
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
Escherichia coli O157:H7, a Shiga toxin-producing E. coli (STEC) is an important pathogen related to foodborne disease that is responsible for a growing number of outbreaks worldwide and has been detected in processed meats, dairy, and fresh vegetables. Although culturing is the gold standard method for detection of this bacteria, molecular methods based on nucleic acid amplification techniques such as PCR are becoming more common because of their rapidity, sensitivity, and specificity. However, to ensure reliable results among the several alternative PCR protocols (e.g., commercial kits and reference methods), different measurement assurance tools, including validated methods, reference materials, and proficiency tests among others, are required. Herein, we present a digital PCR method validation for E. coli O157:H7 detection and quantification using seven gene sequences; this method quantified nucleic acids from different E. coli serotypes, with a detection range between 6.6 to 7,900 copies/µL and a repeatability standard deviation over the concentration range of between 1% to 13.6%. The relative standard uncertainty was 3.5%–14.6%, and the detection limit was 0.27 copies/µL. Subsequently two batches of a candidate reference material based on E. coli O157:H7 genomic DNA were then produced and characterized for evaluation of copy number concentration with the validated dPCR method, with assigned values of 164,770 ± 9,251 and 172 ± 9 copies/μL. Thus, this study demonstrated the development a validated method and reference material for dPCR and qPCR detection of E. coli O157:H7, a key STEC responsible for food poisoning.
Keywords:
E. coli O157:H7
; Reference material
; Method validation
; digital PCR
1. Introduction
Enterohemorrhagic Escherichia coli, a subgroup of Shiga toxin-producing E. coli (STEC), causes intestinal and renal diseases, including hemorrhagic colitis and hemolytic uremic syndrome, and even death. The representative strain is E. coli O157:H7, which was identified in an outbreak of bloody diarrhea in 1983 [1]. This bacterium has subsequently been detected in foods such as processed meats, dairy, and fresh vegetables and is responsible for an increasing number of outbreaks globally [2]. Detection of E. coli O157:H7 and other non-O157 STEC is playing a key role in diagnostics, environmental protection, and food safety.
Microbiological culture is the gold standard method for E. coli O157:H7 detection [3]; however, this method is time-consuming as samples often contain low target cell numbers and high levels of background flora and natural inhibitors that interfere with isolation. To address these issues, several methods for rapid identification based on nucleic acid amplification techniques such as PCR have been developed [4]. PCR has become a powerful diagnostic tool for detection of pathogenic microorganisms in food samples because of advantages such as specificity, sensitivity, and high throughput [5].
PCR detection of STEC is based on the amplification of virulence gene sequences conserved in several serotypes of this group. The most common genes include (i) stx1 and stx2 that encode Shiga-like toxins that have a N-glycosylase RNA activity and thus inhibit protein synthesis [6]. (ii) rfbE that encodes an enzyme associated with membrane lipopolysaccharide biosynthesis in outer membrane biogenesis and is related to O-antigens [6]. (iii) eaeA that encodes an adhesion protein involved in the invasion of E. coli into enterocytes and is an integral component of the membrane [7], and (iv) Z3276, a specific genetic marker of STEC O157:H7 involved in biofilm formation. Other representative genes for genus detection are uidA, which is used to identify E. coli serotypes, as this encodes a β-glucuronidase, although this may be present in Shigella strains, [8] and lacY, which encodes a beta-galactoside transporter that is used to discriminate between E. coli and Shigella strains [9], [10].
PCR analysis methods can be qualitative, semi-qualitative, or quantitative, depending on the analysis purpose, and many of them rely on using calibration curves or threshold value comparisons [11]. One prerequisite to facilitate the development of PCR methods is the availability of DNA standards [12]. Manufacturers usually develop their own standards for use as internal or positive controls. This practice gives rise to systematic error and consequently produces less comparable results between laboratories [13]. In this situation, DNA reference materials (RMs) could help to reduce the within- and among variability in laboratory quantitation [14], allowing a guarantee of the reliability and traceability of PCR results. Digital PCR (dPCR) could be considered as a potential primary method because this can quantify DNA without a standard or internal control [15] and can be used to characterize DNA in the production of RMs [16], [17].
Thus, the purpose of this study was i) to describe a dPCR method validation for E. coli O157:H7 quantification, and ii) the use of this method to characterize a genomic DNA (gDNA) RM. For the validation, the performance characteristics evaluated were working interval, limit of detection (LOD), limit of quantification (LOQ), selectivity, precision, and uncertainty [18]. Subsequently the RM candidate was prepared at two concentration levels in solution. We performed a homogeneity and stability study to assign a copy number concentration value with its uncertainty. This work aimed to contribute to standardization of E. coli O157:H7 detection by PCR assays and improve interlaboratory comparisons toward the strengthening of monitoring processes in agriculture and environmental industries.
2. Materials and Methods
2.1. Reagents and Instruments
The IRMM 449® gDNA of E. coli O157 strain EDL933 certified in identity, was used as a calibrant for PCR amplification [19]. Lyophilized DNA was solubilized in 1X TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) at a nominal concentration of 13.0 ng/µL (approximately 2.14 × 106 genome copies/µL). Six 1:10 serial dilutions were prepared gravimetrically. ERM-AD623-certified RM was used as a quality control to evaluate the performance of the dPCR method [18].
Primers and probes for PCR analysis were synthesized by Integrated DNA Technologies and were solubilized in 1X TE buffer, diluted to 10 mM, and stored at −30°C. Droplet digital PCR (ddPCR) was performed on the QX200 digital PCR system (Bio-Rad 186-40031). The CFX96 touch deep well thermal cycler (Bio-Rad 3600037) was used for both qPCR and dPCR. Large-scale DNA extraction was performed using a Sorvall Lynx 4000 centrifuge with Bioflex HC and TH13 rotors, whereas for small-scale DNA extraction, a Heraus™ Megafuge™ 16R centrifuge with Microliter 30 × 2 rotor was used.
A complete list of bacterial strains used in selectivity study of PCR appears in Table 2. All KWIK-STIK™ strains, purchased from Microbiologics, were preserved at 4 °C until use. E. coli strain O157:H7 derived from ATCC 35150 was used for RM production.
2.2. ddPCR Method Validation
2.2.1. Primers and Probes Design
Seven single-copy, highly conserved genes frequently used for the detection of E. coli spp. and E. coli O157:H7 by PCR were selected based on currently available literature and used as target genes: uidA, lacY, eaeA, rfbE, stx1, stx2, and Z3276. Specific primers and probes were selected or designed for each gene (Supplementary Table S1) using the E. coli K12 (NC_000913.3) and E. coli O157 (NC_002695.1 and NC_002655.2) genomes as references. In silico analysis was performed using SnapGene [20] and Oligoanalizer 3.1 [21] to evaluate dimers or secondary structures between primers and probes. Primer specificity was evaluated using NBLAST [22].
2.2.2. Digital Droplet PCR Validation
A reaction mixture contained 11 μL of 2× ddPCR Supermix for Probes (Bio-Rad 1863024), 800 nM primers, 300 nM probe, and 2 μL of the genomic DNA (gDNA) template (gravimetrically added), with molecular-grade water added to a final volume of 21 μL. A non-template control was included. Both the 21 μL of reaction mix and 70 μL of the droplet generation oil for probes, (Bio-Rad, 1863005) were loaded into 8-well cartridges to generate droplets using the QX200 droplet generator (Bio-Rad 1864002). Then, generated droplets were transferred into a 96-well plate, which was sealed and inserted into a CFX96 touch deep well thermal cycler (Bio-Rad). PCR amplification used the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 58°C for 60 s, and a final step of 10°C for 10 min; all steps employed a heating ramp of 0.5°C/s. The plate was then transferred to a QX200 droplet reader (Bio-Rad, 1864003) to read droplet fluorescence. The intra-laboratory ddPCR method validation was performed in simplex mode, and copy number concentration, validation parameters, and decision criteria are described in Table 1.
2.2.3. Data Analysis
Data acquisition and analysis were performed using Quantasoft software V1.7 (Bio-Rad). Data generated by the QX200 droplet reader were excluded from subsequent analysis when a clog was detected by the Quantasoft software or when a low number of droplets (<10,000) was measured in the PCR mixture. The fluorescence threshold was set manually between the average fluorescence amplitude of positive and negative partitions for each target gene. After exporting, data were analyzed using Microsoft Excel. The mathematical model (Equation (1)) was used to calculate the sample copy number concentration Cm in copies/μL, using a partition volume off 0.773 ± 0.023 nL (measured in laboratory):
where V is the partition volume, d is the total gravimetric dilution, and λ represents the copy number per partition, which was calculated from the negative (N) and total (P) number of partitions according to Equation (2).
2.3. Genomic DNA Reference Material Production
2.3.1. Culture and DNA Extraction
After E. coli O157:H7 (derived from ATCC 35150) cell culture optimization, bacterial cells were harvested from a 200-mL culture via centrifugation at 6,000 × g for 30 min at 4°C in a Sorvall Lynx 4000 centrifuge. The pellet was washed by centrifugation thrice with 200 mL of 1× phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4). gDNA was extracted using the CTAB/chloroform DNA extraction method [26]. Briefly, the bacterial pellet was resuspended in 7.5 mL of 1× TE buffer and transferred to a 30-mL tube. A total of 7.5 mL of CTAB lysis buffer (8% w/v CTAB, 40 mM EDTA, 200 mM Tris-HCl, 2.8 M NaCl, and 6% LiCl) and 20 mg/mL proteinase K were added and mixed and then incubated at 65°C for 1 h. After centrifugation (25,400 × g, 15 min, 4°C in Sorvall Lynx 4000 centrifuge using a swinging rotor), the supernatant (the lysate) was transferred to a new 30-mL tube and extracted twice with 1 volume of 24:1 chloroform: isoamyl alcohol. After centrifugation, RNase (20 mg/mL) was added to the aqueous phase and incubated at 37°C for 1 h with a gentle shaking. Then 10% (w/v) LiCl was added and incubated for 15 min at 4°C and extracted with 1 volume of 24:1 chloroform: isoamyl alcohol. The supernatant was precipitated by 1:1.5 isopropyl alcohol and 2.5 M ammonium acetate at −20°C for 18 h, centrifuged using a swinging rotor at 22,600 × g for 20 min at 4°C, washed twice with 70% ethanol, and dried; the DNA pellet was dissolved in 1 mL of 1× TE buffer and stored at −20°C.
DNA integrity was assessed using 1% agarose gel electrophoresis in 1× TAE buffer (40 mM Tris, 1 mM EDTA, and 20 mM acetic acid, pH 8.5), stained with SYBR® Green DNA dye. DNA purity was assessed using the 260/280 absorbance ratio [27]. The presence of inhibitors was assessed via qPCR amplification, with six gravimetric serial dilutions of gDNA RMs covering four-log concentrations. The acceptability criteria for PCR efficiency were 90%–110% [28].
2.3.2. Material Preparation
Two 40-mL batches of a DNA stock solution, previously measured and quantified using a validated ddPCR method, with a nominal copy number concentration of 100,000 and 100 copies/µL (high and low level, respectively) were prepared using gravimetric serial dilutions with TE 1× pH 8.0 as diluent and total yeast RNA as stabilizer (40 ng/µL) [25]. DNA solutions were homogenized in an orbital shaker for 2 h at room temperature. For each batch, a 500-µL DNA solution was dispensed into 1.2-mL labeled polypropylene cryovials (n = 80) stored at 4 ºC, −20 °C, and −80 °C based on the intended use: homogeneity and stability study, characterization, value assignment and controls, respectively.
2.3.3. Homogeneity Study
dPCR assays in simplex mode with the Z3276 assay gene were used to assess the homogeneity of gDNA RM under repeatability conditions: For each batch, eight units were selected, following a systematic random sampling scheme, three replicas for the high level and six replicas for the low level were measured for every unit. The contribution to the combined uncertainty due to the homogeneity (uhom) was estimated from one-way analysis of variance (ANOVA), according to Equation (3), with vials as the variation factor.
where ubb is the uncertainty between bottles, and is equal to sbb, the standard deviation between bottles, calculated from the mean square between bottles (MSbetween) and the mean squared within bottles (MSwithin) and the number of replicas per bottle (n).
Since the minimum sample volume (5–20 µL) was smaller than the RM unit volume (500 µL), a significant within-unit heterogeneity was checked using six test portions from a unique bottle for each batch via ANOVA, using subsample as the variation factor.
2.3.4. Stability Study
To evaluate the material stability during transport and storage conditions, short- and long-term stability studies were performed at 4 °C and −20 °C, using −80 °C as a reference temperature. For the short-term stability study, following an isochronous design [29], eight units from each batch were stored at 4 ºC and −20 ºC. At 2, 4, 8, and 12 weeks, two units from each temperature were transferred to the reference temperature (−80°C) to be measured in triplicate under repeatability conditions by ddPCR using the Z3276 gene at week 12. The long-term stability study extended the short-term stability study: four different samples previously selected, were stored at 4 °C and −20 °C. Following a classical design, at week 24 and 60, two units from each temperature were removed and measured, with a control sample (stored at −80 °C) via ddPCR in triplicate using Z3276; the data was normalized using the control sample at each time point. Regression analysis was performed in both cases to establish the instability degree, and their contribution to materials uncertainty , was calculated from the slope and its standard deviation according to Equation (4):
The ults, based in the predicted change, is a function of the study time (tm = 60 weeks) and the standard error for the estimated slope s(b).
2.3.5. Material Characterization and Value Assignment
To establish the reference value, nine units were randomly selected: Three units were measured in triplicate using Z3276 and rfbE assays by ddPCR in simplex mode on three different days. The mean value for each gene was determined, and a consensus value ) was established for each batch as the mean of both assays. The value assignment for each batch ) was made following the model described in Equation (5) and is defined for the characterization of the material ), the homogeneity (), and stability ):
where represent the errors associated with homogeneity and stability, which are usually zero, but not their uncertainties. The material uncertainty was calculated from the combination of the characterization, the homogeneity, and the stability uncertainties (Equation (6)) [30].
3. Results
3.1. ddPCR Method Validation
Calibration parameters were evaluated once primer and probe specificities were checked (Supplementary Figure S1).
3.1.1. Selectivity
Eight different E. coli serovars of STEC and non-STEC groups as well as closely related and unrelated species were evaluated via qPCR for the presence of all seven target genes (Table 2). The uidA gene was amplified from E. coli and Shigella strains. The lacY gene was amplified from E. coli and several enterobacteria species. The eaeA, stx1, and stx2 genes were amplified in several STEC strains, whereas the rfbE and Z3276 genes were only amplified in E. coli O157:H7. The results show the amplification selectivity of each gene.

Table 2.
Gene amplification profiles indicating E. coli O157:H7 ddPCR selectivity.
| Group | Species | Reference | uidA | lacY | eaeA | rfbE | Stx1 | Stx2 | Z3276 |
|---|---|---|---|---|---|---|---|---|---|
| Gram (+) | Staphylococcus aureus | ATCC® 6538 | - | - | - | - | - | - | - |
| Staphylococcus aureus | ATCC® 25923 | - | - | - | - | - | - | - | |
| Bacillus cereus | ATCC® 10876 | - | - | - | - | - | - | - | |
| Enterococcus faecalis | ATCC® 14506 | - | - | - | - | - | - | - | |
| Gram (-) | Proteus mirabilis | ATCC® 12453 | - | - | - | - | - | - | - |
| Vibrio parahaemolyticus | ATCC® 17802 | - | - | - | - | - | - | - | |
| Enterobacter aerogenes | ATCC® 13048 | - | - | - | - | - | - | - | |
| Proteus vulgaris | ATCC® 33420 | - | - | - | - | - | - | - | |
| Yersinia enterocolitica | ATCC® 23715 | - | - | - | - | - | - | - | |
| Shigella | Shigella boydii | ATCC® 9207 | + | - | - | - | - | - | - |
| Shigella sonnei | ATCC® 9290 | + | - | - | - | - | - | - | |
| Escherichia coli | Escherichia coli | ATCC® 25922 | + | + | - | - | - | - | - |
| Escherichia coli | Donated1 | + | + | - | - | - | - | - | |
| Escherichia coli | NCTC 10538 | + | + | - | - | - | - | - | |
| Escherichia coli | ATCC® 8739 | + | + | - | - | - | - | - | |
| STEC* | Escherichia coli O104:H4 | ATCC® BAA-2326™ | + | + | - | - | - | + | - |
| Escherichia coli O145:NM | CDC 99-3311 | + | + | + | - | + | + | - | |
| Escherichia coli O157:H7 | ATCC® 700728™ | + | + | + | + | - | - | + | |
| Escherichia coli O157:H7 | ATCC® 35150™ | + | + | + | + | + | + | + | |
| Salmonella | Salmonella Thyphimurium | Donated2 | - | - | - | - | - | - | - |
| Salmonella Thyphi | Donated2 | - | - | - | - | - | - | - | |
| Salmonella enteritidis | Donated2 | - | - | - | - | - | - | - |
1 Donated by the UNAL Medicine Laboratory, 2 Salmonella DNA, donated by the National Institute of Health (INS), Microbiology group. *STEC Shiga Toxin Producing Escherichia coli.
3.1.2. Working Interval
Experimental data fitted a linear regression model in the range of 1.5 to 7,900 copies/µL for the seven genes (Table 3; Supplementary Figure S2); however, not all genes met the precision criteria for RSD in the lowest concentration level; Thus, the working range for all genes by the ddPCR method was established as 6.6–7,900 copies/µL.
3.1.3. Precision
As RSD of repeatability was usually higher than the run-to-run variation for dPCR, and only the first one was calculated as method precision. For this, ANOVA was performed for each DNA target gene at each concentration level to calculate the relative repeatability (Equation (7)).
where is the within-run mean squares, is the number of replicates per run, and is the average copy number concentration calculated over all runs.
From the precision data (Supplementary Tables S2 to S8), the RSD of the repeatability was calculated for the seven DNA target sequences evaluated over the five concentration levels of the working interval (Figure 1). For L1 to L4 concentration levels, the RSD of the intermediate precision was <13.6% (stx2, level 3).
3.1.4. LOQ and LOD
The LOQ was established at 6.6 copies/µL based on the working interval and precision results (Table 3 and Figure 1). The LOD was determined as the lowest concentration where at least nine positive partitions in all replicates were detected, with at least 10,000 total partitions per replica [23]. Six concentration levels under the LOQ were evaluated between 1.60 to 0.08 copies/µL. Figure 2 shows the positive partition distribution related to the concentration level.
Apart from rfbE, with 0.17 copies/µL, the other study genes met the acceptance criteria at 0.27 copies/µL in the reaction; thus, this value was established as the LOD for the E. coli O157:H7 ddPCR method.
3.1.5. Method Uncertainty
The combined standard uncertainty (u) for each gene target and concentration level was calculated based on the uncertainty sources identified for the E. coli O157:H7 ddPCR method (Figure 3 and Equation (8)).
The mathematical model uncertainty component, based in the Equation (1) is described by Equation (9):
The precision uncertainty was calculated as follows (Equation (10)), Where n is days of measurement.
Table 4 shows the relative combined uncertainty calculated for each DNA target sequence and for each concentration level for the E. coli ddPCR method.
3.2. Preparation and Quality Control of E. coli DNA Reference
3.2.1. Culture and DNA Extraction
A total of 1 mL gDNA was obtained from 0.730 g bacterial pellet, with a concentration of 3,728.2 ± 13.7 ng/µg measured via spectrophotometry. The OD260/OD280 and OD260/OD230 ratios of 2.07 and 2.13 indicate high quality DNA.
The amplification efficiencies of the different PCR assays calculated via real-time PCR were 98.5%, 97.7%, 99.3%, 99.3%, 108.8%, 97.8%, and 101.4% based on the slope of standard curves for uidA, lacY, eaeA, rfbE, stx1, stx2, and Z3276 genes, respectively; therefore, no DNA inhibitors were present in the solution. Agarose gel electrophoresis demonstrated that the extracted gDNA had structural integrity. Finally, the estimated concentration of the extracted DNA measured by dPCR was 1.02 × 108 copies/µL.
3.2.2. Homogeneity Study
Once the high and low copy number concentration batches were prepared, a processing run effect was checked. For this, the means of the copy number concentrations were plotted against bottle number in preparation order; no significant trends were detected based on regression analysis (Figure 4). Additionally, no significant difference was detected within the heterogeneity for either study materials from the within-unit homogeneity study. The between-unit homogeneity contribution calculated via Equation (3) from ANOVA (Supplementary Table S9) was 1.83 and 806 copies/µL (11% and 0.5%, respectively) for the low and high copy number concentration levels, respectively.
3.2.3. Stability Study
Based on the regression analysis (and Supplementary Figure S4 and Supplementary Table S10) no significant instability was detected for either the low or high concentration level batches at 4°C or −20°C in the short-term stability study. Similarly, the long-term stability study did not exhibit any significant trend in the material concentration during the 60 weeks evaluated at 4°C or −20°C (Figure 5)
Considering the dispersion of results around the slope of regression analysis over the evaluated time, the contribution to the uncertainty due to the long-term stability , calculated from the regression data (Supplementary information Table S11). For each batch at each temperature is presented in Table 5.
3.3. Material Characterization and Value Assignment
Figure 6 shows the Ishikawa diagram with the identified uncertainty sources that contribute to the material uncertainty for each batch; Z3276, rfbE, and Bias Z3276-rfbE were grouped in the characterization term of Equation (5). Supplementary Figure S4 details the equations used for uncertainty characterization estimation.
The Z3276 and rfbE values and their uncertainties are derived from the experimental results obtained for the nine units evaluated for three days combined with the uncertainty from the mathematical model (Equation (1)).
Figure 7 shows the box plots for rfbE and Z3276 quantification results obtained, for low and high concentration level batches. A t-test for independent samples showed a significative difference between them (Supplementary Information SI1); therefore, the bias between Z3276 and rfbE mean values was included as an uncertainty component (Figure 6), taking the bias as the interval size and assuming a rectangular distribution. The bias uncertainty is derived from the standard deviation from a rectangular distribution between the Z3276 and the rfbE values.
Table 6 presents a summary of the reference values for the batches under study with their associated uncertainty (Supplementary Table S12 to S15 show raw data).
Figure 8 represents the contribution of each source to the uncertainty, over the mean value and over the combined standard uncertainty in the value assignment process.
Figure 8 demonstrates the bias between the rfbE and Z3276 quantification results and homogeneity was not significative for the standard uncertainty of either material. However, the stability appears to be the most important component; and for the low-level concentration, the characterization contributes a further approximately 25% uncertainty.
4. Discussion
4.1. Method Validation
In this study, we validated a ddPCR method to detect, serotyping and quantify E. coli O157:H7 using seven target genes. DNA primers were derived from the reference genomes of E. coli O157 (NC_002695.1 and NC_002655.2) and nonpathogenic E. coli K12 (NC_000913.3) to provide a suitable method for the identification of different E. coli serovars, especially O157:H7, under the same experimental conditions (i.e., primer and probe concentrations, amplification cycles). Although the tests were performed in simplex mode, the selected probes allow the development of duplex PCR assays for improving and optimizing current assays in specialized laboratories.
As indicated by the selectivity analysis, and as expected, the uidA gene was amplified from both E. coli and Shigella strains, although these species can be differentiated based on the amplification of the lactose permease gene lacY, which is negative in Shigella. Although Citrobacter freundii and Klebsiella pneumonia lack lacY, several reports showed that these species harbor sequences similar to those of E. coli lacY; thus, lacY would be useful as a molecular marker only when used in combination with other genes [9]. The eaeA gene should theoretically be amplified in all STEC; however, E. coli O104:H4 tested negative, which may be associated with the multiple variants of this in several STEC serotypes. Thus, degenerate primers should be used for E. coli serotype identification [7]. However, eaeA was positively amplified in the O157:H7 serovar. The rfbE gene was amplified from E. coli O157:H7, demonstrating its selectivity toward the STEC O157 group; however, this gene allows discrimination of serotype O157:H7 only in conjunction with stx1 and stx2 (which are present in STEC strains) and Z3276, which is specific to E. coli O157:H7.
Although this amplification method behaves linearly between 1.6 and 7,900 copies/µL (in PCR master mix) for all target genes based on linear regression analysis, the working interval started at 6.6 copies/µL (LOQ) because of the precision criteria. Only rfbE showed a precision <20% for all evaluated concentrations, whereas uidA and lacY had a precision just above the threshold. For eaeA, stx1, stx2, and Z3276, the precision was >25%. rfbE and Z3276 exhibited an improved precision for the four higher concentrations, indicating that they are the target genes with the highest repeatability. This is important because these genes allow the detection of E. coli O157 and O157:H7 serovars, and this finding implies that the confidence is higher for these target genes than for the other genes. However, eaeA and stx2 exhibited a larger variability for all concentrations tested.
For the LOD, nine positive partitions allowed good differentiation of a low-concentration positive sample from the blank, and this is suitable for the intended use of the method. The detection limit was determined as 0.27 gene copies/µL in the reaction mix, which was equivalent to approximately 3 copies/ reaction.
Method bias was not evaluated because commercially available quantitative RMs are not available. The trueness of the method was evaluated using a certified identity DNA sample (IRMM 449). For each dilution, the concentration in copies/µL was estimated based on the data of the gravimetric dilutions where the working range could be determined by comparing the gravimetrically determined and measured concentrations. The most important factor contributing to uncertainty was derived from the mathematical model; λ was the predominant source of uncertainty. To decrease the uncertainty, some wells could be merged (at least two replicas) in the Quantasoft software to increase the partition number, especially when working with low concentrations near the LOQ.
Based on the results of the validation, this method could quantify the gDNA concentration of E. coli serotypes from 6.6 to 7,900 copies/µL in the reaction, with a detection limit of 0.27 copies/µL. The method had a precision between 1% to 13.6%, with relative uncertainties between 3.5% and 14.6% in the working interval. Due to the selectivity exhibited by the evaluated genes, the method can be used to estimate the DNA concentration in samples during production of RMs of this microorganism and to distinguish between various serotypes of E. coli.
Considering the uncertainty and precision results, the amplification of the Z3276 and rfbE genes was proposed for the characterization of the gDNA of E. coli O157:H7 is a candidate for RM, whereas the other genes can provide additional information. Finally, to obtain concentration data at the lowest uncertainties, it is recommended to prepare the reaction mixture close to 1,000 copies/µL, where the lowest uncertainty and highest precision were apparent.
Key experimental information related to dPCR method can be found in Supplementary Table S16 (dMIQE checklist).
4.2. Reference Material Production
Although the extraction process evaluated allowed us to obtain a large amount of DNA, the preparation of these batches was excessive, so the culture can be reduced to 50 mL to start the extraction process to optimize reactions. Although the DNA extraction results showed several higher values for spectrophotometric analysis, indicating the presence of RNA, the qPCR and electrophoresis analysis (to evaluate inhibitors presence and DNA integrity) demonstrated that the method is adequate for semipreparative extraction of DNA. The small amounts of RNA have no effect on the RM to be prepared since several dilution steps are required to reach the target concentration. Furthermore, during the RM preparation, yeast RNA was added as a stabilizer [26]. Previous studies established that extraction of DNA from solid supports and even solubilization of lyophilized DNA contributes to quantitation variability [31]. Here, we sought to minimize such sources of variability, and the RM candidate of E. coli O157:H7 gDNA was prepared as DNA solutions directly suitable for qPCR and dPCR assays.
Both the high and low RMs prepared here were aqueous solutions of DNA, where the high concentration level can also be considered as diluted and lacked any significative differences among the different subsamples taken for study materials; therefore, the within homogeneity can be considered as zero. Thus, as expected, the maximum degree of heterogeneity found in both RMs was low compared with the assigned value (low level: 1.1% and high level: 0.5%). These values can be established as the target homogeneity uncertainty in a normalized RM production.
For the short-term stability studies, the temperature used for storage did not significantly affect the concentration value (for the evaluated frame of time) at −20 °C and especially at 4 °C. This is crucial if the objective is to use or consume the material rapidly (e.g., in a proficiency test item in an intercomparison study) as this can be transported and kept under refrigeration conditions (4 °C), facilitating the logistics for shipping and preservation of the material.
A similar behavior was observed for both materials at −20 °C and 4 °C for the long-term stability study. Although a significative trend was not observed, the slope and the correlation coefficients based on the regression analysis were higher while the p-values were lower for samples stored at 4 °C. Apparently, this trend was a consequence of high dispersion of results (at week 15) more than the instability degree of the material. Therefore, an intermediate stability point will be required.
For material characterization, seven validated genes could be used to quantify the RMs. However, using a lower number of units, we chose the two most specific genes that exhibited the best performance in the validation study and measured three different units across three different days to complete nine units once we checked the material was homogeneous and stable.
Many laboratories perform qPCR assays and use a logarithmic transformation of concentration (base10), to relate the concentration with Ct values and the expected changes associated with an uncertainty of 10% (as a nominal value) in the high and low-concentration range. This would produce a change in the instrumental response of approximately 1% (Table 7), which could be considered as fit for purpose in measurements by qPCR.
For the uncertainty components, the contributions were similar for both low and high copy number concentration batches, with stability as the higher contributor to combined standard uncertainty at 60% and 75%, respectively. Thus, to improve stability and material uncertainty, it is necessary to change cryovials to avoid evaporation and changes in the assigned value.
The material developed in this work offers several advantages over others on the market. As gDNA, this contains several target genes used to detect not only E. coli O157:H7 but also other serotypes of this genus, which could contribute to the comparability of the results obtained. Once the material is certified, it could be used to quantify microorganisms in samples using calibration curves and to validate various methods that detect serotypes of E. coli. Finally, this RM could be used as a quality control in testing laboratories that detect this E. coli O157:H7 and other strains of this species as well as being used to evaluate the measurement performance of laboratories that use qPCR methods.
5. Conclusions
We developed a simplex ddPCR method using seven targets for the identification and quantification of E. coli O157:H7. The method, when used with the target gene panel proposed here, can distinguish several serotypes of E. coli, and quantify E. coli serotypes from 6.6 to 7,900 copies/µL, with an LOD of 0.27 copies/µL in the PCR mixture; the RSD by repeatability precision was between 1% and 13.6%, and the relative uncertainty was between 3.5% to 14.6% over the working interval. This method can be used to characterize E. coli gDNA RMs to estimate sample DNA concentrations and distinguish serotypes. The method validated here could ensure the traceability and comparability of results obtained by PCR methods for the detection and quantification of E. coli serotypes in food.
Furthermore, this is the first report that describes the production of quantitative RMs of E. coli O157:H7 gDNA for PCR assays and provide guidAnce to produce similar RMs for the quality control of biologicals. In summary, we produced two batches of a gDNA candidate RM, each of which had 80 bottles. The RM had adequate homogeneity and stability for the intended use. This presentation allows application without high manipulation by the user. Such reference material would improve interlaboratory comparisons and contribute toward standardization of methodologies. The validated assay and the candidate RM produced could contribute to improving food safety and to increasing competitiveness in agricultural product markets.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1. Primers and probes of target genes used for E. coli ddPCR study; Figure S1. 2% agarose electrophoresis of amplification products of E. coli O157:H7 gene targets; Figure S2. Linear interval for DNA targets in E. coli O157:H7 gene quantification by ddPCR; Tables S2-S8. Data of precision experiments performed in three different days at five sample concentration levels with three replicas for each sequence target.; Table S9. Analysis of variance of homogeneity between bottles of pilot batches.; Table S10. Regression analysis results for short- and long-term stability study for high (HL) and low (LL) concentration level materials at 4ºC and -20ºC.; Table S11. Regression analysis results for short- and long-term stability study for high (HL) and low (LL) concentration level materials at 4ºC and -20ºC; Table S12. Characterization results of batch 100.000 copies/μL with Z3276; Table S13. Characterization results of batch 100.000 copies/μL with rfbE; Table S14. Characterization results of batch 100 copies/μL with Z3276; Table S15. Characterization results of batch 100 copies/μL with rfbE; Figure S3. Short term stability study results (relative to controls) for High- and low-level copy number concentration batches. Figure S4. Uncertainty estimation candidate for reference material; Table S16. dMIQE checklist.
Author Contributions
Jhon E. Leguizamón: Conceptualization, formal analysis, writing - review & editing. Claudia P. Tere-Peña: Investigation, formal analysis, writing - original draft preparation. Martha N. Calderón: Conceptualization, supervision, project administration, review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Departamento Administrativo de Ciencia, Tecnología e Innovación (Colciencias), project 110174558815, contract 018:2017, and the División de Investigación Sede Bogotá (DIB), Universidad Nacional de Colombia, Call 2016–2018, project 37670. The APC was funded by National Metrology Institute of Colombia (INM).
Acknowledgments
Dr. Magdalena Wisner of Instituto Nacional de Salud for providing Salmonella isolates from clinical samples.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- S. Bouzari, A. Jafari, and M. M. Aslani, “Escherichia coli: A brief review of diarrheagenic pathotypes and their role in diarrheal diseases in Iran,” Iran. J. Microbiol., vol. 4, no. 3, pp. 102–117, 2012.
- J. S. Kim, M. S. Lee, and J. H. Kim, “Recent Updates on Outbreaks of Shiga Toxin-Producing Escherichia coli and Its Potential Reservoirs,” Front. Cell. Infect. Microbiol., vol. 10, no. June, pp. 1–10, 2020. [CrossRef]
- A. Rani, V. B. Ravindran, A. Surapaneni, N. Mantri, and A. S. Ball, “Review: Trends in point-of-care diagnosis for Escherichia coli O157:H7 in food and water,” Int. J. Food Microbiol., vol. 349, no. May, p. 109233, 2021. [CrossRef]
- K. D. A. Pangajam, K. Theyagarajan, “Highly sensitive electrochemical detection of E. coli O157 H7 using conductive carbon dot/ZnO nanorod/PANI composite electrode,” Sens. anf bio-sensing Res., p. 118214, 2019. [CrossRef]
- J. W.-F. Law, N.-S. Ab Mutalib, K.-G. Chan, and L.-H. Lee, “Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations.,” Front. Microbiol., vol. 5, no. January, p. 770, 2015. [CrossRef]
- I. O. for S. ISO, “ISO/TS 13136:2012 - Microbiology of food and animal feed -- Real-time polymerase chain reaction (PCR)-based method for the detection of food-borne pathogens -- Horizontal method for the detection of Shiga toxin-producing Escherichia coli (STEC) and the determination of O157, O111, O26, O103 and O145 serogroups,” 2012. https://www.iso.org/standard/53328.html (accessed Mar. 21, 2019).
- E. M. Nielsen and M. T. Andersen, “Detection and Characterization of Verocytotoxin-Producing Escherichia coli by Automated 5 ′ Nuclease PCR Assay Detection and Characterization of Verocytotoxin-Producing Escherichia coli by Automated 5Ј Nuclease PCR Assay,” J. Clin. Microbiol., vol. 41, no. 7, pp. 2884–2893, 2003. [CrossRef]
- E. T. Gensberg, M. Polt, M. Konrad-k, P. Kinner, A. Sessitsch, and T. Kostic, “Evaluation of quantitative PCR combined with PMA treatment for molecular assessment of microbial water quality,” Water Res., vol. 67, no. 0, pp. 367–376, 2014. [CrossRef]
- S. Das Mitrai et al., “Duplex PCR for specific detection of Escherichia coli and its differentiation from other Enterobacteriaceae,” Indian J. Anim. Sci., vol. 85, no. 8, pp. 16–19, 2015.
- K. Horakova, H. Mlejnkova, and P. Mlejnek, “Specific detection of Escherichia coli isolated from water samples using polymerase chain reaction targeting four genes: Cytochrome bd complex, lactose permease, ??-d-glucuronidase, and ??-d-galactosidase,” J. Appl. Microbiol., vol. 105, no. 4, pp. 970–976, 2008. [CrossRef]
- W. Liang et al., “Quantification of plasmid DNA reference materials for Shiga toxin - producing Escherichia coli based on UV , HR - ICP - MS and digital PCR,” pp. 1–10, 2016. [CrossRef]
- S. Trapmann, P. Catalani, J. Hoorfar, J. Prokisch, P. Van Iwaarden, and H. Schimmel, “Development of a novel approach for the production of dried genomic DNA for use as standards for qualitative PCR testing of food-borne pathogens,” Accredit. Qual. Assur., vol. 9, no. 11–12, pp. 695–699, 2004. [CrossRef]
- L. Wang et al., “Development of a Reference Standard of Escherichia coli DNA for Residual DNA Determination in China,” vol. 8, no. 9, pp. 1–6, 2013. [CrossRef]
- H. J. He, J. L. Almeida, S. P. Lund, C. R. Steffen, S. Choquette, and K. D. Cole, “Development of NIST standard reference material 2373: Genomic DNA standards for HER2 measurements,” Biomol. Detect. Quantif., vol. 8, pp. 1–8, 2016. [CrossRef]
- I. Gutiérrez-Aguirre, N. Rački, T. Dreo, and M. Ravnikar, “Droplet digital PCR for absolute quantification of pathogens,” in Methods in Molecular Biology, Plant Path., vol. 1302, Humana Press, New York, NY, 2015, pp. 331–347. [CrossRef]
- S. Bhat and K. R. Emslie, “Digital polymerase chain reaction for characterization of DNA reference materials,” Biomol. Detect. Quantif., pp. 3–5, 2016. [CrossRef]
- M. H. Cleveland, H. J. He, M. Milavec, Y. K. Bae, P. M. Vallone, and J. F. Huggett, “Digital PCR for the characterization of reference materials,” Mol. Aspects Med., vol. 96, no. January, p. 101256, 2024. [CrossRef]
- L. Deprez et al., “Validation of a digital PCR method for quantification of DNA copy number concentrations by using a certified reference material,” Biomol. Detect. Quantif., vol. 9, pp. 29–39, 2016. [CrossRef]
- P. Van Iwaarden et al., Certification of a Reference Material of Purified Genomic DNA from Escherichia Coli O157 Certified Reference Material IRMM-449, vol. 157, no. Edl 933.
- “SnapGene | Software for everyday molecular biology.” http://www.snapgene.com/ (accessed Mar. 28, 2018).
- R. Owczarzy et al., “IDT SciTools: A suite for analysis and design of nucleic acid oligomers,” Nucleic Acids Res., vol. 36, no. Web Server issue, pp. 163–169, 2008. [CrossRef]
- T. Madden, “The BLAST sequence analysis tool,” in NCBI Handbook, 2nd editio., National Center forBbiotechnology Information (US), Ed. Bethesda, 2013, pp. 1–17. [Online]. Available: http://www.ncbi.nlm.nih.gov/books/NBK153387/.
- D. Morisset, S. Dejan, M. Milavec, K. Gruden, and J. Zel, “Quantitative Analysis of Food and Feed Samples with Droplet Digital PCR,” PLoS ONE, vol. 8, no. 5, 2013. [CrossRef]
- S. L. R. Ellison, M. Rosslein, A. Williams, L. A. Konopelko, and A. V. Garmash, “EURACHEM/CITAC Guide: Quantifying Uncertainty in Analytical Measurement,” Journal of Analytical Chemistry, vol. 58, no. 2. European Federation of National Associations of Analytical Laboratories, p. 191, 2003. [CrossRef]
- B. Magnusson and U. Örnemark, “Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics,” Eurachem Guide. pp. 1–70, 2014. doi: 978-91-87461-59-0.
- C. V. Vallejo, C. P. Tere, M. N. Calderon, M. M. Arias, and J. E. Leguizamon, “Development of a genomic DNA reference material for Salmonella enteritidis detection using polymerase chain reaction,” Mol. Cell. Probes, vol. 55, no. December 2020, p. 101690, 2021. [CrossRef]
- Thermo Scientific, “T042-TECHNICAL BULLETIN NanoDrop Spectrophotometers.” Accessed: Oct. 24, 2019. [Online]. Available: www.nanodrop.com.
- D. Svec, A. Tichopad, V. Novosadova, M. W. Pfaffl, and M. Kubista, “How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments,” Biomol. Detect. Quantif., vol. 3, pp. 9–16, 2015. [CrossRef]
- International Organization for Standardization (ISO), “ISO/Guide 35:2017(en), Reference materials — Guidance for characterization and assessment of homogeneity and stability,” International Organisation for Standardisation, Geneva, Switzerland, 2017. https://www.iso.org/obp/ui/#iso:std:iso:guide:35:ed-4:v1:en (accessed Jan. 14, 2020).
- International Organization for Standardization (ISO), “Evaluation of measurement data — Guide to the expression of uncertainty in measurement,” Int. Organ. Stand. Geneva ISBN, vol. 50, no. September, p. 134, 2008, [Online]. Available: http://www.bipm.org/en/publications/guides/gum.html.
- M. Lauwaars and E. Anklam, “Method validation and reference materials,” Accredit. Qual. Assur., vol. 9, no. 4–5, pp. 253–258, 2004. [CrossRef]
Figure 1.
Precision results for the target genes evaluated in this study. Bars represent the relative standard deviation by repeatability for each gene at 5 levels between 1.3 to 7,900 copies/µL per reaction. L1 (7,920 copies/µL), L2 (718 copies/µL), L3 (66 copies/µL), L4 (6.6 copies/µL), and L5 (1.34 copies/µL).
Figure 1.
Precision results for the target genes evaluated in this study. Bars represent the relative standard deviation by repeatability for each gene at 5 levels between 1.3 to 7,900 copies/µL per reaction. L1 (7,920 copies/µL), L2 (718 copies/µL), L3 (66 copies/µL), L4 (6.6 copies/µL), and L5 (1.34 copies/µL).
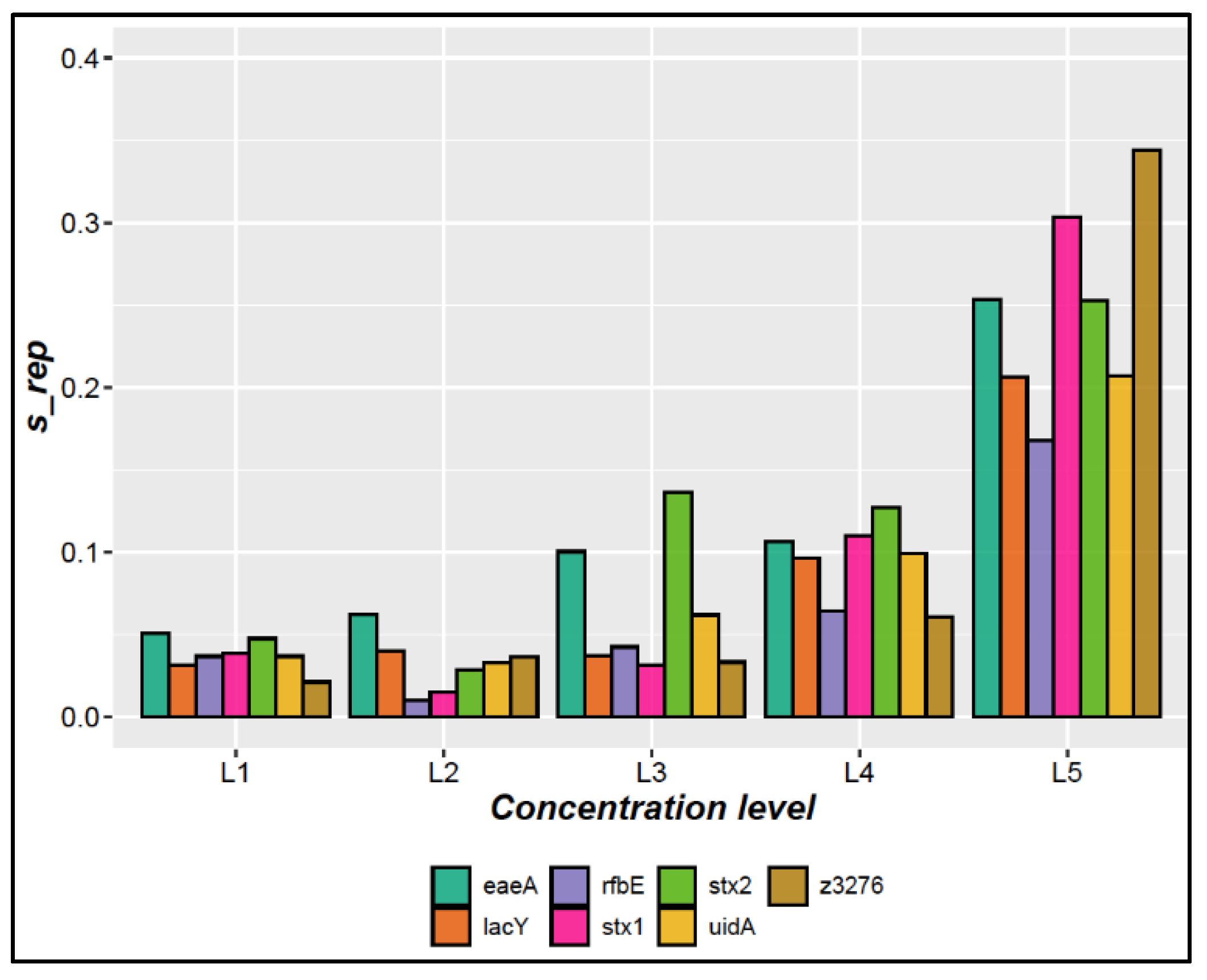
Figure 2.
LOD evaluation for the E. coli O157:H7 ddPCR method. Bars represent the total positive partitions obtained in all replicas for each gene; the red line represents threshold value: nine positive partitions to define LOD.
Figure 2.
LOD evaluation for the E. coli O157:H7 ddPCR method. Bars represent the total positive partitions obtained in all replicas for each gene; the red line represents threshold value: nine positive partitions to define LOD.
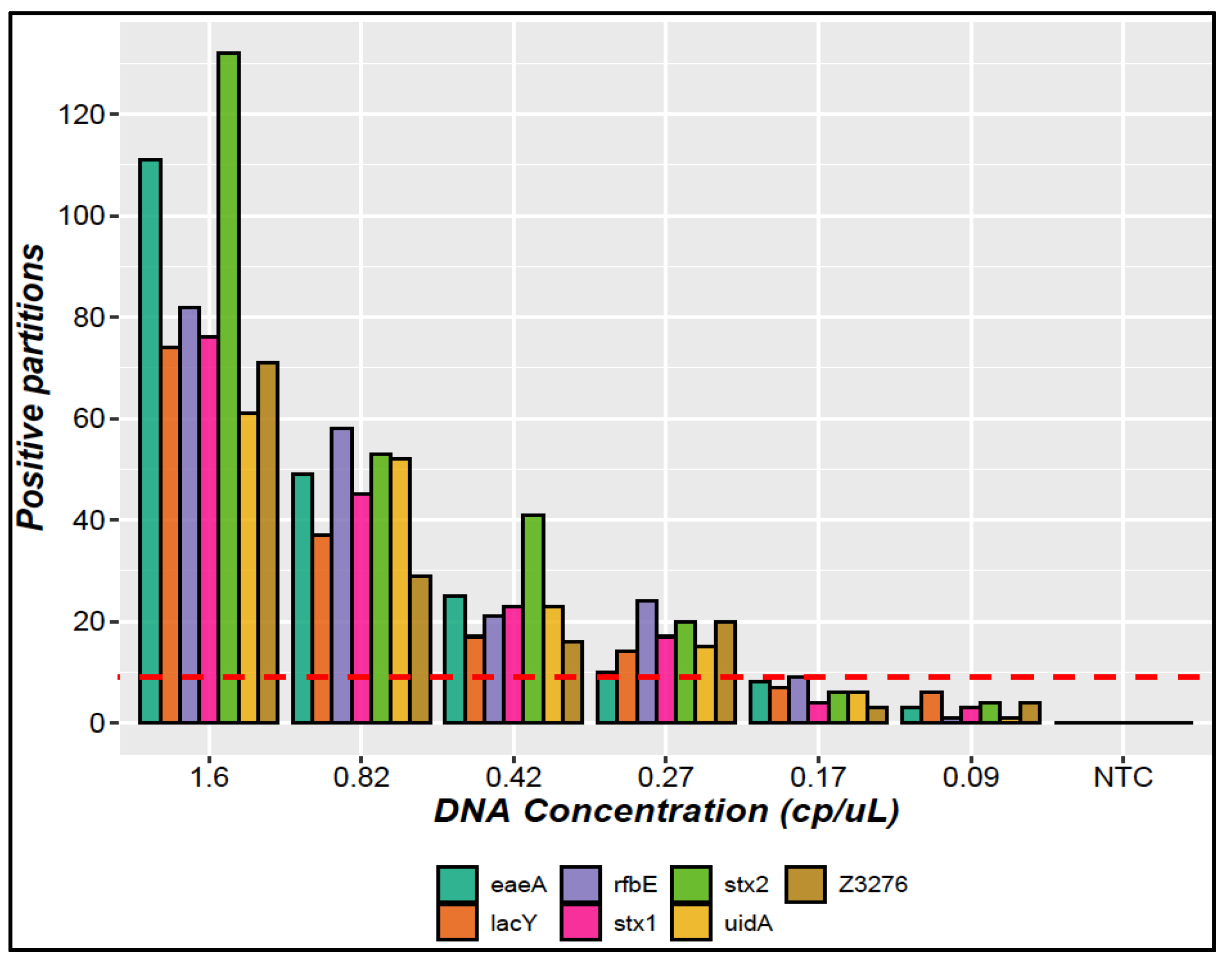
Figure 3.
Schematic representation of the principal factors affecting the measurement value and its uncertainty.
Figure 3.
Schematic representation of the principal factors affecting the measurement value and its uncertainty.
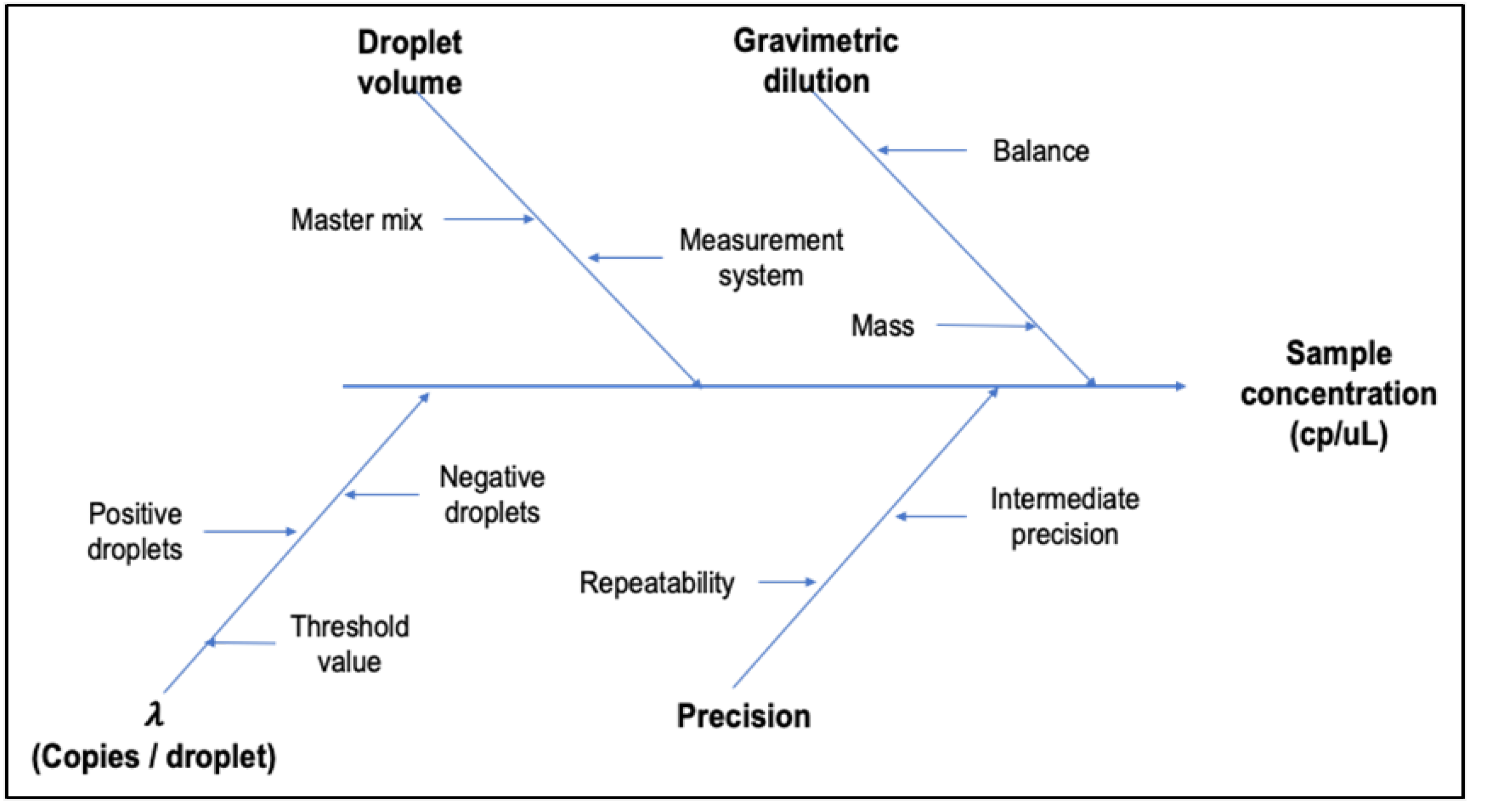
Figure 4.
Study of homogeneity between bottles. Packaging trend evaluation for (a) low level and (b) high level.
Figure 4.
Study of homogeneity between bottles. Packaging trend evaluation for (a) low level and (b) high level.
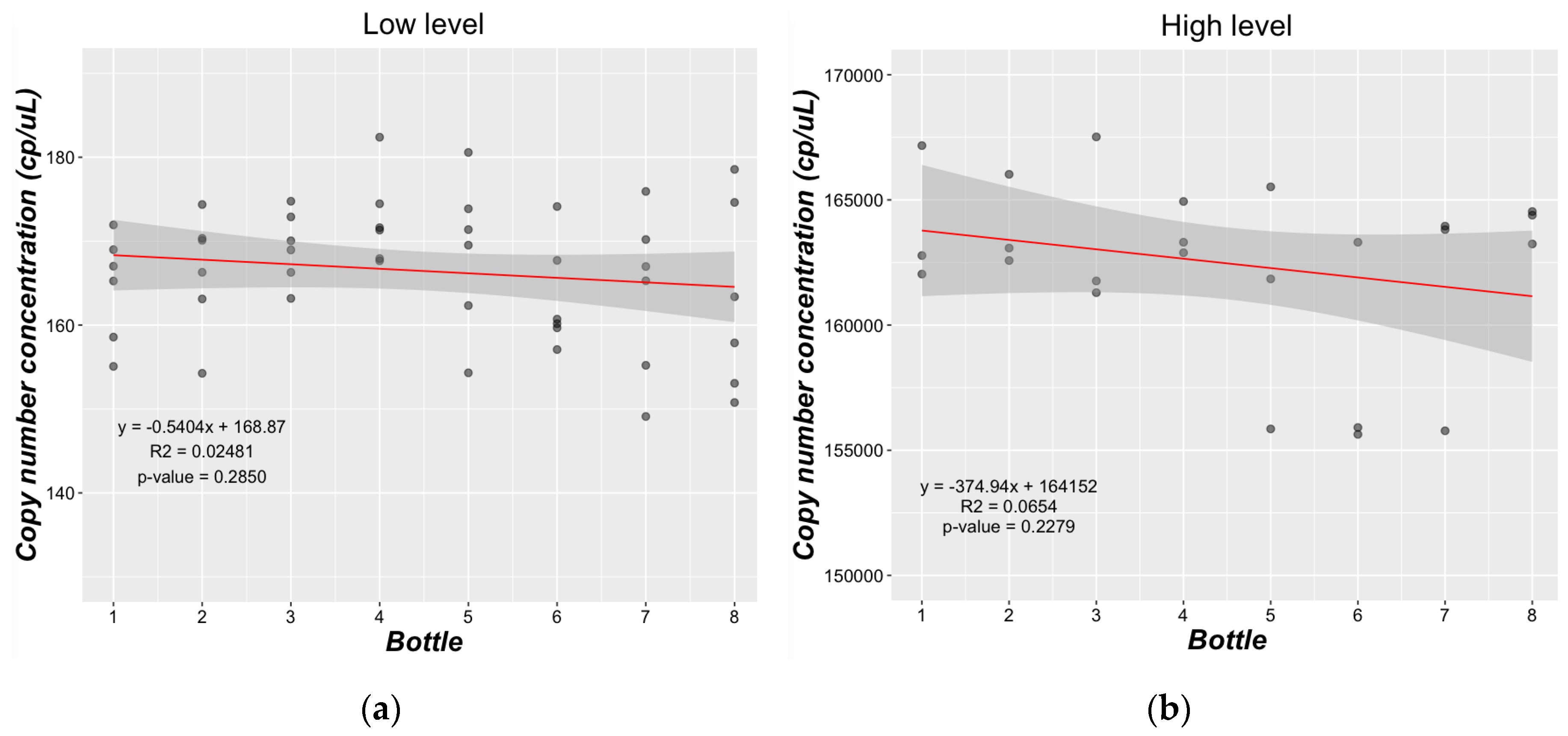
Figure 5.
Long-term stability study. Relative response as a function of time for the (a) high level; and (b) low level. Relative response to sample stored at reference temperature (-70°C).
Figure 5.
Long-term stability study. Relative response as a function of time for the (a) high level; and (b) low level. Relative response to sample stored at reference temperature (-70°C).
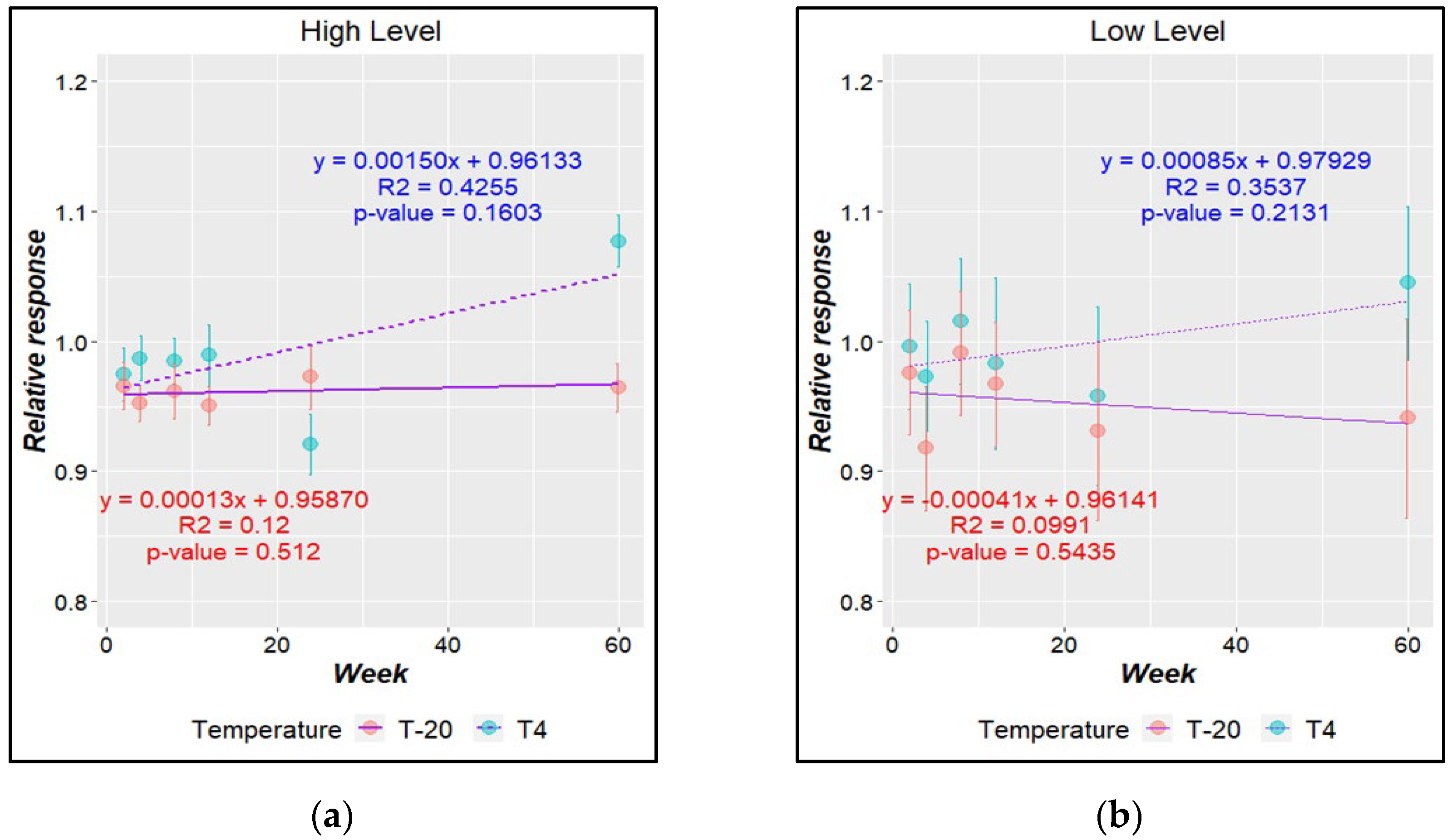
Figure 6.
Schematic representation of the main sources of uncertainty in batches.
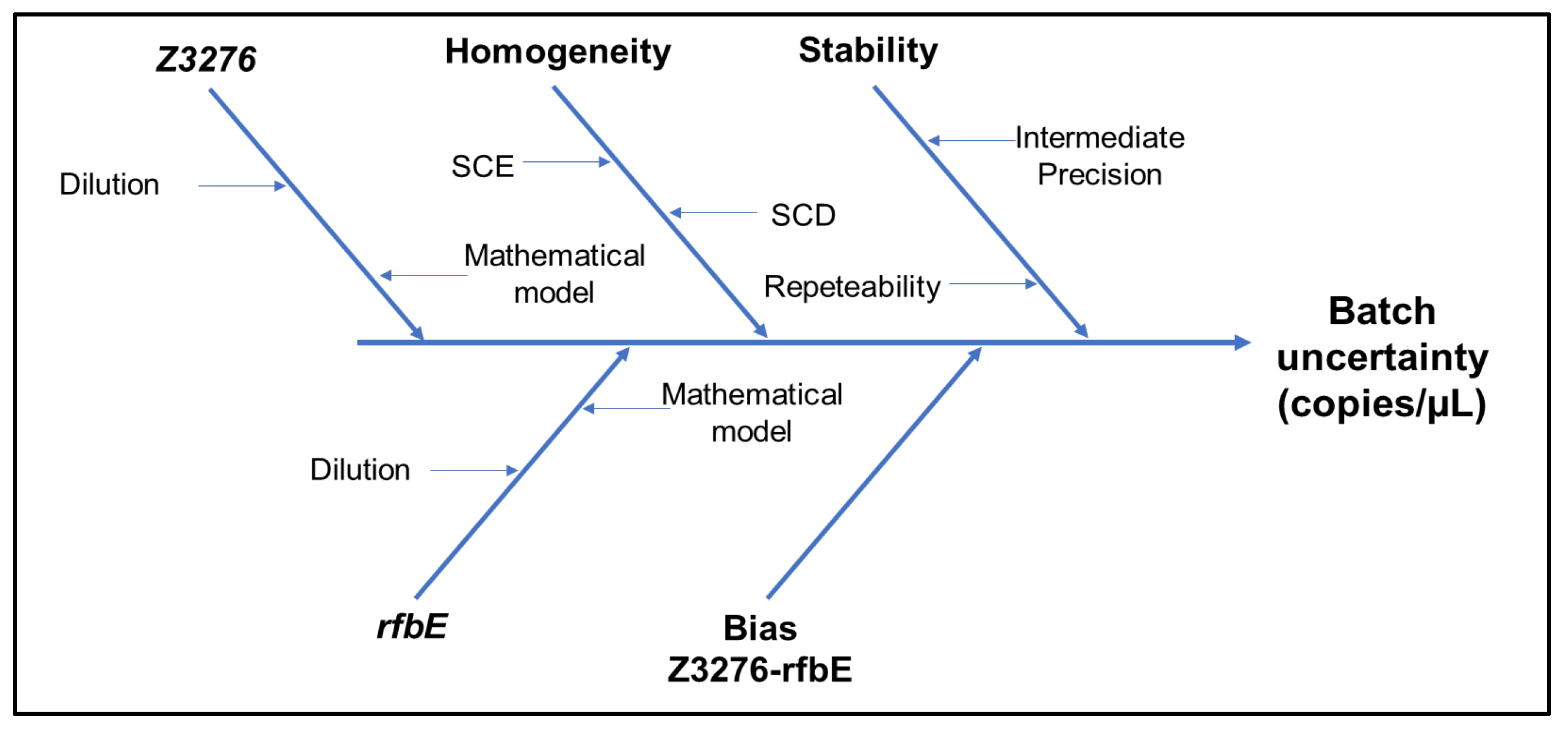
Figure 7.
Comparative results for estimated copy number concentration for low (A) and high level (B) batches with rfbE and Z3276 gene targets.
Figure 7.
Comparative results for estimated copy number concentration for low (A) and high level (B) batches with rfbE and Z3276 gene targets.
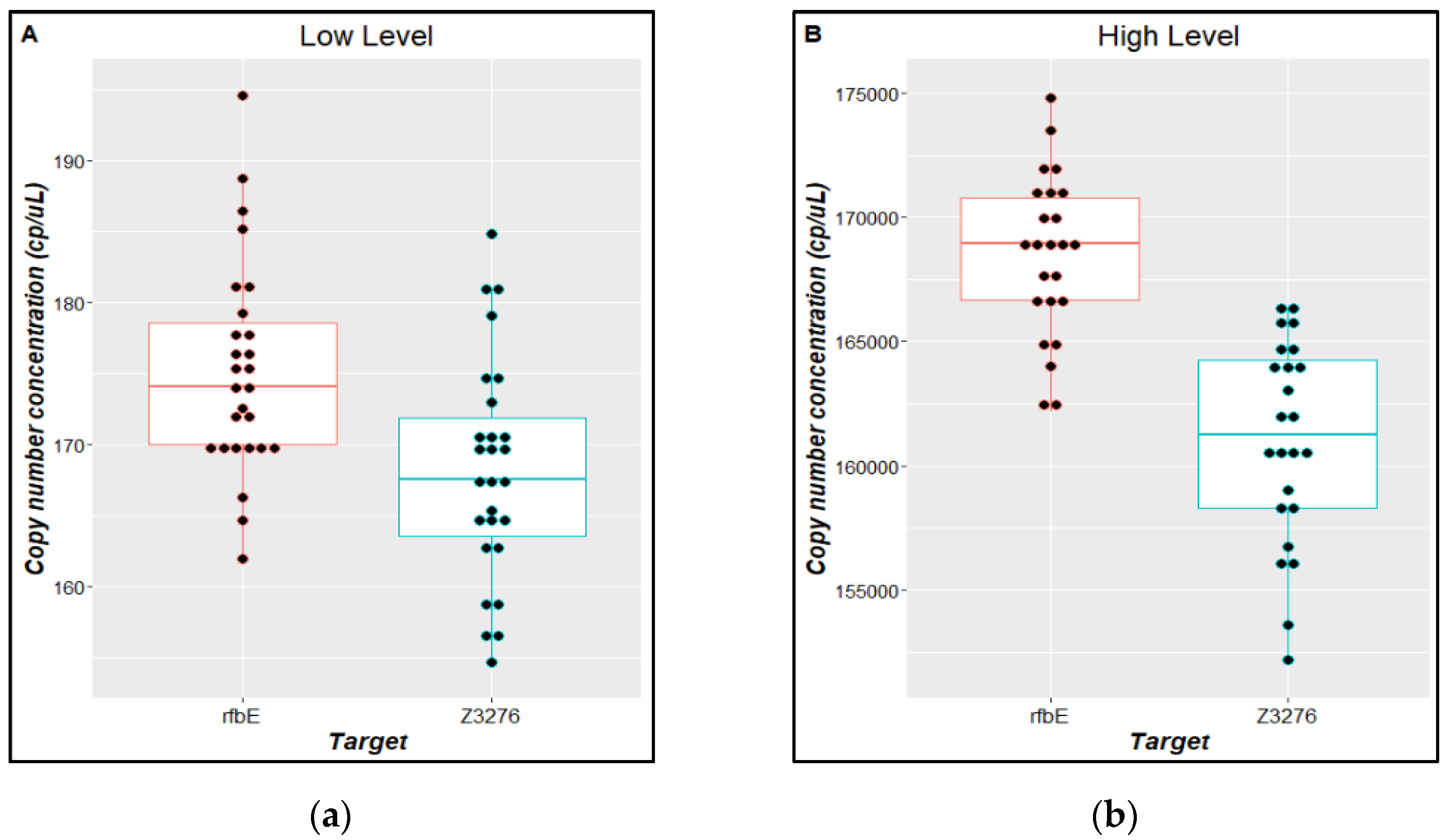
Figure 8.
Comparative results for estimated copy number concentration for low (A) and high level (B) batches with rfbE and Z3276 gene targets.
Figure 8.
Comparative results for estimated copy number concentration for low (A) and high level (B) batches with rfbE and Z3276 gene targets.
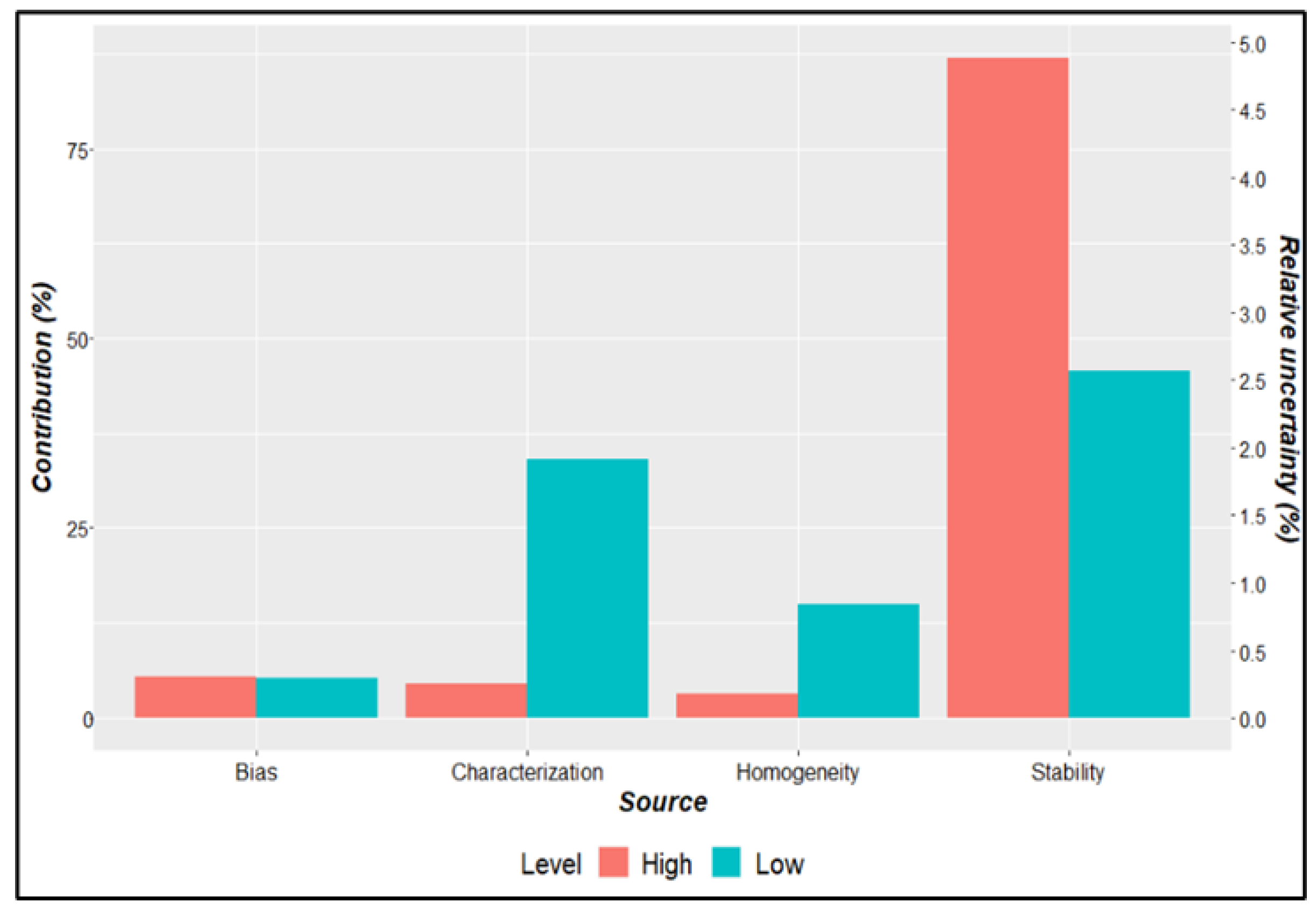
Table 1.
Validation parameters evaluated in ddPCR method validation and acceptance criteria.
| Parameter | Description | Criteria |
|---|---|---|
| Selectivity | Assessed by qPCR, amplifying each gene against a series of related and unrelated bacterial DNA samples. | Positive amplification in E. coli strains. Negative amplification in non-E. coli strains. |
| Working interval | Serial gravimetric dilutions of IRMM 449 over a 5-log DNA concentration range were measured in triplicate for each target gene. Regression analysis was performed to define the working interval. | Correlation coefficient >0.99, a slope significantly (p < 0.05) different from zero, an intercept significantly (p < 0.05) equal to zero, and a precision <25% as relative standard deviation (RSD) |
| Precision | Five concentration levels (L): L1 (7,920 copies/µL), L2 (718 copies/µL), L3 (66 copies/µL), L4 (6.6 copies/µL) and L5 (1.34 copies/µL) were measured in triplicate on three different days for each target gene. | A repeatability RSD <25% was used as acceptance criteria [23]. |
| Limit of quantification (LOQ) |
Defined as the lowest level of the working interval fulfilling linearity and precision criteria. | |
| Limit of detection (LOD) | Six concentration levels below the quantification limit were evaluated in triplicate. | The LOD was established as the lowest copy number concentration level (copies/μL) where three replicates amplify with at least nine positive partitions together [23]. |
| Uncertainty1 | Evaluated for each DNA target in each copy number concentration level, from mathematical model and precision data, according to GUM and EURACHEM guide [24]. |
1 Measurement uncertainty is not a performance characteristic of a particular measurement procedure, but a property of the results obtained using that measurement procedure [25], it was estimated from the validation data.
Table 3.
Linear regression analysis for E. coli O157:H7 quantification by ddPCR.
| Gene | Slope | Intercept | Correlation coefficient (r2) |
|---|---|---|---|
| uidA | 1,516,070 ± 7,553 | −5.3 ± 13.9 | 0.99980 |
| lacY | 1,652,697 ± 14,605 | −17.8 ± 26.5 | 0.99938 |
| eaeA | 1,732,747 ± 15,360 | −21.2 ± 28.8 | 0.99937 |
| rfbE | 1,688,518 ± 19,028 | −12.1 ± 34.8 | 0.99899 |
| stx1 | 1,683,566 ± 19,537 | −23.5 ± 35.4 | 0.99892 |
| stx2 | 1,894,405 ± 11,534 | −22.3 ± 27.3 | 0.99970 |
| Z3276 | 1,612,203 ± 12,551 | −6.5 ± 22.9 | 0.99952 |
Table 4.
Relative standard uncertainty for each gene by the E. coli ddPCR method.
| Concentration level (copies/µL) | uidA | lacY | eaeA | rfbE | stx1 | stx2 | Z3276 |
| 7,920 | 3.5% | 4.1% | 8.4% | 4.1% | 4.1% | 4.6% | 3.4% |
| 718 | 2.7% | 3.0% | 4.2% | 2.5% | 2.0% | 2.2% | 2.9% |
| 66 | 4.8% | 4.2% | 6.6% | 5.0% | 4.2% | 8.0% | 4.4% |
| 6.6 | 13.2% | 12.9% | 12.3% | 7.4% | 13.4% | 13.4% | 11.3% |
Table 5.
Stability uncertainty for candidate RM batches.
| Batch | Storage temperature | |||
|---|---|---|---|---|
| 4°C | −20°C | 4°C | −20°C | |
| ustb | urel (%) | ustb | urel (%) | |
| Low level | 5.95 | 3.4% | 6.37 | 3.7% |
| High level | 8,632 | 5.2% | 1,822 | 1.1% |
ustb stability uncertainty, urel relative uncertainty.
Table 6.
Assigned value and uncertainty for evaluated batches.
| Batch | Gene Z3276 | Gene rfbE | Uncertainty sources | |||||
|---|---|---|---|---|---|---|---|---|
| Value (copies/µL) |
uZ3276 | Value | urfbE | ubias | ucharac. | uhomog. | ustab. | |
| High Level | 16.10 | 2.87 | 168.51 | 2.63 | 2.16 | 1.95 | 824 | 8.63 |
| Low Level | 168 | 7 | 175 | 8 | 2 | 5 | 2 | 6 |
| Value assignment | ||||||||
| Batch | Value | u | Relative u (%) | k | U | Relative U (%) | Batch | Value |
| High level | 164.770 | 9.140 | 5.5 | 2 | 18.280 | 11.1 | High level | 164.770 |
| Low level | 172 | 8 | 4.9 | 2 | 17 | 9.8 | Low level | 172 |
U = expanded uncertainty, u = combined standard uncertainty.
Table 7.
Variation in instrumental response (Ct) as a function of 10% of uncertainty in the assigned value.
Table 7.
Variation in instrumental response (Ct) as a function of 10% of uncertainty in the assigned value.
| Level | Concentration (copies/µL) |
log10 concentration |
Interpolated Ct* | ∆ Ct | ∆ Ct rel (%) |
| High | 100 000 | 5.00 | 23.4 | 0.29 | 1.24 |
| 90 000 | 4.95 | 23.5 | |||
| 110 000 | 5.04 | 23.3 | |||
| Low | 100 | 2 | 33.4 | 0.29 | 0.87 |
| 90 | 1.95 | 33.5 | |||
| 110 | 2.04 | 33.2 |
*Estimated from concentration assuming a 100% efficiency reaction (slope −3.32 and intercept of 40).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



