
Submitted:
16 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
The water-soluble vitamin - vitamin B12, also known as cobalamin, plays a crucial role in cellular metabolism, particularly in DNA synthesis, methylation, and mitochondrial functionality. Its de-ficiency can lead to hematological and neurological disorders, however the manifestation of these clinical outcomes is relatively late. It leads to difficulties in its early diagnosis, often resulting in the development of hidden subclinical cobalamin deficiency. Prolonged lack of vitamin B12 con-dition may have severe consequences including increased morbidity to neurological and cardio-vascular diseases. Beyond inadequate dietary intake, vitamin B12 deficiency might be caused by insufficient bioavailability, blood transport disruptions, or impaired cellular uptake and metabo-lism. Despite nearly 70 years of knowledge since its isolation and characterization, there are still gaps in understanding its metabolic pathways. Thus, this review aims to gather current knowledge about the crucial proteins necessary to efficiently process vitamin B12 in humans, presenting it as a multi-protein mediated network. Based on these findings, we also highlight new potential methods for supporting the evaluation of the risk of epidemiological consequences of vitamin B12 deficiency or clinical warnings of increased risk of vitamin B12 deficiency based on already testing targeting specific moonlighting proteins engaged in vitamin B12 metabolic path-ways.
Keywords:
vitamin B12
; metabolism
; proteins
; anemia
; neurodegeneration
1. Introduction
Vitamin B12 was initially identified in 1948 by two independent scientific teams—Folker's in the USA and Smith's in Great Britain—as an antipernicious anemia factor [1]. Structurally, it possesses an organometallic nature, with the cyanide ion coordinated to the central cobalt atom. The primary Co ligand consists of a corrin macrocycle containing four reduced pyrrole rings and seven amide side chains [2]. The precise structure varies among specific forms of vitamin B12. Hydroxycobalamin and cyanocobalamin are metabolically inactive; however, upon conversion to methylcobalamin and 5-deoxyadenosylcobalamin, they attain full activity [3]. The term "cobalamin" (Cbl) encompasses all chemical derivatives wherein Co3+ is typically coordinated by various ligands on the upper surface of the tetrapyrrolic corrin ring e.g., CN, HO, methyl, or 5-deoxyadenosyl (Figure 1). Among these, HOCbl represents the weakest coordination ligand, while MeCbl exhibits the strongest coordination [4]. Co-C bond dissociation enthalpy (BDE) was measured from kinetic calorimetric methods for the range of 10-30 °C, giving MeCbl the greatest value of 163 ± 21 kJ/mol [2].
Humans cannot endogenously synthesize cobalamin and, consequently, must obtain it through dietary intake, predominantly sourced from meat, fish, dairy products, cheese, eggs, or edible algae. Diminished intake or impaired absorption of B12 commonly manifests in elderly individuals or in vegans adhering to dairy and egg-free diets that are inadequately supplemented [3]. Intestinal incorporation of vitamin B12 is supported by a specific genre of human gut microbiota. Nevertheless, not all forms of cobalamin they produce find their biological function in human metabolism, and might even disrupt the normal metabolism of this vitamin [5]. One of the examples is Coα-[α-(7-adenyl)]-Coβ-cyanocobamide, a corrinoid isolated from Lactobacillus reuteri named commonly pseudovitamin B12. This analog shares a structure resembling cobalamin, except for the lower cobalt ligand. In this modification, adenine substitutes the 5,6-dimethylbenzimidazole [6]. Even though pseudovitamin B12 does not inhibit the uptake of B12 in rats [9], some studies suggest it could potentially disrupt the absorption of a physiological trace concentration of vitamin B12 in the medium through the mediation of transcobalamin II (TCN.2) [10].
Following uptake, it undergoes further transformation as presented in Figure 2. Absorption of dietary vitamin B12 is preceded by realising free cobalamin bound with food protein. First gastric acid and proteases take action in the stomach. Afterward, a salivary protein called haptocorrin (HC) binds to freed vitamin B12 to be subsequently degraded by the alkaline environment of the upper small intestine. Next, in the proximal ileum cobalamin creates a complex with cobalamin binding intrinsic factor (IF), which is further transported and recognized by certain receptors of distal ileum enterocytes. Finally, absorbed vitamin B12 is degraded and captured by transcobalamin II (TCN2) [9]. TCN2 caries about 20% of the vitamin B12 in the circulation [3]. Afterward, cellular uptake of vitamin B12 involves the CD320 receptor, present in all cell types [10]. Next, the B12-TCN2 complex is transferred into lysosomes where it breaks down. ATP Binding Cassette Subfamily D Member (ABCD4) and Lysosomal cobalamin transport escort protein (LMBD1) receptors are responsible for its further transport from the lysosomal lumen to the cytosol. When cobalamin is released into the cytosol, cytoplasmic chaperons – Methylmalonic aciduria and homocystinuria type C protein (MMACHC), and Methylmalonic aciduria and homocystinuria type D protein (MMADHC), whose functions have been not fully established yet, interact with each other and make vitamin B12 available for further processing [11]. In the cytoplasm, vitamin B12 is converted into two active cofactors: methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl). MeCbl acts as a carrier of methyl groups. It is a co-factor of the enzyme methionine synthase (MTR), which is involved in the transformation of homocysteine to methionine in the cytosol. These processes and subsequent metabolic reactions are involved in the synthesis of neurotransmitters, phospholipids, DNA, and RNA. On the other hand, AdoCbl acts as a cofactor of a mitochondrial enzyme, methylmalonyl-coenzyme A mutase (MMUT), which converts methylmalonyl-CoA to succinyl-CoA. This reaction is involved in the catabolism of cholesterol, fatty acids, and several amino acids [10].
Due to its involvement in various cellular pathways and DNA synthesis, alterations in vitamin B12 metabolism strongly correlate with cognitive impairment, hematological disorders, neural tube defects during embryo development, and cardiovascular diseases [12]. However, it is considered that approximately 20% of the gene products associated with investigated cases of inherited vitamin B12 malabsorption remain uncharacterized [13]. Thus, more in-depth research is necessary to fully elucidate the steps and compounds involved in vitamin B12 metabolic pathways.
This review aims to comprehensively summarize all identified proteins essential for the proper metabolism of vitamin B12 in humans and diseases related to its dysfunction. Furthermore, based on the collected data, we propose new diagnostic options as well as new warnings within the field of vitamin B12 deficiency research.
2. Proteins Involved in Vitamin B12 Metabolism
2.1. Gastrointestinal Vitamin B12 Transport
2.1.1. Haptocorrin (HC) and Intrinsic Factor (IF)
As previously elucidated, the absorption and utilization of vitamin B12 within the body require its initial binding to carrier proteins in the gastrointestinal tract. In humans, two proteins facilitating this binding process are haptocorrin (HC) and Intrinsic Factor (IF). Despite their immunological distinctiveness, they exhibit structural similarities characterized by a substantial degree of glycosylation [14,15]. In humans, parietal cells within the gastric mucosa of the ileum produce Intrinsic Factor (IF). The gene responsible for encoding IF is known as Gastric Intrinsic Factor (GIF) or Cobalamin Binding Intrinsic Factor (CBLIF) , and it is recognized on chromosome 11 (11q13) in humans and on chromosome 19 in mice [16,17]. IF is made of 399 amino acid residues [15], and has a two-domain organization, with each domain having a mass of approx. 30 kDa and 20 kDa, respectively, in between vitamin B12 is bound with corrin ring [15].
Initially named the R-binder or transcobalamin I (TCN1), haptocorrin (HC) is the first carrier protein for ingested vitamin B12. Investigation of TCN1 gene sequence and its localization, suggest that HC is an evolutionary product of CBLIF duplication [18]. This glycoprotein of 60 to 70 kDa [18] is present in various body fluids including saliva, breast milk, bile, tears, and blood plasma [16,19]. Upon ingestion, vitamin B12 is bound by HC secreted in saliva, which is responsible for transferring the vitamin to the stomach. HC also binds dietary, released from food carrier proteins vitamin B12 in the stomach [16]. In the acidic pH of the stomach, HC is thought to protect the vitamin from hydrolysis [20]. Vitamin B12 remains bound to HC in the small intestine, although with the increase in the pH, the affinity of vitamin binding by HC decreases [21]. In the duodenum, pancreatic proteases- namely trypsin, chymotrypsin, and elastase- degrade HC, allowing for the release of vitamin B12 and its subsequent binding by IF [10]. Interestingly, deficiencies of pancreatic proteases can lead to defects in vitamin B12 absorption due to the inability to degrade HC and release the vitamin from the vitamin B12-HC complex. Patients with exocrine pancreatic insufficiency or chronic pancreatitis, conditions marked by inadequate delivery of pancreatic digestive enzymes to the intestine, often develop vitamin B12 malabsorption. However, these conditions rarely lead to severe B12 deficiency [22,23,24].
Studies on the exact role of HC in humans are limited due to the lack of homologous genes encoding HC in neither mice nor rats [25]. In these rodents, salivary glands produce another vitamin B12-binding protein transcobalamin (TC), which in humans is responsible for binding the vitamin in blood plasma [10]. This points out that the binding of ingested vitamin B12 varies between species, with some possessing HC and others not, which highlights the need to be cautious when studying HC in in vivo models [25].
After the release of vitamin B12 from HC in the duodenum, the vitamin is taken up by Intrinsic Factor (IF). IF is mostly found in the gastric juice and ileal fluid in humans and is produced and secreted by parietal cells in the stomach. It is responsible for transferring vitamin B12 along the small intestine, to the intestinal epithelial cells, which are mainly composed of enterocytes [15,20]. When vitamin B12 is bound to IF, the domains can be assembled. This mechanism ensures that only the IF-vitamin B12 complex, but not the apoprotein, can be recognized by specific receptors on the mucosal cells along the ileum and absorbed through receptor-mediated endocytosis [14,26,27]
IF deficiency can be caused by the presence of anti-IF or anti-parietal cell autoantibodies, or the loss of parietal cells. The presence of autoantibodies is often linked to Helicobacter pylori (H. pylori) infection. Patients with H. pylori infection have been found to have decreased vitamin B12 levels in serum [28]. Alternatively, in autoimmune gastritis (AIG), an inflammatory disease affecting the stomach and resulting in mucosal atrophy, deficiency of IF arises due to the loss of parietal cells. AIG is also characterized by the presence of anti-IF and anti-parietal cell antibodies that disrupt IF function [29]. The exact cause of AIG is still unknown and there is no treatment available. The wide range of unspecific clinical symptoms, often subtle (many patients are asymptomatic for a long period after the disease onset), result in difficulties in diagnosing the condition promptly [30]. The lack of IF in this condition leads to long-lasting vitamin B12 deficiency due to the inability to deliver vitamin B12 to the cells within the ileum, and subsequent failure to distribute it throughout the body. The consequence of vitamin B12 deficiency in AIG is pernicious anemia (PA) [10,31]. Around 10-15% of patients with AIG develop PA, characterized by both hematologic and neurologic impairments. The hematologic manifestation includes megaloblastic anemia; while the clinical neurologic manifestations of PA include weakness, depression, impaired nerve function, cognitive impairment, and sensory deficits [10,32].
2.1.2. Cubam Receptor
When the vitamin B12-IF complex reaches the intestine, it is internalized into enterocytes by receptor-mediated endocytosis. Cubam, the ileal IF-vitamin B12 receptor, is a multiligand, apical membrane receptor expressed in absorptive epithelia in several tissues. In the kidney, Cubam is expressed in the proximal tubular epithelium, where it mediates the reabsorption of proteins from the renal filtrate, such as albumin, apolipoprotein A-1, hemoglobin, transferrin, and vitamin-D binding protein, leading to reduced proteinuria [33,34]. Cubam is also present in the visceral yolk sac, where it is essential to coordinate maternal nutrient uptake and proper embryo development [35,36]. In the distal ileum, Cubam is expressed in the intestinal epithelium, and its only known function is to facilitate the uptake of the IF-vitamin B12 complex [34].
Cubam is composed of two proteins, cubilin (CUBN), and type-1 transmembrane protein amnionless (AMN). CUBN is a 460 kDa protein composed of three distinct regions: an N-terminal domain, responsible for membrane association; an extracellular fragment formed by epidermal growth factor-like repeats; and 27 domains of complement components C1r/C1s, epidermal growth factor-related protein 1 (Uegf) and bone morphogenetic protein 1 (Bmp1), CUB in short [37]. Four CUB domains, CUB5-8 are responsible for the interaction with IF, as revealed by crystal structures [38]. The remaining domains are involved in the recognizing and binding of other ligands, for example during ultrafiltration and reabsorption in the kidney [39]. To form a receptor, three CUBN subunits combine in a pin-shaped molecule that docks to transmembrane protein AMN. CUBN lacks a transmembrane domain and therefore the interaction with AMN is necessary for the formation of a functional Cubam complex [33].
AMN is a 45-kDa transmembrane protein also composed of three parts: an extracellular domain, a transmembrane helix, and a cytoplasmic domain responsible for clathrin-mediated internalization. In addition to anchoring CUBN to the apical membrane of epithelia, AMN is also required for biosynthetic processing and trafficking of CUBN to the plasma membrane. It also mediates Cubam receptor recycling, and endocytic signaling [40]. The cytoplasmic domain of AMN features two putative FXNPXF endocytic signals that closely resemble the FXNPXY signal found in the low-density lipoprotein receptor superfamily (LDLR). This signaling involves clathrin-associated sorting proteins (CLASPs) and promotes the clathrin-dependent internalization of various cubam ligands in an analogous way to the internalization of LDLRs [34]. LDLR family also includes the protein Megalin, which strongly colocalizes with Cubam, especially in the kidney where it cooperates in ligand recognition. However, in the intestine, Cubam appears to be functioning independently of Megalin, suggesting that only AMN and CUBN are required to form a receptor specific for IF-vitamin B12 internalization [41].
Mutations in either of the CUBN or AMN genes disrupting Cubam receptor functioning result in malabsorption of vitamin B12 and the development of Imerslund-Gräsbeck syndrome (IGS). IGS is a rare autosomal recessive disorder that appears in childhood, characterized by megaloblastic anemia as a consequence of vitamin B12 deficiency [42]. Most IGS patients develop mild proteinuria and neurological impairments. Although the disease may be fatal if left untreated, treatment with parenteral vitamin B12 therapy, typically via regular, intramuscular injections is effective in managing the condition [43,44]. Recent data also demonstrate altered expression of AMN in the kidney and the ileum with aging. This further leads to vitamin B12 deficiency independently of nutritional uptake, and the development of age-related chronic diseases [45].
2.1.3. Multidrug Resistant Protein 1 (MRP1)
Upon the uptake to the enterocyte, the next step in vitamin B12 transport is its distribution to other cells and tissues in the body. To achieve this, vitamin B12 needs to be exported into the blood circulation, and this step is mediated by Multidrug Resistant Protein 1 (MRP1).
For the first time described by Cole et al. in 1992, MRP1 is a 190 kDa glycoprotein that has been shown to transport a wide range of substrates including bioactive compounds, signaling molecules, metabolites, as well as anti-cancer drugs, and xenobiotics, thus conferring to multidrug resistance in cancer (MRP1 is overexpressed in many hematological and solid tumors) [46,47]. Under physiologic conditions, MRP1 is ubiquitously expressed in the body, mostly in the lung, testis, kidney, heart, placenta, small intestine, colon, brain, and peripheral blood mononuclear cells [47,48,49]. In the small intestine, MRP1 is mainly present in the basolateral membranes of the crypt cells [50]. In addition to the plasma membrane localization, MRP1 has also been shown to accumulate in subcellular organelles such as endocytic vesicles, trans-Golgi vesicles, and lysosomes [48,51], although its involvement in the intracellular transport of vitamin B12 has never been demonstrated. MRP1, similarly to lysosomal vitamin B12 exporter ABCD4, also belongs to the ABC transporters family, hence its alternative name ABCC1 [46].
MRP1 has been shown to efflux vitamin B12 from the intestinal epithelium to the blood circulation [52]. Mice deficient for MRP1 ( MRP1(−/−) ) were found with decreased levels of vitamin B12 in their plasma, simultaneously accumulating the vitamin in the ileum and colon. These studies also established that MRP1 transports the vitamin in its protein-free form contrary to the previously hypothesized export of the vitamin in complex with transcobalamin [52,53]. In some patients with recessive hereditary vitamin B12 malabsorption, mutations in MRP1-encoding gene ABCC1 were detected, however, they were ruled out as potential causes of vitamin B12 deficiency, as they appeared either in the intronic gene fragments, did not affect open reading frame, or did not fulfill Mendelian rules for inheritance [54]. Even so, these studies were conducted on a small group (approx. 30) of patients, suggesting a need for large-scale epidemiological investigation to clarify the involvement of ABCC1 mutations in vitamin B12 deficiency. Such investigations could potentially validate the redundancy in vitamin B12 export, as the existence of alternative pathways aside from MRP1 export in humans remains unknown. Among other closely related transporters of the ABC family with wide tissue distribution (ABCB6, ABCG2, MRP3, and MRP5), only MRP1 was shown to be involved in vitamin B12 efflux [52]. Moreover, as presented in Beedholm-Ebsen et al. MRP1(−/−) mice have not presented morphologic and metabolic abnormalities, suggesting that other transporters should compensate for the lack of MRP1, and highlighting the need for further studies to reveal other vitamin B12 exporters.
2.2. Proteins involved in Vitamin B12 Blood Transport
2.2.1. Transcobalamin II (TCN2)
When vitamin B12 is exported into the blood circulation, it is bound by a 43 kDa protein transcobalamin, former transcobalamin II (TCN2) [55]. In humans, TCN2 is synthesized by endothelial cells within the intestinal villi and secreted to blood plasma [56,57]. TCN2 is structurally related to IF and HC, with some conserved regions sharing 60-80% homology, and all three vitamin B12 transporters are derived from a common ancestral gene [53,58]. TCN2 is responsible for the distribution of vitamin B12 into all cells in the body, but only about 25% of total vitamin B12 in plasma is bound by TCN2, while the rest is carried by circulating HC [59,60]. The complex of vitamin B12 and transcobalamin is referred to as holoTC and is the “active” form of vitamin B12 available to cells, whereas the biological function of the complex with HC (holoHC) is not clear [20]. Some studies postulate that holoHC is transferred to the liver, as elevated serum vitamin B12 is observed in patients with various liver injuries (it’s hypothesized that either holoHC is not uptaken by the injured liver, or it possibly leaks into the circulation from the destructed hepatocytes, which corresponds to higher vitamin B12 in plasma) [61,62].
HoloTC levels in serum have been suggested as a specific marker of early vitamin B12 deficiency diagnosis with the use of commercially available immunoassays for holoTC [63]. However, some studies indicate that this is a controversial determinant of vitamin B12 status. Some patients with severe vitamin B12 deficiency resulting in PA displayed falsely normal holoTC levels [64]. It was also hinted that the rationale behind diagnostic holoTC immunoassay is based on a false assumption that holoTC is the first factor to respond to vitamin B12 deficiency, where the level of methylmalonic acid (MMA), a form of substrate for methylmalonyl-CoA mutase which requires vitamin B12 as a cofactor, is more sensitive to vitamin B12 deficits in the early stage [65,66].
Mutations in the TCN2 gene can lead to intracellular cobalamin depletion, causing a rare multisystemic disorder characterized by autosomal recessive inheritance with symptoms such as pancytopenia, megaloblastic anemia, developmental delay, diarrhea, psychomotor impairment, and immune system deficiency [67]. Only a few cases have been investigated so far, showing as a cause of gene alternation either large deletion of the entire exon 8 and premature stop codon (p.E371fsX372) [68] or change in intron 4 leading to the loss of the start codon [77].
2.3. Proteins Involved in Intracellular Vitamin B12 Transport
2.3.1. CD320 Receptor
CD320, also known as the receptor for cobalamin-saturated transcobalamin (TCblR) is a small 282 amino acid protein with 29 kDa calculated molecular weight (observed 58 kDa, probably due to the fact of high glycosylation) [70]. It is one of the LDLR protein family. CD320 is usually localized on the cell surface and contains two domains between the transmembrane helix, C-terminal cytoplasmic region, and extracellular N-terminal fragment with an epidermal growth factor (EGF) separating two LDLR type A sites [71,72]. However, its soluble form (sCD320) might be also found in the bloodstream in human serum fraction [73]. CD320 exhibits high specificity and affinity towards transcobalamin, and TC-saturated cobalamin is uptaken by the process of endocytosis in the presence of Ca2+ ions [74,75]. Initially, holo-TC binds to the N-terminal region of CD320 and it is internalized into an endocytic vesicle. Next, endosomes with holo-320 complexes are fused with a lysosome, where TCN2 and CD320 undergo dissociation and degradation. Subsequently cobalamin is freed into the cytoplasm [76].
During the process of holo-TC uptake receptors are broken down, and the occurrence of CD320 on the cell surface is the direct result of its synthesis. Cobalamin might be introduced into cells when it is needed for DNA synthesis and its excess might be recycled [77]. The level of the CD320 receptor is highly associated with the cell-division cycle and its presence is enhanced in fast-growing cancer cells [76]. The impact of CD320 deficiency was also tested on mice models with CD320 knock-out (KO) fed with a vitamin B12 deficient diet. Those animals showed increased MMA, homocysteine level in serum, and macrocytic anemic phenotype [78]. Neutrophil hypersegmentation is usually also a clinical hallmark of cobalamin uptake failure in patients [79]. Moreover, female mice with CD320 depletion and limited cobalamin intake exhibited infertility. Even though supplementation with cyanocobalamin resulted in a successful pregnancy, all litters died just after birth [78]. Other studies indicate that the knockdown of CD320 caused cognitive and learning disorders along with anxiety behavior responding to symptoms observed in humans, which point out direct changes in the central nervous system [80]. It is suggested that observed alternation of DNA methylation in CD320 KO mice brains might cause neuron demyelination [81].
In humans, very few cases of CD320 receptor defects have been confirmed to be the direct reason for B12-deficiency symptoms. Single nucleotide polymorphism of CD320, rs2336573 leading to glycine-to-arginine change has been identified in patients group with lowered B12 levels in serum [82], and single codon deletion of c.262_264GAG (p.E88del) has been linked with an increased MMA concentration in newborns [83].
2.3.2. ATP Binding Cassette Subfamily D Member (ABCD4) and Lysosomal Cobalamin Transport Escort Protein (LMBD1)
The free B12 vitamin is released into the cytoplasm by ATP (adenosine-5′- triphosphate) Binding Cassette Subfamily D Member protein (ABCD4), a 68.6 kDa exporter located on the lysosomal membrane [84]. ABCD4 consists of a transmembrane domain and a nucleotide-binding domain, which is involved in binding and hydrolysis of ATP [85]. It belongs to the family of ATP-binding cassette (ABC) transporters that mediate the transport of various substrates across the cell membrane against the concentration gradient. Most of the human ABC exporters are localized in such a way that allows the substrate to access from the cytosolic side, in a cis-acting manner. As revealed by cryo-electron microscopy (EM) studies, ABCD4 is a unique exporter, facilitating transfer in a trans-acting manner, thus allowing the transfer of vitamin B12 from the lysosome to the cytoplasm [86].
ABCD4 is an endoplasmic reticulum (ER) protein, hence it requires translocation to the lysosome to facilitate vitamin B12 release. ABCD4 was found to colocalize with 61.4 kDa membrane lysosomal cobalamin transport escort protein (LMBD1), which localizes primarily to lysosomes [84,87]. Initially, LMBD1 was suggested to act as a lysosomal exporter of vitamin B12 [88]. However, later findings demonstrated that LMBD1 acts as an adaptor protein, and mediates the translocation of ABCD4 from ER to the lysosomes [89]. Both proteins function in a coordinated manner and form a complex, as demonstrated by Surface Plasmon Resonance (SPR) [87].
Mutations in ABCD4 and LMBDR1 (the latter encoding LMBD1 protein) underlie disease groups cobalamine J and F diseases (cblJ,cblF), respectively [84,88]. At the cellular level, both errors are marked with a decreased function of methylmalonyl-CoA mutase and methionine synthase and an accumulation of free vitamin B12 in the lysosomes [90]. However, up to date, there is no clear biochemical or clinical distinction between cblJ and cblF. Only molecular genetic testing, either gene-targeted (RNA-sequencing, single-gene testing) or comprehensive (e.g., exome sequencing) was successfully applied to confirm either of the defects [91,92,93]. There is a wide spectrum of clinical presentations in both diseases, which may include retarded growth, developmental delay, developmental abnormalities, progressive hyperpigmentation (found only in cblJ type), cardiac defects, and hematologic impairments (such as macrocytic anemia, neutropenia thrombocytopenia, and pancytopenia). Neurological presentations may include general weakness, dizziness, delayed motor development, and mild intellectual disability [84,88,91,94].
2.3.3. Methylmalonic Aciduria and Homocystinuria Type C Protein (MMACHC)
Cobalamin released from lysosomes into the cytosol is available for two cytoplasmic proteins - Methylmalonic Aciduria and Homocystinuria Type C Protein (MMACHC) and Type D Protein (MMADHC). Analysis of individuals with inborn errors of cobalamin metabolism revealed that the MMACHC gene is localized to the 1p34.1 region of chromosome 1 and consists of five exons. From forty-two different mutations in more than 200 cblC c.271dupA, c.394C4T, and c.331C4T were the most frequent [95]. The MMACHC gene product consists of 282 amino acids and has a mass of 31.7 kDa [96]. The MMACHC gene manifests a substantial degree of evolutionary conservation across mammalian species. Nevertheless, a structural analogy is discerned between the MMACHC model and the monomeric C-terminal domain of bacterial TonB. TonB is instrumental in transducing energy obtained from the proton-motive force to facilitate the transport of both iron and cobalamin across the outer bacterial membrane [96]. MMACHC might operate in a manner akin to TonB, potentially receiving the cobalamin cargo upon its release from the lysosomal compartment. Nevertheless, the identity of the transporter implicated in this process remains elusive [96]. The MMACHC gene exhibits high expression levels in the fetal liver, with its presence also identified in a broad spectrum of tissues, including the spleen, thymus, and bone marrow [96].
MMACHC encodes a protein possessing both chaperone and enzyme functionalities. Its inactivation disrupts the synthesis of two essential cobalamin derivatives, methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl), cofactors for enzymes dependent on vitamin B12 [97]. The defects in MMACHC are presumed to impact a stage occurring after the cellular absorption of cobalamin but preceding the synthesis of AdoCbl and MeCbl. Thus, it occurs before the binding of the cobalamin coenzymes to their respective apoproteins [96] MMACHC stands out among B12 proteins due to its distinctive ability to manipulate the cobalt-carbon bond in B12 through both homolytic and heterolytic mechanisms. It catalyzes the reductive decyanation of cyanocobalamin (CNCbl or vitamin B12 in conjunction with a flavoprotein oxidoreductase [98]. Interestingly, CblC also exhibits the remarkable capability to catalyze the nucleophilic displacement of the alkyl group by glutathione when presented with an alkylcobalamin. The intricate protein-protein interaction between MMACHC and MTR is widely acknowledged for its pivotal role in regulating intracellular cobalamin (Cbl) metabolism. Notably, the interaction of MTR inactive isoforms impedes MMACHC activity [99]. MMCHC is essential also in the conversion of MMA to succinyl-CoA [98].
Mutations in MMACHC gene can result in a rare genetic disorder referred to as methylmalonic aciduria and homocystinuria type C (cblC). They demonstrate deficiencies in both B12-dependent enzymatic functions crucial in mammals—MTR and MMUT [98]. Thus, this disorder is characterized by the accumulation of both MMA and homocysteine (Hcy) in serum [98]. Individuals affected by the cblC disorder manifest severe neurological and systemic metabolic abnormalities [97], such as atypical hemolytic uremic syndromes (aHUS), decline in renal function, idiopathic neuropathies, spinal cord degenerations, ataxias, recurrent thrombosis, visual field defects, maculopathy, optic disc atrophy [100]. Interestingly, recent studies suggest the involvement of cobalamin and MMACHC might have a potential role in the oncogenic transformation of specific tumor subtypes. Melanomas and cell lines derived from melanoma often exhibit a higher occurrence of elevated methylation in the MMACHC promoter compared to other cancer types. Which implicates the inactivation of MMACHC as the causative factor [97,101]
2.3.4. Methylmalonic Aciduria and Homocystinuria Type D Protein (MMADHC)
MMADHC gene is associated with methylmalonic aciduria type D and homocystinuria, and it is classified within the cblD complementation group [102]. The MMADHC gene encodes a protein consisting of 296 amino acids, with a molecular weight of 32.9 kDa. MMADHC protein, similarly to the MMACHC, exhibits a high degree of conservation across various mammalian species. The MMADHC gene encodes a robust N-terminal mitochondrial leader sequence along with a conserved vitamin B12-binding motif located at residues 81–86. [102]. It is present in both the mitochondria and cytoplasm, although the precise mechanisms that lead to this dual localization are not fully understood. Analysis of MMACHC and MMADHC gene expression patterns during mouse development unveils overlapping expression, particularly evident in the developing cardiac, respiratory, and nervous systems. It is suggested that these two proteins are co-expressed in all cells, as there is no discernible specificity for any particular organ, tissue, or cell type exclusively expressing MMADHC. Notably, MMADHC demonstrates expression in all cells of embryos, including those expressing MMACHC [103].
The physiological function of MMADHC is yet to be fully understood, however, it seems to play a role in the production of the two active cobalamin forms, MeCbl and AdoCbl same as MMACHC [104]. Plesa et. al. in 2011 employed a phage display to predict regions on MMADHC that bind to MMACHC. The study identified five distinct regions on MMADHC, all aligning with residues C-terminal to Met116 [105], but not the N-terminus (residues 1–61) [106]. Although MMADHC lacks any recognized enzymatic activity, it is suggested to be the first protein identified to repurpose the nitroreductase fold exclusively for protein-protein interactions [107].
The cblD complementation group can be linked to isolated methylmalonic aciduria (cblD-MMA), isolated homocystinuria (cblD-HC), or a combination of both methylmalonic aciduria and homocystinuria (cblD-MMA/HC) [108]. Individuals characterized with cblD-MMA have at least one altered allele that induces a premature stop codon in the N-terminal region of the protein. In contrast, those with cblD-HC possess at least one missense mutation, resulting in amino acid substitutions located in the C-terminal part of the protein [104]. The clinical presentation indicates that patients with cblD-MMA/HC and cblD-HC exhibit neurological and hematological symptoms. In contrast, cblD-MMA patients experience respiratory distress, hyperammonemia, and neurological symptoms [102,108,109].
2.4. Vitamin B12-Dependent Intracellular Enzymes
2.4.1. Methionine Synthase (MTR)
Methionine synthase (MTR) is a big ~140-kDa monomeric protein found in cytosol, [83] and it consists of four domains. The N-terminal site, dependent on the zinc ions, is binding and activating Hcy while the following domain has a high affinity to methyltetrahydrofolate and might affect its activity [110]. The third module, B12 dependent, is crucial in methyl group transfer from 5-methyltetrahydrofolate to Hcy with the production of tetrahydrofolate and methionine [84], and last, the C-terminal domain binds S-adenosylmethionine for reductive activation of MTR when cobalamin cofactor undergoes oxidation process [85]. The human enzyme displays a 58% identity with methionine synthase from Escherichia coli (E. coli), [111] which is one of the biggest proteins with 1227 residues and a molecular weight of ~136 kDa [112]. The remarkable conservation of amino acid residues across proteins indicates a high degree of similarity in properties and structures between enzymes from both humans and E. coli. Consequently, the information gleaned from the E. coli enzyme is presumed to apply to its human counterpart [111]. However, a crucial divergence is the lack of flavodoxin in mammals, a pivotal component in the reductive reactivation process observed in bacterial models [113].
In mice models, full depletion of the MTR gene caused pregnancy failure [86]. While in proliferating cells of the skin and intestinal epitheliums, as well as in rat intestine crypts and proliferating Caco-2 cells (human epithelial colorectal adenocarcinoma cell line), there is a consistent increase in the transcription, protein expression, and activity of MTR. Additionally, MTR activity correlates with DNA methylation in rat intestine villi [114]. Moreover, in situ hybridization indicated that human MTR is found on the 1q42.3-43 chromosome region [87] and its missense mutation or deletion of three base pairs was identified in the cblG group of patients with cobalamin metabolism disorder [88]. However, also the second form of this disorder has been recognized as "methylcobalamin deficiency cblE type" (cblE), and it is believed to arise from mutations occurring at distinct genetic loci [115]. This rare autosomal disease is associated with homocysteinemia, homocystinuria, and hypomethioninemia [88], and patients usually exhibit symptoms within the initial two years of their lives [116]. Reduced MTR activity also leads to an elevation in the catalytic protein phosphatase 2A (PP2A), resulting in an imbalance in the phosphorylation/methylation of nucleocytoplasmic RNA binding proteins, consequently resulting in compromised energy metabolism and neuroplasticity linked to inborn errors of Cbl metabolism (IECM) [117].
2.4.2. Methylmalonyl-CoA Mutase (MMUT)
Methylmalonyl coenzyme A mutase (MMUT) is an enzymatic protein found in mitochondrial matrix with a ~78 kDa molecular weight [89]. The structure of MMUT is highly conserved evolutionally, thus this AdoCbl-dependent enzyme may be found in both eukaryotes and prokaryotes [91], but not plants [118]. In mammals, it has a homodimeric structure with two active sites. Active α chain consists of a C-terminal, cobalamin-binding domain and TIM-barrel, substrate-binding domain [90,119]. MMUT takes part in converting carbon skeletons of odd-chain fatty acids, cholesterol, and amino acids such as threonine, methionine, valine, and isoleucine into succinyl-CoA [92], for further utilization in the tricarboxylic acid (TCA) cycle [120].
Some studies show that inactivation of MMUT occurs when there is an accumulation of the oxidized form of AdoCbl, known as OH2Cbl, which may occur as the product of the photolysis, especially in an oxygen-high environment [121]. However, the formation of the MMUT-methylmalonic aciduria type A (MMAA) protein complex reduces the rate of oxidized cofactor formation, protecting the human MMUT enzyme [122].
Human MMUT is encoded by the MMUT gene, situated as a lone copy on chromosome 6 (6p21.1), and is composed of 13 exons [123]. Its mutation leads to methylmalonic acidemia – a metabolic autosomal disease. Patients with aciduria suffer from vomiting followed by dehydration, muscle weakness, lethargy, mental incapacity, and rarely, when not treated to death [93].
Two subgroups of MMUT mutation have been recognized so far: mut− defect, characterized by residual activity in the presence of high concentrations of AdoCbl, and mut0 defect with complete loss of MMUT activity [124]. Distinguishing between mut– and mut0 subgroups can be challenging at times. Certain mut– mutations may only impact the apparent Michaelis constant (Km), while others can influence both the apparent Km and the maximum velocity of an enzymatic reaction (Vmax) [125].
3. Vitamin B12 Deficiency
3.1. Epidemiology and Symptoms of Vitamin B12 Deficiency
The prevalence rates of vitamin B12 deficiency in the United States (US) range from 2.5% to 6%, differing by age group, gender, and ethnicity [126]. An analysis of National Health and Nutrition Examination Survey data from 2007 to 2018 revealed that about 3.6% of all adults aged 19 and older have vitamin B12 deficiency (in the US defined as serum vitamin B12 levels below 200 pg/mL (148 pmol/L)). The deficiency rate increases to 3.7% in individuals aged 60 and older. However, vitamin B12 insufficiency (defined as serum levels below 300 pg/mL (221 pmol/L)) is more prevalent, affecting approximately 12.5% of all adults aged 19 and older, and 12.3% of those aged 60 and older [127]. During pregnancy, serum vitamin B12 levels often decline, sometimes dropping to subnormal levels, but typically return to normal after delivery [128]. Furthermore, vitamin B12 deficiency due to pernicious anemia is more common in people of Northern European ancestry than people of African descent [129,130].
Nowadays, food-bound cobalamin malabsorption (FBCM) is a prominent cause of vitamin B12 deficiency [131]. FBCM disrupts the absorption of cobalamin from food sources [132]. Impaired release of cobalamin from transport proteins contributes to FBCM, commonly seen in conditions like gastritis or medication use reducing hydrochloric acid production. Milder symptoms may persist due to IF-dependent transport functionality [133]. Proton pump inhibitors and histamine H2-receptor antagonists heighten the risk of future cobalamin deficiency, with risk reduction following medication cessation. Thus, the widespread use of acid-suppressing medications may lead to undiagnosed cobalamin deficiency [134].
The effects of B12 deficiency are mainly seen in the blood and nervous system. The classic manifestation was first identified as anemia [135]. Since then, the spectrum has shifted considerably, starting with the recognition that neurological manifestations e.g. ataxia, cognitive decline leading to dementia, or psychiatric disorder, often predominate and can occur in the absence of hematological disorders [136]. Anemia is caused by disruption of DNA synthesis in a hematopoietic system, predominantly erythroid precursors. Neurological complications are caused mainly by reduced activity of vitamin B12-dependent enzymes. The increased accumulation of methylmalonic acid (MMA) (product of methylmalonyl-CoA degradation) could contribute to neurological symptoms, due to the formation of odd-chain and methyl-branched-chain fatty acids [137]. Moreover, lack of MeCbl and MTR inhibition leads to hyperhomocysteinemia linked with inflammation, oxidative stress, and microvascular disease that may exacerbate neurological symptoms [138].
In addition to the symptomatic presentation of vitamin B12 deficiency, subclinical cobalamin deficiency (SCCD) presents a condition characterized by biochemical aberrations in the absence of clinical signs [131,139]. SCCD, prevailing more commonly than overt deficiency, often originates from FBCM rather than IF dysfunction. Currently, SCCD is perceived as a transient state without overt cobalamin depletion, necessitating exploration into remediable etiologies. Patients exhibiting serum cobalamin levels between 110 and 148 pmol/L should undergo retesting within one to two months; those exhibiting normalization need not undergo further assessment [140]. Persistent low levels may warrant low-dose oral cobalamin supplementation alongside anti-IF antibody analysis. Neurological manifestations should prompt reassessment by a healthcare provider. Positive anti-IF antibody titers necessitate treatment for pernicious anemia, while negative results mandate reevaluation after three to four months [141]. Additional biochemical assessments are warranted if cobalamin levels remain consistently low.
3.2. Diagnosis and Treatment of Vitamin B12 Deficiency
The assessment of vitamin B12 status typically relies on measurements of its serum or plasma levels. Laboratories often define subnormal values as below 200 or 250 pg/mL (148 or 185 pmol/L). Typically, a value significantly below the lower limit of the reference range suggests probable vitamin B12 deficiency, while a value well above indicates its adequate status. However, exceptions arise in patients with pernicious anemia who have circulating antibodies against IF; their vitamin B12 levels may appear falsely normal despite deficiency [142]. Active vitamin B12 bound to transcobalamin (holotranscobalamin) theoretically offers increased sensitivity for detecting vitamin B12 deficiency compared to total serum vitamin B12 levels. Although the measurement of holotranscobalamin is gradually becoming available in clinical settings, it has demonstrated only marginal improvement over total vitamin B12 levels as a biomarker for deficiency [143,144].
Another valuable and highly sensitive marker of vitamin B12 status is serum MMA level, with a threshold greater than 0.271 µmol/L indicative of deficiency [10]. However, MMA levels can elevate due to renal insufficiency and tend to be higher in older adults. Genetic influences on MMA cutoffs have been also questioned by genome-wide association studies. A common polymorphism (rs291466) in HIBCH, involved in valine catabolism, is prevalent (minor allele frequency 0.43), and homozygotes for the allele show 46% higher plasma MMA levels, that potentially impact its efficacy of this metabolite as a vitamin B12 status indicator [145]. Besides MMA concentration, also total plasma Hcy levels rise rapidly to levels above 15 µmol/L with declining vitamin B12 status. However, Hcy levels lack specificity, being influenced by factors such as folate levels or renal function decline [146].
Optimal management of vitamin B12 deficiency requires identifying its cause, particularly differentiating between its low intake and malabsorption. The Schilling test, once the gold standard, is now rarely used due to the lack of radiolabeled B12 availability and cost constraints. The CobaSorb test, measuring serum holotranscobalamin levels post-vitamin B12 dosing is limited post-treatment [147,148,149]. Promising might be a 14C-labeled vitamin B12 absorption test [150]. Alternative diagnostic approaches include detecting autoantibodies for pernicious anemia and assessing for atrophic gastritis during upper [151] endoscopy. While a negative result for anti-IF autoantibodies does not exclude pernicious anemia, a positive result is highly specific. However, a positive result for anti-H+/K+ ATPase autoantibodies lacks specificity, as it may occur in patients without pernicious anemia [152,153]. In pediatric cases, considering inborn errors of cobalamin metabolism via complementation phenotyping or genetic analysis is crucial for diagnosis. These tests help differentiate between different genetic causes, aiding in tailored treatment approaches for children with vitamin B12 deficiency [154,155].
Treatment for vitamin B12 deficiency varies depending on the underlying cause. In 2021, The British Society for Haematology (BSH) updated guidance on vitamin B12 replacement [156]. For non-diet-related deficiency, such as in pernicious anemia or other conditions, hydroxocobalamin 1 mg intramuscular every 2–3 months for life is recommended. For diet-related deficiency, individuals can either intake orally cyanocobalamin 50–150 micrograms daily between meals or receive a twice-yearly hydroxocobalamin 1 mg injection. In cases of dietary deficiency, treatment duration may vary, with vegans potentially requiring lifelong treatment.
3.3. Long-Distance Consequences of Vitamin B12 Deficiency
Vitamin B12 deficiency, owing to its pivotal role in cellular metabolism, can mimic a diverse array of clinical presentations as listed above. However, a manifestation of these clinical outcomes is relatively late. It results in the development of SCCD, which may have severe consequences including increased risk of neurodegenerative and, cardiovascular diseases or implicate enhanced cancer progression.
In vegetarians, deficiency of vitamin B12 was mostly found to be associated with depression and adverse neurological function. Berkins et al. highlighted that the dietary intake of vitamin B12 might affect brain structure [157]. Interestingly, supplementation of vitamin B12 along with anti-depressant therapy greatly improved depressive symptoms [158]. Other studies conclude that cobalamin deficiency may have some similarities with multiple sclerosis. In both conditions, cerebral neurons impairment is caused by destroyed myelin sheaths, which are more susceptible to the excitatory effects of glutamate [15]. It is well known that cognitive decline often coincides with a high prevalence of vitamin B12 deficiency, although causality in these correlations is not fully established [159]. Furthermore, while the actual percentage of reversible dementia in individuals with vitamin B12 insufficiency is deemed low, impaired vitamin B12 metabolism could potentially serve as one of several factors influencing the onset and progression of cognitive decline and dementia [171]. It could be caused by reduced methylation capacity, elevated Hcy levels, and the role of B vitamins in upholding the integrity of the blood-brain barrier [172]. Some researchers propose that it may act as a predisposing factor for Alzheimer's disease [173]. Additionally, it is associated with various other neurological conditions including Wernicke's encephalopathy, subacute combined degeneration of the spinal cord, and peripheral neuropathy [174].
It is well known that hyperhomocysteinemia, strongly linked with vitamin B12 deficiency is a prominent risk factor for atherosclerotic vascular diseases affecting coronary, cerebral, and peripheral vessels, as well as arterial and venous thromboembolism [175]. The adverse effects of Hcy on the vascular endothelium and coagulation cascade, along with its procoagulant properties, encompass reduced binding of antithrombin III to endothelial heparan sulfate, heightened affinity between lipoprotein(a) and fibrin, increased tissue factor activity in endothelial cells, and inhibition of factor V inactivation by activated protein [176]. Supplementation with vitamin B12 led to the resolution of both clinical and biological abnormalities in all participants [177]. However, whether vitamin B12 deficiency directly influences thrombosis or acts through hyperhomocysteinemia or other factors remains to be fully understood [178]. Lifestyle factors like smoking, BMI, and physical activity may also contribute to the association between hyperhomocysteinemia and thromboembolism [179].
Interestingly, other compounds that are strongly linked with vitamin B12 deficiency- MMA have been recently identified as oncometabolites. MMA is a metabolite of methylmalonyl coenzyme A, and its increased levels result from impaired conversion to succinyl coenzyme A in a vitamin B12-dependent manner [180]. A study published in 2020 by Gomes et al. revealed that MMA levels increase in the serum of older individuals and may have a function as a mediator of tumor progression. Indeed, treatment of adenocarcinoma human alveolar basal epithelial (A549) cells with MMA induced a complete pro-aggressive epithelial to mesenchymal transition-like phenotype with a decline in E-cadherin and a concurrent increase in fibronectin and vimentin [181]. Additionally, two years later Gomes et al. demonstrated that the accumulation of MMA accelerates cancer cell aggressiveness in breast and lung cancers [182]. Intriguingly, in the context of breast cancer, the blockade of other metabolic pathways involving MMA or the activation of MMA production, coupled with vitamin B12 intracellular deficiency, resulted in enhanced tumor metastasis [182]. Recently, MMA was also found to promote metastasis in colon tumor models [183]. However, the direct effect of vitamin B12 deficiency on cancer onset, prognosis, and progression remains unresolved due to many inconsistent studies [173,174,175,176,177,178].
4. Discussion
In this review, we consolidate the existing knowledge regarding the proteins involved in vitamin B12 metabolism and explore their role in the cellular processes as well as the impact of their dysfunction on overall metabolic integrity. Several proteins listed in Table 1, including transcobalamin I and II, intrinsic factor, cubilin, and amnionless, are already utilized in the diagnosis of vitamin B12 deficiency. However, other proteins are primarily investigated at the gene level due to their intracellular localization or low detection levels in serum, limiting their use in routine diagnostics. Nonetheless, the levels of some of these proteins may harbor novel information concerning the risk of complications associated with vitamin B12 deficiency.
In 2018, Tseng et al. established that mice models demonstrated loss of single allele Lmbd1 would result in ventricular hypertrophy which was accompanied by an enhanced myocardial glucose uptake [190]. Moreover, a microarray screening study revealed a 25% reduction in LMBD1 mRNA levels in the septa of 13 heart transplant patients with dilated cardiomyopathy compared to non-failing hearts [191]. This clinical observation, coupled with findings from the mice studies, suggests that diminished expression of LMBD1 protein may play a role in the development of cardiomyopathy. Thus, the single nucleotide polymorphism sites associated with decreased expression of the human LMBRD1 gene evaluation not only may indicate the cause of vitamin B12 deficiency but also prognoses a risk of onset of cardiovascular diseases.
Interestingly, the diminished expression of other proteins involved in vitamin B12- has been noted in the brains of multiple sclerosis (MS) patients highlighting the role of this protein in proper central nervous system function [192]. It was accompanied by worsening the disease by both CD320 genetic deletion and vitamin B12 deficiency [192]. These findings highlight a plausible connection between vitamin B12 deficiency and the progression of pathology in MS. One may conclude that vitamin B12 deficiency caused by CD320 disruption might be linked with an increased risk of neurodegenerative diseases, thus its evaluation might be an interesting prognostic parameter.
Another protein linked with vitamin B12 metabolism that exhibits multifunctional properties is MRP1. MRP1 transports a wide range of therapeutic agents as well as various physiological substrates and may contribute to drug resistance development in several cancers, including lung, breast, and prostate cancers, as well as childhood neuroblastoma [183]. MRP1's role as a glutathione transporter creates a unique vulnerability in cancer cells with high MRP1 expression, providing a potential therapeutic opportunity for targeting cancer. Direct pharmacological inhibition of MRP1 shows promise and is extensively tested in preclinical and clinical studies [184,185,186]. However, non-selective MRP1 inhibition could potentially disrupt vitamin B12 metabolism and lead to its deficiency. Therefore, there is an emerging need to investigate the effects of MRP-1 inhibition on vitamin B12 metabolism during therapy.
Interestingly, it has been noted that the utilization of the common drug – metformin in diabetic patients is linked to potential malabsorption and deficiency of vitamin B12 [197,198]. Numerous observational studies and meta-analyses have underscored a significant correlation between metformin usage and vitamin B12 deficiency [199]. Patients undergoing metformin therapy display reduced absorption of B12, leading to decreased levels of serum total vitamin B12 and transcobalamin II (TCII-B12). This phenomenon is attributed to calcium-dependent antagonism in the ileal membrane, which may be mitigated through calcium supplementation [200]. Given these findings, it is strongly advised that individuals using metformin, undergo regular monitoring of their vitamin B12 levels. This precaution is crucial as prolonged metformin usage may deplete hepatic vitamin B12 reserves [201].
5. Conclusions
Despite the initiation of studies on vitamin B12 as early as the 1960s, our understanding of its biochemical alterations remains incomplete. Over the past decade, there has been a notable surge in research attention directed towards vitamin B12. Surprisingly, some recent studies suggested that vitamin B12 may affect gene expression, cellular plasticity and tissue repair [159,192]. However, the specific molecules and the cellular changes mediated by these alterations are still not fully elucidated.
Besides that vitamin B12 deficiency leads to the presence of serious clinical manifestations, numerous studies have highlighted the correlation between low vitamin B12 levels and the increased risk of civilization diseases. Importantly, some of the proteins involved in vitamin B12 metabolism may act as a moonlighting protein, and their pharmacological modification already used in the treatment of other clinical stages may implicate also proper vitamin B12 metabolism.
Hence, there is a pressing need for more basic science studies to unravel the vitamin B12 metabolism complexity that may lower the risk of vitamin B12 deficiency in clinical studies as well as potentially identify novel diagnostic parameters that may contribute to evaluating the risk of vitamin B12 deficiency consequences.
Author Contributions
Conceptualization, P.M., F.K., M.T.; writing—original draft preparation, P.M., F.K., D.C., M.T.; writing—review and editing, M.T., R.T.S..; supervision, M.T., R.T.S.
Funding
This research was supported by the National Science Centre in Poland (2021/43/D/NZ4/02501).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Folkers, K., D.D., Ed.; J.W.& S. History of Vitamin B12: Pernicious Anemia to Crystalline Cyanocobalamin. In Vitamin B12; NY, USA, 1982; Vol. I.
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins. Molecules 2010, 15, 3228–3259. [Google Scholar] [CrossRef]
- Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press: Washington, D.C., 1998; ISBN 978-0-309-06554-2.
- Obeid, R.; Fedosov, S.N.; Nexo, E. Cobalamin Coenzyme Forms Are Not Likely to Be Superior to Cyano- and Hydroxyl-cobalamin in Prevention or Treatment of Cobalamin Deficiency. Mol. Nutr. Food Res. 2015, 59, 1364–1372. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Cobalamin Deficiency. In; 2012; pp. 301–322.
- Santos, F.; Vera, J.L.; Lamosa, P.; de Valdez, G.F.; de Vos, W.M.; Santos, H.; Sesma, F.; Hugenholtz, J. Pseudovitamin Is the Corrinoid Produced by Lactobacillus Reuteri CRL1098 under Anaerobic Conditions. FEBS Lett. 2007, 581, 4865–4870. [Google Scholar] [CrossRef] [PubMed]
- Latner, A.L.; Raine, L. Effect of Analogues on the Uptake of Vitamin B12 by the Intact Rat. Nature 1957, 180, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Bito, T.; Bito, M.; Hirooka, T.; Okamoto, N.; Harada, N.; Yamaji, R.; Nakano, Y.; Inui, H.; Watanabe, F. Biological Activity of Pseudovitamin B12 on Cobalamin-Dependent Methylmalonyl-CoA Mutase and Methionine Synthase in Mammalian Cultured COS-7 Cells. Molecules 2020, 25, 3268. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F. Vitamin B12 Sources and Bioavailability. Exp. Biol. Med. 2007, 232, 1266–1274. [Google Scholar] [CrossRef]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 Deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef]
- Gherasim, C.; Lofgren, M.; Banerjee, R. Navigating the B12 Road: Assimilation, Delivery, and Disorders of Cobalamin. J. Biol. Chem. 2013, 288, 13186–13193. [Google Scholar] [CrossRef]
- O’Leary, F.; Samman, S. Vitamin B12 in Health and Disease. Nutrients 2010, 2, 299–316. [Google Scholar] [CrossRef]
- Kozyraki, R.; Cases, O. Vitamin B12 Absorption: Mammalian Physiology and Acquired and Inherited Disorders. Biochimie 2013, 95, 1002–1007. [Google Scholar] [CrossRef]
- Wuerges, J.; Geremia, S.; Randaccio, L. Structural Study on Ligand Specificity of Human Vitamin B12 Transporters. Biochem. J. 2007, 403, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mathews, F.S.; Gordon, M.M.; Chen, Z.; Rajashankar, K.R.; Ealick, S.E.; Alpers, D.H.; Sukumar, N. Crystal Structure of Human Intrinsic Factor: Cobalamin Complex at 2.6-Å Resolution. Proc. Natl. Acad. Sci. USA 2007, 104, 17311–17316. [Google Scholar] [CrossRef] [PubMed]
- Alpers, D.H.; Russell-Jones, G. Gastric Intrinsic Factor: The Gastric and Small Intestinal Stages of Cobalamin Absorption. A Personal Journey. Biochimie 2013, 95, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.E.; Gordon, M.M.; Taggart, R.T.; Mohandas, T.K.; Alpers, D.H. Human Gastric Intrinsic Factor: Characterization of CDNA and Genomic Clones and Localization to Human Chromosome 11. Genomics 1991, 10, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Coleman, P.S.; Dupont, B. The Biochemical and Genetic Basis for the Microheterogeneity of Human R-Type Vitamin B12 Binding Proteins. Blood 1982, 59, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Furger, E.; Frei, D.C.; Schibli, R.; Fischer, E.; Prota, A.E. Structural Basis for Universal Corrinoid Recognition by the Cobalamin Transport Protein Haptocorrin. J. Biol. Chem. 2013, 288, 25466–25476. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Rasmussen, M.R.; Andersen, C.B.F.; Nexø, E.; Moestrup, S.K. Vitamin B12 Transport from Food to the Body’s Cells—A Sophisticated, Multistep Pathway. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 345–354. [Google Scholar] [CrossRef]
- Allen, R.H.; Seetharam, B.; Podell, E.; Alpers, D.H. Effect of Proteolytic Enzymes on the Binding of Cobalamin to R Protein and Intrinsic Factor. J. Clin. Investig. 1978, 61, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.T.; Warwick, R.R.G.; Simpson, J.D.; Shearman, D.J.C. Does Malabsorption Of Vitamin B12 Occur In Chronic Pancreatitis ? Lancet 1972, 300, 241–243. [Google Scholar] [CrossRef]
- Guéant, J.-L.; Champigneulle, B.; Gaucher, P.; Nicolas, J.-P. Malabsorption of Vitamin B12 in Pancreatic Insufficiency of the Adult and of the Child. Pancreas 1990, 5, 559–567. [Google Scholar] [CrossRef]
- Forsmark, C.E. Diagnosis and Management of Exocrine Pancreatic Insufficiency. Curr. Treat. Options Gastroenterol. 2018, 16, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Hygum, K.; Lildballe, D.L.; Greibe, E.H.; Morkbak, A.L.; Poulsen, S.S.; Sorensen, B.S.; Petersen, T.E.; Nexo, E. Mouse Transcobalamin Has Features Resembling Both Human Transcobalamin and Haptocorrin. PLoS ONE 2011, 6, e20638. [Google Scholar] [CrossRef] [PubMed]
- Fedosov, S.N.; Fedosova, N.U.; Berglund, L.; Moestrup, S.K.; Nexø, E.; Petersen, T.E. Assembly of the Intrinsic Factor Domains and Oligomerization of the Protein in the Presence of Cobalamin. Biochemistry 2004, 43, 15095–15102. [Google Scholar] [CrossRef] [PubMed]
- Fedosov, S.N.; Fedosova, N.U.; Kräutler, B.; Nexø, E.; Petersen, T.E. Mechanisms of Discrimination between Cobalamins and Their Natural Analogues during Their Binding to the Specific B 12 -Transporting Proteins. Biochemistry 2007, 46, 6446–6458. [Google Scholar] [CrossRef] [PubMed]
- Ayesh, M.H.; Jadalah, K.; Al Awadi, E.; Alawneh, K.; Khassawneh, B. Association between Vitamin B12 Level and Anti-Parietal Cells and Anti-Intrinsic Factor Antibodies among Adult Jordanian Patients with Helicobacter Pylori Infection. Braz. J. Infect. Dis. 2013, 17, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Kulnigg-Dabsch, S. Autoimmune Gastritis. Wien. Med. Wochenschr. 2016, 166, 424–430. [Google Scholar] [CrossRef]
- Lenti, M.V.; Rugge, M.; Lahner, E.; Miceli, E.; Toh, B.-H.; Genta, R.M.; De Block, C.; Hershko, C.; Di Sabatino, A. Autoimmune Gastritis. Nat. Rev. Dis. Primers 2020, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Htut, T.W.; Thein, K.Z.; Oo, T.H. Pernicious Anemia: Pathophysiology and Diagnostic Difficulties. J. Evid. Based Med. 2021, 14, 161–169. [Google Scholar] [CrossRef]
- Qudsiya Z, J.O.D. Subacute Combined Degeneration of the Spinal Cord. StatPearls Publishing 2022.
- Larsen, C.; Etzerodt, A.; Madsen, M.; Skjødt, K.; Moestrup, S.K.; Andersen, C.B.F. Structural Assembly of the Megadalton-Sized Receptor for Intestinal Vitamin B12 Uptake and Kidney Protein Reabsorption. Nat. Commun. 2018, 9, 5204. [Google Scholar] [CrossRef]
- Pedersen, G.A.; Chakraborty, S.; Steinhauser, A.L.; Traub, L.M.; Madsen, M. AMN Directs Endocytosis of the Intrinsic Factor-Vitamin B12 Receptor Cubam by Engaging ARH or Dab2. Traffic 2010, 11, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Strope, S.; Rivi, R.; Metzger, T.; Manova, K.; Lacy, E. Mouse Amnionless, Which Is Required for Primitive Streak Assembly,Mediates Cell-Surface Localization and Endocytic Function of Cubilin on Visceral Endoderm and Kidney Proximal Tubules. Development 2004, 131, 4787–4795. [Google Scholar] [CrossRef] [PubMed]
- Perea-Gomez, A.; Cases, O.; Lelièvre, V.; Pulina, M.V.; Collignon, J.; Hadjantonakis, A.-K.; Kozyraki, R. Loss of Cubilin, the Intrinsic Factor-Vitamin B12 Receptor, Impairs Visceral Endoderm Endocytosis and Endodermal Patterning in the Mouse. Sci. Rep. 2019, 9, 10168. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Verroust, P.J.; Nielsen, R. Receptor-Mediated Endocytosis in Renal Proximal Tubule. Pflug. Arch. 2009, 458, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.B.F.; Madsen, M.; Storm, T.; Moestrup, S.K.; Andersen, G.R. Structural Basis for Receptor Recognition of Vitamin-B12–Intrinsic Factor Complexes. Nature 2010, 464, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, S.; Gburek, J.; Hamard, G.; Nielsen, R.; Willnow, T.E.; Devuyst, O.; Nexo, E.; Verroust, P.J.; Christensen, E.I.; Kozyraki, R. Cubilin Is Essential for Albumin Reabsorption in the Renal Proximal Tubule. J. Am. Soc. Nephrol. 2010, 21, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.C.; Madsen, M.; Højrup, P.; Christensen, E.I.; Tanner, S.M.; de la Chapelle, A.; He, Q.; Moestrup, S.K. The Functional Cobalamin (Vitamin B12)–Intrinsic Factor Receptor Is a Novel Complex of Cubilin and Amnionless. Blood 2004, 103, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.L.; Andersen, R.K.; Hager, H.; Madsen, M. Lack of Megalin Expression in Adult Human Terminal Ileum Suggests Megalin-Independent Cubilin/Amnionless Activity during Vitamin B 12 Absorption. Physiol. Rep. 2014, 2, e12086. [Google Scholar] [CrossRef] [PubMed]
- Gräsbeck, R. Imerslund-Gräsbeck Syndrome (Selective Vitamin B12 Malabsorption with Proteinuria). Orphanet J. Rare Dis. 2006, 1, 17. [Google Scholar] [CrossRef]
- Storm, T.; Zeitz, C.; Cases, O.; Amsellem, S.; Verroust, P.J.; Madsen, M.; Benoist, J.-F.; Passemard, S.; Lebon, S.; Jønsson, I.M.; et al. Detailed Investigations of Proximal Tubular Function in Imerslund-Gräsbeck Syndrome. BMC Med. Genet. 2013, 14, 111. [Google Scholar] [CrossRef]
- Ercan, Z.; Demir, M.E.; Ulas, T.; Ingec, M.; Horoz, M. A Long-Term Follow-up of an Imerslund-Grasbeck Syndrome Patient with Proteinuria. Nefrologia 2013, 33, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Pannérec, A.; Migliavacca, E.; De Castro, A.; Michaud, J.; Karaz, S.; Goulet, L.; Rezzi, S.; Ng, T.P.; Bosco, N.; Larbi, A.; et al. Vitamin B12 Deficiency and Impaired Expression of Amnionless during Aging. J. Cachexia Sarcopenia Muscle 2018, 9, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.C. Multidrug Resistance Protein 1 (MRP1, ABCC1), a “Multitasking” ATP-Binding Cassette (ABC) Transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.; Bhardwaj, G.; Gerlach, J.; Mackie, J.; Grant, C.; Almquist, K.; Stewart, A.; Kurz, E.; Duncan, A.; Deeley, R. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science (1979) 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Bakos, É.; Homolya, L. Portrait of Multifaceted Transporter, the Multidrug Resistance-Associated Protein 1 (MRP1/ABCC1). Pflug. Arch. 2007, 453, 621–641. [Google Scholar] [CrossRef] [PubMed]
- Flens, M.J.; Zaman, G.J.; van der Valk, P.; Izquierdo, M.A.; Schroeijers, A.B.; Scheffer, G.L.; van der Groep, P.; de Haas, M.; Meijer, C.J.; Scheper, R.J. Tissue Distribution of the Multidrug Resistance Protein. Am. J. Pathol. 1996, 148, 1237–1247. [Google Scholar] [PubMed]
- Peng, K.-C.; Cluzeaud, F.; Bens, M.; Duong Van Huyen, J.-P.; Wioland, M.A.; Lacave, R.; Vandewalle, A. Tissue and Cell Distribution of the Multidrug Resistance-Associated Protein (MRP) in Mouse Intestine and Kidney. J. Histochem. Cytochem. 1999, 47, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, A.; Simon, S.M. Subcellular Localization and Activity of Multidrug Resistance Proteins. Mol. Biol. Cell 2003, 14, 3389–3399. [Google Scholar] [CrossRef] [PubMed]
- Beedholm-Ebsen, R.; van de Wetering, K.; Hardlei, T.; Nexø, E.; Borst, P.; Moestrup, S.K. Identification of Multidrug Resistance Protein 1 (MRP1/ABCC1) as a Molecular Gate for Cellular Export of Cobalamin. Blood 2010, 115, 1632–1639. [Google Scholar] [CrossRef]
- Seetharam, B.; Yammani, R.R. Cobalamin Transport Proteins and Their Cell-Surface Receptors. Expert. Rev. Mol. Med. 2003, 5, 1–18. [Google Scholar] [CrossRef]
- Shah, N.P.; Beech, C.M.; Sturm, A.C.; Tanner, S.M. Investigation of the ABC Transporter MRP1 in Selected Patients with Presumed Defects in Vitamin B12 Absorption. Blood 2011, 117, 4397–4398. [Google Scholar] [CrossRef] [PubMed]
- Quadros, E. V.; Rothenberg, S.P.; Pan, Y.C.; Stein, S. Purification and Molecular Characterization of Human Transcobalamin II. J. Biol. Chem. 1986, 261, 15455–15460. [Google Scholar] [CrossRef] [PubMed]
- Quadros, E.V.; Rothenberg, S.P.; Jaffe, E.A. Endothelial Cells from Human Umbilical Vein Secrete Functional Transcobalamin II. Am. J. Physiol. -Cell Physiol. 1989, 256, C296–C303. [Google Scholar] [CrossRef] [PubMed]
- Quadros, E.V.; Regec, A.L.; Khan, K.M.F.; Quadros, E.; Rothenberg, S.P. Transcobalamin II Synthesized in the Intestinal Villi Facilitates Transfer of Cobalamin to the Portal Blood. Am. J. Physiol. -Gastrointest. Liver Physiol. 1999, 277, G161–G166. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Seetharam, S.; Seetharam, B. Genomic Structure of Human Transcobalamin II: Comparison to Human Intrinsic Factor and Transcobalamin I. Biochem. Biophys. Res. Commun. 1995, 208, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Wuerges, J.; Garau, G.; Geremia, S.; Fedosov, S.N.; Petersen, T.E.; Randaccio, L. Structural Basis for Mammalian Vitamin B 12 Transport by Transcobalamin. Proc. Natl. Acad. Sci. 2006, 103, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Morkbak, A.L.; Munz, W.; Nexo, E.; Herrmann, W. The Cobalamin-Binding Proteins Transcobalamin and Haptocorrin in Maternal and Cord Blood Sera at Birth. Clin. Chem. 2006, 52, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.L.; Schneider, R.J.; Mehlman, C.S.; Allen, R.H. Human Plasma R-Type Vitamin B12-Binding Proteins. II. The Role of Transcobalamin I, Transcobalamin III, and the Normal Granulocyte Vitamin B12-Binding Protein in the Plasma Transport of Vitamin B12. J. Biol. Chem. 1975, 250, 7707–7713. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Koda, M.; Okamoto, T.; Miyoshi, K.; Matono, T.; Oyama, K.; Hosho, K.; Okano, J.-I.; Isomoto, H.; Murawaki, Y. Falsely Elevated Serum Vitamin B12 Levels Were Associated with the Severity and Prognosis of Chronic Viral Liver Disease. Yonago Acta Med. 2017, 60, 31–39. [Google Scholar]
- Thorpe, S.J.; Rigsby, P.; Roberts, G.; Lee, A.; Hamilton, M.; Craig, D. An International Standard for Holotranscobalamin (HoloTC): International Collaborative Study to Assign a HoloTC Value to the International Standard for Vitamin B12 and Serum Folate. Clin. Chem. Lab. Med. (CCLM) 2016, 54, 1467–1472. [Google Scholar] [CrossRef]
- Knoepfel, C.; Michel Blanco, M.; Nydegger, U.; Risch, L.; Renz, H.; Risch, M. Failure of the Holotranscobalamin Assay in Vitamin B12-Deficient Patients. LaboratoriumsMedizin 2018, 42, 141–147. [Google Scholar] [CrossRef]
- Golding, P.H. Experimental Vitamin B12 Deficiency in a Human Subject: A Longitudinal Investigation of the Performance of the Holotranscobalamin (HoloTC, Active-B12) Immunoassay. Springerplus 2016, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Golding, P.H. Holotranscobalamin (HoloTC, Active-B12) and Herbert’s Model for the Development of Vitamin B12 Deficiency: A Review and Alternative Hypothesis. Springerplus 2016, 5, 668. [Google Scholar] [CrossRef]
- Trakadis, Y.J.; Alfares, A.; Bodamer, O.A.; Buyukavci, M.; Christodoulou, J.; Connor, P.; Glamuzina, E.; Gonzalez-Fernandez, F.; Bibi, H.; Echenne, B.; et al. Update on Transcobalamin Deficiency: Clinical Presentation, Treatment and Outcome. J. Inherit. Metab. Dis. 2014, 37, 461–473. [Google Scholar] [CrossRef]
- Nissen, P.H.; Nordwall, M.; Hoffmann-Lücke, E.; Sorensen, B.S.; Nexo, E. Transcobalamin Deficiency Caused by Compound Heterozygosity for Two Novel Mutations in the TCN2 Gene: A Study of Two Affected Siblings, Their Brother, and Their Parents. J. Inherit. Metab. Dis. 2010, 33, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.A.M.; Alfaifi, J.; Alqahtani, Y.A.M.; Aljabri, M.F.; Kamal, N.M.; Althopaity, J.; Althobaiti, K.A.; Almalki, A.M.; Abosabie, S.A.S.; Abosabie, S.A.; et al. “Progressive Myoclonic Ataxia and Developmental/Epileptic Encephalopathy Associated with a Novel Homozygous Mutation in <scp> TCN 2 </Scp> Gene. ” Mol. Genet. Genom. Med. 2024, 12. [Google Scholar] [CrossRef]
- Quadros, E.V. Advances in the Understanding of Cobalamin Assimilation and Metabolism. Br. J. Haematol. 2010, 148, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.-J.; Choi, J.-S.; Park, C.-H.; Yoon, S.-O.; Jeoung, D.-I.; Kim, Y.-M.; Choe, J.-S. Expression of CD320 in Human B Cells in Addition to Follicular Dendritic Cells. BMB Rep. 2008, 41, 863–867. [Google Scholar] [CrossRef]
- Jiang, W.; Nakayama, Y.; Sequeira, J.M.; Quadros, E.V. Mapping the Functional Domains of TCblR/ CD320 , the Receptor for Cellular Uptake of Transcobalamin-bound Cobalamin. FASEB J. 2013, 27, 2988–2994. [Google Scholar] [CrossRef]
- Arendt, J.F.B.; Quadros, E.V.; Nexo, E. Soluble Transcobalamin Receptor, SCD320, Is Present in Human Serum and Relates to Serum Cobalamin – Establishment and Validation of an ELISA. Clin. Chem. Lab. Med. 2012, 50. [Google Scholar] [CrossRef]
- Youngdahl-Turner, P.; Mellman, I.S.; Allen, R.H.; Rosenberg, L.E. Protein Mediated Vitamin Uptake. Exp. Cell Res. 1979, 118, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Woo, J.-S.; Schmitz, J.; Prinz, B.; Root, K.; Chen, F.; Bloch, J.S.; Zenobi, R.; Locher, K.P. Structural Basis of Transcobalamin Recognition by Human CD320 Receptor. Nat. Commun. 2016, 7, 12100. [Google Scholar] [CrossRef] [PubMed]
- Gick, G.G.; Arora, K.; Sequeira, J.M.; Nakayama, Y.; Lai, S.-C.; Quadros, E.V. Cellular Uptake of Vitamin B12: Role and Fate of TCblR/CD320, the Transcobalamin Receptor. Exp. Cell Res. 2020, 396, 112256. [Google Scholar] [CrossRef]
- Quadros, E.V.; Jacobsen, D.W. The Dynamics of Cobalamin Utilization in L-1210 Mouse Leukemia Cells: A Model of Cellular Cobalamin Metabolism. Biochim. Et Biophys. Acta (BBA) - General. Subj. 1995, 1244, 395–403. [Google Scholar] [CrossRef]
- Bernard, D.J.; Pangilinan, F.J.; Cheng, J.; Molloy, A.M.; Brody, L.C. Mice Lacking the Transcobalamin-Vitamin B12 Receptor, CD320, Suffer from Anemia and Reproductive Deficits When Fed Vitamin B12-Deficient Diet. Hum. Mol. Genet. 2018, 27, 3627–3640. [Google Scholar] [CrossRef] [PubMed]
- Westerman; Evans; Metz Neutrophil Hypersegmentation in Iron Deficiency Anaemia: A Case-Control Study. Br. J. Haematol. 1999, 107, 512–515. [CrossRef] [PubMed]
- Arora, K.; Sequeira, J.M.; Hernández, A.I.; Alarcon, J.M.; Quadros, E.V. Behavioral Alterations Are Associated with Vitamin B12 Deficiency in the Transcobalamin Receptor/CD320 KO Mouse. PLoS ONE 2017, 12, e0177156. [Google Scholar] [CrossRef] [PubMed]
- Fernàndez-Roig, S.; Lai, S.-C.; Murphy, M.M.; Fernandez-Ballart, J.; Quadros, E. V Vitamin B12 Deficiency in the Brain Leads to DNA Hypomethylation in the TCblR/CD320 Knockout Mouse. Nutr Metab (Lond) 2012, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Kurnat-Thoma, E.L.; Pangilinan, F.; Matteini, A.M.; Wong, B.; Pepper, G.A.; Stabler, S.P.; Guralnik, J.M.; Brody, L.C. Association of Transcobalamin II ( TCN2 ) and Transcobalamin II-Receptor ( TCblR ) Genetic Variations With Cobalamin Deficiency Parameters in Elderly Women. Biol. Res. Nurs. 2015, 17, 444–454. [Google Scholar] [CrossRef]
- Quadros, E.V.; Lai, S.-C.; Nakayama, Y.; Sequeira, J.M.; Hannibal, L.; Wang, S.; Jacobsen, D.W.; Fedosov, S.; Wright, E.; Gallagher, R.C.; et al. Positive Newborn Screen for Methylmalonic Aciduria Identifies the First Mutation in TCblR/CD320, the Gene for Cellular Uptake of Transcobalamin-Bound Vitamin B12. Hum. Mutat. 2010, 31, 924–929. [Google Scholar] [CrossRef]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 Cause a New Inborn Error of Vitamin B12 Metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; van Veen, H.W. Molecular Basis of Multidrug Transport by ABC Transporters. Biochim. Et Biophys. Acta (BBA) - Proteins Proteom. 2009, 1794, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Feng, Z.; Hou, W.-T.; Jiang, Y.-L.; Wang, L.; Sun, L.; Zhou, C.-Z.; Chen, Y. Cryo-EM Structure of Human Lysosomal Cobalamin Exporter ABCD4. Cell Res. 2019, 29, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Deme, J.C.; Hancock, M.A.; Xia, X.; Shintre, C.A.; Plesa, M.; Kim, J.C.; Carpenter, E.P.; Rosenblatt, D.S.; Coulton, J.W. Purification and Interaction Analyses of Two Human Lysosomal Vitamin B 12 Transporters: LMBD1 and ABCD4. Mol. Membr. Biol. 2014, 31, 250–261. [Google Scholar] [CrossRef]
- Rutsch, F.; Gailus, S.; Miousse, I.R.; Suormala, T.; Sagné, C.; Toliat, M.R.; Nürnberg, G.; Wittkampf, T.; Buers, I.; Sharifi, A.; et al. Identification of a Putative Lysosomal Cobalamin Exporter Altered in the CblF Defect of Vitamin B12 Metabolism. Nat. Genet. 2009, 41, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Okamoto, T.; Morita, M.; Imanaka, T. Translocation of the ABC Transporter ABCD4 from the Endoplasmic Reticulum to Lysosomes Requires the Escort Protein LMBD1. Sci. Rep. 2016, 6, 30183. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.; Rosenblatt, D.S. Lessons in Biology from Patients with Inherited Disorders of Vitamin B12 and Folate Metabolism. Biochimie 2016, 126, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; C.N.; A.D.; V.C.P. Disorders of Intracellular Cobalamin Metabolism 2021.
- Kim, J.C.; Lee, N.-C.; Hwu, P.W.-L.; Chien, Y.-H.; Fahiminiya, S.; Majewski, J.; Watkins, D.; Rosenblatt, D.S. Late Onset of Symptoms in an Atypical Patient with the CblJ Inborn Error of Vitamin B12 Metabolism: Diagnosis and Novel Mutation Revealed by Exome Sequencing. Mol. Genet. Metab. 2012, 107, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Watkins, D.; Rosenblatt, D.S. Novel Splice Site Mutations and a Large Deletion in Three Patients with the CblF Inborn Error of Vitamin B12 Metabolism. Mol. Genet. Metab. 2011, 102, 505–507. [Google Scholar] [CrossRef]
- Alfadhel, M.; Lillquist, Y.P.; Davis, C.; Junker, A.K.; Stockler-Ipsiroglu, S. Eighteen-year Follow-up of a Patient with Cobalamin F Disease (CblF): Report and Review. Am. J. Med. Genet. A 2011, 155, 2571–2577. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.P.; Anastasio, N.; Liu, J.; Coelho, D.; Suormala, T.; Stucki, M.; Loewy, A.D.; Gurd, S.; Grundberg, E.; Morel, C.F.; et al. Spectrum of Mutations in MMACHC , Allelic Expression, and Evidence for Genotype–Phenotype Correlations. Hum. Mutat. 2009, 30, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Doré, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the Gene Responsible for Methylmalonic Aciduria and Homocystinuria, CblC Type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.-L.; Chéry, C.; Oussalah, A.; Nadaf, J.; Coelho, D.; Josse, T.; Flayac, J.; Robert, A.; Koscinski, I.; Gastin, I.; et al. A PRDX1 Mutant Allele Causes a MMACHC Secondary Epimutation in CblC Patients. Nat. Commun. 2018, 9, 67. [Google Scholar] [CrossRef]
- Kim, J.; Gherasim, C.; Banerjee, R. Decyanation of Vitamin B12 by a Trafficking Chaperone. Proc. Natl. Acad. Sci. U S A 2008, 105, 14551–14554. [Google Scholar] [CrossRef] [PubMed]
- Bassila, C.; Ghemrawi, R.; Flayac, J.; Froese, D.S.; Baumgartner, M.R.; Guéant, J.-L.; Coelho, D. Methionine Synthase and Methionine Synthase Reductase Interact with MMACHC and with MMADHC. Biochim. Et Biophys. Acta (BBA) - Mol. Basis Dis. 2017, 1863, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, S.; Brezzi, B.; Bracciamà, V.; Barreca, A.; Nozza, P.; Vaisitti, T.; Amoroso, A.; Deaglio, S.; Manganaro, M.; Porta, F.; et al. Adult-Onset CblC Deficiency: A Challenging Diagnosis Involving Different Adult Clinical Specialists. Orphanet J. Rare Dis. 2022, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Sorin, M.; Watkins, D.; Gilfix, B.M.; Rosenblatt, D.S. Methionine Dependence in Tumor Cells: The Potential Role of Cobalamin and MMACHC. Mol. Genet. Metab. 2021, 132, 155–161. [Google Scholar] [CrossRef]
- Coelho, D.; Suormala, T.; Stucki, M.; Lerner-Ellis, J.P.; Rosenblatt, D.S.; Newbold, R.F.; Baumgartner, M.R.; Fowler, B. Gene Identification for the CblD Defect of Vitamin B 12 Metabolism. New Engl. J. Med. 2008, 358, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Pupavac, M.; Garcia, M.A.M.; Rosenblatt, D.S.; Jerome-Majewska, L.A. Expression of Mmachc and Mmadhc during Mouse Organogenesis. Mol. Genet. Metab. 2011, 103, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Coelho, D.; Suormala, T.; Burda, P.; Fowler, B.; Baumgartner, M.R. Molecular Mechanisms Leading to Three Different Phenotypes in the CblD Defect of Intracellular Cobalamin Metabolism. Hum. Mol. Genet. 2012, 21, 1410–1418. [Google Scholar] [CrossRef]
- Plesa, M.; Kim, J.; Paquette, S.G.; Gagnon, H.; Ng-Thow-Hing, C.; Gibbs, B.F.; Hancock, M.A.; Rosenblatt, D.S.; Coulton, J.W. Interaction between MMACHC and MMADHC, Two Human Proteins Participating in Intracellular Vitamin B12 Metabolism. Mol. Genet. Metab. 2011, 102, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Deme, J.C.; Miousse, I.R.; Plesa, M.; Kim, J.C.; Hancock, M.A.; Mah, W.; Rosenblatt, D.S.; Coulton, J.W. Structural Features of Recombinant MMADHC Isoforms and Their Interactions with MMACHC, Proteins of Mammalian Vitamin B12 Metabolism. Mol. Genet. Metab. 2012, 107, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Kopec, J.; Fitzpatrick, F.; Schuller, M.; McCorvie, T.J.; Chalk, R.; Plessl, T.; Fettelschoss, V.; Fowler, B.; Baumgartner, M.R.; et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J. Biol. Chem. 2015, 290, 29167–29177. [Google Scholar] [CrossRef] [PubMed]
- Suormala, T.; Baumgartner, M.R.; Coelho, D.; Zavadakova, P.; Kožich, V.; Koch, H.G.; Berghaüser, M.; Wraith, J.E.; Burlina, A.; Sewell, A.; et al. The CblD Defect Causes Either Isolated or Combined Deficiency of Methylcobalamin and Adenosylcobalamin Synthesis. J. Biol. Chem. 2004, 279, 42742–42749. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Furlan, F.; Brambilla, A.; Codazzi, D.; Vedovati, S.; Corbetta, C.; Fedeli, T.; Merinero, B.; Pérez, B.; Ugarte, M. Severe Neonatal Metabolic Decompensation in Methylmalonic Acidemia Caused by CblD Defect. In; 2013; pp. 133–137.
- Christensen, E.I.; Birn, H. Megalin and Cubilin: Synergistic Endocytic Receptors in Renal Proximal Tubule. Am. J. Physiol. -Ren. Physiol. 2001, 280, F562–F573. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Liu, M.L.; Hwang, H.Y.; Chen, L.S.; Korenberg, J.; Shane, B. Human Methionine Synthase. CDNA Cloning, Gene Localization, and Expression. J. Biol. Chem. 1997, 272, 3628–3634. [Google Scholar] [CrossRef] [PubMed]
- Drennan, C.L.; Matthews, R.G.; Ludwig, M.L. Cobalamin-Dependent Methionine Synthase: The Structure of a Methylcobalamin-Binding Fragment and Implications for Other B12-Dependent Enzymes. Curr. Opin. Struct. Biol. 1994, 4, 919–929. [Google Scholar] [CrossRef]
- Matthews, R.G.; Sheppard, C.; Goulding, C. Methylenetetrahydrofolate Reductase and Methionine Synthase: Biochemistry and Molecular Biology. Eur. J. Pediatr. 1998, 157, S54–S59. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.-L.; Caillerez-Fofou, M.; Battaglia-Hsu, S.; Alberto, J.-M.; Freund, J.-N.; Dulluc, I.; Adjalla, C.; Maury, F.; Merle, C.; Nicolas, J.-P.; et al. Molecular and Cellular Effects of Vitamin B12 in Brain, Myocardium and Liver through Its Role as Co-Factor of Methionine Synthase. Biochimie 2013, 95, 1033–1040. [Google Scholar] [CrossRef]
- Watkin, D.; Rosenblatt, D.S. Functional Methionine Synthase Deficiency (CblE and CblG): Clinical and Biochemical Heterogeneity. Am. J. Med. Genet. 1989, 34, 427–434. [Google Scholar] [CrossRef]
- Watkins, D.; Ru, M.; Hwang, H.-Y.; Kim, C.D.; Murray, A.; Philip, N.S.; Kim, W.; Legakis, H.; Wai, T.; Hilton, J.F.; et al. Hyperhomocysteinemia Due to Methionine Synthase Deficiency, CblG: Structure of the MTR Gene, Genotype Diversity, and Recognition of a Common Mutation, P1173L. Am. J. Human. Genet. 2002, 71, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.-L.; Guéant-Rodriguez, R.-M.; Kosgei, V.J.; Coelho, D. Causes and Consequences of Impaired Methionine Synthase Activity in Acquired and Inherited Disorders of Vitamin B 12 Metabolism. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Iñiguez, T.; García-Hernandez, E.; Arreguín-Espinosa, R.; Flores, M.E. Role of Vitamin B12 on Methylmalonyl-CoA Mutase Activity. J. Zhejiang Univ. Sci. B 2012, 13, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Mancia, F.; Evans, P.R. Conformational Changes on Substrate Binding to Methylmalonyl CoA Mutase and New Insights into the Free Radical Mechanism. Structure 1998, 6, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, K.; Bailey, H.J.; Burda, P.; Chaikuad, A.; Krysztofinska, E.; Suormala, T.; Bürer, C.; Lutz, S.; Fowler, B.; Froese, D.S.; et al. Genetic, Structural, and Functional Analysis of Pathogenic Variations Causing Methylmalonyl-CoA Epimerase Deficiency. Biochim. Et Biophys. Acta (BBA) - Mol. Basis Dis. 2019, 1865, 1265–1272. [Google Scholar] [CrossRef]
- Pratt, J.M. 988. The Chemistry of Vitamin B12. Part II. Photochemical Reactions. J. Chem. Soc. (Resumed) 1964, 5154. [Google Scholar] [CrossRef]
- Takahashi-Iñiguez, T.; González-Noriega, A.; Michalak, C.; Flores, M.E. Human MMAA Induces the Release of Inactive Cofactor and Restores Methylmalonyl-CoA Mutase Activity through Their Complex Formation. Biochimie 2017, 142, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Y.; Liu, T.-T.; Yang, Y.-L.; Chang, Y.-C.; Fan, Y.-L.; Lee, S.-F.; Teng, Y.-T.; Chiang, S.-H.; Niu, D.-M.; Lin, S.-J.; et al. Mutation Profile of the MUT Gene in Chinese Methylmalonic Aciduria Patients. In; 2012; pp. 55–64.
- Willard, H.F.; Rosenberg, L.E. Inherited Methylmalonyl CoA Mutase Apoenzyme Deficiency in Human Fibroblasts. J. Clin. Investig. 1980, 65, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, C.; Benoist, J.; Pereira, S.; Callebaut, I.; Koskas, T.; Porquet, D.; Elion, J. Molecular Basis of Methylmalonyl-CoA Mutase Apoenzyme Defect in 40 European Patients Affected by Mut. ° and Mut. – Forms of Methylmalonic Acidemia: Identification of 29 Novel Mutations in the MUT Gene. Hum. Mutat. 2005, 25, 167–176. [Google Scholar] [CrossRef]
- Bailey, R.L.; Carmel, R.; Green, R.; Pfeiffer, C.M.; Cogswell, M.E.; Osterloh, J.D.; Sempos, C.T.; Yetley, E.A. Monitoring of Vitamin B-12 Nutritional Status in the United States by Using Plasma Methylmalonic Acid and Serum Vitamin B-12. Am. J. Clin. Nutr. 2011, 94, 552–561. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, A.; Yeung, L.F.; Qi, Y.P.; Pfeiffer, C.M.; Crider, K.S. Folate and Vitamin B12 Usual Intake and Biomarker Status by Intake Source in United States Adults Aged ≥19 y: NHANES 2007–2018. Am J Clin Nutr 2023, 118, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Murphy, M.; Solé-Navais, P.; Yajnik, C. Cobalamin Status from Pregnancy to Early Childhood: Lessons from Global Experience. Adv. Nutr. 2017, 8, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, G.; Gütgemann, I.; Kolb, G.; Leischker, A. Klinisch-Hämatologisches Bild Des Vitamin-B12-Mangels Im Alter. Z. Gerontol. Geriatr. 2018, 51, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Devi, A.; Rush, E.; Harper, M.; Venn, B. Vitamin B12 Status of Various Ethnic Groups Living in New Zealand: An Analysis of the Adult Nutrition Survey 2008/2009. Nutrients 2018, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Jajoo, S.S.; Zamwar, U.M.; Nagrale, P. Etiology, Clinical Manifestations, Diagnosis, and Treatment of Cobalamin (Vitamin B12) Deficiency. Cureus 2024. [Google Scholar] [CrossRef] [PubMed]
- Dali-Youcef, N.; Andres, E. An Update on Cobalamin Deficiency in Adults. QJM 2009, 102, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Andres, E. Vitamin B12 (Cobalamin) Deficiency in Elderly Patients. Can. Med. Assoc. J. 2004, 171, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton Pump Inhibitor and Histamine 2 Receptor Antagonist Use and Vitamin B 12 Deficiency. JAMA 2013, 310, 2435. [Google Scholar] [CrossRef] [PubMed]
- Green, R. Vitamin B12 Deficiency from the Perspective of a Practicing Hematologist. Blood 2017, 129, 2603–2611. [Google Scholar] [CrossRef]
- Lindenbaum, J.; Healton, E.B.; Savage, D.G.; Brust, J.C.M.; Garrett, T.J.; Podell, E.R.; Margell, P.D.; Stabler, S.P.; Allen, R.H. Neuropsychiatric Disorders Caused by Cobalamin Deficiency in the Absence of Anemia or Macrocytosis. New Engl. J. Med. 1988, 318, 1720–1728. [Google Scholar] [CrossRef]
- Green, R.; Kinsella, L.J. Current Concepts in the Diagnosis of Cobalamin Deficiency. Neurology 1995, 45, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Troen, A.M.; Shea-Budgell, M.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. B-Vitamin Deficiency Causes Hyperhomocysteinemia and Vascular Cognitive Impairment in Mice. Proc. Natl. Acad. Sci. 2008, 105, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R. Diagnosis and Management of Clinical and Subclinical Cobalamin Deficiencies: Why Controversies Persist in the Age of Sensitive Metabolic Testing. Biochimie 2013, 95, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R. Subclinical Cobalamin Deficiency. Curr. Opin. Gastroenterol. 2012, 28, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Devalia, V.; Hamilton, M.S.; Molloy, A.M. Guidelines for the Diagnosis and Treatment of Cobalamin and Folate Disorders. Br. J. Haematol. 2014, 166, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R.; Agrawal, Y.P. Failures of Cobalamin Assays in Pernicious Anemia. New Engl. J. Med. 2012, 367, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Garrod, M.G.; Rockwood, A.L.; Kushnir, M.M.; Allen, L.H.; Haan, M.N.; Green, R. Measurement of Total Vitamin B12 and Holotranscobalamin, Singly and in Combination, in Screening for Metabolic Vitamin B12 Deficiency. Clin. Chem. 2006, 52, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Nexo, E.; Hoffmann-Lücke, E. Holotranscobalamin, a Marker of Vitamin B-12 Status: Analytical Aspects and Clinical Utility. Am. J. Clin. Nutr. 2011, 94, 359S–365S. [Google Scholar] [CrossRef] [PubMed]
- Molloy, A.M.; Pangilinan, F.; Mills, J.L.; Shane, B.; O’Neill, M.B.; McGaughey, D.M.; Velkova, A.; Abaan, H.O.; Ueland, P.M.; McNulty, H.; et al. A Common Polymorphism in HIBCH Influences Methylmalonic Acid Concentrations in Blood Independently of Cobalamin. Am. J. Human. Genet. 2016, 98, 869–882. [Google Scholar] [CrossRef]
- Allen, L.H.; Miller, J.W.; de Groot, L.; Rosenberg, I.H.; Smith, A.D.; Refsum, H.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND): Vitamin B-12 Review. J. Nutr. 2018, 148, 1995S–2027S. [Google Scholar] [CrossRef]
- Hvas, A.-M.; Morkbak, A.L.; Hardlei, T.F.; Nexo, E. The Vitamin B12 Absorption Test, CobaSorb, Identifies Patients Not Requiring Vitamin B12 Injection Therapy. Scand. J. Clin. Lab. Invest. 2011, 71, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Bor, M.V.; Çetin, M.; Aytaç, S.; Altay, C.; Nexo, E. Nonradioactive Vitamin B12 Absorption Test Evaluated in Controls and in Patients with Inherited Malabsorption of Vitamin B12. Clin. Chem. 2005, 51, 2151–2155. [Google Scholar] [CrossRef]
- Hvas, A.-M.; Morkbak, A.L.; Nexo, E. Plasma Holotranscobalamin Compared with Plasma Cobalamins for Assessment of Vitamin B12 Absorption; Optimisation of a Non-Radioactive Vitamin B12 Absorption Test (CobaSorb). Clin. Chim. Acta 2007, 376, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Garrod; Rossow; Calvert; Miller; Green; Buchholz; Allen 14C-Cobalamin Absorption from Endogenously Labeled Chicken Eggs Assessed in Humans Using Accelerator Mass Spectrometry. Nutrients 2019, 11, 2148. [CrossRef]
- Vitamin B 12 Deficiency. New Engl. J. Med. 2013, 368, 2040–2042. [CrossRef]
- Toh, B.-H. Diagnosis and Classification of Autoimmune Gastritis. Autoimmun. Rev. 2014, 13, 459–462. [Google Scholar] [CrossRef]
- Lahner, E.; Norman, G.L.; Severi, C.; Encabo, S.; Shums, Z.; Vannella, L.; Fave, G.D.; Annibale, B. Reassessment of Intrinsic Factor and Parietal Cell Autoantibodies in Atrophic Gastritis With Respect to Cobalamin Deficiency. Am. J. Gastroenterol. 2009, 104, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, C.; Lofgren, M.; Banerjee, R. Navigating the B12 Road: Assimilation, Delivery, and Disorders of Cobalamin. J. Biol. Chem. 2013, 288, 13186–13193. [Google Scholar] [CrossRef]
- Froese, D.S.; Gravel, R.A. Genetic Disorders of Vitamin B 12 Metabolism: Eight Complementation Groups – Eight Genes. Expert. Rev. Mol. Med. 2010, 12, e37. [Google Scholar] [CrossRef]
- British Society for Haematology BSH Guidance on B12 Replacement during the COVID-19 Pandemic.
- Berkins, S.; Schiöth, H.B.; Rukh, G. Depression and Vegetarians: Association between Dietary Vitamin B6, B12 and Folate Intake and Global and Subcortical Brain Volumes. Nutrients 2021, 13, 1790. [Google Scholar] [CrossRef]
- Sangle, P.; Sandhu, O.; Aftab, Z.; Anthony, A.T.; Khan, S. Vitamin B12 Supplementation: Preventing Onset and Improving Prognosis of Depression. Cureus 2020. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.R.; Di Matteo, G.; La Rosa, P.; Barbati, S.A.; Mannina, L.; Moreno, S.; Tata, A.M.; Cavallucci, V.; Fidaleo, M. Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2024, 25, 590. [Google Scholar] [CrossRef] [PubMed]
- Alruwaili, M.; Basri, R.; AlRuwaili, R.; Albarrak, A.M.; Ali, N.H. Neurological Implications of Vitamin B12 Deficiency in Diet: A Systematic Review and Meta-Analysis. Healthcare 2023, 11, 958. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Quan, M.; Li, T.; Jia, J. Serum Homocysteine, Vitamin B12, Folate, and Their Association with Mild Cognitive Impairment and Subtypes of Dementia. J. Alzheimer’s Dis. 2022, 90, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Mander, A.; Ames, D.; Carne, R.; Sanders, K.; Watters, D. Cognitive Impairment and Vitamin B12: A Review. Int. Psychogeriatr. 2012, 24, 541–556. [Google Scholar] [CrossRef]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the Nervous System: Current Knowledge of the Biochemical Modes of Action and Synergies of Thiamine, Pyridoxine, and Cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Alam, S.F. Role of Homocysteine in the Development of Cardiovascular Disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- COPPOLA, A.; DAVI, G.; DE STEFANO, V.; MANCINI, F.P.; CERBONE, A.M.; DI MINNO, G. Homocysteine, Coagulation, Platelet Function, and Thrombosis. Semin. Thromb. Hemost. 2000, Volume 26, 243–254. [Google Scholar] [CrossRef]
- Ammouri, W.; Tazi, Z.M.; Harmouche, H.; Maamar, M.; Adnaoui, M. Venous Thromboembolism and Hyperhomocysteinemia as First Manifestation of Pernicious Anemia: A Case Series. J. Med. Case Rep. 2017, 11, 250. [Google Scholar] [CrossRef]
- Azzini, E.; Raguzzini, A.; Polito, A. A Brief Review on Vitamin B12 Deficiency Looking at Some Case Study Reports in Adults. Int. J. Mol. Sci. 2021, 22, 9694. [Google Scholar] [CrossRef]
- Ospina-Romero, M.; Cannegieter, S.C.; den Heijer, M.; Doggen, C.J.M.; Rosendaal, F.R.; Lijfering, W.M. Hyperhomocysteinemia and Risk of First Venous Thrombosis: The Influence of (Unmeasured) Confounding Factors. Am. J. Epidemiol. 2018, 187, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Çoker, M.; de Klerk, J.B.C.; Poll-The, B.T.; Huijmans, J.G.M.; Duran, M. Plasma Total Odd-chain Fatty Acids in the Monitoring of Disorders of Propionate, Methylmalonate and Biotin Metabolism. J. Inherit. Metab. Dis. 1996, 19, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Ilter, D.; Low, V.; Endress, J.E.; Fernández-García, J.; Rosenzweig, A.; Schild, T.; Broekaert, D.; Ahmed, A.; Planque, M.; et al. Age-Induced Accumulation of Methylmalonic Acid Promotes Tumour Progression. Nature 2020, 585, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Ilter, D.; Low, V.; Drapela, S.; Schild, T.; Mullarky, E.; Han, J.; Elia, I.; Broekaert, D.; Rosenzweig, A.; et al. Altered Propionate Metabolism Contributes to Tumour Progression and Aggressiveness. Nat. Metab. 2022, 4, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Ye, M.; Bai, J.; Liu, P.; Lu, F.; Chen, J.; Yu, P.; Chen, T.; Shi, X.; Tang, Q. Methylmalonic Acid Promotes Colorectal Cancer Progression via Activation of Wnt/β-Catenin Pathway Mediated Epithelial–Mesenchymal Transition. Cancer Cell Int. 2023, 23, 131. [Google Scholar] [CrossRef] [PubMed]
- Loedin, A.K.; Speijer, D. Is There a Carcinogenic Risk Attached to Vitamin B 12 Deficient Diets and What Should We Do About It? Reviewing the Facts. Mol. Nutr. Food Res. 2021, 65. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R. High Plasma Vitamin B12 and Cancer in Human Studies: A Scoping Review to Judge Causality and Alternative Explanations. Nutrients 2022, 14, 4476. [Google Scholar] [CrossRef] [PubMed]
- Haghighat, G.; Khajeh-Mehrizi, A.; Ranjbar, H. Evaluation of Serum Vitamin B12 Levels in Patients with Colon and Breast Cancer: A Case-Control Study. Int. J. Hematol. Oncol. Stem Cell Res. 2023. [Google Scholar] [CrossRef]
- Zhang, S.M.; Willett, W.C.; Selhub, J.; Hunter, D.J.; Giovannucci, E.L.; Holmes, M.D.; Colditz, G.A.; Hankinson, S.E. Plasma Folate, Vitamin B6, Vitamin B12, Homocysteine, and Risk of Breast Cancer. JNCI J. Natl. Cancer Inst. 2003, 95, 373–380. [Google Scholar] [CrossRef]
- Wu, W.; Kang, S.; Zhang, D. Association of Vitamin B6, Vitamin B12 and Methionine with Risk of Breast Cancer: A Dose–Response Meta-Analysis. Br. J. Cancer 2013, 109, 1926–1944. [Google Scholar] [CrossRef]
- Matejcic, M.; de Batlle, J.; Ricci, C.; Biessy, C.; Perrier, F.; Huybrechts, I.; Weiderpass, E.; Boutron-Ruault, M.C.; Cadeau, C.; His, M.; et al. Biomarkers of Folate and Vitamin B12 and Breast Cancer Risk: Report from the EPIC Cohort. Int. J. Cancer 2017, 140, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Mathews, F.S.; Gordon, M.M.; Chen, Z.; Rajashankar, K.R.; Ealick, S.E.; Alpers, D.H.; Sukumar, N. Crystal Structure of Human Intrinsic Factor: Cobalamin Complex at 2.6-Å Resolution. Proc. Natl. Acad. Sci. 2007, 104, 17311–17316. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.T.-L.; Lin, C.-L.; Pan, K.-H.; Tzen, K.-Y.; Su, M.-J.; Tsai, C.-T.; Li, Y.-H.; Li, P.-C.; Chiang, F.-T.; Chang, S.C.; et al. Single Allele Lmbrd1 Knockout Results in Cardiac Hypertrophy. J. Formos. Med. Assoc. 2018, 117, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.S.; Kuner, R.; Buness, A.; Ruschhaupt, M.; Merk, S.; Zwermann, L.; Kääb, S.; Kreuzer, E.; Steinbeck, G.; Mansmann, U.; et al. Identification of a Common Gene Expression Signature in Dilated Cardiomyopathy Across Independent Microarray Studies. J. Am. Coll. Cardiol. 2006, 48, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, D.; Kihara, Y.; Groves, A.; Ray, M.; Saha, A.; Ellington, C.; Lee-Okada, H.-C.; Furihata, T.; Yokomizo, T.; Quadros, E.V.; et al. FTY720 Requires Vitamin B12-TCN2-CD320 Signaling in Astrocytes to Reduce Disease in an Animal Model of Multiple Sclerosis. Cell Rep. 2023, 42, 113545. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Henderson, M.; Haber, M.; Norris, M. Role of the MRP1/ABCC1 Multidrug Transporter Protein in Cancer. IUBMB Life 2007, 59, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, C.; Yeh, S.; Xin, J.; Wang, P.; Shi, Q.; Ming, X. Combinatory Therapy of MRP1-Targeted Photoimmunotherapy and Liposomal Doxorubicin Promotes the Antitumor Effect for Chemoresistant Small Cell Lung Cancer. Int. J. Pharm. 2022, 625, 122076. [Google Scholar] [CrossRef] [PubMed]
- Pietz, H.L.; Abbas, A.; Johnson, Z.L.; Oldham, M.L.; Suga, H.; Chen, J. A Macrocyclic Peptide Inhibitor Traps MRP1 in a Catalytically Incompetent Conformation. Proc. Natl. Acad. Sci. 2023, 120. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, K.M.; Haber, M.; Fletcher, J.I. Targeting Multidrug Resistance-Associated Protein 1 (MRP1)-Expressing Cancers: Beyond Pharmacological Inhibition. Drug Resist. Updates 2021, 59, 100795. [Google Scholar] [CrossRef]
- Alhaji, J.H. Vitamin B12 Deficiency in Patients with Diabetes on Metformin: Arab Countries. Nutrients 2022, 14, 2046. [Google Scholar] [CrossRef]
- Bell, D.S.H. Metformin-induced Vitamin B12 Deficiency Can Cause or Worsen Distal Symmetrical, Autonomic and Cardiac Neuropathy in the Patient with Diabetes. Diabetes Obes. Metab. 2022, 24, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-Term Metformin Therapy and Vitamin B12 Deficiency: An Association to Bear in Mind. World J. Diabetes 2021, 12, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Bauman, W.A.; Shaw, S.; Jayatilleke, E.; Spungen, A.M.; Herbert, V. Increased Intake of Calcium Reverses Vitamin B12 Malabsorption Induced by Metformin. Diabetes Care 2000, 23, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Out, M.; Kooy, A.; Lehert, P.; Schalkwijk, C.A.; Stehouwer, C.D.A. Long-Term Treatment with Metformin in Type 2 Diabetes and Methylmalonic Acid: Post Hoc Analysis of a Randomized Controlled 4.3 Year Trial. J. Diabetes Complicat. 2018, 32, 171–178. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Melendez, E.; Chondronasiou, D.; Pietrocola, F.; Bernad, R.; Caballe, A.; Junza, A.; Capellades, J.; Holguín-Horcajo, A.; Prats, N.; et al. Vitamin B12 Is a Limiting Factor for Induced Cellular Plasticity and Tissue Repair. Nat. Metab. 2023, 5, 1911–1930. [Google Scholar] [CrossRef]
Figure 1.
Skeletal formula of cobalamin. R= CN, HO, methyl, or 5-deoxyadenosyl.
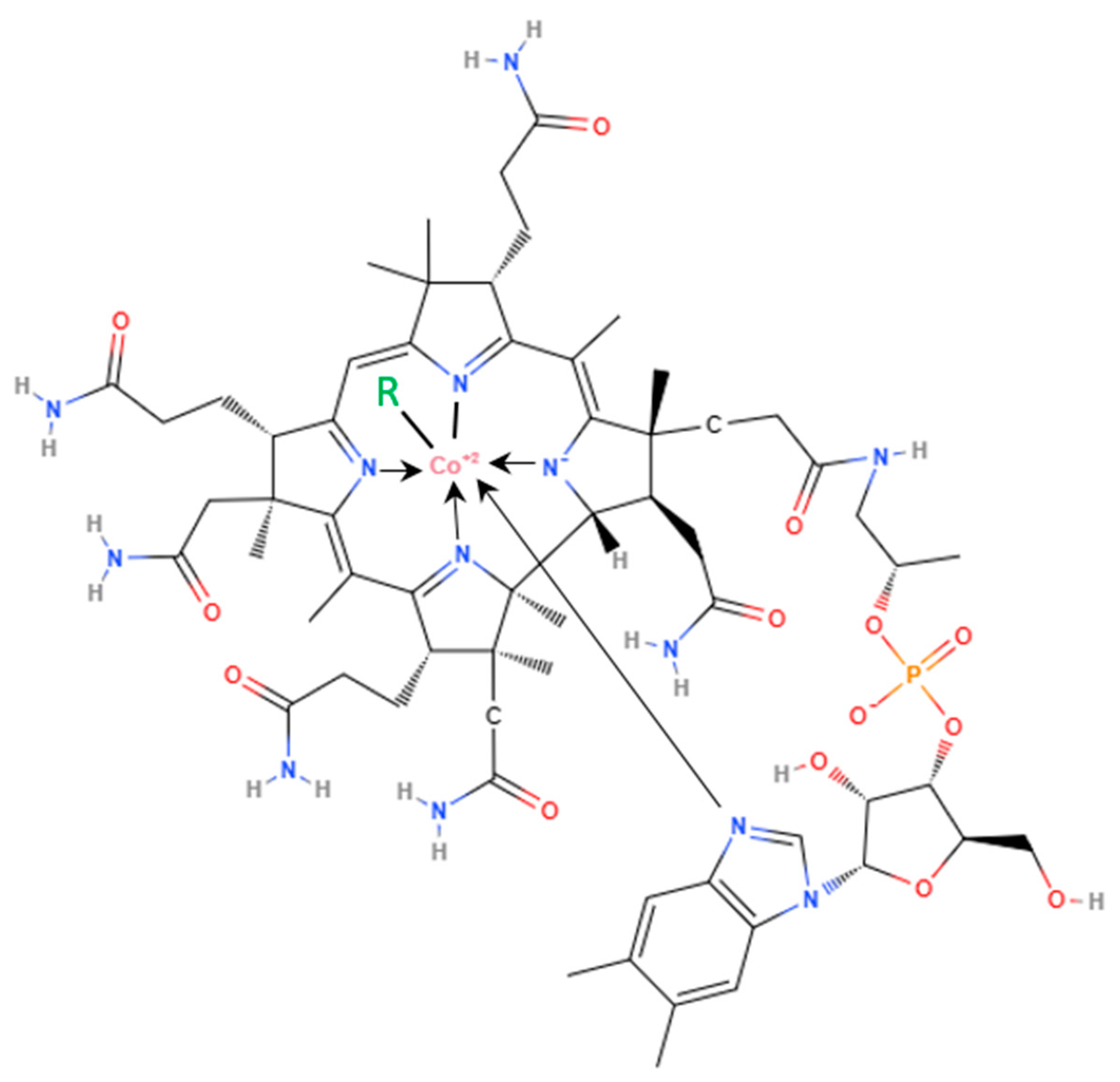
Figure 1.
Adsorption, blood transport, and intracellular metabolism of vitamin B12. IF- intrinsic factor, TCN2- transcobalamin II, AdoCl- adenosylcobalamin, MeCbl- methylcobalamin, MMUT- methylmalonyl CoA mutase, MTR- methionine syntethase. Created with BioRender.com.
Figure 1.
Adsorption, blood transport, and intracellular metabolism of vitamin B12. IF- intrinsic factor, TCN2- transcobalamin II, AdoCl- adenosylcobalamin, MeCbl- methylcobalamin, MMUT- methylmalonyl CoA mutase, MTR- methionine syntethase. Created with BioRender.com.
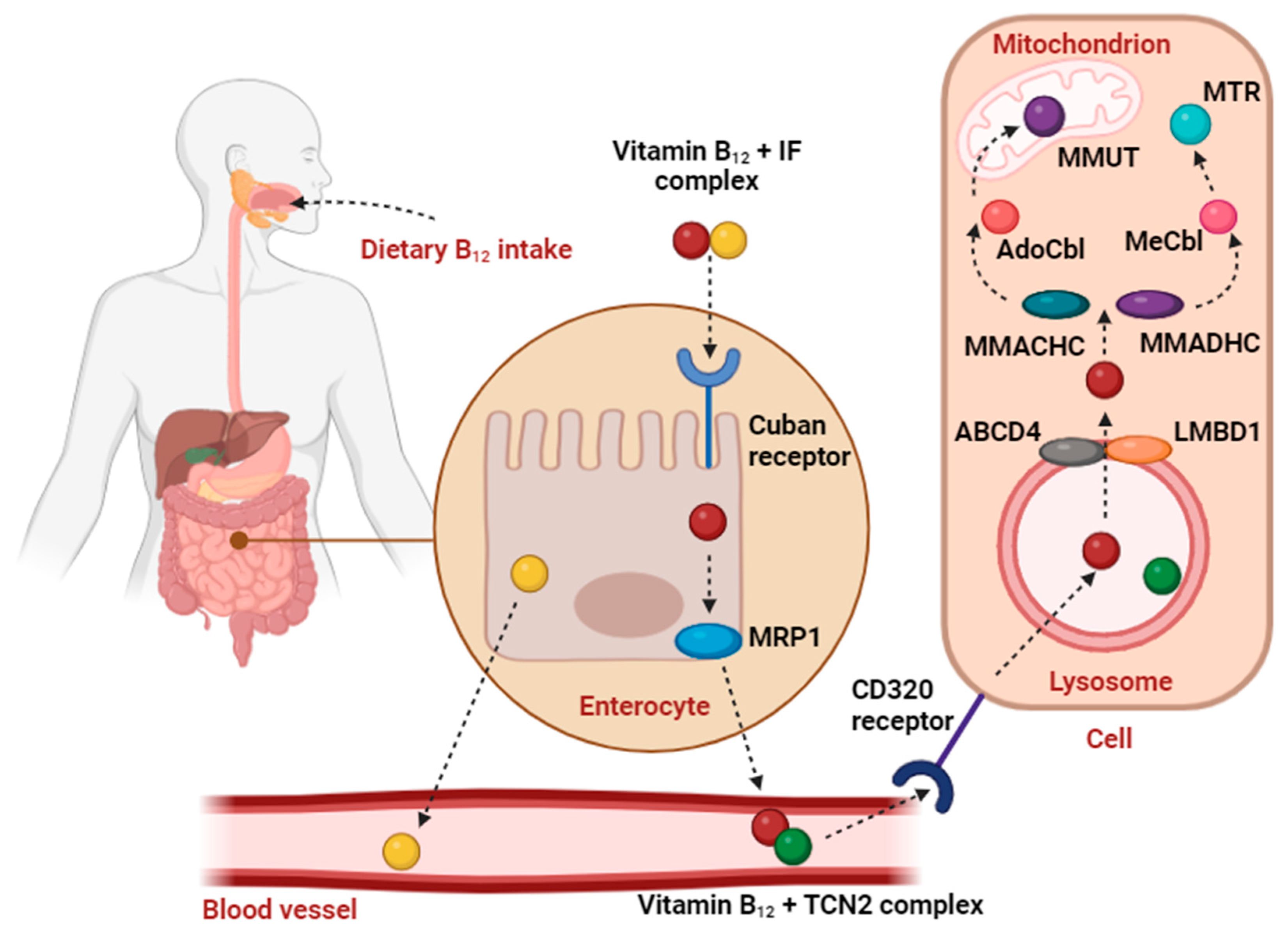

Table 1.
Proteins involved in vitamin B12 metabolism.
| PROTEIN | SHORTNAME | GENE | SIZE | TYPE | FUNCTION | PATHOLOGY | REF. | |
|---|---|---|---|---|---|---|---|---|
| Haptocorrin, Transcobalamin I | HC, TCN1 | TCN1 | 60 to 70 kDa | Glycoprotein | Binding vitamin B12 in saliva, transferring the vitamin to the stomach; Protection of B12 from hydrolysis in the acidic pH of the stomach. | The inability of HC degradation and release of the vitamin from the vitamin B12-HC complex (caused by deficiency of pancreatic proteases.) may led to vitamin B12 malabsorption | [14,179] | |
| Intrinsic Factor | IF | GIF or CBLIF | Two domains of approx. 30 kDa and 20 kDa (399 amino acid residues) | Glycoprotein | Binding vitamin B12 in the duodenum. | The lack of IF in the condition of H. pylori infection or AIG leads to long-lasting vitamin B12 deficiency due to the inability to deliver vitamin B12 to the cells within the ileum. | [14,15,20,179,179] | |
|
Cubam receptor |
Cubilin | CUBN | CUBN | 460 kDa | Multiligand, apical membrane receptor |
Interaction with IF; Docking to transmembrane protein AMN; Forming with AMN a receptor specific for IF-vitamin B12 internalization. | Disrupting this Cubam results in malabsorption of vitamin B12 and the development of Imerslund-Gräsbeck syndrome (IGS), a rare autosomal recessive disorder that appears in childhood, characterized by megaloblastic anemia | [33,34,37,38] |
| Type-1 transmembrane protein Amnionless | AMN | AMN | 45 kDa | Clathrin-mediated internalization; Anchoring Cubilin; Forming with CUBN a receptor specific for IF-vitamin B12 internalization. |
||||
|
Multidrug Resistant Protein 1 |
MRP1, ABCC1 | ABCC1 | 190 kDa | ATP-binding cassette (ABC) transporters family | Effluxing vitamin B12 from the intestinal epithelium to the blood circulation. | Mutations of gene ABCC1 were detected in some patients with vitamin B12 malabsorption. | [46,47,52,54] |
|
| Transcobalamin II | TCN2 | TCN2 | 43 kDa | β-globulin protein | Binding vitamin B12 in the bloodstream; Distribution of vitamin B12 into all cells in the body. | Mutations in the gene can lead to intracellular cobalamin depletion, causing a rare multisystemic disorder characterized by autosomal recessive inheritance. | [55,59,60,67,68,69] |
|
| Receptor for cluster of differentiation 320, Receptor for cobalamin-saturated transcobalamin | CD320, TCblR | TCblR or CD320 | 29 kDa (282 amino acids) (observed 58 kDa) | Low-density Lipoprotein Receptor protein family | Endocytosis of TCN2-saturated cobalamin. | In humans, very few cases of CD320 receptor defects have been confirmed to be the direct reason for lowered B12 levels in serum and increased MMA concentration in newborns. | [70,71,72,74,75,82,83] |
|
| ATP-binding cassette sub-family D member 4 | ABCD4 | ABCD4 | 68.6 kDa | Exporter protein | Transport free vitamin B12 into the cytoplasm. | Mutations are related to disease groups cblJ and cblF. At the cellular level, both errors are marked with a decreased function of MMUT and MTR and an accumulation of free vitamin B12 in the lysosomes. | [84,84,84,87,88,88,88,89,91,91,94] | |
| Lysosomal cobalamin transport escort protein | LMBD1 | LMBDR1 | 61.4 kDa | Adaptor protein, chaperone | Mediates the translocation of ABCD4 from ER to the lysosomes. | |||
| Methylmalonic aciduria and homocystinuria type C protein | MMACHC | MMACHC | 31.7 kDa (282 amino acids) | Chaperone, enzyme | Potentially receiving the Cbl cargo upon its release from the lysosomal compartment; Catalyzing the reductive decyanation of cyanocobalamin. | Mutations result in a rare genetic disorder referred to as methylmalonic aciduria and homocystinuria type C (cblC); deficiencies in both B12-dependent enzymatic functions MTR and MMUT - accumulation of both MMA and Hcy in serum. | [95,96,97,98,100] | |
| Methylmalonic aciduria and homocystinuria type D protein | MMADHC | MMADHC | 32.9 kDa (296 amino acids) | Probably enzyme | Play a role in the production of the two active cobalamin forms, MeCbl and AdoCb. | The cblD complementation group can be linked to isolated methylmalonic aciduria (cblD-MMA), isolated homocystinuria (cblD-HC), or a combination of both methylmalonic aciduria and homocystinuria (cblD-MMA/HC). | [102,103,104,107,108,109] |
|
| Methionine synthase | MTR, MS | MTR | 140 kDa | Cytoplasmic enzyme | Vitamin B12-dependent transfer of methyl group from 5-methyltetrahydrofolate to homocysteine with the production of tetrahydrofolate and methionine. | Missense mutation or deletion of three base pairs was identified in the cblG group of patients with cobalamin metabolism disorder and methylcobalamin deficiency cblE type, rare autosomal diseases associated with homocysteinemia, homocystinuria, and hypomethioninemia. | [83,84,85,87,88,110,115,116] |
|
| Methylmalonyl-CoA mutase | MMUT, MCM, MUT | MMUT | 78 kDa | Mitochondrial enzyme | Vitamin B12-dependent conversion methylmalonyl-CoA to succinyl-CoA. | Mutation leads to methylmalonic acidemia. | [89,90,91,93,120,122] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



