
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
Atopic dermatitis and psoriasis are prevalent inflammatory skin conditions with diverse treatment options. Despite advances in understanding their underlying mechanisms, recent research highlights the significance of interleukins IL-18 and IL-37 as potential therapeutic targets. This review synthesizes knowledge on these interleukins, their roles in atopic dermatitis and psoriasis, and emerging treatment strategies. Findings of a literature search up to May 30, 2024 underscore a research gap in IL-37-targeted therapies. Conversely, IL-18-focused treatments have demonstrated promise in adult-onset Still’s Disease, warranting further exploration for their potential efficacy in psoriasis and atopic dermatitis.
Keywords:
psoriasis
; atopic dermatitis
; IL-18
; IL-37
1. Introduction
Interleukin (IL)-18 and IL-37 are constituents of the IL-1 cytokine family, which is divided into three subgroups: IL-1 (including IL-1α, IL-1β, IL-33, and IL-1Rα), IL-18 (comprising IL-18 and IL-37), and IL-36 (composed of IL-36α, IL-36β, IL-36γ, IL-36Rα, and IL-38) [1,2,3]. IL-18, known for its inflammatory properties, is notably abundant in epithelial barrier cells of the skin, upper airway, and gastrointestinal tract [4,5,6,7]. Recent research has underscored its emerging role in pathogenesis and clinical contexts [8]. Conversely, IL-37, a more recently identified cytokine, possesses anti-inflammatory properties [9]. Its synthesis is stimulated by pro-inflammatory agents, acting as a safeguard against excessive inflammation and subsequent tissue damage, thereby modulating both innate and adaptive immune responses [10]. It is noteworthy that, unlike other IL-1 family members, there is no homologous gene for IL-37 in mice, necessitating the use of transgenic mouse models for its study [9]. Both IL-18 and IL-37 have shown abnormal expression levels in various inflammatory and autoimmune disorders, including skin diseases such as atopic dermatitis (AD) and psoriasis.
2. Material and Methods
An electronic literature search was conducted on the Medline/PubMed database up to May 2024, using Medical Subject Headings (MeSH) terms and relevant medical terminology. The search criteria included the terms: 'IL-18', 'IL-37', 'psoriasis', and 'atopic dermatitis'. We considered original studies, reviews, systematic reviews, and meta-analyses that specifically investigated the role of IL-18 and IL-37 in AD and psoriasis. Manuscripts written in English were eligible for inclusion, while letters to the editor, editorials, expert opinions, conference proceedings, and studies exclusively involving patients with psoriatic arthritis were excluded. The selection of publications was performed by two independent researchers (LR, LP), and any disagreements were resolved through consensus.
3. Atopic Dermatitis and Psoriasis Pathogenesis
AD is one of the most prevalent chronic cutaneous immune-mediated inflammatory disease, affecting approximately 10% of adults and 20% of children globally [11]. Its pathogenesis is multifaceted, involving genetic susceptibility, epidermal dysfunction, and T-cell-mediated inflammation [12,13,14,15,16]. Both innate and adaptive immune responses play significant roles in AD etiology [12,14]. Traditionally viewed as primarily driven by T-helper 2 (Th2) responses, recent research reveals a more complex immunological landscape, involving activation of Th2, Th22, Th17, and Th1 subsets [12,13,14]. Notably, a dynamic transition from Th2 to Th1 responses occurs in both acute and chronic AD lesions. Key cytokines such as interleukin-4 (IL-4) and interleukin-13 (IL-13) are central to AD pathophysiology, promoting Th2 cell differentiation and IgE production [17,18]. Elevated levels of Th2-related cytokines exacerbate inflammation in acute AD lesions and contribute to epidermal barrier dysfunction by downregulating terminal differentiation proteins [17]. Understanding the intricate interplay between cytokine dysregulation, immune activation, and barrier dysfunction is crucial for elucidating AD mechanisms and guiding therapeutic strategies to improve patient outcomes.
Psoriasis, a chronic immune-mediated inflammatory skin disorder, has a prevalence ranging from 1% to 3% in Western populations. The pathogenesis of psoriasis is characterized by a complex interplay between keratinocytes and the innate and adaptive immune systems [19,20,21]. Various hypotheses have been proposed to elucidate the underlying mechanisms driving psoriasis pathogenesis [20,22,23]. The prevailing theory posits an initial triggering event followed by a sustained chronic inflammatory phase orchestrated by a positive feedback loop mediated by cytokine-induced keratinocyte activation and proliferation [24,25]. In genetically predisposed individuals, exposure to triggers such as trauma, infection, or certain medications precipitates keratinocyte apoptosis or necrosis, leading to the release of nucleic acids (DNA or RNA). These nucleic acids can activate a type I interferon-mediated autoinflammatory response, along with potential autoantigens including cathelicidin (LL37), ADAMTSL-5, and phospholipase A2 (PLA2G4D). LL37 interacts with self-DNA or -RNA to trigger innate immune responses via Toll-like receptors (TLR), notably TLR7 and TLR8 [26,27]. Activation of these receptors induces the production of interferon (IFN)-α and IFN-β by keratinocytes and plasmacytoid dendritic cells (pDCs), as well as interleukin (IL)-6 and tumor necrosis factor (TNF) by myeloid dendritic cells (mDCs).IL-6 promotes the differentiation of CD4+ naïve T-cells into T helper (Th)-17 cells [28,29], while type I IFNs and TNF facilitate the secretion of IL-12 and IL-23 by mDCs [30,31]. IL-12 and IL-23 play pivotal roles in orchestrating the immune cascade underlying psoriasis pathogenesis. IL-12 drives the differentiation of CD4+ naïve T-cells into Th1 cells in conjunction with tumor growth factor (TGF)-β and IL-6, while IL-23 promotes Th17 cell development and activates αβ T cells [32,33,34,35,36]. Th1 and Th17 activated cells exhibit distinct cytokine secretion profiles, with Th1 cells producing IFN-γ and TNF, and Th17 cells releasing IL-17, IL-22, and TNF [37]. These cytokines collectively induce keratinocyte proliferation, differentiation, and inflammatory activation, with IL-17A serving as the principal effector cytokine driving psoriasis pathogenesis and being essential for the development and maintenance of psoriatic plaques [37]. Additionally, stimulated keratinocytes produce inflammatory cytokines such as IL-1, IL-6, and TNF, as well as chemokines including CXCL1, CXCL2, and CXCL3, and antimicrobial peptides (AMPs) such as S100A7/8/9, human β-defensin 2, and LL-37 [35,38,39]. These mediators attract and activate immune cells, exacerbating psoriatic inflammation.
4. Biology of IL-18
As constituents of the IL-1 family, IL-18 and IL-37 are synthesized and initially present in the cytoplasm in an inactive precursor form, awaiting subsequent activation through structural modifications [8]. The IL-18 gene is located on chromosome 11 [40]. Various inflammatory stimuli, such as lipopolysaccharide (LPS), interferons (IFNs), and IL-22, prompt the transcription and translation of the IL-18 gene [41,42,43]. This results in the production of pro-IL-18, which is subsequently released into the cytoplasm, where it undergoes activation. Eventually, IL-18 is transported to the extracellular space through cellular pores [44].
The process of IL-18 activation is intricate and remains incompletely understood. Pro-IL-18, which lacks a well-defined biological function, undergoes cleavage mediated by inflammasome-mediated activation of caspase-1, a process also involved in the activation of IL-1β [45,46,47]. Notably, several inflammasomes, including NLRP3, pyrin, NLRC4, NLRP6, and NLRP9B, play roles in IL-18 activation [48,49,50]. Genetic associations have been established between systemic IL-18 levels and NLRC4 activity [49]. Additionally, non-inflammasome proteases can activate IL-18; for instance, Fas ligand stimulates macrophages, leading to the release of active IL-18, while granzyme-B from cytotoxic granules cleaves keratinocyte-derived pro-IL-18 [51,52]. Furthermore, caspase-1 can cleave gasdermin-D, creating additional pores through which IL-18 can exit [53,54,55]. Among its diverse functions, IL-18 mediates IFN-γ production [56].
Extracellular IL-18 exhibits a half-life of approximately 16 hours [57]. Its activity is counteracted by the presence of the IL-18 binding protein (IL-18BP), which has a high affinity for IL-18 and solely functions to inhibit IL-18 activity [58]. In healthy individuals, over 95% of circulating IL-18 is bound to soluble IL-18BP [58,59]. Given the strength of this interaction, it is unlikely that IL-18BP serves as a reservoir for cleaved IL-18 [8,60]. Another cytokine that interacts with extracellular IL-18, thereby dampening its inflammatory effects, is IL-37 [8,60]. Notably, IFN-γ serves as a potent inducer of IL-18BP, leading to an indirect negative-feedback loop involving IL-18, IFN-γ, and IL-18BP [61,62]. Furthermore, immune evasion and active suppression by neoplasms and various viruses, including poxviruses, Chikungunya, and hepatitis C, involve the induction of IL-18BP expression [58,63,64,65].
IL-18 signaling is facilitated through the IL-18 receptor (IL-18R), composed of two chains referred to as α and β, encoded by the IL18R1 and IL18RAP genes, respectively [66]. Initially, IL-18 binds to the α chain with modest affinity, followed by the recruitment of the β chain, leading to the assembly of a high-affinity receptor capable of initiating signaling cascades [67,68,69,70,71] (Figure 1). The cytosolic region of the IL-18R includes a Toll/IL-1 receptor (TIR) domain, where myeloid differentiation primary response 88 (Myd88) binds, leading to the recruitment and phosphorylation of IL-1 receptor-associated kinases (IRAKs) [72,73,74]. Notably, IRAK1 and IRAK4 have been implicated in IL-18 signaling in Th1 and NK cells. These IRAKs further associate with the adaptor molecule TNF receptor-associated factor 6 (TRAF6), which in turn binds to the kinase TAK1 (transforming growth factor-b-activated kinase 1) [44]. TAK1 phosphorylates nuclear factor kappa B (NF-κB)-induced kinase (NIK), leading to NIK activation. Subsequently, NIK activates the inhibitor of nuclear kappa B (IkB) kinase (IKK), which phosphorylates IkB, resulting in IkB ubiquitination and rapid degradation. The freed transcription factor NF-κB translocates into the nucleus, where it binds to specific regulatory sequences (κB sites) in the promoter regions of various inflammatory genes (e.g., IFN-γ, IL-8, IL-1β, TNF-α) [75,76,77,78,79]. Moreover, TRAF6 phosphorylation also associates with apoptosis signal-regulating kinase 1 (ASK1) through TAK1, activating mitogen-activated protein kinases (MAPK)-related signaling downstream, including p38 mitogen-activated protein kinase (p38MAPK), Jun kinase (JNK), phosphoinositide 3-kinase (PI3K)/AKT, and extracellular regulated protein kinases (ERK), ultimately leading to the activation of the AP-1 transcription factor [78,80] (Figure 2).
The expression of genes encoding the α and β chains of IL-18R is predominantly confined to immune cells, particularly natural killer (NK) cells, NK-like innate lymphoid cells (ILCs), and activated CD4+ and CD8+ T cells [8]. While other immune cell types such as B cells, γδ-T cells, basophils, dendritic cells (DCs), synovial macrophages, mast cells, neutrophils, and skin-resident type 2 innate lymphoid (ILC2) cells also express IL18R1 and IL18RAP, their functional significance remains unclear [8,81,82,83,84,85,86,87].
According to Hu et al., naïve T cells exhibit minimal expression of IL-18R genes initially. However, following antigen presentation or stimulation with inflammatory cytokines such as IL-2, IL-12, and IL-15, the expression of IL18R1 and IL18RAP is induced [88]. Additionally, TCR signaling is essential for IL-18 to interact with CD8+ T cells, indicating that the effects of IL-18 on these cells are context-dependent [89]. Conversely, NK cells, NK T cells, and NK-like ILCs constitutively express both IL18R1 and IL18RAP genes, highlighting the significant role of those cells in contributing to the alarmin function of IL-18.
IL-18 signaling has numerous functions, primarily supporting the initiation and maintenance of cellular states associated with IFN-γ production [8]. IL-18BP inhibits the biological activity of IL-18, thereby reducing IFN-γ production and dampening Th1 immune responses. An imbalance in the IL-18/IL-18BP ratio may contribute to the perpetuation of chronic inflammation driven by T helper type I cytokines [44,90,91].
IL-18 has contrasting effects on the balance between Th1 and Th2 activation. When in conjunction with IL-12 or IL-15, IL-18 promotes the differentiation of naive T cells into Th1 cells. However, IL-18 increases IgE levels and the production of IL-4 and IL-13 by mast cells, basophils, NK cells, and CD4+ T cells, thereby promoting a Th2 response [92,93,94]. IL-18, along with IL-2, enhances IL-13 production in NK cells and T cells [95]. Furthermore, IL-18 synergizes with IL-13 to trigger histamine release from basophils. IL-18 also enhances the expression of Fas Ligand on NK cells, promoting Fas Ligand-mediated NK cytotoxicity [96].
IL-18 can stimulate allergic inflammation by increasing the production of key factors contributing to atopic inflammation through mast cells and basophils [97]. This evidence underscores the pleiotropic nature of IL-18, playing a pivotal role in fostering immune responses and inflammation, thereby serving as a crucial link between innate and adaptive immunity.
4.1. IL-18 in Keratinocytes
Keratinocytes constitute approximately 95% of the epidermal layer and, beyond their structural role, significantly contribute to skin inflammation and immune responses [44]. Compared to monocytes, peripheral blood mononuclear cells (PBMCs), or leukocytes, keratinocytes synthesize substantial amounts of pro-IL-18 [98]. Cho et al. demonstrated the induction of IL-18 production in the human keratinocyte cell line HaCaT following ultraviolet B (UVB) irradiation [99]. This induction occurred in a time- and dose-dependent manner, facilitated by the generation of reactive oxygen intermediates (ROIs) and the activation of activator protein-1 (AP-1). Notably, keratinocytes express the IL-18 receptor (IL-18R), suggesting that IL-18 may exert its effects in an autocrine or paracrine fashion on neighboring keratinocytes [100]. According to Fenini et al., NRLP1 inflammasomes play a crucial role in sensing UVB radiation and subsequently facilitating the secretion of IL-1β and IL-18 from keratinocytes [101].
IFN-γ induces the expression of chemokines such as CXCL9 (chemokine C-X-C motif ligand 9), CXCL10, and CXCL11, which recruit Th1 cells by binding to CXCR3 (chemokine C-X-C motif receptor 3) on the cell surface, resulting in significant infiltration of Th1 cells observed in inflammatory skin conditions [102]. In keratinocytes, IL-18 amplifies the mRNA expression of CXCL9, CXCL10, and CXCL11 induced by IFN-γ [95]. Additionally, IFN-γ activates signal transducer and activator of transcription 1 (STAT1) via Janus kinase 1 (JAK1)/JAK2 and/or p38MAPK pathways, leading to the production of CXCL9, CXCL10, and CXCL11. IFN-γ may also influence the activation of interferon regulatory factor-1 (IRF-1), responsible for CXCL11 expression, primarily through the p38MAPK pathway, while IL-18 enhances this activation via PI3K/AKT and MEK/ERK pathways. IL-18 induces NF-kB activity through MEK/ERK and PI3K/AKT pathways, thereby augmenting IFN-γ -induced CXCL9 secretion [95].
Furthermore, IL-18 has been shown to possess antifibrotic effects on human dermal fibroblasts [103]. IL-18 suppresses the activation of the transcription factor Ets-1 through the ERK pathway, resulting in decreased collagen expression and inhibition of collagen production induced by transforming growth factor-beta (TGF-β).
4.2. IL-18 in Immune Cells
The skin, serving as the primary barrier of the body, is continually exposed to various external stimuli, leading to irritation of keratinocytes and subsequent release of IL-18. This cytokine not only interacts with neighboring keratinocytes but also reaches immune cells present in the skin, including DCs, Langerhans cells (LCs), and macrophages [44]. Upon stimulation by diverse triggers, LCs undergo activation into mature DCs. Specifically, IL-18 stimulation of LCs induces the accumulation of DCs and the migration of LCs to local lymph nodes.
Within the skin-draining lymph nodes, LCs present antigens to CD4+ T cells, thereby mediating the adaptive immune response. Although IL-18 alone does not induce Th1 cell differentiation, it promotes the expression of the interleukin-12 receptor β (IL-12Rβ), thereby facilitating IL-12-mediated Th1 cell differentiation [104,105]. Additionally, the combined action of IL-18 and IL-12 synergistically stimulates Th1 cells to produce IFN-γ.
IL-18 also interacts with neutrophils and PBMCs, eliciting the release of various inflammatory cytokines in neutrophils and stimulating the production of IL-8 in PBMCs [94]. Conversely, IL-18BP inhibits the IL-12-induced production of IFN-γ in PBMCs.
5. Biology of IL-37
IL-37, a member of the IL-1 family, was first identified in 2000 and its gene is located on chromosome 2q12-13 [106]. Unlike other cytokines within the IL-1 family, the murine homologue gene for IL-37 has yet to be discovered [9], necessitating the use of transgenic mice models for functional studies.
The IL-37 gene proximity to the regulatory regions of the IL-1α and IL-1β genes suggests a potential relationship with the anti-inflammatory role of IL-37 [106]. Functionally, IL-37 serves as an anti-inflammatory cytokine that responds to pro-inflammatory stimuli [10]. It plays a crucial role in mitigating excessive inflammation and preventing severe tissue damage, thereby regulating both the innate and adaptive immune responses. Elevated levels of IL-37 have been consistently observed in various inflammatory and autoimmune conditions [10], and significant attention has been directed towards its potential inhibitory effects on cancer initiation and progression [107].
Despite structural and production process similarities between IL-37 and other IL-1 family cytokines, particularly IL-18, notable differences exist. IL-37 is secreted in an inactive precursor form, activated by caspase-1-mediated cleavage [108]. Unlike IL-18 and other members of the IL-1 family, IL-37 is not constitutively expressed in healthy individuals’ cells; its expression is upregulated in response to pro-inflammatory stimuli [106]. Consequently, IL-37 is hypothesized to orchestrate a negative-feedback mechanism to regulate and mitigate excessive inflammation, maintaining immunological homeostasis.
IL-37 can act both intracellularly and extracellularly, though the specific conditions dictating its predominant mechanism of action remain unclear [109]. No specific IL-37 receptor has been identified [9], but IL-37 binds to the α chain of the IL-18 receptor (IL-18R), with about 50 times lower affinity than IL-18 [67] leading to the recruitment of the decoy receptor IL-1R8, encoded by SIGIRR. This extracellular mechanism blocks the IL-18 inflammatory pathway [110]. Additionally, IL-37 can bind to IL-18BP, forming a complex that binds to the β chain of IL-18R [9], preventing the formation of the functional (heterodimeric) IL-18 receptor (Figure 3).
Regarding intracellular anti-inflammatory effects, the absence of a nuclear localization sequence suggests that the transportation of IL-37 to the nucleus likely occurs through association with Smad3 [109]. IL-37 enhances the anti-inflammatory functions of Smad3 rather than directly engaging in DNA binding. Upon exposure to pro-inflammatory stimuli, the intracellular concentration of IL-37 precursor increases. Subsequently, activated caspase-1 cleaves the precursor, facilitating the binding of the C-terminal domain of IL-37 to Smad3 [109]. Following phosphorylation, this complex translocates to the nucleus, where it contributes to the regulation of gene expression [109], suppressing inflammatory pathways such as mammalian target of rapamycin (mTOR), mitogen-activated protein kinase (MAPK), NF-kB, and various transcription factors [111,112]. IL-37 also enhances the activation of STAT3, protein phosphatase and tensin homolog (PTEN), and 5’AMP-activated protein kinase (AMPK) [111].
Interestingly, at elevated concentrations, the inhibitory effect of IL-37 on inflammatory cytokines diminishes due to the formation of IL-37 homodimers, initiating an auto-regulatory mechanism that curtails its immunosuppressive effect [109].
Several stimuli have been identified as triggers for IL-37 expression or inhibition [113]. Toll-like receptor (TLR) ligands such as lipopolysaccharides (LPS) and Pam3CysSerLys4 (Pam3CSK4), as well as TGF-β1, induce IL-37 production. Conversely, Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and IL-4 inhibit its expression. IL-37 appears to be stored in monocytes for rapid release following stimulation by LPS, for example [114].
5.1. IL-37 in the Skin
While there remains a considerable need for further research on the role of IL-37 in skin biology, recent studies have provided new insights. IL-37 is predominantly expressed in mature and differentiated keratinocytes [115,116]. Multiple studies have reported decreased expression of IL-37 in the epidermis of patients with AD [115,117,118], although findings have been inconsistent [119]. Specifically, chronic AD skin lesions have shown the most significant reduction in IL-37 levels [115].
Zhou et al. established a positive association between the expression of IL-37 and that of filaggrin (FLG), FLG2, and involucrin (IVL) [115]. Additionally, using an in vitro 3D human skin model (EpidermFT), they demonstrated that IL-4, IL-13, and IL-31 can decrease IL-37 levels. Conversely, IL-37 increased the expression of FLG and FLG2 but had no impact on IVL. In a separate study, Hou et al. observed a reduction in epidermal thickness and scratching frequency in IL-37-transgenic mice [117].
According to Tsuji et al. [120], IL-33 plays a well-established role in the pathogenesis of AD by inducing a type 2 immune response, which directly stimulates nerves, leading to pruritus. IL-37 was found to inhibit the expression of keratinocyte-derived IL-33, possibly through the inhibition of MAPK and the prevention of STAT1 activation in keratinocytes [120,121]. Therefore, it is reasonable to hypothesize that IL-37 may modulate epidermal barrier function [115]. However, a significant proportion of patients with AD harbor an inherent mutation in the FLG gene, suggesting that the administration of IL-37 alone may be insufficient for these patients [122].
5.2. IL-37 in Immune Cells
IL-37 levels are decreased in conditions such as AD, asthma, and allergic rhinitis [117,123,124,125]. In allergic patients, reduced IL-37 levels may lead to ineffective immune suppression and dysregulated responses to allergen stimulation [117].
The Th2 immune response appears to inhibit IL-37 production [126], while IL-37 itself can influence Th2 cells [127]. STAT6 activation, facilitated by IL-4, promotes the differentiation of CD4+ T cells into Th2 cells by upregulating GATA3. In animals treated with IL-37, both STAT6 phosphorylation and IL-4 levels were decreased, indicating that IL-37 potentially inhibits GATA3 expression by disrupting the IL-4/STAT6 signaling pathway. This inhibitory effect consequently reduces the proliferation, differentiation, and activation of Th2 cells, as observed in the IL-37-transgenic mouse model of allergic rhinitis [127]. Additionally, in vivo studies have shown that IL-37 can inhibit the function of not only Th2 cells but also Th1 and Th17 cells through peripheral blood mononuclear cells (PBMCs), M1 macrophages, and dendritic cells (DCs) [128]. Human DCs when cultured with CD4+ T cells and treated with IL-37 obtain a tolerogenic phenotype and promote Tregs expansion and suppression of Th1 and Th17 populations [129].
Th2-secreted cytokines (IL-4 and GM-CSF) suppress IL-37 in CD4+ T cells, while Th1-secreted cytokines (IFN-γ and IL-17) induce IL-37 expression in these cells [130].
IL-37 also mediates Treg cell activity, contributing to their production of TGF-β1 and/or IL-10 [10,131,132,133]. IL-37 expression level is consistently low in freshly isolated human CD4+CD25+ Tregs that are not stimulated [10]. Silencing IL-37 in these Tregs significantly reduces their suppressive capability [132]. Stimulation with IL-37 enhances the suppressive function of CD4+CD25+ Tregs by upregulating CTLA-4 and FOXP3 and increasing TGF- β1 levels, although it does not IL-10 levels [131].
IL-2 enhances the proliferative responses of effector T cells (Teff) and NK cells while maintaining immune homeostasis by promoting the proliferation, differentiation, and function of regulatory Tregs. Wang et al. studied the effect of IL-37 and IL-2 in mice, comparing groups pre-treated with recombinant IL-37 to non-pre-treated groups. No disparity in IL-2 levels has been found between IL-37-stimulated and control groups [131], although reduced IL-37 levels contribute to increased IL-2 secretion and exacerbated inflammation [132].
IL-37 also interacts with macrophages, inhibiting Notch1 and NF-kB activation and thereby attenuating M1 (proinflammatory) polarization; furthermore, recombinant IL-37 enhance M2 macrophages and their secretion of anti-inflammatory cytokines [134].
The expression of IL-37 in dendritic cells prevents their maturation via the IL-1R8–TLR4–NF-kB pathway and function, resulting in semi-mature tolerogenic dendritic cells that inhibit Teff activation and promote Treg development [135,136].
Eosinophils are involved in AD pathogenesis and contribute to the transition from a Th2-like immune response seen in acute AD lesions to a Th1-like immune response seen in chronic AD lesions [137]. IL-37 has shown efficacy in reducing eosinophilic inflammation [127,131,138].
Basophils, activated and recruited by inflammatory mediators like thymic stromal lymphopoietin (TSLP) produced by epithelial cells, release IL-4, promoting the differentiation of naïve T cells into Th2 cells and contributing to pruritus in AD [138,139,140]. IL-37b suppresses the expression of the TSLP receptor on basophils [138].
Mast cells play a significant role in AD pathogenesis by suppressing production of IL-12 in dendritic cells, prompting a shift in T cell response towards a Th2 profile [137]. IL-33, a potent activator of mast cells, promotes their migration, maturation, adhesion, survival, and secretion of inflammatory cytokines [141]. IL-33-induced mast cell inflammation is mediated through the NF-kB and p38MAPK pathways [142]. IL-37, in conjunction with Smad3, inhibits NF-kB activation and p38MAPK phosphorylation, attenuating mast cell inflammation in AD [142].
In the presence of IL-4 and IL-13, B cells undergo immunoglobulin class-switch recombination, facilitated by IL-4Rα and STAT6 [143]. This event prompts a transition from producing IgM to IgE antibodies. In an experimental murine model of allergic rhinitis, IL-37 administration led to decreased concentrations of IL-4 and IL-13 in serum and nasal lavage fluid, resulting in lower IgE levels [144]. However, several studies have found no statistically significant correlation between IL-37 and IgE levels in AD [117,126].
6. IL-18 and IL-37 in Inflammatory Skin Diseases
Despite there being much ground to cover, accumulating evidence highlights the involvement of IL-18 and IL-37 in the pathogenesis of inflammatory diseases such as AD and psoriasis.
Elevated levels of IL-18 have been detected in both the serum and lesional skin of individuals with psoriasis compared to healthy controls [145]. These heightened levels correlate with the Psoriasis Area and Severity Index (PASI) values, suggesting IL-18 as a potential biomarker for psoriasis [146,147,148]. A 2021 study by Verma et al. demonstrated increased plasma levels of IL-18 and IL-1β in psoriasis patients, which normalized with anti-TNF therapies [149]. In a mouse model of imiquimod-induced psoriasis, IL-18 exacerbated inflammation and contributed to micro-abscess formation of scale development by upregulating pro-inflammatory cytokines and reducing anti-inflammatory cytokines [150].
A recent investigation discovered that the combination of recombinant mouse (rm) IL-18 with rmIL-23 exacerbates inflammation, upregulates IFN-γ and CXCL9, and enhances psoriasis-like epidermal hyperplasia [151]. This suggests a potential cooperation between IL-18 and IL-23 in triggering a Th1 immune response, thereby worsening psoriatic inflammation. Additionally, IL-18 promotes the generation and persistence of Th17 cells [152]. Zhang et al. demonstrated that an IL-18-neutralizing antibody could impede the Th17 immune response in a psoriasis-like mouse model, indicating that the IL-18-mediated T cell response plays a crucial role in the pathogenesis of psoriasis and that inhibiting IL-18 could be a promising therapeutic strategy [152,153].
IL-18 levels are also elevated in the serum and stratum corneum of AD patients compared to healthy individuals [154,155,156,157]. These stratum corneum IL-18 levels strongly associate with barrier function alterations and AD severity [158]. In a transgenic mice study IL-18 was essential for developing AD-like dermatitis [159]. Chen et al. observed that IL-18 knockout mice exhibited lower inflammatory cell infiltration in AD-like lesions induced by MC903 compared to wild-type mice, although IgE levels were upregulated in the knockouts [160]. Additionally, IL-18 activates mast cells and basophils, inducing the expression of IL-4, IL-13, and histamine [160,161]. These data highlight the critical role of IL-18 in AD pathogenesis and its potential as a therapeutic target.
IL-37, in contrast, plays an important role reducing the inflammatory reaction in contact hypersensitivity. Luo et al. showed that increased IL-37 expression diminished auricular swelling and mitigated contact hypersensitivity in mice [162]. IL-37 expression in dendritic cells inhibits their antigen presentation and maturation ability while promoting regulatory T cells, thus suppressing contact hypersensitivity. In a murine model of AD induced by MC903, human IL-37b expression suppressed auricular swelling, pruritus, and the production of inflammatory cytokines and chemokines during AD progression [133,138]. Subcutaneous injections of human IL-37b reduced basophil infiltration and suppressed IL-4 production by basophils following TSLP stimulation, suggesting that IL-37 might improve AD through basophil modulation [138].
IL-37 serum levels in AD patients have shown controversial results. Fujita et al. reported elevated IL-37 levels in AD patients that correlated with AD severity [119]. However, some patients with severe AD had reduced IL-37 levels, suggesting different AD phenotypes regarding IL-37 synthesis [119]. IL-37 levels in the skin were reduced in AD lesions compared to non-lesional skin [115,117]. Immunohistochemical analysis in healthy individuals identified IL-37 expression in the granular layer of the epidermis, along with loricrin [163] and filaggrin [115]. In AD patients, reduced IL-37 expression correlated with decreased filaggrin expression [115]. IL-33, a major cytokine AD pathogenesis, may reduce IL-37 through two pathways: a) downregulating loricrin and filaggrin expression either directly or through IL-33-induced IL-4 and IL-13 downregulation, and subsequently downregulating IL-37 [164,165,166], or b) stimulating keratinocyte production of chemokines such as CXCL1 and CXCL8, promoting neutrophil infiltration, and CCL20, facilitating Th17 cell recruitment [167]. IL-17, whose release and production by neutrophils and Th17 cells is induced by IL-33 [120], would reduce filaggrin and loricrin expression and lead to IL-37 downregulation [164] (Figure 4).
IL-37 has also been investigated in psoriasis. In a mouse model, administering a plasmid containing human IL-37 improved skin lesions and decreased TNF expression [168]. Teng et al. reported that human IL-37 can inhibit the production of CXCL8, IL-6, and S100A7, all implicated in psoriasis pathogenesis. However, intradermal injection of IL-37 in a mouse model with imiquimod-induced psoriasis showed a protective effect without statistical significance, and lower levels of IL-37 in human psoriasis lesions compared to healthy skin [169]. Key cytokines involved in the pathogenesis of psoriasis, including IL-17A, IL-17C, IL-17F, and IL-22, may contribute to this phenomenon [169]. In randomized phase 2 trial, cutaneous levels of IL-37 in psoriasis lesions rapidly increased following treatment with tofacitinib [170].
6.1. IL-18 and IL-37 as Therapeutic Targets
Given their involvement in various inflammatory pathways, IL-18 and IL-37 have emerged as promising therapeutic targets. Multiple drugs targeting IL-18 are currently in development, though no specific drugs targeting IL-37 are under investigation at this time.
6.2. IL-18 as a Therapeutic Target
As IL-18 plays a pro-inflammatory role, drug development for inflammatory diseases typically focuses on inhibiting IL-18 [8]. Interestingly, inducers of IL-18 have also been studied as potential treatments for oncological conditions.
Currently, up to six different molecules targeting IL-18 are under development. Among these, three –Tadekinig alfa, CMK-389 and AMP18P1RA/IL-18bp-Fc-IL-1ra– are being specifically studied for dermatological conditions [44].
6.3. Tadekinig Alfa
Tadekinig alfa is a recombinant human IL-18 binding protein (IL-18BP) that inhibits IL-18. It is being studied for several conditions, including Adult-Onset Still’s Disease, lymphoproliferative disorders, immune system diseases, and macrophage activation syndrome [44]. In 2006, a phase 2 trial evaluated Tadekinig alfa in healthy volunteers and patients with psoriasis or rheumatoid arthritis. Unfortunately, it failed to demonstrate any efficacy in blocking IL-18 for these conditions [171].
6.4. CMK-389
CMK-389, an IL-18 inhibitor, was evaluated in a phase 2 randomized, placebo-controlled trial (NCT04836858) involving 71 patients with moderate to severe AD. Patients received either intravenous CMK389 10mg/kg (n=34), or subcutaneous CMK389 300mg (n=17), or placebo (intravenous, n=8, or subcutaneous, n=8) every four weeks for a 12-week period. The endpoint was assessed at week 16 and an observational period followed, lasting approximately 12 weeks. The primary outcome, defined by the Investigator Global Assessment (IGA) response (clear or almost clear skin with at least a 2 point-reduction from baseline at week 16), was achieved by 14.7% of the intravenous CMK-389 group and 11.8% of the subcutaneous group, compared to none in the placebo groups. Serious adverse events, including AD worsening and heavy menstrual bleeding, were reported but did not lead to study discontinuation. Common adverse events included nasopharyngitis, COVID-19 infection, headache, and diarrhea.
6.5. AMP18P1RA/IL-18bp-Fc-IL-1ra
AMP18P1RA/IL-18bp-Fc-IL-1ra is a fusion protein consisting of IL-18BP and IL-1 receptor antagonist (IL-1ra) domains linked to the Fc fragment of human IgG1 [172]. The amino-terminal IL-18BP domain binds to and inhibits the activity of IL-18, whereas the carboxy-terminal IL-1ra binds to the IL-1 receptor (IL-1R) and antagonizes the effects of IL-1α and IL-1β, thus preventing their role in immune activation [173]. Currently, AMP18P1RA/IL-18bp-Fc-IL-1ra is in preclinical phases.
6.6. IL-37 as a Therapeutic Target
While no specific drugs targeting IL-37 are in development, the role of IL-37 in reducing inflammatory reactions in contact hypersensitivity and other conditions suggests potential therapeutic applications. In murine models, IL-37 has shown promise in suppressing inflammatory responses, suggesting the potential benefit of its modulation in treating inflammatory skin diseases.
7. Conclusions
This review highlights the complex involvement of IL-18 and IL-37 in the immune innate and adaptative systems and their roles in AD and psoriasis. While significant advances have been made in understanding the pathogenesis of these diseases, particularly concerning IL-18, much remains unknown about IL-37. Despite the disappointing results of the first clinical trial evaluating Tadekinig alfa for psoriasis, a phase 2 trial of CMK-389 showed promising outcomes. Additionally, AMP18P1RA/IL-18bp-Fc-IL-1ra, with its unique mechanism of action, may also represent a promising future therapy.
8. Future Directions
Further research is needed to explore the therapeutic potential of targeting IL-18 and IL-37 in inflammatory skin diseases, with ongoing and future studies likely to provide deeper insights and new treatment avenues.
Author Contributions
All authors declare to have contributed to the conception and design of the article, the writing of the manuscript, the critical review of the intellectual content and the final approval of the version presented.
Conflicts of interest
L. Puig has perceived consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Abbvie, Almirall, Amgen, Biogen, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Fresenius-Kabi, Gebro, Janssen, JS BIOCAD, Leo-Pharma, Lilly, Novartis, Pfizer, Samsung-Bioepis, and UCB. L. Rusiñol declares no conflicts of interest for the related topic.
References
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The Family of the Interleukin-1 Receptors. Immunol Rev 2018, 281, 197–232. [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev 2018, 281, 8–27. [CrossRef]
- Matarazzo, L.; Hernandez Santana, Y.E.; Walsh, P.T.; Fallon, P.G. The IL-1 Cytokine Family as Custodians of Barrier Immunity. Cytokine 2022, 154, 155890. [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.D.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [CrossRef]
- Pechkovsky, D. V; Goldmann, T.; Vollmer, E.; Müller-Quernheim, J.; Zissel, G. Interleukin-18 Expression by Alveolar Epithelial Cells Type II in Tuberculosis and Sarcoidosis. FEMS Immunol Med Microbiol 2006, 46, 30–38. [CrossRef]
- Companjen, A.R.; van der Velden, V.H.; Vooys, A.; Debets, R.; Benner, R.; Prens, E.P. Human Keratinocytes Are Major Producers of IL-18: Predominant Expression of the Unprocessed Form. Eur Cytokine Netw 2000, 11, 383–390.
- Wittmann, M.; Macdonald, A.; Renne, J. IL-18 and Skin Inflammation. Autoimmun Rev 2009, 9, 45–48. [CrossRef]
- Landy, E.; Carol, H.; Ring, A.; Canna, S. Biological and Clinical Roles of IL-18 in Inflammatory Diseases. Nat Rev Rheumatol 2024, 20, 33–47. [CrossRef]
- Cavalli, G.; Dinarello, C.A. Suppression of Inflammation and Acquired Immunity by IL-37. Immunol Rev 2018, 281, 179–190. [CrossRef]
- Osborne, D.G.; Domenico, J.; Fujita, M. Expression of IL-37 Induces a Regulatory T-Cell-like Phenotype and Function in Jurkat Cells. Cells 2022, 11. [CrossRef]
- Carrascosa-Carrillo, J.M.; Aterido, A.; Li, T.; Guillén, Y.; Martinez, S.; Marsal, S.; Julià, A. Toward Precision Medicine in Atopic Dermatitis Using Molecular-Based Approaches. Actas Dermosifiliogr 2023. [CrossRef]
- Guo, Y.; Luo, L.; Zhu, J.; Li, C. Multi-Omics Research Strategies for Psoriasis and Atopic Dermatitis. Int J Mol Sci 2023, 24.
- Sroka-Tomaszewska, J.; Trzeciak, M. Molecular Mechanisms of Atopic Dermatitis Pathogenesis. Int J Mol Sci 2021, 22. [CrossRef]
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic Dermatitis: Pathophysiology. Adv Exp Med Biol 2017, 1027, 21–37. [CrossRef]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat Rev Dis Primers 2018, 4, 1. [CrossRef]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic Dermatitis. The Lancet 2020, 396, 345–360. [CrossRef]
- Oyoshi, M.K.; He, R.; Kumar, L.; Yoon, J.; Geha, R.S. Cellular and Molecular Mechanisms in Atopic Dermatitis. Adv Immunol 2009, 102, 135–226. [CrossRef]
- Çetinarslan, T.; Kümper, L.; Fölster-Holst, R. The Immunological and Structural Epidermal Barrier Dysfunction and Skin Microbiome in Atopic Dermatitis-an Update. Front Mol Biosci 2023, 10, 1159404. [CrossRef]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int J Mol Sci 2019, 20.
- Ni, X.; Lai, Y. Crosstalk between Keratinocytes and Immune Cells in Inflammatory Skin Diseases. Exploration of Immunology 2021, 418–431. [CrossRef]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of Psoriasis. Annu Rev Immunol 2014, 32, 227–255. [CrossRef]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [CrossRef]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N Engl J Med 2009, 361, 496–509. [CrossRef]
- Ben Abdallah, H.; Johansen, C.; Iversen, L. Key Signaling Pathways in Psoriasis: Recent Insights from Antipsoriatic Therapeutics. Psoriasis: Targets and Therapy 2021, Volume 11, 83–97. [CrossRef]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis Pathogenesis and the Development of Novel Targeted Immune Therapies. J Allergy Clin Immunol 2017, 140, 645–653. [CrossRef]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-Antimicrobial Peptide Complexes Activate Human Dendritic Cells through TLR7 and TLR8. J Exp Med 2009, 206, 1983–1994. [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.-H.; Homey, B.; Cao, W.; Wang, Y.-H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled with Antimicrobial Peptide. Nature 2007, 449, 564–569. [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol 2005, 6, 1133–1141. [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct from the T Helper Type 1 and 2 Lineages. Nat Immunol 2005, 6, 1123–1132. [CrossRef]
- Nakajima, A.; Matsuki, T.; Komine, M.; Asahina, A.; Horai, R.; Nakae, S.; Ishigame, H.; Kakuta, S.; Saijo, S.; Iwakura, Y. TNF, but Not IL-6 and IL-17, Is Crucial for the Development of T Cell-Independent Psoriasis-like Dermatitis in Il1rn-/- Mice. J Immunol 2010, 185, 1887–1893. [CrossRef]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The Antimicrobial Peptide LL37 Is a T-Cell Autoantigen in Psoriasis. Nat Commun 2014, 5, 5621. [CrossRef]
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J Biol Chem 2003, 278, 1910–1914. [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [CrossRef]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.; Wang, T.; Zheng, J.; et al. Pivotal Role of Dermal IL-17-Producing Γδ T Cells in Skin Inflammation. Immunity 2011, 35, 596–610. [CrossRef]
- Bielecki, P.; Riesenfeld, S.J.; Hütter, J.-C.; Torlai Triglia, E.; Kowalczyk, M.S.; Ricardo-Gonzalez, R.R.; Lian, M.; Amezcua Vesely, M.C.; Kroehling, L.; Xu, H.; et al. Skin-Resident Innate Lymphoid Cells Converge on a Pathogenic Effector State. Nature 2021, 592, 128–132. [CrossRef]
- Matos, T.R.; O’Malley, J.T.; Lowry, E.L.; Hamm, D.; Kirsch, I.R.; Robins, H.S.; Kupper, T.S.; Krueger, J.G.; Clark, R.A. Clinically Resolved Psoriatic Lesions Contain Psoriasis-Specific IL-17-Producing Aβ T Cell Clones. J Clin Invest 2017, 127, 4031–4041. [CrossRef]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin Rev Allergy Immunol 2018, 55, 379–390. [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J Invest Dermatol 2011, 131, 677–687. [CrossRef]
- Nolan, K.F.; Greaves, D.R.; Waldmann, H. The Human Interleukin 18 Gene IL18 Maps to 11q22.2-Q22.3, Closely Linked to the DRD2 Gene Locus and Distinct from Mapped IDDM Loci. Genomics 1998, 51, 161–163. [CrossRef]
- Chen, G.; Deutsch, G.H.; Schulert, G.S.; Zheng, H.; Jang, S.; Trapnell, B.; Lee, P.Y.; Macaubas, C.; Ho, K.; Schneider, C.; et al. Identification of Distinct Inflammatory Programs and Biomarkers in Systemic Juvenile Idiopathic Arthritis and Related Lung Disease by Serum Proteome Analysis. Arthritis Rheumatol 2022, 74, 1271–1283. [CrossRef]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoité, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.-Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [CrossRef]
- Heng, T.S.P.; Painter, M.W.; Immunological Genome Project Consortium The Immunological Genome Project: Networks of Gene Expression in Immune Cells. Nat Immunol 2008, 9, 1091–1094. [CrossRef]
- Wang, X.; Wang, L.; Wen, X.; Zhang, L.; Jiang, X.; He, G. Interleukin-18 and IL-18BP in Inflammatory Dermatological Diseases. Front Immunol 2023, 14, 955369. [CrossRef]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 Processes IFN-Gamma-Inducing Factor and Regulates LPS-Induced IFN-Gamma Production. Nature 1997, 386, 619–623. [CrossRef]
- Fantuzzi, G.; Puren, A.J.; Harding, M.W.; Livingston, D.J.; Dinarello, C.A. Interleukin-18 Regulation of Interferon Gamma Production and Cell Proliferation as Shown in Interleukin-1beta-Converting Enzyme (Caspase-1)-Deficient Mice. Blood 1998, 91, 2118–2125.
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of Interferon-Gamma Inducing Factor Mediated by Interleukin-1beta Converting Enzyme. Science 1997, 275, 206–209. [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.M.; Sanchez, G.A.M.; Kim, H.; et al. An Activating NLRC4 Inflammasome Mutation Causes Autoinflammation with Recurrent Macrophage Activation Syndrome. Nat Genet 2014, 46, 1140–1146. [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [CrossRef]
- Bossaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.K.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1β and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J Immunol 2012, 189, 5508–5512. [CrossRef]
- Akeda, T.; Yamanaka, K.; Tsuda, K.; Omoto, Y.; Gabazza, E.C.; Mizutani, H. CD8+ T Cell Granzyme B Activates Keratinocyte Endogenous IL-18. Arch Dermatol Res 2014, 306, 125–130. [CrossRef]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D Pore Acts as a Conduit for IL-1β Secretion in Mice. Eur J Immunol 2018, 48, 584–592. [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res 2015, 25, 1285–1298. [CrossRef]
- Tapia, V.S.; Daniels, M.J.D.; Palazón-Riquelme, P.; Dewhurst, M.; Luheshi, N.M.; Rivers-Auty, J.; Green, J.; Redondo-Castro, E.; Kaldis, P.; Lopez-Castejon, G.; et al. The Three Cytokines IL-1β, IL-18, and IL-1α Share Related but Distinct Secretory Routes. J Biol Chem 2019, 294, 8325–8335. [CrossRef]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 Regulates Both Th1 and Th2 Responses. Annu Rev Immunol 2001, 19, 423–474. [CrossRef]
- Hosohara, K.; Ueda, H.; Kashiwamura, S.-I.; Yano, T.; Ogura, T.; Marukawa, S.; Okamura, H. Interleukin-18 Induces Acute Biphasic Reduction in the Levels of Circulating Leukocytes in Mice. Clin Diagn Lab Immunol 2002, 9, 777–783. [CrossRef]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 Binding Protein: A Novel Modulator of the Th1 Cytokine Response. Immunity 1999, 10, 127–136. [CrossRef]
- Ha, C.T.; Li, X.; Fu, D.; Xiao, M. Circulating IL-18 Binding Protein (IL-18BP) and IL-18 as Dual Biomarkers of Total-Body Irradiation in Mice. Radiat Res 2016, 185, 375–383. [CrossRef]
- Bufler, P.; Azam, T.; Gamboni-Robertson, F.; Reznikov, L.L.; Kumar, S.; Dinarello, C.A.; Kim, S.-H. A Complex of the IL-1 Homologue IL-1F7b and IL-18-Binding Protein Reduces IL-18 Activity. Proc Natl Acad Sci U S A 2002, 99, 13723–13728. [CrossRef]
- Prencipe, G.; Bracaglia, C.; De Benedetti, F. Interleukin-18 in Pediatric Rheumatic Diseases. Curr Opin Rheumatol 2019, 31, 421–427. [CrossRef]
- Harel, M.; Girard-Guyonvarc’h, C.; Rodriguez, E.; Palmer, G.; Gabay, C. Production of IL-18 Binding Protein by Radiosensitive and Radioresistant Cells in CpG-Induced Macrophage Activation Syndrome. J Immunol 2020, 205, 1167–1175. [CrossRef]
- Kaser, A.; Novick, D.; Rubinstein, M.; Siegmund, B.; Enrich, B.; Koch, R.O.; Vogel, W.; Kim, S.H.; Dinarello, C.A.; Tilg, H. Interferon-Alpha Induces Interleukin-18 Binding Protein in Chronic Hepatitis C Patients. Clin Exp Immunol 2002, 129, 332–338. [CrossRef]
- Michels, M.; de Mast, Q.; Netea, M.G.; Joosten, L.A.; Dinarello, C.A.; Rudiman, P.I.F.; Sinarta, S.; Wisaksana, R.; Alisjahbana, B.; van der Ven, A.J.A.M. Normal Free Interleukin-18 (IL-18) Plasma Levels in Dengue Virus Infection and the Need to Measure Both Total IL-18 and IL-18 Binding Protein Levels. Clin Vaccine Immunol 2015, 22, 650–655. [CrossRef]
- Chirathaworn, C.; Poovorawan, Y.; Lertmaharit, S.; Wuttirattanakowit, N. Cytokine Levels in Patients with Chikungunya Virus Infection. Asian Pac J Trop Med 2013, 6, 631–634. [CrossRef]
- Hoshino, K.; Tsutsui, H.; Kawai, T.; Takeda, K.; Nakanishi, K.; Takeda, Y.; Akira, S. Cutting Edge: Generation of IL-18 Receptor-Deficient Mice: Evidence for IL-1 Receptor-Related Protein as an Essential IL-18 Binding Receptor. J Immunol 1999, 162, 5041–5044.
- Tsutsumi, N.; Kimura, T.; Arita, K.; Ariyoshi, M.; Ohnishi, H.; Yamamoto, T.; Zuo, X.; Maenaka, K.; Park, E.Y.; Kondo, N.; et al. The Structural Basis for Receptor Recognition of Human Interleukin-18. Nat Commun 2014, 5, 5340. [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int J Mol Sci 2019, 20. [CrossRef]
- Kim, S.H.; Reznikov, L.L.; Stuyt, R.J.; Selzman, C.H.; Fantuzzi, G.; Hoshino, T.; Young, H.A.; Dinarello, C.A. Functional Reconstitution and Regulation of IL-18 Activity by the IL-18R Beta Chain. J Immunol 2001, 166, 148–154. [CrossRef]
- Wu, C.; Sakorafas, P.; Miller, R.; McCarthy, D.; Scesney, S.; Dixon, R.; Ghayur, T. IL-18 Receptor Beta-Induced Changes in the Presentation of IL-18 Binding Sites Affect Ligand Binding and Signal Transduction. J Immunol 2003, 170, 5571–5577. [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front Immunol 2013, 4, 289. [CrossRef]
- Suzuki, N.; Chen, N.-J.; Millar, D.G.; Suzuki, S.; Horacek, T.; Hara, H.; Bouchard, D.; Nakanishi, K.; Penninger, J.M.; Ohashi, P.S.; et al. IL-1 Receptor-Associated Kinase 4 Is Essential for IL-18-Mediated NK and Th1 Cell Responses. J Immunol 2003, 170, 4031–4035. [CrossRef]
- Kanakaraj, P.; Ngo, K.; Wu, Y.; Angulo, A.; Ghazal, P.; Harris, C.A.; Siekierka, J.J.; Peterson, P.A.; Fung-Leung, W.P. Defective Interleukin (IL)-18-Mediated Natural Killer and T Helper Cell Type 1 Responses in IL-1 Receptor-Associated Kinase (IRAK)-Deficient Mice. J Exp Med 1999, 189, 1129–1138. [CrossRef]
- Carroll, H.P.; Paunovic, V.; Gadina, M. Signalling, Inflammation and Arthritis: Crossed Signals: The Role of Interleukin-15 and -18 in Autoimmunity. Rheumatology 2008, 47, 1269–1277. [CrossRef]
- Puren, A.J.; Fantuzzi, G.; Gu, Y.; Su, M.S.; Dinarello, C.A. Interleukin-18 (IFNgamma-Inducing Factor) Induces IL-8 and IL-1beta via TNFalpha Production from Non-CD14+ Human Blood Mononuclear Cells. J Clin Invest 1998, 101, 711–721. [CrossRef]
- Robinson, D.; Shibuya, K.; Mui, A.; Zonin, F.; Murphy, E.; Sana, T.; Hartley, S.B.; Menon, S.; Kastelein, R.; Bazan, F.; et al. IGIF Does Not Drive Th1 Development but Synergizes with IL-12 for Interferon-Gamma Production and Activates IRAK and NFkappaB. Immunity 1997, 7, 571–581. [CrossRef]
- Murphy, J.E.; Robert, C.; Kupper, T.S. Interleukin-1 and Cutaneous Inflammation: A Crucial Link between Innate and Acquired Immunity. J Invest Dermatol 2000, 114, 602–608. [CrossRef]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 Is a Unique Cytokine That Stimulates Both Th1 and Th2 Responses Depending on Its Cytokine Milieu. Cytokine Growth Factor Rev 2001, 12, 53–72. [CrossRef]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front Med (Lausanne) 2021, 8, 639103. [CrossRef]
- Virtue, A.; Wang, H.; Yang, X. MicroRNAs and Toll-like Receptor/Interleukin-1 Receptor Signaling. J Hematol Oncol 2012, 5, 66. [CrossRef]
- Ricardo-Gonzalez, R.R.; Van Dyken, S.J.; Schneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.-E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue Signals Imprint ILC2 Identity with Anticipatory Function. Nat Immunol 2018, 19, 1093–1099. [CrossRef]
- Leung, B.P.; Culshaw, S.; Gracie, J.A.; Hunter, D.; Canetti, C.A.; Campbell, C.; Cunha, F.; Liew, F.Y.; McInnes, I.B. A Role for IL-18 in Neutrophil Activation. J Immunol 2001, 167, 2879–2886. [CrossRef]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, Although Antiallergic When Administered with IL-12, Stimulates IL-4 and Histamine Release by Basophils. Proc Natl Acad Sci U S A 1999, 96, 13962–13966. [CrossRef]
- Gracie, J.A.; Forsey, R.J.; Chan, W.L.; Gilmour, A.; Leung, B.P.; Greer, M.R.; Kennedy, K.; Carter, R.; Wei, X.Q.; Xu, D.; et al. A Proinflammatory Role for IL-18 in Rheumatoid Arthritis. J Clin Invest 1999, 104, 1393–1401. [CrossRef]
- Tucci, M.; Quatraro, C.; Dammacco, F.; Silvestris, F. Increased IL-18 Production by Dendritic Cells in Active Inflammatory Myopathies. Ann N Y Acad Sci 2007, 1107, 184–192. [CrossRef]
- Gutzmer, R.; Langer, K.; Mommert, S.; Wittmann, M.; Kapp, A.; Werfel, T. Human Dendritic Cells Express the IL-18R and Are Chemoattracted to IL-18. J Immunol 2003, 171, 6363–6371. [CrossRef]
- Airoldi, I.; Gri, G.; Marshall, J.D.; Corcione, A.; Facchetti, P.; Guglielmino, R.; Trinchieri, G.; Pistoia, V. Expression and Function of IL-12 and IL-18 Receptors on Human Tonsillar B Cells. J Immunol 2000, 165, 6880–6888. [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep 2017, 20, 3025–3033. [CrossRef]
- Landy, E.; Varghese, J.; Dang, V.; Szymczak-Workman, A.; Kane, L.P.; Canna, S.W. Complementary HLH Susceptibility Factors Converge on CD8 T-Cell Hyperactivation. Blood Adv 2023, 7, 6949–6963. [CrossRef]
- Xu, D.; Trajkovic, V.; Hunter, D.; Leung, B.P.; Schulz, K.; Gracie, J.A.; McInnes, I.B.; Liew, F.Y. IL-18 Induces the Differentiation of Th1 or Th2 Cells Depending upon Cytokine Milieu and Genetic Background. Eur J Immunol 2000, 30, 3147–3156. [CrossRef]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444-456.e14. [CrossRef]
- Hoshino, T.; Yagita, H.; Ortaldo, J.R.; Wiltrout, R.H.; Young, H.A. In Vivo Administration of IL-18 Can Induce IgE Production through Th2 Cytokine Induction and up-Regulation of CD40 Ligand (CD154) Expression on CD4+ T Cells. Eur J Immunol 2000, 30, 1998–2006. [CrossRef]
- Son, Y.I.; Dallal, R.M.; Mailliard, R.B.; Egawa, S.; Jonak, Z.L.; Lotze, M.T. Interleukin-18 (IL-18) Synergizes with IL-2 to Enhance Cytotoxicity, Interferon-Gamma Production, and Expansion of Natural Killer Cells. Cancer Res 2001, 61, 884–888.
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-Regulates IL-18 Receptor Expression on T Cells, Th1 Cells, and B Cells: Synergism with IL-18 for IFN-Gamma Production. J Immunol 1998, 161, 3400–3407.
- Kanda, N.; Shimizu, T.; Tada, Y.; Watanabe, S. IL-18 Enhances IFN-Gamma-Induced Production of CXCL9, CXCL10, and CXCL11 in Human Keratinocytes. Eur J Immunol 2007, 37, 338–350. [CrossRef]
- Pourcet, B.; Gage, M.C.; León, T.E.; Waddington, K.E.; Pello, O.M.; Steffensen, K.R.; Castrillo, A.; Valledor, A.F.; Pineda-Torra, I. The Nuclear Receptor LXR Modulates Interleukin-18 Levels in Macrophages through Multiple Mechanisms. Sci Rep 2016, 6, 25481. [CrossRef]
- Wang, J.; Zhang, H.; Zheng, W.; Xie, H.; Yan, H.; Lin, X.; He, S. Correlation of IL-18 with Tryptase in Atopic Asthma and Induction of Mast Cell Accumulation by IL-18. Mediators Inflamm 2016, 2016, 4743176. [CrossRef]
- Companjen, A.R.; van der Velden, V.H.; Vooys, A.; Debets, R.; Benner, R.; Prens, E.P. Human Keratinocytes Are Major Producers of IL-18: Predominant Expression of the Unprocessed Form. Eur Cytokine Netw 2000, 11, 383–390.
- Cho, D.; Seung Kang, J.; Hoon Park, J.; Kim, Y.-I.; Hahm, E.; Lee, J.; Yang, Y.; Jeon, J.; Song, H.; Park, H.; et al. The Enhanced IL-18 Production by UVB Irradiation Requires ROI and AP-1 Signaling in Human Keratinocyte Cell Line (HaCaT). Biochem Biophys Res Commun 2002, 298, 289–295. [CrossRef]
- Koizumi, H.; Sato-Matsumura, K.C.; Nakamura, H.; Shida, K.; Kikkawa, S.; Matsumoto, M.; Toyoshima, K.; Seya, T. Distribution of IL-18 and IL-18 Receptor in Human Skin: Various Forms of IL-18 Are Produced in Keratinocytes. Arch Dermatol Res 2001, 293, 325–333. [CrossRef]
- Fenini, G.; Grossi, S.; Contassot, E.; Biedermann, T.; Reichmann, E.; French, L.E.; Beer, H.-D. Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J Invest Dermatol 2018, 138, 2644–2652. [CrossRef]
- Flier, J.; Boorsma, D.M.; Bruynzeel, D.P.; Van Beek, P.J.; Stoof, T.J.; Scheper, R.J.; Willemze, R.; Tensen, C.P. The CXCR3 Activating Chemokines IP-10, Mig, and IP-9 Are Expressed in Allergic but Not in Irritant Patch Test Reactions. J Invest Dermatol 1999, 113, 574–578. [CrossRef]
- Kim, H.J.; Song, S.B.; Choi, J.M.; Kim, K.M.; Cho, B.K.; Cho, D.H.; Park, H.J. IL-18 Downregulates Collagen Production in Human Dermal Fibroblasts via the ERK Pathway. J Invest Dermatol 2010, 130, 706–715. [CrossRef]
- Wang, B.; Feliciani, C.; Howell, B.G.; Freed, I.; Cai, Q.; Watanabe, H.; Sauder, D.N. Contribution of Langerhans Cell-Derived IL-18 to Contact Hypersensitivity. J Immunol 2002, 168, 3303–3308. [CrossRef]
- Chang, J.T.; Segal, B.M.; Nakanishi, K.; Okamura, H.; Shevach, E.M. The Costimulatory Effect of IL-18 on the Induction of Antigen-Specific IFN-Gamma Production by Resting T Cells Is IL-12 Dependent and Is Mediated by up-Regulation of the IL-12 Receptor Beta2 Subunit. Eur J Immunol 2000, 30, 1113–1119. [CrossRef]
- Sharaf, N.; Nicklin, M.J.; di Giovine, F.S. Long-Range DNA Interactions at the IL-1/IL-36/IL-37 Gene Cluster (2q13) Are Induced by Activation of Monocytes. Cytokine 2014, 68, 16–22. [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 Regulatory Pathways in Cancer Progression and Therapy. Immunol Rev 2018, 281, 57–61. [CrossRef]
- Jia, H.; Liu, J.; Han, B. Reviews of Interleukin-37: Functions, Receptors, and Roles in Diseases. Biomed Res Int 2018, 2018, 3058640. [CrossRef]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front Immunol 2021, 12, 696605. [CrossRef]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 Requires the Receptors IL-18Rα and IL-1R8 (SIGIRR) to Carry out Its Multifaceted Anti-Inflammatory Program upon Innate Signal Transduction. Nat Immunol 2015, 16, 354–365. [CrossRef]
- Pan, Y.; Wen, X.; Hao, D.; Wang, Y.; Wang, L.; He, G.; Jiang, X. The Role of IL-37 in Skin and Connective Tissue Diseases. Biomed Pharmacother 2020, 122, 109705. [CrossRef]
- Mei, Y.; Liu, H. IL-37: An Anti-Inflammatory Cytokine with Antitumor Functions. Cancer Rep (Hoboken) 2019, 2, e1151. [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 Is a Fundamental Inhibitor of Innate Immunity. Nat Immunol 2010, 11, 1014–1022. [CrossRef]
- Rudloff, I.; Cho, S.X.; Lao, J.C.; Ngo, D.; McKenzie, M.; Nold-Petry, C.A.; Nold, M.F. Monocytes and Dendritic Cells Are the Primary Sources of Interleukin 37 in Human Immune Cells. J Leukoc Biol 2017, 101, 901–911. [CrossRef]
- Zhou, J.; Gemperline, D.C.; Turner, M.J.; Oldach, J.; Molignano, J.; Sims, J.T.; Stayrook, K.R. Transcriptomic Analysis of Healthy and Atopic Dermatitis Samples Reveals the Role of IL-37 in Human Skin. Immunohorizons 2021, 5, 830–843. [CrossRef]
- Lachner, J.; Mlitz, V.; Tschachler, E.; Eckhart, L. Epidermal Cornification Is Preceded by the Expression of a Keratinocyte-Specific Set of Pyroptosis-Related Genes. Sci Rep 2017, 7, 17446. [CrossRef]
- Hou, T.; Tsang, M.S.-M.; Chu, I.M.-T.; Kan, L.L.-Y.; Hon, K.-L.; Leung, T.-F.; Lam, C.W.-K.; Wong, C.-K. Skewed Inflammation Is Associated with Aberrant Interleukin-37 Signaling Pathway in Atopic Dermatitis. Allergy 2021, 76, 2102–2114. [CrossRef]
- Guttman-Yassky, E.; Diaz, A.; Pavel, A.B.; Fernandes, M.; Lefferdink, R.; Erickson, T.; Canter, T.; Rangel, S.; Peng, X.; Li, R.; et al. Use of Tape Strips to Detect Immune and Barrier Abnormalities in the Skin of Children With Early-Onset Atopic Dermatitis. JAMA Dermatol 2019, 155, 1358–1370. [CrossRef]
- Fujita, H.; Inoue, Y.; Seto, K.; Komitsu, N.; Aihara, M. Interleukin-37 Is Elevated in Subjects with Atopic Dermatitis. J Dermatol Sci 2013, 69, 173–175. [CrossRef]
- Tsuji, G.; Yamamura, K.; Kawamura, K.; Kido-Nakahara, M.; Ito, T.; Nakahara, T. Regulatory Mechanism of the IL-33-IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int J Mol Sci 2023, 24. [CrossRef]
- Yan, X.; Tsuji, G.; Hashimoto-Hachiya, A.; Furue, M. Galactomyces Ferment Filtrate Potentiates an Anti-Inflammaging System in Keratinocytes. J Clin Med 2022, 11. [CrossRef]
- Mesjasz, A.; Trzeciak, M.; Gleń, J.; Jaskulak, M. Potential Role of IL-37 in Atopic Dermatitis. Cells 2023, 12. [CrossRef]
- Ewald, D.A.; Malajian, D.; Krueger, J.G.; Workman, C.T.; Wang, T.; Tian, S.; Litman, T.; Guttman-Yassky, E.; Suárez-Fariñas, M. Meta-Analysis Derived Atopic Dermatitis (MADAD) Transcriptome Defines a Robust AD Signature Highlighting the Involvement of Atherosclerosis and Lipid Metabolism Pathways. BMC Med Genomics 2015, 8, 60. [CrossRef]
- Schröder, A.; Lunding, L.P.; Zissler, U.M.; Vock, C.; Webering, S.; Ehlers, J.C.; Orinska, Z.; Chaker, A.; Schmidt-Weber, C.B.; Lang, N.J.; et al. IL-37 Regulates Allergic Inflammation by Counterbalancing pro-Inflammatory IL-1 and IL-33. Allergy 2022, 77, 856–869. [CrossRef]
- Ahmed, M.B.; Ad’hiah, A.H. Reduced Levels of Interleukin-37 in Serum of Patients with Allergic Rhinitis or Asthma. Rev Fr Allergol 2021, 61, 410–414. [CrossRef]
- Shilovskiy, I.P.; Dyneva, M.E.; Kurbacheva, O.M.; Kudlay, D.A.; Khaitov, M.R. The Role of Interleukin-37 in the Pathogenesis of Allergic Diseases. Acta Naturae 2019, 11, 54–64. [CrossRef]
- Wang, J.; Shen, Y.; Li, C.; Liu, C.; Wang, Z.-H.; Li, Y.-S.; Ke, X.; Hu, G.-H. IL-37 Attenuates Allergic Process via STAT6/STAT3 Pathways in Murine Allergic Rhinitis. Int Immunopharmacol 2019, 69, 27–33. [CrossRef]
- Zeng, H.; Zhou, K.; Ye, Z. Biology of Interleukin-37 and Its Role in Autoimmune Diseases (Review). Exp Ther Med 2022, 24, 495. [CrossRef]
- Mao, X.; Zhu, R.; Zhang, F.; Zhong, Y.; Yu, K.; Wei, Y.; Sun, H.; Xu, W.; Luo, Q.; Wang, Y.; et al. IL-37 Plays a Beneficial Role in Patients with Acute Coronary Syndrome. Mediators Inflamm 2019, 2019, 9515346. [CrossRef]
- Ye, L.; Jiang, B.; Deng, J.; Du, J.; Xiong, W.; Guan, Y.; Wen, Z.; Huang, K.; Huang, Z. IL-37 Alleviates Rheumatoid Arthritis by Suppressing IL-17 and IL-17-Triggering Cytokine Production and Limiting Th17 Cell Proliferation. J Immunol 2015, 194, 5110–5119. [CrossRef]
- Wang, D.-W.; Dong, N.; Wu, Y.; Zhu, X.-M.; Wang, C.-T.; Yao, Y.-M. Interleukin-37 Enhances the Suppressive Activity of Naturally Occurring CD4+CD25+ Regulatory T Cells. Sci Rep 2016, 6, 38955. [CrossRef]
- Shuai, X.; Wei-min, L.; Tong, Y.; Dong, N.; Sheng, Z.; Yao, Y. Expression of IL-37 Contributes to the Immunosuppressive Property of Human CD4+CD25+ Regulatory T Cells. Sci Rep 2015, 5, 14478. [CrossRef]
- Hou, T.; Sun, X.; Zhu, J.; Hon, K.-L.; Jiang, P.; Chu, I.M.-T.; Tsang, M.S.-M.; Lam, C.W.-K.; Zeng, H.; Wong, C.-K. IL-37 Ameliorating Allergic Inflammation in Atopic Dermatitis Through Regulating Microbiota and AMPK-MTOR Signaling Pathway-Modulated Autophagy Mechanism. Front Immunol 2020, 11, 752. [CrossRef]
- Zhou, P.; Li, Q.; Su, S.; Dong, W.; Zong, S.; Ma, Q.; Yang, X.; Zuo, D.; Zheng, S.; Meng, X.; et al. Interleukin 37 Suppresses M1 Macrophage Polarization Through Inhibition of the Notch1 and Nuclear Factor Kappa B Pathways. Front Cell Dev Biol 2020, 8, 56. [CrossRef]
- Luo, Y.; Cai, X.; Liu, S.; Wang, S.; Nold-Petry, C.A.; Nold, M.F.; Bufler, P.; Norris, D.; Dinarello, C.A.; Fujita, M. Suppression of Antigen-Specific Adaptive Immunity by IL-37 via Induction of Tolerogenic Dendritic Cells. Proc Natl Acad Sci U S A 2014, 111, 15178–15183. [CrossRef]
- Liu, T.; Liu, J.; Lin, Y.; Que, B.; Chang, C.; Zhang, J.; Liang, Z.; Gao, X.; Liu, S.; Liu, L.; et al. IL-37 Inhibits the Maturation of Dendritic Cells through the IL-1R8-TLR4-NF-ΚB Pathway. Biochim Biophys Acta Mol Cell Biol Lipids 2019, 1864, 1338–1349. [CrossRef]
- Liu, F.-T.; Goodarzi, H.; Chen, H.-Y. IgE, Mast Cells, and Eosinophils in Atopic Dermatitis. Clin Rev Allergy Immunol 2011, 41, 298–310. [CrossRef]
- Hou, T.; Tsang, M.S.-M.; Kan, L.L.-Y.; Li, P.; Chu, I.M.-T.; Lam, C.W.-K.; Wong, C.-K. IL-37 Targets TSLP-Primed Basophils to Alleviate Atopic Dermatitis. Int J Mol Sci 2021, 22. [CrossRef]
- Mali, S.S.; Bautista, D.M. Basophils Add Fuel to the Flame of Eczema Itch. Cell 2021, 184, 294–296. [CrossRef]
- Yamanishi, Y.; Mogi, K.; Takahashi, K.; Miyake, K.; Yoshikawa, S.; Karasuyama, H. Skin-Infiltrating Basophils Promote Atopic Dermatitis-like Inflammation via IL-4 Production in Mice. Allergy 2020, 75, 2613–2622. [CrossRef]
- Saluja, R.; Khan, M.; Church, M.K.; Maurer, M. The Role of IL-33 and Mast Cells in Allergy and Inflammation. Clin Transl Allergy 2015, 5, 33. [CrossRef]
- Li, W.; Ding, F.; Zhai, Y.; Tao, W.; Bi, J.; Fan, H.; Yin, N.; Wang, Z. IL-37 Is Protective in Allergic Contact Dermatitis through Mast Cell Inhibition. Int Immunopharmacol 2020, 83, 106476. [CrossRef]
- Kader, H.A.; Azeem, M.; Jwayed, S.A.; Al-Shehhi, A.; Tabassum, A.; Ayoub, M.A.; Hetta, H.F.; Waheed, Y.; Iratni, R.; Al-Dhaheri, A.; et al. Current Insights into Immunology and Novel Therapeutics of Atopic Dermatitis. Cells 2021, 10. [CrossRef]
- Lei, H.; Sun, Y.; Quan, S. IL-37 Relieves Allergic Inflammation by Inhibiting the CCL11 Signaling Pathway in a Mouse Model of Allergic Rhinitis. Exp Ther Med 2020, 20, 3114–3121. [CrossRef]
- Arican, O.; Aral, M.; Sasmaz, S.; Ciragil, P. Serum Levels of TNF-Alpha, IFN-Gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in Patients with Active Psoriasis and Correlation with Disease Severity. Mediators Inflamm 2005, 2005, 273–279. [CrossRef]
- Forouzandeh, M.; Besen, J.; Keane, R.W.; de Rivero Vaccari, J.P. The Inflammasome Signaling Proteins ASC and IL-18 as Biomarkers of Psoriasis. Front Pharmacol 2020, 11, 1238. [CrossRef]
- Gangemi, S.; Merendino, R.A.; Guarneri, F.; Minciullo, P.L.; DiLorenzo, G.; Pacor, M.; Cannavò, S.P. Serum Levels of Interleukin-18 and s-ICAM-1 in Patients Affected by Psoriasis: Preliminary Considerations. J Eur Acad Dermatol Venereol 2003, 17, 42–46. [CrossRef]
- Flisiak, I.; Klepacki, A.; Chodynicka, B. Plasma and Scales Levels of Interleukin 18 in Comparison with Other Possible Clinical and Laboratory Biomarkers of Psoriasis Activity. Biomarkers 2006, 11, 194–200. [CrossRef]
- Verma, D.; Fekri, S.Z.; Sigurdardottir, G.; Bivik Eding, C.; Sandin, C.; Enerbäck, C. Enhanced Inflammasome Activity in Patients with Psoriasis Promotes Systemic Inflammation. J Invest Dermatol 2021, 141, 586-595.e5. [CrossRef]
- Niu, X.-L.; Huang, Y.; Gao, Y.-L.; Sun, Y.-Z.; Han, Y.; Chen, H.-D.; Gao, X.-H.; Qi, R.-Q. Interleukin-18 Exacerbates Skin Inflammation and Affects Microabscesses and Scale Formation in a Mouse Model of Imiquimod-Induced Psoriasis. Chin Med J (Engl) 2019, 132, 690–698. [CrossRef]
- Shimoura, N.; Nagai, H.; Fujiwara, S.; Jimbo, H.; Yoshimoto, T.; Nishigori, C. Interleukin (IL)-18, Cooperatively with IL-23, Induces Prominent Inflammation and Enhances Psoriasis-like Epidermal Hyperplasia. Arch Dermatol Res 2017, 309, 315–321. [CrossRef]
- Zhang, W.; Guo, S.; Li, B.; Liu, L.; Ge, R.; Cao, T.; Wang, H.; Gao, T.; Wang, G.; Li, C. Proinflammatory Effect of High-Mobility Group Protein B1 on Keratinocytes: An Autocrine Mechanism Underlying Psoriasis Development. J Pathol 2017, 241, 392–404. [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [CrossRef]
- Kou, K.; Aihara, M.; Matsunaga, T.; Chen, H.; Taguri, M.; Morita, S.; Fujita, H.; Yamaguchi, Y.; Kambara, T.; Ikezawa, Z. Association of Serum Interleukin-18 and Other Biomarkers with Disease Severity in Adults with Atopic Dermatitis. Arch Dermatol Res 2012, 304, 305–312. [CrossRef]
- Lyubchenko, T.; Collins, H.K.; Goleva, E.; Leung, D.Y.M. Skin Tape Sampling Technique Identifies Proinflammatory Cytokines in Atopic Dermatitis Skin. Ann Allergy Asthma Immunol 2021, 126, 46-53.e2. [CrossRef]
- Andersson, A.M.; Sølberg, J.; Koch, A.; Skov, L.; Jakasa, I.; Kezic, S.; Thyssen, J.P. Assessment of Biomarkers in Pediatric Atopic Dermatitis by Tape Strips and Skin Biopsies. Allergy 2022, 77, 1499–1509. [CrossRef]
- Inoue, Y.; Aihara, M.; Kirino, M.; Harada, I.; Komori-Yamaguchi, J.; Yamaguchi, Y.; Nagashima, Y.; Ikezawa, Z. Interleukin-18 Is Elevated in the Horny Layer in Patients with Atopic Dermatitis and Is Associated with Staphylococcus Aureus Colonization. Br J Dermatol 2011, 164, 560–567. [CrossRef]
- McAleer, M.A.; Jakasa, I.; Hurault, G.; Sarvari, P.; McLean, W.H.I.; Tanaka, R.J.; Kezic, S.; Irvine, A.D. Systemic and Stratum Corneum Biomarkers of Severity in Infant Atopic Dermatitis Include Markers of Innate and T Helper Cell-Related Immunity and Angiogenesis. Br J Dermatol 2019, 180, 586–596. [CrossRef]
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.-I.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 Contributes to the Spontaneous Development of Atopic Dermatitis-like Inflammatory Skin Lesion Independently of IgE/Stat6 under Specific Pathogen-Free Conditions. Proc Natl Acad Sci U S A 2002, 99, 11340–11345. [CrossRef]
- Chen, J.-L.; Niu, X.-L.; Gao, Y.-L.; Ma, L.; Gao, X.-H.; Chen, H.-D.; Qi, R.-Q. IL-18 Knockout Alleviates Atopic Dermatitis-like Skin Lesions Induced by MC903 in a Mouse Model. Int J Mol Med 2020, 46, 880–888. [CrossRef]
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 Induction of IgE: Dependence on CD4+ T Cells, IL-4 and STAT6. Nat Immunol 2000, 1, 132–137. [CrossRef]
- Luo, Y.; Cai, X.; Liu, S.; Wang, S.; Nold-Petry, C.A.; Nold, M.F.; Bufler, P.; Norris, D.; Dinarello, C.A.; Fujita, M. Suppression of Antigen-Specific Adaptive Immunity by IL-37 via Induction of Tolerogenic Dendritic Cells. Proc Natl Acad Sci U S A 2014, 111, 15178–15183. [CrossRef]
- Li, B.; Tsoi, L.C.; Swindell, W.R.; Gudjonsson, J.E.; Tejasvi, T.; Johnston, A.; Ding, J.; Stuart, P.E.; Xing, X.; Kochkodan, J.J.; et al. Transcriptome Analysis of Psoriasis in a Large Case-Control Sample: RNA-Seq Provides Insights into Disease Mechanisms. J Invest Dermatol 2014, 134, 1828–1838. [CrossRef]
- Furue, M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. Int J Mol Sci 2020, 21. [CrossRef]
- Dai, X.; Shiraishi, K.; Muto, J.; Utsunomiya, R.; Mori, H.; Murakami, M.; Sayama, K. Nuclear IL-33 Plays an Important Role in IL-31‒Mediated Downregulation of FLG, Keratin 1, and Keratin 10 by Regulating Signal Transducer and Activator of Transcription 3 Activation in Human Keratinocytes. J Invest Dermatol 2022, 142, 136-144.e3. [CrossRef]
- Dai, X.; Utsunomiya, R.; Shiraishi, K.; Mori, H.; Muto, J.; Murakami, M.; Sayama, K. Nuclear IL-33 Plays an Important Role in the Suppression of FLG, LOR, Keratin 1, and Keratin 10 by IL-4 and IL-13 in Human Keratinocytes. J Invest Dermatol 2021, 141, 2646-2655.e6. [CrossRef]
- Zeng, F.; Chen, H.; Chen, L.; Mao, J.; Cai, S.; Xiao, Y.; Li, J.; Shi, J.; Li, B.; Xu, Y.; et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J Invest Dermatol 2021, 141, 596-606.e7. [CrossRef]
- Teng, X.; Hu, Z.; Wei, X.; Wang, Z.; Guan, T.; Liu, N.; Liu, X.; Ye, N.; Deng, G.; Luo, C.; et al. IL-37 Ameliorates the Inflammatory Process in Psoriasis by Suppressing Proinflammatory Cytokine Production. J Immunol 2014, 192, 1815–1823. [CrossRef]
- Rønholt, K.; Nielsen, A.L.-L.; Johansen, C.; Vestergaard, C.; Fauerbye, A.; López-Vales, R.; Dinarello, C.A.; Iversen, L. IL-37 Expression Is Downregulated in Lesional Psoriasis Skin. Immunohorizons 2020, 4, 754–761. [CrossRef]
- Krueger, J.; Clark, J.D.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Cueto, I.; Wang, C.Q.; Tan, H.; Wolk, R.; Rottinghaus, S.T.; Whitley, M.Z.; et al. Tofacitinib Attenuates Pathologic Immune Pathways in Patients with Psoriasis: A Randomized Phase 2 Study. J Allergy Clin Immunol 2016, 137, 1079–1090. [CrossRef]
- Tak, P.P.; Bacchi, M.; Bertolino, M. Pharmacokinetics of IL-18 Binding Protein in Healthy Volunteers and Subjects with Rheumatoid Arthritis or Plaque Psoriasis. Eur J Drug Metab Pharmacokinet 2006, 31, 109–116. [CrossRef]
- Leung K 99mTc-Interleukin-18-Binding Protein-Fc-Interlukin-1 Receptor Antagonist Available online: https://www.ncbi.nlm.nih.gov/books/NBK138562/ (accessed on 12 May 2024).
- Liu, Z.; Wyffels, L.; Barber, C.; Wan, L.; Xu, H.; Hui, M.M.; Furenlid, L.R.; Woolfenden, J.M. Characterization of 99mTc-Labeled Cytokine Ligands for Inflammation Imaging via TNF and IL-1 Pathways. Nucl Med Biol 2012, 39, 905–915. [CrossRef]
Figure 1.
The interaction between IL-18, IL-18R and IL-18BP. Top: IL-18 initially binds to IL-18Rα, forming an inactive IL-18/IL-18Rα complex, which subsequently recruits the IL-18Rβ chain to establish a high-affinity complex that initiates the IL-18-dependent signaling pathway. Bottom: IL-18 binding protein (IL-18BP) binds to IL-18 and subsequently forms an inhibitory complex with IL-18Rα, thereby preventing IL-18Rβ from activating cells. Created with biorender.com. Abbreviations: IL-18, interleukin-18; IL-18BP, Interleukin-18 binding protein; IL-18Rα, Interleukin-18 receptor alpha; IL-18Rβ; interleukin-18 receptor beta.
Figure 1.
The interaction between IL-18, IL-18R and IL-18BP. Top: IL-18 initially binds to IL-18Rα, forming an inactive IL-18/IL-18Rα complex, which subsequently recruits the IL-18Rβ chain to establish a high-affinity complex that initiates the IL-18-dependent signaling pathway. Bottom: IL-18 binding protein (IL-18BP) binds to IL-18 and subsequently forms an inhibitory complex with IL-18Rα, thereby preventing IL-18Rβ from activating cells. Created with biorender.com. Abbreviations: IL-18, interleukin-18; IL-18BP, Interleukin-18 binding protein; IL-18Rα, Interleukin-18 receptor alpha; IL-18Rβ; interleukin-18 receptor beta.
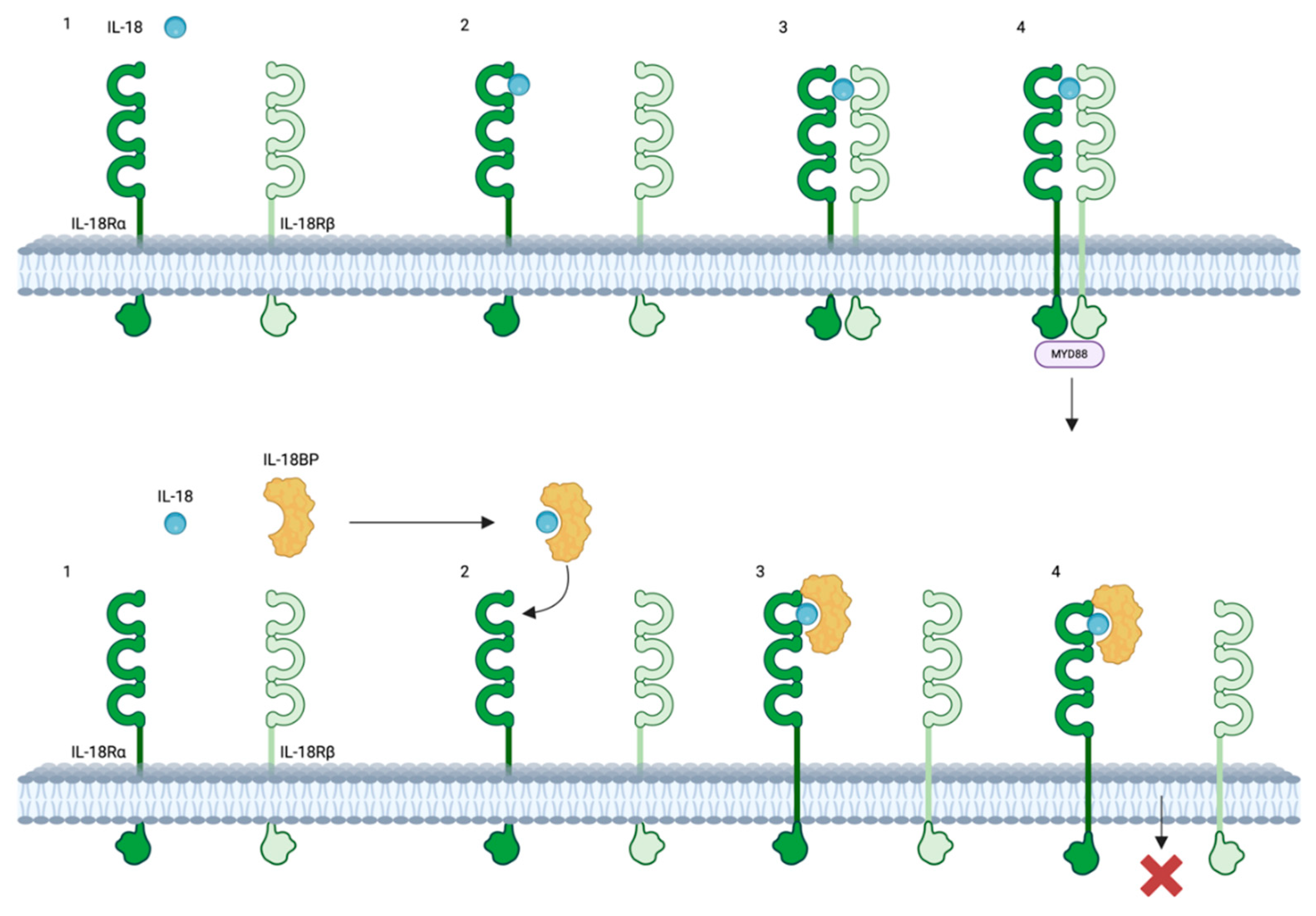
Figure 2.
IL-18 synthesis and signaling. Top: Lipopolysaccharide and IFN induce the synthesis of pro-IL-18 and IL-18BP. Concurrently, the inflammasome activates pro-caspase-1 into caspase-1, which cleaves pro-IL-18 into IL-18. Additionally, caspase-1 cleaves gasdermin D (GSDMD) into N-GSDMD, forming pores through which IL-18 is released from the cells. Bottom: Once secreted into the extracellular space, IL-18 can bind to its receptor (IL-18R) or to IL-18 binding protein (IL-18BP). Binding to IL-18BP inhibits IL-18 signaling, whereas binding to IL-18R initiates the signaling cascade. The cytosolic domain of IL-18R contains a Toll/IL-1 receptor (TIR) domain that recruits Myd88, facilitating the recruitment of IL-1 receptor-associated kinase (IRAK). IRAK then associates with TNF receptor-associated factor 6 (TRAF6), which in turn binds to the kinase TAK1. TAK1 phosphorylates and activates NF-κB-inducing kinase (NIK). Activated NIK then activates the IκB kinase (IKK) complex, which phosphorylates IkB, leading to IkB ubiquitination and rapid degradation. This process liberates the NF-κB transcription factor, allowing it to translocate to the nucleus, where it binds to κB sites in the promoter regions of inflammatory genes. Moreover, TRAF6 phosphorylation also connects with apoptosis signal-regulating kinase 1 (ASK1) through TAK1, activating downstream mitogen-activated protein kinase (MAPK) signaling pathways, including p38 MAPK, c-Jun N-terminal kinase (JNK), phosphoinositide 3-kinase/protein kinase B (PI3K/AKT), and extracellulart signal-regulated kinase (ERK). This cascade culminates in the activation of the AP-1 transcription factor. Created with biorender.com. Abbreviations: LPS, lipopolysaccharide; IFN, interferon; GSDMD, gasdermin D; N-GSDMD, N-terminal gasdermin D, TIR, Toll/IL-1 receptor; Myd88, Myeloid differentiation primary response 88; IRAK, interleukin-1 receptor associated kinase; TRAF 6, TNF receptor-associated factor 6; TAK1, transforming growth factor-b-activated kinase 1; NIK, nuclear factor kappa B (NF-κB)-induced kinase; IKK, inhibitor of nuclear kappa B kinase; IkB, inhibitor of nuclear kappa B; NF-kB, nuclear factor kappa B; ASK1, apoptosis signal-regulating kinase 1; p38MAPK, p38 mitogen-activated protein kinase; JNK, Jun kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; ERK extracellular regulated protein kinases; AP-1, activator protein 1.
Figure 2.
IL-18 synthesis and signaling. Top: Lipopolysaccharide and IFN induce the synthesis of pro-IL-18 and IL-18BP. Concurrently, the inflammasome activates pro-caspase-1 into caspase-1, which cleaves pro-IL-18 into IL-18. Additionally, caspase-1 cleaves gasdermin D (GSDMD) into N-GSDMD, forming pores through which IL-18 is released from the cells. Bottom: Once secreted into the extracellular space, IL-18 can bind to its receptor (IL-18R) or to IL-18 binding protein (IL-18BP). Binding to IL-18BP inhibits IL-18 signaling, whereas binding to IL-18R initiates the signaling cascade. The cytosolic domain of IL-18R contains a Toll/IL-1 receptor (TIR) domain that recruits Myd88, facilitating the recruitment of IL-1 receptor-associated kinase (IRAK). IRAK then associates with TNF receptor-associated factor 6 (TRAF6), which in turn binds to the kinase TAK1. TAK1 phosphorylates and activates NF-κB-inducing kinase (NIK). Activated NIK then activates the IκB kinase (IKK) complex, which phosphorylates IkB, leading to IkB ubiquitination and rapid degradation. This process liberates the NF-κB transcription factor, allowing it to translocate to the nucleus, where it binds to κB sites in the promoter regions of inflammatory genes. Moreover, TRAF6 phosphorylation also connects with apoptosis signal-regulating kinase 1 (ASK1) through TAK1, activating downstream mitogen-activated protein kinase (MAPK) signaling pathways, including p38 MAPK, c-Jun N-terminal kinase (JNK), phosphoinositide 3-kinase/protein kinase B (PI3K/AKT), and extracellulart signal-regulated kinase (ERK). This cascade culminates in the activation of the AP-1 transcription factor. Created with biorender.com. Abbreviations: LPS, lipopolysaccharide; IFN, interferon; GSDMD, gasdermin D; N-GSDMD, N-terminal gasdermin D, TIR, Toll/IL-1 receptor; Myd88, Myeloid differentiation primary response 88; IRAK, interleukin-1 receptor associated kinase; TRAF 6, TNF receptor-associated factor 6; TAK1, transforming growth factor-b-activated kinase 1; NIK, nuclear factor kappa B (NF-κB)-induced kinase; IKK, inhibitor of nuclear kappa B kinase; IkB, inhibitor of nuclear kappa B; NF-kB, nuclear factor kappa B; ASK1, apoptosis signal-regulating kinase 1; p38MAPK, p38 mitogen-activated protein kinase; JNK, Jun kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; ERK extracellular regulated protein kinases; AP-1, activator protein 1.
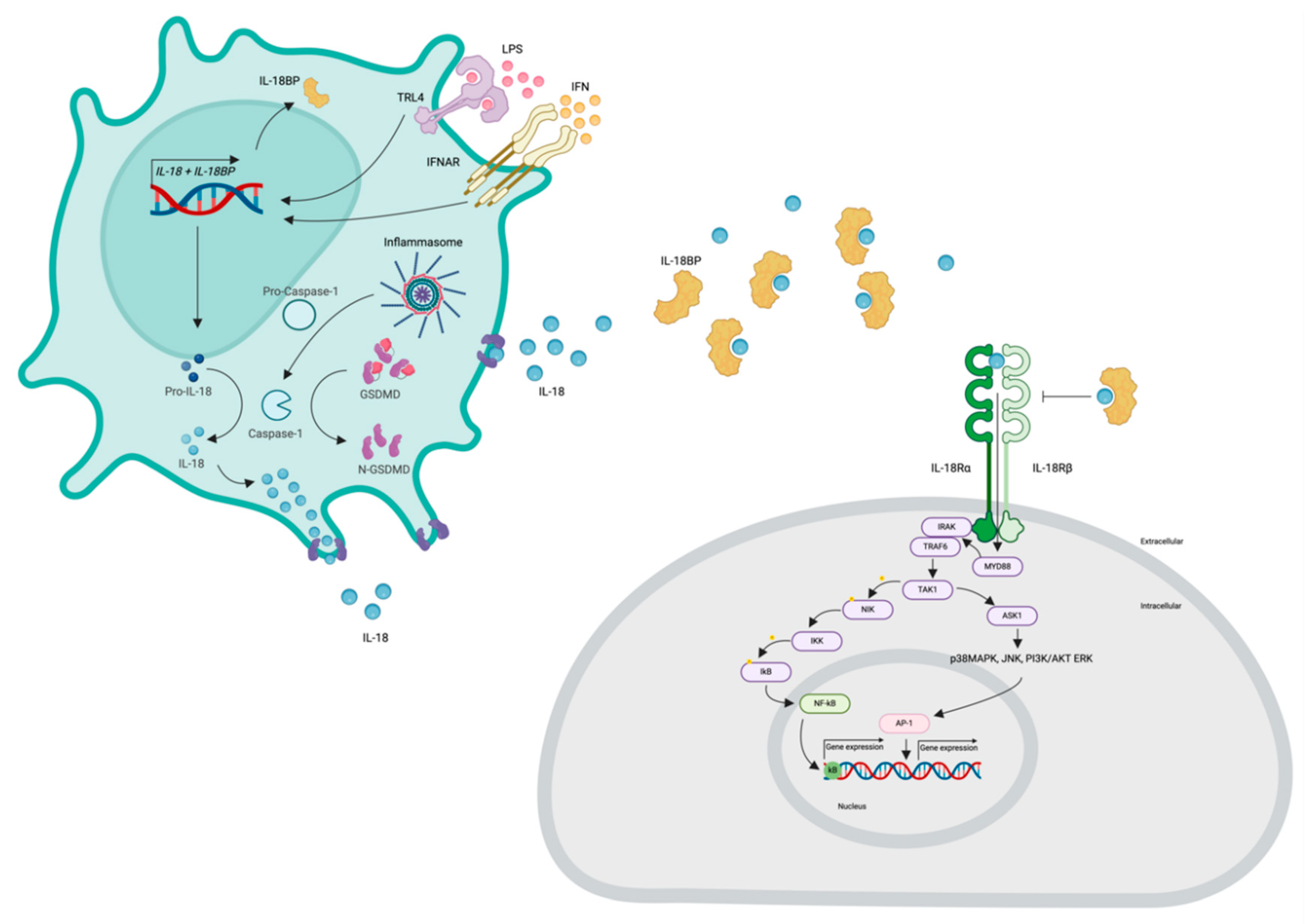
Figure 3.
IL-37 mechanism of action. Top: IL-37 binds to IL-18Rα, causing the loss of recruitment with IL-18Rβ. Bottom: IL-37 binding to IL-18BP forms a complex with IL-18Rb, thereby enhancing the inhibition of IL-18 activity. Created with biorender.com. Abbreviations: IL-37, interleukin-37; IL-18BP, Interleukin-18 binding protein; IL-18Rα, Interleukin-18 receptor alpha; IL-18Rβ; Interleukin-18 receptor beta.
Figure 3.
IL-37 mechanism of action. Top: IL-37 binds to IL-18Rα, causing the loss of recruitment with IL-18Rβ. Bottom: IL-37 binding to IL-18BP forms a complex with IL-18Rb, thereby enhancing the inhibition of IL-18 activity. Created with biorender.com. Abbreviations: IL-37, interleukin-37; IL-18BP, Interleukin-18 binding protein; IL-18Rα, Interleukin-18 receptor alpha; IL-18Rβ; Interleukin-18 receptor beta.
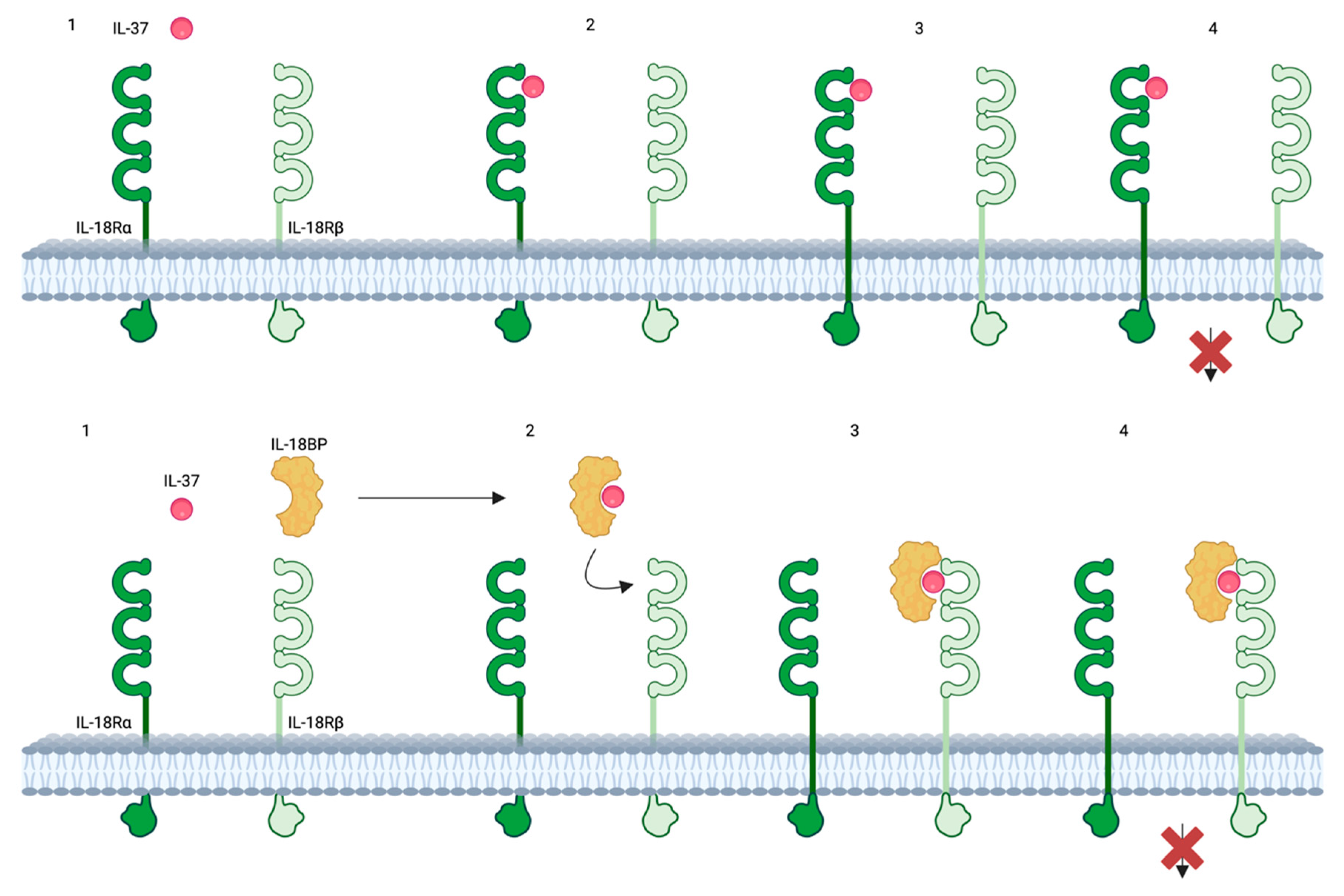
Figure 4.
IL-33 and IL-37 in the pathogenesis of psoriasis and AD. Psoriasis and AD patients experiencing pruritus exhibit increased IL-33 production within keratinocytes following scratch injury. IL-33 subsequently activates pruritogenic sensory nerves, thereby promoting pruritus. Furthermore, IL-33 mediates a Th2 immune response in both psoriasis and AD through the activation of ILC2 cells, T cells, and dendritic cells. These Th2 cells produce IL-4 and IL-13, which downregulate filaggrin and loricrin, consequently reducing IL-37 expression in keratinocytes. Additionally, IL-33 induces the production of CCL20, which facilitates the recruitment of Th17 cells, and CXCL8, which promotes neutrophil influx into the epidermis. The accumulation of Th17 cells and neutrophils significantly contributes to psoriasis pathogenesis. Moreover, IL-33, along with IL-17 and IL-22, collectively downregulates filaggrin and loricrin expression, further reducing IL-37 expression in keratinocytes. Created with biorender.com. Abbreviations: IL-33, interleukin-33; ILC2 cells, innate lymphoid cells type 2; Th2 cell, T helper 2 cell; IL-4, interleukin-4; IL-13, interleukin-13; CCL20, C-C Motif Chemokine Ligand 20; Th17, T helper 17 cell; CXCL8, interleukin-8; IL-22, interleukin-22; TNF, tumor necrosis factor.
Figure 4.
IL-33 and IL-37 in the pathogenesis of psoriasis and AD. Psoriasis and AD patients experiencing pruritus exhibit increased IL-33 production within keratinocytes following scratch injury. IL-33 subsequently activates pruritogenic sensory nerves, thereby promoting pruritus. Furthermore, IL-33 mediates a Th2 immune response in both psoriasis and AD through the activation of ILC2 cells, T cells, and dendritic cells. These Th2 cells produce IL-4 and IL-13, which downregulate filaggrin and loricrin, consequently reducing IL-37 expression in keratinocytes. Additionally, IL-33 induces the production of CCL20, which facilitates the recruitment of Th17 cells, and CXCL8, which promotes neutrophil influx into the epidermis. The accumulation of Th17 cells and neutrophils significantly contributes to psoriasis pathogenesis. Moreover, IL-33, along with IL-17 and IL-22, collectively downregulates filaggrin and loricrin expression, further reducing IL-37 expression in keratinocytes. Created with biorender.com. Abbreviations: IL-33, interleukin-33; ILC2 cells, innate lymphoid cells type 2; Th2 cell, T helper 2 cell; IL-4, interleukin-4; IL-13, interleukin-13; CCL20, C-C Motif Chemokine Ligand 20; Th17, T helper 17 cell; CXCL8, interleukin-8; IL-22, interleukin-22; TNF, tumor necrosis factor.
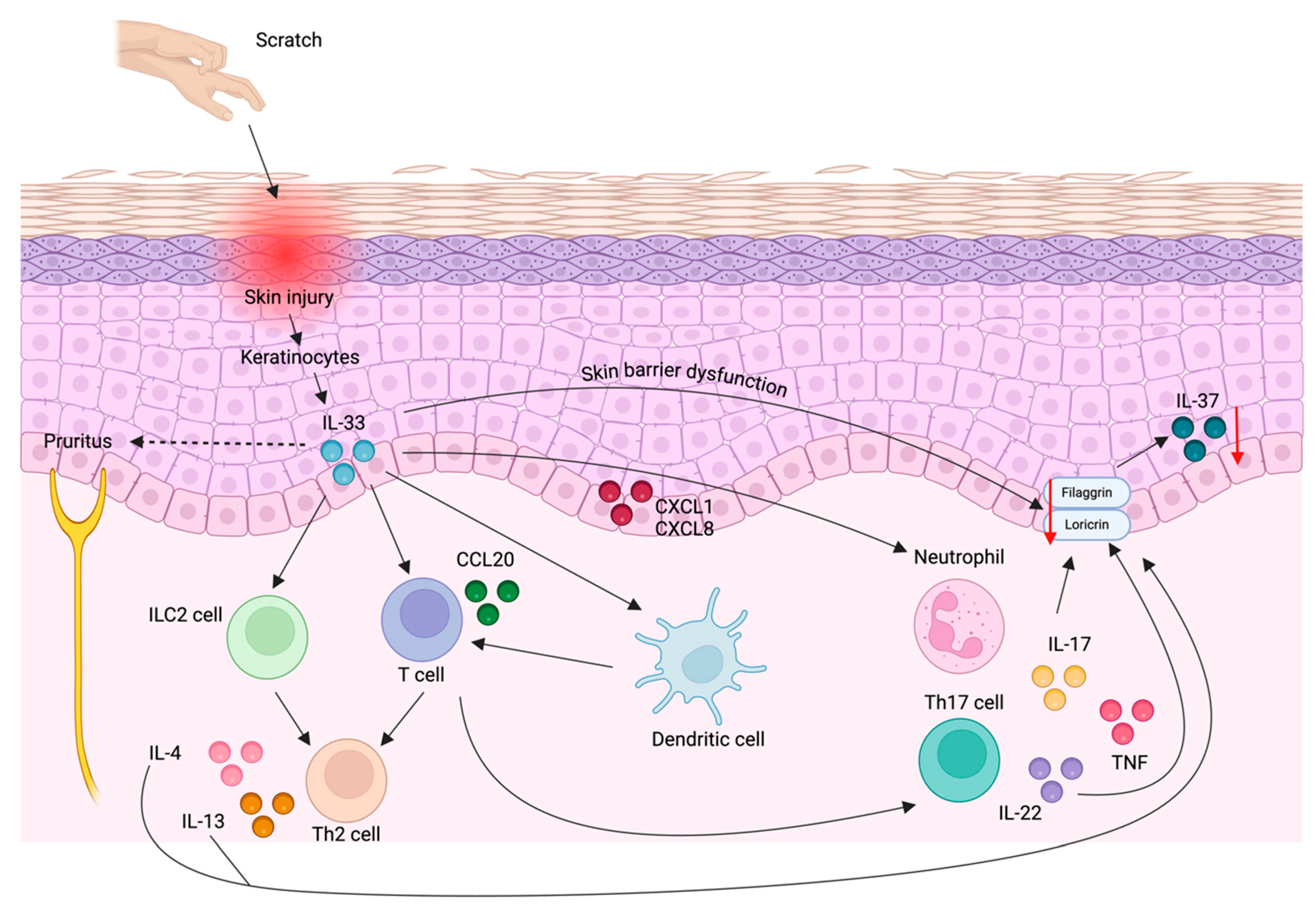
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



