
Submitted:
14 June 2024
Posted:
17 June 2024
You are already at the latest version
Abstract
A fully automated bacteria whole genome sequencing (WGS) assay was evaluated to characterize Mycobacterium tuberculosis (MTB) and non-tuberculosis Mycobacterium (NTM) clinical isolates. Results generated were highly reproducible with 100% concordance in species and sub-lineage classification, and 92% concordance between antimicrobial resistance (AMR) genotypic and phenotypic profiles. These findings demonstrate that a fully automated WGS assay, with a short turnaround time of 24.5 hours, provides timely and valuable insights into MTB outbreak investigation while providing reliable genotypic AMR profiling consistent with traditional antimicrobial susceptibility tests (AST). This study establishes a favorable proposition in the adoption of an end-to-end fully automated WGS solutions for decentralized TB diagnostics, thereby aiding in WHO’s vision of tuberculosis eradication.
Keywords:
Mycobacterium tuberculosis
; Whole Genome Sequencing
; Automation
; Antimicrobial Resistance
; Decentralized Diagnostics
1. Introduction
Mycobacterium tuberculosis (MTB) is a preventable and usually curable disease, and yet it causes tens of millions of new cases of tuberculosis (TB) resulting in more than one (1) million deaths every year worldwide [1]. Multidrug-resistant (MDR) tuberculosis is a major health problem and seriously threatens worldwide TB control and prevention initiatives. Since 2020, there have been an estimated 465,000 Rifampicin-resistant (RR) tuberculosis cases, with 78% of these cases classified as multidrug-resistant (MDR). This indicates that the MTB strains causing these cases are resistant to both Rifampicin and Isoniazid. [2]. There are two main challenges with MDR TB: underdiagnosis of clinical cases and poor treatment outcomes, both of which lead to more serious clinical complications. Collectively, the failure to identify and successfully treat these cases leads to further exacerbation and transmission of MDR TB strains.
The WHO has emphasized that accurate diagnosis and characterization of TB is the key strategy for successful treatment and eradication of complicated TB cases [3]. Rapid genotypic-based diagnostic tools, such as GeneXpert, have been widely adopted as they are faster and cheaper than traditional culture-based diagnostic susceptibility testing. However, outbreaks caused by MDR strains with novel antimicrobial resistance (AMR) profiles may not be picked up by these assays, which focus on a limited number of targets. This underscores the importance of employing assays with a genome-wide approach that cover a broader range of resistance determinants [4]. Although molecular targeted assays have shown great value in TB AMR characterization [2], these methods are still dependent on known genotypic and mutation profiles [1]. Failure to detect novel AMR genotypes and mutations within the MTB genome can lead to inaccurate prescription of patient treatment regimes, posing a risk of treatment failure. [3]. Whole genome sequencing (WGS) is an agnostic approach to screen the entire pathogen genome for the detection of specific single nucleotide polymorphisms (SNPs) to discriminate known MTB complex (MTBC) (sub)lineages. This approach not only enables presence/absence detection of AMR genes but also the agnostic screening and deep characterization of AMR loci. WGS has significant potential to replace traditional culture-based phenotypic diagnostic tests, which can typically take weeks to months [5]. By reducing turnaround time to less than two (2) weeks, it will improve antibiotic stewardship for first-line drugs in TB treatment.
Widespread adoption and implementation of this methodology remains challenging due to a lack of necessary infrastructure, the high cost of entry and the requirement for technical expertise to process the samples and analyze the generated sequencing data [4]. Integrating next generation (NGS) technologies with automation will alleviate these challenges and has been proven successful for application in the food safety and infectious diseases space [6,7,8,9]. Automation imparts multiple benefits to existing manual workflows. It can improve results consistency and reproducibility by reducing the number of human touch points, eliminating human/random errors in the process. Researcher efficiency can also be improved via the removal of the need for manual and routine tasks. Automation enables the performance of workflows round the clock without human supervision and intervention, further reducing turnaround time from sample input to results output. And finally, it aids in improving workplace safety by eliminating the exposure of laboratorians to potentially harmful chemicals [10]. In this study, we evaluated the accuracy and specificity of a fully automated bacteria WGS assay for the characterization of MDR TB isolates from reference collections as well as clinical isolates.
2. Results
2.1. WGS Characterization of MTB and NTM Isolates
A total of 86 samples (44 unique isolates) were sequenced in this study using the fully automated bacteria WGS assay. MTBs and NTMs isolates were assembled, with their taxon correctly identified down to the species level and their sub-lineages characterized (Table 1). AMR genotypic profiles and associated drug resistance predictions were determined and summarized in Table 2.
2.2. Robustness and Reproducibility of the Fully Automated Bacteria WGS Assay
Three (3) strains of clinical MTB isolates provided by NSPHL, together with four (4) strains each of MTB and NTM isolates procured from reference repositories (ATCC and BEI Resources), were used to assess the reproducibility and robustness of the fully automated bacteria WGS assay. Replicates were sequenced on three (3) different systems by two (2) different operators and results are summarized in Table 3. Lyve-SET analysis of NSPHL Strains 8, 12 and 17 reported no SNP distance between replicates (see Table S1).
2.3. Study Site-Specific Clinical Isolate Relatedness Analysis
Whole genome phylogenetic analysis of clinical isolates was performed to predict MTB clusters from NSPHL and SFPHL (Figure 2a and Figure 2b, respectively). Phylogenetic groupings for the genomes within each study site were consistent with pairwise SNP distances as computed by Lyve-SET for the same genomes (Tables S1 and S2). Isolates within the same cluster shared the same node and showed lower SNP distances, while isolates from different clusters had higher SNP distances. For example, NSPHL Strains 10 and 17 that were within a single cluster had a SNP distance of 229, while NSPHL Strains 10 and 13 that were between clusters had a SNP distance of 1,979 (see Table S1). SFPHL clinical isolates 36359772, 36360344 and Z008267 that were within the same cluster had zero (0) SNP distance, while isolates Z008272 and Z008273 that were from different clusters had a SNP distance of 2,013 (see Table S2).
2.4. Comparison of Genotypic and Phenotypic AMR Results
Genotypic AMR profiles from the WGS data of the clinical isolates were compared with phenotypic data from AST provided by the study sites. The predicted AMR drug classes, based on the AMR gene profiles, perfectly matched the TB-specific AST phenotypic results from NSPHL clinical isolates (Table 4). The vast majority of clinical MTB isolates from SFPHL were sensitive to Streptomycin, Isoniazid, Rifampicin, Ethambutol, and Pyrazinamide, aligning perfectly with the genotypic AMR profiles. Three (3) isolates exhibited non-concordance between their genotypic AMR profiles and AST results. Isoniazid resistance was predicted for 36360377 while Pyrazinamide resistance was predicted for 36360355 and Z008272. AST results indicated that all three (3) isolates were sensitive to all TB first-line drugs (data not shown).
3. Discussion
Although bacteriological confirmation of TB is still the gold standard and necessary for the testing of resistance to anti-TB drugs, genetic sequencing at reference-level laboratories is slowly gaining popularity [11]. WGS is, without a doubt, a more powerful tool and can play a major role in the diagnosis of MTB drug resistance when compared with PCR-based methods. It has the advantage of enabling the identification of “off targets” or new candidate resistance mutations when facing AST discrepancies or resistance to new and/or repurposed drugs, for which resistance catalogs are still being developed [12]. In recent years, application of WGS for TB diagnosis has rapidly progressed from academic research only perspective to routine patient care in the clinics, population-level surveillance, and formulation of public health intervention strategies [13,14,15,16]. These strategies in turn help with more effective antibiotic stewardship, as well as the implementation of pathogen-based precision medicine treatments for TB [17].
Integrating WGS with automation will help reduce the complexity and hands-on time required for the experimental workflow, further reducing turnaround time for diagnosis to increase actionability. This would open up the potential of providing a turnkey solution for widespread adoption, as well as democratizing WGS technology for TB diagnosis to laboratories, hospitals, and clinics with limited personnel and technical expertise. Here, we showed that a fully automated bacteria WGS assay, using extracted DNA from isolates as starting material, is capable of generating highly robust and reproducible results with a turnaround time of approximately 24.5 hours as compared to the 6 - 10 days needed for manual workflow. The fully automated assay was able to achieve the short turnaround time by enabling around-the-clock operation without the need of human supervision and intervention, which is not the case for manual workflows restricted by stipulated laboratorian operating hours. Sample registrations, and the loading of samples and consumables took approximately 30 minutes in total. Data from sequencing the entire MTB genome provided high resolution information, conferring unique insights into AMR genotypic profiles of individual isolates, as well as how they were related to one another.
Greater depth of information helped tease out the intricate relationships of isolates classified within the same sub-lineage. For example, in the case of SFPHL clinical isolates, although isolates 36360342, 36360361, 36360364, Z008274, Z008277 and Z008278 are all from sub-lineage 1.1.1.1, isolates 36360342, 36360361 and 36360364 sub-clustered together and had a SNP distance of zero (0) (see Figure 2b and Table S2). Therefore, we hypothesized that these three (3) isolates might be the same strain or from the same origin. Isolates 36360347, 36360388 and Z008270 had a SNP distance of five (5) when compared to one another while isolate Z008279 had a SNP distance of between 173 and 194 when compared against these three (3) isolates (see Figure 2b and Table S2). We therefore hypothesized that isolates 36360347, 36360388 and Z008270 might be derived from the same outbreak while isolate Z008279 is from a separate event, even though they share the same AMR profile and 4.6.2.2 sub-lineage classification.
Correlations between genotypic and phenotypic profiles of AMR have been widely debated, with varying concordances reported dependent on drug classes and organisms tested [18,19,20,21]. Similarly high, but imperfect concordance has been reported for MTB [12,22]. Findings in this study are consistent with those in the literature. Clinical isolates from NSPHL revealed 100% concordance between WGS genotypic AMR profiles and AST phenotypes, further supporting the use case of applying WGS for comprehensive TB diagnosis under clinical settings. Out of the 24 clinical isolates from SFPHL, 21 showed agreement between genotypic and phenotypic AMR profiles, while three (3) isolates had some discrepancies. These three (3) isolates had AMR genotypes predicting resistance against Isoniazid or Pyrazinamide, despite AST data indicating susceptibility to both drugs in all three cases. The authors emphasize that achieving consistent data without human error is a significant achievement, far outweighing the few discrepancies. This outcome strongly endorses the use of a fully automated WGS assay in TB diagnosis. While the results from the genotypic AMR profile may impact treatment options, in this case, the “false positive” results would encourage clinicians to proceed with caution in treatment selection. This cautious approach further promotes antibiotic stewardship efforts in choosing and tailoring appropriate first-line drug treatments for specific TB cases.
4. Conclusion
In conclusion, the present study highlighted the suitability and robustness of a fully automated bacteria WGS assay for seamless, accurate detection and in-depth characterization of MTB clinical isolates and NTM reference isolates with high consistency. The assay demonstrated 100% concordance in species and sub-lineage classification, and 92% (35 out of 38 isolates) concordance in genotypic vs phenotypic AMR characterization. The WGS data generated will further propel research in the investigation of different MTB virulence factor mechanisms, such as secretion factors, cell surface components, enzymes involved in cellular metabolism, and transcriptional regulators [23]. This fully automated WGS solution, with a short turnaround time, can further aid in the surveillance and resolution of TB outbreaks in a timely manner. A significantly shorter (less than 2-day) turnaround time will increase actionability and patient safety by reducing and preventing MDR TB transmission and related fatalities, as well as reducing healthcare costs [24,25]. A fully automated bacteria WGS assay will be a valuable technology and tool to enhance the efficiency of diagnostic laboratories while supporting WHO’s vision in the elimination of this deadly disease. This study has implications for both government and nongovernmental organization resource allocation decisions and policy formulation, thereby establishing a favorable proposition in the adoption of a fully automated turnkey WGS solution for the decentralization of precision medicine and TB diagnostics.
5. Materials and Methods
5.1. Sample Source
Bacterial isolates or extracted DNA from reference MTB and non-tuberculosis Mycobacterium (NTM) were procured from ATCC or BEI Resources, NIAID, NIH. DNA extracted from de-identified clinical isolates were obtained from two (2) study sites: the Nevada State Public Health Laboratory (NSPHL) and the San Francisco Public Health Laboratory (SFPHL). Details of isolates and strains used in this study are summarized in Table 5.
5.2. Isolate Cultures and DNA Extraction
Non-tuberculosis Mycobacterium (NTM) and an attenuated MTB strain (NR-122) extracted at Clear Labs, Inc. were first grown on Löwenstein–Jensen (LJ) Agar slants (Hardy Diagnostics, Santa Maria, CA) incubated at 35°C ± 2°C for 7-10 days. Bacterial isolates were resuspended in Clear Labs’ Resuspension Buffer v2.0 (Clear Labs, Inc., San Carlos, CA) and heat inactivated at 95°C for 30 minutes. MTB cell suspensions were lysed and DNA extracted using a proprietary Clear Labs inhouse-developed extraction protocol. Extracted DNA was stored at -80°C until use.
MTB clinical samples provided by NSPHL were grown on LJ agar slants, resuspended in HPLC water and inactivated by boiling for 30 minutes. DNA was then extracted from heat inactivated cultures using the Maxwell RSC Cultured Cell Kit (Promega, Madison, WI) or DNeasy Blood & Tissue QIAcube Kit (QIAGEN, Venlo, Netherlands). Extracted DNA was stored at -80°C until use.
MTB clinical samples provided by SFPHL were grown on LJ agar slants, resuspended in HPLC water and inactivated by boiling for one (1) hour. DNA extractions were then performed using the MagNA Pure 24 Total NA Isolation Kit (Roche Life Sciences, Indianapolis, IN) or the EZ1&2 Virus Mini Kit v2.0 (Qiagen, Germantown, MD) as detailed in Table 1. Extracted DNA was stored at -80°C until use.
5.3. DNA Quantification
Extracted DNA was quantified on a Qubit™ 4 Fluorometer using the Qubit 1x dsDNA High Sensitivity Kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA).
5.4. Workflow of a Fully Automated Platform for WGS of Extracted DNA from MTB and NTM
Extracted DNA from all isolates were first diluted to a concentration of 0.5 ng/µL in 30 µl volume (total DNA input yield of 15 ng). Samples were prepared, sequenced and analyzed using the “Low DNA Input” protocol of the Clear Dx™ Microbial Surveillance WGS v2.0 application on the fully automated and integrated Clear Dx™ platform, which comprised liquid handling robotics, thermal cycler and sequencers (Figure 1). Briefly, samples, reagents, and consumables were first loaded onto the instrument according to the manufacturer’s instructions (Clear Labs, San Carlos, CA). Up to 12 samples per run were then registered using the ClearView App with “Mycobacterium (4.4 MB)” selected as “Organism” and “30 – 40x” selected as “Coverage”. Upon pushing the “Start Run” button, the fully automated system performs library preparation using the 2 x 150 bp chemistry, followed by the loading of sequencing cartridges onto the Illumina iSeq100 sequencers (Illumina, Inc., San Diego, CA) as a part of the end-to-end workflow. After completion of sequencing, raw sequencing data was automatically uploaded onto the cloud for bioinformatics analysis. Final output generated by the workflow comprised compressed FASTQ files, sample identity (genus and/or species level), and quality metrics for each isolate. The fully automate end-to-end workflow took approximately 24.5 hours in total.
5.5. Data Analysis
The TheiaProk bioinformatics pipeline was used for bioinformatics analysis [26]. Briefly, paired-end Illumina reads in FASTQ format generated by the fully automated bacteria WGS assay were first trimmed and quality filtered using Trimmomatic and fastq_scan pipelines. Cleaned reads were then de novo assembled using the Shovill algorithm, assembly quality control (QC) checked by QUAST and BUSCO pipelines, and taxonomic assignment performed using GAMBIT. Further downstream analysis includes genome annotation with Prokka, AMR characterization with AMRFinderPlus, and TB-specific analysis using TBProfiler. MashTree was used for site-specific phylogenetic analysis of isolates while Lyve-SET [27] was used for SNP analysis with M. tuberculosis H37Rv (NC_000962.3) set as the reference genome. Antimicrobial susceptibility test (AST) results of MTB isolates were provided by NSPHL and SFPHL.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Lyve-SET data from NSPHL clinical isolates. Table S2: Lyve-SET data from SFPHL clinical isolates.
Author Contributions
Conceptualization: R.K., G.M., M.P. and J.H.J.N.; Methodology: A.G., D.S., E.W, and A.L.; Formal analysis: A.A., S.S. and J.H.J.N.; Project administration: R.K. and J.H.J.N.; Resources: A.G., D.S., E.W, and A.L.; Supervision: R.K., G.M. and M.P.; Visualization: A.A. and J.H.J.N.; Writing — original draft: S.S., A.L. and J.H.J.N.; Writing — review and editing: R.K., G.M., M.P., D.S., A.A., S.S., A.L. and J.H.J.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We would like to thank Michael Balamotis, Derreck Carter-House, Norma Ordaz-Yuan, Rick Kim, Akshay Paropkari, Samuel Hoeffel, David Tran and Alexander Jorjorian from Clear Labs, Inc. for contributing to this study. We would also like to thank Biodefense and Emerging Infections Research Resource Repository (BEI Resources), established by the National Institute of Allergy and Infectious Diseases (NIAID), and American Type Culture Collection (ATCC) for sharing MTB and NTM strains and genomic material.
Conflicts of Interest
The following authors are employee of Clear Labs, Inc.: Justin H. J. Ng, Adam Allred, Andrew Lin, Shadi Shokralla and Ramin Khaksar. Despite this affiliation, the authors declare that they have conducted the research impartially and have adhered to rigorous scientific standards to ensure the integrity and objectivity of the data analysis. The findings and conclusions in this publication are solely those of the authors and are not influenced by the interests of their employer.
References
- Tuberculosis. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 30 May 2024).
- Reported Tuberculosis in the United States, 2021. Drug-Resistant TB. Available online: https://www.cdc.gov/tb/statistics/reports/2021/drug_resistant.htm (accessed on 30 May 2024).
- Dohál, M.; Porvazník, I.; Solovič, I.; Mokrý, J. Advancing tuberculosis management: the role of predictive, preventive, and personalized medicine. Front. Microbiol. 2023, 14, 1225438. [Google Scholar] [CrossRef] [PubMed]
- Pongpeeradech, N.; Kasetchareo, Y.; Chuchottaworn, C.; Lawpoolsri, S.; Silachamroon, U.; Kaewkungwal, J. Evaluation of the use of GeneXpert MTB/RIF in a zone with high burden of tuberculosis in Thailand. PLoS One 2022, 17, e0271130. [Google Scholar] [CrossRef] [PubMed]
- Drug Resistant Tuberculosis: The Next Global Health Crisis? CDC Congressional Testimony. Available online: https://archive.cdc.gov/www_cdc_gov/washington/testimony/2015/t20151208.htm (accessed on 30 May 2024).
- Lin, A.; Singh, A.; Allred, A.; Allard, M.; Waltman, D.; Imanian, B.; Ng, J.H.J.; Sanahmadi, Y.; Khaksar, R. Targeted Next-Generation Sequencing Assay for Direct Detection and Serotyping of Salmonella from Enrichment. J. Food Prot. 2024, 87, 100256. [Google Scholar] [CrossRef] [PubMed]
- Imanian, B.; Donaghy, J.; Jackson, T.; Gummalla, S.; Ganesan, B.; Baker, R.C.; Henderson, M.; Butler, E.K.; Hong, Y.; Ring, B.; Thorp, C.; Khaksar, R.; Samadpour, M.; Lawless, K.A.; MacLaren-Lee, I.; Carleton, H.A.; Tian, R.; Zhang, W.; Wan, J. The power, potential, benefits, and challenges of implementing high-throughput sequencing in food safety systems. NPJ Sci. Food. 2022, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, K.; Iwen, P.C.; Abdalhamid, B. Applications of Clear Dx whole genome sequencing system in SARS-CoV-2 diagnostics. J. Infect. Public Health. 2022, 15, 894–895. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Khubbar, M.; Akinyemi, K.; Bauer, A.; Carranza, F.; Weiner, J.; Bhattacharyya, S.; Payne, D.; Balakrishnan, N. Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA. Viruses. 2023, 15, 1940. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.; Davies, J.A. Automation in the Life Science Research Laboratory. Front. Bioeng. Biotechnol. 2020, 8, 571777. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2023. Available online: https://www.who.int/publications/i/item/9789240083851 (accessed on 30 May 2024).
- Cloutier Charette, W.; Rabodoarivelo, M.S.; Point, F.; Knoblauch, A.M.; Andrianomanana, F.R.; Hall, M.B.; Iqbal, Z.; Supply, P.; Martin, A.; Rakotosamimanana, N.; Grandjean Lapierre, S. Concordance of targeted and whole genome sequencing for Mycobacterium tuberculosis genotypic drug susceptibility testing. Diagn. Microbiol. Infect. Dis. 2024, 109, 116249. [Google Scholar] [CrossRef] [PubMed]
- Genestet, C.; Hodille, E.; Berland, J.L.; Ginevra, C.; Bryant, J.E.; Ader, F.; Lina, G.; Dumitrescu, O. ; Lyon TB Study Group. Whole-genome sequencing in drug susceptibility testing of Mycobacterium tuberculosis in routine practice in Lyon, France. Int. J. Antimicrob. Agents. 2020, 55, 105912. [Google Scholar] [CrossRef]
- Zignol, M.; Cabibbe, A.M.; Dean, A.S.; Glaziou, P.; Alikhanova, N.; Ama, C.; Andres, S.; Barbova, A.; Borbe-Reyes, A.; Chin, D.P.; Cirillo, D.M.; Colvin, C.; Dadu, A.; Dreyer, A.; Driesen, M.; Gilpin, C.; Hasan, R.; Hasan, Z.; Hoffner, S.; Hussain, A.; Ismail, N.; Kamal, S.M.M.; Khanzada, F.M.; Kimerling, M.; Kohl, T.A.; Mansjö, M.; Miotto, P.; Mukadi, Y.D.; Mvusi, L.; Niemann, S.; Omar, S.V.; Rigouts, L.; Schito, M.; Sela, I.; Seyfaddinova, M.; Skenders, G.; Skrahina, A.; Tahseen, S.; Wells, W.A.; Zhurilo, A.; Weyer, K.; Floyd, K.; Raviglione, M.C. Genetic sequencing for surveillance of drug resistance in tuberculosis in highly endemic countries: a multi-country population-based surveillance study. Lancet Infect. Dis. 2018, 18, 675–683. [Google Scholar] [CrossRef]
- Arnold, A.; Witney, A.A.; Vergnano, S.; Roche, A.; Cosgrove, C.A.; Houston, A.; Gould, K.A.; Hinds, J.; Riley, P.; Macallan, D.; Butcher, P.D.; Harrison, T.S. XDR-TB transmission in London: Case management and contact tracing investigation assisted by early whole genome sequencing. J. Infect. 2016, 73, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; Ofori-Anyinam, B.; Dreyer, V.; Supply, P.; Suresh, A.; Utpatel, C.; van Soolingen, D.; Zhou, Y.; Ashton, P.M.; Brites, D.; Cabibbe, A.M.; de Jong, B.C.; de Vos, M.; Menardo, F.; Gagneux, S.; Gao, Q.; Heupink, T.H.; Liu, Q.; Loiseau, C.; Rigouts, L.; Rodwell, T.C.; Tagliani, E.; Walker, T.M.; Warren, R.M.; Zhao, Y.; Zignol, M.; Schito, M.; Gardy, J.; Cirillo, D.M.; Niemann, S.; Comas, I.; Van Rie, A. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, M.I.; Walker, T.M.; van der Werf, T.S.; Lange, C.; Niemann, S.; Merker, M. Pathogen-based precision medicine for drug-resistant tuberculosis. PLoS Pathog. 2018, 14, e1007297. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Nolan, D.J.; Ashcraft, D.; Feehan, A.K.; Velez-Climent, L.; Huston, C.; Lain, B.; Rosenthal, S.; Miele, L.; Fogel, G.B.; Pankey, G.; Garcia-Diaz, J.; Lamers, S.L. Comparing antimicrobial resistant genes and phenotypes across multiple sequencing platforms and assays for Enterobacterales clinical isolates. BMC Microbiol. 2023, 23, 225. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Liu, C.; Xu, Y.; Wang, J.; Jin, D. Concordance between Genotypic and Phenotypic Drug-Resistant Profiles of Shigella Isolates from Taiyuan City, Shanxi Province, China, 2005 to 2016. Microbiol. Spectr. 2023, 11, e0011923. [Google Scholar] [CrossRef]
- Schwan, C.L.; Lomonaco, S.; Bastos, L.M.; Cook, P.W.; Maher, J.; Trinetta, V.; Bhullar, M.; Phebus, R.K.; Gragg, S.; Kastner, J.; Vipham, J.L. Genotypic and Phenotypic Characterization of Antimicrobial Resistance Profiles in Non-typhoidal Salmonella enterica Strains Isolated From Cambodian Informal Markets. Front. Microbiol. 2021, 12, 711472. [Google Scholar] [CrossRef]
- Moura, A.; Leclercq, A.; Vales, G.; Tessaud-Rita, N.; Bracq-Dieye, H.; Thouvenot, P.; Madec, Y.; Charlier, C.; Lecuit, M. Phenotypic and genotypic antimicrobial resistance of Listeria monocytogenes: an observational study in France. Lancet Reg. Health Eur. 2023, 37, 100800. [Google Scholar] [CrossRef]
- Vīksna, A.; Sadovska, D.; Berge, I.; Bogdanova, I.; Vaivode, A.; Freimane, L.; Norvaiša, I.; Ozere, I.; Ranka, R. Genotypic and phenotypic comparison of drug resistance profiles of clinical multidrug-resistant Mycobacterium tuberculosis isolates using whole genome sequencing in Latvia. BMC Infect. Dis. 2023, 23, 638. [Google Scholar] [CrossRef] [PubMed]
- Bhuwan, M.; Arora, N.; Sharma, A.; Khubaib, M.; Pandey, S.; Chaudhuri, T.K.; Hasnain, S.E.; Ehtesham, N.Z. Interaction of Mycobacterium tuberculosis Virulence Factor RipA with Chaperone MoxR1 Is Required for Transport through the TAT Secretion System. mBio. 2016, 7, e02259. [Google Scholar] [CrossRef]
- Gordon, L.G.; Elliott, T.M.; Forde, B.; Mitchell, B.; Russo, P.L.; Paterson, D.L.; Harris, P.N.A. Budget impact analysis of routinely using whole-genomic sequencing of six multidrug-resistant bacterial pathogens in Queensland, Australia. BMJ Open. 2021, 11, e041968. [Google Scholar] [CrossRef]
- Sundermann, A.J.; Chen, J.; Kumar, P.; Ayres, A.M.; Cho, S.T.; Ezeonwuka, C.; Griffith, M.P.; Miller, J.K.; Mustapha, M.M.; Pasculle, A.W.; Saul, M.I.; Shutt, K.A.; Srinivasa, V.; Waggle, K.; Snyder, D.J.; Cooper, V.S.; Van Tyne, D.; Snyder, G.M.; Marsh, J.W.; Dubrawski, A.; Roberts, M.S.; Harrison, L.H. Whole-Genome Sequencing Surveillance and Machine Learning of the Electronic Health Record for Enhanced Healthcare Outbreak Detection. Clin. Infect. Dis. 2022, 75, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Libuit, K.G.; Doughty, E.L.; Otieno, J.R.; Ambrosio, F.; Kapsak, C.J.; Smith, E.A.; Wright, S.M.; Scribner, M.R.; Petit Iii, R.A.; Mendes, C.I.; Huergo, M.; Legacki, G.; Loreth, C.; Park, D.J.; Sevinsky, J.R. Accelerating bioinformatics implementation in public health. Microb. Genom. 2023, 9, mgen001051. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Griswold, T.; Williams-Newkirk, A.J.; Wagner, D.; Petkau, A.; Sieffert, C.; Van Domselaar, G.; Deng, X.; Carleton, H.A. A Comparative Analysis of the Lyve-SET Phylogenomics Pipeline for Genomic Epidemiology of Foodborne Pathogens. Front. Microbiol. 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the fully automated bacterial WGS workflow using extracted DNA as sample input.
Figure 1.
Schematic representation of the fully automated bacterial WGS workflow using extracted DNA as sample input.

Figure 2.
Phylogeny of Clinical Isolates from (a) Nevada State Public Health Laboratory and (b) San Francisco Public Health Laboratory with Corresponding AMR Genotypic Profile.
Figure 2.
Phylogeny of Clinical Isolates from (a) Nevada State Public Health Laboratory and (b) San Francisco Public Health Laboratory with Corresponding AMR Genotypic Profile.
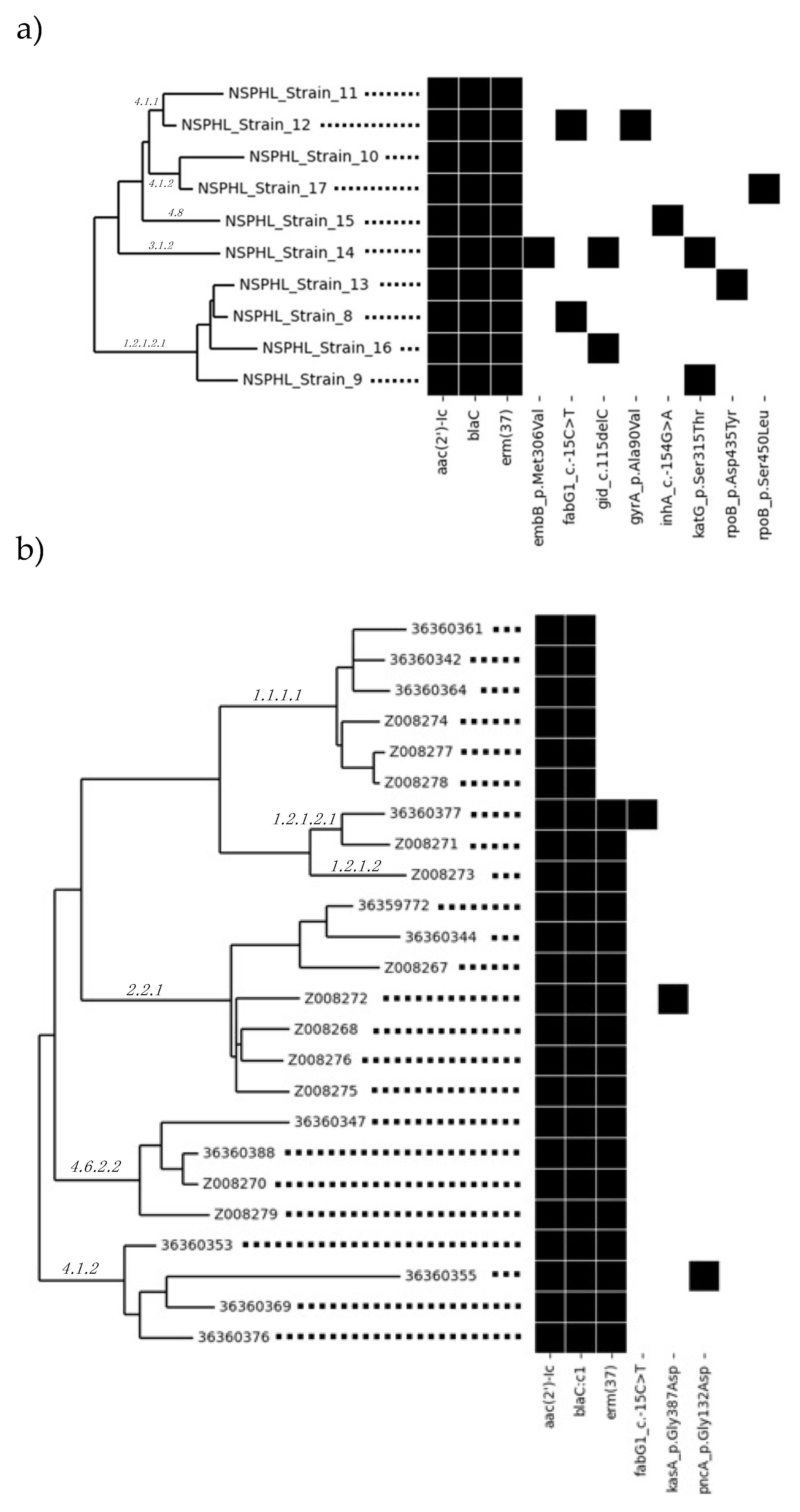

Table 1.
Automated Bacterial WGS Assay Results of MTBs and NTMs.
| Sample ID | Predicted Taxon | Sub-Lineage | Assembly Length (bp) | Number of Contigs | EstimatedDepth of Coverage |
|---|---|---|---|---|---|
| NSPHL_Strain_8 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,353,471 | 127 | 119.63 |
| NSPHL_Strain_9 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,372,144 | 122 | 60.37 |
| NSPHL_Strain_10 | Mycobacterium tuberculosis | 4.1.2.1 | 4,316,704 | 142 | 33.78 |
| NSPHL_Strain_11 | Mycobacterium tuberculosis | 4.1.1.1 | 4,344,545 | 118 | 100.73 |
| NSPHL_Strain_12 | Mycobacterium tuberculosis | 4.1.1.3 | 4,335,708 | 124 | 58.77 |
| NSPHL_Strain_13 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,356,191 | 139 | 62.57 |
| NSPHL_Strain_14 | Mycobacterium tuberculosis | 3.1.2 | 4,343,589 | 125 | 114.74 |
| NSPHL_Strain_15 | Mycobacterium tuberculosis | 4.8 | 4,335,730 | 115 | 52.41 |
| NSPHL_Strain_16 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,326,334 | 239 | 20.66 |
| NSPHL_Strain_17 | Mycobacterium tuberculosis | 4.1.2.1 | 4,326,395 | 159 | 34.55 |
| 36359772 | Mycobacterium tuberculosis | 2.2.1 | 4,288,293 | 189 | 45.65 |
| 36360342 | Mycobacterium tuberculosis | 1.1.1.1 | 4,312,674 | 222 | 33.05 |
| 36360344 | Mycobacterium tuberculosis | 2.2.1 | 4,257,211 | 278 | 33.06 |
| 36360347 | Mycobacterium tuberculosis | 4.6.2.2 | 4,258,952 | 267 | 32.12 |
| 36360353 | Mycobacterium tuberculosis | 4.1.2 | 4,322,326 | 233 | 34.83 |
| 36360355 | Mycobacterium tuberculosis | 4.1.2 | 4,203,142 | 593 | 21.84 |
| 36360361 | Mycobacterium tuberculosis | 1.1.1.1 | 4,306,182 | 247 | 34.21 |
| 36360364 | Mycobacterium tuberculosis | 1.1.1.1 | 4,314,878 | 195 | 45.81 |
| 36360369 | Mycobacterium tuberculosis | 4.1.2 | 4,292,556 | 272 | 36.49 |
| 36360376 | Mycobacterium tuberculosis | 4.1.2 | 4,300,124 | 293 | 31.64 |
| 36360377 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,360,550 | 150 | 100.91 |
| 36360388 | Mycobacterium tuberculosis | 4.6.2.2 | 4,314,959 | 121 | 98.35 |
| Z008267 | Mycobacterium tuberculosis | 2.2.1 | 4,280,868 | 298 | 25.63 |
| Z008268 | Mycobacterium tuberculosis | 2.2.1 | 4,322,819 | 159 | 43.21 |
| Z008270 | Mycobacterium tuberculosis | 4.6.2.2 | 4,309,779 | 118 | 78.37 |
| Z008271 | Mycobacterium tuberculosis | 1.2.1.2.1 | 4,355,604 | 136 | 58.3 |
| Z008272 | Mycobacterium tuberculosis | 2.2.1 | 4,314,821 | 181 | 36.07 |
| Z008273 | Mycobacterium tuberculosis | 1.2.1.2 | 4,341,999 | 153 | 50.4 |
| Z008274 | Mycobacterium tuberculosis | 1.1.1.1 | 4,332,833 | 166 | 37.56 |
| Z008275 | Mycobacterium tuberculosis | 2.2.1 | 4,319,340 | 196 | 35.28 |
| Z008276 | Mycobacterium tuberculosis | 2.2.1.1 | 4,305,996 | 191 | 29.81 |
| Z008277 | Mycobacterium tuberculosis | 1.1.1.1 | 4,348,679 | 120 | 92.59 |
| Z008278 | Mycobacterium tuberculosis | 1.1.1.1 | 4,347,657 | 114 | 156.87 |
| Z008279 | Mycobacterium tuberculosis | 4.6.2.2 | 4,321,629 | 139 | 38.43 |
| ATCC_35734 | Mycobacterium tuberculosis | La1.2.BCG | 4,229,722 | 138 | 46.66 |
| ATCC_35822D-2 | Mycobacterium tuberculosis | 4.9 | 4,285,606 | 122 | 60.58 |
| NR-122 | Mycobacterium tuberculosis | 4.9 | 4,348,364 | 124 | 54.23 |
| NR-59207 | Mycobacterium tuberculosis | La1.8.1 | 4,242,260 | 183 | 24.48 |
| NR-44263 | Mycobacteroides abscessus | - | 5,133,647 | 21 | 47.31 |
| NR-44274 | Mycobacteroides abscessus | - | 5,188,746 | 30 | 37.27 |
| NR-49658 | Mycobacterium canettii | - | 4,341,653 | 223 | 28.38 |
| ATCC_6841 | Mycolicibacterium fortuitum | - | 6,279,033 | 47 | 43.01 |
| ATCC_14470 | Mycobacterium gordonae | - | 7,368,456 | 278 | 27.74 |
| NR-49070 | Mycobacterium palustre | - | 5,768,069 | 204 | 42.5 |
Table 2.
Antimicrobial Resistance (AMR) Genotypic and Drug Class Profiles of MTB Isolates.
| Sample ID | Sub-Lineage | Predicted AMR Drug Classes | Identified AMR Genes / Mutations | TB DR Type* |
|---|---|---|---|---|
| NSPHL_Strain_8 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethionamide | aac(2')-Ic, blaC, erm(37), fabG1_c.-15C>T | HR-TB |
| NSPHL_Strain_9 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid | aac(2')-Ic, blaC, erm(37), katG_p.Ser315Thr | HR-TB |
| NSPHL_Strain_10 | 4.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| NSPHL_Strain_11 | 4.1.1.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| NSPHL_Strain_12 | 4.1.1.3 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Fluoroquinolones, Ethionamide | aac(2')-Ic, blaC, erm(37), fabG1_c.-15C>T, gyrA_p.Ala90Val | HR-TB |
| NSPHL_Strain_13 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Rifampicin | aac(2')-Ic, blaC, erm(37), rpoB_p.Asp435Tyr | RR-TB |
| NSPHL_Strain_14 | 3.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethambutol, Streptomycin | aac(2')-Ic, blaC, erm(37), embB_p.Met306Val, gid_c.115delC, katG_p.Ser315Thr | HR-TB |
| NSPHL_Strain_15 | 4.8 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethambutol | aac(2')-Ic, blaC, erm(37), inhA_c.-154G>A | HR-TB |
| NSPHL_Strain_16 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Streptomycin | aac(2')-Ic, blaC, erm(37), gid_c.115delC | Other |
| NSPHL_Strain_17 | 4.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Rifampicin | aac(2')-Ic, blaC, erm(37), rpoB_p.Ser450Leu | RR-TB |
| 36359772 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360342 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| 36360344 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360347 | 4.6.2.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360353 | 4.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360355 | 4.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Pyrazinamide | aac(2')-Ic, blaC, erm(37), pncA_p.Gly132Asp | Other |
| 36360361 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| 36360364 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| 36360369 | 4.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360376 | 4.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| 36360377 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid | aac(2')-Ic, blaC, erm(37), fabG1_c.-15C>T | HR-TB |
| 36360388 | 4.6.2.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008267 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008268 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008270 | 4.6.2.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008271 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008272 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Pyrazinamide | aac(2')-Ic, blaC, erm(37), kasA_p.Gly387Asp | HR-TB |
| Z008273 | 1.2.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008274 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| Z008275 | 2.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008276 | 2.2.1.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| Z008277 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| Z008278 | 1.1.1.1 | Aminoglycoside, Beta-Lactam | aac(2')-Ic, blaC | Sensitive |
| Z008279 | 4.6.2.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| ATCC_35734 | La1.2.BCG | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Pyrazinamide | aac(2')-Ic, blaC, erm(37), pncA_p.His57Asp | Other |
| ATCC_35822D-2 | 4.9 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid | aac(2')-Ic, blaC, erm(37), ahpC_c.-39C>T, ahpC_c.-54C>T, katG_c.-11139_*36437del | HR-TB |
| NR-122 | 4.9 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | aac(2')-Ic, blaC, erm(37) | Sensitive |
| NR-59207 | La1.8.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Pyrazinamide | aac(2')-Ic, blaC, erm(37), pncA_p.His57Asp | Other |
* DR: Drug Resistance; HR-TB: Isoniazid-Resistant TB; RR-TB: Rifampicin-Resistant TB.
Table 3.
Reproducibility Study Validated the Robustness of the Fully Automated Bacteria WGS Assay.
| Sample ID | Sample Size | Assembly Length (bp)Mean ± S.D. | Number of ContigsMean ± S.D. | Mean Q ScoresMean ± S.D. | Mean Read Length (bp)Mean ± S.D. |
|---|---|---|---|---|---|
| NSPHL_Strain_8 (MTB) | 4 | 4,335,605 ± 2,228 | 172.67 ± 8.74 | 36.12 ± 0.06 | 147.43 ± 0.78 |
| NSPHL_Strain_12 (MTB) | 3 | 4,305,569 ± 5,272 | 201.00 ± 8.28 | 36.19 ± 0.01 | 147.46 ± 0.34 |
| NSPHL_Strain_17 (MTB) | 3 | 4,324,000 ± 5,569 | 166.67 ± 2.42 | 36.08 ± 0.04 | 147.49 ± 0.81 |
| ATCC_35734 (MTB) | 3 | 4,224,952 ± 4,199 | 141.67 ± 5.51 | 35.56 ± 0.05 | 145.82 ± 0.95 |
| ATCC_35822D-2 (MTB) | 5 | 4,282,387 ± 3,431 | 129.00 ± 4.95 | 35.49 ± 0.05 | 144.77 ± 1.93 |
| NR-122 (MTB) | 16 | 4,336,706 ± 7,704 | 136.47 ± 10.11 | 35.49 ± 0.34 | 147.21 ± 1.33 |
| NR-59207 (MTB) | 3 | 4,238,013 ± 6,183 | 192.67 ± 8.50 | 34.71 ± 0.00 | 145.99 ± 0.57 |
| ATCC_6841 (NTM) | 3 | 6,274,061 ± 4,522 | 56.67 ± 9.07 | 35.73 ± 0.09 | 146.60 ± 1.18 |
| NR-44263 (NTM) | 4 | 5,130,504 ± 3,015 | 29.25 ± 8.88 | 35.73 ± 0.08 | 144.11 ± 1.68 |
| NR-44274 (NTM) | 4 | 5,189,594 ± 3.121 | 31.25 ± 7.09 | 35.84 ± 0.09 | 147.98 ± 0.11 |
| NR-49070 (NTM) | 2 | 5,768,681 ± 865 | 205.00 ± 1.41 | 36.03 ± 0.61 | 144.26 ± 3.20 |
Table 4.
Concordance Between Genotypic and Phenotypic AMR Profiles of MTB Isolates from NSPHL.
| Sample ID | Sub-Lineage | Predicted AMR Drug Classes | Antimicrobial Susceptibility Test (AST) | ||||
|---|---|---|---|---|---|---|---|
| Ethambutol 5.0 ug/mL | Isoniazid 0.1 ug/mL | Isoniazid 0.4 ug/mL | Rifampin 1.0 ug/mL | Pyrazinamide 100 ug/mL | |||
| NSPHL_Strain_8 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethionamide | Sensitive | Resistant | Resistant | Sensitive | Sensitive |
| NSPHL_Strain_9 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid | Sensitive | Resistant | Resistant | Sensitive | Sensitive |
| NSPHL_Strain_10 | 4.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | Not Available | ||||
| NSPHL_Strain_11 | 4.1.1.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide | Sensitive | Sensitive | Sensitive | Sensitive | Sensitive |
| NSPHL_Strain_12 | 4.1.1.3 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Fluoroquinolones, Ethionamide | Sensitive | Resistant | Sensitive | Sensitive | Sensitive |
| NSPHL_Strain_13 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Rifampicin | Sensitive | Sensitive | Sensitive | Resistant | Sensitive |
| NSPHL_Strain_14 | 3.1.2 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethambutol, Streptomycin | Sensitive | Resistant | Resistant | Sensitive | Sensitive |
| NSPHL_Strain_15 | 4.8 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Isoniazid, Ethambutol | Sensitive | Resistant | Sensitive | Sensitive | Sensitive |
| NSPHL_Strain_16 | 1.2.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Streptomycin | Not Available | ||||
| NSPHL_Strain_17 | 4.1.2.1 | Aminoglycoside, Beta-Lactam, Lincosamide/Macrolide, Rifampicin | Not Available | ||||
Table 5.
Bacterial Strains Used in this Study.
| Organism | Sample ID | Source |
|---|---|---|
| Mycobacterium tuberculosis strains | ||
| Mycobacterium tuberculosis3 | NSPHL Strain 8 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 9 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 10 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 11 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 12 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 13 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 14 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 15 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 16 | NSPHL |
| Mycobacterium tuberculosis3 | NSPHL Strain 17 | NSPHL |
| Mycobacterium tuberculosis4 | 36359772 | SFPHL |
| Mycobacterium tuberculosis4 | 36360342 | SFPHL |
| Mycobacterium tuberculosis4 | 36360344 | SFPHL |
| Mycobacterium tuberculosis4 | 36360347 | SFPHL |
| Mycobacterium tuberculosis4 | 36360353 | SFPHL |
| Mycobacterium tuberculosis4 | 36360355 | SFPHL |
| Mycobacterium tuberculosis4 | 36360361 | SFPHL |
| Mycobacterium tuberculosis4 | 36360364 | SFPHL |
| Mycobacterium tuberculosis4 | 36360369 | SFPHL |
| Mycobacterium tuberculosis4 | 36360376 | SFPHL |
| Mycobacterium tuberculosis4 | 36360377 | SFPHL |
| Mycobacterium tuberculosis4 | 36360388 | SFPHL |
| Mycobacterium tuberculosis5 | Z008267 | SFPHL |
| Mycobacterium tuberculosis5 | Z008268 | SFPHL |
| Mycobacterium tuberculosis5 | Z008270 | SFPHL |
| Mycobacterium tuberculosis5 | Z008271 | SFPHL |
| Mycobacterium tuberculosis5 | Z008272 | SFPHL |
| Mycobacterium tuberculosis5 | Z008273 | SFPHL |
| Mycobacterium tuberculosis5 | Z008274 | SFPHL |
| Mycobacterium tuberculosis5 | Z008275 | SFPHL |
| Mycobacterium tuberculosis5 | Z008276 | SFPHL |
| Mycobacterium tuberculosis5 | Z008277 | SFPHL |
| Mycobacterium tuberculosis5 | Z008278 | SFPHL |
| Mycobacterium tuberculosis5 | Z008279 | SFPHL |
| Mycobacterium tuberculosis, H37Ra1 | NR-122 | BEI Resources |
| Mycobacterium tuberculosis, Strain TMC 3032 | ATCC_35822D-2 | ATCC |
| Mycobacterium tuberculosis variant bovis BCG1 | ATCC_35734 | ATCC |
| Mycobacterium bovis, Strain 95-13152 | NR-59207 | BEI Resources |
| Non-tuberculosis Mycobacterium strains | ||
| Mycobacteroides abscessus, 45301 | NR-44274 | BEI Resources |
| Mycobacteroides abscessus bolletii, MA 19481 | NR-44263 | BEI Resources |
| Mycobacterium canettii, Strain NLA0000171202 | NR-49658 | BEI Resources |
| Mycobacterium gordonae1 | ATCC_14470 | ATCC |
| Mycobacterium palustre, FI-050881 | NR-49070 | BEI Resources |
| Mycolicibacterium fortuitum, Strain TMC 15291 | ATCC_6841 | ATCC |
1 DNA was extracted by Clear Labs, Inc. using proprietary inhouse-developed extraction protocol. 2 Genomic DNA obtained from the culture collections (ATCC or BEI Resources). 3 DNA was extracted by Nevada State Public Health Laboratory (NSPHL) using Promega Maxwell RSC Cultured Cell Kit. 4DNA was extracted by San Francisco Public Health Laboratory (SFPHL) using Roche MagNA Pure 24 Total NA Isolation Kit 5 DNA was extracted by San Francisco Public Health Laboratory (SFPHL) using QIAGEN EZ1&2 Virus Mini Kit.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



