
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
The Chikungunya virus (CHIKV) poses a significant global public health concern, especially in Africa. Since its first isolation in Tanzania in 1953, CHIKV has caused recurrent outbreaks, challenging healthcare systems in low-resource settings. Recent outbreaks in Africa highlight the dynamic nature of CHIKV transmission and the challenges of underreporting and underdiagnosis. Here, we review literature and analyze publicly available cases, outbreaks, and genomic data providing insights into the epidemiology, genetic diversity, and transmission dynamics of CHIKV in Africa. Our analyses reveal the circulation of geographically distinct CHIKV genotypes, with certain regions experiencing a disproportionate burden of disease. Phylogenetic analysis of sporadic outbreaks in West Africa suggests repeated emergence of the virus through enzootic spillover, which is markedly different from inferred transmission dynamics in East Africa, where the virus is often introduced from Asian outbreaks, including the recent reintroduction of the Indian Ocean lineage from the Indian subcontinent to east Africa. Furthermore, there is limited evidence of viral movement between these two regions. Understanding the history and transmission dynamics of outbreaks is crucial for public health planning. Despite advances in surveillance and research, diagnostic and surveillance challenges persist. This review and secondary analysis highlight the importance of ongoing surveillance, research, and collaboration to mitigate the burden of CHIKV in Africa and improve public health outcomes.
Keywords:
Chikungunya virus
; Africa
; transmission dynamics
; epidemiology
; genomic distribution
; genomic surveillance
1. Introduction
Chikungunya virus (CHIKV) is an arthritogenic, enveloped, positive-strand RNA Alphavirus, belonging to the family Togaviridae, that is responsible for the febrile Chikungunya illness. Clinical studies from the eighteenth and nineteenth, along with molecular clock analyses of contemporary CHIKV genomes suggest that this virus existed for 300 to 500 years before the first isolation in 1953 [1,2]. The virus is globally widespread in tropical and subtropical regions and is transmitted by Aedes mosquito species; Ae. albopictus and Ae. aegypti being the primary vectors for transmitting CHIKV to humans [3,4,5,6]. CHIKV is classified into four lineages: East-Central-South Africa (ECSA), Asian, Indian Ocean lineage (IOL), and West African (WA). Recent investigations have shown the distribution of Ae. albopictus has expanded to locations with lower temperatures, associated with the concerning potential for CHIKV transmission in new climatic environments [5,6].
The Chikungunya fever outbreak within the Newala district of Tanzania in 1952 and 1953 carries notable significance as one of the earliest recorded human cases of Chikungunya fever and the first isolation of the virus [4,7,8]. Chikungunya is a term derived from the Kimakonde language signifying “to bend up” or “to become contorted,” which encapsulates the severe joint pain and altered posture, hallmark symptoms of the disease [6,8]. The outbreak was characterized by a high prevalence of cases, accompanied by reports of febrile illness and incapacitating joint pain of varying severity [6,7]. Nineteenth-century records document an epidemic termed Kidenga pepo in Zanzibar during 1823, a Swahili term denoting “a sudden cramp-like seizure caused by an evil spirit”, spreading to the Americas and West Indies in 1827 [9]. Prior to this, evidence points to an earlier epidemic spanning from 1779–1785, affecting Egypt, the Arabian Peninsula, India, and Southeast Asia [9,10].
CHIKV infections vary widely in severity, from mild to severe symptoms, with 3%–28% of cases being asymptomatic [11]. The principal indicators include acute fever and profound joint pain [12]. Concomitant symptoms during the acute phase of the disease encompass myalgia, headache, fatigue, and gastrointestinal disturbances. Within a few days following the onset of fever, individuals may develop a maculopapular rash spanning the trunk, limbs, and face [5,13]. Although rare, severe complications such as neurological and cardiovascular manifestations may occur. While the majority of cases recover within weeks, some individuals, particularly those with underlying health conditions may exhibit self-limiting prolonged joint pain and fatigue [5]. The arthritogenic symptoms of Chikungunya fever can persist for several years [3]. Factors like age, duration of infection, joint pain, and swelling adversely impact the health and quality of life leading to pervasive absenteeism and economic losses. For example, in the Réunion Island outbreak of 2005–2006, the total medical expenditure of the chikungunya epidemic was estimated to be €43.9 million, with direct healthcare expenses and the loss of productivity estimated at €26.5 million and €17.4 million, respectively [14,15].
Clinical presentation of CHIKV infection may differ depending on the geographic location of the epidemic. This variability may be attributed to factors such as genetics, comorbidities, pre-existing immunity to CHIKV, and socioeconomic status of the affected communities [13]. For example, Paixåo et al, 2018 investigated the association of CHIKV genotype heterogeneity with disease severity. The study found considerable variability in the prevalence of self-reported chronic symptoms with CHIKV lineages. The prevalence of chronic disease was significantly higher in IOL (52%) followed by the Asian (39%) and ECSA (14%) lineage.
In low-resource socio-economic settings, the prevalence of undifferentiated febrile illness (UFI) outbreaks poses a significant diagnostic challenge [16]. Over the last decade, many African nations, including, Gabon, Nigeria, Kenya, the Democratic Republic of Congo, and Sudan have witnessed substantial outbreaks of febrile diseases [17]. These epidemics are frequently associated with long-term disabilities and high case fatality rates [18,19]. A systematic review and meta-analysis by Nooh et al, (2023) revealed that the etiology of febrile illness is unknown in more than 60% of febrile patients in East Africa and among the analyzed studies CHIKV is the most prevalent causative pathogen.
Understanding the epidemiology, clinical impact, and socio-economic implications of CHIKV in Africa is vital for effective disease management and control. The Climate Amplified Diseases and Epidemics (CLIMADE) consortium, which this work stems from, has been established to develop approaches to predict, track, and control diseases such as CHIKV in the regions most impacted by climate change (https://climade.health/africa). The initiative with global collaborators aims to form a pan-African coalition to create localized knowledge and technologies to fill crucial knowledge gaps in disease transmission, improve the genomic representation of climate-amplified diseases, and develop long-term genomic sequencing capacity [20].
The transmission dynamics and burden of CHIKV in low-resourced regions in Africa remain poorly understood. Our study combined a literature review with phylogenetic and phylodynamic analyses of publicly available genomic data to provide insights into the spatiotemporal movement within and out of the African continent. Here, we review and synthesize the current knowledge on CHIKV in Africa, aiming to elucidate the genetic diversity and evolutionary dynamics of the virus on the continent, while identifying factors that contribute to the spread of infection, and highlighting existing burdens and knowledge gaps. We hypothesize that heterogeneous transmission dynamics across different regions in Africa have driven the widespread circulation of CHIKV.
2. Cases, Outbreaks, and Current Burden in Africa
Since its discovery in 1953, Africa has experienced numerous CHIKV outbreaks, with an estimated 70% of the population residing in high-risk areas for arboviral infections [21]. Suspected CHIKV cases are defined by the presence of acute high fever (>38.5°C) and severe joint pain. Laboratory confirmation requires CHIKV isolation, RNA detection by RT-PCR, CHIKV-specific IgM antibodies, or an increase in IgG antibodies over time [22]. Outbreaks can be substantial in areas with high mosquito vector activity and favorable environmental conditions for viral transmission [23].
Figure 1 illustrates the distribution and surveillance of CHIKV in Africa based on the following data sources: World Health Organization African Regional Office (WHO-AFRO) CHIKV reported cases since 2018 (https://www.afro.who.int/health-topics/disease-outbreaks/outbreaks-and-other-emergencies-updates), publicly available genomic data (https://www.ncbi.nlm.nih.gov/nuccore), and case and outbreak data from published literature. Figure 1 highlights the disparities between reported cases and genomic data. Ethiopia and Chad have reported over 30,000 suspected cases with no genomic data available for the outbreaks that occurred in 2019 and 2020, respectively, indicating surveillance gaps. Additionally, the distinct differences in genotype circulation in west, central and east Africa become immediately apparent.
In Central Africa, CHIKV was first detected in 1958 in the Doruma region in the DRC [64]. Significant outbreaks occurred in 1999 and 2000 in Brazzaville and Kinshasa, resulting in an estimated 50,000 reported cases [31]. Outbreaks in 2011 and 2018 saw cases reaching 8,000 and 11,000, respectively. From 2002 to 2006, isolated CHIKV outbreaks were recorded in Equatorial Guinea, Sudan, and Cameroon. Cameroon reported cases throughout this period [27,45,65]. In 2007, Gabon experienced a CHIKV outbreak that impacted at least 20,000 individuals in Libreville, the country’s capital and largest city. This outbreak lasted 3 years and extended to southern areas of the country [45].
The West African region emerges as a potential hotspot for arboviral disease transmission with significant outbreaks in Senegal, Nigeria and intermittent cases in Guinea in 1992 and 2002, Sierra Leone in 2012-2013, Mali and Burkina Faso in 2023-2024 (Figure 1) [6,53,66,67,68,69,70,71]. Two genomes were isolated from Cote d’Ivoire in 1981 and 1993 [72].
In East Africa, Kenya witnessed an unprecedented epidemic in 2004 spreading to Mombasa and Lamu Island. Lamu Island reported a 75% attack rate (AR) [73]. By 2005, the virus spread to the islands of Comoros, Mayotte, Seychelles, Réunion, and Mauritius. Réunion Island alone reported an estimated 250,000 cases, affecting about 32% of the island’s population [34]. After a 12-year hiatus in Kenya, unprecedented CHIKV outbreaks occurred in Mandera City in 2016 and Mombasa in 2017-2018 [6,34]. In neighboring East African countries, Tanzania reported CHIKV infections between 2007 and 2008. Somalia reported its first cases in 2016 and Ethiopia reported more than 50,000 suspected cases in 2019 [6]. During the nine-month epidemic in Sudan from 2017-2018, Kassala State and Red Sea State collectively reported 48,763 cases [35].
Efforts were made after the 2005 outbreak on Réunion Island to differentiate between the two epidemic waves. The second wave exhibited a notably higher AR compared to the initial wave and a higher proportion of symptomatic cases relative to total infections. The severity of symptoms and incidence of neuro-chikungunya was also greater during the second wave, with a higher proportion of children discharged with disabilities. These findings underscore the evolving nature and severity of the epidemic across distinct phases, highlighting the need for comprehensive surveillance and response strategies within regions of high transmission risk and their neighboring countries [74].
3. Genotype Distribution
Over 110 countries have reported CHIKV outbreaks since the 1950s, with the virus spreading from Sub-Saharan Africa to new territories, such as Europe and the Pacific, facilitated by global travel and trade [11,36,54,75] (Figure 2). This dissemination has led to the emergence of three monophyletic and geographically distinct genotypes: the ancestral ECSA genotype, the Asian genotype, and the WA genotype. Three distinct CHIKV lineages circulate in Africa: WA, ECSA, and the recent IOL [5].
CHIKV is a single-stranded, positive-sense RNA virus, which has evolved as a quasispecies with geographically distinct variants [76]. Since the first identification in East Africa in 1953, the ECSA genotype initially circulated in east, southern, and central regions of Africa and has since spread to cause significant outbreaks in Asia and the Americas [77]. The ECSA genotype comprises three clades; Clade 1 (ancestral east and southern Africa strains), Clade 2 (Central African strains), and Clade 3 (Indian Ocean lineage) which has disseminated from Eastern Africa to Indian Ocean Islands, subsequently spreading to southeast Asia, Oceania, and Europe (Figure 2. a) [6].
The re-emergence of Chikungunya in 2004 triggered outbreaks in the Indian Ocean basin, giving rise to the IOL which is characterized by higher vector competence in Ae. albopictus [78]. Genomic data suggests that IOL spread to the Comoros and Réunion islands and expanded and diversified rapidly, resulting in outbreaks in the Indian Ocean Islands, later to India, Sri Lanka, Thailand, Malaysia, and Italy [34,78,79].
In 2013, a significant milestone in CHIKV transmission occurred when the Asian lineage was imported into St. Martin’s Island, triggering ongoing outbreaks across the Americas [80]. The virus subsequently spread to neighboring countries driven by international travel, trade, and the presence of suitable mosquito vectors [81,82]. Additionally, the ECSA lineage was introduced in the Bahia state of Brazil in 2014, resulting in over 5,000 reported cases. Since then, over 3.6 million cases have been reported in the Americas [80,83,84]. The co-circulation of the ECSA and Asian genotypes have led to outbreaks in South America and the Caribbean islands since 2014, highlighting the global spread and adaptability of CHIKV genotypes [84].
The WA lineage of CHIKV was first identified in 1964 in Ibadan, Nigeria, following its isolation from a mosquito sample obtained in 1963 [70]. Subsequently, it was discovered in Rufisque, Senegal in 1966 [66]. The ECSA lineage has also been detected in West Africa, isolated from a bat sample in Senegal in 1963, suggesting dissemination beyond its geographical origin and possible spatial overlap of the two lineages. The potential coexistence of the WA and ECSA genotypes in West Africa emphasizes the region’s complex CHIKV dynamics however, this was a singular occurrence with no ECSA lineage strains detected since [77].
4. Importance of Genomic and Epidemiological Surveillance
Genomic surveillance is crucial for understanding the transmission dynamics, evolution, and epidemiology of vector-borne diseases. Traditionally, public health officials relied extensively on epidemiological data, but advancements in whole genome sequencing and phylogenetic methods have enhanced the ability to map transmission, detect viral strain, and characterize lineages [85]. Integrating clinical, demographic, and epidemiological metadata with sequencing data helps to identify variables that influence transmission patterns, viral competence, lineage prevalence, and epidemic potential of emergence strains [37,81,86,87]. For example, during the SARS-CoV-2 pandemic, global genomic and epidemiological surveillance revolutionized variant tracking and transmission across spatial and temporal scales and elucidated the multifactorial nature of virus evolution [87].
Urbanization, human mobility, and climate change have facilitated the movement of arboviruses to new regions, increasing the risk of outbreaks within previously unexposed populations, in turn highlighting the need for effective surveillance systems [86]. Comprehensive genetic data from each outbreak and inter-epidemic strains is essential for tracking the movement of viral strains and identifying mutations that may contribute to future outbreaks [73,87].
Since 2016, a flagship arbovirus genomic and epidemiological surveillance program in South America has characterized CHIKV outbreaks and epidemics in Brazil [88], Uruguay [89], Paraguay [90], and Argentina [80], demonstrating the crucial role of genomic surveillance for vector-borne diseases. Unfortunately, Africa lacked such a program and genomic data from African cases are limited despite the virus being endemic. CLIMADE’s strategy for Africa includes forming a consortium to develop locally relevant knowledge and technologies, thereby addressing major knowledge gaps in disease transmission. Key goals include forming a pan-African coalition to assess arbovirus susceptibility, improving genomic representation of climate-amplified diseases, and developing long-term genomic capability and collaboration with global partners [20].
Currently, there are 393 CHIKV genomes sampled from Africa that are publicly available, with 93 being whole genomes and 300 being partial gene sequences (Figure 3). Advances in sequencing technologies have shifted from Sanger sequencing to Next Generation sequencing platforms such as Illumina and Oxford Nanopore for whole genome sequencing (Supplementary Table S1).
Figure 3 highlights the evolving landscape of CHIKV sequencing in Africa, showcasing the steady increase in genomic data generation. CHIKV sequencing efforts began in the early 1950s with a noticeable increase in sequencing from the early 2000s. Aedes mosquito vectors and humans are predominant sources for both partial and whole genomes, additionally, border surveillance efforts are noted by genomes isolated from bats and mice. The ECSA genotype appears consistently over decades, with the WA genotype appearing less frequently and sporadically since the 1960s. A notable increase in IOL genomes between 2010-2020 reflects the increase in resources directed to sequencing efforts after major outbreaks. Continued investment in routine genomic surveillance is crucial for the characterization and tracking of epidemic strains. As a result, providing important information for the implementation of effective control and preventive measures for CHIKV and other vector-borne diseases in Africa.
5. Genetic Diversity and Transmission Dynamics in Africa
The transmission dynamics of CHIKV in Africa are shaped by entomological, environmental, and sociodemographic factors [2]. Our analysis (see supplementary materials for a description of the methods) reveals geographically distinct lineages, including the WA, the ECSA, and the IOL (Figure 4) exhibiting distinct genetic fingerprints and transmission patterns [81]. Estimated evolutionary rates for each lineage varied considerably. The highest rate was observed in the IOL of 5.31 × 10−4 substitutions per nucleotide per year (subs/nt/yr) (Figure 4. a-c), followed by the ECSA, Asian, and WA lineage of 4.44 × 10−4, 4.178 × 10−4 and 2.84 × 10−4 subs/nt/yr, respectively. A study conducted by Volks et al in 2010 obtained comparable evolutionary rates, which showed a notable difference between enzootic lineages having a lower evolutionary rate than epidemic lineages [77]. Here, we have included South American ECSA strains in the phylogenetic analysis, which likely contributed to the difference in rates observed in the aforementioned study.
Geographic and environmental factors such as seasonality, vector abundance, and human mobility, significantly influence CHIKV evolution [2,5,91]. For instance, ECSA sublineages 1 and 2 observed in east, southern, and central African clades (Figure 4. a), show long-standing regional diversity suggestive of limited genomic exchange and localized transmission [6,77]. Similarly, the monophyletic clustering of WA sequences (Figure 4. c) and long branching is presumptively driven by sylvatic spillover seeding sporadic outbreaks followed by endemic circulation [78].
Genetic variation among emerging CHIKV strains may influence vector competence, viral fitness, and adaptation to varied settings, as was evident in the 2005-2006 Réunion Island outbreak with the acquisition of the E1:A226V mutation [81]. Initially, the Réunion strains had E1:226A and E1:226V later in the outbreak [37,92]. Independent convergent evolution led to the emergence of the E1:A226V amino acid substitution resulting in higher vector competence in Ae. albopictus [39]. Similarly, sequences from Madagascar, Seychelles, Mayotte, and Mauritius showed variations in the E1-226 amino acid, impacting local transmission and epidemic peaks [37]. The strain responsible for the Indian Ocean outbreak is believed to have originated from the Kenyan Mombasa outbreak in 2004. The most recent common ancestor (TMRCA) of the 2004 Kenyan genomes emerged in mid-2002, eventually diverging into the two variants responsible for the Lamu and the Mombasa outbreak [34].
Strains collected in Central Africa between 2006–2019 belong to ECSA Central African clade (Figure 4. a) containing the E1:A226V mutation. Isolates from the 2020 outbreak in Chad clustered with the outbreak in DRC in 2019 but lacked the E1 226 mutation [93]. The presence of the E1:A226V mutation isolated from a French traveller returning from Madagascar in 2006, suggests the circulation of Ae. albopictus-associated-virus strains alongside Ae. aegypti-associated strains in Madagascar as early as 2006. Additionally, adaptive mutations for transmission by Ae. albopictus, such as the E1:A98T and the epistatic K211E mutations and several mutations in the E2 gene (D60G, R198Q, L210Q, I211T, K233E, K252Q), enhance viral fitness [39]. The E2:L210Q has been found to enhance CHIKV dissemination in Ae. albopictus present within sequences from the 2016–2019 outbreaks in Cameroon of the Central African clade [45]. Phylogenetic analysis suggests the Central African Clade containing the E1:A226V and E2:I211T mutations emerged around the same time as the IOL via convergent evolution [45]. At low prevalence in ECSA viruses, the E2:I211T is consistently present in viruses harbouring E1:A226V, with in vitro studies suggesting it works epistatically to facilitate the effects of E1:A226V on infectivity [39].
Genomic analyses of CHIKV strains have also identified mutations such as E1:K211E and E2:V264A within the IOL, increasing infectivity and transmission in Ae. aegypti vectors. These mutations emerged between 2005 and 2008, likely originating in India [46]. Detected in the 2016 and 2018 Kenyan outbreaks, these mutations are believed to enhance the virus fitness for Ae. aegypti with no effect on Ae. albopictus vector competence [34].
To understand CHIKV transmission heterogeneity, we examined the spatial and temporal dispersal of CHIKV strains in and out of Africa using ancestral-state reconstruction (Figure 5). Our results show notable transmission patterns observed both within Africa and across continents. Within Africa, CHIKV spread from East Africa to Angola, South Africa, and Central Africa. In the late 1980s and mid-1990s, the virus exhibited transcontinental movement, moving from central Africa to North America and from Angola to Brazil approximately two decades later. Concurrently, transmission from East Africa spread into the Indian subcontinent, notably highlighting the expansion of the ECSA genotype. Further analysis of the WA genotype showed no viral exchange between regions.
Our analysis of the IOL (Figure 5. b) reveals initial exportation from East Africa to Comoros Island, followed by circulation to and among Réunion Island, Mauritius, Madagascar, and Mayotte Island. The international movement included transmission from East Africa into North America and the IOL to France, with subsequent spread to India between 2010 and 2015. A re-introduction event was observed from the Indian subcontinent into East Africa, which is also supported by the literature [46], followed by circulation into northeast Africa after 2015.
The movement of infected individuals across borders has played a documented role in the global dissemination of CHIKV [2]. The spread of the ECSA genotype highlights the impact of travel, trade, and human migration on the CHIKV viral movement [94]. The first importation of the ECSA genotype into Brazil in September 2014 was closely related to the Angolan isolate from 1962 [95].
The IOL is thought to have evolved from the Mombasa strain of CHIKV [37], which is associated with morbidity and mortality, later spreading to the Indian Ocean basin and Asia causing widespread epidemics [96,97]. The IOL outbreak led to several travel-associated cases and the first imported case of CHIKV into France from a traveller returning from the Comoros Islands [98]. In March 2005, the onset of the outbreak in Réunion Island was linked to a patient returning from Comoros Island, where the outbreak had been ongoing since January 2005. The travel-related strains shared identical polymorphic sites with the ancestral ECSA lineage, suggesting early 2006 outbreak sequences likely represent the ancestral genotype of the Réunion outbreak [37].
In the present study, genomic analysis shows an independent introduction of CHIKV from India to Kenya, with the 2016 outbreak genetically similar to Indian genomes. This is supported by the temporal introduction of the IOL from India to Kenya between 2010 and 2015, as seen in Figure 5b. Literature suggests a potential importation event across the Somali border [34], however, without genomic data, we cannot correctly induce the likely route of introductions. We uncover evidence of re-introductions of the ECSA lineage (Figure 5. a) into Africa from the Indian subcontinent. The 2018–2019 CHIKV epidemic that occurred in the eastern states of Sudan was caused by an independent introduction of CHIKV into the region from the Indian subcontinent [35]. The 2017-2018 Mombasa outbreaks indicate separate introductions [36]. These outbreaks spread to Eastern and Northern African countries, including Sudan (Figure 5. b) and Djibouti in 2019. Our findings are consistent with the prior genomic investigations suggesting distinct introduction events with separate strains and temporal origins [46].
CHIKV transmission in west Africa typically occurs within an enzootic cycle involving primatophilic mosquito vectors and NHP as a presumed natural reservoir [66,99], allowing viruses to persist in the natural environment [81]. However, the precise origin and dynamics of transmission of this cycle remain incompletely understood, necessitating further investigation [99]. The two circulating genotypes in Africa showed distinct separation [100], except for a single ECSA isolate from Senegal (Figure 4. a). This separation highlights the complex interplay of ecological factors, socioeconomic situations, and viral transmission dynamics, supporting the studies hypothesis that the heterogeneous nature of CHIKV transmission drives the ongoing circulation of the virus across Africa.
6. Vectors and Transmission
To reliably interpret CHIKV transmission dynamics, it is important to consider transmission routes, including the role of vectors. CHIKV primarily spreads through two main transmission cycles: the sylvatic and urban cycles [2,101]. In the sylvatic cycle, the virus is transmitted between non-human primates (NHP) and forest-dwelling mosquitoes, occasionally spilling over into humans in close proximity to the forest [101,102,103,104]. This cycle is said to represent the ancestral state associated with periods of silence between outbreaks, attributed to the generational susceptibility of NHP. Various mosquito species as vectors in this cycle, including Ae. aegypti formous, Ae. africanus, Ae. luteocephalus, Ae. neoafricanus, Ae. furcifer-taylori, Ae. dalzieli, Ae. vittatus and Culex quinquefasciatus [6,105,106,107]. Spillover of CHIKV from sylvatic cycles occurs via vector species that possess the ability to feed on both humans and NHP, known as bridge vectors [101,104]. Primatophilic Ae. africanus and Ae. furcifer have been widely linked to the sylvatic transmission cycle in east-central, western, and southern Africa [81,101]. During the 1976 epidemic in South Africa, Ae. furcifer was implicated as the primary vector of transmission with baboons as the likely NHP host [108]. Aedes furcifer was also implicated as a significant bridge vector for CHIKV transmission in West Africa [101,104]. In more rural settings, outbreaks are occasionally detected through the implementation of adequate routine surveillance and are related to the increase in sylvatic mosquito populations after periods of seasonal rainfall [65].
The adaptation of the virus to domesticated Aedes mosquito species, particularly Ae. aegypti, has provided an alternate vector and facilitated the introduction of the disease to previously unexposed human populations [109]. Aedes aegypti, also known as the yellow fever mosquito, is a container-breeding species primarily found in urban areas. This domesticated form is commonly found in tropical and subtropical regions in 167 countries globally [110]. The behavioral adaptation and ecology of Ae. aegypti to urbanization and deforestation make the species highly conducive for epidemic transmission [111,112].
The tropical ecoregions of the Afrotropical realm provide an ideal wet-dense ecosystem for both sylvatic and urban populations of both the peridomestic Ae. aegypti and, to a lesser extent, Ae. albopictus [45]. Aedes albopictus originating from Asia has spread across central Africa; the first documented introduction into Africa occurred in 1989 via imported tires from Tokyo, Japan to Cape Town, South Africa [67,113]. Its distribution has expanded to 126 countries worldwide and into a wide range of environments due to the species’ resilience in more temperate regions [110]. Aedes albopictus was the main vector responsible for CHIKV transmission in the 2007 Gabonese and 2011 CAR outbreaks [112]. Aedes albopictus, known for its invasive nature, exhibits a combination of zoophilic and anthropophilic behaviors that enable it to colonize new ecological niches and adapt to seasonal fluctuations. This adaptability is evident in its diverse breeding habitats, opportunistic feeding behaviors, and the ability to outcompete native species regardless of temperature conditions [21,114].
The global surge in CHIKV outbreaks and transmission has been strongly associated with the appearance of mutations in the virus permitting enhanced viral adaptation and fitness to existing and novel vectors, leading to increased transmission [93,115]. An important example of this adaptation is the E1:A226V mutation on the envelope 1 gene, facilitating the transmission of CHIKV by the highly invasive Ae. albopictus mosquitoes. The E1:A226V mutation has been detected in recent central African CHIKV outbreaks including the 2019 outbreak in the DRC [79]. The scenario of convergent evolution advocates for the colonization by Ae. albopictus in recent years, indicating significant implications for CHIKV transmission patterns [39]. This additionally suggests that CHIKV is adapting to the increased presence of Ae. albopictus in Africa, leading to new territories being colonized by this vector species and the potential for more frequent outbreaks in naive human populations [39].
Entomological surveillance yields valuable data on vector species prevalence, abundance, and behavior, aiding in the understanding of arboviral transmission dynamics [116]. CHIKV transmission is closely associated with the passive dispersal of vectors via trade and migration, alongside increased environmental suitability, facilitating global vector spread [110,111]. In recent decades, these vectors have expanded to several areas previously devoid of Aedes species, with most new introductions attributed to vegetative eggs contained in timber and tire exportation [36]. Surveillance efforts have declined, with a 70% decrease in Aedes prevalence studies from 2009 to 2018 when compared to the previous decade [67]. Limited ecological and entomological studies exist for Ae. aegypti and Ae. albopictus in Guinea, Guinea-Bissau, Liberia, Sierra Leone, and Togo. Ongoing surveillance systems are crucial in detecting and controlling outbreaks, ensuring opportune public health responses [117]. Focus on routine surveillance systems and occurrence data should be prioritized especially within countries with limited or no available data such as Guinea Bissau, Togo, Chad, South Sudan, Ethiopia, Eritrea, and Somalia [115,118].
7. Challenges and Gaps in CHIKV Surveillance in Africa
Under-reporting and under-diagnosis of CHIKV in Africa leads to an incomplete understanding of the true burden of the disease, particularly in regions of co-circulation of arboviruses [35,37]. Socioeconomic factors and inadequate diagnostic infrastructure further exacerbate these challenges [35]. Inconsistencies and lack of standardization in case and outbreak reporting across different regions reduce the reliability, accuracy, and comparability of data, hindering the assessment of CHIKV transmission dynamics.
The study identifies significant gaps in surveillance and diagnosis, emphasizing the need for enhanced genomic surveillance to better understand epidemiological patterns of CHIKV and interactions with other arboviruses [33,87,119]. Prospective studies should prioritize the identification of CHIKV isolation in potential host reservoir species, as an enhanced understanding of the enzootic, sylvatic cycle of the virus to prevent future outbreaks and re-emergence in regions where sylvatic transmission regularly occurs [120].
The lack of effective vaccines for CHIKV necessitates increased interventions from public health systems in tracking and controlling outbreaks. The immunological state significantly impacts CHIKV transmission patterns. Periodic outbreaks may develop as a result of waning immunity or exposure to immunologically naive populations [71].
In Africa, neglected arboviruses like CHIKV can cause significant epidemics, yet our understanding of its impact, distribution, and viral heterogeneity remains limited. Historically, public health responses and resources have predominantly been directed toward malaria prevention and control [121]. Understanding the genetic diversity of CHIKV in Africa is essential for advising effective public health strategies for disease surveillance and outbreak control tailored to the continent and its regions. An assessment by the Resilience Against Future Threats through Vector Control (RAFT) research consortium in September 2022 revealed that most African countries have inadequate capacities for arbovirus surveillance and control [121]. To effectively address future pandemics, global genome sequencing equity is essential, requiring targeted funding, distribution of sequencing technologies, increased training, networking, and public health policy decisions [87,119,122]. Effective leadership, collaboration, and adaption of existing surveillance systems are all essential for successful cross-border and continental disease control [87].
8. Conclusions
Urbanization and the rapidly increasing human population provide a suitable environment for the heightened risk of CHIKV transmission. Our findings underscore the complex dynamics of CHIKV transmission, highlighting the heterogeneous nature of CHIKV transmission across the continent. Efforts to monitor and mitigate the movement of infected individuals are crucial in containing the spread of CHIKV and preventing further outbreaks. However, to successfully address the CHIKV burden, it is critical to prioritize support for susceptible populations within epidemic regions. This includes providing access to healthcare, effective vector control, and diagnostics measures to prevent the emergence and re-emergence of CHIKV.
Increased data collection is essential to better understand the epidemiological background and prevalence of CHIKV in Africa. Investing in research and surveillance systems enables us to generate comprehensive data, driving regional-based prevention and control measures. Global collaboration and data sharing are essential in addressing the burden of disease. Through working together, we can strengthen the public health impact of CHIKV in communities at risk, generate data, and promote research that effectively combats chikungunya in Africa. The CLIMADE consortium plays a pivotal role in achieving this vision, by fostering partnerships and collaborations, enhancing genomic surveillance, and developing localized capacity within Africa. Through CLIMADE we can ensure the coordinated action to help mitigate the impact of CHIKV and other emerging infectious diseases across the continent.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Summary of Studies on Chikungunya Virus Genomic and Case Surveillance in Africa; Table S2: List of Accession numbers; Supplementary Material 1: Methods and Materials; Supplementary Material 2: CLIMADE Consortium Author list.
Author Contributions
Conceptualization, H.T., M.M., and Y.R.; methodology, Y.R., H.T., and M.M.; software, Y.R.; validation, Y.R., formal analysis, Y.R.; investigation, Y.R.; resources, Y.R., J.E.S., M.R., M.H., M.M., E.W., and H.T.; data curation, Y.R., and J.E.S.; writing original draft preparation, Y.Y.; writing review and editing, M.M., H.T., C.B., T.O., E.W., J.E.S., M.H., and M.R. visualization, Y.R.; supervision, H.T., M.M., T.O.; project administration, C.B.; funding acquisition, C.B., and T.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by CLIMADE (Climate Amplified Diseases and Epidemics), which is funded in part by grants from the Rockefeller Foundation (HTH 017), the Abbott Pandemic Defense Coalition (APDC), the National Institute of Health USA (U01 AI151698) for the United World Antivirus Research Network (UWARN), the SAMRC South African mRNA Vaccine Consortium (SAMVAC), the Health Emergency Preparedness and Response Umbrella Program (HEPR Program), managed by the World Bank Group (TF0B8412), the Medical Research Foundation (MRF-RG-ICCH-2022-100069), and the Wellcome Trust for the Global.health project (228186/Z/23/Z). The content and findings reported herein are the sole deduction, view, and responsibility of the researcher/s and do not reflect the official position and sentiments of the funding agencies.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data utilized in this study was sourced from publicly available repositories and publications. Genomic data was obtained from GenBank and Bacterial and Viral Bioinformatics Resource Center (BV-BRC) (https://www.bv-brc.org/), Outbreak and case data were retrieved from relevant scientific literature and the World Health Organisation Regional Office in Africa (WHO-AFRO) (https://www.afro.who.int/health-topics/disease-outbreaks/outbreaks-and-other-emergencies-updates).
Acknowledgments
We extend our gratitude and appreciation to the originating and sequencing laboratories for their invaluable contributions to generating and sharing the chikungunya sequences analyzed in this study. We also acknowledge the efforts of the CLIMADE consortium authors, whose collaborative work made this research possible. A detailed list of contributors is provided in the Supplementary Material.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Phadungsombat, J.; Imad, H.; Rahman, M.; Nakayama, E.E.; Kludkleeb, S.; Ponam, T.; Rahim, R.; Hasan, A.; Poltep, K.; Yamanaka, A.; et al. A Novel Sub-Lineage of Chikungunya Virus East/Central/South African Genotype Indian Ocean Lineage Caused Sequential Outbreaks in Bangladesh and Thailand. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Deeba, F.; Haider, M.S.H.; Ahmed, A.; Tazeen, A.; Faizan, M.I.; Salam, N.; Hussain, T.; Alamery, S.F.; Parveen, S. Global Transmission and Evolutionary Dynamics of the Chikungunya Virus. Epidemiol Infect 2020. [Google Scholar] [CrossRef] [PubMed]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.-C.; Lavenir, R.; Pardigon, N.; Reynes, J.-M.; Pettinelli, F.; et al. Genome Microevolution of Chikungunya Viruses Causing the Indian Ocean Outbreak. PLoS Med 2006, 3, 1058–1070. [Google Scholar] [CrossRef]
- Silva, J.V.J.; Ludwig-Begall, L.F.; de Oliveira-Filho, E.F.; Oliveira, R.A.S.; Durães-Carvalho, R.; Lopes, T.R.R.; Silva, D.E.A.; Gil, L.H.V.G. A Scoping Review of Chikungunya Virus Infection: Epidemiology, Clinical Characteristics, Viral Co-Circulation Complications, and Control. Acta Trop 2018, 188, 213–224. [Google Scholar]
- Bartholomeeusen, K.; Daniel, M.; LaBeaud, D.A.; Gasque, P.; Peeling, R.W.; Stephenson, K.E.; Ng, L.F.P.; Ariën, K.K. Chikungunya Fever. Nat Rev Dis Primers 2023, 9, 1–21. [Google Scholar] [CrossRef]
- Russo, G.; Subissi, L.; Rezza, G. Chikungunya Fever in Africa: A Systematic Review. Pathog Glob Health 2020, 114, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mcintosh, B.M.; Harwin, R.M.; Paterson, H.E.; Westwatert, M.L. An Epidemic of Chikungunya in South-Eastern Southern Rhodesia. Cent Afr J Med 1963, 9, 351–359. [Google Scholar]
- Lumsden, W.H.R. An Epidemic of Virus Disease in Southern Province, Tanganyika Territory, in 1952–1953 II. General Description and Epidemiology. Trans R Soc Trop Med Hyg 1955, 49, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Reappearance of Chikungunya, Formerly Called Dengue, in the Americas. Emerg Infect Dis 2015, 21, 557–561. [Google Scholar] [CrossRef]
- Carey, D.E. Chikungunya and Dengue: A Case of Mistaken Identity? J Hist Med Allied Sci 1971, XXVI, 243–262. [Google Scholar] [CrossRef]
- de Lima Cavalcanti, T.Y.V.; Pereira, M.R.; de Paula, S.O.; Franca, R.F.d.O. A Review on Chikungunya Virus Epidemiology, Pathogenesis and Current Vaccine Development. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Leroy, E.M.; Nkoghe, D.; Ollomo, B.; Nze-Nkogue, C.; Becquart, P.; Grard, G.; Pourrut, X.; Charrel, R.; Moureau, G.; Ndjoyi-Mbiguino, A.; et al. Concurrent Chikungunya and Dengue Virus Infections during Simultaneous Outbreaks, Gabon, 2007. Emerg Infect Dis 2009, 15, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Thiberville, S.D.; Boisson, V.; Gaudart, J.; Simon, F.; Flahault, A.; de Lamballerie, X. Chikungunya Fever: A Clinical and Virological Investigation of Outpatients on Reunion Island, South-West Indian Ocean. PLoS Negl Trop Dis 2013, 7. [Google Scholar] [CrossRef]
- Costa, L.B.; Barreto, F.K.d.A.; Barreto, M.C.A.; dos Santos, T.H.P.; de Andrade, M.d.M.O.; Farias, L.A.B.G.; de Freitas, A.R.R.; Martinez, M.J.; Cavalcanti, L.P.d.G. Epidemiology and Economic Burden of Chikungunya: A Systematic Literature Review. Trop Med Infect Dis 2023, 8. [Google Scholar] [CrossRef]
- Soumahoro, M.K.; Boelle, P.Y.; Gaüzere, B.A.; Atsou, K.; Pelat, C.; Lambert, B.; La Ruche, G.; Gastellu-Etchegorry, M.; Renault, P.; Sarazin, M.; et al. The Chikungunya Epidemic on La Réunion Island in 2005-2006: A Cost-of-Illness Study. PLoS Negl Trop Dis 2011, 5. [Google Scholar] [CrossRef]
- Tam, P.Y.I.; Obaro, S.K.; Storch, G. Challenges in the Etiology and Diagnosis of Acute Febrile Illness in Children in Low- and Middle- Income Countries. J Pediatric Infect Dis Soc 2016, 5, 190–205. [Google Scholar] [CrossRef]
- Fenollar, F.; Mediannikov, O. Emerging Infectious Diseases in Africa in the 21st Century. New Microbes New Infect 2018, 26, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Maze, M.J.; Bassat, Q.; Feasey, N.A.; Mandomando, I.; Musicha, P.; Crump, J.A. The Epidemiology of Febrile Illness in Sub-Saharan Africa: Implications for Diagnosis and Management. Clinical Microbiology and Infection 2018, 24, 808–814. [Google Scholar] [CrossRef]
- Nooh, F.; Chernet, A.; Reither, K.; Okuma, J.; Brattig, N.W.; Utzinger, J.; Probst-Hensch, N.; Paris, D.H.; Dreyfus, A. Prevalence of Fever of Unidentified Aetiology in East African Adolescents and Adults: A Systematic Review and Meta-Analysis. Infect Dis Poverty 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- CLIMADE. Available online: https://climade.health/africa (accessed on 5 June 2024).
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan-a Web and Mobile App for Systematic Reviews. Syst Rev 2016, 5. [Google Scholar] [CrossRef]
- Padane, A.; Tegally, H.; Ramphal, Y.; Seyni, N.; Sarr, M.; Diop, M.M.; Diedhiou, C.K.; Mboup, A.; Diouf, N.D.; Souaré, A.; et al. An Emerging Clade of Chikungunya West African Genotype Discovered in Real-Time during 2023 Outbreak in Senegal. medRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.-R.N.; Sangare, A.K.; et al. A Year of Genomic Surveillance Reveals How the SARS-CoV-2 Pandemic Unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Egid, B.R.; Coulibaly, M.; Dadzie, S.K.; Kamgang, B.; McCall, P.J.; Sedda, L.; Toe, K.H.; Wilson, A.L. Review of the Ecology and Behaviour of Aedes Aegypti and Aedes Albopictus in Western Africa and Implications for Vector Control. Current Research in Parasitology and Vector-Borne Diseases 2022, 2. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Technical Guidelines for Integrated Disease Surveillance and Response in the African Region: Third edition. Available online: https://www.afro.who.int/publications/technical-guidelines-integrated-disease-surveillance-and-response-african-region-third (accessed on 15 May 2024).
- Mascarenhas, M.; Garasia, S.; Berthiaume, P.; Corrin, T.; Greig, J.; Ng, V.; Young, I.; Waddell, L. A Scoping Review of Published Literature on Chikungunya Virus. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation African Region Outbreaks and Emergencies Bulletin. Available online: https://www.afro.who.int/health-topics/disease-outbreaks/outbreaks-and-other-emergencies-updates (accessed on 28 April 2024).
- Edwards, C.J.; Welch, S.R.; Chamberlain, J.; Hewson, R.; Tolley, H.; Cane, P.A.; Lloyd, G. Molecular Diagnosis and Analysis of Chikungunya Virus. J Clin Virol 2007, 39, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Fourié, T.; Dia, A.; Savreux, Q.; Pommier de Santi, V.; de Lamballerie, X.; Leparc-Goffart, I.; Simon, F. Emergence of Indian Lineage of ECSA Chikungunya Virus in Djibouti, 2019. Int J Infect Dis 2021, 108, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Agbodzi, B.; Yousseu, F.B.S.; Simo, F.B.N.; Kumordjie, S.; Yeboah, C.; Mosore, M.T.; Bentil, R.E.; Prieto, K.; Colston, S.M.; Attram, N.; et al. Chikungunya Viruses Containing the A226V Mutation Detected Retrospectively in Cameroon Form a New Geographical Subclade. Int J Infect Dis 2021, 113, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, A.; Ciccozzi, M.; Cella, E.; Lai, A.; Simonetti, F.R.; Galli, M.; Zehender, G.; Rezza, G. Origin, Evolution, and Phylogeography of Recent Epidemic CHIKV Strains. Infect Gene Evol 2012, 12, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Mudurangaplar, B. Molecular Characterisation of Clinical Isolates of Chikungunya Virus: A Study from Tertiary Care Hospitals in Southern India. J Clin Diagn Res 2016. [Google Scholar] [CrossRef] [PubMed]
- Thiberville, S.-D.; Boisson, V.; Gaudart, J.; Simon, F.; Flahault, A.; de Lamballerie, X. Chikungunya Fever: A Clinical and Virological Investigation of Outpatients on Reunion Island, South-West Indian Ocean. PLoS Negl Trop Dis 2013, 7, e2004. [Google Scholar] [CrossRef]
- Pastorino, B.; Muyembe-Tamfum, J.J.; Bessaud, M.; Tock, F.; Tolou, H.; Durand, J.P.; Peyrefitte, C.N. Epidemic Resurgence of Chikungunya Virus in Democratic Republic of the Congo: Identification of a New Central African Strain. J Med Virol 2004, 74, 277–282. [Google Scholar] [CrossRef]
- Peyrefitte, C.N.; Rousset, D.; Pastorino, B.A.M.; Pouillot, R.; Bessaud, M.; Tock, F.; Mansaray, H.; Merle, O.L.; Pascual, A.M.; Paupy, C.; et al. Chikungunya Virus, Cameroon, 2006. Emerg Infect Dis 2007, 13, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Takaya, S.; Kutsuna, S.; Nakayama, E.; Taniguchi, S.; Tajima, S.; Katanami, Y.; Yamamoto, K.; Takeshita, N.; Hayakawa, K.; Kato, Y.; et al. Chikungunya Fever in Traveler from Angola to Japan, 2016. Emerg Infect Dis 2017, 23, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Eyase, F.; Pollett, S.; Konongoi, S.L.; Joyce, M.G.; Figueroa, K.; Ofula, V.; Koka, H.; Koskei, E.; Nyunja, A.; et al. Global Outbreaks and Origins of a Chikungunya Virus Variant Carrying Mutations Which May Increase Fitness for Aedes Aegypti: Revelations from the 2016 Mandera, Kenya Outbreak. Am J Trop Med Hyg 2019, 100, 1249–1257. [Google Scholar] [CrossRef]
- Bower, H.; el Karsany, M.; Adam, A.A.A.H.; Idriss, M.I.; Alzain, M.A.; Alfakiyousif, M.E.A.; Mohamed, R.; Mahmoud, I.; Albadri, O.; Mahmoud, S.A.A.; et al. “Kankasha” in Kassala: A Prospective Observational Cohort Study of the Clinical Characteristics, Epidemiology, Genetic Origin, and Chronic Impact of the 2018 Epidemic of Chikungunya Virus Infection in Kassala, Sudan. PLoS Negl Trop Dis 2021, 15, e0009387. [Google Scholar] [CrossRef] [PubMed]
- Eyase, F.; Langat, S.; Berry, I.M.; Mulwa, F.; Nyunja, A.; Mutisya, J.; Owaka, S.; Limbaso, S.; Ofula, V.; Koka, H.; et al. Emergence of a Novel Chikungunya Virus Strain Bearing the E1:V80A Substitution, out of the Mombasa, Kenya 2017-2018 Outbreak. PLoS ONE 2020, 15, e0241754. [Google Scholar] [CrossRef]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.-C.; Lavenir, R.; Pardigon, N.; Reynes, J.-M.; Pettinelli, F.; et al. Genome Microevolution of Chikungunya Viruses Causing the Indian Ocean Outbreak. PLoS Med 2006, 3, e263. [Google Scholar] [CrossRef]
- Moyen, N.; Thiberville, S.-D.; Pastorino, B.; Nougairede, A.; Thirion, L.; Mombouli, J.-V.; Dimi, Y.; Leparc-Goffart, I.; Capobianchi, M.R.; Lepfoundzou, A.D.; et al. First Reported Chikungunya Fever Outbreak in the Republic of Congo, 2011. PLoS ONE 2014, 9, e115938. [Google Scholar] [CrossRef]
- Selhorst, P.; Makiala-Mandanda, S.; De Smet, B.; Mariën, J.; Anthony, C.; Binene-Mbuka, G.; De Weggheleire, A.; Ilombe, G.; Kinganda-Lusamaki, E.; Pukuta-Simbu, E.; et al. Molecular Characterization of Chikungunya Virus during the 2019 Outbreak in the Democratic Republic of the Congo. Emerg Microbes Infect 2020, 9, 1912–1918. [Google Scholar] [CrossRef]
- Moyen, N.; Thiberville, S.-D.; Pastorino, B.; Nougairede, A.; Thirion, L.; Mombouli, J.-V.; Dimi, Y.; Leparc-Goffart, I.; Capobianchi, M.R.; Lepfoundzou, A.D.; et al. First Reported Chikungunya Fever Outbreak in the Republic of Congo, 2011. PLoS ONE 2014, 9, e115938. [Google Scholar] [CrossRef]
- Konongoi, S.L.; Nyunja, A.; Ofula, V.; Owaka, S.; Koka, H.; Koskei, E.; Eyase, F.; Langat, D.; Mancuso, J.; Lutomiah, J.; et al. Human and Entomologic Investigations of Chikungunya Outbreak in Mandera, Northeastern Kenya, 2016. PLoS ONE 2018, 13, e0205058. [Google Scholar] [CrossRef]
- Parola, P.; de Lamballerie, X.; Jourdan, J.; Rovery, C.; Vaillant, V.; Minodier, P.; Brouqui, P.; Flahault, A.; Raoult, D.; Charrel, R. Novel Chikungunya Virus Variant in Travelers Returning from Indian Ocean Islands. Emerg Infect Dis 2006, 12, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Masia, M.; Sánchez-Seco, M.-P.; de Ory, F.; Benito, A.; Cano, J.; Tenorio, A.; Collao, X.; Negredo, A.I. Different Lineages of Chikungunya Virus in Equatorial Guinea in 2002 and 2006. Am J Trop Med Hyg 2010, 82, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Paupy, C.; Grard, G.; Becquart, P.; Mombo, I.; Nso, B.B.B.; Kassa Kassa, F.; Nkoghe, D.; Leroy, E.M. Recent Introduction and Rapid Dissemination of Chikungunya Virus and Dengue Virus Serotype 2 Associated With Human and Mosquito Coinfections in Gabon, Central Africa. Clinical Infectious Diseases 2012, 55, e45–e53. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Nkoghe, D.; Ollomo, B.; Nze-Nkogue, C.; Becquart, P.; Grard, G.; Pourrut, X.; Charrel, R.; Moureau, G.; Ndjoyi-Mbiguino, A.; et al. Concurrent Chikungunya and Dengue Virus Infections during Simultaneous Outbreaks, Gabon, 2007. Emerg Infect Dis 2009, 15, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Phadungsombat, J.; Imad, H.A.; Nakayama, E.E.; Leaungwutiwong, P.; Ramasoota, P.; Nguitragool, W.; Matsee, W.; Piyaphanee, W.; Shioda, T. Spread of a Novel Indian Ocean Lineage Carrying E1-K211E/E2-V264A of Chikungunya Virus East/Central/South African Genotype across the Indian Subcontinent, Southeast Asia, and Eastern Africa. Microorganisms 2022, 10, 354. [Google Scholar] [CrossRef]
- Simo Tchetgna, H.; Sem Ouilibona, R.; Nkili-Meyong, A.A.; Caron, M.; Labouba, I.; Selekon, B.; Njouom, R.; Leroy, E.M.; Nakoune, E.; Berthet, N. Viral Exploration of Negative Acute Febrile Cases Observed during Chikungunya Outbreaks in Gabon. Intervirology 2018, 61, 174–184. [Google Scholar] [CrossRef]
- Vairo, F.; Aimè Coussoud-Mavoungou, M.; Ntoumi, F.; Castilletti, C.; Kitembo, L.; Haider, N.; Carletti, F.; Colavita, F.; Gruber, C.; Iannetta, M.; et al. Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation. Viruses 2020, 12, 1020. [Google Scholar] [CrossRef]
- Onoja, A.B.; Omatola, A.C.; Maiga, M.; Gadzama, I.S. Recurrent Episodes of Some Mosquito-Borne Viral Diseases in Nigeria: A Systematic Review and Meta-Analysis. Pathogens 2022, 11, 1162. [Google Scholar] [CrossRef]
- Delisle, E.; Rousseau, C.; Broche, B.; Leparc-Goffart, I.; L’Ambert, G.; Cochet, A.; Prat, C.; Foulongne, V.; Ferré, J.B.; Catelinois, O.; et al. Chikungunya Outbreak in Montpellier, France, September to October 2014. Eurosurveillance 2015, 20. [Google Scholar] [CrossRef]
- Chinedu Eneh, S.; Uwishema, O.; Nazir, A.; El Jurdi, E.; Faith Olanrewaju, O.; Abbass, Z.; Mustapha Jolayemi, M.; Mina, N.; kseiry, l.; Onyeaka, H. Chikungunya Outbreak in Africa: A Review of the Literature. Annals of Medicine & Surgery 2023, 85, 3545–3552. [Google Scholar] [CrossRef] [PubMed]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Global Expansion of Chikungunya Virus: Mapping the 64-Year History. Intl J Infect Dis 2017, 58, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Collao, X.; Negredo, A.I.; Cano, J.; Tenorio, A.; De Ory, F.; Benito, A.; Masia, M.; Sánchez-Seco, M.P. Short Report: Different Lineages of Chikungunya Virus in Equatorial Guinea in 2002 and 2006. Am J Trop Med Hyg 2010, 82, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Bettis, A.A.; L’Azou Jackson, M.; Yoon, I.K.; Breugelmans, J.G.; Goios, A.; Gubler, D.J.; Powers, A.M. The Global Epidemiology of Chikungunya from 1999 to 2020: A Systematic Literature Review to Inform the Development and Introduction of Vaccines. PLoS Negl Trop Dis 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Desdouits, M.; Kamgang, B.; Berthet, N.; Tricou, V.; Ngoagouni, C.; Gessain, A.; Manuguerra, J.C.; Nakouné, E.; Kazanji, M. Genetic Characterization of Chikungunya Virus in the Central African Republic. Infect Gene Evol 2015, 33, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ratsitorahina, M.; Harisoa, J.; Ratovonjato, J.; Biacabe, S.; Reynes, J.M.; Zeller, H.; Raoelina, Y.; Talarmin, A.; Richard, V.; Soares, J.L. Outbreak of Dengue and Chikungunya Fevers, Toamasina, Madagascar, 2006. Emerg Infect Dis 2008, 14, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Beesoon, S.; Funkhouser, E.; Kotea, N.; Spielman, A.; Robich, R.M. Chikungunya Fever, Mauritius, 2006. Emerg Infect Dis 2008, 14, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Sergon, K.; Njuguna, C.; Kalani, R.; Ofula, V.; Onyango, C.; Konongoi, L.S.; Bedno, S.; Burke, H.; Dumilla, A.M.; Konde, J.; et al. Seroprevalence of Chikungunya Virus (CHIKV) Infection on Lamu Island, Kenya, October 2004. Am J Trop Med Hyg 2008, 78, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Nkoghe, D.; Kassa, R.F.; Caron, M.; Grard, G.; Mombo, I.; Bikié, B.; Paupy, C.; Becquart, P.; Bisvigou, U.; Leroy, E.M. Clinical Forms of Chikungunya in Gabon, 2010. PLoS Negl Trop Dis 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Suhana, O.; Nazni, W.A.; Apandi, Y.; Farah, H.; Lee, H.L.; Sofian-Azirun, M. Insight into the Origin of Chikungunya Virus in Malaysian Non-Human Primates via Sequence Analysis. Heliyon 2019, 5, e02682. [Google Scholar] [CrossRef]
- Nyamwaya, D.K.; Otiende, M.; Omuoyo, D.O.; Githinji, G.; Karanja, H.K.; Gitonga, J.N.; R. de Laurent, Z.; Otieno, J.R.; Sang, R.; Kamau, E.; et al. Endemic Chikungunya Fever in Kenyan Children: A Prospective Cohort Study. BMC Infect Dis 2021, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, Y.; Abe, H.; Mbadinga, M.J.V.M.; Ondo, G.N.; Bikangui, R.; Agnandji, S.T.; Lell, B.; Yasuda, J. Re-Emergence of Dengue, Chikungunya, and Zika Viruses in 2021 after a 10-Year Gap in Gabon. IJID Regions 2022, 5, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Bacterial and Viral Bioinformatics Resource Center (BV-BRC). Available online: https://www.bv-brc.org/ (accessed on 12 January 2024).
- De Weggheleire, A.; Nkuba-Ndaye, A.; Mbala-Kingebeni, P.; Mariën, J.; Kindombe-Luzolo, E.; Ilombe, G.; Mangala-Sonzi, D.; Binene-Mbuka, G.; De Smet, B.; Vogt, F.; et al. A Multidisciplinary Investigation of the First Chikungunya Virus Outbreak in Matadi in the Democratic Republic of the Congo. Viruses 2021, 13, 1988. [Google Scholar] [CrossRef] [PubMed]
- Demanou, M.; Antonio-Nkondjio, C.; Ngapana, E.; Rousset, D.; Paupy, C.; Manuguerra, J.-C.; Zeller, H. Chikungunya Outbreak in a Rural Area of Western Cameroon in 2006: A Retrospective Serological and Entomological Survey. BMC Res Notes 2010, 3, 128. [Google Scholar] [CrossRef]
- Buchwald, A.G.; Hayden, M.H.; Dadzie, S.K.; Paull, S.H.; Carlton, E.J. Aedes-Borne Disease Outbreaks in West Africa: A Call for Enhanced Surveillance. Acta Trop 2020, 209, 105468. [Google Scholar] [CrossRef]
- Ansumana, R.; Jacobsen, K.H.; Leski, T.A.; Covington, A.L.; Bangura, U.; Hodges, M.H.; Lin, B.; Bockarie, A.S.; Lamin, J.M.; Bockarie, M.J.; et al. Reemergence of Chikungunya Virus in Bo, Sierra Leone. Emerg Infect Dis 2013, 19, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.T.; Tytheridge, S.J.; Smith, S.J.; Levine, R.S.; Hodges, M.H.; Ansumana, R.; Wulff, S.; Whitworth, J.; Logan, J.G. The Threat of Vector-Borne Diseases in Sierra Leone. Am J Trop Med Hyg 2023, 109, 10–21. [Google Scholar] [CrossRef]
- Moore, D.L.; Reddy, S.; Akinkugbe, F.M.; Lee, V.H.; David-West, T.S.; Causey, O.R.; Carey, D.E. An Epidemic of Chikungunya Fever at Ibadan, Nigeria, 1969. Ann Trop Med Parasitol 1974, 68, 59–68. [Google Scholar] [CrossRef]
- Powers, A.M.; Logue, C.H. Changing Patterns of Chikungunya Virus: Re-Emergence of a Zoonotic Arbovirus. Journal of General Virology 2007, 88, 2363–2377. [Google Scholar] [CrossRef]
- Sow, A.; Faye, O.; Diallo, M.; Diallo, D.; Chen, R.; Faye, O.; Diagne, C.T.; Guerbois, M.; Weidmann, M.; Ndiaye, Y.; et al. Chikungunya Outbreak in Kedougou, Southeastern Senegal in 2009-2010. Open Forum Infect Dis 2018, 5. [Google Scholar] [CrossRef]
- Njenga, M.K.; Nderitu, L.; Ledermann, J.P.; Ndirangu, A.; Logue, C.H.; Kelly, C.H.L.; Sang, R.; Sergon, K.; Breiman, R.; Powers, A.M. Tracking Epidemic Chikungunya Virus into the Indian Ocean from East Africa. J Gen Virol 2008, 89, 2754–2760. [Google Scholar] [CrossRef] [PubMed]
- Gérardin, P.; Guernier, V.; Perrau, J.; Fianu, A.; Le Roux, K.; Grivard, P.; Michault, A.; de Lamballerie, X.; Flahault, A.; Favier, F. Estimating Chikungunya Prevalence in La Réunion Island Outbreak by Serosurveys: Two Methods for Two Critical Times of the Epidemic. BMC Infect Dis 2008, 8, 99. [Google Scholar] [CrossRef]
- Kang, H.; Auzenbergs, M.; Clapham, H.; Maure, C.; Kim, J.-H.; Salje, H.; Taylor, C.G.; Lim, A.; Clark, A.; Edmunds, W.J.; et al. Chikungunya Seroprevalence, Force of Infection, and Prevalence of Chronic Disability after Infection in Endemic and Epidemic Settings: A Systematic Review, Meta-Analysis, and Modelling Study. Lancet Infect Dis 2024, 24, 488–503. [Google Scholar] [CrossRef]
- Cottis, S.; Blisnick, A.A.; Failloux, A.-B.; Vernick, K.D. Determinants of Chikungunya and O’nyong-Nyong Virus Specificity for Infection of Aedes and Anopheles Mosquito Vectors Determinants of Chikungunya and O’nyong-Nyong Virus Specificity for Infection of Aedes and Anopheles Mosquito. Viruses 2023, 15, 589. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-Scale Phylogenetic Analyses of Chikungunya Virus Reveal Independent Emergences of Recent Epidemics and Various Evolutionary Rates. J Virol 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive Genome Scale Phylogenetic Study Provides New Insights on the Global Expansion of Chikungunya Virus. J Virol 2016, 90, 10600–10611. [Google Scholar] [CrossRef]
- Galán-Huerta, K.A.; Rivas-Estilla, A.M.; Fernández-Salas, I.; Farfan-Ale, J.A.; Ramos-Jiménez, J. Chikungunya Virus: A General Overview. Medicina Universitaria 2015, 17, 175–183. [Google Scholar] [CrossRef]
- de Souza, W.M.; Ribeiro, G.S.; de Lima, S.T.S.; de Jesus, R.; Moreira, F.R.R.; Whittaker, C.; Anice Sallum, M.M.; Carrington, C.V.F.; Sabino, E.C.; Kitron, U.; et al. Chikungunya: A Decade of Burden in the Americas. Lancet Reg Health -Am 2024, 30, 100673. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Sherman, M.B.; Weaver, S.C. Chikungunya Virus: Evolution and Genetic Determinants of Emergence. Curr Opin Virol 2011, 1, 310–317. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Weaver, S.C. Interspecies Transmission and Chikungunya Virus Emergence. Curr Opin Virol 2016, 16, 143–150. [Google Scholar] [CrossRef]
- De Almeida Di Maio Ferreira, F.C.P.; Da Silva, A.S.V.; Recht, J.; Guaraldo, L.; Moreira, M.E.L.; De Siqueira, A.M.; Gerardin, P.; Brasil, P. Vertical Transmission of Chikungunya Virus: A Systematic Review. PLoS ONE 2021, 16. [Google Scholar]
- Faria, N.R.; Lourenço, J.; Marques de Cerqueira, E.; Maia de Lima, M.; Carlos Junior Alcantara, L. Epidemiology of Chikungunya Virus in Bahia, Brazil, 2014-2015. PLoS Curr 2016. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking Virus Outbreaks in the Twenty-First Century. Nat Microbiol 2019, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wallau, G.L.; Abanda, N.N.; Abbud, A.; Abdella, S.; Abera, A.; Ahuka-Mundeke, S.; Falconi-Agapito, F.; Alagarasu, K.; Ariën, K.K.; Ayres, C.F.J.; et al. Arbovirus Researchers Unite: Expanding Genomic Surveillance for an Urgent Global Need. Lancet Glob Health 2023, 11, e1501–e1502. [Google Scholar] [CrossRef]
- Zeghbib, S.; Kemenesi, G.; Jakab, F. The Importance of Equally Accessible Genomic Surveillance in the Age of Pandemics. Biol Futur 2023, 74, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Iani, F.C.d.M.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, Epidemiological and Digital Surveillance of Chikungunya Virus in the Brazilian Amazon. PLoS Negl Trop Dis 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Burgueño, A.; Giovanetti, M.; Fonseca, V.; Morel, N.; Lima, M.; Castro, E.; Guimarães, N.R.; Iani, F.C.M.; Bormida, V.; Cortinas, M.N.; et al. Genomic and Eco-Epidemiological Investigations in Uruguay Reveal Local Chikungunya Virus Transmission Dynamics during Its Expansion across the Americas in 2023. Emerg Microbes Infect 2024, 13. [Google Scholar] [CrossRef]
- Giovanetti, M.; Vazquez, C.; Lima, M.; Castro, E.; Rojas, A.; Gomez de la Fuente, A.; Aquino, C.; Cantero, C.; Fleitas, F.; Torales, J.; et al. Rapid Epidemic Expansion of Chikungunya Virus East/Central/South African Lineage, Paraguay. Emerg Infect Dis 2023, 9, 1859–1863. [Google Scholar] [CrossRef]
- Huber, J.H.; Childs, M.L.; Caldwell, J.M.; Mordecai, E.A. Seasonal Temperature Variation Influences Climate Suitability for Dengue, Chikungunya, and Zika Transmission. PLoS Negl Trop Dis 2018, 12. [Google Scholar] [CrossRef]
- Vazeille, M.; Moutailler, S.; Coudrier, D.; Rousseaux, C.; Khun, H.; Huerre, M.; Thiria, J.; Dehecq, J.S.; Fontenille, D.; Schuffenecker, I.; et al. Two Chikungunya Isolates from the Outbreak of La Reunion (Indian Ocean) Exhibit Different Patterns of Infection in the Mosquito, Aedes Albopictus. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Yonga, M.G.; Yandai, F.H.; Sadeuh-Mba, S.; Abdallah, A.H.; Ouapi, D.; Gamougam, K.; Abanda, N.N.; Endengue-Zanga, M.C.; Demanou, M.; Njouom, R. Molecular Characterization of Chikungunya Virus from the First Cluster of Patients during the 2020 Outbreak in Chad. Arch Virol 2022, 167, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, R.; Fouque, F.; Mahdy, M.A.K.; Zhong, Q.; Al-Eryani, S.M.A.; Alkwri, A.; Beier, J.C. Multisectoral Approach to Address Chikungunya Outbreaks Driven by Human Mobility: A Systematic Review and Meta-Analysis. Journal of Infectious Diseases 2020, 222, S709–S716. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.G.; de Oliveira, L.F.; da Silva Azevedo, R.d.S.; da Silva, D.E.A.; da Silva, E.V.P.; da Silva, S.P.; et al. Emergence and Potential for Spread of Chikungunya Virus in Brazil. BMC Med 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Panella, A.J.; Velez, J.O.; Lambert, A.J.; Campbell, G.L. Chikungunya Virus in US Travelers Returning from India, 2006. Emerg Infect Dis 2007, 13, 764–767. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog 2007, 3, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Mourad, O.; Makhani, L.; Chen, L.H. Chikungunya: An Emerging Public Health Concern. Curr Infect Dis Rep 2022, 24, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Yergolkar, P.; Tandale, B.; Arankalle, V.; Sathe, P.; Gandhe, S.; Gokhle, M.; Jacob, G.; Hundekar, S.; Mishra, A. Chikungunya Outbreaks Caused by African Genotype, India. Emerg Infect Dis 2006, 12, 1580–1583. [Google Scholar] [CrossRef]
- Powers, A.M.; Brault, A.C.; Tesh, R.B.; Weaver, S.C. Re-Emergence of Chikungunya and o’nyong-Nyong Viruses: Evidence for Distinct Geographical Lineages and Distant Evolutionary Relationships. Microbiology (N Y) 2000, 81, 471–479. [Google Scholar] [CrossRef]
- Weaver, S.C.; Chen, R.; Diallo, M. Chikungunya Virus: Role of Vectors in Emergence from Enzootic Cycles. Annu Rev Entomol 2020, 65, 313–332. [Google Scholar] [CrossRef]
- Valentine, M.J.; Murdock, C.C.; Kelly, P.J. Sylvatic Cycles of Arboviruses in Non-Human Primates. Parasit Vectors 2019, 12, 463. [Google Scholar] [CrossRef]
- Diallo, M.; Thonnon, J.; ~aore-Lamizana, M. Vectors of Chikungunya Virus in Senegal: Current Data and Transmission Cycles. Am J Trop Med Hyg 1999, 60, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Diallo, D.; Sall, A.A.; Buenemann, M.; Chen, R.; Faye, O.; Diagne, C.T.; Faye, O.; Ba, Y.; Dia, I.; Watts, D.; et al. Landscape Ecology of Sylvatic Chikungunya Virus and Mosquito Vectors in Southeastern Senegal. PLoS Negl Trop Dis 2012, 6. [Google Scholar] [CrossRef]
- Pialoux, G.; Gaüzère, B.-A.; Jauréguiberry, S.; Strobel, M. Chikungunya, an Epidemic Arbovirosis. Lancet Infect Dis 2007, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Richman, R.; Diallo, D.; Diallo, M.; Sall, A.A.; Faye, O.; Diagne, C.T.; Dia, I.; Weaver, S.C.; Hanley, K.A.; Buenemann, M. Ecological Niche Modeling of Aedes Mosquito Vectors of Chikungunya Virus in Southeastern Senegal. Parasit Vectors 2018, 11, 255. [Google Scholar] [CrossRef]
- Weaver, S.C.; Lecuit, M. Chikungunya Virus and the Global Spread of a Mosquito-Borne Disease. New England Journal of Medicine 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Mcintosh, B.M.; Jupp, P.G.; Dos Santos, I. Rural Epidemic of Chikungunya in South Africa with Involvement of Aedes (Diceromyia) Furcifer (Edwards) and Baboons. S Afr J Sci 1977, 73. [Google Scholar]
- Powell, J.R.; Tabachnick, W.J. History of Domestication and Spread of Aedes Aegypti - A Review. Mem Inst Oswaldo Cruz 2013, 108, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Laporta, G.Z.; Potter, A.M.; Oliveira, J.F.A.; Bourke, B.P.; Pecor, D.B.; Linton, Y.M. Global Distribution of Aedes Aegypti and Aedes Albopictus in a Climate Change Scenario of Regional Rivalry. Insects 2023, 14. [Google Scholar] [CrossRef]
- Pfeffer, M.; Zöller, G.; Essbauer, S.; Tomaso, H.; Behrens-Riha, N.; Löscher, T.; Dobler, G. Clinical and Virological Characterization of Imported Cases of Chikungunya Fever. Wien Klin Wochenschr 2008, 120, 95–100. [Google Scholar] [CrossRef]
- Longbottom, J.; Walekhwa, A.W.; Mwingira, V.; Kijanga, O.; Mramba, F.; Lord, J.S. Aedes Albopictus Invasion across Africa: The Time Is Now for Cross-Country Collaboration and Control. Lancet Glob Health 2023, 11, e623–e628. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.J.; Hunt, R.H. Aedes Albopictus in Africa? First Records of Live Specimens in Imported Tires in Capetown. J Am Mosq Control Assoc 1991, 7, 107–108. [Google Scholar]
- Bonizzoni, M.; Gasperi, G.; Chen, X.; James, A.A. The Invasive Mosquito Species Aedes Albopictus: Current Knowledge and Future Perspectives. Trends Parasitol 2013, 29, 460–468. [Google Scholar] [CrossRef]
- Weetman, D.; Kamgang, B.; Badolo, A.; Moyes, C.L.; Shearer, F.M.; Coulibaly, M.; Pinto, J.; Lambrechts, L.; McCall, P.J. Aedes Mosquitoes and Aedes-Borne Arboviruses in Africa: Current and Future Threats. Int J Environ Res Public Health 2018, 15. [Google Scholar] [CrossRef]
- Tajudeen, Y.A.; Oladunjoye, I.O.; Bajinka, O.; Oladipo, H.J. Zoonotic Spillover in an Era of Rapid Deforestation of Tropical Areas and Unprecedented Wildlife Trafficking: Into the Wild. Challenges 2022, 13, 41. [Google Scholar] [CrossRef]
- Tajudeen, Y.A.; Oladipo, H.J.; Oladunjoye, I.O.; Mustapha, M.O.; Mustapha, S.T.; Abdullahi, A.A.; Yusuf, R.O.; Abimbola, S.O.; Adebayo, A.O.; Ikebuaso, J.G.; et al. Preventing the Next Pandemic through a Planetary Health Approach: A Focus on Key Drivers of Zoonosis. Challenges 2022, 13, 50. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Sinka, M.E.; Duda, K.A.; Mylne, A.; Shearer, F.M.; Brady, O.J.; Messina, J.P.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; et al. The Global Compendium of Aedes Aegypti and Ae. Albopictus Occurrence. Sci Data 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Alcantara, L.C.J.; Dorea, A.S.; Ferreira, Q.R.; Marques, W.d.A.; Franca de Barros, J.; Adelino, T.E.R.; Tosta, S.; Fritsch, H.; Iani, F.C. de M.; et al. Promoting Responsible Research and Innovation (RRI) During Brazilian Activities of Genomic and Epidemiological Surveillance of Arboviruses. Front Public Health 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Althouse, B.M.; Guerbois, M.; Cummings, D.A.T.; Diop, O.M.; Faye, O.; Faye, A.; Diallo, D.; Sadio, B.D.; Sow, A.; Faye, O.; et al. Role of Monkeys in the Sylvatic Cycle of Chikungunya Virus in Senegal. Nat Commun 2018, 9. [Google Scholar] [CrossRef]
- Braack, L.; Wulandhari, S.A.; Chanda, E.; Fouque, F.; Merle, C.S.; Nwangwu, U.; Velayudhan, R.; Venter, M.; Yahouedo, A.G.; Lines, J.; et al. Developing African Arbovirus Networks and Capacity Strengthening in Arbovirus Surveillance and Response: Findings from a Virtual Workshop. Parasit Vectors 2023, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Salgado, A.; De Souza Fonseca, V.; Tosta, F.D.O.; Xavier, J.; De Jesus, J.G.; Iani, F.C.M.; Adelino, T.E.R.; Barreto, F.K.; Faria, N.R.; et al. Pan-Genomics of Virus and Its Applications. Pan-genomics: Applications, Challenges, and Future Prospects 2020, 237–250. [Google Scholar] [CrossRef]
Figure 1.
Distribution and Surveillance of Genome, Cases, and Outbreaks of CHIKV in Africa. The colour ramp of the map illustrates the geographic distribution of CHIKV Cumulative and Suspected cases from 2018–2024 obtained from the WHO-AFRO weekly reports [24], overlaid markers in yellow indicate the reported year of outbreaks and reported cases in parenthesis obtained from literature[6,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] and the coloured circles show the lineages and the number of genomes generated in each country received from Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63] and NCBI’s GenBank databases.
Figure 1.
Distribution and Surveillance of Genome, Cases, and Outbreaks of CHIKV in Africa. The colour ramp of the map illustrates the geographic distribution of CHIKV Cumulative and Suspected cases from 2018–2024 obtained from the WHO-AFRO weekly reports [24], overlaid markers in yellow indicate the reported year of outbreaks and reported cases in parenthesis obtained from literature[6,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] and the coloured circles show the lineages and the number of genomes generated in each country received from Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63] and NCBI’s GenBank databases.
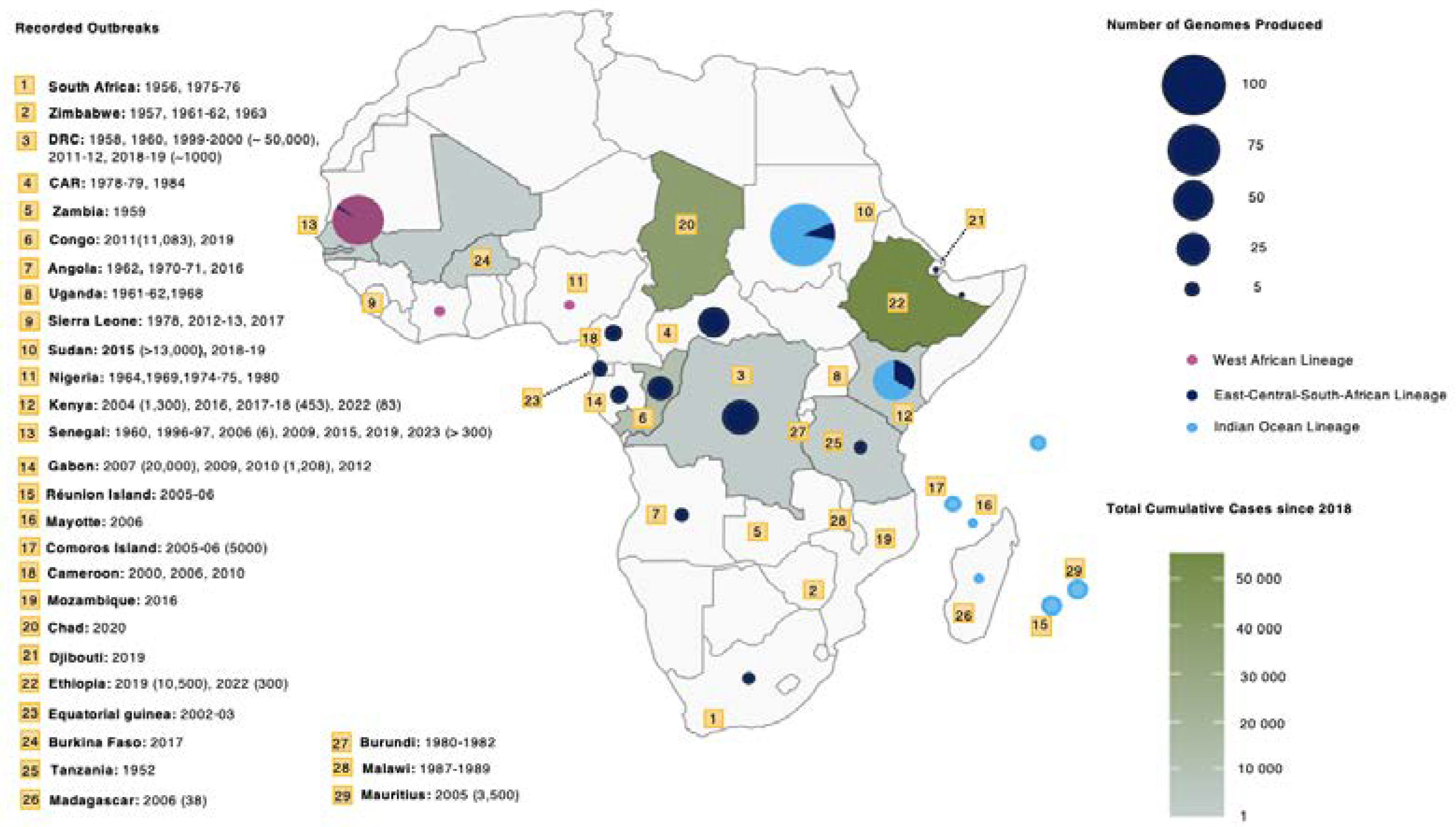
Figure 2.
Global Distribution of CHIKV Lineages. a) World map depicting the distribution and number of genomes generated for each CHIKV lineage. Circle sizes represent the number of CHIKV genomes produced per country and colours represent the proportion of different CHIKV lineages in each country; b) Global map coloured by the date of first genome isolation for each CHIKV lineage, ranging from 1953 to 2023. Data source: Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63].
Figure 2.
Global Distribution of CHIKV Lineages. a) World map depicting the distribution and number of genomes generated for each CHIKV lineage. Circle sizes represent the number of CHIKV genomes produced per country and colours represent the proportion of different CHIKV lineages in each country; b) Global map coloured by the date of first genome isolation for each CHIKV lineage, ranging from 1953 to 2023. Data source: Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63].
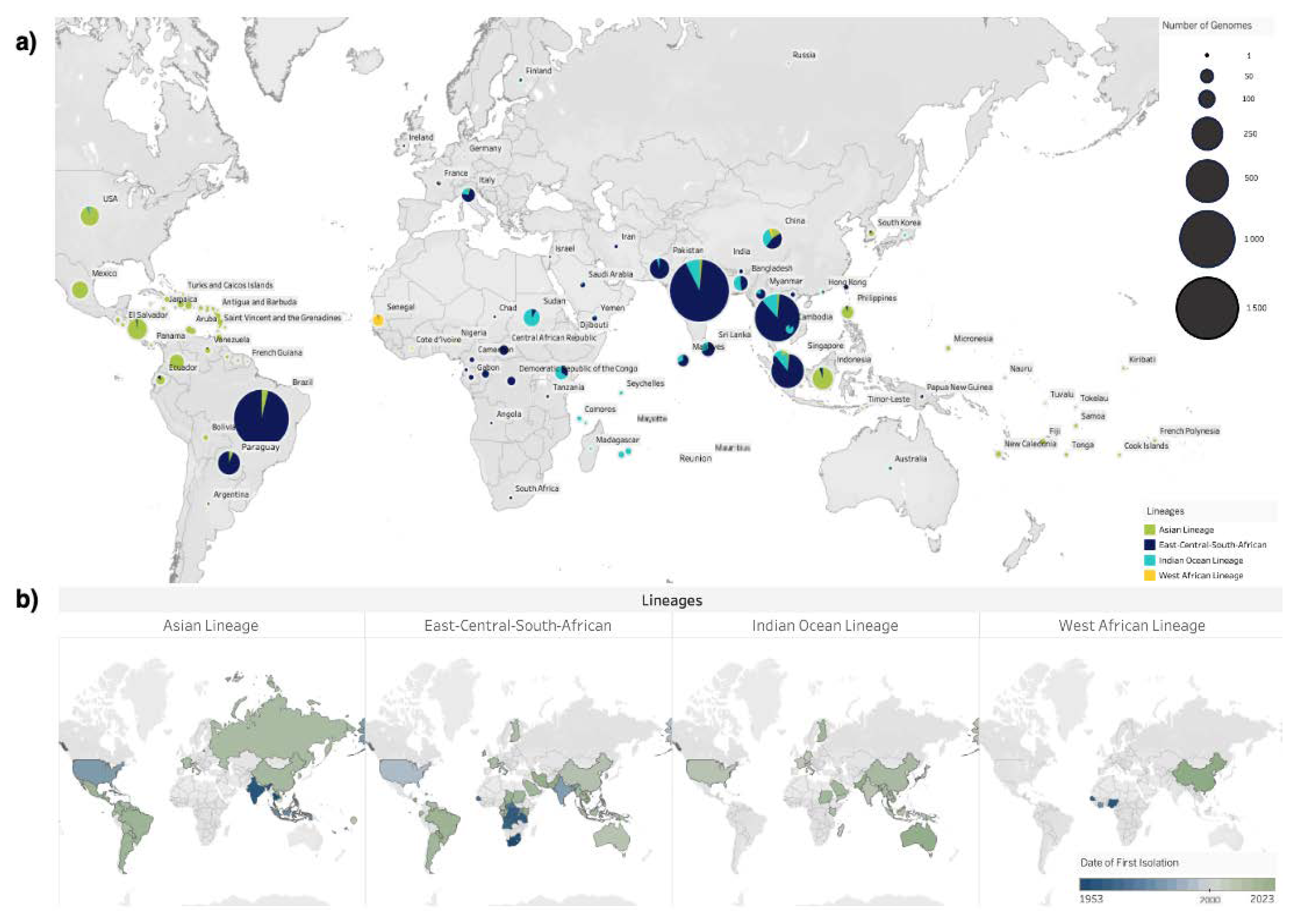
Figure 3.
Overview of genomic sequencing efforts in Africa since the 1950s. Shapes are used to illustrate the different CHIKV genotypes: the circle represents the East-Central-South African genotype, the diamond represents the Indian Ocean Lineage (IOL) and the square represents the WA genotype. The shapes’ sizes show the number of genomes produced for each isolation source in each African country, which is represented by a different color. The figure also depicts the type of genomes produced for each host species. Data source: Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63] and NCBI GenBa.nk
Figure 3.
Overview of genomic sequencing efforts in Africa since the 1950s. Shapes are used to illustrate the different CHIKV genotypes: the circle represents the East-Central-South African genotype, the diamond represents the Indian Ocean Lineage (IOL) and the square represents the WA genotype. The shapes’ sizes show the number of genomes produced for each isolation source in each African country, which is represented by a different color. The figure also depicts the type of genomes produced for each host species. Data source: Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [63] and NCBI GenBa.nk
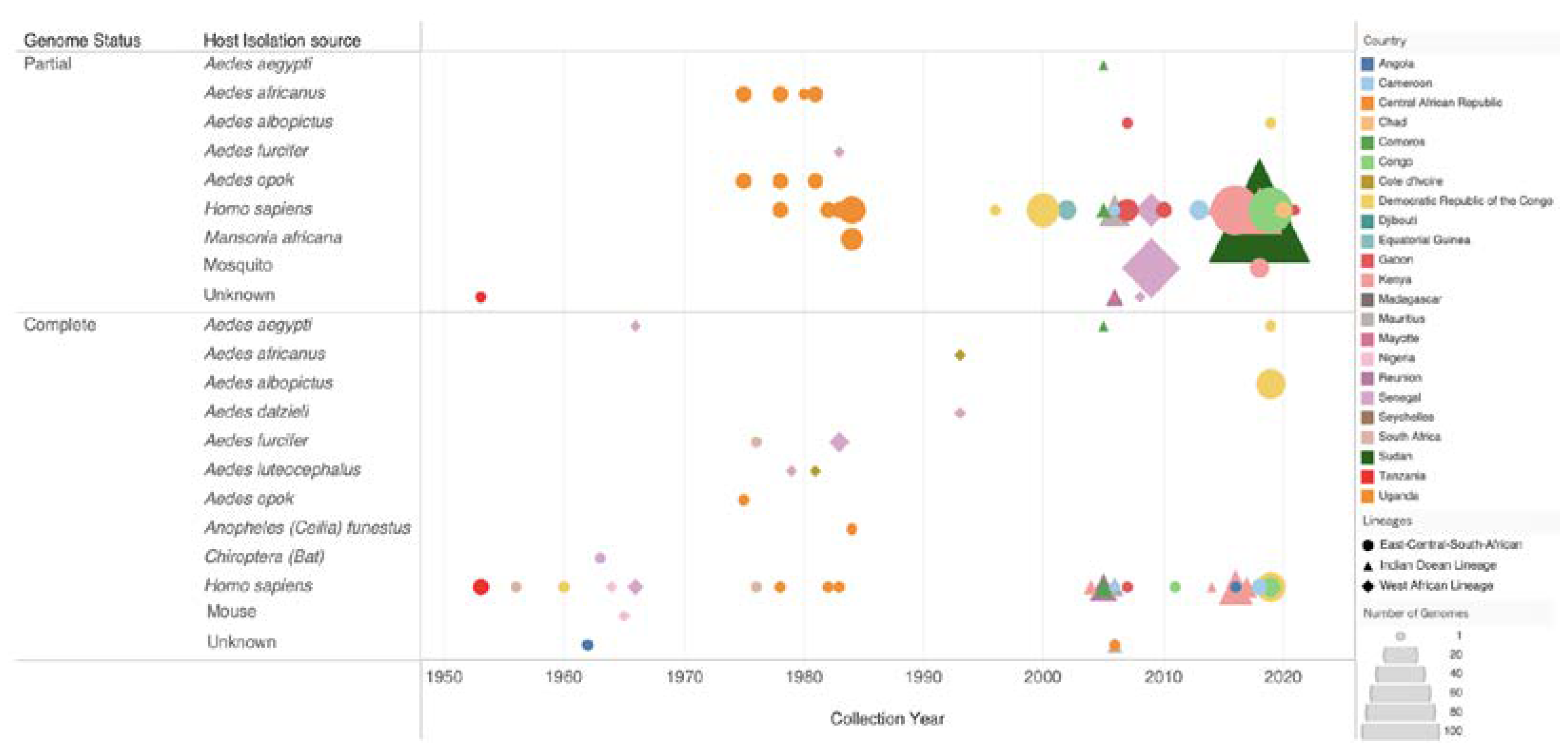
Figure 4.
Maximum likelihood time-scaled tree illustrating the evolutionary relationships among the different circulating lineages in Africa, providing insights into the temporal aspects of CHIKV evolution and dispersal. a) ECSA Genotype, this genotype is depicted as showing two geographically distinct clades. b) WA Genotype circulates within the West African region. c) Indian Ocean Lineage–showing recent outbreaks in Africa and the initial outbreaks on Indian Ocean islands, aggregated by continent.
Figure 4.
Maximum likelihood time-scaled tree illustrating the evolutionary relationships among the different circulating lineages in Africa, providing insights into the temporal aspects of CHIKV evolution and dispersal. a) ECSA Genotype, this genotype is depicted as showing two geographically distinct clades. b) WA Genotype circulates within the West African region. c) Indian Ocean Lineage–showing recent outbreaks in Africa and the initial outbreaks on Indian Ocean islands, aggregated by continent.
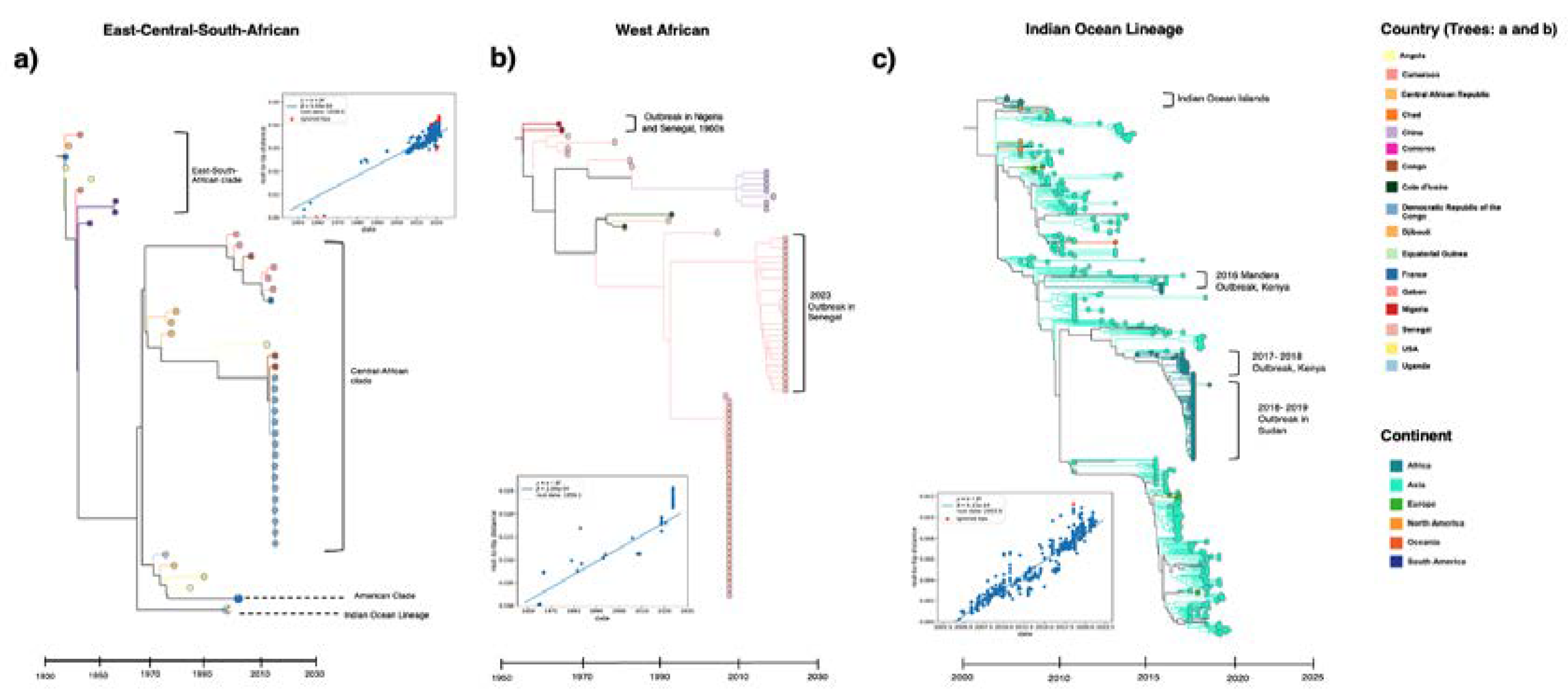
Figure 5.
Transmission dynamics of CHIKV lineages worldwide and in Africa. World map illustrating the temporal and spatial CHIKV dissemination patterns in, out of, and within Africa, a) The ECSA genotype, and b) IOL lineage.
Figure 5.
Transmission dynamics of CHIKV lineages worldwide and in Africa. World map illustrating the temporal and spatial CHIKV dissemination patterns in, out of, and within Africa, a) The ECSA genotype, and b) IOL lineage.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



