
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
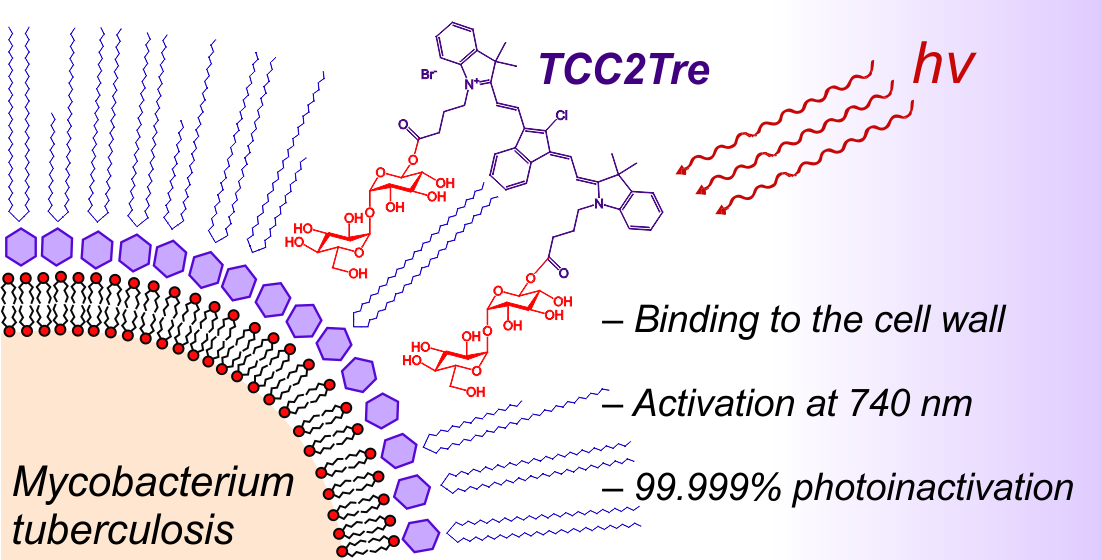
The spread of multidrug-resistant mycobacterium strains requires the development of new approaches to combat diseases caused by these pathogens. For that, photodynamic inactivation (PDI) is a promising approach. In this study, a tricarbocyanine (TCC) is used for the first time as a near-infrared (740 nm) activatable PDI photosensitizer to kill mycobacteria with deeper light penetration. For better targeting, a novel tricarbocyanine dye functionalized with two trehalose units (TCC2Tre) is developed. The photodynamic effect of the conjugate against mycobacteria, including Mycobacterium tuberculosis, is evaluated. Under irradiation, TCC2Tre causes more effective killing of mycobacteria compared to the photosensitizer without trehalose conjugation, with 99.99% dead vegetative cells of M. tuberculosis and M. smegmatis. In addition, effective photoinactivation of dormant forms of M. smegmatis is observed after incubation with TCC2Tre. Mycobacteria treated with TCC2Tre are more sensitive to 740 nm light than the Gram-positive Micrococcus luteus and the Gram-negative Escherichia coli. For the first time, this study demonstrates the possibility of in vitro PDI of mycobacteria including the fast-growing M. smegmatis and the slow-growing M. tuberculosis using near-infrared activatable photosensitizers. These findings are useful for the development of new efficient alternatives to antibiotic therapy.
Keywords:
Mycobacterium tuberculosis
; mycobacteria
; antibacterial photodynamic inactivation
; trehalose
; dormant mycobacteria
; tricarbocyanine dye
1. Introduction
Most pathogenic mycobacteria cause lung infections, although in some cases lymph nodes, bones and joints, skin and wounds are affected [1]. However, the most concerning disease caused by mycobacteria is tuberculosis (TB), caused by Mycobacterium tuberculosis. The situation with this disease is complicated by the high and increasing levels of antibiotic-resistant strains of M. tuberculosis and multidrug-resistant tuberculosis resistant to first-line antibiotics (isoniazid and rifampicin). Chemotherapy treatment for such infections has extremely limited effectiveness [2].
A special place in this problem is occupied by the ability of pathogenic mycobacteria to form dormant forms to survive under stress conditions, including in the host body. In this state, bacteria stop all biosynthetic processes, including those that are targeted by antibiotics, and thus become invulnerable to the action of antimicrobial drugs. M. tuberculosis in a dormant state is the cause of latent tuberculosis – a widespread, asymptomatic, and difficult-to-treat form [3]. Such forms of mycobacteria are able to remain viable for a long time in the human body and enter an active state, causing the resumption of the disease, for example, in the treatment of coronavirus inflammation using corticosteroids [4].
The traditional direction of combating drug resistance is associated with the search for new antibiotic substances. Modern methods of identifying new targets in pathogen cells, deciphering their structures and selecting active substances using screening systems have significantly increased the efficiency of such studies. However, within a very short period of time from the start of using a new antibiotic in the clinic, resistant strains arise. Such cycles of developing new drugs and the subsequent emergence of resistant strains can be repeated indefinitely.
The combination of these challenges necessitates the development of fundamentally novel approaches to combat genetically determined resistant strains and dormant forms of mycobacteria.
Antimicrobial photodynamic therapy (aPDT), or photodynamic inactivation (PDI), represents an interesting alternative or adjunct to chemotherapy for microbiological control [5]. This method reduces systemic toxicity, is non-invasive, minimizes antibiotic use, and mitigates the risk of resistance due to its broad action on biomolecules. APDT is a promising method for addressing various bacterial infections, particularly those that are antibiotic-resistant [6]. Currently, methylene blue, radachlorin and various porphyrin derivatives are used as exogenously photosensitizers (PSs) for the inactivation of mycobacteria [5]. Mycobacterial cells with endogenously accumulated porphyrines also exhibited a great sensitivity to illumination [7,8,9]. Such PSs are activated by light with a wavelength not exceeding 690 nm, which prevents their widespread use for inactivating the pathogen in the deep tissues of the macroorganism. While the application of PDI is not as widespread for mycobacteria as for other bacterial species, studies have shown that it can effectively deactivate various mycobacterial strains, especially those that cause skin infections, in both laboratory settings and animal models [5]. Despite the fact that the inclusion of exogenous PS leads to a high inactivation effect in vitro, the use of chemically synthesized PSs is complicated by their transportation to localized foci of bacterial infections. This obviously limits the use of PDI to superficial (skin) diseases.
Consequently, a promising avenue of research is the application of new PSs activated in the near-infrared range to kill mycobacteria, which should increase the depth of the effective action of light. The rationale for this assumption is the use of tricarbocyanine (TCC) dyes activated in the near-infrared range (780 nm), which was demonstrated to increase the depth of tumor damage [10]. However, until now, such PSs have not been used for the PDI of mycobacteria.
The second challenge when using exogenous PSs to inactivate mycobacteria located inside the host body is the potential for side effects on host tissues. When conducting aPDT using hydroxyaluminum phthalocyanine (Photosens) as a PS in an experimental model of tuberculosis infection in mice, it was demonstrated that the tissues of the experimental animal retain a significant amount of light energy. Nevertheless, the residual light transmission was sufficient to provide a pronounced therapeutic effect, manifested in a significant reduction of the mycobacterial load and specific inflammatory process in all examined internal organs [11]. However, side effects on the host organism were observed when using light with a high dose density (above 100 J/cm2). With effective inactivation of M. tuberculosis, this illumination regimen is unacceptable for clinical purposes, as it causes severe photonecrosis of photosensitized tissues [12].
To enhance the specificity of PSs targeting bacteria, the conjugation of fluorescent molecules with antibiotics, peptides, and antibodies was employed [13]. In particular, trehalose-related PSs for the PDI of mycobacteria were synthesized and studied [14,15].
Trehalose is an essential component for mycobacteria [16] and serves as a precursor for various glycolipids in their cell envelopes [17]. Mycobacteria are capable of producing trehalose internally, but can also use trehalose from external sources. Indeed, mycobacteria have an effective trehalose transporter, which is localized in the cell wall. This transporter can transport trehalose and trehalose analogues, including conjugates with PSs, from the external environment into the bacterium [18,19]. Because trehalose is not produced by mammals, it establishes the basis for the selectivity of the uptake of trehalose-linked PSs by bacterial cells within a macroorganism [19]. It was shown that 14C-trehalose can effectively enter and be absorbed by mouse macrophages, and is subsequently taken up by M. tuberculosis bacteria within those cells [20]. This result illustrates the potential of trehalose-functionalized probes for in vivo applications.
In this study, we report a synthetic strategy to prepare a conjugate of a tricarbocyanine (heptamethine cyanine) dye with trehalose and evaluate its photodynamic effect against mycobacteria (slow-growing M. tuberculosis and fast-growing M. smegmatis) in different physiological states.
2. Results
The conjugate with trehalose was obtained using a previously synthesized tricarbocyanine dye (TCC) in the form of a bromide salt bearing 4-meso-chloride and an indene moiety in the polymethine chain (Figure 1) [21]. To prepare the conjugate of TCC with trehalose (TCC2Tre), both carboxyl groups of TCC were esterified with trehalose (Figure 1) using 1-methyl-2-bromo-1-methylpyridiniummethanesulfonate as an analogue of the Mukaiyama reagent [22]. The synthesized TCC2Tre was a mixture of six isomers due to the ability of trehalose to form esters at different carbon atoms. The predominant isomer (27% in the mixture) was the diester bonded at the sixth carbon atom in both trehalose units (for more details and analytical data, see Section 4.2 and Figures S1-S3).
2.1. The Binding of TCC2Tre and TCC with Mycobacterium smegmatis Cells
The interaction of TCC2Tre and TCC with M. smegmatis was investigated by analyzing the spectral properties of the dyes accumulated in mycobacterial cells. M. smegmatis is a non-pathogenic relative of the causative agent of tuberculosis. The species was chosen for these experiments as it is a generally accepted model for the study of mycobacteria including M. tuberculosis [23].
First, TCC2Tre and TCC were compared with respect to their binding with M. smegmatis. The mycobacterial cells were incubated with the dyes in PBS for 1 h and then washed to remove unbound dye molecules. As illustrated in Figure 2a, the absorbance of TCC2Tre bound with mycobacteria exceeds that of TCC by an order of magnitude. As anticipated, the incorporation of trehalose into the molecular structure of TCC markedly enhances its binding with mycobacterial cells. The absorbance of TCC2Tre bound with M. smegmatis is almost directly proportional to the concentration of the dye between 10 and 100 µM in the incubation medium (Figure 2b).
Tween-80 is frequently added to growth media for mycobacteria to prevent their aggregation [24]. The impact of this detergent on the binding of TCC2Tre with M. smegmatis was also examined. As shown in Figure 3a, the presence of 0.05 wt% Tween-80 during the incubation period results in a significant decrease in the absorbance of TCC2Tre accumulated in M. smegmatis, by approximately one order of magnitude. The detergent probably facilitates the solubilization of the dye in PBS, thus reducing the affinity of the dye for the cell wall.
In addition to PBS, M. smegmatis were also incubated with TCC2Tre in 7H9 mineral (broth base) medium and in rich nutrient broth (NB). After the incubation period, the mycobacteria were washed from unbound dye and resuspended in PBS. While the binding of TCC2Tre with M. smegmatis is similar in PBS and 7H9 medium, it is improved approximately 2-fold in the NB medium (Figure 3b).
2.2. The Comparison of the Photoinactivation of M. smegmatis Cells with TCC and TCC2Tre
2.2.1. Conditions for Achieving the Maximum Photodynamic Effect of Dyes against Vegetative M. smegmatis Cells
The effectiveness of TCC and TCC2Tre (Figure 1) for the photoinactivation of M. smegmatis cells using 740 nm light was compared. It was found that the use of the hydrophobic TCC results in almost zero effectiveness of M. smegmatis photoinactivation (Table 1). However, the incubation of the cells with TCC in the presence of BSA improved photodynamic activity. Presumably, the adsorption on BSA prevented the aggregation of the hydrophobic TCC thus allowing the dye to maintain its properties. A similar effect was produced by the addition of Tween-80 (0.05%) during the incubation of the cells with TCC (Table 1).
Therefore, the photodynamic effects of the two dyes were compared under identical conditions, where BSA was added to NB medium for binding. The results of these experiments are presented in Figure 4.
TCC demonstrated a rather weak photodynamic effect against M. smegmatis. At a starting concentration of 40 μM TCC and a light dose of 468 J/cm2, the number of dead M. smegmatis cells was 97%. At the same concentration and a light dose, the photoactivity of TCC2Tre was substantially higher, with the number of dead M. smegmatis cells reaching 99.999% (Figure 4a). These data on the photoinactivation of M. smegmatis are consistent with the data on the binding of these PSs with mycobacteria (Figure 2). At a dye concentration of 40 μM, the dark toxicity of these compounds against M. smegmatis was completely absent (Table 1). Furthermore, the illumination of vegetative cells of M. smegmatis at a maximum irradiance of 240 mW/cm2 for 30 min without prior incubation with the dyes did not result in a decrease in bacterial viability (Figure 4a).
At an irradiance of 240 mW/cm2, the maximum inactivation of M. smegmatis cells with TCC2Tre was achieved after 15 min of illumination. Further increase in the exposure time did not significantly improve the photoinactivation effect (Figure 4b). With TCC and at the same irradiance, noticeable mycobacterial death was observed after 30 min of illumination (Figure 4b).
In contrast to TCC, the presence of BSA or Tween-80 significantly reduced the photoactivity of TCC2Tre against M. smegmatis cells (Figure 5a). This is consistent with the negative influence of Tween-80 on the binding of TCC2Tre with the cells (Figure 3a). Thus, the maximum photodynamic effect was observed when TCC2Tre was used in the absence of both BSA and Tween-80 in the medium (Figure 5a). Nevertheless, even in the presence of BSA, the photoinactivation of M. smegmatis using TCC2Tre was more effective than using TCC (Figure 4).
To assess the influence of environmental components on the photoactivity of TCC2Tre for M. smegmatis cells, two media were tested: rich NB and mineral medium 7H9. The incubation of the bacteria with TCC2Tre in the NB medium resulted in an improved photodynamic effect compared to the mineral medium (Figure 5b).
Thus, the optimal conditions for achieving the maximum photodynamic effect of the water-soluble TCC2Tre against M. smegmatis cells are as follows: incubation in a rich medium, without Tween-80 or BSA. In contrast, for the hydrophobic TCC, the maximum photodynamic effect requires the presence of BSA or Tween-80 in the incubation medium.
The influence of the incubation time of M. smegmatis cells with the PSs on the photoinactivation was studied (Figure 6a). The photodynamic effect was found to be proportional to the incubation time with TCC2Tre in the NB medium. More specifically, a 3 h incubation period resulted in a decrease in the CFU number by almost 3 orders of magnitude. TCC exhibited low activity even when incubated with BSA under the same conditions and for the same duration.
2.2.2. The Photoactivity of TCC2Tre against Bacteria with Different Cell Wall Structures and against Dormant M. smegmatis
The effect of the new TCC2Tre against bacteria with different cell wall structures was assessed. Micrococcus luteus was used as a representative of Gram-positive bacteria, Escherichia coli was employed as a representative of Gram-negative bacteria, and M. smegmatis was selected as a representative of mycobacteria. It was found that TCC2Tre is more active against M. smegmatis, and TCC is more active against M. luteus (Figure 6a, b). Both dyes were almost ineffective against E. coli (Figure 6c).
The effectiveness of these PSs was assessed in a mixed 1:1 culture consisting of M. smegmatis and M. luteus. The distinct morphology and coloration of the colonies in conjunction with the low bacterial concentration of both species allowed us to evaluate the effect of the PSs on each bacterial specie simultaneously. TCC was found to be more effective against M. luteus, while TCC2Tre was more active against mycobacteria, even in the context of a mixed culture comprising the two microbial species (Figure 7).
The effectiveness of TCC2Tre against the dormant form of M. smegmatis was also assessed. The dormant form was obtained under conditions of gradual acidification of the external environment [25]. As in the case of vegetative cells of M. smegmatis, incubation of dormant cells (6 months of dormancy) of M. smegmatis with TCC2Tre or TCC allowed photodynamic inactivation of these forms of bacteria whilst in the lesser extent then for viable, vegetative cells (Figure 8).
2.2.3. Determination of the Optimal Light Dose for Effective Photoinactivation of M. smegmatis Incubated with TCC2Tre
The minimum light dose required for effective photoinactivation of M. smegmatis using TCC2Tre was determined. At the same irradiance (240 mW/cm2) and different exposure times, the optimal light dose for the inactivation of M. smegmatis was between 50 and 180 J/cm2 (Figure 9). A further increase in the light dose did not greatly affect the number of viable mycobacteria. At a light dose of 50 J/cm2, significant inactivation of viable bacteria was observed, while a light dose of approximately 12 J/cm2 was ineffective (Figure 9). For comparison, a light dose of 100 J/cm2 was previously demonstrated to be well tolerated by mouse tissues during photodynamic therapy with an aluminum phthalocyanine PS [12].
2.3. Efficiency of the TCC2Tre for the Photoinactivation of Mycobacterium tuberculosis
The sensitivity of the M. tuberculosis H37Rv strain to PDI in the presence of tricarbocyanine PSs was assessed. Similar to the experiments with M. smegmatis (Figure 4), TCC2Tre was more effective against M. tuberculosis compared to TCC (Figure 10a). In the absence of BSA, the activity of TCC2Tre increased (Figure 10b). The PDI of M. tuberculosis in the presence of TCC2Tre is more pronounced compared to the PDI of M. smegmatis under the same conditions (Figure 4 and Figure 10). With increasing illumination time at an irradiance of 240 mW/cm2, an increase in the number of dead cells in the M. tuberculosis culture was observed. At a light dose of 468 J/cm2, complete inactivation of M. tuberculosis cells was revealed when using TCC2Tre incubated without BSA (Figure 10b).
The effect of irradiance on the photoinactivation of M. tuberculosis cells was assessed at a constant light dose of 100 J/cm2. The most effective inactivation of M. tuberculosis was observed at irradiances above 100 mW/cm2 (Figure 11).
3. Discussion
In the present study, we sought to combine the spectral advantages of TCC for deeper light penetration of tissues upon aPDT with trehalose conjugation (Figure 1) for improved targeting of mycobacteria. To this end, we synthesized a novel trehalose conjugate of TCC (TCC2Tre), which is activated by light at a wavelength of 740 nm. As expected, TCC2Tre demonstrated superior binding with M. smegmatis cells in comparison with unconjugated TCC (Figure 2). Correspondingly, the PDI of M. smegmatis with TCC2Tre was more pronounced than that with TCC (Figure 4, 6a). The specificity of TCC2Tre for mycobacteria was evident from the comparison of its photoactivity against bacteria with different cell wall structures (Figure 6). This was confirmed in the experiment with a mixed culture, where TCC2Tre was substantially more effective against M. smegmatis than against M. luteus (Figure 7).
In two previous publications, the photoactivity of different PSs conjugated with trehalose towards mycobacteria was demonstrated. To improve the performance of photodynamic therapy for M. smegmatis and M. abscessus, PSs linked to trehalose (2AT2AT-PPIX; 6AT-I-BODIPY) were specifically designed. These compounds are able to penetrate the mycomembrane more effectively, resulting in a stronger photodynamic effect when exposed to light at a wavelength of 550-650 nm. These compounds exhibited minimal dark toxicity and were well tolerated by mammalian cells [14]. In another study, a trehalose-porphyrin conjugate with a two-carbon linker (C2) was shown to display increased phototoxicity against M. smegmatis following exposure to 415 nm light in comparison to the conjugate with a six-carbon linker (C6) [26].
Trehalose conjugates of some fluorescent compounds were also used for the detection of M. tuberculosis. In particular, the fluorogenic dye DMN-Tre, a conjugate of 4-N,N-Dimethylamino-1,8-naphthalimide and trehalose, selectively labels the mycobacterial cell wall, making it a valuable tool for studying M. tuberculosis physiology both in vitro and intracellularly [27]. A novel trehalose probe based on a 3-hydroxychromone (3HC-3) dye was developed. When attached to trehalose, this dye exhibits a 10-fold increase in fluorescence intensity compared to DMN-Tre [28]. A fluorescent probe cephalosphorinase-dependent green trehalose (CDG-Tre) was constructed to specifically label single live Bacille Calmette-Guérin (BCG) cells within macrophages. CDG-Tre demonstrated high selectivity for mycobacteria compared to other species in the Corynebacterineae suborder [29]. Selective targeting of mycobacteria was also achieved using trehalose derivatized carbazole (Tre-Cz). The presence of trehalose in the conjugate with carbazole allowed for the detection of mycobacteria in mixed cultures and in patient sputum [30].
In one of the aforementioned experiments, a trehalose conjugate with BODIPY was found to have a minimum inhibitory concentration of 0.50.8 µM for its photoactivity against M. smegmatis and M. abscessus according to the resazurin method for estimation of viability [14]. However, for the trehalose-porphyrin conjugate, only one concentration of the compound was used (10 mM), which resulted in a discernible inhibition of M. smegmatis growth according to the resazurin method [26]. In our experiments, we determined the effect of PDI by counting cell viability via CFU, which directly estimates the bactericidal effect in contrast to the resazurin method. We found that 20 μM TCC2Tre was sufficient for photoinactivating 99.9% of mycobacteria (Figure 4a). The discrepancy in the effective concentrations in the current work and previous publications may be attributed to different efficiency of PSs and the methodologies employed for phototoxicity and cell viability assessment.
TCC2Tre at a concentration of 40 µM produced significant killing effect on M. tuberculosis cells (Figure 10). To the best of our knowledge, this represents the first instance of the application of a trehalose-conjugated PS for the PDI of the pathogenic M. tuberculosis.
In the present study, the incubation periods of mycobacteria with the PSs were relatively short (2–3 h). It is evident that in mineral media such as PBS or Middlebrook base, in the absence of essential nutrients (e.g., glycerol, glucose, etc.), only a limited amount of TCC2Tre is capable of binding to the cell wall without being consumed by the cells during this period (Figure 3b). At the same time, a significant increase in the quantity of bound TCC2Tre in the rich NB medium in comparison with the mineral media may be attributed to the uptake of the PS by cells within the first hour of incubation. Similarly, an increase in cell sensitivity to light during incubation with TCC2Tre but not TCC in the NB medium (Figure 6a, Figure 7b) may also reflect active uptake of TCC2Tre by cells. Active accumulation of trehalose-bound substances by mycobacterial cells and their incorporation in mycolic acids via a specific trehalose transporter over a 24-h incubation period were experimentally proven [14,27,28,29,30].
The significantly lesser sensitivity of dormant M. smegmatis cells for the PSs and the lesser difference in sensitivity between TCC2Tre and TCC could be due to the sufficiently depressed metabolism in dormant cells [25] and suppression of transport processes. Nevertheless, the dormant cells still exhibited photosensitivity (Figure 8), which could be due to the hydrophobic interaction of the dye molecules with the cell wall structure.
In conclusion, our work demonstrates the possibility of using PSs targeted to the cell wall of mycobacteria and activated in the near-infrared range, which expands the horizons for the aPDT of intracellular pathogens such as Mycobacterium tuberculosis. The application and study of the new PSs for in vivo PDT using laboratory animals are currently underway in our laboratory.
4. Materials and Methods
4.1. Bacterial Strains, Growth Media, and Culture Conditions
Strains Mycobacterium smegmatis mc2 155, Escherichia coli K-12 and Micrococcus luteus NCIMB 13267 (previously described as Fleming strain 2665) were grown aerobically in 20 mL of the peptide-rich nutrient broth (NB) medium (Himedia, India) for 20 h at 37 °C under constant shaking at 110 rpm. M. smegmatis were grown with Tween-80 (0.05%) added to the medium.
Strain Mycobacterium tuberculosis H37Rv was grown in Middlebrook 7H9 liquid medium (Himedia, India) with supplements (ADC and 0.05% Tween-80) for 7 days at 37°C, under constant shaking at 180 rpm.
4.2. Synthesis of a Tricarbocyanine Dye Functionalized with Trehalose (TCC2Tre) and Its Characterization
TCC is a bromide salt of a hydrophobic 4-meso-chloro-substituted indotricarbocyanine dye with an indene moiety in the polymethine chain (Figure 1). TCC was synthesized as described in Ref. [21]. To obtain TCC2Tre, both carboxyl groups of TCC were esterified with trehalose (Figure 1) using 1-methyl-2-bromo-1-methylpyridiniummethanesulfonate as an analogue of the Mukaiyama reagent [22]. Anhydrous cesium fluoride (5 equiv.) and 1-methyl-2-bromo-1-methylpyridinium (2 equiv.) were added to a dimethylformamide solution of TCC. A dimethylformamide solution of anhydrous trehalose (4 equiv.) was then added to this mixture. After allowing the reaction mixture to stand at room temperature for 10 h, it was diluted with ethyl acetate, and the resulting precipitate was filtered through filter paper and washed with deionized water to remove cesium salts. After filtration, the precipitate was washed with acetone to remove TCC and pyridine compounds and then with ethanol.
The resulting compound was characterized by high-performance liquid chromatography (HPLC) coupled to electrospray ionization mass spectrometry (ESI-MS). HPLC was performed on an Agilent 1200 LC system equipped with a Zorbax Eclipse XDB-C-18 (4.6x150 mm, 5 µm). Mobile phase A was 0.05 vol% trifluoroacetic acid and mobile phase B was MeCN. The linear gradient was 40–95% phase B in 0–10 min. Chromatograms were recorded at 710 nm with a photodiode detector. After the chromatographic separation, absorption spectra were recorded using a diode-array detector. ESI-MS analysis was then performed on an Agilent 6410 Triple Quadrupole LC/MS system in positive ion detection mode with the fragmentor voltage set at 135 V.
The chromatogram for the synthesized compound shows six peaks with retention times of 4.939, 5.385, 5.565, 5.672, 5.832, and 5.985 min (Figure S1). The mass spectra for all these peaks contain species with m/z = 1309.5 corresponding to the esters of TCC with two trehalose units, i.e., TCC2Tre (Figure S2). The additional peaks in the mass spectra with lower m/z values can be assigned to the fragments of the molecular ion of TCC2Tre that appear during electrospray ionization. The six species with m/z = 1309.5 correspond to esters formed between the carboxyl groups of TCC and various hydroxyl groups of trehalose. The primary hydroxyl group located at the sixth carbon atom (see Figure 1) of trehalose is known to have the greatest relative reactivity [31]. Therefore, the most intense HPLC peak with a retention time of 5.565 min should correspond to a diester bonded at the sixth carbon atom in both trehalose units. The HPLC peaks with retention times of 5.385 and 5.832 min are more intense compared to the rest of the peaks and can therefore be assigned to TCC esters with one of the trehalose units bonded at the sixth carbon atom and the other trehalose unit bonded at a different carbon atom. The absorption spectra corresponding to the six HPLC peaks are almost identical in position and shape (Figure S3).
4.3. Binding of Tricarbocyanine Dyes with Mycobacterial Cells
An overnight culture of M. smegmatis was centrifuged (5 min at 8000 rpm), and the supernatant was discarded. The pellet was resuspended in PBS (50 mM, pH 7.45, Gibco), the NB medium, or Middlebrook 7H9 mineral medium. The absorbance of the resulting cell suspensions at 590 nm was 0.2, which corresponded to ~107 cells/mL. M. smegmatis were incubated at 37 ℃ with different concentrations (5, 10, 20, 40, and 100 µM) of TCC2Tre and TCC. The dyes were introduced into cell suspensions via a DMSO solution, with the final concentration of DMSO in cell suspensions being 5 vol%. Following a 1, 2, or 3-hour incubation period, the mycobacterial cells were washed with fresh PBS and centrifuged at 8000 rpm for 5 min three times. Following this, the cells were resuspended in PBS to a concentration of 107 cells/mL and transferred to 1 cm semi-micro polystyrene cuvettes (Fisher Scientific, catalog No. 11547692). Absorption spectra of the cell suspensions were acquired using a SOLAR PV1251 spectrophotometer.
4.4. Preparation of the Dormant Forms of M. smegmatis upon the Medium Self-Acidification
Mycobacterium smegmatis mc2155 cells were initially grown in the NB medium at 37°C for 24 h, with shaking at 200 rpm. The inoculum (1 mL) was added to 100 mL of Sauton medium, containing (g/L) K2HPO4, 0.5; MgSO4·7H2O, 1.4; L-Asparagine, 4.0; ferric ammonium citrate, 0.05; Na3(citrate), 2.0; ZnSO4, 0.001 (Sigma Aldrich), and 0.2 vol% glycerol (Panreac, Spain). The initial pH value was 6.0 (instead of pH 7.0 typical of the usual Sauton composition). To prevent aggregation of the cells, Tween 80-was added (0.05%). The culture was grown at 37°C under constant stirring (200 rpm) for 18 days until a constant pH value (ca. 6.0) of the medium was established [25]. Dormant cells were stored for 6 months without agitation in 15 -mL capped tubes in the dark.
4.6. Preparation of Bacteria for Photoinactivation
The bacterial cell cultures were diluted with sterile culture medium to the desired absorbance. The absorbance was measured in 1.5 mL plastic cuvettes with an optical path length of 10 mm using a photoelectric colorimeter (Biochrom WPA CO7500 Colourwave, England). The culture medium was used as a control. One mL of cell suspension (0.1 absorbance at 590 nm) and 10 µL of a 4 mM TCC (or TCCTre) solution were added to a sterile 2 mL tube. Bacteria were incubated with the dyes for different time intervals at 37°C under constant stirring. Following the incubation period, the cells were centrifuged for 10 min at 10,000 rpm. The precipitate was washed two times with PBS for 10 min at 10,000 rpm.
4.7. Antibacterial Photoinactivation
Suspensions of bacteria with 0.1 absorbance at 590 nm, which corresponds to ca. 107 bacteria per mL, were used for light inactivation experiments. Aliquots (100 µL) of the bacterial suspensions were placed in the wells of a 96-well plate (Nunc). Illumination was carried out using a 740 nm LED SOLIS-740C (Thorlabs, USA) with a bandwidth of 45 nm (full width at half maximum). The light beam was collimated to a diameter of 5 mm, corresponding to the diameter of the wells of a 96-well plate. Illumination coupling MT-CON with optimized optics for bright bottom homogenous illumination of a suspension in the wells was used. The irradiance in each mode of the LED was measured with a Newport 2936-c power meter (Table 2). In most cases, a light dose of 468 J/cm2 (240 mW/cm2 irradiance and 30 min illumination time) was used. For some experiments, a light dose of 100 J/cm2 was used, which was obtained with a different irradiance (Table 2). The temperature in was controlled with a Fluke 80BK Type K Multimeter Thermocouple Temperature Probe directly in the microwells before and after illumination and in the presence and without dyed M. smegmatis suspension with ± 0.2°C precision. The temperature was lower than 40°C in the wells during all the experiments.
After illuminating the samples, serial tenfold dilutions (10−1 to 10−7) were prepared in the NB medium with 0.05 % Tween-80, and aliquots (3 × 20 μL) were plated on NB agar plates for CFU (for vegetative bacteria) or MPN (for dormant bacteria) determination.
4.8. Viability Estimation by CFU
For M. smegmatis, E. coli, and M. luteus, bacterial suspensions were serially diluted in fresh NB medium, and then three replicate 10 μL samples from each dilution were spotted on NB (Himedia, India) agar. The plates were incubated at 37°C for 3-5 days, then the number of colony forming units (CFUs) was counted.
For M. tuberculosis, bacterial suspensions were serially diluted in fresh 7H9 medium (Himedia, India) containing 0.05% Tween-80, and then three replicate 10 μL samples from each dilution were spotted on 7H10 (Himedia, India) agar. Plates were incubated at 37 °C for 21 days, and then the number of colony forming units (CFUs) was counted. The limit of detection was 10 CFU/mL.
4.9. MPN Assay
The MPN procedure was performed for dormant M. smegmatis cells in 48-well plastic plates (Corning) filled with a 1:1 mixture of Sauton medium and the NB medium. Appropriate serial dilutions of M. smegmatis cells (100 μL) were added to each well. The plates were incubated at 37°C with agitation (130 rpm) for 14 days. Wells with the visible bacterial growth were counted as positive, and the MPN values were calculated using standard statistical methods [32]. These experiments were performed two times in three technical replicas.
4.10. Statistics
Statistical processing was carried out via the analysis of the standard deviation or relative error within each data group. All the data are presented as the mean values of three replicate determinations. The MPN values were determined using de Man’s tables calculated on the basis of a Poisson distribution [32]. For the MPN assay, (95%) confidence limits were calculated. The MPN values were considered statistically different if low and high confidence limits were not overlapping.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Chromatogram for the synthesized TCC2Tre. The six HPLC peaks that are referred to in text are highlighted in grey; Figure S2: Mass spectra for the six HPLC peaks shown in Figure S1.; Figure S3: Absorption spectra for the six HPLC peaks shown in Figure S1.
Author Contributions
Conceptualization, M.P.S., A.S.K., A.P.S. and M.O.S.; methodology, M.P.S., M.O.S. and A.P.S.; software, N.V.K.; validation, A.S.K., A.P.S., M.P.S. and M.O.S.; formal analysis, A.S.K and A.P.S.; investigation, N.V.K., M.P.S., A.P.L., N.V.B. and D.S.T.; resources, M.O.S. and M.P.S.; data curation, N.V.K., N.V.B. and D.S.T.; writing—original draft preparation, M.O.S. and N.V.B.; writing—review and editing, M.P.S., N.V.B., A.S.K., A.P.S and M.O.S.; visualization, N.V.B. and M.O.S.; supervision, M.O.S.; project administration, M.O.S.; funding acquisition, M.O.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation grant 24-15-00221.
Data Availability Statement
Data is contained within the article or Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gonzalez-Santiago, T.M.; Drage, L.A. Nontuberculous Mycobacteria Skin and Soft Tissue Infections. Dermatologic Clinics 2015, 33, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Klopper, M.; Warren, R.M.; Hayes, C.; van Pittius, N.C.G.; Streicher, E.M.; Müller, B.; Sirgel, F.A.; Chabula-Nxiweni, M.; Hoosain, E.; Coetzee, G.; et al. Emergence and Spread of Extensively and Totally Drug-Resistant Tuberculosis, South Africa. Emerging Infectious Diseases 2013, 19, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Shleeva, M.O.; Kaprelyants, A.S. Hypobiosis of Mycobacteria: Biochemical Aspects. Biochemistry (Moscow) 2023, 88, S52–S74. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.; Batabyal, S.; Mandal, K.; Ghose, D.; Sarkar, J. Tuberculosis and COVID-19: A Combined Global Threat to Human Civilization. Clinical Epidemiology and Global Health 2022, 15, 101031. [Google Scholar] [CrossRef] [PubMed]
- Shleeva, M.; Savitsky, A.; Kaprelyants, A. Photoinactivation of Mycobacteria to Combat Infection Diseases: Current State and Perspectives. Appl Microbiol Biotechnol. 2021, 105, 4099–4109. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, R.; Zaat, S.A.J.; Breukink, E.; Heger, M. Antibacterial Photodynamic Therapy: Overview of a Promising Approach to Fight Antibiotic-Resistant Bacterial Infections. Journal of clinical and translational research 2015, 1, 140–167. [Google Scholar] [PubMed]
- Shleeva, M.O.; Savitsky, A.P.; Nikitushkin, V.D.; Solovyev, I.D.; Kazachkina, N.I.; Perevarov, V.V.; Kaprelyants, A.S. Photoinactivation of Dormant Mycobacterium Smegmatis Due to Its Endogenous Porphyrins. Applied Microbiology and Biotechnology 2019, 103, 9687–9695. [Google Scholar] [CrossRef]
- Shleeva, M.O.; Savitsky, A.P.; Nikitushkin, V.D.; Soloviev, I.D.; Trutneva, K.A. Effect of Photodynamic Inactivation against Dormant Forms and Active Growing Cells of Mycobacterium Smegmatis. Applied Biochemistry and Microbiology 2020, 56, 242–249. [Google Scholar] [CrossRef]
- Shleeva, M.O.; Linge, I.A.; Gligonov, I.A.; Vostroknutova, G.N.; Shashin, D.M.; Tsedilin, A.M.; Apt, A.S.; Kaprelyants, A.S.; Savitsky, A.P. Acquiring of Photosensitivity by Mycobacterium Tuberculosis in Vitro and inside Infected Macrophages Is Associated with Accumulation of Endogenous Zn–Porphyrins. Scientific Reports 2024, 14, 846. [Google Scholar] [CrossRef] [PubMed]
- Lugovski, A.A.; Samtsov, M.P.; Kaplevsky, K.N.; Tarasau, D.; Voropay, E.S.; Petrov, P.T.; Istomin, Y.P. Novel Indotricarbocyanine Dyes Covalently Bonded to Polyethylene Glycol for Theranostics. Journal of Photochemistry and Photobiology A: Chemistry 2016, 316, 31–36. [Google Scholar] [CrossRef]
- Belogorodtsev, S.N.; Lykov, A.P.; Nikonov, S.D.; Bredikhin, D.A.; Schwartz, Y.A.S.H. Efficacy of Photodynamic Therapy in an Experimental Mice Tuberculosis Model. Tuberculosis and Lung Diseases 2023, 101, 51–56. [Google Scholar] [CrossRef]
- Nikonov, S.D.; Bredikhin, D.A.; Belogorodtsev, S.N.; Schwartz, Y.S. Photodynamic Inactivation of Mycobacterium Tuberculosis Using Alluminium Phthalocyanine. Bulletin of Experimental Biology and Medicine 2023, 175, 367–370. [Google Scholar] [CrossRef]
- Grimmeisen, M.; Jessen-Trefzer, C. Increasing the Selectivity of Light-Active Antimicrobial Agents – Or How To Get a Photosensitizer to the Desired Target. ChemBioChem 2023, 24, e202300177. [Google Scholar] [CrossRef]
- Dutta, A.K.; Choudhary, E.; Wang, X.; Záhorszka, M.; Forbak, M.; Lohner, P.; Jessen, H.J.; Agarwal, N.; Kordulákova, J.; Jessen-Trefzer, C. Trehalose Conjugation Enhances Toxicity of Photosensitizers against Mycobacteria. ACS Central Science 2019, 5, 644–650. [Google Scholar] [CrossRef]
- Liang, J.; Liu, F.; Xu, P.; Shangguan, W.; Hu, T.; Wang, S.; Yang, X.; Xiong, Z.; Yang, X.; Guddat, L.W.; et al. Molecular Recognition of Trehalose and Trehalose Analogues by Mycobacterium Tuberculosis LpqY-SugABC. Proceedings of the National Academy of Sciences 2017, 120, 2017. [Google Scholar] [CrossRef]
- Woodruff, P.J.; Carlson, B.L.; Siridechadilok, B.; Pratt, M.R.; Senaratne, R.H.; Mougous, J.D.; Riley, L.W.; Williams, S.J.; Bertozzi, C.R. Trehalose Is Required for Growth of Mycobacterium Smegmatis. Journal of Biological Chemistry 2004, 279, 28835–28843. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.; Alarico, S.; Maranha, A.; Mendes, V.; Empadinhas, N. The Molecular Biology of Mycobacterial Trehalose in the Quest for Advanced Tuberculosis Therapies. Microbiology (United Kingdom) 2014, 160, 1547–1570. [Google Scholar] [CrossRef]
- Liang, J.; Liu, F.; Xu, P.; Shangguan, W.; Hu, T.; Wang, S.; Yang, X.; Xiong, Z.; Yang, X.; Guddat, L.W.; et al. Molecular Recognition of Trehalose and Trehalose Analogues by Mycobacterium Tuberculosis LpqY-SugABC. Proceedings of the National Academy of Sciences 2023, 120, e2307625120. [Google Scholar] [CrossRef]
- Devulder, G.; de Montclos, M.P.; Flandrois, J.P. A Multigene Approach to Phylogenetic Analysis Using the Genus Mycobacterium as a Model. International Journal of Systematic and Evolutionary Microbiology 2005, 55, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Boshoff, H.L.; Barry, C.S.; Boutureira, O.; Patel, M.K.; D’Hooge, F.; Lee, S.S.; Via, L.E.; Tahlan, K.; Barry, C.E., III; et al. Uptake of Unnatural Trehalose Analogs as a Reporter for Mycobacterium Tuberculosis. Nat Chem Biol. 2011, 7, 228–235. [Google Scholar] [CrossRef]
- Lugovski, A.A.; Samtsov, M.P.; Kaplevsky, K.N.; Tarasau, D.; Voropay, E.S.; Petrov, P.T.; Istomin, Y.P. Novel Indotricarbocyanine Dyes Covalently Bonded to Polyethylene Glycol for Theranostics. Journal of Photochemistry and Photobiology A: Chemistry 2016, 316, 31–36. [Google Scholar] [CrossRef]
- Novosjolova, I. The Mukaiyama Reagent: An Efficient Condensation Agent. Synlett 2013, 24, 135–136. [Google Scholar] [CrossRef]
- Sparks, I.L.; Derbyshire, K.M.; Jacobs, W.R.; Morita, Y.S. Mycobacterium Smegmatis: The Vanguard of Mycobacterial Research. Journal of Bacteriology 2023, 205, e0033722. [Google Scholar] [CrossRef] [PubMed]
- Connell, N.D. Chapter 6: Mycobacterium: Isolation, Maintenance, Transformation, and Mutant Selection. Methods in Cell Biology 1994, 45, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Kudykina, Y.K.; Shleeva, M.O.; Artsabanov, V.Y.; Suzina, N.E.; Kaprelyants, A.S. Generation of Dormant Forms by Mycobacterium Smegmatis in the Poststationary Phase during Gradual Acidification of the Medium. Microbiology 2011, 80, 638–649. [Google Scholar] [CrossRef]
- Dixon, C.F.; Nottingham, A.N.; Lozano, A.F.; Sizemore, J.A.; Russell, L.A.; Valiton, C.; Newell, K.L.; Babin, D.; Bridges, W.T.; Parris, M.R.; et al. Synthesis and Evaluation of Porphyrin Glycoconjugates Varying in Linker Length: Preliminary Effects on the Photodynamic Inactivation of Mycobacterium Smegmatis. RSC Advances 2021, 11, 7037–7042. [Google Scholar] [CrossRef] [PubMed]
- Sahile, H.A.; Rens, C.; Shapira, T.; Andersen, R.J.; Av-Gay, Y. DMN-Tre Labeling for Detection and High-Content Screening of Compounds against Intracellular Mycobacteria. ACS Omega 2020, 5, 3661–3669. [Google Scholar] [CrossRef] [PubMed]
- Kamariza, M.; Keyser, S.G.L.; Utz, A.; Knapp, B.D.; Ealand, C.; Ahn, G.; Cambier, C.J.; Chen, T.; Kana, B.; Huang, K.C.; et al. Toward Point-of-Care Detection of Mycobacterium Tuberculosis: A Brighter Solvatochromic Probe Detects Mycobacteria within Minutes. JACS Au 2021, 1, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Xie, J.; Zhu, Q.; Kamariza, M.; Jiang, K.; Bertozzi, C.R.; Rao, J. A Fluorogenic Trehalose Probe for Tracking Phagocytosed Mycobacterium Tuberculosis. J Am Chem Soc. 2020, 142, 15259–15264. [Google Scholar] [CrossRef]
- Liyanage, S.H.; Raviranga, N.G.H.; Ryan, J.G.; Shell, S.S.; Ramström, O.; Kalscheuer, R.; Yan, M. Azide-Masked Fluorescence Turn-On Probe for Imaging Mycobacteria. JACS Au 2023, 3, 1017–1028. [Google Scholar] [CrossRef]
- Dimakos, V.; Taylor, M.S. Site-Selective Functionalization of Hydroxyl Groups in Carbohydrate Derivatives. Chemical Reviews 2018, 118, 11457–11517. [Google Scholar] [CrossRef] [PubMed]
- de Man, J.C. The Probability of Most Probable Numbers. European Journal of Applied Microbiology 1974, 1, 67–78. [Google Scholar] [CrossRef]
Figure 1.
The structural formulae of TCC and TCC2Tre. The numbers of carbon atoms in trehalose are indicated in red. The molar masses for TCC and TCC2Tre are 742 and 1389 g/mol, respectively.
Figure 1.
The structural formulae of TCC and TCC2Tre. The numbers of carbon atoms in trehalose are indicated in red. The molar masses for TCC and TCC2Tre are 742 and 1389 g/mol, respectively.
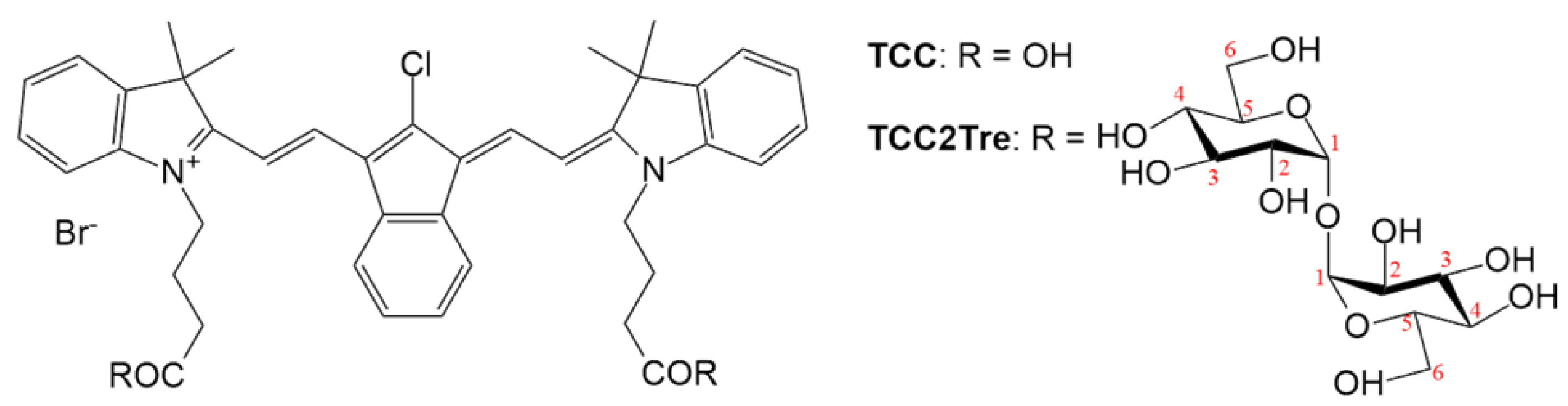
Figure 2.
Absorption spectra for the suspensions of M. smegmatis cells in PBS incubated with TCC2Tre and TCC. A. Equal amounts of bacterial cells (with the final concentration of 107 cells/mL corresponding to the absorbance of 0.2 at 590 nm) were incubated with 40 µM TCC2Tre (red curve) and TCC (blue). After washing, the cells were resuspended in PBS to the concentration of 107 per ml, and the spectra were recorded. B. The cells were incubated with 10 µM (black curve), 20 µM (red curve), 40 µM (blue curve), and 100 µM (magenta curve) TCC2Tre for 1 h in PBS. The inset shows peak absorbance as a function of dye concentration.
Figure 2.
Absorption spectra for the suspensions of M. smegmatis cells in PBS incubated with TCC2Tre and TCC. A. Equal amounts of bacterial cells (with the final concentration of 107 cells/mL corresponding to the absorbance of 0.2 at 590 nm) were incubated with 40 µM TCC2Tre (red curve) and TCC (blue). After washing, the cells were resuspended in PBS to the concentration of 107 per ml, and the spectra were recorded. B. The cells were incubated with 10 µM (black curve), 20 µM (red curve), 40 µM (blue curve), and 100 µM (magenta curve) TCC2Tre for 1 h in PBS. The inset shows peak absorbance as a function of dye concentration.
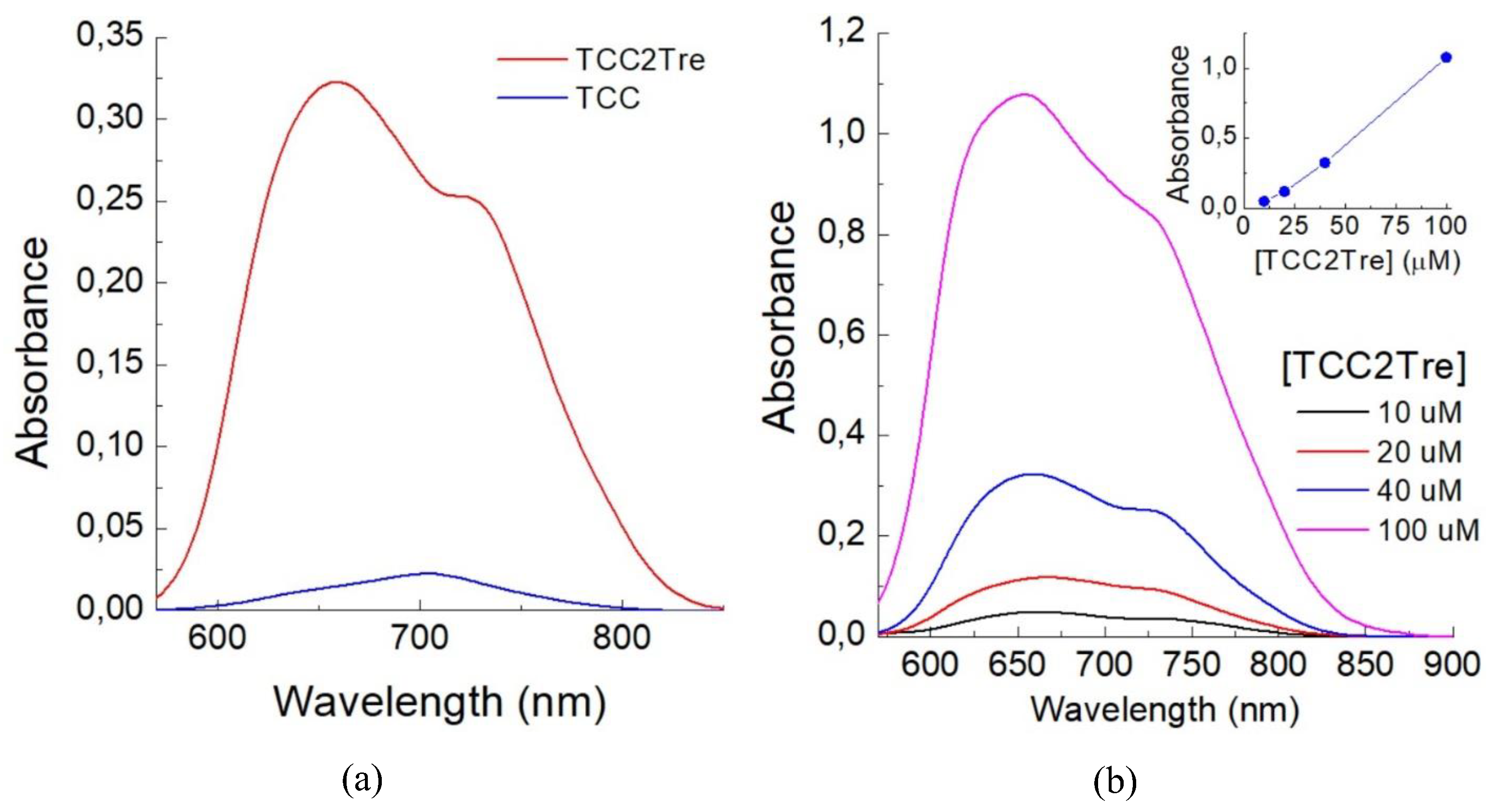
Figure 3.
Absorption spectra for the suspensions of M. smegmatis in PBS incubated with TCC2Tre in different media. A. The cells were incubated with 40 µM TCC2Tre for 1 h in PBS (red curve) and in PBS with 0.05 wt% Tween-80 added (blue curve). B. The cells were incubated with 40 µM TCC2Tre for 1 h in PBS (black curve), 7H9 mineral medium (red curve), and the peptide-rich NB medium (blue curve). For more details, see the caption of Figure 2.
Figure 3.
Absorption spectra for the suspensions of M. smegmatis in PBS incubated with TCC2Tre in different media. A. The cells were incubated with 40 µM TCC2Tre for 1 h in PBS (red curve) and in PBS with 0.05 wt% Tween-80 added (blue curve). B. The cells were incubated with 40 µM TCC2Tre for 1 h in PBS (black curve), 7H9 mineral medium (red curve), and the peptide-rich NB medium (blue curve). For more details, see the caption of Figure 2.
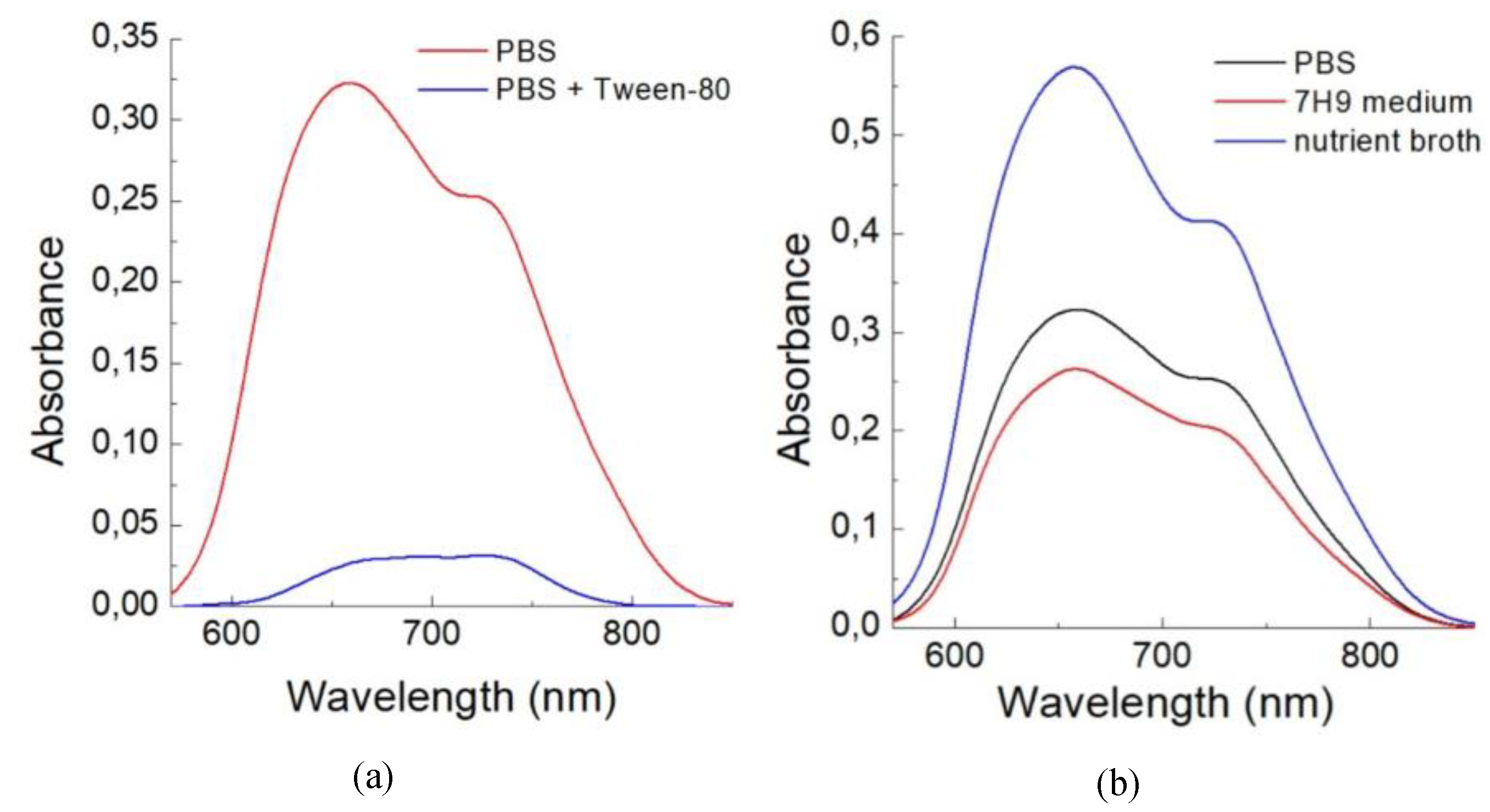
Figure 4.
The survival of M. smegmatis cells after light exposure. M. smegmatis cells were cultured and subjected to PDI as described in Section 4. The incubation of bacteria with the dyes was performed in the NB medium in the presence of 5% BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspension was plated on agar plates for the CFU estimation. A. A 2 h incubation period with various concentrations of the dyes was followed by illumination for 30 min; 100% corresponds to the viability of non-illuminated bacteria. B. The cells were incubated with 20 µM of the dyes followed by illumination under static conditions for various time intervals. Each experiment was repeated three times. The bars represent ± standard deviation.
Figure 4.
The survival of M. smegmatis cells after light exposure. M. smegmatis cells were cultured and subjected to PDI as described in Section 4. The incubation of bacteria with the dyes was performed in the NB medium in the presence of 5% BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspension was plated on agar plates for the CFU estimation. A. A 2 h incubation period with various concentrations of the dyes was followed by illumination for 30 min; 100% corresponds to the viability of non-illuminated bacteria. B. The cells were incubated with 20 µM of the dyes followed by illumination under static conditions for various time intervals. Each experiment was repeated three times. The bars represent ± standard deviation.
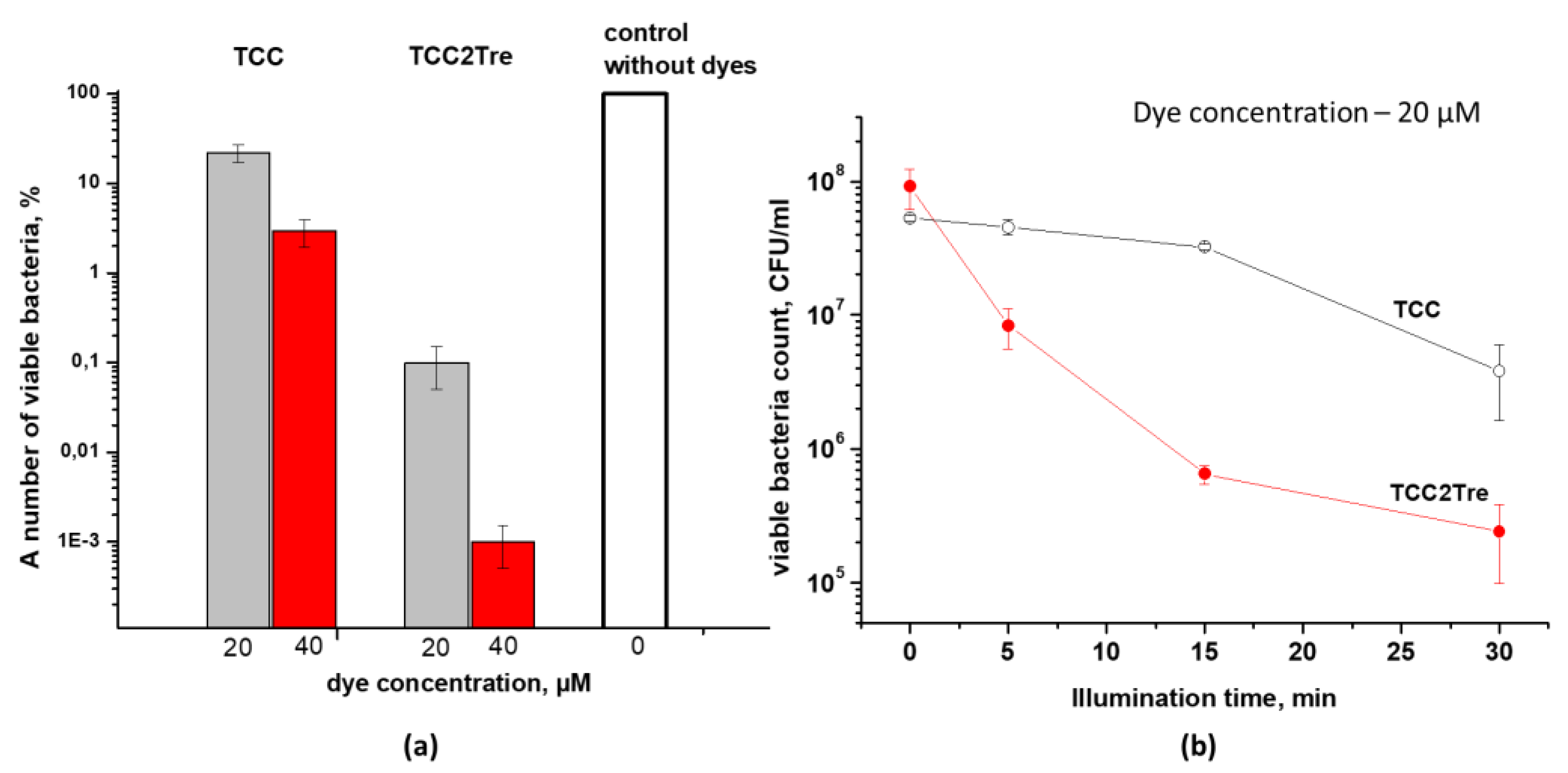
Figure 5.
The influence of medium composition on TCC2Tre effectivity. M. smegmatis cells were cultured and subjected to PDI as described in Section 4. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions was plated on agar plates for CFU estimation. A. Incubation with 20 µM TCC2Tre in presence or absence of 5% BSA and 0.05% Tween-80 for 2 h at 37℃ in the NB medium. B. A 2 h incubation with 20 µM TCC2Tre in different media without Tween-80 and BSA was followed by illumination for 30 min. Each experiment was repeated two times. The bars represent ± standard deviation.
Figure 5.
The influence of medium composition on TCC2Tre effectivity. M. smegmatis cells were cultured and subjected to PDI as described in Section 4. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions was plated on agar plates for CFU estimation. A. Incubation with 20 µM TCC2Tre in presence or absence of 5% BSA and 0.05% Tween-80 for 2 h at 37℃ in the NB medium. B. A 2 h incubation with 20 µM TCC2Tre in different media without Tween-80 and BSA was followed by illumination for 30 min. Each experiment was repeated two times. The bars represent ± standard deviation.
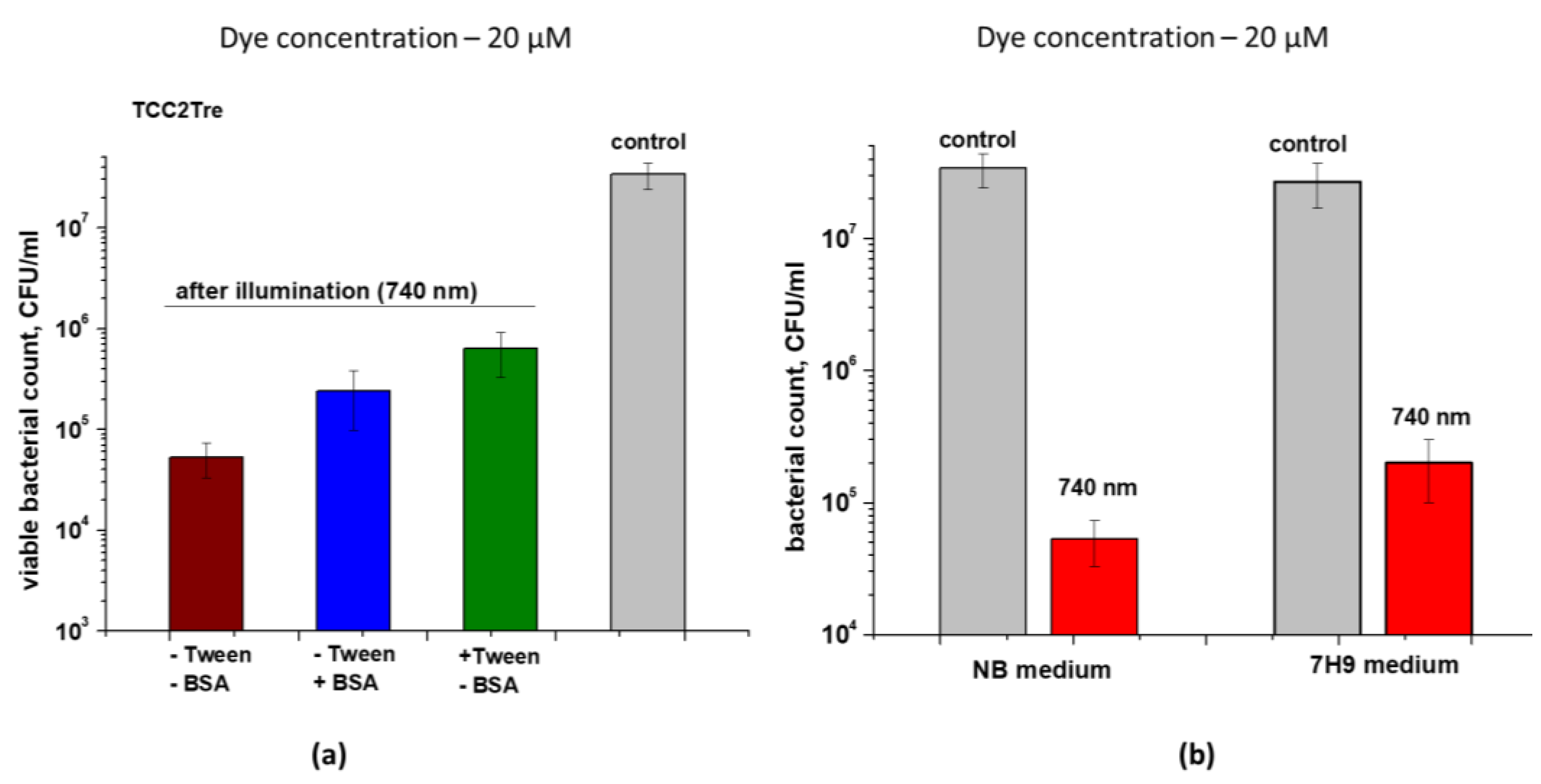
Figure 6.
The photoinactivation of bacteria with different cell wall composition after binding with TCC or TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with 20 µM of the dyes was performed in the NB medium. The cells were incubated with TCC in the presence of BSA and with TCC2Tre in the absence of BSA. Before illumination, the bacteria were washed two times in PBS. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions were plated on agar plates for CFU estimation. A. Mycobacterium smegmatis; B. Micrococcus luteus (Gram-positive); C. Escherichia coli (Gram-negative). The control samples were non-illuminated cells incubated with the PSs. Each experiment was repeated three times. The bars represent ± standard deviation.
Figure 6.
The photoinactivation of bacteria with different cell wall composition after binding with TCC or TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with 20 µM of the dyes was performed in the NB medium. The cells were incubated with TCC in the presence of BSA and with TCC2Tre in the absence of BSA. Before illumination, the bacteria were washed two times in PBS. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions were plated on agar plates for CFU estimation. A. Mycobacterium smegmatis; B. Micrococcus luteus (Gram-positive); C. Escherichia coli (Gram-negative). The control samples were non-illuminated cells incubated with the PSs. Each experiment was repeated three times. The bars represent ± standard deviation.
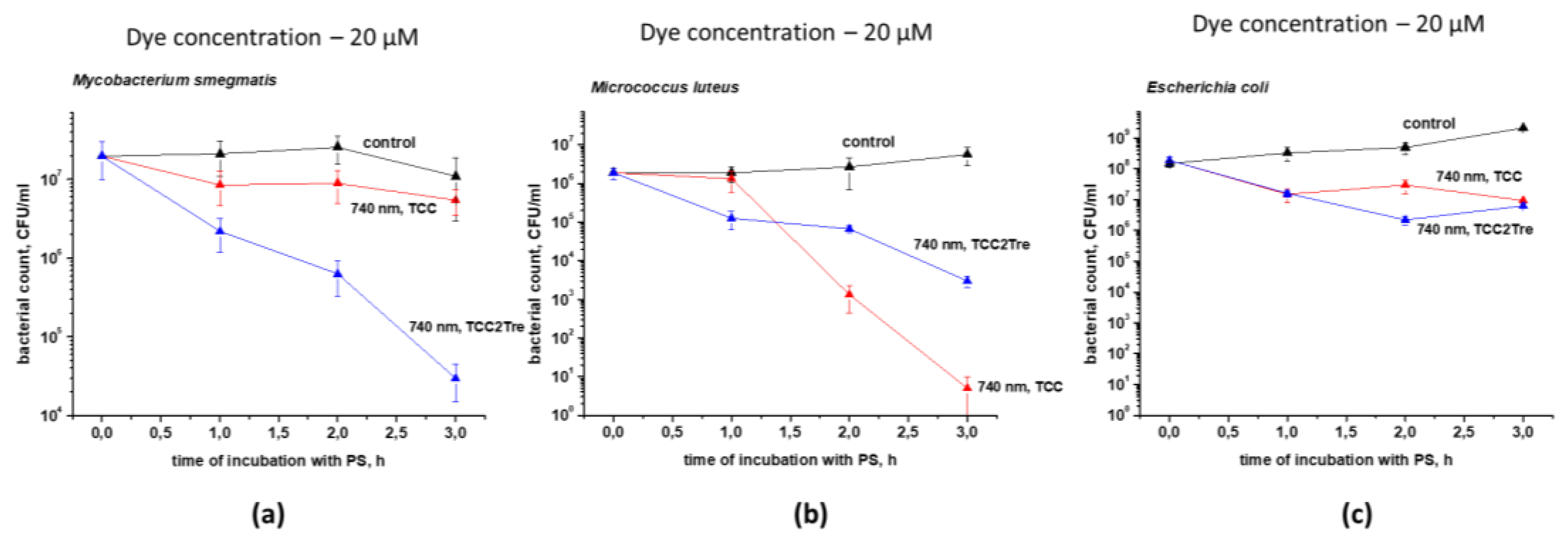
Figure 7.
The photoactivity of TCC and TCC2Tre in a 1:1 mixed culture containing Micrococcus luteus and Mycobacterium smegmatis. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with 40 µM of the dyes was performed in the NB medium without Tween-80 for 1, 2, and 3 h. The incubation medium for TCC contained BSA, while the incubation medium for TCC2Tre did not contain BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions were plated on agar plates for CFU estimation. A. Micrococcus luteus; B. Mycobacterium smegmatis. The control samples were non-illuminated cells incubated with the PSs. Each experiment was repeated two times. The bars represent ± standard deviation.
Figure 7.
The photoactivity of TCC and TCC2Tre in a 1:1 mixed culture containing Micrococcus luteus and Mycobacterium smegmatis. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with 40 µM of the dyes was performed in the NB medium without Tween-80 for 1, 2, and 3 h. The incubation medium for TCC contained BSA, while the incubation medium for TCC2Tre did not contain BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 under static conditions, 100 μL of the cell suspensions were plated on agar plates for CFU estimation. A. Micrococcus luteus; B. Mycobacterium smegmatis. The control samples were non-illuminated cells incubated with the PSs. Each experiment was repeated two times. The bars represent ± standard deviation.
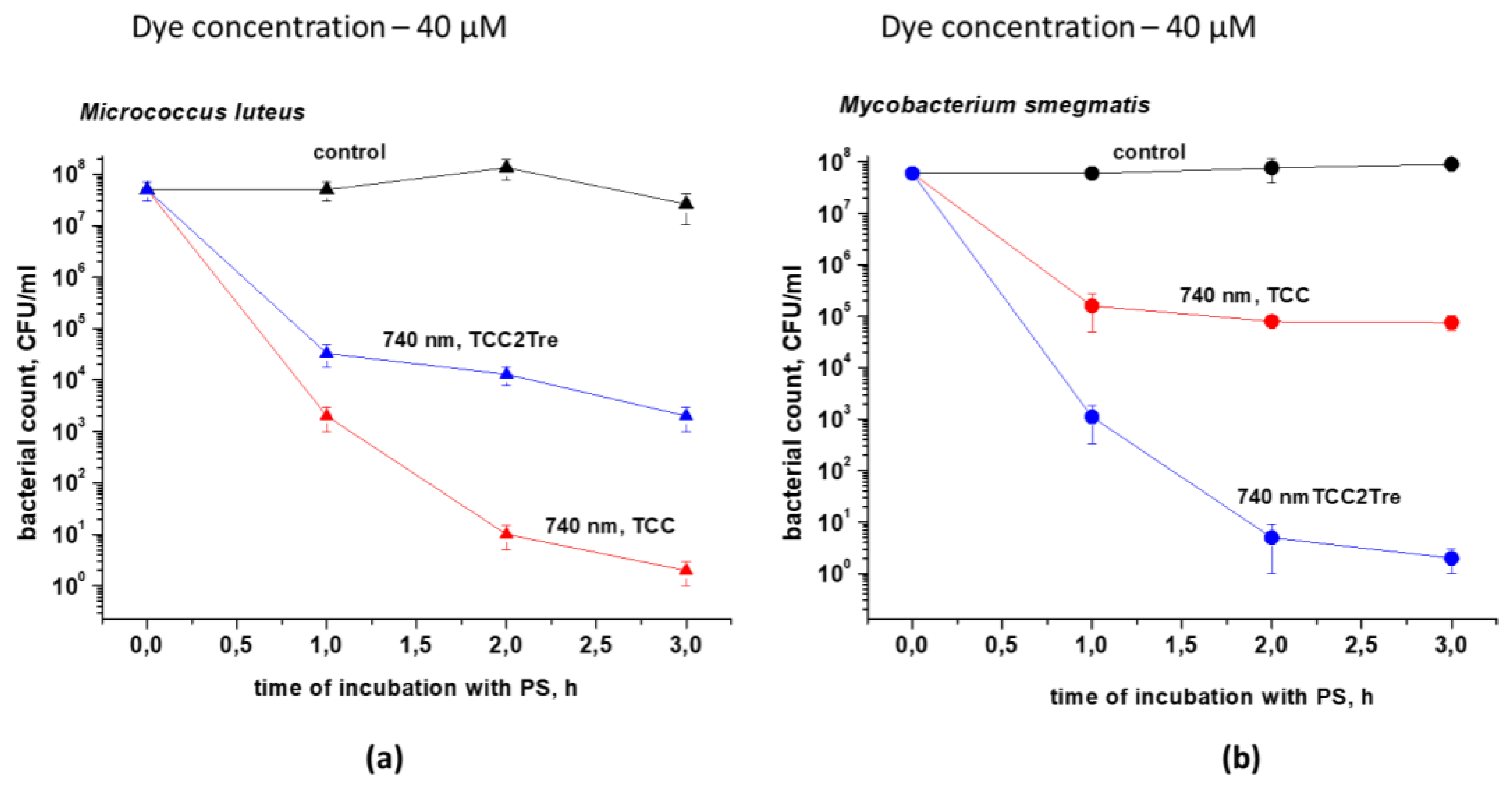
Figure 8.
PDI of cultures of M. smegmatis cells in a dormant state (6 months). Dormant M. smegmatis cells were obtained and subjected to PDI as described in Section 4. The bacteria were incubated with 40 µM of the dyes in Sauton medium in the presence of Tween-80 for 2 h. The incubation medium for TCC contained BSA, while the incubation medium for TCC2Tre did not contain BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 for 30 min under static conditions, the viability of the bacteria was estimated by the MPN assay in liquid medium. The MPN method was performed for two biological replicates, with three series of dilutions made within each. The bars represent ± standard deviation.
Figure 8.
PDI of cultures of M. smegmatis cells in a dormant state (6 months). Dormant M. smegmatis cells were obtained and subjected to PDI as described in Section 4. The bacteria were incubated with 40 µM of the dyes in Sauton medium in the presence of Tween-80 for 2 h. The incubation medium for TCC contained BSA, while the incubation medium for TCC2Tre did not contain BSA. Following exposure to 740 nm light at an irradiance of 240 mW/cm2 for 30 min under static conditions, the viability of the bacteria was estimated by the MPN assay in liquid medium. The MPN method was performed for two biological replicates, with three series of dilutions made within each. The bars represent ± standard deviation.
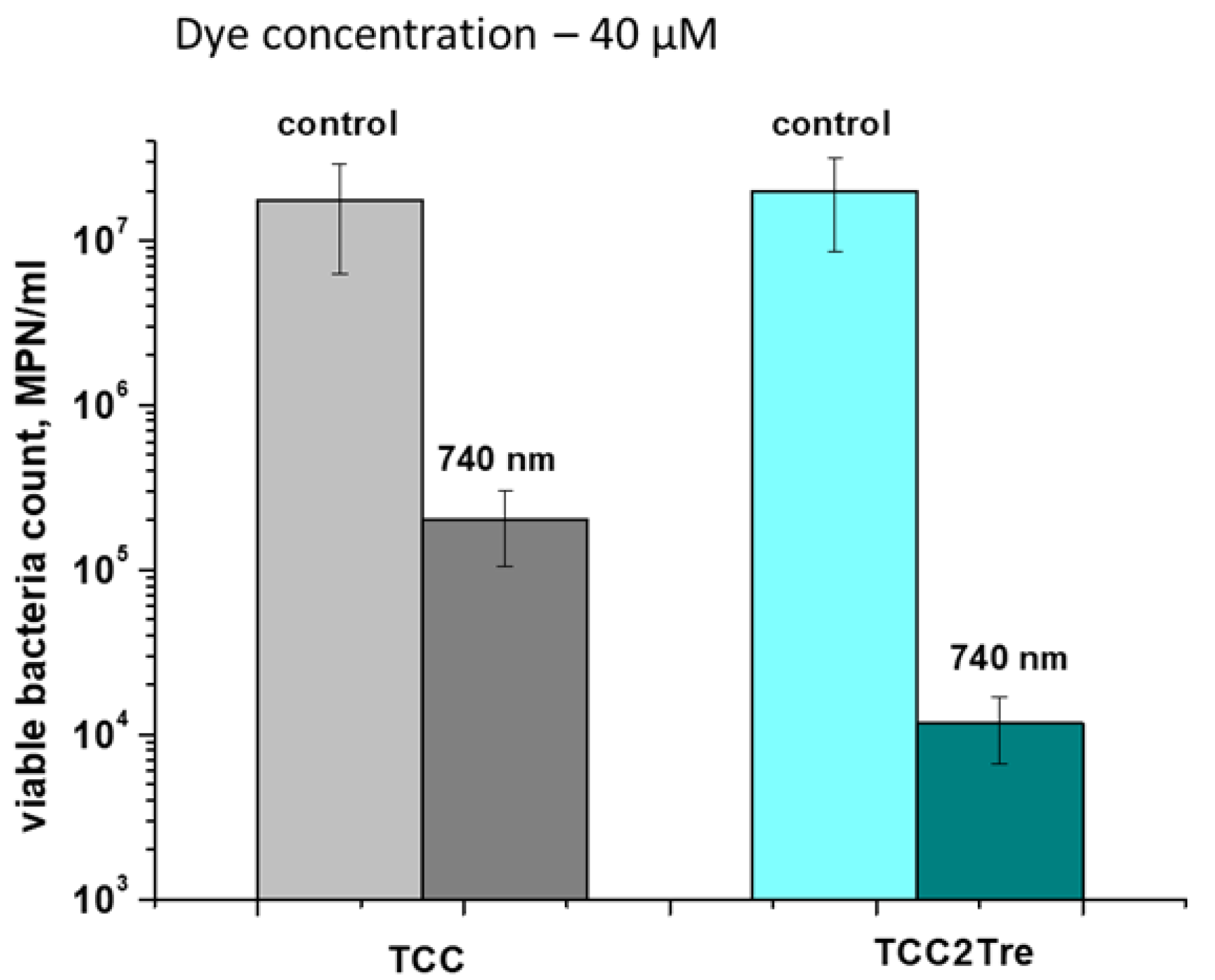
Figure 9.
The effect of light dose on the efficiency of photoinactivation of M. smegmatis in the presence of TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The bacteria were incubated with 20 µM TCC2Tre in the NB medium without Tween-80 for 2 h at 37℃, washed, and exposed to 740 nm light at an irradiance of 240 mW/cm2 for varying periods of time. Each experiment was repeated three times. Typical experiment is shown. The bars represent ± standard deviation.
Figure 9.
The effect of light dose on the efficiency of photoinactivation of M. smegmatis in the presence of TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The bacteria were incubated with 20 µM TCC2Tre in the NB medium without Tween-80 for 2 h at 37℃, washed, and exposed to 740 nm light at an irradiance of 240 mW/cm2 for varying periods of time. Each experiment was repeated three times. Typical experiment is shown. The bars represent ± standard deviation.
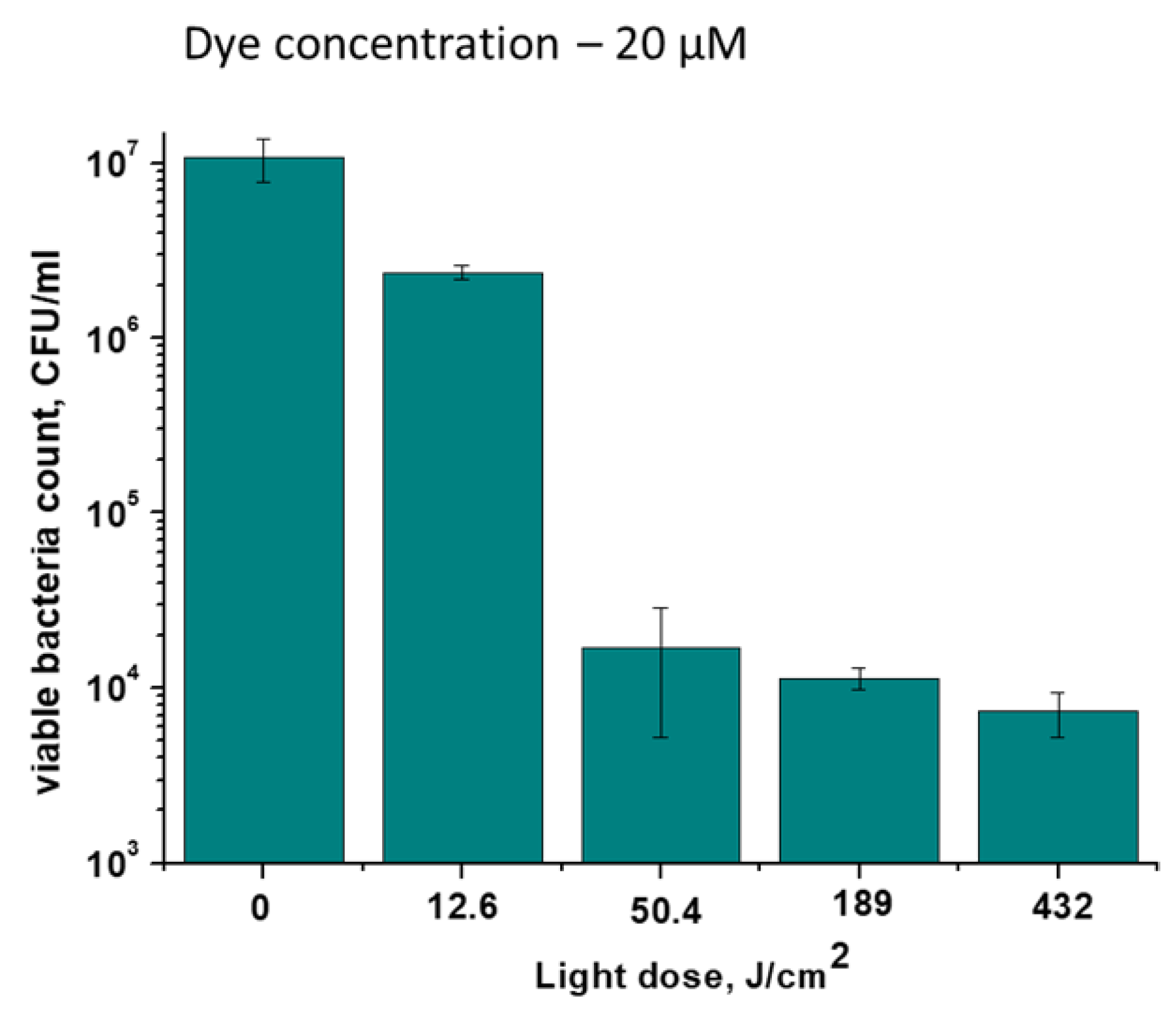
Figure 10.
Photoinactivation of M. tuberculosis cells after their incubation with TCC or TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with the dyes was performed in the 7H9 medium without Tween-80 for 2 h at 37℃. TCC was prepared with BSA, while TCC2Tre was prepared with and without BSA. Before illumination, the bacteria were washed two times in PBS. A. The viability of M. tuberculosis cells incubated with 40 µM of the dyes after exposure to 740 nm light at a light dose 468 J/cm2 (240 mW/cm2 for 30 min). TCC was incubated with BSA, while TCC2Tre was incubated without BSA. B. The viability of M. tuberculosis cells incubated for 2 h with 40 µM of the dyes and exposed to 740 nm light at an irradiance of 240 mW/cm2 for varying periods of time. Each experiment was repeated three times. The bars represent ± standard deviation.
Figure 10.
Photoinactivation of M. tuberculosis cells after their incubation with TCC or TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. The incubation with the dyes was performed in the 7H9 medium without Tween-80 for 2 h at 37℃. TCC was prepared with BSA, while TCC2Tre was prepared with and without BSA. Before illumination, the bacteria were washed two times in PBS. A. The viability of M. tuberculosis cells incubated with 40 µM of the dyes after exposure to 740 nm light at a light dose 468 J/cm2 (240 mW/cm2 for 30 min). TCC was incubated with BSA, while TCC2Tre was incubated without BSA. B. The viability of M. tuberculosis cells incubated for 2 h with 40 µM of the dyes and exposed to 740 nm light at an irradiance of 240 mW/cm2 for varying periods of time. Each experiment was repeated three times. The bars represent ± standard deviation.
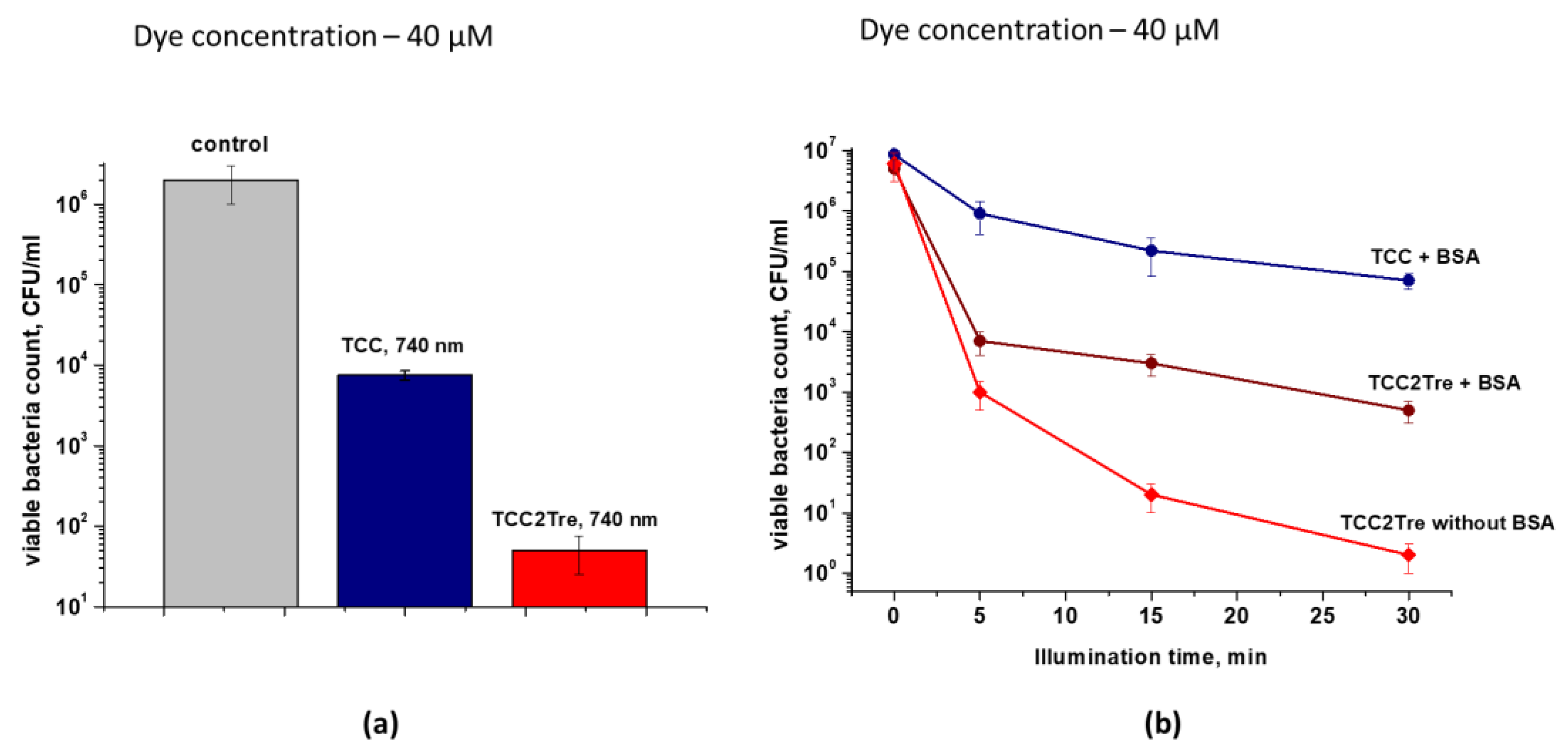
Figure 11.
The effect of the irradiance value on the efficiency of photoinactivation of M. tuberculosis in the presence of TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. Cells were incubated in the 7H9 medium for 2 h with 20 µM TCC2Tre without Tween-80 and BSA. Then, the bacteria were washed and exposed to 740 nm light at a light dose of 100 J/cm2 and different irradiances (mW/cm2) as described in Section 4.7. The plates were incubated at 37 ℃ for 22 days. Each experiment was repeated two times.
Figure 11.
The effect of the irradiance value on the efficiency of photoinactivation of M. tuberculosis in the presence of TCC2Tre. Bacterial cells were cultured and subjected to PDI as described in Section 4. Cells were incubated in the 7H9 medium for 2 h with 20 µM TCC2Tre without Tween-80 and BSA. Then, the bacteria were washed and exposed to 740 nm light at a light dose of 100 J/cm2 and different irradiances (mW/cm2) as described in Section 4.7. The plates were incubated at 37 ℃ for 22 days. Each experiment was repeated two times.

Table 1.
The influence of the composition of the incubation medium on the photodynamic activity of TCC2Tre and TCC against M. smegmatis.
Table 1.
The influence of the composition of the incubation medium on the photodynamic activity of TCC2Tre and TCC against M. smegmatis.
| Incubation conditions | TCC | TCC2Tre |
|---|---|---|
| NB medium | (2.6 ± 1.5) x 107* | (4 ± 2) x 104* |
| NB medium + 5% BSA | (3 ± 2) x 105* | (1.6 ± 1.5) x 105* |
| NB medium + 0.05% Tween 80 | (5 ± 3) x 105* | (5 ± 2) x 105* |
| control with dye without illumination | (3 ± 1.8) x 107* | (4.6 ± 2) x 107* |
| control without dye and without illumination | (2 ± 1.2) x 107* | (3.2 ± 2) x 107* |
A 2-h incubation period with 20 µM TCC2Tre or TCC was followed by a 30-min illumination at 740 nm at an irradiance of 240 mW/cm2. * CFU (cells per mL) for cells after illumination are shown.
Table 2.
Irradiance and illumination time used to obtain a constant light dose.
| Irradiance (mW/cm2) | Illumination time (min) | Light dose (J/cm2) |
|---|---|---|
| 240 105 28 14 |
7 16 60 120 |
100 100 100 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



