC tumors and cell lines. In this tumor type, DEPTOR is transcriptionally suppressed by both HIF-1 and HIF-2 mediated by the HIF target gene, BHLHe40 [110] (Figure 7b left).
Finally, a study revealed a new mechanism for the deregulation of mTORC1 in ccRCC. The report showed that pVHL represses the regulatory-associated protein of mTOR (RAPTOR), inhibiting mTORC1 signaling. Therefore, the loss of pVHL function in ccRCC is consistent with the hyperactivation of mTORC1 signaling. This mechanism describes a novel pVHL-mediated regulation of mTORC1 by targeted ubiquitination and degradation independent of HIF [111] (Figure 7b right).
pVHL is a critical tumor suppressor in ccRCCs. On the other hand, most patients with ccRCC are drug-resistant to therapies. Although targeted therapies that inhibit angiogenesis and mTOR pathways can lead to initial tumor control, most patients develop resistance [112,113,114]. Therefore, the identification of additional pVHL substrates could improve therapeutic options for ccRCC. Next, we review some pVHL substrates and possible therapeutic targets in ccRCCs.
Most reports on pVHL/HIF transcriptional activation have focused on HIF-bound promoters. However, evidence suggests distal enhancer elements in pVHL/HIF transcriptional control. In this sense, a study identified a master regulator crucial for the pathogenesis of ccRCC, ZNF395. pVHL loss stabilizes HIF2α occupancy in tumor-specific gained enhancers. HIF2α recruits, histone acetyltransferase p300 to maintain H3K27 acetylation, upregulating the expression of ccRCC-specific genes such as ZNF395. ZNF395 has a functional role in ccRCC tumorigenesis in vitro and in vivo [115] (Figure 8a-1).
A genome-wide in vitro expression strategy to identify proteins that bind to pVHL when hydroxylated determined that zinc fingers and homeoboxes 2 (ZHX2) are a novel pVHL substrate transcription factor. Analysis of tumors from ccRCC patients and pVHL loss-of-function mutations confirmed pVHL loss usually increases the abundance and nuclear levels of ZHX2 in ccRCC tumors. Mechanically, this study revealed that ZHX2 could promote NF-kB activation and carcinogenesis of ccRCC [116]. Another study showed the downstream signaling pathway of ZHX2. ZHX2 facilitated proliferation and migration in ccRCC cell lines by activating the MEK1/ERK1/2 signaling pathway. Furthermore, ZHX2 overexpression could induce Sunitinib resistance by activating autophagy through the MAPK / ERK signal pathway [117] (Figure 8a-2 up).
Another novel genome-wide in vitro expression strategy coupled with a GST-binding screen for pVHL substrates identified Scm-like with four malignant brain tumor domains 1 (SFMBT1) as a direct target of pVHL. In renal tumors compared to adjacent normal tissue, the levels of SFMBT1 are high. On the other hand, the functional characterization of SFMBT1 showed that it promotes ccRCC cell proliferation, anchorage-independent growth, and tumor xenograft growth. The analysis also identified sphingosine kinase 1 (SPHK1). SPHK1 is an SFMBT1 target gene that contributes to the oncogenic phenotype of renal tumors [118]. SFMBT1 and its downstream target gene SPHK1 could represent a new therapeutic strategy for patients with ccRCC treatment (Figure 8a-2 below).
Finally, mRNA expression levels of both transcription factors (ZHX2 and SFMBT1) in a tissue microarray constructed using 97 ccRCC samples showed an association with overall survival (OS) and disease-free survival (DFS) analyses. In this sense, survival analysis demonstrated that poor clinical outcomes in patients with ccRCC were associated with combined high expression levels of SFBMT1 and ZHX2. These results suggest that the coexpression of these two targets could be a promising biomarker for predicting the outcome of patients with ccRCC [119].
However, a study describes the pathologic angiogenic phenotype of missense pVHL missense mutations in ccRCC. In this case, the SUMO Enhancer (RSUME) sumoylates pVHL mutants. This post-translational modification promotes HIF-2α stabilization and leads to enhanced VEGF action. Therefore, it promotes more vascularized tumors. Regarding pathology, RSUME levels are higher in tumor patients with pVHL mutations and are associated with a poor prognosis in RCC tumors. These findings suggest that RSUME could be a biomarker of the outcomes of renal cell carcinoma [120] (Figure 8a-3).
Finally, tumors from ccRCC patients with pVHL loss show elevated TANK binding kinase 1 (TBK1). A study discovered that loss of pVHL or hypoxia in cancer hyperactivates TBK1. As a result of TBK1 hyperactivation, TBK1 phosphorylates p62 on the Ser366 residue and promotes renal tumorigenesis [121] (Figure 8a-4).
Regarding SNC hemangioblastomas, the analysis of tissues with mutations in the expressions of VHL gene showed the JAK2 and STAT3. The results of this study indicate that pVHL binds to JAK2 and STAT3 and mediates its ubiquitination. Furthermore, the study suggests that hemangioblast progenitor cells can differentiate into neoplastic cells by activating the JAK-STAT signaling pathway [122] (Figure 8b-1).
6. Animal Models for the Study of VHL
Current animal models for VHL disease can partially capture the disease by showing the involvement of a particular organ; therefore, much work is still needed to develop a model that exhibits several of the clinical manifestations. Other manifestations of VHL disease, such as tumors of the endolymphatic sac, middle ear, pheochromocytoma, cerebellar, and cervical hemangioblastomas, are not seen in almost any of the models. Additionally, phenotypes not associated with VHL disease can also arise from the product of the mutated VHL protein.[123] [124]
The first animal model for VHL disease led to the development of an aberrant placenta and thus was lethal to the embryo. Mice died in utero at embryonic days 10.5 and 12.5 [125]. However, the use of conditional VHL knockouts in mice has been shown to be an effective approach to delineate the role of VHL in individual organ systems.
Eyes. To date, there is still no adequate model to study the CNS and retinal hemangioblastomas. A zebrafish model has been developed expressing retinal neovascularization from vascular leakage, edema, and retinal detachment has been developed[126]. Studies have shown an increase in VEGF and CXCR4A in the CNS in these zebrafish. In fact, this model manifests certain aspects of age-related macular degeneration, diabetic retinopathy, and some cases of VHL [127]. However, zebrafish do not develop hemangioblastomas, van Rooijen and colleagues were able to use this zebrafish model to demonstrate inhibition of angiogenesis through the administration of VEGF receptor tyrosine kinase inhibition.
Kidney. CCRCC is a malignant kidney neoplasm that arises sporadically or is inherited through inactivation of VHL, Rankin et al. were the first group to successfully create a model that generated renal microcysts and macrocysts with similar morphological and molecular characteristics found in VHL-associated kidney disease[128]. Using Cre-loxp, they eliminated VHL expression from the proximal tubule using the phosphoenolpyruvate carboxykinase (PEPCK) promoter to drive Cre expression. Other groups have managed to obtain phenotypes that manifest acute nephritis with hematuria, proteinuria, and renal failure; characteristic features of pauciimmune RPGN (glomerulonephritis crescent) with prominent segmental fibrin deposition and fibrinoid necrosis [129].
Pancreas. Shen et al. produced a model using the insulin promoter factor 1 (Pdx) promoter to drive Cre expression. Survivors secondary to incomplete penetrance expressed highly vascularized cysts and microcystic adenomas, eliminating VHL expression throughout the pancreas [130].
Reproductive system. Ksp1.3-Cre mice were crossed with Vhlhfl/fl mice and Ptenfl/fl mice. The modified mice were then bred to generate a mouse model with dual VHL and Pten deficiencies specific for genital tract epithelium. These mice were able to recapitulate clear cell cystadenoma of the genital tract. in both males and females [131].
Liver. A mouse model in which VHL was acutely inactivated in utero exhibited embryonic lethality with liver necrosis and vascular defects [132]. A mouse model that conditionally inactivated VHL in hepatocytes led to hepatic hemangiomas [133]. Similarly, another model that conditionally inactivated VHL in a mosaic pattern in multiple organs showed liver hemangiomas, as well as angiectasias in the pancreas, heart, lung, and kidney [134]. Some other models showed growth deficiency, angiectasias, hemangiomas, endothelial cell proliferation, severe liver steatosis (accumulation of neutral fat in hepatocytes), and inflammatory cell infiltration [128].
In recent years, various animal models have been proposed in which the main objective is inactivation of the VHL gene product in various organs. In these models, the mechanisms associated with HIF and its link with tumorigenesis. However, these models are generally considered incapable of recapitulation of the most common characteristics of human VHL disease. To date, there are no models that develop retinal hemangioblastomas, the most common clinical manifestation of the disease [135].
7. Biomarkers in VHL Disease
Biomarkers for VHL disease are scarce. However, some studies propose several. For example, monitoring plasma levels of HIF-dependent molecules would allow monitoring of disease activity in VHL patients [84]. Although a report confirmed that elevated plasma VEGF levels are associated with an increased risk of dying from ccRCC, plasma VEGF levels are also significantly increased in tumors without VHL alteration. It suggests that VHL-independent mechanisms are involved in up-regulation of VEGF in ccRCC [136]. On the other hand, in serum from patients with VHL, no correlation was found between VEGF levels and the presence of manifestations of VHL disease [137]. Finally, in another study, the presence of abnormalities in VHL did not correlate with overall survival (OS), disease-specific survival (DSS), and progression or recurrence-free survival (PFS), and the expression of VEGF had no prognostic value. However, this study showed an association with a poorer prognosis in patients with no expression of VHL and HIF1-α expression and patients with overexpression of ERK5 [138].
The remarkable phenotypic heterogeneity in organ involvement and tumor onset age between and within VHL families has not allowed reliable markers to predict the age-related tumor risks in VHL patients. In this sense, the information shown in Table 3 is a compilation of several molecules proposed as biomarkers. The samples are mainly renal tissues. The recruited subjects are patients with VHL disease with ccRCC, while some patients have VHL disease with hemangioblastoma or pancreatic lesions. The heterogeneity of the disease concerning clinical and molecular issues manifests itself in the diversity of the molecules described in the table. Last, several molecules are prognostic biomarkers, which means those that indicate the probability of a change in a future clinical event, disease recurrence, or progression in an identified population [139].
The main limitation of these studies is the relatively small number of patients, but they provide an approximation to standard clinical and pathological data that are still essential in the development of biomarker panels for Von Hippel-Lindau disease addition to the molecular mechanisms that underlie this disease.
8. Targeted Therapy in VHL Disease
Clinical research on VHL disease is summarized in the next table (Table 4).
10. Discussion
The current review provides a comprehensive overview of Von Hippel-Lindau (VHL) disease, a rare genetic disorder characterized by multiple organ neoplastic syndrome. The disease is caused by deletions or mutations in the VHL gene [163], resulting in the development of cysts and tumors in various organs, including the brain, spine, eyes, kidneys, pancreas, adrenal glands, inner ears, reproductive tract, liver, and lung [92]. The prevalence of VHL disease is estimated to be between 1 in 39,000 and 1 in 91,000 individuals in different regional populations [13,14]. The penetrance of the disease is high, with a significant risk of developing clinical manifestations throughout life, emphasizing the importance of thoroughly understanding the underlying genetics and pathophysiology. Here we discuss the etiology, epidemiology, pathophysiology, genetics, clinical manifestations, diagnosis, and current treatments, as well as the molecular aspects of the disease. VHL is inherited in an autosomal dominant manner and is associated with mutations in the VHL gene. It discusses the role of Von Hippel–Lindau protein (pVHL) as a tumor suppressor known for regulating hypoxia-inducible factor (HIF) activity [8]. We provide insights into the inheritance pattern, mutations, and animal models used to study this disease, emphasizing the need for more comprehensive models that capture the diverse clinical manifestations of the disease. Furthermore, this review explores the biomarkers for VHL disease (Table 3). We discuss the proposed biomarkers for VHL disease that include monitoring plasma levels of HIF-dependent molecules, such as VEGF, which may allow for the monitoring of disease activity in VHL patients. However, it's important to note that elevated plasma VEGF levels are also significantly increased in tumors without VHL alteration, suggesting the involvement of VHL-independent mechanisms in up-regulating VEGF in clear cell renal cell carcinoma (ccRCC) [84,136,137,164]. Additionally, studies have shown that the presence of abnormalities in VHL does not correlate with overall survival, disease-specific survival, and progression or recurrence-free survival. Telomere length is another potential biomarker for risk assessment in VHL patients. It is reported shorted blood telomers in VHL patients [140]. The limitations of using blood telomere length as a biomarker for VHL disease include the relatively small number of patients studied, which may impact the generalizability of the findings. Additionally, the heterogeneity of the disease in terms of clinical and molecular aspects may affect the reliability of telomere length as a predictive marker for age-related tumor risks in VHL patients. Furthermore, it is argued that while shorter blood telomere length is associated with higher age-related risks of VHL-associated central nervous system hemangioblastomas, renal cell carcinoma, pancreatic cysts, and neuroendocrine tumors, the correlation may not be consistent across all patients with VHL disease. Therefore, the use of blood telomere length as a biomarker for VHL disease may have limitations in accurately predicting tumor risks and disease progression in all individuals with VHL.
Therefore, while these biomarkers show promise, further research is needed to establish their reliability and clinical utility in predicting tumor risks and disease progression in VHL patients.
The identification of mutations in the VHL gene has been crucial for the diagnosis and management of VHL [165]. Mutations in this gene have been observed to predispose affected individuals to develop a variety of tumors. Understanding how these mutations lead to tumor formation is critical for the development of more specific and effective therapeutic approaches.
In terms of pathophysiology, it has been demonstrated that the inactivation of the wild-type allele of the VHL gene is an initial event in the development of the disease, followed by the loss of function of the second allele, triggering tumorigenesis. This "two-hit" model proposed by Knudson has been fundamental in understanding tumor progression in VHL and has led to deeper investigations into the molecular mechanisms involved in this disease.
Furthermore, the review highlights the importance of clinical heterogeneity in VHL, with significant variations in disease presentation even within the same family. This underscores the need for an individualized approach in the diagnosis and management of patients with VHL, considering both genetic and clinical aspects.
The current review discusses the various manifestations of VHL disease, such as retinal hemangioblastomas, renal cell carcinomas, pheochromocytomas, and pancreatic cysts, and the corresponding diagnostic modalities and treatments. Furthermore, it sheds light on the molecular basis of VHL disease, elucidating the role of the pVHL protein in regulating hypoxia-inducible factor-alpha (HIF-α) protein levels through degradation under normoxic conditions. It also explores the association between VHL loss and the activation of the mTOR pathway [104], highlighting the significance of various pVHL substrates, such as ZNF395, ZHX2, SFMBT1, RSUME, and TBK1, in the pathogenesis of clear-cell type renal cell carcinomas [120,121,122].
In conclusion, ongoing research in the genetics and pathophysiology of Von Hippel-Lindau disease is crucial for improving early diagnosis, clinical management, and the development of more effective therapies. Understanding the underlying molecular mechanisms of VHL will not only expand our knowledge of this disease but also open up new opportunities for more precise and personalized therapeutic interventions in the future.
9. Conclusions
The genetic basis of VHL disease, characterized by mutations in the VHL gene, plays a central role in predisposing individuals to a spectrum of tumors and cysts in various organs. Further research into the genetic mechanisms underlying VHL is essential for advancing diagnostic and therapeutic strategies.
The pathophysiology of VHL syndrome, involving the loss of function of the VHL tumor suppressor protein and subsequent activation of hypoxia-inducible factors, provides valuable insights into the molecular pathways driving tumorigenesis in this condition. Understanding these pathways is crucial for developing targeted therapies.
The clinical heterogeneity observed in VHL underscores the need for personalized approaches to diagnosis and management. Tailoring treatment strategies to individual patients based on their genetic and clinical profiles can optimize outcomes and quality of life.
Advances in the understanding of VHL disease at the genetic and molecular levels hold promise for the development of more effective and personalized therapeutic interventions. Targeted therapies that address the specific molecular alterations in VHL-associated tumors could revolutionize treatment outcomes.
Future Directions: Continued research into the genetics, pathophysiology, and clinical management of VHL disease is essential for improving patient outcomes and quality of life. Collaborative efforts across disciplines, including genetics, oncology, and molecular biology, will be crucial for advancing our understanding of VHL and translating this knowledge into innovative therapeutic approaches.
In summary, the present review underscores the importance of a comprehensive understanding of the genetic, molecular, and clinical aspects, biomarkers, and animal models of Von Hippel-Lindau disease contributing to a deeper understanding of this rare genetic disorder and its clinical implications, ultimately improving outcomes for individuals affected by this complex genetic syndrome.
Author Contributions
M.C.C.A., L.G.V., M.V.P., M.C.S.L. designed the review, wrote the review, and contributed to the design of the figures. L.C.F., J.B.V., J.A.D.H. and F.A.J.O. performed the literature search, wrote, and contributed to the design and creation of the figures. M.C.C.A, conceived the review, supervised its development, and performed final paper revision writing and discussion.
Funding
This work was supported by grant number: 319578 from CONAHCYT, “Ciencia Básica, Ciencia de Frontera Modalidad: Paradigmas y Controversias de la Ciencia 2022.”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank the postdoctoral program from DGAPA/UNAM (2018-2020) and CONACYT for the postdoctoral fellowship (from 2020 to 2024) granted to Dr. Maria del Carmen Silva-Lucero co-first author of this work. We are grateful to the postdoctoral program from DGAPA/UNAM for the postdoctoral fellowship granted to Dr. Laura Gomez-Virgilio (2022-2024), who is the co-first author of this work and granted to Dr. Juan Ramón Padilla-Mendoza (2023-2024).
Conflicts of Interest
The authors declare that this review article was prepared in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
References
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A Review of Von Hippel-Lindau Syndrome. J Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef]
- Choyke, P.L.; Glenn, G.M.; Walther, M.M.; Patronas, N.J.; Linehan, W.M.; Zbar, B. von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 1995, 194, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Melmon, K.L.; Rosen, S.W. Lindau's Disease. Review of the Literature and Study of a Large Kindred. Am J Med 1964, 36, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Seizinger, B.R.; Rouleau, G.A.; Ozelius, L.J.; Lane, A.H.; Farmer, G.E.; Lamiell, J.M.; Haines, J.; Yuen, J.W.; Collins, D.; Majoor-Krakauer, D.; et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988, 332, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat Rev Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Schofield, P.N.; Fleming, S.; Maher, E.R. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum Mol Genet 1996, 5, 639–644. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Glasker, S.; Bender, B.U.; Apel, T.W.; van Velthoven, V.; Mulligan, L.M.; Zentner, J.; Neumann, H.P. Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry 2001, 70, 644–648. [Google Scholar] [CrossRef]
- Glasker, S.; Sohn, T.S.; Okamoto, H.; Li, J.; Lonser, R.R.; Oldfield, E.H.; Vortmeyer, A.O.; Zhuang, Z. Second hit deletion size in von Hippel-Lindau disease. Ann Neurol 2006, 59, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Maddock, I.R.; Moran, A.; Maher, E.R.; Teare, M.D.; Norman, A.; Payne, S.J.; Whitehouse, R.; Dodd, C.; Lavin, M.; Hartley, N.; et al. A genetic register for von Hippel-Lindau disease. J Med Genet 1996, 33, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 2010, 152A, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Iselius, L.; Yates, J.R.; Littler, M.; Benjamin, C.; Harris, R.; Sampson, J.; Williams, A.; Ferguson-Smith, M.A.; Morton, N. Von Hippel-Lindau disease: a genetic study. J Med Genet 1991, 28, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Wiestler, O.D. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 1991, 337, 1052–1054. [Google Scholar] [CrossRef]
- Binderup, M.L.; Galanakis, M.; Budtz-Jorgensen, E.; Kosteljanetz, M.; Luise Bisgaard, M. Prevalence, birth incidence, and penetrance of von Hippel-Lindau disease (vHL) in Denmark. Eur J Hum Genet 2017, 25, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Semin Cell Dev Biol 2005, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A 1996, 93, 10595–10599. [Google Scholar] [CrossRef]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Vortmeyer, A.O.; Falke, E.A.; Glasker, S.; Li, J.; Oldfield, E.H. Nervous system involvement in von Hippel-Lindau disease: pathology and mechanisms. Acta Neuropathol 2013, 125, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Albanyan, S.; Giles, R.H.; Gimeno, E.M.; Silver, J.; Murphy, J.; Faghfoury, H.; Morel, C.F.; Machado, J.; Kim, R.H. Characterization of VHL promoter variants in patients suspected of Von Hippel-Lindau disease. Eur J Med Genet 2019, 62, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Le, V.H.; Oyama, T.; Ricketts, C.J.; Ho, T.H.; Cheng, E.H. Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol 2018, JCO2018792549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, J.; Li, L.; Yi, Z.; Lu, R.; Li, C.; Gong, K. Intronic mutation of the VHL gene associated with central nervous system hemangioblastomas in two Chinese families with Von Hippel-Lindau disease: case report. BMC Med Genet 2020, 21, 191. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front Endocrinol (Lausanne) 2020, 11, 586857. [Google Scholar] [CrossRef] [PubMed]
- Tarade, D.; He, S.; St-Germain, J.; Petroff, A.; Murphy, A.; Raught, B.; Ohh, M. The long form of pVHL is artifactually modified by serine protease inhibitor AEBSF. Protein Sci 2020, 29, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Calsina, B.; Flores, S. Germline mutations in the new E1' cryptic exon of the VHL gene in patients with tumours of von Hippel-Lindau disease spectrum or with paraganglioma. J Med Genet 2020, 57, 752–759. [Google Scholar] [CrossRef]
- Xie, H.; Ma, K.; Zhang, J.; Hong, B.; Zhou, J.; Li, L.; Zhang, K.; Gong, K.; Cai, L. Novel genetic characterisation and phenotype correlation in von Hippel-Lindau (VHL) disease based on the Elongin C binding site: a large retrospective study. J Med Genet 2020, 57, 744–751. [Google Scholar] [CrossRef]
- Reich, M.; Jaegle, S.; Neumann-Haefelin, E.; Klingler, J.H.; Evers, C.; Daniel, M.; Bucher, F.; Ludwig, F.; Nuessle, S.; Kopp, J.; et al. Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol 2021. [Google Scholar] [CrossRef] [PubMed]
- van der Horst-Schrivers, A.N.A.; Sluiter, W.J.; Kruizinga, R.C.; van Leeuwaarde, R.S.; Giles, R.; Olderode-Berends, M.J.W.; Links, T.P. The incidence of consecutive manifestations in Von Hippel-Lindau disease. Fam Cancer 2019, 18, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Vocke, C.D.; Ricketts, C.J.; Schmidt, L.S.; Ball, M.W.; Middelton, L.A.; Zbar, B.; Linehan, W.M. Comprehensive characterization of Alu-mediated breakpoints in germline VHL gene deletions and rearrangements in patients from 71 VHL families. Hum Mutat 2021. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Krzystolik, K.; Murgia, A.; Gorski, B.; Debniak, T.; Jakubowska, A.; Martella, M.; Kurzawski, G.; Prost, M.; Kojder, I.; et al. Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J Med Genet 2002, 39, E38. [Google Scholar] [CrossRef] [PubMed]
- Tabaro, F.; Minervini, G.; Sundus, F.; Quaglia, F.; Leonardi, E.; Piovesan, D.; Tosatto, S.C. VHLdb: A database of von Hippel-Lindau protein interactors and mutations. Sci Rep 2016, 6, 31128. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.R.; Woodward, E.R.; Killick, P.; Lim, C.; Macdonald, F.; Maher, E.R. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum Mutat 2007, 28, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Zbar, B.; Kishida, T.; Chen, F.; Schmidt, L.; Maher, E.R.; Richards, F.M.; Crossey, P.A.; Webster, A.R.; Affara, N.A.; Ferguson-Smith, M.A.; et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996, 8, 348–357. [Google Scholar] [CrossRef]
- Chen, F.; Kishida, T.; Yao, M.; Hustad, T.; Glavac, D.; Dean, M.; Gnarra, J.R.; Orcutt, M.L.; Duh, F.M.; Glenn, G.; et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 1995, 5, 66–75. [Google Scholar] [CrossRef]
- Kanno, H.; Shuin, T.; Kondo, K.; Ito, S.; Hosaka, M.; Torigoe, S.; Fujii, S.; Tanaka, Y.; Yamamoto, I.; Kim, I.; et al. Molecular genetic diagnosis of von Hippel-Lindau disease: analysis of five Japanese families. Jpn J Cancer Res 1996, 87, 423–428. [Google Scholar] [CrossRef]
- Moore, P.S.; Antonello, D.; Martignoni, G.; Racchini, C.; Ventrucci, M.; Scarpa, A. Identification of a novel mutation (c279delC) and a polymorphism (c291C>G) in the von Hippel-Lindau gene in Italian patients. Hum Mutat 2000, 15, 582. [Google Scholar] [CrossRef]
- Crossey, P.A.; Richards, F.M.; Foster, K.; Green, J.S.; Prowse, A.; Latif, F.; Lerman, M.I.; Zbar, B.; Affara, N.A.; Ferguson-Smith, M.A.; et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet 1994, 3, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Stolle, C.; Glenn, G.; Zbar, B.; Humphrey, J.S.; Choyke, P.; Walther, M.; Pack, S.; Hurley, K.; Andrey, C.; Klausner, R.; et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat 1998, 12, 417–423. [Google Scholar] [CrossRef]
- Rechsteiner, M.P.; von Teichman, A.; Nowicka, A.; Sulser, T.; Schraml, P.; Moch, H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Cancer Res 2011, 71, 5500–5511. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Llorente, S.; Bravo, J.; Cebrian, A.; Cascon, A.; Pollan, M.; Telleria, D.; Leton, R.; Urioste, M.; Rodriguez-Lopez, R.; de Campos, J.M.; et al. Genetic characterization and structural analysis of VHL Spanish families to define genotype-phenotype correlations. Hum Mutat 2004, 23, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.R.; Maher, E.R.; Moore, A.T. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol 1999, 117, 371–378. [Google Scholar] [CrossRef] [PubMed]
- van Houwelingen, K.P.; van Dijk, B.A.; Hulsbergen-van de Kaa, C.A.; Schouten, L.J.; Gorissen, H.J.; Schalken, J.A.; van den Brandt, P.A.; Oosterwijk, E. Prevalence of von Hippel-Lindau gene mutations in sporadic renal cell carcinoma: results from The Netherlands cohort study. BMC Cancer 2005, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; Silva, R.L.; Mendonca, B.B.; Marui, S.; Simpson, A.J.; Camargo, A.A. High frequency of novel germline mutations in the VHL gene in the heterogeneous population of Brazil. J Med Genet 2003, 40, e31. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom-O'Brien, M.; van der Luijt, R.B.; van Rooijen, E.; van den Ouweland, A.M.; Majoor-Krakauer, D.F.; Lolkema, M.P.; van Brussel, A.; Voest, E.E.; Giles, R.H. Genetic analysis of von Hippel-Lindau disease. Hum Mutat 2010, 31, 521–537. [Google Scholar] [CrossRef]
- Cho, H.J.; Ki, C.S.; Kim, J.W. Improved detection of germline mutations in Korean VHL patients by multiple ligation-dependent probe amplification analysis. J Korean Med Sci 2009, 24, 77–83. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, N.; Wang, X.; Ning, X.; Li, T.; Bu, D.; Gong, K. Family history of von Hippel-Lindau disease was uncommon in Chinese patients: suggesting the higher frequency of de novo mutations in VHL gene in these patients. J Hum Genet 2012, 57, 238–243. [Google Scholar] [CrossRef]
- Kang, H.C.; Kim, I.J.; Park, J.H.; Shin, Y.; Jang, S.G.; Ahn, S.A.; Park, H.W.; Lim, S.K.; Oh, S.K.; Kim, D.J.; et al. Three novel VHL germline mutations in Korean patients with von Hippel-Lindau disease and pheochromocytomas. Oncol Rep 2005, 14, 879–883. [Google Scholar] [CrossRef]
- Germline mutations in the von Hippel-Lindau disease (VHL) gene in Japanese VHL. Clinical Research Group for VHL in Japan. Hum Mol Genet 1995, 4, 2233–2237. [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ashida, S.; Kondo, K.; Kobayashi, K.; Kanno, H.; Shinohara, N.; Shitara, N.; Kishida, T.; Kawakami, S.; Baba, M.; et al. Germ-line mutation analysis in patients with von Hippel-Lindau disease in Japan: an extended study of 77 families. Jpn J Cancer Res 2000, 91, 204–212. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Olschwang, S.; Richard, S.; Boisson, C.; Giraud, S.; Laurent-Puig, P.; Resche, F.; Thomas, G. Germline mutation profile of the VHL gene in von Hippel-Lindau disease and in sporadic hemangioblastoma. Hum Mutat 1998, 12, 424–430. [Google Scholar] [CrossRef]
- Glavac, D.; Neumann, H.P.; Wittke, C.; Jaenig, H.; Masek, O.; Streicher, T.; Pausch, F.; Engelhardt, D.; Plate, K.H.; Hofler, H.; et al. Mutations in the VHL tumor suppressor gene and associated lesions in families with von Hippel-Lindau disease from central Europe. Hum Genet 1996, 98, 271–280. [Google Scholar] [CrossRef]
- Maher, E.R.; Webster, A.R.; Richards, F.M.; Green, J.S.; Crossey, P.A.; Payne, S.J.; Moore, A.T. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996, 33, 328–332. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Lubensky, I.A.; Fogt, F.; Linehan, W.M.; Khettry, U.; Zhuang, Z. Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am J Pathol 1997, 151, 951–956. [Google Scholar]
- Bauters, C.; Vantyghem, M.C.; Leteurtre, E.; Odou, M.F.; Mouton, C.; Porchet, N.; Wemeau, J.L.; Proye, C.; Pigny, P. Hereditary phaeochromocytomas and paragangliomas: a study of five susceptibility genes. J Med Genet 2003, 40, e75. [Google Scholar] [CrossRef]
- Ritter, M.M.; Frilling, A.; Crossey, P.A.; Hoppner, W.; Maher, E.R.; Mulligan, L.; Ponder, B.A.; Engelhardt, D. Isolated familial pheochromocytoma as a variant of von Hippel-Lindau disease. J Clin Endocrinol Metab 1996, 81, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Hes, F.J.; van der Luijt, R.B.; Janssen, A.L.; Zewald, R.A.; de Jong, G.J.; Lenders, J.W.; Links, T.P.; Luyten, G.P.; Sijmons, R.H.; Eussen, H.J.; et al. Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clin Genet 2007, 72, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, N.; Ning, X.; Li, T.; Wu, P.; Peng, S.; Fan, Y.; Bu, D.; Gong, K. Higher prevalence of novel mutations in VHL gene in Chinese Von Hippel-Lindau disease patients. Urology 2014, 83, 675–e671. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Matias-Guiu, X.; Cabezas, R.; Chico, A.; Prat, J.; Baiget, M.; De Leiva, A. Molecular diagnosis of von Hippel-Lindau disease in a kindred with a predominance of familial phaeochromocytoma. Clin Endocrinol (Oxf) 1997, 46, 359–363. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, J.H.; Lee, K.E.; Kim, J.H.; Hong, J.M.; Ra, E.K.; Seo, S.H.; Lee, S.J.; Kim, M.J.; Park, S.S.; et al. Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-alpha binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med Genet 2016, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Crossey, P.A.; Eng, C.; Ginalska-Malinowska, M.; Lennard, T.W.; Wheeler, D.C.; Ponder, B.A.; Maher, E.R. Molecular genetic diagnosis of von Hippel-Lindau disease in familial phaeochromocytoma. J Med Genet 1995, 32, 885–886. [Google Scholar] [CrossRef]
- Gross, D.J.; Avishai, N.; Meiner, V.; Filon, D.; Zbar, B.; Abeliovich, D. Familial pheochromocytoma associated with a novel mutation in the von Hippel-Lindau gene. J Clin Endocrinol Metab 1996, 81, 147–149. [Google Scholar] [CrossRef]
- Bar, M.; Friedman, E.; Jakobovitz, O.; Leibowitz, G.; Lerer, I.; Abeliovich, D.; Gross, D.J. Sporadic phaeochromocytomas are rarely associated with germline mutations in the von Hippel-Lindau and RET genes. Clin Endocrinol (Oxf) 1997, 47, 707–712. [Google Scholar] [CrossRef]
- Tisherman, S.E.; Gregg, F.J.; Danowski, T.S. Familial pheochromocytoma. JAMA 1962, 182, 152–156. [Google Scholar] [CrossRef]
- Murgia, A.; Martella, M.; Vinanzi, C.; Polli, R.; Perilongo, G.; Opocher, G. Somatic mosaicism in von Hippel-Lindau Disease. Hum Mutat 2000, 15, 114. [Google Scholar] [CrossRef]
- Mete, T.; Berker, D.; Yilmaz, E.; Ozgen, G.; Yalcin, Y.; Tuna, M.; Ciliz, D.; Onen, M.; Aydin, Y.; Guler, S. Clinical presentation of Von Hippel Lindau syndrome type 2B associated with VHL p.A149S mutation in a large Turkish family. Endocrine 2014, 45, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Atuk, N.O.; Stolle, C.; Owen, J.A., Jr.; Carpenter, J.T.; Vance, M.L. Pheochromocytoma in von Hippel-Lindau disease: clinical presentation and mutation analysis in a large, multigenerational kindred. J Clin Endocrinol Metab 1998, 83, 117–120. [Google Scholar] [CrossRef]
- Brookes, C.; Prosser, D.O.; Love, J.M.; Gardner, R.J.; Love, D.R. Diagnostic genetics at a distance: von hippel-lindau disease and a novel mutation. Genet Res Int 2013, 2013, 189196. [Google Scholar] [CrossRef]
- Ganeshan, D.; Menias, C.O.; Pickhardt, P.J.; Sandrasegaran, K.; Lubner, M.G.; Ramalingam, P.; Bhalla, S. Tumors in von Hippel-Lindau Syndrome: From Head to Toe-Comprehensive State-of-the-Art Review. Radiographics 2018, 38, 849–866. [Google Scholar] [CrossRef] [PubMed]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb Clin Neurol 2015, 132, 139–156. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, K.; Ma, K.; Zhou, J.; Gong, Y.; Cai, L.; Gong, K. The Genotype-Phenotype Association of Von Hipple Lindau Disease Based on Mutation Locations: A Retrospective Study of 577 Cases in a Chinese Population. Front Genet 2020, 11, 532588. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef]
- Domene, C.; Illingworth, C.J. Effects of point mutations in pVHL on the binding of HIF-1alpha. Proteins 2012, 80, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Razafinjatovo, C.; Bihr, S.; Mischo, A.; Vogl, U.; Schmidinger, M.; Moch, H.; Schraml, P. Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC Cancer 2016, 16, 638. [Google Scholar] [CrossRef]
- Pastore, Y.D.; Jelinek, J.; Ang, S.; Guan, Y.; Liu, E.; Jedlickova, K.; Krishnamurti, L.; Prchal, J.T. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood 2003, 101, 1591–1595. [Google Scholar] [CrossRef]
- Zhou, M.I.; Wang, H.; Foy, R.L.; Ross, J.J.; Cohen, H.T. Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res 2004, 64, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Cervio, A.; Villalonga, J.F.; Mormandi, R.; Alcorta, S.C.; Sevlever, G.; Salvat, J. Surgical treatment of cerebellar hemangioblastomas. Surg Neurol Int 2017, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Klingler, J.H.; Glasker, S.; Bausch, B.; Urbach, H.; Krauss, T.; Jilg, C.A.; Steiert, C.; Puzik, A.; Neumann-Haefelin, E.; Kotsis, F.; et al. Hemangioblastoma and von Hippel-Lindau disease: genetic background, spectrum of disease, and neurosurgical treatment. Childs Nerv Syst 2020, 36, 2537–2552. [Google Scholar] [CrossRef] [PubMed]
- Glasker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. Onco Targets Ther 2020, 13, 5669–5690. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.; Arabi, A.; Shahraki, T.; Safi, S. Von Hippel-Lindau Disease and the Eye. J Ophthalmic Vis Res 2020, 15, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Shields, C.L.; Shields, J.A. von Hippel-Lindau disease. Surv Ophthalmol 2001, 46, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Findeis-Hosey, J.J.; McMahon, K.Q.; Findeis, S.K. Von Hippel-Lindau Disease. J Pediatr Genet 2016, 5, 116–123. [Google Scholar] [CrossRef]
- Lin, G.; Zhao, Y.; Zhang, Z.; Zhang, H. Clinical diagnosis, treatment and screening of the VHL gene in three von Hippel-Lindau disease pedigrees. Exp Ther Med 2020, 20, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Jabi, F. Subclinical Pheochromocytoma and Paraganglioma in an Elderly Patient: A Case Report. JSM Clinical Reports 2014, 2, 1036. [Google Scholar]
- van Asselt, S.J.; de Vries, E.G.; van Dullemen, H.M.; Brouwers, A.H.; Walenkamp, A.M.; Giles, R.H.; Links, T.P. Pancreatic cyst development: insights from von Hippel-Lindau disease. Cilia 2013, 2, 3. [Google Scholar] [CrossRef]
- Blansfield, J.A.; Choyke, L.; Morita, S.Y.; Choyke, P.L.; Pingpank, J.F.; Alexander, H.R.; Seidel, G.; Shutack, Y.; Yuldasheva, N.; Eugeni, M.; et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 2007, 142, 814–818, discussion 818 e811-812. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Neumann, H.P.; Richard, S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011, 19, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Zanoletti, E.; Girasoli, L.; Borsetto, D.; Opocher, G.; Mazzoni, A.; Martini, A. Endolymphatic sac tumour in von Hippel-Lindau disease: management strategies. Acta Otorhinolaryngol Ital 2017, 37, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Butman, J.A.; Kim, H.J.; Baggenstos, M.; Ammerman, J.M.; Dambrosia, J.; Patsalides, A.; Patronas, N.J.; Oldfield, E.H.; Lonser, R.R. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA 2007, 298, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gomella, P.T.; Shin, P.; Srinivasan, R.; Linehan, W.M.; Ball, M.W. Obstructive azoospermia secondary to bilateral epididymal cystadenomas in a patient with von Hippel-Lindau. Urol Case Rep 2019, 27, 100922. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, G.Z.; Millo, C.; Sadowski, S.M.; Bagci, U.; Patronas, N.J. Epididymal Cystadenomas in von Hippel-Lindau Disease Showing Increased Activity on 68Ga DOTATATE PET/CT. Clin Nucl Med 2016, 41, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.D.; Cunha, T.M.; Tereso, A. Tumors of the broad ligament: what and when to suspect such rare location. Radiol Bras 2020, 53, 349–355. [Google Scholar] [CrossRef]
- Nogales, F.F.; Goyenaga, P.; Preda, O.; Nicolae, A.; Vieites, B.; Ruiz-Marcellan, M.C.; Pedrosa, A.; Merino, M.J. An analysis of five clear cell papillary cystadenomas of mesosalpinx and broad ligament: four associated with von Hippel-Lindau disease and one aggressive sporadic type. Histopathology 2012, 60, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci U S A 2006, 103, 9814–9819. [Google Scholar] [CrossRef]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 2003, 1, E83. [Google Scholar] [CrossRef]
- Schodel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 2019, 15, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int 2006, 69, 1302–1307. [Google Scholar] [CrossRef]
- Robb, V.A.; Karbowniczek, M.; Klein-Szanto, A.J.; Henske, E.P. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol 2007, 177, 346–352. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev 2001, 15, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Kucejova, B.; Pena-Llopis, S.; Yamasaki, T.; Sivanand, S.; Tran, T.A.; Alexander, S.; Wolff, N.C.; Lotan, Y.; Xie, X.J.; Kabbani, W.; et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res 2011, 9, 1255–1265. [Google Scholar] [CrossRef]
- Doan, H.; Parsons, A.; Devkumar, S.; Selvarajah, J.; Miralles, F.; Carroll, V.A. HIF-mediated Suppression of DEPTOR Confers Resistance to mTOR Kinase Inhibition in Renal Cancer. iScience 2019, 21, 509–520. [Google Scholar] [CrossRef]
- Ganner, A.; Gehrke, C.; Klein, M.; Thegtmeier, L.; Matulenski, T.; Wingendorf, L.; Wang, L.; Pilz, F.; Greidl, L.; Meid, L.; et al. VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma. Sci Rep 2021, 11, 14827. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Flaherty, K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol Oncol 2008, 26, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Bellmunt, J.; Pons, F.; Foreshew, A.; Fay, A.P.; Powles, T.; Porta, C.; Bracarda, S.; Lampron, M.E.; Cerbone, L.; Sternberg, C.N.; et al. Sequential targeted therapy after pazopanib therapy in patients with metastatic renal cell cancer: efficacy and toxicity. Clin Genitourin Cancer 2014, 12, 262–269. [Google Scholar] [CrossRef]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov 2017, 7, 1284–1305. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, R.; Yan, H.; Zhang, J.; Lin, Z. ZHX2 drives cell growth and migration via activating MEK/ERK signal and induces Sunitinib resistance by regulating the autophagy in clear cell Renal Cell Carcinoma. Cell Death Dis 2020, 11, 337. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol Cell 2020, 77, 1294–1306. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, L.; Xue, S.; Shi, J.; He, C.; Zhang, Q. Novel VHL substrate targets SFMBT1 and ZHX2 may be important prognostic predictors in patients with ccRCC. Oncol Lett 2021, 21, 379. [Google Scholar] [CrossRef]
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Garcia Marchinena, P.A.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis 2019, 10, 266. [Google Scholar] [CrossRef]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov 2020, 10, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Yoshizumi, T.; Shinonaga, M.; Kubo, A.; Murata, H.; Yao, M. Role of VHL-JAK-STAT signaling pathway in central nervous system hemangioblastoma associated with von Hippel-Lindau disease. J Neurooncol 2020, 148, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, S.; Nobili, B.; Ferraro, M.; Migliaccio, C.; Borriello, A.; Cucciolla, V.; Martinelli, V.; Rossi, F.; Punzo, F.; Cirillo, P.; et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood 2006, 107, 514–519. [Google Scholar] [CrossRef]
- Bento, M.C.; Chang, K.T.; Guan, Y.; Liu, E.; Caldas, G.; Gatti, R.A.; Prchal, J.T. Congenital polycythemia with homozygous and heterozygous mutations of von Hippel-Lindau gene: five new Caucasian patients. Haematologica 2005, 90, 128–129. [Google Scholar] [PubMed]
- Gnarra, J.R.; Ward, J.M.; Porter, F.D.; Wagner, J.R.; Devor, D.E.; Grinberg, A.; Emmert-Buck, M.R.; Westphal, H.; Klausner, R.D.; Linehan, W.M. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A 1997, 94, 9102–9107. [Google Scholar] [CrossRef] [PubMed]
- van Rooijen, E.; Voest, E.E.; Logister, I.; Bussmann, J.; Korving, J.; van Eeden, F.J.; Giles, R.H.; Schulte-Merker, S. von Hippel-Lindau tumor suppressor mutants faithfully model pathological hypoxia-driven angiogenesis and vascular retinopathies in zebrafish. Dis Model Mech 2010, 3, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y. Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc 2005, 103, 495–511. [Google Scholar] [PubMed]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 2006, 66, 2576–2583. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Cui, S.; Li, C.; Jothy, S.; Haase, V.; Steer, B.M.; Marsden, P.A.; Pippin, J.; Shankland, S.; Rastaldi, M.P.; et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nature Medicine 2006, 12, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Adem, A.; Ylaya, K.; Wilson, A.; He, M.; Lorang, D.; Hewitt, S.M.; Pechhold, K.; Harlan, D.M.; Lubensky, I.A.; et al. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PLoS One 2009, 4, e4897. [Google Scholar] [CrossRef]
- Frew, I.J.; Minola, A.; Georgiev, S.; Hitz, M.; Moch, H.; Richard, S.; Vortmeyer, A.O.; Krek, W. Combined VHLH and PTEN mutation causes genital tract cystadenoma and squamous metaplasia. Mol Cell Biol 2008, 28, 4536–4548. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.B.; Furihata, M.; Baba, M.; Zbar, B.; Schmidt, L.S. Vascular defects and liver damage by the acute inactivation of the VHL gene during mouse embryogenesis. Lab Invest 2006, 86, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H.; Glickman, J.N.; Socolovsky, M.; Jaenisch, R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A 2001, 98, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Tessarollo, L.; Hong, S.B.; Baba, M.; Southon, E.; Back, T.C.; Spence, S.; Lobe, C.G.; Sharma, N.; Maher, G.W.; et al. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res 2003, 63, 5320–5328. [Google Scholar] [PubMed]
- Park, S.; Chan, C.C. Von Hippel-Lindau disease (VHL): a need for a murine model with retinal hemangioblastoma. Histol Histopathol 2012, 27, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Patard, J.J.; Rioux-Leclercq, N.; Masson, D.; Zerrouki, S.; Jouan, F.; Collet, N.; Dubourg, C.; Lobel, B.; Denis, M.; Fergelot, P. Absence of VHL gene alteration and high VEGF expression are associated with tumour aggressiveness and poor survival of renal-cell carcinoma. Br J Cancer 2009, 101, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Aarsman, C.J.; Terpstra, L.; Wittebol-Post, D.; Lips, C.J.; Blijham, G.H.; Voest, E.E. Elevated ocular levels of vascular endothelial growth factor in patients with von Hippel-Lindau disease. Ann Oncol 1997, 8, 1015–1022. [Google Scholar] [CrossRef]
- Salinas-Sanchez, A.S.; Serrano-Oviedo, L.; Nam-Cha, S.Y.; Roche-Losada, O.; Sanchez-Prieto, R.; Gimenez-Bachs, J.M. Prognostic Value of the VHL, HIF-1alpha, and VEGF Signaling Pathway and Associated MAPK (ERK1/2 and ERK5) Pathways in Clear-Cell Renal Cell Carcinoma. A Long-Term Study. Clin Genitourin Cancer 2017, 15, e923–e933. [Google Scholar] [CrossRef]
- In BEST (Biomarkers, EndpointS, and other Tools) Resource; Silver Spring (MD), 2016.
- Wang, J.Y.; Peng, S.H.; Ning, X.H.; Li, T.; Liu, S.J.; Liu, J.Y.; Hong, B.A.; Qi, N.N.; Peng, X.; Zhou, B.W.; et al. Shorter telomere length increases age-related tumor risks in von Hippel-Lindau disease patients. Cancer Med 2017, 6, 2131–2141. [Google Scholar] [CrossRef]
- Maroto, P.; Esteban, E.; Parra, E.F.; Mendez-Vidal, M.J.; Domenech, M.; Perez-Valderrama, B.; Calderero, V.; Perez-Gracia, J.L.; Grande, E.; Algaba, F. HIF pathway and c-Myc as biomarkers for response to sunitinib in metastatic clear-cell renal cell carcinoma. Onco Targets Ther 2017, 10, 4635–4643. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, Y.; Peng, Z.; Wang, L.; Wang, K.; Feng, D.; He, J.; Zheng, J. SERPINH1 overexpression in clear cell renal cell carcinoma: association with poor clinical outcome and its potential as a novel prognostic marker. J Cell Mol Med 2018, 22, 1224–1235. [Google Scholar] [CrossRef]
- Hong, B.; Cai, L.; Wang, J.; Liu, S.; Zhou, J.; Ma, K.; Zhang, J.; Zhou, B.; Peng, X.; Zhang, N.; et al. Differential Expression of PD-L1 Between Sporadic and VHL-Associated Hereditary Clear-Cell Renal Cell Carcinoma and Its Correlation With Clinicopathological Features. Clin Genitourin Cancer 2019, 17, 97–104 e101. [Google Scholar] [CrossRef] [PubMed]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Forsch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol Lett 2019, 18, 2654–2660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yan, A.; Cao, W.; Shi, H.; Cao, K.; Liu, X. Development and validation of a VHL-associated immune prognostic signature for clear cell renal cell carcinoma. Cancer Cell Int 2020, 20, 584. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhou, J.; Zhang, K.; Li, L.; Xu, Y.; Ma, K.; Xie, H.; Cai, L.; Gong, Y.; Gong, K. Identification and validation of the clinical roles of the VHL-related LncRNAs in clear cell renal cell carcinoma. J Cancer 2021, 12, 2702–2714. [Google Scholar] [CrossRef]
- Oosting, S.F.; van Asselt, S.J.; Brouwers, A.H.; Bongaerts, A.H.; Steinberg, J.D.; de Jong, J.R.; Lub-de Hooge, M.N.; van der Horst-Schrivers, A.N.; Walenkamp, A.M.; Hoving, E.W.; et al. 89Zr-Bevacizumab PET Visualizes Disease Manifestations in Patients with von Hippel-Lindau Disease. J Nucl Med 2016, 57, 1244–1250. [Google Scholar] [CrossRef]
- Shell, J.; Tirosh, A.; Millo, C.; Sadowski, S.M.; Assadipour, Y.; Green, P.; Patel, D.; Nilubol, N.; Kebebew, E. The utility of (68)Gallium-DOTATATE PET/CT in the detection of von Hippel-Lindau disease associated tumors. Eur J Radiol 2019, 112, 130–135. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Jacobs-El, N.; Wong, W.T.; Wiley, H.E.; Cukras, C.A.; Meyerle, C.B.; Chew, E.Y. Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol Retina 2017, 1, 181–187. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol 2011, 22, 2661–2666. [Google Scholar] [CrossRef]
- Hwang, C.K.; Chew, E.Y.; Cukras, C.A.; Keenan, T.D.L.; Wong, W.T.; Linehan, W.M.; Chittiboina, P.; Pacak, K.; Wiley, H.E. Intravitreous treatment of severe ocular von Hippel-Lindau disease using a combination of the VEGF inhibitor, ranibizumab, and PDGF inhibitor, E10030: Results from a phase 1/2 clinical trial. Clin Exp Ophthalmol 2021. [Google Scholar] [CrossRef]
- Batist, G.; Patenaude, F.; Champagne, P.; Croteau, D.; Levinton, C.; Hariton, C.; Escudier, B.; Dupont, E. Neovastat (AE-941) in refractory renal cell carcinoma patients: report of a phase II trial with two dose levels. Ann Oncol 2002, 13, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Choueiri, T.K.; Oudard, S.; Szczylik, C.; Negrier, S.; Ravaud, A.; Chevreau, C.; Venner, P.; Champagne, P.; Croteau, D.; et al. Prognostic factors of metastatic renal cell carcinoma after failure of immunotherapy: new paradigm from a large phase III trial with shark cartilage extract AE 941. J Urol 2007, 178, 1901–1905. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, B.; Villar Gomez de Las Heras, K.; Aguirre, D.T.; Rodriguez-Padial, L.; Albinana, V.; Recio-Poveda, L.; Cuesta, A.M.; Botella, L.M.; Jimenez-Escribano, R.M. Evaluation of the safety and effectiveness of oral propranolol in patients with von Hippel-Lindau disease and retinal hemangioblastomas: phase III clinical trial. BMJ Open Ophthalmol 2019, 4, e000203. [Google Scholar] [CrossRef] [PubMed]
- Panakis, N.; Offiah, C.; Azizan, A.; Roy, A.; Plowman, P.N. Sorafenib therapy and CNS hemangioblastomas in individuals with Von Hippel Lindau syndrome. Journal of Clinical Oncology 2008, 26, 2065–2065. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol 2018, 19, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. Journal of Clinical Oncology 2020, 38, 5003–5003. [Google Scholar] [CrossRef]
- Hasanov, E.; Jonasch, E. MK-6482 as a potential treatment for von Hippel-Lindau disease-associated clear cell renal cell carcinoma. Expert Opin Investig Drugs 2021, 30, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Matin, S.F.; Woodson, A.H.; Marcott, V.D.; Bird, S.; Slack, R.; Fuller, G.; McCutcheon, I.E.; Jonasch, E. Pilot study of dovitinib in patients with VHL disease. Journal of Clinical Oncology 2016, 34, 587–587. [Google Scholar] [CrossRef]
- Wong, W.T.; Liang, K.J.; Hammel, K.; Coleman, H.R.; Chew, E.Y. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology 2008, 115, 1957–1964. [Google Scholar] [CrossRef]
- Stamatakis, L.; Shuch, B.; Singer, E.A.; Nix, J.; Truong, H.; Friend, J.C.; Fowler, S.; Bratslavsky, G.; Metwalli, A.R.; Shih, J.H.; et al. Phase II trial of vandetanib in Von Hippel-Lindau-associated renal cell carcinoma. Journal of Clinical Oncology 2013, 31, 4584–4584. [Google Scholar] [CrossRef]
- Ma, K.; Hong, B.; Zhou, J.; Gong, Y.; Wang, J.; Liu, S.; Peng, X.; Zhou, B.; Zhang, J.; Xie, H.; et al. The Efficacy and Safety of Tyrosine Kinase Inhibitors for Von Hippel-Lindau Disease: A Retrospective Study of 32 Patients. Front Oncol 2019, 9, 1122. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.A. Von Hippel-Lindau syndrome. A pleomorphic condition. Cancer 1999, 86, 2478–2482. [Google Scholar] [CrossRef]
- Nagai, Y.; Ando, H.; Nohara, E.; Yamashita, H.; Takamura, T.; Kobayashi, K. Plasma levels of vascular endothelial growth factor in patients with acromegaly. Horm Metab Res 2000, 32, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Struckmann, K.; Hatz, F.; Sonnet, S.; Kully, C.; Gasser, T.; Sauter, G.; Mihatsch, M.J.; Moch, H. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol 2002, 196, 186–193. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Localization of the VHL gene in Chromosome 3. The VHL gene site is indicated with an arrow. Created with BioRender.com.
Figure 1.
Localization of the VHL gene in Chromosome 3. The VHL gene site is indicated with an arrow. Created with BioRender.com.
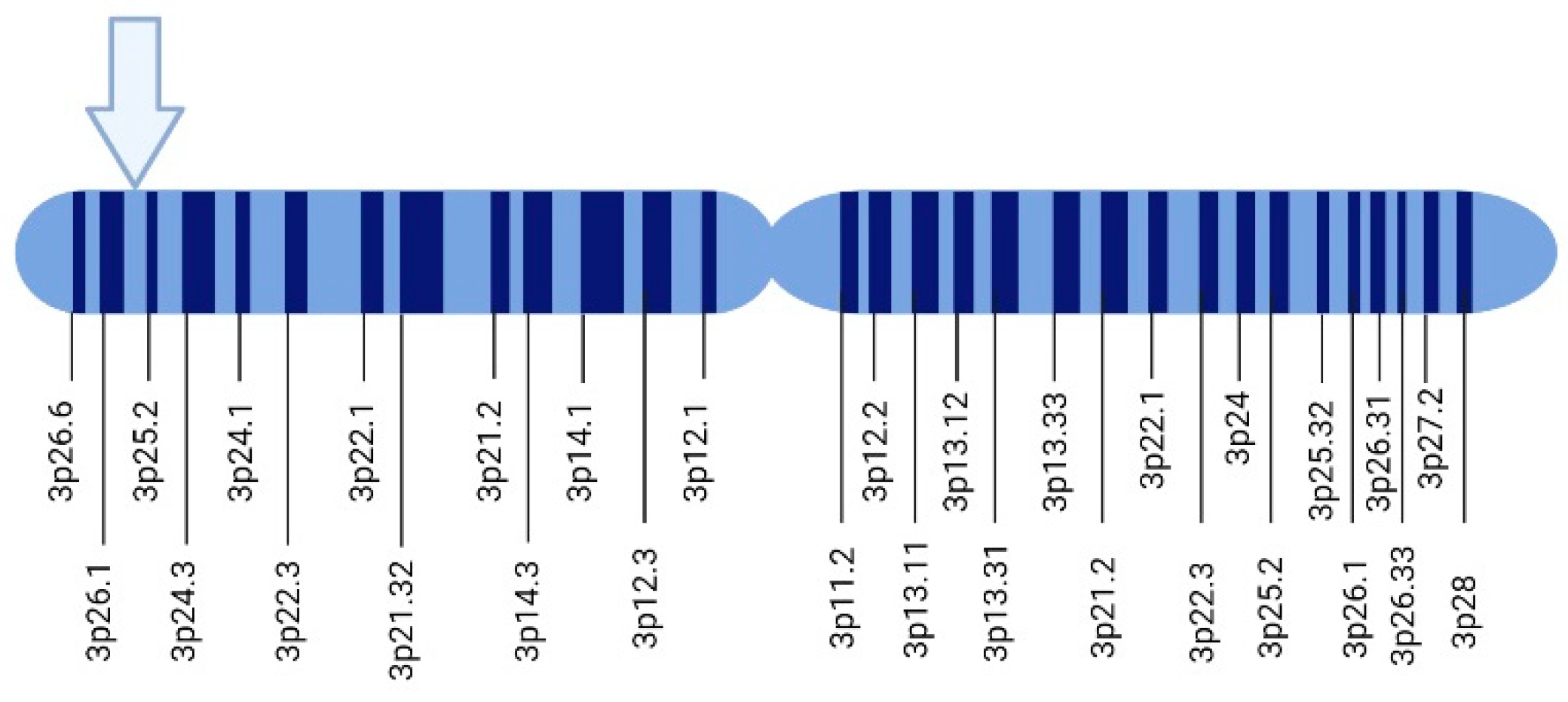
Figure 2.
Structure of the VHL gene. Created with BioRender.com.
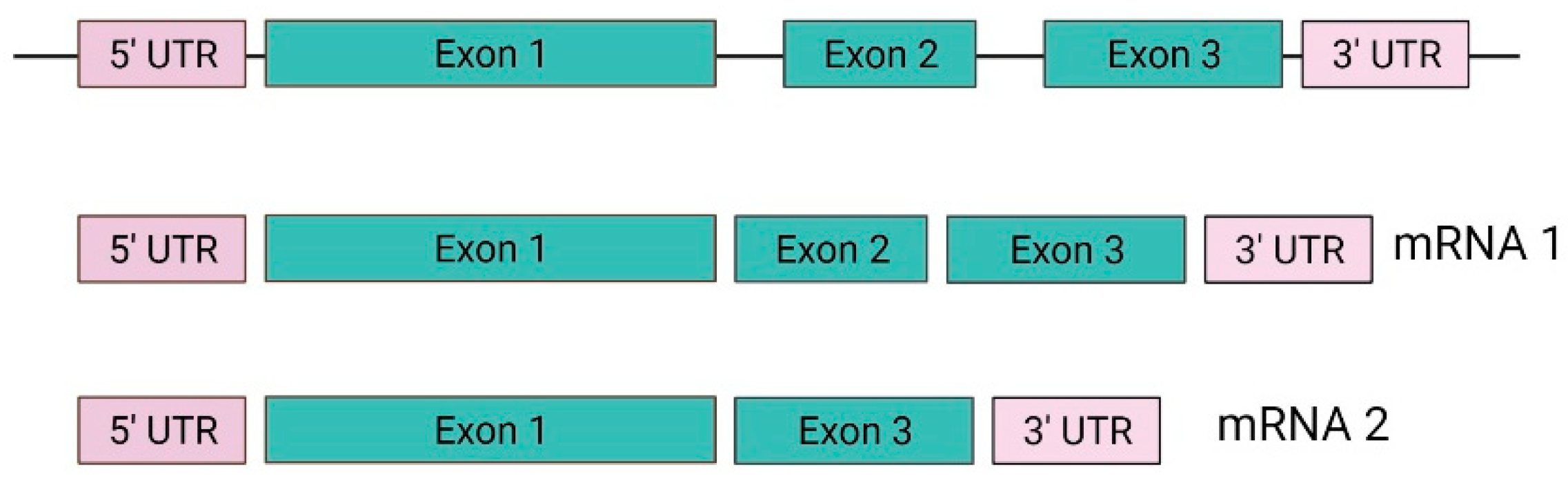
Figure 3.
The structure of the pVHL protein has two domains: the β domain that ranges from 63 to 155 aa and the α domain that includes amino acids 156 to 193. Created with BioRender.com.
Figure 3.
The structure of the pVHL protein has two domains: the β domain that ranges from 63 to 155 aa and the α domain that includes amino acids 156 to 193. Created with BioRender.com.
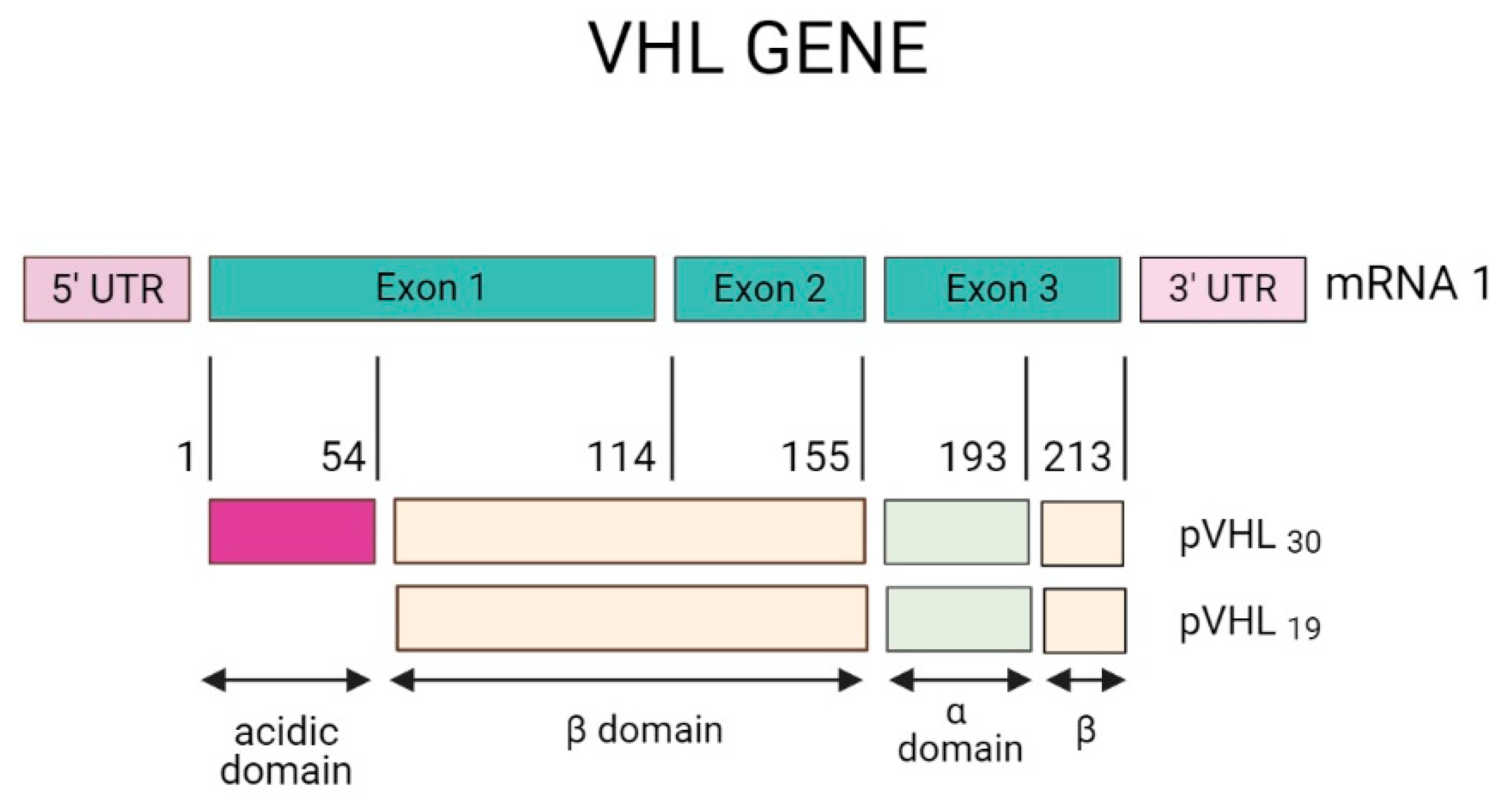
Figure 4.
Inheritance of a dominant gene. Created with BioRender.com.
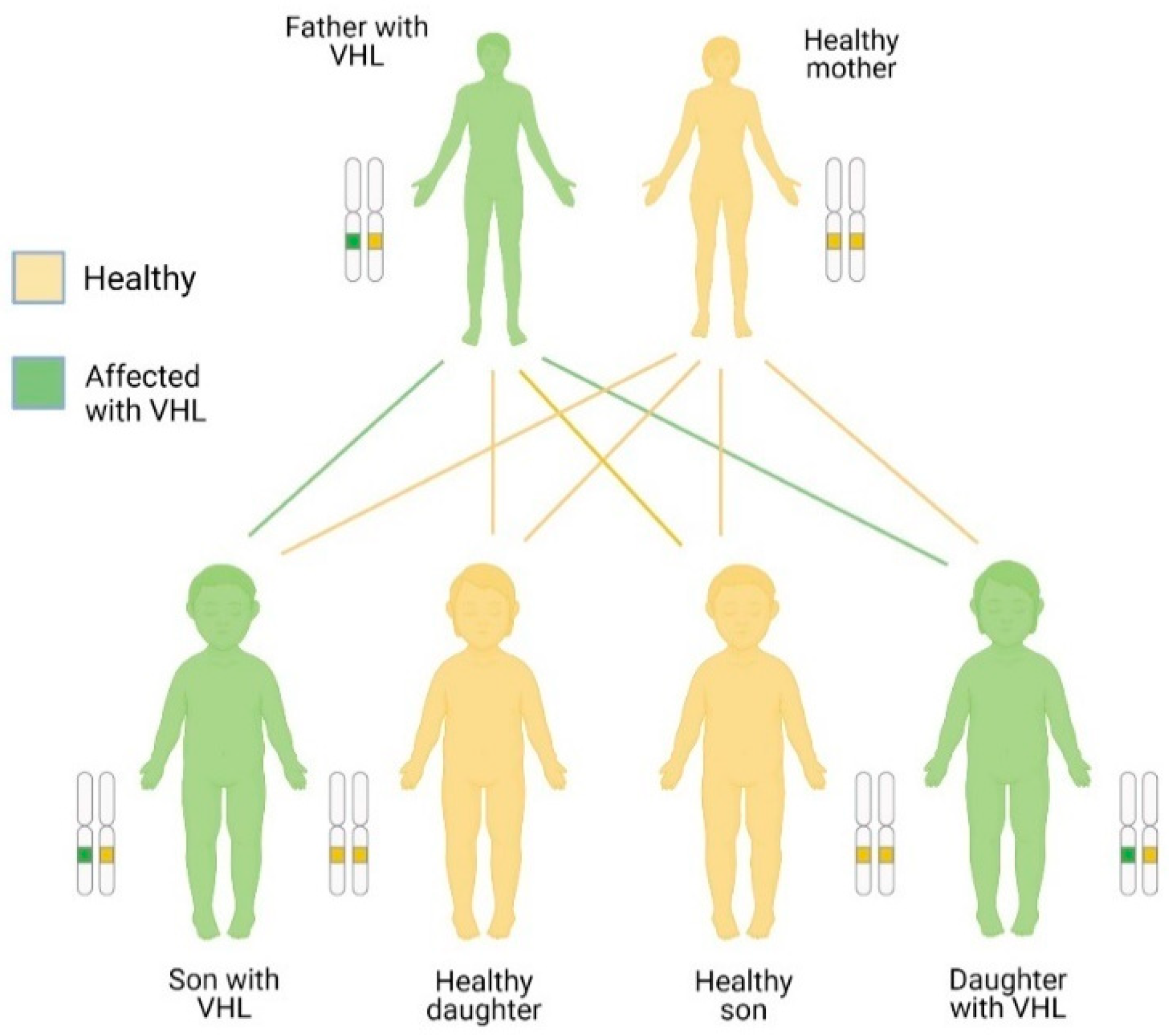
Figure 5.
Scheme explaining tumor formation in VHL. Created with BioRender.com.
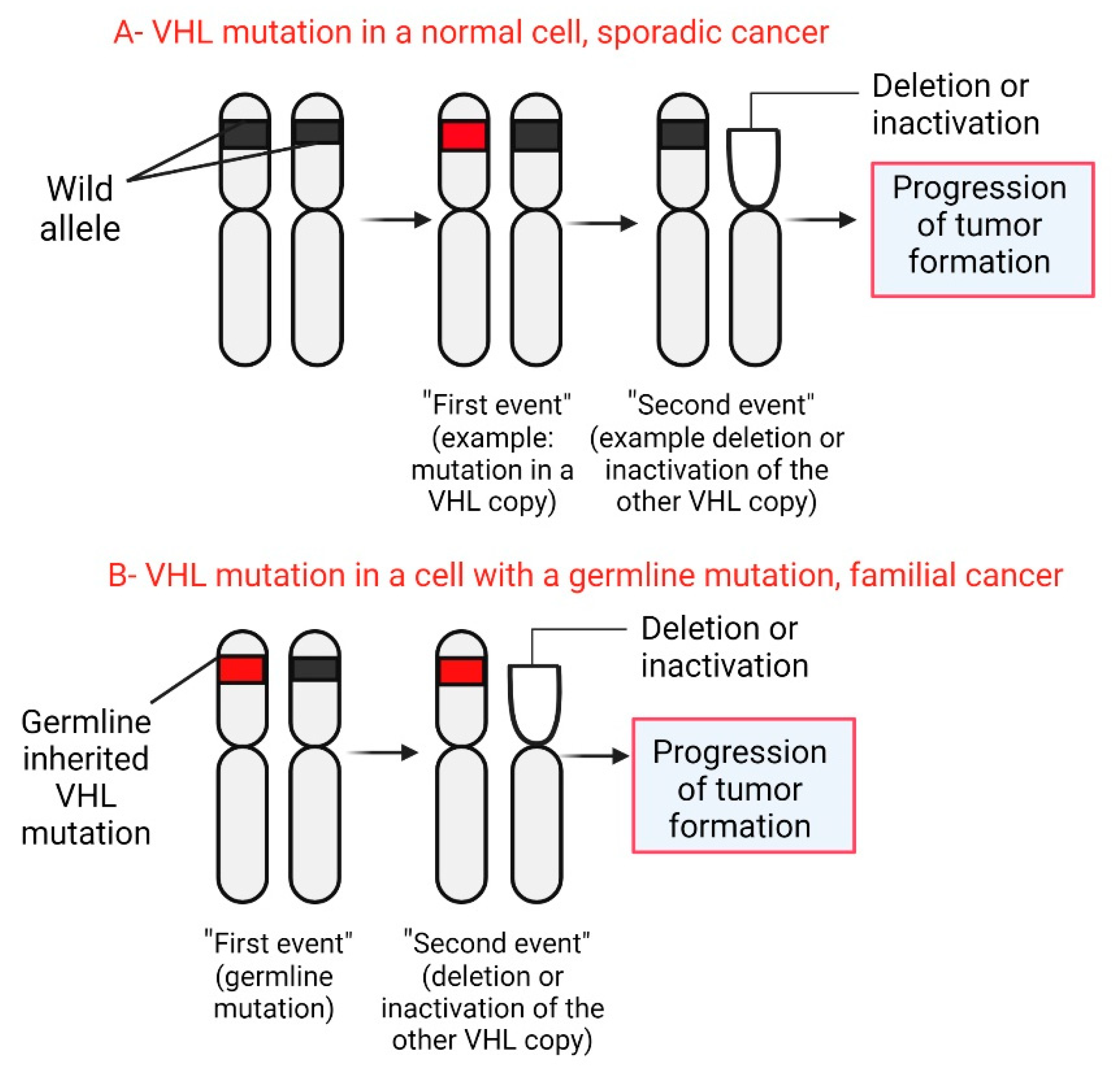
Figure 6.
Major pVHL mutations associated with the development of tumorigenesis in VHL disease. The VHL protein has a beta, alpha, beta domain, 2 isoforms are known according to the molecular weight of pVHL30 and pVHL18/19, both proteins are tumor suppressor. Created with BioRender.com.
Figure 6.
Major pVHL mutations associated with the development of tumorigenesis in VHL disease. The VHL protein has a beta, alpha, beta domain, 2 isoforms are known according to the molecular weight of pVHL30 and pVHL18/19, both proteins are tumor suppressor. Created with BioRender.com.
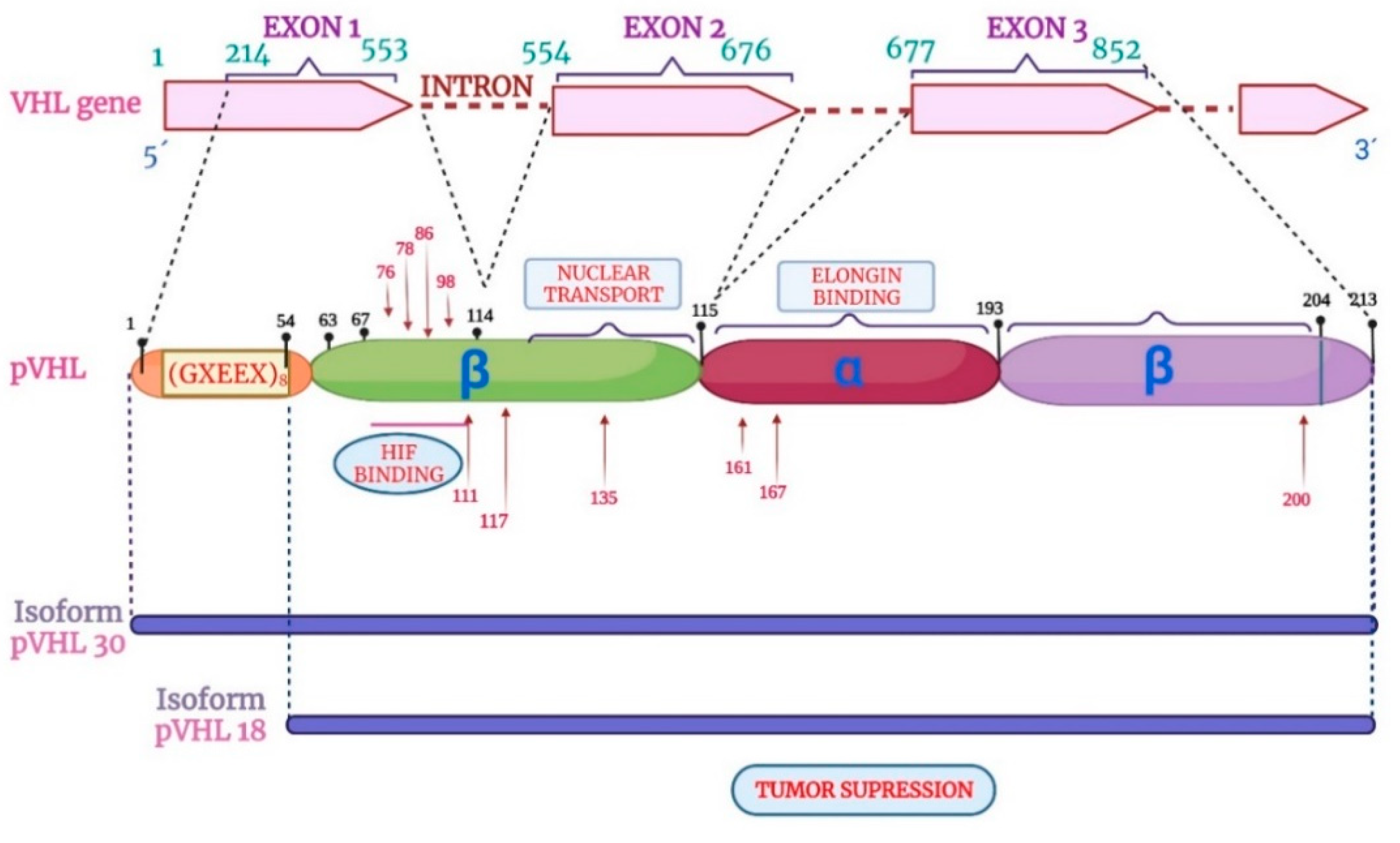
Figure 7.
pVHL function under different conditions. a) Normaloxic and hypoxic conditions. Under normoxic conditions, pVHL interacts with HIF-α hydroxylated. HIF-α is ubiquitylated and degraded via the proteasome. In hypoxic conditions, pVHL does not recognize HIF-α. Then, HIF-α dimerizes with HIF-β. The complex translocates to the nucleus and promotes the transcription of many genes such as VEGF, PDGF, TGF-α, EGFR, and VEGFR. Another HIF- activated gene activated by HIF-α is ReDD1 and negatively regulates mTORC1 through Tuberous Sclerosis Complex 2 (TSC2). b) In renal cell carcinoma conditions, the pVHL mutant does not bind to HIF-α and promotes the transcription of genes involved in tumor progression. Another gene upregulated by HIF is ReDD1, but mTORC1 is activated. Thus, another HIF-dependent mechanism that activates mTORC1 is DEPTOR down-regulation. However, pVHL also regulates the deregulation of mTORC1 through the repression of RAPTOR in renal cell carcinoma. RAPTOR activates mTORC1 by destroying pVHL function.
Figure 7.
pVHL function under different conditions. a) Normaloxic and hypoxic conditions. Under normoxic conditions, pVHL interacts with HIF-α hydroxylated. HIF-α is ubiquitylated and degraded via the proteasome. In hypoxic conditions, pVHL does not recognize HIF-α. Then, HIF-α dimerizes with HIF-β. The complex translocates to the nucleus and promotes the transcription of many genes such as VEGF, PDGF, TGF-α, EGFR, and VEGFR. Another HIF- activated gene activated by HIF-α is ReDD1 and negatively regulates mTORC1 through Tuberous Sclerosis Complex 2 (TSC2). b) In renal cell carcinoma conditions, the pVHL mutant does not bind to HIF-α and promotes the transcription of genes involved in tumor progression. Another gene upregulated by HIF is ReDD1, but mTORC1 is activated. Thus, another HIF-dependent mechanism that activates mTORC1 is DEPTOR down-regulation. However, pVHL also regulates the deregulation of mTORC1 through the repression of RAPTOR in renal cell carcinoma. RAPTOR activates mTORC1 by destroying pVHL function.
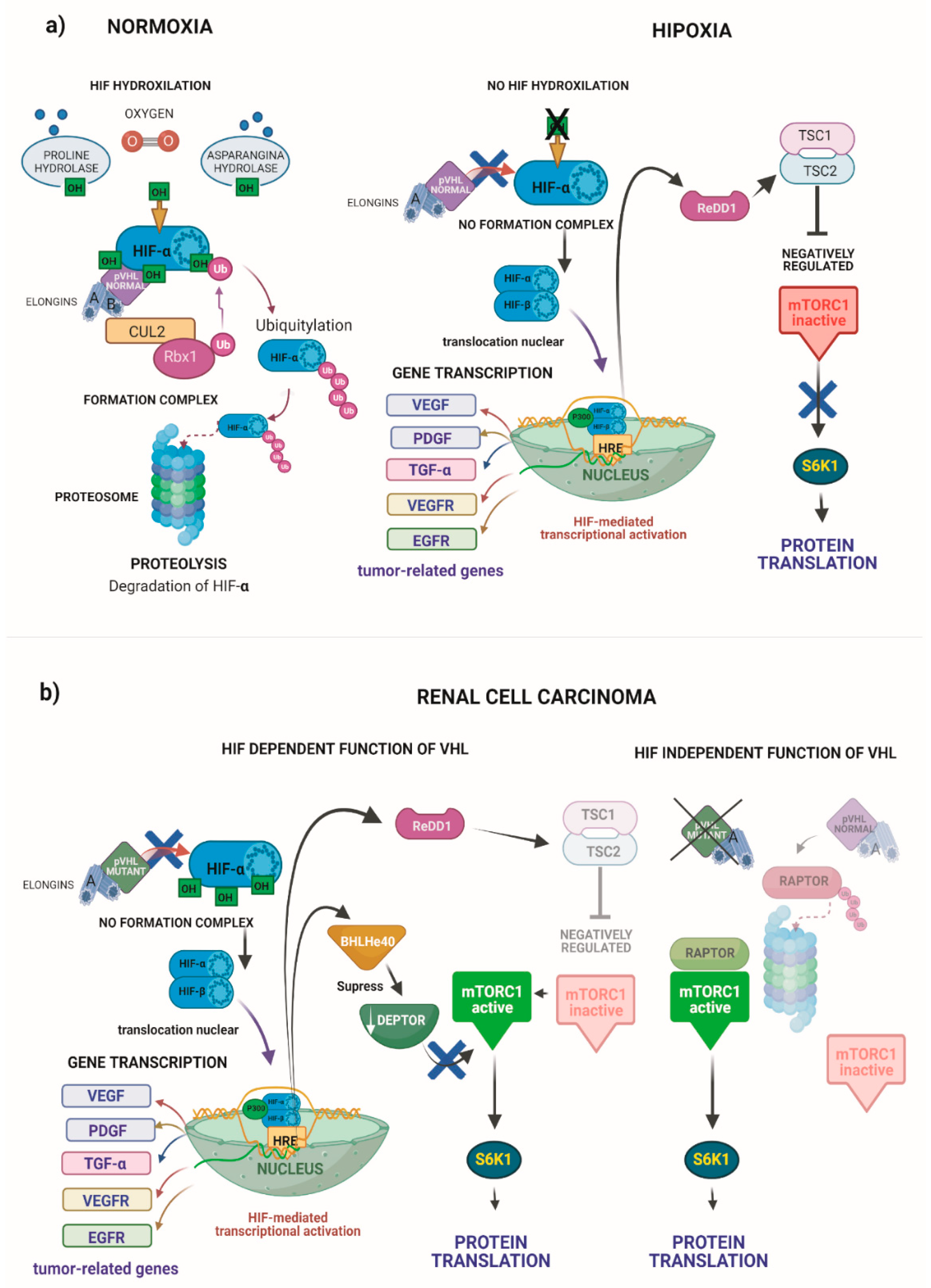
Figure 8.
Other targets of pVHL in ccRCC and SNC hemangioblastomas. a) ccRCC. 1) The loss of pVHL stabilizes HIF-α at enhancers, recruits p300 to maintain H3K27 acetylation and up-regulates ZNF395. ZNF395 promotes the tumorigenesis of ccRCC. 2) ZHX2 and SFMBT1 are novel pVHL substrates. ZHX2 promotes NF-kB activation, and thus cell growth in ccRCC. On the other hand, ZHX2 also activates the MAPK / ERK signal pathway, facilitating several cell functions in addition to causing drug resistance. SFMBT1 promotes SPHK1 expression that contributes to the oncogenic phenotype. 3) RSUME sumoylates pVHL mutants. This post-translational modification stabilizes HIF-α and thus enhances VEGF action promoting vascularized tumors. 4) Loss of pVHL hyperactivates TBK1 phosphorylating p62 and promotes renal tumorigenesis. b) SNC hemangioblastomas. 1) The pVHL mutant does not ubiquitinate STAT and JAK2. It activates the JAK-STAT signaling pathway, thus differentiating hemangioblast progenitor cells into neoplastic cells.
Figure 8.
Other targets of pVHL in ccRCC and SNC hemangioblastomas. a) ccRCC. 1) The loss of pVHL stabilizes HIF-α at enhancers, recruits p300 to maintain H3K27 acetylation and up-regulates ZNF395. ZNF395 promotes the tumorigenesis of ccRCC. 2) ZHX2 and SFMBT1 are novel pVHL substrates. ZHX2 promotes NF-kB activation, and thus cell growth in ccRCC. On the other hand, ZHX2 also activates the MAPK / ERK signal pathway, facilitating several cell functions in addition to causing drug resistance. SFMBT1 promotes SPHK1 expression that contributes to the oncogenic phenotype. 3) RSUME sumoylates pVHL mutants. This post-translational modification stabilizes HIF-α and thus enhances VEGF action promoting vascularized tumors. 4) Loss of pVHL hyperactivates TBK1 phosphorylating p62 and promotes renal tumorigenesis. b) SNC hemangioblastomas. 1) The pVHL mutant does not ubiquitinate STAT and JAK2. It activates the JAK-STAT signaling pathway, thus differentiating hemangioblast progenitor cells into neoplastic cells.
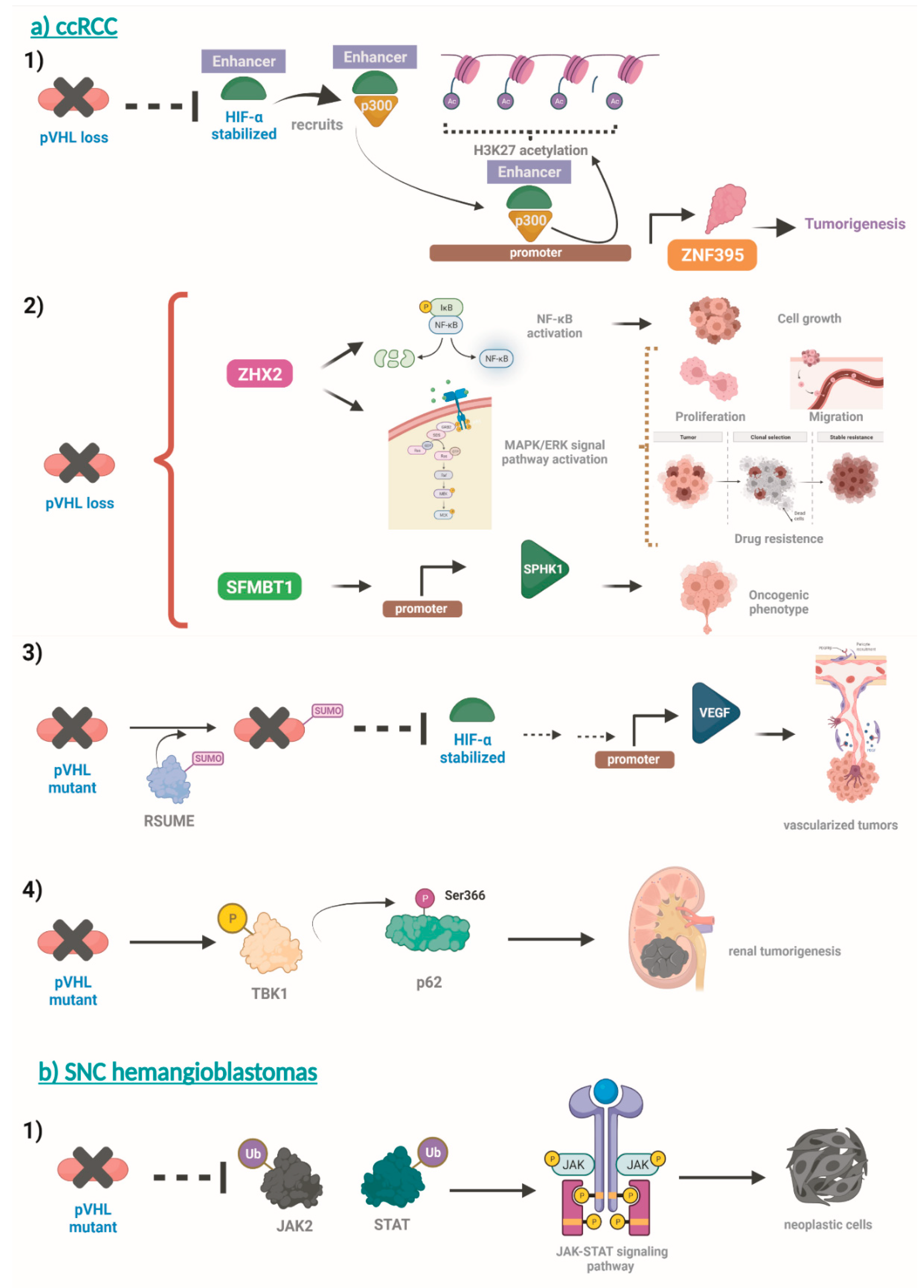

Table 1.
Clinical classification of VHL and associated mutations. Information obtained from VHLdb database (https://vhldb.bio.unipd.it/mutations) according to [35]. Database was revised in September 30, 2021.
Table 1.
Clinical classification of VHL and associated mutations. Information obtained from VHLdb database (https://vhldb.bio.unipd.it/mutations) according to [35]. Database was revised in September 30, 2021.
| Clinical classification | Associated mutations | References |
|---|---|---|
| Von Hippel-Lindau syndrome Type 1 | p.Asn78Ser | [36] |
| p.Pro81Ser | [37] | |
| p.Trp117Cys | [38] | |
| p.Phe136Ser | [36] | |
| In-frame del | [39] | |
| Stop at 158 | [40] | |
| p.Asn78His | [41] | |
| p.Ser65Trp | [42] | |
| p.Leu89Pro | [43] | |
| p.Ser111Asn | [44] | |
| p.Ser111Arg | [45] | |
| p.Ser80Arg | [46] | |
| p.Pro86Leu | [38] | |
| p.Ser65Leu | [44] | |
| p.Trp88Arg | [47] | |
| In-frame del Phe76 | [37] | |
| p.Gln96Pro | [48] | |
| p.Trp88Ser | [37] | |
| p.Gln73X | [49] | |
| p.Asn90Ile | [50] | |
| p.Leu184Pro | [37] | |
| p.Ile180Val | [43] | |
| p.Cys162Arg | [37] | |
| Von Hippel-Lindau syndrome Type 2 | p.Phe119Leu | [51] |
| Von Hippel-Lindau syndrome Type 1 and Type 2 | Frameshift | [36,52] |
| p.Leu118Pro | [36,48] | |
| p.Leu178Pro | [37,38] | |
| p.Ser80Asn | [43,53] | |
| p.Gly93Asp | [38,54] | |
| p.Leu158Pro | [37,55] | |
| p.His115Gln | [37,56] | |
| p.Gly114Cys | [36,41] | |
| Von Hippel-Lindau syndrome Type 1 and Type 2B | p.Arg113X | [42,54] |
| Von Hippel-Lindau syndrome Type 1, Type 2, and Type 2A | p.Tyr98His | [38,57] |
| Von Hippel-Lindau syndrome Type 1, Type 2, and Type 2B | p.Arg161X | [42,58,59] |
| p.Cys162Tyr | [54,60] | |
| p.Val74Gly | [37,57] | |
| p.Cys162Trp | [37,42,56] | |
| Von Hippel-Lindau syndrome Type 1, Type 2, and Type 2C | p.Leu188Val | [42,57,61] |
| p.Gly93Ser | [37,53,62] | |
| Von Hippel-Lindau syndrome Type 1, Type 2A and Type 2B | p.Pro86Ser | [48] |
| Von Hippel-Lindau syndrome Type 1, Type 2, Type 2A and Type 2B | p.Arg167Gln | [53,56,57,63] |
| p.Arg167Trp | [42,63,64,65] | |
| Von Hippel-Lindau syndrome Type 1, Type 2, Type 2A, Type 2B and Type 2C | p.Arg167Trp | [37,42,56,66] |
| Von Hippel-Lindau syndrome Type 2 and Type 2A | p.Val166Phe | [56,67] |
| p.Arg161Gln | [56,68] | |
| p.Tyr98His | [53,57] | |
| p.Tyr112His | [38,69] | |
| Von Hippel-Lindau syndrome Type 2 and Type 2B | p.Arg161Gly | [57,70] |
| Von Hippel-Lindau syndrome Type 2, Type 2A and Type 2B | p.Thr157Ile | [37,54,56] |
| Von Hippel-Lindau syndrome Type 2A and Type 2B | p.Ala149Ser | [71,72] |
Table 2.
Summary of the lesions, symptoms, diagnosis, imaging, and treatment of VHL patients.
| Lesion/frequency in patients by age | Symptoms |
Diagnosis (MR,Magnetic Resonance; CT, Computed Tomography) |
Imagen | Treatment and management | Refs. |
|---|---|---|---|---|---|
|
CNS hemangioblastomas (HBs) cerebellum and spinal cord. Early third decade (ages 22–26 years). Hemangioblastomas occur in approximately 60 to 80% of patients with VHL. |
Headache, gait imbalance, ataxia, abnormal head position, nausea, vomiting, and papilledema. | MR- brain |
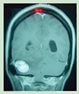 [82] http://creativecommons.org/licenses/by-nc-nd/3.0/. |
Followed by repeated MRI scans in asymptomatic patients. In patients, symptomatic tumors should be surgically removed. | [82,83] |
| Neurological impairment, urinary or bowel abnormalities, singultus, dysphagia, myelopathic disorders, syringomyelia, and polyglobulia. | MR-spinal cord |
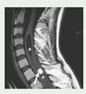 [84] http://creativecommons.org/licenses/by-nc-nd/2.0/. |
Depending on the size of the tumor, surgical removal is recommended. | [83,84] | |
|
Retinal hemangioblastoma (RH). Median age 21 and 25 years old, with a frecuency from 49% to 85%. |
Gradual loss of vision | Angiography Ultrasonography |
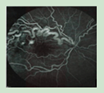 [85] http://creativecommons.org/licenses/by/4.0/. |
Ablative treatment: thermal laser photocoagulation, cryotherapy, radiation, and transpupillary thermotherapy | [85,86,87] |
|
Renal cysts. Commonly it occurs at the fourth decade of life, but this variant of VHL disease, could occurrs as young as 16 years old. 60%-70% of the lesions were carcinomas, all with clear cell features (RCCs, Renal Cysts Clear Cells). |
Mostly asymptomatic. flank pain or hematuria. | Ultrasound, Abdominal MRI or CT |
 [1] http://creativecommons.org/licenses/by/4.0/. |
Partial nephrectomy is the option for tumors that have reached 3 cm reduce the risk of metastasis while maintaining kidney function. RCCs -VHL were treated with radical nephrectomies | [1,74,75,84,87] |
|
Pheochromocytoma (PCC). Diagnosticated arround the third decade of life with a frecuency of 33%. |
Headache, sweating, palpitation, and hypertension | TC o RM |
 [88] http://creativecommons.org/licenses/by-nc-nd/4.0/. |
Initial treatment is with alpha adrenergic blockers such as phenoxybenzamine for the control of hypertension. However, surgical resection remains the definitive treatment. | [27,84,87,88,89] |
|
Pancreatic cysts and pancreatic neuroendocrine tumors (NETs). Is diagnosis already fourth decade, 35-75% of patients with VHL disease will develop simple pancreatic cysts. |
Jaundice, abdominal pain, pruritis, vomiting and Abdominal swelling | MRI, CT |
 [1] http://creativecommons.org/licenses/by/4.0/. |
Pancreatic cysts do not require surgical intervention; PNETs with a potential for metastatic disease are resected with enucleation by Whipple’s procedure or partial pancreatectomy depending on location and size greater than 3 cm. | [1,84,90,91,92] |
|
Endolymphatic sac tumors. (ELTS). 10 to 15% of patients with VHL disease develop ELSTs. The mean age of onset is 22 years old (range, 12-50 years) and they may be bilateral in 30% of cases. |
Facial nerve palsy, vestibulocochlear impairments. Tinnitus, vertigo, disequilibrium, and hearing loss. |
CT |
 [93] http://creativecommons.org/licenses/by-nc-nd/3.0/. |
Treatment of ELTS requires extensive surgery with adequate bone removal around the area of the macroscopically evident tumor. | [93,94] |
|
Epididymal cystadenomas. 16-80 years old. Occur in 25–60% of affected men |
Mostly asymptomatic | Ultrasonography PET/CT images |
 [95] http://creativecommons.org/licenses/by/4.0/. |
Surgery is rarely performed in epididymal cystadenomas. routinely followed by a physical exam and ultrasonography. | [75,95,96] |
|
Broad ligament cystadenomas. Diagnosticated about the second decade with 10% frecuency. Women unilateral presentation |
Mostly asymptomatic | Transvaginal ultrasound |
 [97] http://creativecommons.org/licenses/by/4.0/. |
Being benign lesions, they are usually managed conservatively. without surgery. | [97,98] |
Table 3.
Biomarkers in Von Hippel-Lindau disease.
| Biomarker | Sample | Cohort | Results/Remarks | Ref. |
|---|---|---|---|---|
| Telomere length | Blood |
|
|
[140] |
| c-Myc/HIF-2α | Formalin-fixed, paraffin-embedded primary tumor samples. |
|
|
[141] |
| Serpin peptidase inhibitor clade H member 1 (SERPINH1) | Primary ccRCC and adjacent normal kidney tissues. |
|
|
[142] |
| Programmed death ligand-1 (PD-L1) | Formalin-fixed and paraffin-embedded surgical RCC simples. |
|
|
[143] |
| Lamin-B1 | Sample tissues (primary tumors and normal tissues samples). |
|
|
[144] |
| Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) and PD-1 | Formalin-fixed paraffin embedded (FFPE) specimens collected from radical surgery. |
|
|
[145] |
| lncRNA FGD5-AS1 | Renal tissue (adjacent normal renal tissue and ccRCC tissue). |
|
|
[146] |
| SFMBT1 and ZHX2 | Tumor tissues and adjacent normal tissues. |
|
|
[119] |
| 89Zr-bevacizumab | Imaging |
|
|
[147] |
| 68Gallium-DOTATATE PET/CT | Imaging |
|
|
[148] |
Table 4.
Clinical trials in Von Hippel-Lindau disease.
| Clinical Trial Identifier & Therapeutic Agent | Clinical Phase | Status | Condition or disease | Molecular Target | Participants | Outcome | Refs. |
|---|---|---|---|---|---|---|---|
|
NCT00673816 Sunitinib |
II | Terminated | Advanced Ocular Disease of von Hippel-Lindau Syndrome | Inhibition of multiple receptor tyrosine kinases (RTK), including the vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). | 5 patients receive 9 months of sunitinib malate therapy administered in 6 cycles. Each cycle consisted of a daily oral dose of 50 mg sunitinib malate for 4 weeks followed by a 2-week rest period. (Only one completed treatment) | Change in Best-Correction Visual Acuity (BCVA), Retinal Thickness, and Retinal Angioma Leakage from Baseline to Week 36. The recruitment goal was to enroll five participants; however, the study was terminated after only two participants had been enrolled due to slow recruitment and adverse events. | [149] |
|
NCT00330564 Sunitinib |
II | Terminated | Von Hippel-Lindau syndrome: Renal cell carcinoma and / or Hemangioblastoma |
Treatment with SU011248/sunitinib malate (50 mg daily dose for 4 weeks, then 2 weeks off) for 6 months in 15 patients with Von Hippel-Lindau Syndrome (VHL) who have a measurable lesion undergoing surveillance. | Nine of the 15 patients completed all four cycles of therapy, and the expected toxic effects were responsible for the necessary dosage reductions and discontinuation of treatment. Renal cell carcinomas responded better to sunitinib therapy than other VHL related lesions using the RECIST measure. The study ended early due to slow accrual. | [150] | |
|
NCT02859441 Ranibizumab E10030 |
I/II | Completed | Von-Hippel-Lindau (VHL)-Retinal Capillary Hemangiomas (RCH) | E10030, a PDGF-B antagonist, and ranibizumab, a VEGF-A antagonist | This was a single-arm open-label phase 1/2 study, consisting of 3 adults with VHL-associated RH associated with VHL and vision loss. Intravitreous injections of ranibizumab (0.5 mg) and E10030 (1.5 mg) were administered unilaterally and each received 9 injections prior to week 52 and were followed for 104 weeks. | One participant manifested mild episodic ocular hypertension in the study eye. The change in BCVA in the study eye at week 52 for the three participants was –5, –12, and +2 letters. No reduction in RH size was measured at 52 weeks. Variable mild improvements in exudation in two participants at week 16 were not sustained through week 52. Intranavitreous injection with ranibizumab and E10030 demonstrated a reasonable preliminary safety profile, but limited treatment effect. | [151] |
|
Protocol number CT/AE-941/002 Neovastat (Canadá) |
II | Completed | Renal Cell Carcinoma (RCC) | Inhibition of angiogenesis | 22 patients with a primary diagnosis of refractory CCR. They were treated with Neovastat 240 ml/day (n = 14) compared to patients receiving 60 ml/day (n = 8) | The higher dose of Neovastat administered in this trial is associated with a survival benefit in RCC. Neovastat is well tolerated by advanced cancer patients at doses of 60 and 240 ml/day. | [152] |
|
Not definite. Neovastat (Canadá) |
III | Completed | Metastatic renal cell carcinoma in whom immunotherapy failed | 300 patients from 48 international centers were randomized to receive 120 ml twice daily of oral Neovastat or placebo Neovastat. The 300 patients who received at least 1 dose of study medication were included in this analysis. | The study of metastatic CRC provides a prognostic model that has a significant impact on risk-adjusted survival. Although external validation in an independent data set is lacking, the results of this trial may lead to a new paradigm for clinical trial design and risk stratification when considering future investigations of patients with metastatic CRC in whom immunotherapy has failed. | [153] | |
|
EudraCT Number: 2014-003671-30 Propanolol (Spain) |
III | Completed | Von Hippel-Lindau disease and retinal hemangioblastomas | VEGF inhibitor | 7 patients were included. All patients received a daily dose of 120 mg propranolol for 1 year. Clinical variables were evaluated at baseline, and at 1, 3, 6, 9 and 12 months. | The number and size of retinal hemangioblastomas remained stable in all patients. The only adverse effect reported was hypotension in one patient. The results suggest that propranolol could be useful for the treatment of retinal hemangioblastomas in patients with VHL, especially when there are retinal exudates. The results of this clinical trial allowed propranolol designation to treat von Hippel-Lindau disease, granted by the European Medicines Agency (EMA). | [154] |
|
EudraCT Number: 2007-002132-29 Sorafenib (UK) |
II | Terminated | Renal cancer associated Renal Cancer VHL | Tyrosine kinase inhibitor | In 4 patients with VHL syndrome who had therapy for advanced RCC, oral received whom sorafenib (400 mg twice a day) for up to six months. | This study concludes that over a 6-month period of sorafenib, at the standard dose used in RCC, there was no response effect in CNS hemangioblastomas in this population of patients. | [155] |
|
NCT01436227 Pazopanib |
II | Active, not recruiting | von Hippel-Lindau disease genetically confirmed or one disease-related lesion. | Vascular endothelial growth factor receptors (VEGFR) -1, -2, and -3, c-kit and platelet-derived growth factor receptor (PDGF-R) inhibitors | Thirty-one eligible patients were treated with pazopanib 800 mg by mouth daily for 24 weeks. |
To ensure timely dissemination of data, the decision was made to close the trial after 31 evaluable patients were accrued. Pazopanib induces a reduction in the burden of the disease in von Hippel-Lindau disease patients. Efficacy data indicate benefit in individuals with renal cell carcinomas and pancreatic lesions, and some potential efficacy signals in hemangioblastomas as well. Pazopanib could be considered in patients with von Hippel-Lindau disease and growing lesions where surgical resection may be required in the relatively near future, or in patients with unresectable lesions where a decrease in tumor size is desired. | [156] |
|
NCT03401788 Belzutifan |
II | Active, not recruiting | Von Hippel Lindau Disease-Associated Renal Cell Carcinoma | Inhibitor of HIF-2α | 61 patients receive 120 mg of belzutifan orally once a day until progression, intolerable toxicity, or the investigator / patient’s decision to withdraw. | Of 61 patients, 53 (86.9%) had a decrease in the size of the target lesions. Responses were also observed in CNS, retinal, and pancreatic lesions. MK-6482 showed promising efficacy and tolerability in patients with VHL-associated ccRCC and responses in other VHL-related lesions. | [157,158] |
|
NCT03108066 PT2385 |
II | Active, not recruiting | Von Hippel Lindau Disease-Associated Renal Cell Carcinoma | Inhibitor of HIF-2α | 4 patients will be enrolled in each stage of a two-stage design. PT2385 was administered orally at a dose of 800 mg twice daily, follow-up of 19 weeks | All patients had stable disease (SD) as their best response at the latest assessment. PT2385 demonstrated stabilization of disease in VHL-associated clear cell RCC and other tumors and showed an acceptable safety profile. | See below |
|
NCT01266070 Dovitinib |
II | Terminated | VHL-related hemangioblastoma | A multityrosine kinase that inhibits FGFR, VEGFR, and PDGFR. | Six participants received 500 mg/day (5 days in / 2 days out of dosing). Completed at least two cycles of therapy. | The trial was stopped after six patients due to the activation of the toxicity stopping rule. The lack of response in HBs in this population treated with dovitinib is surprising, and molecular profiling of HB tissue would be extremely useful to help understand the biologic underpinnings of this lack of efficacy. | [159] |
|
NCT00089765 Ranibizumab |
I | Completed | Angiomas (blood vessel tumors) in patients with Von Hippel-Lindau syndrome (VHL) | VEGF-neutralizing agent | 5 patients with retinal capillary hemangioblastomas (RCH) associated with VHL with exudative changes and visual loss. Monthly intravitreal injections of ranibizumab (0.5mg) were administered over a 6 month course for a total of 7 injections, with additional injections considered until week 52. | The primary outcome was the change in best-corrected visual acuity (BCVA). Secondary outcomes included change in lesion size, change in retinal thickness, and adverse event assessments. Intravitreal ranibizumab, administered as monotherapy every 4 weeks, had minimal beneficial effects on most RCHs related to VHL. Future studies are needed to determine a combination with other therapies for the treatment of ocular tumors associated with VHL. | [160] |
|
NCT02108002 Vorinostat |
I | Completed | VHL-related hemangioblastoma | Histone deacetylase inhibitor (HDACi) | 7 germline missense VHL patients with symptomatic CNS hemangioblastomas, received 400 mg of Vorinostat by mouth daily for seven days prior to surgery and subsequently underwent surgical resection. | Vorinostat is well tolerated by patients with symptomatic CNS hemangioblastomas in the context of germline missense VHL disease and shows results in mutated stabilization of the pVHL protein. This suggests that Vorinostat may be a promising treatment for patients with a germline mutation. | See below |
|
NCT00566995 Vandetanib |
II | Completed | VHL-associated renal cell carcinoma | Dual VEGFR2/EGFR inhibitor | 34 participants received 300 mg/day (starting dose) oral dose of vandetanib once a day for 28 days. | Vandetanib demonstrated antitumor activity. However, the poor tolerability required drug withdrawal in a significant proportion of patients. Newer agents that selectively target VEGF receptors may offer a more tolerable alternative and could optimize clinical benefits in this population. | [161] |
|
Medical Ethics Committee of Peking University First Hospital (Beijing, China) TKIs |
ND | Completed | Von Hippel Lindau Disease | Tyrosine Kinase Inhibitor (TKI) |
32 patients receiving TKIs were recruited. For sunitinib, a dosage of 50 mg/day was administered orally for 28 days, followed by a 14-day break per cycle for several cycles. For sorafenib, a dose of 800 mg/day divided into two doses was administered orally. For axitinib, a dose of 10 mg/day divided into two doses was administered orally. For pazopanib, a dose of 800 mg/day was administered orally. | A partial response was observed in 11 (31%) of 36 renal cell carcinomas, 4 (27%) of 15 pancreatic lesions, and 1 (20%) of five central nervous system (CNS) hemangioblastomas. The mean tumor size decreased significantly for renal cell carcinomas (P=0.0001), renal cysts (P=0.027), and pancreatic lesions (P=0.003) after TKI therapy. Finally, the side effects were acceptable. | [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



