
Submitted:
19 June 2024
Posted:
20 June 2024
You are already at the latest version
Abstract
Non-Vitamin K Oral Anticoagulants (NOACs) have revolutionized anticoagulant therapy, offering improved safety and efficacy over traditional agents like warfarin. This review comprehensively examines the dual roles of NOACs—Apixaban, Rivaroxaban, Edoxaban, and Dabigatran—not only as anticoagulants but also as modulators of inflammation via Protease-Activated Receptor (PAR) signaling. We highlight the unique pharmacotherapeutic properties of each NOAC, supported by key clinical trials demonstrating their effectiveness in preventing thromboembolic events. Beyond their established anticoagulant roles, emerging research suggests that NOACs influence inflammation through PAR signaling pathways, implicating factors such as FXa and Thrombin in the modulation of inflammatory responses. This review synthesizes current evidence on the anti-inflammatory potential of NOACs, exploring their impact on inflammatory markers and conditions like atherosclerosis and diabetes. By delineating the mechanisms by which NOACs mediate anti-inflammatory effects, this work aims to expand their therapeutic utility, offering new perspectives for managing inflammatory diseases. Our findings underscore the broader clinical implications of NOACs, advocating for their consideration in therapeutic strategies aimed at addressing inflammation-related pathologies. This comprehensive synthesis not only enhances understanding of NOACs' multifaceted roles but also paves the way for future research and clinical applications in inflammation and cardiovascular health
Keywords:
NOAC
; Anti-Inflammation
; Protease-Activated-Receptor Signaling
; Factor Xa
; Thrombin.
1. Introduction
Blood coagulation is an innate response to vascular injury that results from a series of amplified reactions in which zymogens of serine proteases circulating in the plasma are sequentially activated by limited proteolysis, leading to the formation of blood clot, preventing loss of blood [1]. It is initiated through the extrinsic pathway [1]. As shown in Figure 1, membrane-bound tissue factor (TF), exposed during vascular injury, interacts with factor VIIa (FVIIa), which pre-exists in the plasma (at 1–2% of total factor VII) [2], and forms the extrinsic tenase complex.
This complex activates factor X (FX) to FXa. In association with its cofactor factor Va (FVa), FXa performs a proteolytic cleavage of prothrombin to thrombin. Thrombin cleaves fibrinogen to fibrin, promoting the formation of a fibrin clot, and activates platelets for inclusion in the clot. The TF·FVIIa complex can also activate factor IX (FIX) to factor IXa (FIXa), thus facilitating the propagation of the coagulation cascade through the intrinsic pathway [3]. The coagulation cascade is under tight regulation. Regulation of this cascade involves several natural anticoagulants, such as antithrombin, protein C, and protein S, which inhibit various clotting factors, as well as fibrinolytic mechanisms that degrade fibrin clots, maintaining a delicate balance between clot formation and dissolution.Any imbalance in its regulation could lead to either unclottable blood, resulting in excessive bleeding during injuries, or unwanted clot formation, resulting in death and debilitation due to vascular occlusion with the consequence of myocardial infarction, stroke, pulmonary embolism, or venous thrombosis [4].
Anticoagulants are pivotal for the prevention and treatment of thromboembolic disorders. The community pharmacy oral anticoagulant safety audit in the UK found that the three most frequently prescribed anticoagulants were apixaban (41.4%), rivaroxaban (25.2%) and warfarin (20.8%) [5]. Another study analysing trends in oral anticoagulant use among 436,864 patients with atrial fibrillation in the US found that overall anticoagulation rates increased from 56.3% in 2011 to 64.7% in 2020, suggesting that a majority of high-risk patients with atrial fibrillation in the US are receiving anticoagulant therapy [6].
For decades, warfarin, heparin, and its low molecular weight derivatives such as enoxaparin have held the reins as the mainstay of oral anticoagulant therapy, for the management of thromboembolic events. However, despite their effectiveness, these anticoagulants have pitfalls that need to be considered. Heparin can lead to heparin-induced thrombocytopenia [7], a potentially serious condition where the body attacks its own platelets, increasing the risk of blood clots. Warfarin, known for its narrow therapeutic window and inter-individual variability within patient response, requires careful monitoring to avoid bleeding complications [8,9]. Additionally, warfarin interacts with many drugs and foods, necessitating close attention to prevent adverse effects [10,11]. Enoxaparin, while effective, can also lead to bleeding complications, especially if not dosed correctly in obese patients [12]. These pitfalls have fueled the quest for safer alternatives [13]. This pursuit culminated in the emergence of Non-Vitamin K Oral Anticoagulants (NOACs), also known as Direct Oral Anticoagulants (DOACs), fundamentally transforming the anticoagulation landscape [14].
In contrast to warfarin’s method of indirectly inhibiting vitamin K-dependent clotting factors, NOACs directly target specific enzymes within the coagulation cascade (either FXa or thrombin), offering a series of significant advantages [15]. This direct approach enables predictable dosing, eliminating the need for the routine monitoring that is mandatory with warfarin therapy, thus simplifying treatment protocols and enhancing medication compliance [16,17]. Furthermore, most NOACs have minimal food interactions, providing patients with a much-appreciated dietary freedom. In comparison to warfarin, which has a wide range of drug interactions, NOACs have a notably reduced profile of interactions with other medications [18]. Additionally, certain NOACs come with the option of specific antidotes for rapid reversal in the event of bleeding complications, offering an extra layer of safety in emergency situations [19]. NOACs targeting Factor Xa include the drugs: rivaroxaban, apixaban, edoxaban, and betrixaban. These drugs work by inhibiting the cleavage of prothrombin to thrombin, a key step in the coagulation cascade (Figure 1). Thrombin targeting NOAC, include dabigatran, which directly binds to both clot-bound and free thrombin, to prevent blood clotting (Figure 1). It’s important to note that NOACs are inhibitors of coagulation factors rather than inactivators. They compete with substrates for binding at the active site of the target protease, and that binding is reversible [20]. This reversible binding mechanism of NOACs provides the advantage of a predictable and controlled anticoagulant effect, which can be easily managed in the event of bleeding or when surgical procedures are necessary, as the anticoagulation can be quickly reversed [21].
Beyond their critical roles in blood coagulation, FXa and thrombin orchestrate a myriad of physiological processes, predominantly mediated through a family of G-protein coupled receptors Protease-Activated Receptors (PARs). These include influencing inflammation, cell proliferation, and tissue repair mechanisms, highlighting their importance in both hemostasis and broader cellular and systemic functions [22]. In line, recent research has identified potential off-label uses of NOACs beyond traditional anticoagulation, particularly highlighting their anti-inflammatory and anti-atherosclerotic effects [23]. The molecular underpinnings of these off-label therapeutic or ‘Pleiotropic Effects’ effects seem to reside in the interactions between NOACs, the coagulation protease they bind to, with the concomitant effect of this binding on the activation of PARs. PARs, particularly PAR-1 and PAR-2, are known to play crucial roles in the pathophysiology of inflammation and atherosclerosis. The activation of these receptors’ triggers signaling pathways implicated in vascular inflammation, endothelial dysfunction, and the promotion of atherogenesis; as well augmenting the inflammatory milieu in specific diseases, such as osteoarthritis (OA). Case in point, in a study by Ito et al rivaroxaban, has been demonstrated to attenuate inflammation and atherosclerosis, not merely by inhibiting coagulation but potentially via modulating PAR-2 signaling [24]. It was observed that there is a significant reduction atherosclerotic area in the aorta of ApoE–/– mice fed with a high-fat diet upon administration of an appropriate dose of rivaroxaban. It was also shown that FXa significantly suppressed macrophage autophagy, an effect that was relieved by rivaroxaban. However, in PAR2 knockout macrophages, FXa was unable to prevent 7-ketocholesterol-induced autophagy and inflammasome activation. In ApoE–/– mice, genetic deletion of PAR2 substantially reduced the atherosclerotic area, an effect partially reversed by chloroquine (a drug that reverses this effect, not because it directly influences atherosclerosis through PAR2, but because of its role in modulating inflammatory responses and autophagy, processes that are deeply intertwined with atherosclerotic development). This study suggests that rivaroxaban can reduce atherosclerosis by impeding the FXa-PAR2-mediated inhibition of macrophage autophagy and inflammasome activity. Building on this premise, this review critically evaluates the dual roles of FXa and thrombin as both coagulants and mediators of inflammation via PAR-signaling, alongside the emerging evidence supporting the anti-inflammatory effects of NOACs, focusing on rivaroxaban, apixaban, edoxaban, and dabigatran.
By synthesizing current research, it posits that NOACs, traditionally used for their anticoagulant properties, may also modulate inflammation through PAR-signaling pathways, suggesting a broader clinical utility in managing inflammatory conditions and atherosclerosis. This concise exploration not only provides a fresh perspective on NOACs beyond anticoagulation but also underscores the potential for innovative therapeutic strategies in disease treatment. The review aims to inform and inspire ongoing research and clinical practice, making it a significant contribution to the existing body of literature.
2. Exploring NOACs: From Pharmacokinetics to Clinical Efficacy and Pivotal Trials
Building on our exploration of NOACs, Table 1 offers a comprehensive summary of NOACs: rivaroxaban, apixaban, edoxaban, and dabigatran. It covers essential aspects such as pharmacology, the necessity and approach to laboratory monitoring, guidelines for peri-operative management, and their clinical profiles. Furthermore, the table delves into the advantages and potential challenges associated with each NOAC, providing a well-rounded view that complements our discussion on their impact in clinical practice.
It is pertinent to note that, all NOACs underwent evaluation in large-scale, Phase III clinical trials against warfarin as the control arm. Table 2 succinctly outlines the clinical trials under discussion, highlighting the unique aspects and key distinctions of each study. A 2014 meta-analysis by Ruff et al., involving 71,683 participants across the RE-LY, ROCKET AF, ARISTOTLE, and ENGAGE AF–TIMI 48 trials, highlighted the superior efficacy of NOACs over warfarin in reducing stroke or systemic embolic events by 19%, with this benefit primarily driven by a significant decrease in hemorrhagic stroke [25]. Notably, NOACs also demonstrated a marked reduction in all-cause mortality and intracranial hemorrhage, albeit with an observed increase in gastrointestinal bleeding. This pivotal analysis offers a comprehensive evaluation of rivaroxaban, apixaban, edoxaban, and dabigatran, emphasizing their favorable risk-benefit profile, consistent efficacy across diverse patient subgroups, and potential to significantly mitigate the risk of stroke in patients with atrial fibrillation. Additionally, the analysis underscores the importance of NOACs in providing a safer alternative to warfarin, with similar major bleeding rates but reduced instances of intracranial hemorrhage, thus reaffirming their role in the current therapeutic landscape for atrial fibrillation.
Dabigatran etexilate, was the inaugural thrombin-targeting NOAC to receive regulatory approval. Table 1 compiles the essential pharmacokinetic attributes and clinical profiles of dabigatran, offering an in-depth examination of its therapeutic properties, efficacy, and safety parameters. A pooled analysis of two trials published (RE-NOVATE and RE-NOVATE II) demonstrated that dabigatran was at least as effective as enoxaparin for thromboprophylaxis after knee or hip replacement [26]. The RE-COVER I and II trials showed that dabigatran was non-inferior to warfarin for the treatment of acute venous thromboembolism [27]. In patients with atrial fibrillation at risk of stroke, the 2009 RE-LY study reported that dabigatran was non-inferior to warfarin with respect to the primary efficacy outcome of stroke and systemic embolism in patients with atrial fibrillation, (Table 2). Dabigatran received FDA approval for this indication in the subsequent year. More recent studies have compared dabigatran with other anticoagulants. A 2023 multicenter cross-sectional study in Spain assessed the usage of NOACs among elderly patients (≥75 years) with atrial fibrillation, focusing on dabigatran and rivaroxaban among others [28]. The study, encompassing 500 patients, revealed a nuanced landscape where dabigatran and rivaroxaban not only provided effective stroke prevention but also navigated the complex balance of dosing appropriateness in a demographic challenged by frailty and varied comorbidities. It highlighted that while 76% of NOAC prescriptions adhered to recommended dosing guidelines, a significant portion of the elderly population was still either underdosed or overdosed, underscoring the critical need for meticulous patient assessment to optimize therapeutic outcomes. Particularly, the study illuminated how dabigatran and rivaroxaban, despite their higher costs, offer a compelling blend of efficacy and safety for heart protection in this vulnerable cohort, thereby enriching the therapeutic arsenal against atrial fibrillation-induced cardiovascular events in the elderly. Additionally, a 2018 meta-analysis found no significant difference between 110 mg dabigatran and warfarin on the rate of stroke and systemic embolism, but the 110 mg dabigatran was associated with a lower incidence of bleeding compared to warfarin [29]. However, in a comprehensive 2023 nationwide cohort study, a detailed analysis revealed that dabigatran was associated with more than a twofold increase in the incidence of myocardial infarction compared with the Factor Xa inhibitors apixaban and rivaroxaban [30]. Specifically, dabigatran demonstrated a rate of 1.4 myocardial infarction events per 100 person-years, which starkly contrasts with the lower rates of 0.7 events per 100 person-years observed for both apixaban and rivaroxaban. Despite this significant difference in myocardial infarction rates, the all-cause mortality remained comparable across all three drugs, suggesting that the elevated myocardial infarction risk associated with dabigatran does not translate into increased mortality within the study period. The observed higher myocardial infarction rates with dabigatran may stem from its unique pharmacological action. Unlike apixaban, edoxaban and rivaroxaban, which inhibit FXa, dabigatran directly inhibits thrombin. This distinction in mechanism could influence the coagulation cascade and platelet activation differently, potentially contributing to the increased myocardial infarction risk. Previous research has suggested that dabigatran may enhance platelet activation, a pivotal process in the pathogenesis of myocardial infarction, thereby providing a plausible explanation for the observed elevated risk [31]. This finding is particularly relevant for clinical practice, highlighting the necessity for clinicians to consider the differential risk profiles of NOACs when selecting the most appropriate anticoagulant therapy, especially for patients at higher risk of myocardial infarction. The study underscores the importance of personalized medicine, where the benefits and risks of each anticoagulant are carefully balanced against the individual patient’s risk factors and clinical history to optimize therapeutic outcomes.
Hence, dabigatran emerges as a potent anticoagulant, demonstrating efficacy that matches or surpasses conventional anticoagulants such as warfarin and enoxaparin across diverse clinical settings. Nevertheless, akin to all anticoagulants, dabigatran is associated with a bleeding risk. Its application necessitates a tailored approach, taking into consideration the unique risk profile and clinical attributes of each patient to ensure optimal safety and effectiveness.
Rivaroxaban, a direct FXa inhibitor, has been extensively studied and utilized in the prevention and treatment of thromboembolic disorders. The key pharmacokinetic and clinical profiles of rivaroxaban are summarized in Table 1. Following the RECORD I–IV trials conducted between 2008 and 2009, rivaroxaban emerged as a favorable alternative to enoxaparin for VTE prophylaxis after total hip or knee arthroplasty [32]. The EINSTEIN-DVT and EINSTEIN-PE trials, published in 2010 and 2012, further established rivaroxaban’s noninferiority to enoxaparin or warfarin in the treatment of VTE [33]. In the realm of AF, the ROCKET-AF trial of 2011 demonstrated rivaroxaban’s noninferiority to warfarin for stroke prevention in patients with moderate-to-high stroke risk, leading to FDA approval for both stroke prevention in AF and VTE prophylaxis that same year, summarized in Table 2 [34]. Recent studies have continued to evaluate rivaroxaban’s efficacy and safety profile. The PREVENT-HD trial, a multicenter, randomized, double-blind, placebo-controlled study, assessed the efficacy and safety of prophylactic rivaroxaban, although specific results from this trial are not detailed in the search results [35]. A 2021 study examining the comparative risks associated with rivaroxaban and apixaban in patients with atrial fibrillation unveiled significant findings regarding their safety profiles [36]. Conducted on a large cohort of 581,451 Medicare beneficiaries aged 65 years or older, the research revealed that rivaroxaban treatment was associated with a higher incidence of major ischemic events, including ischemic and hemorrhagic strokes, compared to apixaban. Specifically, the adjusted incidence rates for major ischemic events were 8.6 per 1000 person-years for rivaroxaban versus 7.6 per 1000 person-years for apixaban. Additionally, hemorrhagic stroke rates were notably higher for rivaroxaban at 2.5 per 1000 person-years, compared to 1.7 per 1000 person-years for apixaban. A plausible explanation for this increased risk lies in the distinct pharmacokinetic characteristics of rivaroxaban, particularly its once-daily dosing schedule which results in pronounced peak-trough fluctuations in drug concentration. These fluctuations could lead to periods of under-anticoagulation, heightening the risk of thromboembolic events, and periods of over-anticoagulation, increasing the risk of bleeding events. On the other hand, apixaban’s twice-daily regimen achieves more stable plasma concentrations, potentially reducing the risks of both ischemic and hemorrhagic complications. Furthermore, a comprehensive 2023 randomized controlled trial, aimed to determine the optimal duration of rivaroxaban treatment for patients with symptomatic isolated distal DVT [37]. This study randomized participants who had already completed an initial six weeks of standard rivaroxaban therapy to either continue with the medication or switch to a placebo for an additional six weeks, with outcomes observed over a 24-month follow-up. The primary efficacy outcome focused on the recurrence of VTE, including progression or recurrence of isolated distal DVT, proximal DVT, symptomatic pulmonary embolism, or fatal pulmonary embolism. The findings highlighted that extending rivaroxaban treatment beyond the initial six weeks significantly reduced the risk of recurrent VTE, mainly through the prevention of recurrent isolated distal DVT, without increasing hemorrhage risks. This significant contribution to our understanding of rivaroxaban’s therapeutic efficacy underscores the value of tailored anticoagulant regimens in preventing VTE recurrence, especially in isolated distal DVT cases. However, the study did not address variations in stroke risk associated with rivaroxaban, as seen in other research. Differences in stroke risk may be attributed to rivaroxaban’s unique pharmacokinetic and pharmacodynamic properties, including its once-daily dosing, which could influence its safety profile. This suggests a need for further investigation to elucidate the mechanisms driving these differences and to refine treatment approaches that balance efficacy with minimizing adverse effects such as stroke. The FDA has also expanded the approved indications for rivaroxaban over the years, including its use in reducing the continued risk of VTE, secondary prevention after acute coronary syndrome, and in hospitalized adult patients with an acute medical illness at risk of thromboembolic complications [38]. Additionally, in 2021, rivaroxaban was approved for use in certain pediatric populations to treat and help prevent types of blood clots [39]. Furthermore, in a 2024 OSCAR-UK study, rivaroxaban was found to be a reasonable alternative compared to LMWHs in patients with cancer-associated venous thromboembolism [40]. A 2024 meta-analysis concluded that rivaroxaban to be a better option as compared to warfarin, due to its association with significantly lower risks of stroke and bleeding outcomes in obese patients with atrial fibrillation [41].
Therefore, rivaroxaban remains a key player in the management of thromboembolic diseases, with ongoing research contributing to a nuanced understanding of its role in various clinical scenarios. Its approval for a range of indications reflects its versatility and effectiveness as an anticoagulant, although recent findings highlight the importance of individualized patient assessment when considering its use.
Apixaban, the second direct FXa inhibitor to reach the market, boasts a broad clinical profile as summarized in Table 1. Numerous trials have established its efficacy and safety across various thromboembolic and cardiovascular conditions. In the realm of VTE prevention, the ADVACE-2 trial (2010) demonstrated apixaban’s superiority over enoxaparin for preventing VTE after total knee replacement surgery without increasing bleeding risk [42]. The AMPLIFY trial (2013) confirmed alone a fixed dose of apixaban to be non-inferior to conventional therapy (subcutaneous enoxaparin, followed by warfarin) for treating acute VTE [43]; with a 2015 analysis further showing significant reductions in all-cause hospitalization and length of hospital stay [44]. For atrial fibrillation management, the AVERROES trial (2011) established apixaban’s superiority over aspirin in preventing strokes for patients unsuitable for VKA therapy, without significant increased risk of major bleeding [45]. The AVERROES-MRI assessment study (2016) showed a nonsignificant trend towards reduced ischemic stroke and covert embolic-pattern infarction with apixaban compared to aspirin, with no increase in microbleeds [46]. The ARISTOTLE trial (2012) further expounded apixaban’s position by demonstrating its superiority over warfarin in AF patients, translating to a lower risk of stroke and systemic embolism with a better bleeding profile (details in Table 2) [47]. Apixaban’s efficacy extends to cancer-associated thrombosis. The AVERT trial (2019) demonstrated that apixaban significantly reduced VTE for thromboprophylaxis compared to placebo in ambulatory cancer patients who were at intermediate-to-high risk for VTE [(Khorana score, ≥2); The Khorana score, ranging from 0 to 6, is a risk assessment tool used to predict cancer-associated VTE in chemotherapy patients, with a higher score indicating a greater risk and the potential need for preventative anticoagulation], and were initiating chemotherapy [48]. The CARAVAGGIO trial (2020) established oral apixaban’s non-inferiority to subcutaneous dalteparin (a LMWH) for treating cancer-associated VTE, again without increased major bleeding [49]. Additionally, a 2020 randomized trial demonstrated oral apixaban’s non-inferiority to subcutaneous enoxaparin in terms of safety and efficacy, for thromboprophylaxis after gynecologic cancer surgery, suggesting oral apixaban as a potentially safe, less painful, and more convenient alternative [50]. Recent studies have explored apixaban’s use in special populations. The AXADIA-AFNET 8 (2022), an investigator-initiated PROBE (prospective randomized open blinded end point) outcome assessment trial, showed no apparent differences in safety or efficacy between oral apixaban and VKA therapy in atrial fibrillation patients on chronic hemodialysis [51]. A 2023 Phase 2, open label trial in children with heart disease requiring thromboprophylaxis found infrequent bleeding and thromboembolic events with both oral apixaban and current standard care (VKA or LMWH). These findings support the use of oral apixaban as an alternative to Standard-of-Care (SOC) thromboprophylaxis in pediatric heart disease [52]. The PREVAPIX-ALL study, a pioneering phase 3, open-label, randomized, controlled trial conducted across 74 pediatric hospitals in 9 countries, evaluated the efficacy and safety of oral apixaban for primary thromboprophylaxis in pediatric patients with acute lymphoblastic leukemia or lymphoma [53]. This trial, unique in its assessment of a direct oral anticoagulant for preventing VTE in this high-risk group, randomly assigned participants to receive either SOC without systemic anticoagulation or weight-adjusted twice-daily apixaban during induction chemotherapy. Despite no statistically significant reduction in VTE with apixaban, major and clinically relevant non-major (CRNM) bleeding events were uncommon, though CRNM bleeding, mainly mild epistaxis, was more frequent in the apixaban group. With a total of 512 participants analyzed, the study’s findings suggest that for pediatric patients considered at particularly high risk of thrombosis, apixaban presents an encouraging safety profile, indicating its potential utility in clinical scenarios where the benefits may outweigh the risks of bleeding.
Overall, apixaban emerges as a versatile and effective anticoagulant, with a robust body of evidence supporting its use across a spectrum of thromboembolic and cardiovascular conditions, demonstrating not only its efficacy and safety in adults but also its potential in pediatric populations, marking it as a pivotal option in the evolving landscape of anticoagulation therapy.
Edoxaban, the fourth NOAC approved by the FDA on January 9, 2015 (details on its pharmacokinetics and clinical properties are provided in Table 1), has established itself as a valuable addition to the treatment armamentarium. Numerous trials have confirmed its efficacy and safety across various thromboembolic and cardiovascular conditions. The landmark trial of ENGAGE AF-TIMI 48 study (2013) evaluated edoxaban’s long-term effectiveness and safety compared to warfarin in patients with AF. Edoxaban demonstrated non-inferiority to warfarin in preventing stroke or systemic embolism, with the added benefit of significantly lower bleeding and cardiovascular death rates (details in Table 2) [54]. Edoxaban’s utility extends to VTE treatment. The Hokusai-VTE trial (2013) assessed its efficacy as an alternative to warfarin in patients with acute VTE following initial heparin therapy. Edoxaban, administered once daily after initial heparin administration, proved non-inferior to warfarin while causing significantly less bleeding across a broad spectrum of VTE patients, including those with severe pulmonary embolism [55]. A 2016 subgroup analysis of this trial suggested edoxaban might be as effective as warfarin for treating VTE in cancer patients, with a reduced risk of clinically relevant bleeding [56]. This was further confirmed by the Hokusai VTE Cancer trial (2018), which demonstrated the non-inferiority of oral edoxaban to subcutaneous dalteparin for preventing recurrent VTE or major bleeding in patients with cancer [57]. Edoxaban’s effectiveness in preventing thromboembolic complications following surgery has also been established. In 2014, a Japanese study compared edoxaban to subcutaneous enoxaparin for preventing thromboembolic events after hip fracture surgery, demonstrating similar safety and efficacy [58]. Another 2014 trial, STARS E-3, showed oral edoxaban to be more effective than subcutaneous enoxaparin for thromboprophylaxis following total knee arthroplasty, with a similar bleeding incidence [59]. Studies have explored edoxaban’s safety and efficacy in special populations. A 2015 study in Japanese patients with non-valvular atrial fibrillation (NVAF) and severe renal impairment found similar safety, plasma concentration, and biomarker profiles for edoxaban compared to doses used in patients with normal or mild renal function [60]. The ENSURE-AF trial (2016) compared edoxaban to enoxaparin-warfarin therapy in patients with AF undergoing cardioversion. Edoxaban displayed similar rates of major bleeding and thromboembolism compared with patients on well-managed, optimized enoxaparin– warfarin therapy [61]. Recent years have witnessed continued investigation into edoxaban’s potential. The ENNOBLE-ATE in Children at Risk Because of Cardiac Disease trial (2022) suggests edoxaban as a promising alternative thromboprophylaxis option for children with cardiac disease, with low bleeding rates and advantages like once-daily dosing and less frequent monitoring compared to SOC (VKA or LMWH) [62]. The ENAVLE trial (2023) compared edoxaban with warfarin in patients following surgical bioprosthetic valve implantation or valve repair. Edoxaban showed non-inferiority to warfarin in preventing thromboembolism and a potentially comparable risk of major bleeding during the initial 3 months [63]. Finally, the KABUKI trial (2024) explored edoxaban in patients with chronic thromboembolic pulmonary hypertension (CTEPH). The study demonstrated edoxaban’s non-inferiority to warfarin in preventing worsening pulmonary vascular resistance (PVR) with a similar Incidence of clinically relevant bleeding [64].
Through a diverse range of pivotal trials, edoxaban has solidified its status as a key player in the NOAC landscape, demonstrating broad efficacy and safety in preventing and treating thromboembolic and cardiovascular conditions,
Having delineated the individual NOACs and their pivotal roles in managing thromboembolic and cardiovascular conditions, we now transition to examine the PARs, exploring their intricate involvement in inflammation and the broader implications for therapy.
3. Exploring Protease-Activated Receptors (PARs): Unveiling Their Role in Inflammation and Disease
PARs are a unique class of G protein-coupled receptors activated by proteolytic cleavage. This cleavage unveils a tethered ligand that binds to a specific region within the receptor, triggering a conformational shift and subsequent cellular signaling. Interestingly, synthetic peptides mimicking the tethered ligand can directly stimulate PARs, even in the absence of protease activity [65]. There are four known PAR subtypes: PAR1, PAR2, PAR3, and PAR4, each with distinct characteristics like the number of amino acid residues, tethered ligands, and preferred activating proteases/synthetic ligands (refer to Table 3 for details). Notably, the cellular responses elicited by PAR activation are highly variable, depending on the specific PAR subtype and the activating protease involved. Even with the same PAR, different proteases can trigger diverse cellular responses. This intricate interplay between proteases, PARs, and resulting cellular responses paints a complex and evolving picture in the niche of cell-signaling. The complex signal transduction mechanisms in PAR-mediated inflammation involve factors like Ca2+, extracellular signal-regulated kinase (ERK)1/2, and cAMP signaling, ultimately leading to a cascade of cellular changes (as illustrated in Figure 2 and Figure 3). Interestingly, activation of PAR3 requires the involvement of PAR1, PAR2, or PAR4 [66,67].
Recent evidence suggests FXa exerts non-hemostatic effects, primarily mediated through PAR-1 and PAR-2 [68,69]. These effects contribute to pathophysiological conditions like atherosclerosis, inflammation, and fibrosis. FXa initiates diverse non-hemostatic effects across different cell types, bridging coagulation with inflammatory conditions. While thrombin production primarily occurs on platelets and subendothelial vascular walls, its presence has also been detected extraluminally, in areas like synovial fluid [70] and around tumors [71]. Thrombin plays a central role in platelet activation [72]. PAR1, PAR3, and PAR4 can all be activated by thrombin, but each through distinct mechanisms [73].
Notably, it has been observed that PARs play a dynamic role in modulating cellular signaling across different inflammatory zones in the human body. They can both promote and suppress inflammatory reactions, indicating a dualistic function in the regulation of inflammation. This principle and its significance in diseases linked to inflammation are detailed in Table 4 and explored in the subsequent discussion.
3.1. Exploring the Role of PARs in Neuroinflammation and Hyperalgesia
PARs are extensively expressed in central and peripheral neurons, implicating them not only in physiological and somatosensory functions but also in progression of neuroinflammatory conditions, with the capacity to both induce neurodegenerative and neuroprotective effects [74]. Among the PARs family, PAR1 and PAR2 emerge as key contributors in nerve-related inflammation. Granzyme B (a serine protease released by cytotoxic T-lymphocytes and natural killer cells, instrumental in inducing apoptosis in target cells as part of the immune response) has been observed to mediate neurodegeneration through PAR1 activation, triggering diverse downstream signaling cascades. Given its ability to induce neural apoptosis, inhibiting PAR1 is hypothesized to offer a neuroprotective effect in the context of neuroinflammatory diseases [75]. PAR2, found on glia, neurons, and mast cells, is noted for enhancing neuroinflammation in the CNS by activating pathways like ERK1/2, c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinases (MAPKs). Moreover, its interaction with glial maturation factor and mast cell proteases, including mouse mast cell protease (MMCP) 6 and MMCP-7, results in heightened PAR2 expression in primary astrocytes and neurons, further intensifying the neuroinflammatory condition [76]. However, it’s crucial to note that PAR1 activation through low concentrations of thrombin has exhibited therapeutic potential, showcasing neuroprotective effects that enhance cell viability and microarchitecture in primary cortical neurons, safeguarding them against excitotoxic damage caused by glutamate exposure [74].
The PARs play a role in hyperalgesia by directly activating nociceptors and/or sensitizing them. This process involves the activation of receptors such as transient receptor potential vanilloid subfamily 1 (TRPV1), TRPA1, TRPV4, and P2X3, which are crucial in mediating neurogenic inflammation and inflammatory pain [77]. Additionally, PAR2 coupling with TRPV4 has been reported to amplify pro-inflammatory state via calcium release, leading to a chronic hyperexcitability of nociceptors, especially implicated in irritable bowel syndrome (IBS) [78,79]. Furthermore, it is observed that PAR2 agonist signal primary spinal efferent neurons, meditated by central activation of neurokinin (NK) 1-receptors and release of prostaglandins to cause a pronounced and chronic hyperalgesia [80]. In contrast, it was demonstrated that topical administration of selective PAR1 agonists resulted in attenuation of the nociceptive response elicited by noxious mechanical and thermal stimuli. Furthermore, a significant reduction in inflammatory mechanical and thermal hyperalgesia was observed [81]. Overall, the active participation of the PARs, especially PAR2, in the pathogenesis of hyperalgesia and neuroinflammation denotes them as a promising therapeutic target.
3.2. Chronic Gastrointestinal Inflammation
Overall, the active participation of the PARs, especially PAR2, in the pathogenesis of hyperalgesia and neuroinflammation denotes them as a promising therapeutic target.
Chronic inflammation in the gastrointestinal tract is closely linked to the activity of both human and bacterial proteases within the gut’s lumen. These proteases, through PARs, notably PAR1 and PAR2, regulate the intestinal barrier’s permeability, mirroring their role in endothelial barriers. The presence of all four PARs in the gastrointestinal lining underscores their significant role in driving the complex mechanisms behind chronic gastrointestinal conditions via PAR-mediated inflammation.
Inflammatory bowel disease (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic and recurrent inflammatory condition of the gastrointestinal (GI) tract. PARs have been implicated in IBD pathogenesis through a biphasic regulatory mechanism depending on the cell type and the pathogen in question. For instance, PAR1 exhibits a pro-inflammatory role in bacterial-induced colitis, as evidenced by Citrobacter rodentium infection and the enhancement of T helper 17 cell (Th17) type immune responses in Crohn’s disease patients [82]. On the flip side, PAR1 displays an anti-inflammatory effect in oxazolone-induced colitis, which involves a T helper 2 cell (Th2) mediated immune response, showcasing its multifaceted role in modulating inflammation within the gastrointestinal tract [83]. Furthermore, the function of PAR1 in colitis is found to vary based on cell type. Boucher et al. demonstrated that PAR1 expression on specific effector cells within bacterial-derived colitis differentially modulates pro-inflammatory cytokine expression profiles [84], highlighting the nuanced role of PAR1 in influencing the inflammatory response.
PAR2 exhibits widespread distribution within the GI cells, particularly at apical and basolateral membranes of epithelial cells [85]. PAR2 deficiency has been reported to significantly attenuate C. rodentium-induced inflammation, suggesting a proinflammatory effect similar to PAR1 [86]. Furthermore, increased mast cell tryptase secretion, which can activate the PAR2/Protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway, can contribute to intestinal fibrosis, a hallmark of IBD [87]. Alternatively, within high-fat diet-fed murine colitis model, PAR2 seems to play a protective role in inflammatory processes. This protective effect appears to be mediated by promoting autophagy and inhibiting apoptosis in intestinal cells. This recent study underscores the potential of PAR2 agonism as a novel therapeutic avenue for patients suffering from IBD who also present with comorbid metabolic syndrome. This interventional strategy could address the complex pathophysiological nexus between inflammatory and metabolic pathways in such cohorts. Notably, the therapeutic implication draws from the multifaceted role that PARs play in modulating gastrointestinal inflammation and metabolic homeostasis, further bolstering the case for PAR2-targeted treatments in dual-pathology contexts such as IBD with concurrent metabolic syndrome [88]. This evidence coalesces into a compelling narrative, advocating for the broader exploration of PARs’ duality in IBD management paradigms.
3.3. Inflammatory Disorders of Respiratory System
The role of PARs in the pathophysiology of respiratory disorders, including asthma and pneumonia, represents a focal point of medical research due to the critical insights it provides into pulmonary inflammatory mechanisms. The heightened research interest is propelled by clinical observations that demonstrate significantly increased concentrations of proteases, such as thrombin and tryptase, which activate PARs in the bronchoalveolar lavage fluid of patients suffering from pulmonary inflammation [89,90]. These proteases are implicated in the exacerbation of airway remodeling and hyperresponsiveness, which are pivotal factors in the progression of these respiratory conditions [91,92,93]. The upregulation of key inflammatory mediators, including matrix metalloproteinase (MMP)-9, interleukin (IL)-1β, IL-6 and substance P (SP), through PAR activation, delineates a pathogenic pathway that contributes to the severity and chronicity of respiratory diseases [91]. This evidence highlights the potential of targeting PARs in the therapeutic management of pathological airway responses.
Direct activation of PAR1 is implicated in the initiation of asthma symptoms through mechanisms involving transforming growth factor-beta (TGF-β), which in turn fosters a Th2-skewed immune response characteristic of allergic asthma [94]. Conversely, the inhibition of PAR2 has shown efficacy in reducing allergen-induced airway hyperresponsiveness and the infiltration of inflammatory cells [95]. This highlights the mechanistic role of PAR2 in mediating the pathophysiology of allergic airway inflammation, offering a clearer understanding of its potential as a therapeutic target in asthma management. Furthermore, non-mammalian PAR-activating proteases, found in sources like house dust mites and cockroaches, suggest the significance of PARs in respiratory health [96]. In fact, selective PAR2 antagonists have shown efficacy in reducing allergen-induced asthma symptoms in animal models [97]. The influence of PAR2 on bronchoconstriction or dilation presents a complex picture, colored by interspecies differences and the nuanced responses of diverse tissues [98,99]. Despite these complexities, human studies suggest that both PAR1 and PAR2 agonists—activated by thrombin, trypsin, and tryptase—have the capacity to induce bronchoconstriction by catalyzing Ca2+ signaling pathways within airway smooth muscle cells [100,101]. Additionally, persistent activation of PAR1 and PAR2 is associated with the development of pulmonary fibrosis, suggesting a deleterious role in prolonged inflammatory responses [102,103,104]. Intriguingly, a low thrombin dose has been observed to mitigate inflammatory signaling in the context of allergic bronchial asthma, indicating a potential therapeutic window where careful modulation of PAR1 may exert anti-inflammatory effects [105].
Paralleling their role in asthma, accumulating evidence suggests pro-fibrotic and pro-inflammatory effects of PARs in the pathogenesis of pneumonia. Streptococcal pneumonia appears to exploit PAR1 activation to exacerbate neutrophil infiltration and viral co-infections through downstream mediators such as IL-1β, chemokine (C-X-C motif) ligand (CXCL) 1, and chemokine (C-C motif) ligand (CCL) 7 and CCL2 [106]. Conversely, PAR1 inhibition has been shown to ameliorate excessive neutrophilic inflammation and alveolar barrier disruption [107]. While PAR2 activation by proteases facilitates Streptococcus pneumoniae colonization in the respiratory tract followed by systemic dissemination most likely due to impairment of the lung-blood barrier integrity [108]. On the flip side, evidence suggests that PAR2 deletion has the capacity to attenuate TRPV4-dependent Ca2+ signaling which primarily promotes persistent lung inflammation [109]. Interestingly, PAR4 appears to contribute to host defense by enhancing the systemic cytokine response during late-stage pneumococcal infection in murine models [110]. Despite PAR4’s immunomodulatory role, PAR1 and PAR2 remain critical modulators in pneumococcal pneumonia. The data suggests that combined inhibition of PAR1 and PAR2 might be a superior therapeutic strategy compared to targeting them individually.
3.4. Cardiovascular Inflammatory Disorders
The multifaceted expression profile, enzymatic activity, and availability of selective antagonists position PARs as attractive molecular targets for therapeutic intervention in cardiovascular diseases. Furthermore, evidence suggests that PAR signaling disrupts the normal homeostatic functions of cardiac fibroblasts and cardiomyocytes, contributing to the structural remodeling th myocardium [111,112,113]. Elucidating the specific roles of PARs in these processes holds significant promise for improving our understanding and treatment of cardiovascular system disorders.
Mirroring the observed functional dichotomy of PAR1 and PAR2 discussed previously, these receptors appear to mediate opposing effects on inflammatory responses within viral myocarditis, specifically in ssRNA viruses such as coxsackievirus group B (CVB) 3 and influenza A virus [114]. Studies have demonstrated that upregulation of PAR1 and PAR4 protected cardiomyocytes by attenuating viral myocarditis [115,116]. This protective effect appears to be mediated by enhanced TLR3 signaling, and subsequent increased expression of Interferon (IFN) β and CXCL10, which was associated with reduced viral load and decreased expression of pro-inflammatory mediators, such as Tumor Necrosis Factor (TNF)-α, monocyte chemoattractant protein (MCP) 1 and CXCL1. Notably, PAR1, is primarily expressed by non-hematopoietic cells, which can potentially infiltrate the heart and further limit viral replication by augmenting the cytotoxic activity of natural killer (NK) cells [117]. Conversely, PAR2 activation was observed to reduce the antiviral response mediated by TLR3-dependent expression of IFN-β, promoting viral load and replication, potentially leading to severe myocarditis [118,119]. Interestingly however, both PAR1 and PAR2 activation have been implicated in the pathogenesis of cardiac remodeling, heart dysfunction, and myocardial infarction [111,112].
These findings collectively suggest that broad-spectrum PAR inhibition might not be an optimal therapeutic strategy for myocarditis. Instead, a more nuanced approach utilizing PAR-specific ligands tailored to their individual effects may be a more promising avenue for therapeutic development.
3.5. Inflammatory Disorders of Urinary System
PARs emerge as key regulators of inflammatory processes within the urinary system, encompassing the kidneys, bladder, and urethra, under both physiological and pathological conditions. In cystitis, a condition frequently associated with urothelial dysfunction and persistent bladder inflammation, PAR signaling appears to play a pivotal role. While PAR activation, particularly by PAR1 and PAR4, is not directly causative of cystitis in the urothelium, it contributes to the disease progression through the mediation of macrophage migration inhibitory factor (MIF), at least in part through activation of CXCR4 receptors [120]. Furthermore, PAR4 activation in urothelial cells has been shown to induce the release of high-mobility group box 1 protein (HMGB1) via a MIF-dependent mechanism, potentially contributing to bladder pain [121]. Notably, PAR1 expression appears to be upregulated in bladder following cystitis induced by classical mediators such as LPS, SP and cyclophosphamide (CYP), potentially exacerbating the bladder inflammatory response, thereby impacting bladder sensation and function during cystitis [122,123]. Similarly, PAR2 activation in primary cultures of human urothelial cells and mouse urothelium/suburothelium is associated with increased COX-2 expression, augmenting the inflammatory processes in cystitis primarily mediated by the ERK1/2 MAPK pathway [124].
Within the renal compartment, thrombin-mediated PAR activation has been implicated in both autocrine and paracrine signaling pathways, thereby linking PARs and proteases to nephritis [125]. Renal inflammation, a key driver of chronic kidney disease (CKD) and often accompanied by renal tubulointerstitial fibrosis (RTIF), exhibits a direct correlation between FXa activity and the severity of fibrosis. FXa-meditated PAR1signaling has been demonstrated to enhance the production and proliferation of procollagen by fibroblasts [126]. Similarly, PAR2 activation by FXa in these cells appears to stimulate both fibrotic and pro-inflammatory responses [127]. Additionally, PAR2 plays a critical role in the formation of glomerular crescents (a hallmark of rapidly progressive forms of glomerulonephritis) by promoting macrophage accumulation, activation of parietal epithelial cells and glomerular thrombosis [128]. Collectively, these findings suggest a pro-inflammatory role for PARs, particularly PAR1 and PAR2, in the urinary system.
3.6. Rheumatoid Arthritis and Osteoarthritis
PAR1 and PAR2, have emerged as intriguing players in the intricate world of arthritis. While arthritis encompasses various joint disorders, this review focuses on their involvement in rheumatoid arthritis (RA), an autoimmune systemic inflammatory disorder, and osteoarthritis (OA), degenerative joint disease with inflammation as its hallmark.
Initial evidence for PAR2’s direct role in rheumatic diseases came from a 2003 study by Ferrell et al. [129]. Their work demonstrated that activating PAR2 with a specific peptide triggered robust inflammation in wild type mice, including joint swelling and increased blood flow to the synovium. In addition, studies since have shown that spontaneous release of TNF-alpha and IL-1beta from RA synovium is significantly inhibited by a PAR2 antagonist in a dose-dependent manner [130]. Further solidifying PAR2’s role as a key pro-inflammatory mediator, a study by Russell et al. [131] demonstrated that PAR2 plays an acute inflammatory role in the knee joint. This effect is mediated by TRPV1 and NK1 receptors, suggesting involvement of both PAR2-mediated neuronal sensitization and leukocyte trafficking. Supporting this notion, another study examined the differential expression of mast cell tryptase and PAR-2 in the synovium and synovial fluid of patients with rheumatoid arthritis (RA) and osteoarthritis (OA) [132]. The study revealed that mast cell tryptase in synovial fluid stimulates the proliferation of synovial fibroblasts (SFCs) and the release of pro-inflammatory cytokines via PAR-2. This suggests a potential role for PAR-2 in the exacerbation of synovitis in arthritis. Further substantiating this link, murine studies assessed the capacity of mast cell tryptase to mediate synovial pro-inflammatory responses via PAR-2 and investigated whether degranulating mast cells induced synovial hyperemia by PAR-2 activation [133].
Clinical relevance was established by studies revealing elevated PAR2 expression in circulating leukocytes from RA patients compared to healthy individuals [134].
Further studies using murine models solidified PAR2’s pro-inflammatory function. Mice deficient in PAR2 exhibited reduced inflammatory response during acute arthritis. Additionally, inflamed joints showed increased PAR2 protein expression, and silencing PAR2 using siRNA effectively dampened inflammation [135].
PAR1, unlike PAR2, appears to have opposing effects in RA. While uPA-activated PAR1 demonstrably attenuates inflammatory osteoclastogenesis, a process essential for bone destruction in RA [136], the picture with thrombin-PAR1 interaction is more complex. Thrombin can activate PAR1 on human osteoblasts, leading to increased CCL2 expression, which promotes macrophage aggregation, potentially contributing to inflammation [137]. Similarly, thrombin-PAR1 signaling induces RANTES mRNA expression, possibly facilitating the migration of inflammatory cells to the synovium in RA patients [138].
However, thrombin-PAR1 signaling in synovial fibroblasts appears to have a dual effect. While it stimulates their proliferation [139], it also promotes the secretion of MMP-2 and inhibits the invasion of these fibroblasts, potentially limiting tissue damage. Additionally, thrombin-PAR1 activation reduces the production of pro-inflammatory cytokines IL-17 and TNF-alpha by synovial fibroblasts. In contrast, PAR2 activation in synovial fibroblasts appears to be predominantly detrimental, promoting their growth, invasion, and TNF-alpha production [140]. This suggests that while PAR1 has some anti-inflammatory properties in synovial fibroblasts, PAR2 seems to be a key driver of their aggressive behavior, contributing to the pathological progression of RA. Within the context of OA, an in vitro model using LPS-induced inflammation in chondrocytes revealed upregulation of PAR2 expression [141]. Interestingly, this study also showed that subsequent downregulation of PAR2 expression was associated with a decrease in inflammatory markers, including TNF-alpha, IL-6, IL-8, and IFN-gamma.
PAR1 and PAR2 play intricate roles in the development of RA and OA. While PAR2 appears to be a key pro-inflammatory mediator in arthritis, PAR1 has both pro-inflammatory and anti-inflammatory effects, while PAR3 and PAR4 seem to have minimal involvement. These findings suggest that PAR2 antagonist may hold promise as therapeutic strategies for the treatment of arthritis.
Having established the critical role of PARs in inflammatory processes, we now shift our focus back to individual NOACs. Now we will delve into the complex interplay between NOACs and inflammatory pathways, exploring their potential anti-inflammatory properties and the broader therapeutic implications.
4. Anti-Inflammatory Potentials of NOACs
4.1. Apixaban’s Anti-Inflammatory Profile
In a review of the anti-inflammatory effects of apixaban (Table 5), the data present a multifaceted picture. Studies stemming from the ARISTOTLE trial in atrial fibrillation (AF) patients revealed limited impact on conventional inflammatory markers. Specifically, apixaban did not significantly influence Growth Differentiation Factor 15 (GDF-15), high-sensitivity C-reactive protein (hs-CRP), or IL-6 levels in these patients [142,143]. However, intriguingly, obesity, a condition often associated with chronic inflammation, was linked to better survival outcomes in apixaban-treated AF patients, hinting at a potential, yet indirect, anti-inflammatory role for the drug [144]. In contrast to these neutral to ambiguous findings in AF, more targeted research has offered promising results. An in vitro and in vivo study demonstrated that apixaban, especially when used in combination therapy, increased the specificity of Activated Protein C (APC), a molecule with known anti-inflammatory effects [145]. Similarly, in a clinical setting of coagulopathy associated with extracorporeal membrane oxygenation (ECMO), apixaban impaired plasmatic coagulation and platelet aggregation, though the direct anti-inflammatory impact was not assessed [146]. Furthermore, in a study related to chronic kidney disease (CKD), apixaban significantly reduced markers like VCAM-1, ICAM-1, and VWF while normalizing ROS levels in endothelial cells exposed to uremic serum [147]. Collectively, while the anti-inflammatory effects of apixaban appear to be condition- and context-dependent, these findings open doors for further investigations into its role beyond anticoagulation.
The heterogenous data concerning apixaban’s anti-inflammatory effects points toward an exigent need for more granular future studies. In vitro cell line studies employing endothelial and glial cells could be particularly informative, with assays such as Western blot and RT-PCR focusing on key signaling pathways like NF-κB or MAPK. Moreover, transgenic mouse models could offer crucial in vivo insights. These mice, engineered through advanced genetic manipulation techniques such as CRISPR to either amplify or suppress inflammatory responses, can serve as valuable models to assess apixaban’s systemic and localized impact. One particularly promising model is the L2-IL-1β transgenic mice, which exhibit chronic inflammation along with a stepwise development of esophageal conditions including Barrett’s esophagus-like metaplasia, dysplasia, and adenocarcinoma [148]. These mice also display squamous cell carcinoma (SCC) in the esophagus and tongue, with T cell-predominant inflammatory profiles and significantly increased levels of various pro-inflammatory markers. Importantly, this model could serve as an excellent system for studying the anti-inflammatory potential of apixaban in inflammation-driven esophageal and oral cancers. Researchers could administer apixaban to these mice at different developmental stages and then employ immunohistochemistry and cytokine profiling to assess whether apixaban mitigates the inflammation and related carcinoma progression. This specific model, validated for both Germ Free and Specific Pathogen Free conditions, could provide nuanced insights into how apixaban might interact with IL-1β-mediated inflammation pathways and whether it has a role in halting or reversing the progression of SCC and adenocarcinoma. In addition to classical cell lines and animal models, specialized in vitro systems can offer invaluable insights. For instance, the recent development of a 3D culture system to generate chondrocytes from bone marrow-derived mesenchymal stem cells (BMSCs) has opened new avenues in cartilage biology and inflammation research [149]. Given the key role of chondrocytes in maintaining cartilage integrity and their susceptibility to inflammation, this model could serve as an ideal platform for investigating FXa’s role in inflammation associated with OA. In this system, a pro-inflammatory environment is simulated by treating chondrocytes with lipopolysaccharide, creating a setup that can be leveraged to evaluate the anti-inflammatory effects of apixaban. Specifically, the impact of apixaban on tumor necrosis factor-α levels and other inflammatory markers could be quantified using enzyme-linked immunosorbent assay (ELISA). Moreover, the 3D architecture of this model more closely mimics the in vivo cellular environment compared to 2D monolayers, offering a more physiologically relevant assessment of apixaban’s potential anti-inflammatory effects. Such an approach not only expands the toolkit for studying cartilage-related disorders but also offers a more nuanced understanding of how apixaban could mitigate inflammation in a highly tissue-specific context.
In the same vein, 3D organoid cultures [150], representing different human organs, offer a more physiologically relevant platform to study apixaban’s effects on specific tissues. These organoids could be treated with various concentrations of apixaban, followed by immunohistochemical and transcriptomic analyses to evaluate anti-inflammatory mechanisms. Case in point, multicellular 3D neurovascular unit organoids present a promising platform for studying apixaban’s anti-inflammatory mechanisms in the context of BBB dysfunction. These organoids, comprising human brain microvascular endothelial cells, pericytes, astrocytes, microglia, oligodendrocytes, and neurons, mimic the complex cellular interactions and environment of the native BBB [151]. Dysfunction of the BBB is evident in many neurological disorders, such as ischemic stroke and chronic neurodegenerative diseases, and is often associated with hypoxia and neuroinflammation. Utilizing this organoid model to simulate hypoxic conditions—by culturing the organoids in a hypoxic chamber with 0.1% O2 for 24 hours—could offer an invaluable system for investigating the anti-inflammatory effects of apixaban. Specifically, the model enables quantification of changes in permeability, pro-inflammatory cytokine production, and oxidative stress markers in response to apixaban treatment. Preliminary experiments with other anti-inflammatory agents like secoisolariciresinol diglucoside and 2-arachidonoyl glycerol have already shown promising results in reducing inflammatory cytokine levels under hypoxic conditions. Therefore, this 3D organoid model not only recapitulates key characteristics of BBB dysfunction but also offers a platform for assessing how apixaban may counteract these changes at the cellular and molecular levels. It’s an ideal system for high-throughput screening of drug candidates and elucidating the mechanistic pathways through which apixaban exerts its anti-inflammatory effects.
As for human research, future cohort studies should focus on specialized sub-populations, such as those with chronic inflammatory conditions like chronic kidney disease [152] or autoimmune disorders [153], to investigate real-world anti-inflammatory efficacy. Statistical comparisons can then be made to measure apixaban’s impact on specific markers and outcomes. These multi-pronged, detailed experimental approaches would provide a comprehensive understanding of apixaban’s anti-inflammatory potential and could pave the way for its broader clinical applications beyond anticoagulation. Also, in the section on specific human populations, the utility of mass spectrometry for deeply understanding the anti-inflammatory effects of apixaban can be highlighted by drawing upon insights from studies investigating metabolic changes in coronary artery disease (CAD) patients. By leveraging widely targeted plasma metabolomic and lipidomic profiling via mass spectrometry in CAD patients, researchers have successfully mapped the metabolic disturbances that accompany systemic immune inflammation [154]. A comprehensive panel of 860 metabolites was assessed, offering a robust framework for defining general and low-grade inflammatory states. Mass spectrometry enabled precise quantification of associations between multiple metabolites and seven established inflammatory markers, including high-sensitivity C-reactive protein and neutrophil-lymphocyte ratio. Techniques like least absolute shrinkage and selection operator (LASSO) logistic-based classifiers were deployed to further hone in on the significant biomarkers and metabolic pathways involved, such as glycerophospholipid metabolism and arginine and proline metabolism. In particular, β-pseudouridine emerged as an inflammation-associated metabolite linked to arteriosclerosis indicators. Importantly, mass spectrometry facilitated the development of predictive models with high discriminatory power (area under the curve: 0.81 to 0.88) for various inflammatory states. Adding mass spectrometry to the investigation of apixaban’s effects in specific populations, such as CAD patients, could offer unparalleled depth of insight. Not only does it allow for the precise quantification of a wide array of metabolites and lipids, but it also enables the elucidation of the intricate metabolic pathways that may be impacted by apixaban’s anti-inflammatory mechanisms. The detailed metabolic profiles generated could serve as robust markers for assessing drug efficacy and understanding the broader metabolic context in which apixaban operates. This adds a layer of sophistication to therapeutic monitoring, paving the way for individualized treatment approaches that are tailored to the patient’s unique inflammatory and metabolic landscape.
4.2. Edoxaban’s Anti-Inflammatory Profile
Edoxaban has showcased diverse anti-inflammatory effects across various pathophysiological conditions (A comprehensive review of these effects is depicted in Table 6). In models of diabetic nephropathy, Edoxaban has been shown to reduce the expression of proinflammatory and profibrotic genes such as TGF-β, PAI-1, Collagen Type I/IV, TNF-α, MCP-1, IL-8, and COX-2. It also decreased the expression of PAR1 and PAR2, which are implicated in the pathogenesis of diabetic nephropathy, suggesting a potential therapeutic role for Edoxaban in this condition [155]. Similarly, in a model of renal tubulointerstitial injury, Edoxaban suppressed elevated levels of FX, PAR-1, and PAR-2, reduced fibrosis and extracellular matrix expression, and attenuated the upregulation of inflammatory molecules and macrophage [127]. Furthermore, the anti-inflammatory effects of Edoxaban were also observed in an immortalized proximal tubule epithelial cell line, where it alleviated oxidative stress induced by FXa through its FXa inhibitory and direct radical scavenging activity. This is particularly relevant as oxidative stress is closely interrelated with inflammation. In a transient middle cerebral artery occlusion (tMCAO) model, Edoxaban reduced infarct volumes, improved neurological outcomes, enhanced blood-brain barrier function, and attenuated brain tissue inflammation, suggesting potential protective effects in ischemic stroke [156]. However, not all studies have shown significant anti-inflammatory effects. For instance, in a murine model of vascular remodeling and arteriogenesis, Edoxaban did not exhibit a significant effect on the mRNA expression of inflammatory cytokines such as IL6, MCP-1, IL1b, and TNF-alpha [157]. Similarly, in an LPS-induced microvascular thrombosis model, Edoxaban did not affect the levels of inflammatory cytokines [158]. Furthermore, its efficacy in vascular remodeling, thrombosis during infection, and systemic inflammation, especially in HIV-positive populations, has been met with equivocal outcomes and did not manifest significant reductions in specific inflammatory markers [157,158,159].
The potential pleiotropic effects of Edoxaban in inflammation, atherosclerosis, and stroke have been experimentally tested in various models, with key findings suggesting that Edoxaban is a promising target for inhibition of the PAR-2 pathway [160]. Despite these promising observations, there are gaps that need to be addressed through further research. The exact molecular mechanisms by which Edoxaban exerts its anti-inflammatory effects are not fully understood. Clinical trials are needed to confirm the anti-inflammatory benefits of Edoxaban observed in preclinical models, and the long-term safety and efficacy of Edoxaban in the context of inflammation need to be evaluated. It is also important to explore the potential differential effects of Edoxaban on various types of inflammation and in different patient populations. In conclusion, Edoxaban has demonstrated significant anti-inflammatory effects in various models, suggesting its potential utility in treating inflammatory conditions. However, further research is necessary to fully elucidate its mechanisms of action and to validate these effects in clinical settings. The studies summarized in Table 6 provide a strong foundation for future investigations into the anti-inflammatory potential of Edoxaban.
Building on the aforementioned findings, it is pivotal to acknowledge the limitations that accompany them. A substantial proportion of the investigations are rooted in animal models. While these models are undeniably instrumental, they might not mirror human pathophysiological conditions in their entirety. This becomes especially pertinent when considering the effects of edoxaban on certain populations, such as HIV-positive individuals, where the anticipated anti-inflammatory outcomes were not significantly realized [159], thereby raising questions regarding the drug’s broad-spectrum applicability. Moreover, a noteworthy segment of the research, specifically those marked with (***) [157,158,159], was unable to demonstrate a pronounced impact of edoxaban on particular inflammatory markers. This underscores the imperative for more refined and nuanced analyses, to truly decipher the drug’s therapeutic breadth and limitations in diverse clinical scenarios.
Understanding the intricacies of edoxaban’s mechanisms remains a continuously unfolding domain. A salient aspect of this domain is the drug’s potential impact on oxidative stress markers, especially in the context of CKD. The alleviation of oxidative stress induced by FXa in HK-2 cells has emphasized the pivotal relationship between oxidative stress and inflammation [161]. Future endeavors could delve deeper into this aspect by leveraging patient-derived kidney organoids [162] or advanced cellular models like induced pluripotent stem cell (iPSC)-derived renal cells. Contrastingly, in vascular remodeling studies with ApoE-/- mice, edoxaban didn’t display significant influence on specific inflammatory markers [157]. This deviation from the general positive anti-inflammatory narrative suggests that further research is warranted. Explorations into alternative animal models, such as LDLr-/- mice or Zebrafish models — renowned for their insights into vascular development — might prove enlightening.
Another discernible research gap pertains to the population diversity in existing studies. Though preliminary research on HIV-positive individuals [159] and atrial fibrillation patients from the ENGAGE AF-TIMI 48 trial [163] has been undertaken, the ambit of these studies needs broadening. Future research should factor in a wider range of populations, spanning diverse age brackets, ethnicities, and other comorbid conditions. Such an approach can ensure a comprehensive grasp of edoxaban’s therapeutic spectrum across varying clinical scenarios
4.3. Rivaroxaban’s Anti-Inflammatory Profile
Rivaroxaban demonstrates a significant anti-inflammatory effect, as outlined in various studies summarized in the attached Table 7. For instance, in Japanese subjects with NVAF, rivaroxaban was associated with an increase in thrombomodulin levels and a decrease in MMP-9 compared to warfarin, indicating its potent anti-inflammatory action [164]. Further examination of ex vivo abdominal aortic aneurysm samples revealed that rivaroxaban notably reduced markers of inflammation and oxidative stress, such as IL-6 and MMP-9, underscoring its potential in mitigating vascular inflammation [165]. An ancillary analysis of the X-TRA study also demonstrated rivaroxaban’s efficacy in reducing hsCRP, D-dimer, vWF, and TAT complex levels in AF patients, further supporting its anti-inflammatory capabilities [166]. Moreover, in a direct comparison between rivaroxaban and warfarin users with NVAF, rivaroxaban users showed decreased levels of inflammatory cytokines, highlighting a less evident inflammatory profile in these patients [167].
These findings are complemented by studies across various models, including ex vivo AAA models, clinical trials, and animal studies, consistently showing rivaroxaban’s role in modulating key inflammatory markers and pathways. The table provides a comprehensive overview of rivaroxaban’s anti-inflammatory effects (Table 7), showcasing its therapeutic potential beyond its conventional anticoagulant role.
4.4. Dabigatran’s Anti-Inflammatory Profile
Dabigatran, exhibits diverse anti-inflammatory effects across different models and conditions, as delineated in provided Table 8. For instance, in patients undergoing elective orthopaedic surgical procedures, dabigatran demonstrated comparable efficacy to LMWH in managing post-operative inflammation, with no significant differences in the incidence of local or systemic inflammatory complications [168]. In vitro analyses using human Peripheral Blood Mononuclear Cells (PBMCs) and whole blood revealed that dabigatran significantly reduced levels of pro-inflammatory cytokines, growth factors, and chemokines, suggesting potential anti-inflammatory effects by dampening pro-inflammatory stimuli [169].
Furthermore, in experimental models, such as the bleomycin-induced interstitial lung disease in female C57BL/6 mice, dabigatran markedly decreased thrombin activity, TGF-β1 levels, and the number of inflammatory cells in bronchoalveolar lavage fluid, alongside a reduction in histologically evident lung inflammation and fibrosis [170]. Similar anti-inflammatory and antifibrotic effects were observed in models of atherosclerosis and nonalcoholic fatty liver disease (NAFLD), where dabigatran attenuated hepatic fibrin deposition, inflammation, hepatocellular injury, and steatosis in mice fed a high-fat diet, suggesting that thrombin may drive NAFLD pathogenesis by altering expression of genes associated with lipid metabolism and bile acid synthesis [171].
These observations are supported by studies across various models, including ex vivo and in vivo models of pulmonary fibrosis, atherosclerosis, and liver injury, consistently demonstrating dabigatran’s role in modulating key inflammatory markers and pathways. Table 8 provides a detailed overview of dabigatran’s anti-inflammatory effects, underscoring its therapeutic potential beyond anticoagulation by reducing inflammation in cardiovascular diseases, interstitial lung disease, NAFLD, and potentially other inflammatory conditions.
4.5. Comparative Anti-Inflammatory Potential of NOACs
The comparative analysis, as detailed in the Table 9, reveals the nuanced anti-inflammatory - dabigatran, rivaroxaban, apixaban, and edoxaban - across various models, including sickle cell disease (SCD), endothelial cell inflammation, acute ischemic stroke, and more. For example, in the Berkeley mouse model of SCD, rivaroxaban attenuated systemic inflammation by decreasing plasma levels of IL-6, whereas dabigatran did not significantly affect IL-6 levels but impacted neutrophil infiltration in the lung, delineating their specific pathways of action [172]. Another study in human umbilical vascular endothelial cells (HUVECs) underscored that both dabigatran and rivaroxaban suppressed inflammatory gene expression post-thrombin stimulation, indicating their potential anti-inflammatory capabilities [173].
A critical observation from the studies is the differential impact of these anticoagulants on specific inflammatory markers, suggesting a complex interplay between coagulation and inflammation pathways. For instance, dabigatran seemed to reduce hs-CRP and increase pentraxin-3 levels more prominently than apixaban in acute ischemic stroke patients, highlighting potential variances in their anti-inflammatory profiles [174]. Moreover, while both dabigatran and rivaroxaban were observed to reduce aortic clamping-induced renal tissue oxidation and inflammation in a Wistar rat model, further research is warranted to delineate their distinct mechanistic pathways and the clinical relevance of these findings [175].
Critically, while the Table 9 provides valuable insights into the anti-inflammatory effects of these NOACs, it also underscores the necessity for further investigation. Notably, the variability in the effects observed across different inflammatory markers, conditions, and models calls for more comprehensive studies to fully understand the therapeutic potential and limitations of DOACs in modulating inflammation. Additionally, the potential impact of NOACs on the progression of conditions like colorectal cancer, as seen with edoxaban’s superior ability to suppress tumor growth and mitigate elevated levels of inflammatory markers including PAR2, signal transducer and activator of transcrip- tion-3 (STAT3), cyclin D1, and Ki67 warrants further exploration to elucidate the broader implications of these findings for clinical practice [176].
5. Novel Revelations of Apixaban in Osteoarthritis
As discussed previously, PAR2 is a key player in OA pathophysiology, significantly contributing to the characteristic inflammatory cascade of the disease. The intricate interplay between coagulation and immune-inflammatory systems, particularly FXa activating PAR2, underscores a crucial pathway contributing to OA inflammation. NOACs like apixaban, a selective FXa inhibitor, emerge as potential modulators of this pathway. This is further supported by accumulating evidence that suggests apixaban might possess anti-inflammatory properties (Refer above for details).
Building upon the established role of PAR2 in OA inflammation and the critical role of FXa in PAR2 activation, our team has investigated the potential of apixaban to mitigate FXa-PAR2 mediated inflammation in OA. We specifically aim to expand apixaban’s therapeutic utility beyond anticoagulation to potentially influence OA’s inflammatory processes. This could pave the way for novel OA treatment strategies through targeted modulation of key inflammatory pathways essential for disease progression.
The core of our research involved a meticulously constructed in vitro model to dissect the anti-inflammatory potential of apixaban in a controlled environment mimicking the inflammatory state of OA. The OA model was created via an FXa-induced inflammatory state in BMSC-derived human chondrocytes. Our model successfully establishes a solid foundation for understanding the induced inflammatory response, as evidenced by the increased expression of TNF-alpha within our in vitro OA system. The model’s precision is further validated by its ability to replicate essential inflammatory markers in chondrocytes, thereby providing robust insights into the inflammatory mechanisms at play.
Significantly, treatment with apixaban led to a notable decrease in PAR-2 levels, suggesting its ability to alter this receptor’s activity and potentially dampen the downstream inflammatory cascade. Detailed analysis is further required to investigate this effect, mapping out the intricate inflammatory pathways within chondrocytes and how they are influenced by FXa and PAR2 interaction. While we are not presenting new data, the hypothetical scheme depicted in Figure 4 illustrates the potential consequences of this downregulation.
The analysis suggests that apixaban’s inhibition of FXa curtails PAR-2 expression, potentially impacting the NF-κB signaling pathway, monocyte recruitment, and osteoclastogenesis. These factors are crucial contributors to reactive oxygen species (ROS) generation and chronic pain associated with OA. Additionally, the analysis visualizes the downstream modulation of inflammatory cytokines like TNF-α and IL-1β, and signaling proteins ERK1/2, which contribute to the deterioration of chondrocyte integrity. The involvement of ADAMTS5 and aggrecan in chondrocyte structure, alongside SOX4’s role in integrity loss, underscores the comprehensive nature of the inflammatory pathway. Collectively, these findings encapsulate the disease pathogenesis and the potential therapeutic modulation by apixaban, highlighting its anti-inflammatory capacity within the chondrocyte model.
In summary, our preliminary data alludes to apixaban’s effectiveness in modulating the key inflammatory mediator PAR2. This sets a promising stage for further in-depth investigation of the pathway outlined. As we delve deeper into this research, we aim to elucidate apixaban’s pleiotropic effects, which could significantly contribute to advancements in the management of OA, a condition desperately in need of novel therapeutic approaches. Utilizing multi-omics approaches, such as genomics, transcriptomics, proteomics, and metabolomics, can provide a holistic understanding of the systemic effects of NOACs. This integrative approach can help identify novel biomarkers and therapeutic targets, as well as elucidate the complex interactions between NOACs and inflammatory pathways. For instance, a hypothetical study could analyze the transcriptomic and proteomic profiles of patients with rheumatoid arthritis before and after apixaban treatment, identifying changes in gene expression and protein levels associated with inflammation. Metabolomic analysis could further reveal shifts in metabolic pathways, providing insights into how NOACs influence inflammatory processes at a molecular level. By integrating these diverse data sets, researchers can map out comprehensive signaling networks and identify potential points of therapeutic intervention, ultimately leading to more targeted and effective treatments for inflammatory diseases.
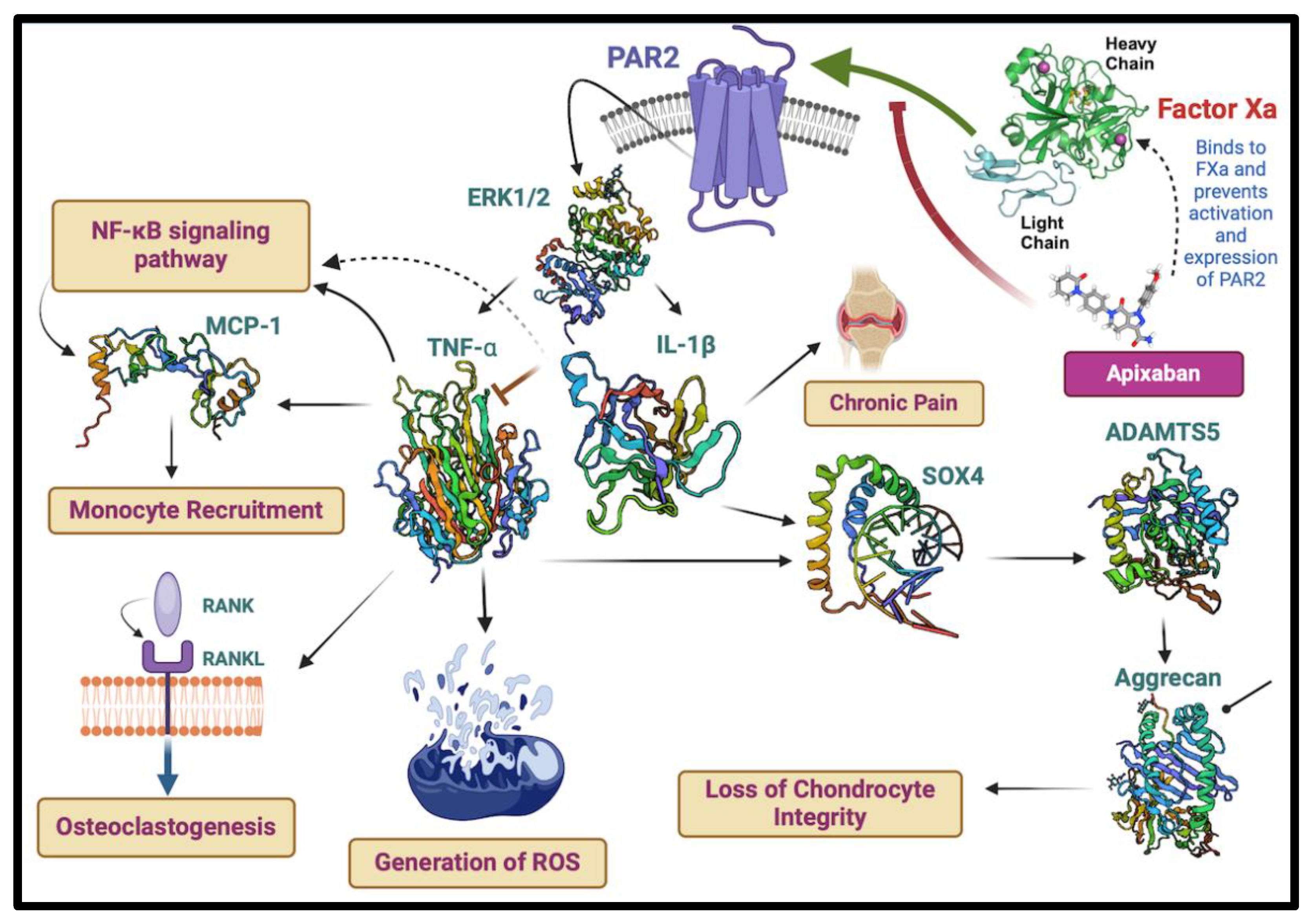
- 1.
- AR2 Inhibition: The blockage of PAR2 activation by apixaban may ameliorate the downstream signalling events mediated by ERK1/2 that led to the production of pro-inflammatory cytokines, such as TNF-α and IL-1β, potentially alleviating chronic pain associated with OA.
- 2.
- Cytokine Modulation: The expected reduction in TNF-α and IL-1β due to apixaban’s action on FXa mitigates the upregulation of molecules like MCP-1, which are involved in monocyte recruitment and the NF-κBsignaling pathway, both key contributors to inflammation and osteoclastogenesis.
- 3.
- Protein Expression: The illustration also indicates the potential effects of apixaban on the expression of regulatory proteins, including SOX4 and ADAMTS5, and their impact on critical components like aggrecan, which is essential for cartilage integrity.
- 4.
- Chondrocyte Integrity and Bone Health: By modulating these inflammatory and catabolic pathways, apixaban may help preserve chondrocyte integrity, mitigate the generation of reactive oxygen species (ROS), and contribute to maintaining joint health by potentially impacting the RANK/RANKL pathway, which is crucial for osteoclast activity and bone resorption
6. Unexplored Aspects of NOACs in Inflammation
Despite the extensive research and numerous clinical trials surrounding NOACs), certain aspects of their roles in inflammation/anti-inflammation remain inadequately explored. This section aims to identify the existing gaps in our understanding and suggest methodologies and future directions to address these gaps effectively. The focus will be on the potential anti-inflammatory mechanisms of NOACs, their broader therapeutic applications, and the experimental models and techniques that could be utilized to further investigate these properties.
6.1. Identified Gaps in Current Research
6.1.1. Limited Understanding of Molecular Mechanisms
While it is established that NOACs, such as rivaroxaban and apixaban, can modulate inflammation via PAR signaling pathways, the precise molecular mechanisms remain unclear. For instance, how these drugs interact with different PARs (PAR1, PAR2, etc.) and influence downstream signaling pathways like NF-κB and MAPK is not fully understood. The current studies have predominantly focused on the broad effects without delving into the specific intracellular events triggered by NOACs.
6.1.2. Inconsistencies in Anti-Inflammatory Effects Across Models
There are inconsistencies in the anti-inflammatory effects of NOACs across different experimental models and clinical studies. For example, while rivaroxaban has shown promise in reducing markers of inflammation in animal models of atherosclerosis and AAA (abdominal aortic aneurysm), similar effects are not always observed in human studies [177]. This discrepancy suggests a need for more standardized and comparable research protocols.
6.1.3. Lack of Longitudinal Human Studies
Most of the existing research is based on short-term studies. Longitudinal studies examining the long-term effects of NOACs on inflammation and related conditions are scarce. Understanding how chronic administration of NOACs influences inflammation over extended periods is crucial for assessing their potential therapeutic roles in chronic inflammatory diseases.
6.1.4. Population Diversity in Research
The majority of studies have focused on specific patient populations, such as those with atrial fibrillation or undergoing orthopedic surgery. There is a need to explore the anti-inflammatory effects of NOACs in a broader range of conditions, including autoimmune disorders, chronic kidney disease, and metabolic syndromes.
6.1.5. Comparative Effectiveness of Different NOACs
While individual NOACs have been studied, comparative research on their relative effectiveness in reducing inflammation is limited. This is particularly important for understanding which NOAC might be more beneficial for specific inflammatory conditions
6.2. Proposed Methodologies and Future Directions
6.2.1. Advanced Molecular and Cellular Studies
To elucidate the molecular mechanisms of NOACs in inflammation, advanced molecular biology techniques such as CRISPR-Cas9 gene editing can be used to create knockout models for specific PARs. These models can help determine the exact role of each PAR in mediating the anti-inflammatory effects of NOACs. Additionally, utilizing techniques like RNA sequencing and proteomics can provide a comprehensive view of the changes in gene and protein expression in response to NOAC treatment. In fact, currently, we are analyzing the transcriptomic data obtained following apixaban treatment in our pro-inflammatory chondrocyte model. This ongoing research aims to identify specific gene expression changes induced by apixaban, providing insights into its molecular mechanisms of action in inflammation. Preliminary results suggest modulation of key inflammatory pathways (Patnaik, R; Jannati, S et al.; unpublished data), and further analysis will help elucidate the broader impact of apixaban on chondrocyte function and viability.
6.2.2. Standardized Experimental Models
Developing standardized experimental models across different laboratories can help address the inconsistencies observed in the effects of NOACs. For example, using uniform protocols for inducing inflammation and measuring outcomes in animal models and in vitro systems can provide more reliable and comparable data.
6.2.3. Longitudinal Clinical Studies
Conducting longitudinal studies in diverse human populations can provide valuable insights into the long-term anti-inflammatory effects of NOACs. These studies should include regular monitoring of inflammatory biomarkers, clinical outcomes, and potential side effects over extended periods. This approach can help identify the sustained benefits and risks associated with chronic NOAC therapy.
6.2.4. Population Specific Research
Expanding research to include diverse populations, such as those with chronic inflammatory diseases, autoimmune disorders, and metabolic syndromes, can help determine the broader applicability of NOACs. Stratified analysis based on demographic factors (age, gender, ethnicity) and comorbid conditions can provide a more nuanced understanding of how different populations respond to NOAC therapy.
6.2.5. Comparative Effectiveness of Research
Conducting head-to-head trials comparing the anti-inflammatory effects of different NOACs can help identify the most effective agents for specific conditions. These studies should include a comprehensive assessment of both efficacy and safety, considering various inflammatory markers and clinical endpoints.
6.2.6. Integration of Multi-Omics Approaches
Utilizing multi-omics approaches, such as genomics, transcriptomics, proteomics, and metabolomics, can provide a holistic understanding of the systemic effects of NOACs. This integrative approach can help identify novel biomarkers and therapeutic targets, as well as elucidate the complex interactions between NOACs and inflammatory pathways. For instance, a hypothetical study could analyze the transcriptomic and proteomic profiles of patients with rheumatoid arthritis before and after Apixaban treatment, identifying changes in gene expression and protein levels associated with inflammation. Metabolomic analysis could further reveal shifts in metabolic pathways, providing insights into how NOACs influence inflammatory processes at a molecular level. By integrating these diverse data sets, researchers can map out comprehensive signaling networks and identify potential points of therapeutic intervention, ultimately leading to more targeted and effective treatments for inflammatory diseases.
6.2.7. Innovative Experimental Models
Leveraging advanced experimental models, such as 3D organoid cultures and patient-derived organoids, can offer more physiologically relevant platforms for studying the anti-inflammatory effects of NOACs. For example, 3D neurovascular unit organoids can be used to investigate the impact of NOACs on blood-brain barrier function and neuroinflammation in conditions like stroke and neurodegenerative diseases.
6.2.8. Designing Better Thrombin and Factor Xa Using AI
The advancements in artificial intelligence (AI) have revolutionized protein engineering, enabling the design of optimized thrombin and FXa molecules that can be used to develop peptides targeting PARs and conjugated with NOACs for enhanced anti-inflammatory properties.
Utilizing AI-based approaches such as generative adversarial networks (GANs), transformer models, and specifically, tools like AlphaFold [178], ESM2 [179], and ProteinMPNN [180] researchers can predict and design thrombin and FXa inhibitors that mimic natural proteins but with optimized functionality for laboratory use, a strategy as predicted by Johnson et ِal. [181].
AlphaFold plays a crucial role in accurately predicting protein structures, which helps in understanding the potential interactions of these inhibitors with their targets. ESM2 leverages extensive protein sequence datasets to generate potential thrombin and FXa inhibitors optimized for stability and function, while ProteinMPNN refines these sequences to ensure they are structurally feasible and functionally active in laboratory conditions. Once optimized thrombin and FXa molecules are designed, these sequences can be used to derive peptides specifically targeting PARs. This involves optimizing peptide sequences for high binding affinity and specificity to PARs, validating their binding conformations using structural prediction tools like AlphaFold, and conducting in vitro assays to test their binding efficacy.
Subsequently, these AI-derived peptides can be conjugated to NOACs, using a strategy similar to Martin et al [182] to combine their anticoagulant properties with targeted PAR inhibition, potentially enhancing therapeutic efficacy and reducing side effects. The resulting NOAC-peptide conjugates will then have to undergo rigorous pharmacokinetic and pharmacodynamic testing. Finally, the anti-inflammatory activity of these conjugates can be evaluated through comparative studies against NOACs alone, measuring the reduction in pro-inflammatory markers such as TNF-alpha and MCP-1 using techniques like ELISA, RT-PCR, and Western blotting. This approach aims to demonstrate that the conjugates offer superior anti-inflammatory benefits compared to NOACs alone, showcasing the power of AI in protein engineering and promising more effective and targeted anticoagulant therapies with added anti-inflammatory properties.
6.2.9. Designing NOAC-Antibody Conjugates for Enhanced Anti-Inflammatory Activity
The strategic development of NOAC-antibody conjugates, inspired by the successful design of antibody–peptide conjugates (APICs) for targeting cysteine proteases [183], promises to enhance the anti-inflammatory properties of NOACs. This approach integrates advanced AI-driven design of optimized thrombin and FXa inhibitors and their conjugation with antibodies to ensure targeted delivery and enhanced therapeutic efficacy. Utilizing AI-based techniques such as generative adversarial networks (GANs), transformer models, AlphaFold, ESM2, and ProteinMPNN, we can predict and design thrombin and FXa inhibitors that are both structurally feasible and functionally active. These inhibitors serve as the foundation for deriving peptides that specifically target PARs.
The process begins with the rational design of thrombin and FXa inhibitors using AI to predict sequences that exhibit high binding affinity and specificity to PARs. AlphaFold’s structural predictions and ESM2’s sequence optimization play crucial roles in refining these peptides to ensure their functionality in biological systems. Once the optimized sequences are validated, they are chemically modified to enhance their binding capabilities to the PARs. Following peptide optimization, the next step involves conjugating these peptides to antibodies. This conjugation is designed to leverage the natural targeting and internalization properties of antibodies, thereby ensuring that the NOAC-peptide conjugates accumulate in specific cell types where PAR is expressed. By targeting these specific cells, the conjugates can deliver the NOACs more precisely, reducing systemic side effects and increasing local therapeutic efficacy.
The final evaluation phase involves rigorous testing of the NOAC-antibody conjugates for their anti-inflammatory activity compared to NOACs alone. This includes in vitro and in vivo assays to measure the reduction in pro-inflammatory markers such as TNF-alpha and MCP-1. Techniques like ELISA, RT-PCR, and Western blotting will quantify these effects, providing comparative data to demonstrate the enhanced efficacy of the conjugates. The targeted delivery mechanism is expected to exhibit superior anti-inflammatory benefits by ensuring higher concentrations of the therapeutic agent at the site of inflammation, thus maximizing efficacy and minimizing off-target effects.
This innovative strategy of designing NOAC-antibody conjugates using AI-driven approaches and targeted delivery systems exemplifies the potential of integrating computational tools with advanced bioconjugation techniques. It holds promise for developing more effective and targeted anti-inflammatory therapies, offering significant improvements over traditional NOAC treatments.
6.2.10. Design of Aptameric NOAC Conjugates to Attenuate PAR-Mediated Inflammation
Aptamers are single-stranded oligonucleotides that fold into specific three-dimensional shapes, allowing them to bind with high affinity and specificity to various target molecules, including proteins, small molecules, and even cells [184]. The unique properties of aptamers make them suitable for therapeutic applications, including the development of aptameric NOAC conjugates designed to attenuate PAR-mediated inflammation.
To design aptameric NOAC conjugates targeting FXa/thrombin and PAR interactions, we can employ a strategy inspired by the creation of EXACT inhibitors, which combine exosite-binding aptamers with active site inhibitors to enhance potency and selectivity [185]. The process involves several key steps:
Selection and Optimization of Aptamers: The first step is to identify and optimize aptamers that specifically bind to FXa/thrombin and PAR. This can be achieved using a selection process such as SELEX (Systematic Evolution of Ligands by EXponential enrichment), where a large library of random oligonucleotide sequences is screened for binding to the target proteins [185]. Selected aptamers are then optimized for binding affinity and specificity through iterative rounds of selection and amplification [Once optimized aptamers are obtained, they are conjugated to NOACs. This involves chemically linking the aptamer to the active site inhibitor, thereby ensuring that the conjugate retains both the aptamer’s binding affinity and the inhibitory activity of the NOAC. The design must consider the spatial arrangement and flexibility of the linker to allow for effective binding to FXa, thrombin, and PAR simultaneously. The linker length and composition are critical parameters that need to be optimized to prevent steric hindrance and ensure synergistic binding [185]
The aptamer-NOAC conjugates are then to be tested for their ability to bind to FXa or thrombin and PAR and inhibit their activity. This involves in vitro assays such as surface plasmon resonance (SPR) and ELISA to measure binding affinity and specificity. Additionally, functional assays such as coagulation assays and cell-based assays are used to evaluate the inhibitory effects on FXa or thrombin and the attenuation of PAR.
7. Addressing Specific Research Gaps
7.1. Molecular Mechanism of NOACs
Investigating the interaction of NOACs with PARs at a molecular level using techniques like co-immunoprecipitation and fluorescence resonance energy transfer (FRET) can help map out the specific signaling pathways involved. Additionally, exploring the role of NOACs in modulating epigenetic changes associated with inflammation can provide new insights into their long-term effects.
7.2. Comparative Studies in Diverse Models
Conducting parallel studies in both animal models and human cell lines can help bridge the gap between preclinical and clinical findings. Using transgenic animal models that mimic human inflammatory conditions can provide more relevant data on the efficacy of NOACs.
7.3. Long-Term Impacts of NOACs
Implementing longitudinal cohort studies with regular follow-up intervals can help assess the cumulative impact of NOACs on inflammation. These studies should include comprehensive health assessments, biomarker analysis, and quality of life evaluations to capture the full spectrum of effects.
7.3. Broadening Research Populations
Including underrepresented populations in clinical trials and observational studies can ensure that the findings are generalizable. Collaborating with international research centers can facilitate the inclusion of diverse cohorts, enhancing the robustness of the data.
7.4. Integration of Multi-Omics Data
Creating integrated databases that combine multi-omics data with clinical information can help identify patterns and correlations that are not apparent from single-layer analyses. Machine learning algorithms can be employed to analyze these complex datasets and generate predictive models for the anti-inflammatory effects of NOACs.
8. Materials and Methods
The methodology for this review on Non-Vitamin K Oral Anticoagulants (NOACs) and their role in inflammation and Protease-Activated Receptor (PAR) signaling involved a systematic literature search in PubMed, using terms such as "Non-Vitamin K Oral Anticoagulants," "NOACs," "Direct Oral Anticoagulants," "inflammation," "Protease-Activated Receptor," "PAR signaling," "rivaroxaban," "apixaban," "edoxaban," and "dabigatran," limited to articles published from January 1996 to May 2024 in English and peer-reviewed journals. Two independent reviewers (SJ and YB), screened titles and abstracts for relevance, with full texts assessed based on inclusion criteria: studies focusing on NOACs and their effects on inflammation and PAR signaling. Exclusion criteria included non-relevant studies, reviews, and editorials.
Data extraction, performed by two reviewers (SJ and YB), captured study design, sample size, NOACs investigated, outcomes, and key findings. Data synthesis involved narrative synthesis, with grouping by NOAC type and investigated outcomes. Discrepancies between reviewers were resolved through discussion. Ethical considerations were minimal as the review used publicly available data. Limitations included potential language bias and heterogeneity of the studies, which might affect generalizability.
9. Conclusions
The potential anti-inflammatory effects of NOACs represent a promising yet underexplored area of research. By addressing the identified gaps through advanced methodologies and comprehensive studies, we can unlock new therapeutic applications for these drugs. The integration of molecular, cellular, and clinical approaches, along with a focus on diverse populations and long-term effects, will provide a deeper understanding of how NOACs can modulate inflammation. This knowledge will pave the way for innovative treatments for chronic inflammatory diseases, enhancing patient outcomes and expanding the therapeutic utility of NOACs beyond anticoagulation.
Author Contributions
Y.B. conceptualized the study. Information curation was managed by S.J., R.P., and Y.B. Funding was acquired by Y.B. The investigation was conducted by S.J. and Y.B. Y.B. provided supervision. The original draft was written by S.J. and Y.B., with both also handling the review and editing of the manuscript. All authors have made significant contributions to the work. All authors have read and agreed to the published version of the manuscript.
Funding
No funding is available for this study. APC for this article will be defrayed by suitable emoluments from the office of Provost at MBRU, Dubai Health, UAE.
Institutional Review Board Statement
The experimental procedures in this research were carried out in vitro and did not entail the utilization of animal models, patient samples, or human subjects. As a result, the study presented minimal risk and falls within one of the exempt review categories outlined by the institutional review board (IRB) regulations at Mohammed Bin Rashid University of Medicine and Health Sciences (MBRU). For additional details and clarification, inquiries can be directed to the MBRU IRB at irb@mbru.ac.ae. The study exclusively utilized in vitro experiments with readily available cell lines, eliminating the involvement of human subjects. It is essential to highlight that no individuals under the age of 18 were part of the study, rendering parental or guardian consent unnecessary. The experiments conducted did not include any form of direct or indirect interaction with human subjects. It’s crucial to emphasize that our research did not require access to medical records or archived samples. In summary, our study focused on in vitro experiments using commercially acquired cell lines and did not entail the participation of human subjects. Consequently, participant consent was not deemed necessary, and the requirement for consent was waived.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available but are accessible from the corresponding author upon reasonable request. Interested researchers may contact the corresponding author for data access inquiries, subject to compliance with any applicable privacy or confidentiality obligations.
Acknowledgments
The authors extend their deep gratitude to the Al Jalila Foundation for their magnanimous support, which was crucial in providing the necessary laboratory space and state-of-the-art equipment needed to carry out this research.
Conflicts of Interest
The authors declare that they have no competing financial interests or personal relationships which could have influenced the work reported in this paper.
References
- Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58(5):515-23. [CrossRef]
- Bernardi F, Mariani G. Biochemical, molecular and clinical aspects of coagulation factor VII and its role in hemostasis and thrombosis. Haematologica. 2021;106(2):351-62. [CrossRef]
- Owens AP, 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. 2010;104(3):432-9.
- Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015;29(1):17-24. [CrossRef]
- England NHSN. Community pharmacy oral anticoagulant safety audit 2021/22: NHS England; 2023 [Available from: https://www.england.nhs.uk/long-read/community-pharmacy-oral-anticoagulant-safety-audit-2021-22/#about-this-report.
- Navar AM, Kolkailah AA, Overton R, Shah NP, Rousseau JF, Flaker GC, et al. Trends in Oral Anticoagulant Use Among 436 864 Patients With Atrial Fibrillation in Community Practice, 2011 to 2020. J Am Heart Assoc. 2022;11(22):e026723. [CrossRef]
- Arepally GM, Cines DB. Pathogenesis of heparin-induced thrombocytopenia. Transl Res. 2020;225:131-40. [CrossRef]
- Kimmel SE. Warfarin therapy: in need of improvement after all these years. Expert Opin Pharmacother. 2008;9(5):677-86. [CrossRef]
- Shields LBE, Fowler P, Siemens DM, Lorenz DJ, Wilson KC, Hester ST, et al. Standardized warfarin monitoring decreases adverse drug reactions. BMC Fam Pract. 2019;20(1):151. [CrossRef]
- Holbrook AM, Pereira JA, Labiris R, McDonald H, Douketis JD, Crowther M, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165(10):1095-106.
- Tan CSS, Lee SWH. Warfarin and food, herbal or dietary supplement interactions: A systematic review. Br J Clin Pharmacol. 2021;87(2):352-74. [CrossRef]
- Chilbert MR, Zammit K, Ahmed U, Devlin A, Radparvar S, Schuler A, et al. A systematic review of therapeutic enoxaparin dosing in obesity. J Thromb Thrombolysis. 2024;57(4):587-97. [CrossRef]
- Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139-51.
- Al Said S, Ellscheid M, Beltsios ET, Frey N. Non-Vitamin K Antagonist Oral Anticoagulants in Coronary Artery Disease. Hamostaseologie. 2022;42(3):201-9. [CrossRef]
- Kemkes-Matthes, B. [Anticoagulation-direct oral anticoagulants]. Internist (Berl). 2017;58(6):585-97.
- Mueck W, Schwers S, Stampfuss J. Rivaroxaban and other novel oral anticoagulants: pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb J. 2013;11(1):10. [CrossRef]
- Eek AK, Oie E, Granas AG. Prescribing of NOACs has outnumbered warfarin: exploring how physicians choose anticoagulant treatments. Eur J Clin Pharmacol. 2018;74(3):323-30. [CrossRef]
- Di Minno A, Frigerio B, Spadarella G, Ravani A, Sansaro D, Amato M, et al. Old and new oral anticoagulants: Food, herbal medicines and drug interactions. Blood Rev. 2017;31(4):193-203. [CrossRef]
- Udayachalerm S, Rattanasiri S, Angkananard T, Attia J, Sansanayudh N, Thakkinstian A. The Reversal of Bleeding Caused by New Oral Anticoagulants (NOACs): A Systematic Review and Meta-Analysis. Clin Appl Thromb Hemost. 2018;24(9_suppl):117S-26S. [CrossRef]
- Hoffman M, Monroe DM. Impact of Non-Vitamin K Antagonist Oral Anticoagulants From a Basic Science Perspective. Arterioscler Thromb Vasc Biol. 2017;37(10):1812-8. [CrossRef]
- Ruff CT, Giugliano RP, Antman EM. Management of Bleeding With Non-Vitamin K Antagonist Oral Anticoagulants in the Era of Specific Reversal Agents. Circulation. 2016;134(3):248-61. [CrossRef]
- Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111(1):25-7.
- van Gorp RH, Dijkgraaf I, Broker V, Bauwens M, Leenders P, Jennen D, et al. Off-target effects of oral anticoagulants - vascular effects of vitamin K antagonist and non-vitamin K antagonist oral anticoagulant dabigatran etexilate. J Thromb Haemost. 2021;19(5):1348-63. [CrossRef]
- Ito Y, Maejima Y, Nakagama S, Shiheido-Watanabe Y, Tamura N, Sasano T. Rivaroxaban, a Direct Oral Factor Xa Inhibitor, Attenuates Atherosclerosis by Alleviating Factor Xa-PAR2-Mediated Autophagy Suppression. JACC Basic Transl Sci. 2021;6(12):964-80.
- Ruff CT, Giugliano RP, Braunwald E, Hoffman EB, Deenadayalu N, Ezekowitz MD, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-62. [CrossRef]
- Eriksson BI, Dahl OE, Rosencher N, Clemens A, Hantel S, Feuring M, et al. Oral dabigatran etexilate versus enoxaparin for venous thromboembolism prevention after total hip arthroplasty: pooled analysis of two phase 3 randomized trials. Thromb J. 2015;13:36. [CrossRef]
- Goldhaber SZ, Schulman S, Eriksson H, Feuring M, Fraessdorf M, Kreuzer J, et al. Dabigatran versus Warfarin for Acute Venous Thromboembolism in Elderly or Impaired Renal Function Patients: Pooled Analysis of RE-COVER and RE-COVER II. Thromb Haemost. 2017;117(11):2045-52. [CrossRef]
- Diez-Villanueva P, Cosin-Sales J, Roldan-Schilling V, Barrios V, Riba-Artes D, Gavin-Sebastian O, et al. Use of Direct Acting Oral Anticoagulants in Elderly Patients with Atrial Fibrillation: A Multicenter, Cross-Sectional Study in Spain. J Clin Med. 2023;12(3). [CrossRef]
- Yu YB, Liu J, Fu GH, Fang RY, Gao F, Chu HM. Comparison of dabigatran and warfarin used in patients with non-valvular atrial fibrillation: Meta-analysis of random control trial. Medicine (Baltimore). 2018;97(46):e12841. [CrossRef]
- Grymonprez M, De Backer TL, Bertels X, Steurbaut S, Lahousse L. Long-term comparative effectiveness and safety of dabigatran, rivaroxaban, apixaban and edoxaban in patients with atrial fibrillation: A nationwide cohort study. Front Pharmacol. 2023;14:1125576. [CrossRef]
- Achilles A, Mohring A, Dannenberg L, Grandoch M, Hohlfeld T, Fischer JW, et al. Dabigatran enhances platelet reactivity and platelet thrombin receptor expression in patients with atrial fibrillation. J Thromb Haemost. 2017;15(3):473-6. [CrossRef]
- Russell RD, Hotchkiss WR, Knight JR, Huo MH. The efficacy and safety of rivaroxaban for venous thromboembolism prophylaxis after total hip and total knee arthroplasty. Thrombosis. 2013;2013:762310. [CrossRef]
- Prins MH, Lensing AW, Bauersachs R, van Bellen B, Bounameaux H, Brighton TA, et al. Oral rivaroxaban versus standard therapy for the treatment of symptomatic venous thromboembolism: a pooled analysis of the EINSTEIN-DVT and PE randomized studies. Thromb J. 2013;11(1):21. [CrossRef]
- Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883-91. [CrossRef]
- Piazza G, Spyropoulos AC, Hsia J, Goldin M, Towner WJ, Go AS, et al. Rivaroxaban for Prevention of Thrombotic Events, Hospitalization, and Death in Outpatients With COVID-19: A Randomized Clinical Trial. Circulation. 2023;147(25):1891-901. [CrossRef]
- Ray WA, Chung CP, Stein CM, Smalley W, Zimmerman E, Dupont WD, et al. Association of Rivaroxaban vs Apixaban With Major Ischemic or Hemorrhagic Events in Patients With Atrial Fibrillation. JAMA. 2021;326(23):2395-404. [CrossRef]
- Ageno W, Bertu L, Bucherini E, Camporese G, Dentali F, Iotti M, et al. Rivaroxaban treatment for six weeks versus three months in patients with symptomatic isolated distal deep vein thrombosis: randomised controlled trial. BMJ. 2022;379:e072623. [CrossRef]
- Ajmal M, Friedman J, Sipra Q, Lassar T. Rivaroxaban: Expanded Role in Cardiovascular Disease Management-A Literature Review. Cardiovasc Ther. 2021;2021:8886210. [CrossRef]
- Young G, Lensing AWA, Monagle P, Male C, Thelen K, Willmann S, et al. Rivaroxaban for treatment of pediatric venous thromboembolism. An Einstein-Jr phase 3 dose-exposure-response evaluation. J Thromb Haemost. 2020;18(7):1672-85. [CrossRef]
- Cohen AT, Wallenhorst C, Rivera M, Ay C, Schaefer B, Abdelgawwad K, et al. Comparison of Clinical Outcomes in Patients with Active Cancer Receiving Rivaroxaban or Low-Molecular-Weight Heparin: The OSCAR-UK Study. Thromb Haemost. 2024. [CrossRef]
- Adelkhanova A, Oli PR, Shrestha DB, Shtembari J, Jha V, Shantha G, et al. Safety and efficacy of direct oral anticoagulants in comparison to warfarin in obese patients with atrial fibrillation: A systematic review and meta-analysis. Health Sci Rep. 2024;7(4):e2044. [CrossRef]
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P, et al. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375(9717):807-15. [CrossRef]
- Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799-808. [CrossRef]
- Liu X, Johnson M, Mardekian J, Phatak H, Thompson J, Cohen AT. Apixaban Reduces Hospitalizations in Patients With Venous Thromboembolism: An Analysis of the Apixaban for the Initial Management of Pulmonary Embolism and Deep-Vein Thrombosis as First-Line Therapy (AMPLIFY) Trial. J Am Heart Assoc. 2015;4(12). [CrossRef]
- Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806-17.
- O’Donnell MJ, Eikelboom JW, Yusuf S, Diener HC, Hart RG, Smith EE, et al. Effect of apixaban on brain infarction and microbleeds: AVERROES-MRI assessment study. Am Heart J. 2016;178:145-50. [CrossRef]
- Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-92. [CrossRef]
- Carrier M, Abou-Nassar K, Mallick R, Tagalakis V, Shivakumar S, Schattner A, et al. Apixaban to Prevent Venous Thromboembolism in Patients with Cancer. N Engl J Med. 2019;380(8):711-9. [CrossRef]
- Agnelli G, Becattini C, Meyer G, Munoz A, Huisman MV, Connors JM, et al. Apixaban for the Treatment of Venous Thromboembolism Associated with Cancer. N Engl J Med. 2020;382(17):1599-607. [CrossRef]
- Guntupalli SR, Brennecke A, Behbakht K, Tayebnejad A, Breed CA, Babayan LM, et al. Safety and Efficacy of Apixaban vs Enoxaparin for Preventing Postoperative Venous Thromboembolism in Women Undergoing Surgery for Gynecologic Malignant Neoplasm: A Randomized Clinical Trial. JAMA Netw Open. 2020;3(6):e207410.
- Reinecke H, Engelbertz C, Bauersachs R, Breithardt G, Echterhoff HH, Gerss J, et al. A Randomized Controlled Trial Comparing Apixaban With the Vitamin K Antagonist Phenprocoumon in Patients on Chronic Hemodialysis: The AXADIA-AFNET 8 Study. Circulation. 2023;147(4):296-309. [CrossRef]
- Payne RM, Burns KM, Glatz AC, Male C, Donti A, Brandao LR, et al. Apixaban for Prevention of Thromboembolism in Pediatric Heart Disease. J Am Coll Cardiol. 2023;82(24):2296-309. [CrossRef]
- O’Brien SH, Rodriguez V, Lew G, Newburger JW, Schultz CL, Orgel E, et al. Apixaban versus no anticoagulation for the prevention of venous thromboembolism in children with newly diagnosed acute lymphoblastic leukaemia or lymphoma (PREVAPIX-ALL): a phase 3, open-label, randomised, controlled trial. Lancet Haematol. 2024;11(1):e27-e37. [CrossRef]
- Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369(22):2093-104. [CrossRef]
- Hokusai VTEI, Buller HR, Decousus H, Grosso MA, Mercuri M, Middeldorp S, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406-15.
- Raskob GE, van Es N, Segers A, Angchaisuksiri P, Oh D, Boda Z, et al. Edoxaban for venous thromboembolism in patients with cancer: results from a non-inferiority subgroup analysis of the Hokusai-VTE randomised, double-blind, double-dummy trial. Lancet Haematol. 2016;3(8):e379-87.
- Raskob GE, van Es N, Verhamme P, Carrier M, Di Nisio M, Garcia D, et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N Engl J Med. 2018;378(7):615-24.
- Fuji T, Fujita S, Kawai Y, Nakamura M, Kimura T, Kiuchi Y, et al. Safety and efficacy of edoxaban in patients undergoing hip fracture surgery. Thromb Res. 2014;133(6):1016-22. [CrossRef]
- Fuji T, Wang CJ, Fujita S, Kawai Y, Nakamura M, Kimura T, et al. Safety and efficacy of edoxaban, an oral factor Xa inhibitor, versus enoxaparin for thromboprophylaxis after total knee arthroplasty: the STARS E-3 trial. Thromb Res. 2014;134(6):1198-204. [CrossRef]
- Koretsune Y, Yamashita T, Kimura T, Fukuzawa M, Abe K, Yasaka M. Short-Term Safety and Plasma Concentrations of Edoxaban in Japanese Patients With Non-Valvular Atrial Fibrillation and Severe Renal Impairment. Circ J. 2015;79(7):1486-95. [CrossRef]
- Goette A, Merino JL, Ezekowitz MD, Zamoryakhin D, Melino M, Jin J, et al. Edoxaban versus enoxaparin-warfarin in patients undergoing cardioversion of atrial fibrillation (ENSURE-AF): a randomised, open-label, phase 3b trial. Lancet. 2016;388(10055):1995-2003.
- Portman MA, Jacobs JP, Newburger JW, Berger F, Grosso MA, Duggal A, et al. Edoxaban for Thromboembolism Prevention in Pediatric Patients With Cardiac Disease. J Am Coll Cardiol. 2022;80(24):2301-10. [CrossRef]
- Shim CY, Seo J, Kim YJ, Lee SH, De Caterina R, Lee S, et al. Efficacy and safety of edoxaban in patients early after surgical bioprosthetic valve implantation or valve repair: A randomized clinical trial. J Thorac Cardiovasc Surg. 2023;165(1):58-67 e4.
- Hosokawa K, Watanabe H, Taniguchi Y, Ikeda N, Inami T, Yasuda S, et al. A Multicenter, Single-Blind, Randomized, Warfarin-Controlled Trial of Edoxaban in Patients With Chronic Thromboembolic Pulmonary Hypertension: KABUKI Trial. Circulation. 2024;149(5):406-9. [CrossRef]
- Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153 Suppl 1(Suppl 1):S263-82. [CrossRef]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394(6694):690-4. [CrossRef]
- Lin H, Liu AP, Smith TH, Trejo J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev. 2013;65(4):1198-213. [CrossRef]
- Ruf, W. Roles of factor Xa beyond coagulation. J Thromb Thrombolysis. 2021;52(2):391-6. [CrossRef]
- Bukowska A, Zacharias I, Weinert S, Skopp K, Hartmann C, Huth C, et al. Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue. Eur J Pharmacol. 2013;718(1-3):114-23. [CrossRef]
- Ohba T, Takase Y, Ohhara M, Kasukawa R. Thrombin in the synovial fluid of patients with rheumatoid arthritis mediates proliferation of synovial fibroblast-like cells by induction of platelet derived growth factor. J Rheumatol. 1996;23(9):1505-11.
- Itsekson-Hayosh Z, Shavit-Stein E, Last D, Goez D, Daniels D, Bushi D, et al. Thrombin Activity and Thrombin Receptor in Rat Glioblastoma Model: Possible Markers and Targets for Intervention? J Mol Neurosci. 2015;56(3):644-51.
- Ofosu, FA. Protease activated receptors 1 and 4 govern the responses of human platelets to thrombin. Transfus Apher Sci. 2003;28(3):265-8. [CrossRef]
- Coughlin, SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96(20):11023-7.
- Garcia PS, Ciavatta VT, Fidler JA, Woodbury A, Levy JH, Tyor WR. Concentration-Dependent Dual Role of Thrombin in Protection of Cultured Rat Cortical Neurons. Neurochem Res. 2015;40(11):2220-9. [CrossRef]
- Lee PR, Johnson TP, Gnanapavan S, Giovannoni G, Wang T, Steiner JP, et al. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1beta. J Neuroinflammation. 2017;14(1):131.
- Kempuraj D, Selvakumar GP, Thangavel R, Ahmed ME, Zaheer S, Kumar KK, et al. Glia Maturation Factor and Mast Cell-Dependent Expression of Inflammatory Mediators and Proteinase Activated Receptor-2 in Neuroinflammation. J Alzheimers Dis. 2018;66(3):1117-29. [CrossRef]
- Takayama Y, Derouiche S, Maruyama K, Tominaga M. Emerging Perspectives on Pain Management by Modulation of TRP Channels and ANO1. Int J Mol Sci. 2019;20(14). [CrossRef]
- Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, et al. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem. 2013;288(8):5790-802. [CrossRef]
- Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, et al. Role for protease activity in visceral pain in irritable bowel syndrome. J Clin Invest. 2007;117(3):636-47.
- Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, et al. Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway. Nat Med. 2001;7(7):821-6. [CrossRef]
- Asfaha S, Brussee V, Chapman K, Zochodne DW, Vergnolle N. Proteinase-activated receptor-1 agonists attenuate nociception in response to noxious stimuli. Br J Pharmacol. 2002;135(5):1101-6. [CrossRef]
- Saeed MA, Ng GZ, Dabritz J, Wagner J, Judd L, Han JX, et al. Protease-activated Receptor 1 Plays a Proinflammatory Role in Colitis by Promoting Th17-related Immunity. Inflamm Bowel Dis. 2017;23(4):593-602. [CrossRef]
- Cenac N, Cellars L, Steinhoff M, Andrade-Gordon P, Hollenberg MD, Wallace JL, et al. Proteinase-activated receptor-1 is an anti-inflammatory signal for colitis mediated by a type 2 immune response. Inflamm Bowel Dis. 2005;11(9):792-8. [CrossRef]
- Boucher AA, Rosenfeldt L, Mureb D, Shafer J, Sharma BK, Lane A, et al. Cell type-specific mechanisms coupling protease-activated receptor-1 to infectious colitis pathogenesis. J Thromb Haemost. 2020;18(1):91-103.
- Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol. 2011;300(1):C113-23. [CrossRef]
- Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci U S A. 2005;102(23):8363-8. [CrossRef]
- Liu B, Yang MQ, Yu TY, Yin YY, Liu Y, Wang XD, et al. Mast Cell Tryptase Promotes Inflammatory Bowel Disease-Induced Intestinal Fibrosis. Inflamm Bowel Dis. 2021;27(2):242-55. [CrossRef]
- Her JY, Lee Y, Kim SJ, Heo G, Choo J, Kim Y, et al. Blockage of protease-activated receptor 2 exacerbates inflammation in high-fat environment partly through autophagy inhibition. Am J Physiol Gastrointest Liver Physiol. 2021;320(1):G30-G42. [CrossRef]
- Majewski S, Zhou X, Milkowska-Dymanowska J, Bialas AJ, Piotrowski WJ, Malinovschi A. Proteomic profiling of peripheral blood and bronchoalveolar lavage fluid in interstitial lung diseases: an explorative study. ERJ Open Res. 2021;7(1). [CrossRef]
- Bhargava M, Viken K, Wang Q, Jagtap P, Bitterman P, Ingbar D, et al. Bronchoalveolar Lavage Fluid Protein Expression in Acute Respiratory Distress Syndrome Provides Insights into Pathways Activated in Subjects with Different Outcomes. Sci Rep. 2017;7(1):7464. [CrossRef]
- Lambrecht BN, Hammad H. Asthma and coagulation. N Engl J Med. 2013;369(20):1964-6. [CrossRef]
- de Boer JD, Majoor CJ, van ’t Veer C, Bel EH, van der Poll T. Asthma and coagulation. Blood. 2012;119(14):3236-44.
- Deng Z, Xie H, Cheng W, Zhang M, Liu J, Huo Y, et al. Dabigatran ameliorates airway smooth muscle remodeling in asthma by modulating Yes-associated protein. J Cell Mol Med. 2020;24(14):8179-93.
- Yue M, Hu M, Fu F, Ruan H, Wu C. Emerging Roles of Platelets in Allergic Asthma. Front Immunol. 2022;13:846055. [CrossRef]
- Gandhi VD, Shrestha Palikhe N, Vliagoftis H. Protease-activated receptor-2: Role in asthma pathogenesis and utility as a biomarker of disease severity. Front Med (Lausanne). 2022;9:954990. [CrossRef]
- Ouyang X, Reihill JA, Douglas LEJ, Martin SL. Airborne indoor allergen serine proteases and their contribution to sensitisation and activation of innate immunity in allergic airway disease. Eur Respir Rev. 2024;33(172). [CrossRef]
- Rivas CM, Yee MC, Addison KJ, Lovett M, Pal K, Ledford JG, et al. Proteinase-activated receptor-2 antagonist C391 inhibits Alternaria-induced airway epithelial signalling and asthma indicators in acute exposure mouse models. Br J Pharmacol. 2022;179(10):2208-22. [CrossRef]
- Ricciardolo FL, Steinhoff M, Amadesi S, Guerrini R, Tognetto M, Trevisani M, et al. Presence and bronchomotor activity of protease-activated receptor-2 in guinea pig airways. Am J Respir Crit Care Med. 2000;161(5):1672-80. [CrossRef]
- Carr MJ, Schechter NM, Undem BJ. Trypsin-induced, neurokinin-mediated contraction of guinea pig bronchus. Am J Respir Crit Care Med. 2000;162(5):1662-7. [CrossRef]
- Schmidlin F, Amadesi S, Vidil R, Trevisani M, Martinet N, Caughey G, et al. Expression and function of proteinase-activated receptor 2 in human bronchial smooth muscle. Am J Respir Crit Care Med. 2001;164(7):1276-81. [CrossRef]
- Hauck RW, Schulz C, Schomig A, Hoffman RK, Panettieri RA, Jr. alpha-Thrombin stimulates contraction of human bronchial rings by activation of protease-activated receptors. Am J Physiol. 1999;277(1):L22-9. [CrossRef]
- Lin C, von der Thusen J, Daalhuisen J, ten Brink M, Crestani B, van der Poll T, et al. Protease-activated receptor (PAR)-2 is required for PAR-1 signalling in pulmonary fibrosis. J Cell Mol Med. 2015;19(6):1346-56.
- Howell DC, Laurent GJ, Chambers RC. Role of thrombin and its major cellular receptor, protease-activated receptor-1, in pulmonary fibrosis. Biochem Soc Trans. 2002;30(2):211-6. [CrossRef]
- Lin C, von der Thusen J, Daalhuisen J, ten Brink M, Crestani B, van der Poll T, et al. Pharmacological Targeting of Protease-Activated Receptor 2 Affords Protection from Bleomycin-Induced Pulmonary Fibrosis. Mol Med. 2015;21(1):576-83. [CrossRef]
- Miyake Y, D’Alessandro-Gabazza CN, Takagi T, Naito M, Hataji O, Nakahara H, et al. Dose-dependent differential effects of thrombin in allergic bronchial asthma. J Thromb Haemost. 2013;11(10):1903-15. [CrossRef]
- Jose RJ, Williams AE, Mercer PF, Sulikowski MG, Brown JS, Chambers RC. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol. 2015;194(12):6024-34. [CrossRef]
- Jose R, Williams A, Sulikowski M, Brealey D, Brown J, Chambers R. Regulation of neutrophilic inflammation in lung injury induced by community-acquired pneumonia. Lancet. 2015;385 Suppl 1:S52. [CrossRef]
- van den Boogaard FE, Brands X, Duitman J, de Stoppelaar SF, Borensztajn KS, Roelofs J, et al. Protease-Activated Receptor 2 Facilitates Bacterial Dissemination in Pneumococcal Pneumonia. J Infect Dis. 2018;217(9):1462-71. [CrossRef]
- Rayees S, Joshi JC, Tauseef M, Anwar M, Baweja S, Rochford I, et al. PAR2-Mediated cAMP Generation Suppresses TRPV4-Dependent Ca(2+) Signaling in Alveolar Macrophages to Resolve TLR4-Induced Inflammation. Cell Rep. 2019;27(3):793-805 e4. [CrossRef]
- de Stoppelaar SF, Van’t Veer C, van den Boogaard FE, Nieuwland R, Hoogendijk AJ, de Boer OJ, et al. Protease activated receptor 4 limits bacterial growth and lung pathology during late stage Streptococcus pneumoniae induced pneumonia in mice. Thromb Haemost. 2013;110(3):582-92. [CrossRef]
- Sabri A, Muske G, Zhang H, Pak E, Darrow A, Andrade-Gordon P, et al. Signaling properties and functions of two distinct cardiomyocyte protease-activated receptors. Circ Res. 2000;86(10):1054-61. [CrossRef]
- Ide J, Aoki T, Ishivata S, Glusa E, Strukova SM. Proteinase-activated receptor agonists stimulate the increase in intracellular Ca2+ in cardiomyocytes and proliferation of cardiac fibroblasts from chick embryos. Bull Exp Biol Med. 2007;144(6):760-3. [CrossRef]
- Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116(20):2298-306. [CrossRef]
- Weithauser A, Witkowski M, Rauch U. The Role of Protease-Activated Receptors for the Development of Myocarditis: Possible Therapeutic Implications. Curr Pharm Des. 2016;22(4):472-84. [CrossRef]
- Antoniak S, Owens AP, 3rd, Baunacke M, Williams JC, Lee RD, Weithauser A, et al. PAR-1 contributes to the innate immune response during viral infection. J Clin Invest. 2013;123(3):1310-22.
- Tatsumi K, Schmedes CM, Houston ER, Butler E, Mackman N, Antoniak S. Protease-activated receptor 4 protects mice from Coxsackievirus B3 and H1N1 influenza A virus infection. Cell Immunol. 2019;344:103949. [CrossRef]
- Naldini A, Carney DH. Thrombin modulation of natural killer activity in human peripheral lymphocytes. Cell Immunol. 1996;172(1):35-42. [CrossRef]
- Mackman N, Antoniak S. Roles of PAR1 and PAR2 in viral myocarditis. Thromb Res. 2014;133 Suppl 1(0 1):S18-20. [CrossRef]
- Pan HY, Yano M, Kido H. Effects of inhibitors of Toll-like receptors, protease-activated receptor-2 signalings and trypsin on influenza A virus replication and upregulation of cellular factors in cardiomyocytes. J Med Invest. 2011;58(1-2):19-28. [CrossRef]
- Kouzoukas DE, Meyer-Siegler KL, Ma F, Westlund KN, Hunt DE, Vera PL. Macrophage Migration Inhibitory Factor Mediates PAR-Induced Bladder Pain. PLoS One. 2015;10(5):e0127628. [CrossRef]
- Ma F, Hunt DE, Leng L, Bucala R, Meyer-Siegler KL, Vera PL. Protease activated-receptor 4 activation as a model of persistent bladder pain: Essential role of macrophage migration inhibitory factor and high mobility group box 1. Int J Urol. 2018;25(10):887-93. [CrossRef]
- Saban R, D’Andrea MR, Andrade-Gordon P, Derian CK, Dozmorov I, Ihnat MA, et al. Mandatory role of proteinase-activated receptor 1 in experimental bladder inflammation. BMC Physiol. 2007;7:4. [CrossRef]
- Monjotin N, Gillespie J, Farrie M, Le Grand B, Junquero D, Vergnolle N. F16357, a novel protease-activated receptor 1 antagonist, improves urodynamic parameters in a rat model of interstitial cystitis. Br J Pharmacol. 2016;173(14):2224-36. [CrossRef]
- Wang ZY, Wang P, Bjorling DE. Role of mast cells and protease-activated receptor-2 in cyclooxygenase-2 expression in urothelial cells. Am J Physiol Regul Integr Comp Physiol. 2009;297(4):R1127-35. [CrossRef]
- Madhusudhan T, Kerlin BA, Isermann B. The emerging role of coagulation proteases in kidney disease. Nat Rev Nephrol. 2016;12(2):94-109. [CrossRef]
- Blanc-Brude OP, Archer F, Leoni P, Derian C, Bolsover S, Laurent GJ, et al. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res. 2005;304(1):16-27. [CrossRef]
- Horinouchi Y, Ikeda Y, Fukushima K, Imanishi M, Hamano H, Izawa-Ishizawa Y, et al. Renoprotective effects of a factor Xa inhibitor: fusion of basic research and a database analysis. Sci Rep. 2018;8(1):10858. [CrossRef]
- Han Y, Tian L, Ma F, Tesch G, Vesey DA, Gobe GC, et al. Pharmacological inhibition of protease-activated receptor-2 reduces crescent formation in rat nephrotoxic serum nephritis. Clin Exp Pharmacol Physiol. 2019;46(5):456-64. [CrossRef]
- Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111(1):35-41.
- Kelso EB, Ferrell WR, Lockhart JC, Elias-Jones I, Hembrough T, Dunning L, et al. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium: ex vivo studies using a novel proteinase-activated receptor 2 antagonist. Arthritis Rheum. 2007;56(3):765-71. [CrossRef]
- Russell FA, Schuelert N, Veldhoen VE, Hollenberg MD, McDougall JJ. Activation of PAR(2) receptors sensitizes primary afferents and causes leukocyte rolling and adherence in the rat knee joint. Br J Pharmacol. 2012;167(8):1665-78. [CrossRef]
- Nakano S, Mishiro T, Takahara S, Yokoi H, Hamada D, Yukata K, et al. Distinct expression of mast cell tryptase and protease activated receptor-2 in synovia of rheumatoid arthritis and osteoarthritis. Clin Rheumatol. 2007;26(8):1284-92. [CrossRef]
- Palmer HS, Kelso EB, Lockhart JC, Sommerhoff CP, Plevin R, Goh FG, et al. Protease-activated receptor 2 mediates the proinflammatory effects of synovial mast cells. Arthritis Rheum. 2007;56(11):3532-40. [CrossRef]
- Crilly A, Burns E, Nickdel MB, Lockhart JC, Perry ME, Ferrell PW, et al. PAR(2) expression in peripheral blood monocytes of patients with rheumatoid arthritis. Ann Rheum Dis. 2012;71(6):1049-54. [CrossRef]
- Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, et al. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316(3):1017-24. [CrossRef]
- Kanno Y, Ishisaki A, Kawashita E, Kuretake H, Ikeda K, Matsuo O. uPA Attenuated LPS-induced Inflammatory Osteoclastogenesis through the Plasmin/PAR-1/Ca(2+)/CaMKK/AMPK Axis. Int J Biol Sci. 2016;12(1):63-71.
- Huang CY, Chen SY, Tsai HC, Hsu HC, Tang CH. Thrombin induces epidermal growth factor receptor transactivation and CCL2 expression in human osteoblasts. Arthritis Rheum. 2012;64(10):3344-54. [CrossRef]
- Hirano F, Kobayashi A, Hirano Y, Nomura Y, Fukawa E, Makino I. Thrombin-induced expression of RANTES mRNA through protease activated receptor-1 in human synovial fibroblasts. Ann Rheum Dis. 2002;61(9):834-7. [CrossRef]
- Furuhashi I, Abe K, Sato T, Inoue H. Thrombin-stimulated proliferation of cultured human synovial fibroblasts through proteolytic activation of proteinase-activated receptor-1. J Pharmacol Sci. 2008;108(1):104-11. [CrossRef]
- Xue M, Chan YK, Shen K, Dervish S, March L, Sambrook PN, et al. Protease-activated receptor 2, rather than protease-activated receptor 1, contributes to the aggressive properties of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2012;64(1):88-98. [CrossRef]
- Patnaik R, Riaz S, Sivani BM, Faisal S, Naidoo N, Rizzo M, et al. Evaluating the potential of Vitamin D and curcumin to alleviate inflammation and mitigate the progression of osteoarthritis through their effects on human chondrocytes: A proof-of-concept investigation. PLoS One. 2023;18(12):e0290739. [CrossRef]
- Hijazi Z, Aulin J, Andersson U, Alexander JH, Gersh B, Granger CB, et al. Biomarkers of inflammation and risk of cardiovascular events in anticoagulated patients with atrial fibrillation. Heart. 2016;102(7):508-17. [CrossRef]
- Wallentin L, Hijazi Z, Andersson U, Alexander JH, De Caterina R, Hanna M, et al. Growth differentiation factor 15, a marker of oxidative stress and inflammation, for risk assessment in patients with atrial fibrillation: insights from the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial. Circulation. 2014;130(21):1847-58.
- Sandhu RK, Ezekowitz JA, Hijazi Z, Westerbergh J, Aulin J, Alexander JH, et al. Obesity paradox on outcome in atrial fibrillation maintained even considering the prognostic influence of biomarkers: insights from the ARISTOTLE trial. Open Heart. 2018;5(2):e000908. [CrossRef]
- Golderman V, Gofrit SG, Maggio N, Gera O, Gerasimov A, Laks D, et al. A Novel Highly Sensitive Method for Measuring Inflammatory Neural-Derived APC Activity in Glial Cell Lines, Mouse Brain and Human CSF. Int J Mol Sci. 2020;21(7). [CrossRef]
- Granja T, Hohenstein K, Schussel P, Fischer C, Prufer T, Schibilsky D, et al. Multi-Modal Characterization of the Coagulopathy Associated With Extracorporeal Membrane Oxygenation. Crit Care Med. 2020;48(5):e400-e8. [CrossRef]
- Torramade-Moix S, Palomo M, Vera M, Jerez D, Moreno-Castano AB, Zafar MU, et al. Apixaban Downregulates Endothelial Inflammatory and Prothrombotic Phenotype in an In Vitro Model of Endothelial Dysfunction in Uremia. Cardiovasc Drugs Ther. 2021;35(3):521-32. [CrossRef]
- Muthupalani S, Annamalai D, Feng Y, Ganesan SM, Ge Z, Whary MT, et al. IL-1beta transgenic mouse model of inflammation driven esophageal and oral squamous cell carcinoma. Sci Rep. 2023;13(1):12732.
- Patnaik R, Jannati S, Sivani BM, Rizzo M, Naidoo N, Banerjee Y. Efficient Generation of Chondrocytes From Bone Marrow-Derived Mesenchymal Stem Cells in a 3D Culture System: Protocol for a Practical Model for Assessing Anti-Inflammatory Therapies. JMIR Res Protoc. 2023;12:e42964. [CrossRef]
- Suarez-Martinez E, Suazo-Sanchez I, Celis-Romero M, Carnero A. 3D and organoid culture in research: physiology, hereditary genetic diseases and cancer. Cell Biosci. 2022;12(1):39. [CrossRef]
- Nzou G, Wicks RT, VanOstrand NR, Mekky GA, Seale SA, El-Taibany A, et al. Author Correction: Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci Rep. 2020;10(1):20384. [CrossRef]
- Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J Immunol Res. 2018;2018:2180373. [CrossRef]
- Duan L, Rao X, Sigdel KR. Regulation of Inflammation in Autoimmune Disease. J Immunol Res. 2019;2019:7403796. [CrossRef]
- Zhu Q, Wu Y, Mai J, Guo G, Meng J, Fang X, et al. Comprehensive Metabolic Profiling of Inflammation Indicated Key Roles of Glycerophospholipid and Arginine Metabolism in Coronary Artery Disease. Front Immunol. 2022;13:829425. [CrossRef]
- Oe Y, Hayashi S, Fushima T, Sato E, Kisu K, Sato H, et al. Coagulation Factor Xa and Protease-Activated Receptor 2 as Novel Therapeutic Targets for Diabetic Nephropathy. Arterioscler Thromb Vasc Biol. 2016;36(8):1525-33. [CrossRef]
- Bieber M, Foerster KI, Haefeli WE, Pham M, Schuhmann MK, Kraft P. Treatment with Edoxaban Attenuates Acute Stroke Severity in Mice by Reducing Blood-Brain Barrier Damage and Inflammation. Int J Mol Sci. 2021;22(18). [CrossRef]
- Millenaar D, Bachmann P, Bohm M, Custodis F, Schirmer SH. Effects of edoxaban and warfarin on vascular remodeling: Atherosclerotic plaque progression and collateral artery growth. Vascul Pharmacol. 2020;127:106661. [CrossRef]
- Morishima Y, Shibutani T, Noguchi K, Ito Y, Honda Y. Edoxaban, a direct oral factor Xa inhibitor, ameliorates coagulation, microvascular thrombus formation, and acute liver injury in a lipopolysaccharide-induced coagulopathy model in rats. J Thromb Thrombolysis. 2021;52(1):9-17. [CrossRef]
- Baker JV, Wolfson J, Peterson T, Mooberry M, Gissel M, Mystakelis H, et al. Factor Xa Inhibition Reduces Coagulation Activity but Not Inflammation Among People With HIV: A Randomized Clinical Trial. Open Forum Infect Dis. 2020;7(2):ofaa026. [CrossRef]
- Goette A, Mollenhauer M, Rudolph V, Lamparter M, Meier M, Bohm M. Pleiotropic effects of NOACs with focus on edoxaban: scientific findings and potential clinical implications. Herzschrittmacherther Elektrophysiol. 2023;34(2):142-52.
- Narita Y, Hamamura K, Kashiyama M, Utsumi S, Kakizoe Y, Kondo Y, et al. Edoxaban Exerts Antioxidant Effects Through FXa Inhibition and Direct Radical-Scavenging Activity. Int J Mol Sci. 2019;20(17). [CrossRef]
- Romero-Guevara R, Ioannides A, Xinaris C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front Physiol. 2020;11:563981. [CrossRef]
- Fagundes A, Jr., Ruff CT, Morrow DA, Murphy SA, Palazzolo MG, Chen CZ, et al. Neutrophil-lymphocyte ratio and clinical outcomes in 19,697 patients with atrial fibrillation: Analyses from ENGAGE AF- TIMI 48 trial. Int J Cardiol. 2023;386:118-24. [CrossRef]
- Chan MY, Lin M, Lucas J, Moseley A, Thompson JW, Cyr D, et al. Plasma proteomics of patients with non-valvular atrial fibrillation on chronic anti-coagulation with warfarin or a direct factor Xa inhibitor. Thromb Haemost. 2012;108(6):1180-91. [CrossRef]
- Monux G, Zamorano-Leon JJ, Marques P, Sopena B, Garcia-Garcia JM, Laich de Koller G, et al. FXa inhibition by rivaroxaban modifies mechanisms associated with the pathogenesis of human abdominal aortic aneurysms. Br J Clin Pharmacol. 2017;83(12):2661-70. [CrossRef]
- Miyazawa K, Pastori D, Hammerstingl C, Cappato R, Meng IL, Kramer F, et al. Left atrial thrombus resolution in non-valvular atrial fibrillation or flutter: biomarker substudy results from a prospective study with rivaroxaban (X-TRA). Ann Med. 2018;50(6):511-8. [CrossRef]
- Martins GL, Duarte RCF, Vieira ELM, Rocha NP, Figueiredo EL, Silveira FR, et al. Comparison of Inflammatory Mediators in Patients With Atrial Fibrillation Using Warfarin or Rivaroxaban. Front Cardiovasc Med. 2020;7:114. [CrossRef]
- Mrozik D, Jackiewicz A, Krzeminski M. Dabigatran vs. low molecular weight heparin in preventing venous thromboembolism after elective hip and knee arthroplasty: evaluation of selected clinical parameters. Pol Orthop Traumatol. 2012;77:111-4.
- Paar V, Jirak P, Gruber S, Prodinger C, Cadamuro J, Wernly B, et al. Influence of dabigatran on pro-inflammatory cytokines, growth factors and chemokines - Slowing the vicious circle of coagulation and inflammation. Life Sci. 2020;262:118474. [CrossRef]
- Bogatkevich GS, Ludwicka-Bradley A, Nietert PJ, Akter T, van Ryn J, Silver RM. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011;63(5):1416-25. [CrossRef]
- Kopec AK, Joshi N, Towery KL, Kassel KM, Sullivan BP, Flick MJ, et al. Thrombin inhibition with dabigatran protects against high-fat diet-induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014;351(2):288-97. [CrossRef]
- Sparkenbaugh EM, Chantrathammachart P, Mickelson J, van Ryn J, Hebbel RP, Monroe DM, et al. Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood. 2014;123(11):1747-56. [CrossRef]
- Ellinghaus P, Perzborn E, Hauenschild P, Gerdes C, Heitmeier S, Visser M, et al. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb Res. 2016;142:44-51. [CrossRef]
- Nakase T, Moroi J, Ishikawa T. Anti-inflammatory and antiplatelet effects of non-vitamin K antagonist oral anticoagulants in acute phase of ischemic stroke patients. Clin Transl Med. 2018;7(1):2. [CrossRef]
- Durmaz S, Kurtoglu T, Rahman OF, Tataroglu C, Yilmaz M, Barbarus E, et al. Direct oral anticoagulant agents attenuate temporary aortic occlusion-induced renal oxidative and inflammatory responses in rats. Turk Gogus Kalp Damar Cerrahisi Derg. 2022;30(2):184-91. [CrossRef]
- Hiramoto K, Akita N, Nishioka J, Suzuki K. Edoxaban, a Factor Xa-Specific Direct Oral Anticoagulant, Significantly Suppresses Tumor Growth in Colorectal Cancer Colon26-Inoculated BALB/c Mice. TH Open. 2023;7(1):e1-e13. [CrossRef]
- Christopoulou EC, Filippatos TD, Elisaf MS. Non-hemorrhage-related adverse effects of rivaroxaban. Arch Med Sci Atheroscler Dis. 2017;2:e108-e12. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583-9. [CrossRef]
- Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. 2023;379(6637):1123-30. [CrossRef]
- Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49-56. [CrossRef]
- Johnson SR, Fu X, Viknander S, Goldin C, Monaco S, Zelezniak A, et al. Computational scoring and experimental evaluation of enzymes generated by neural networks. Nat Biotechnol. 2024.
- Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat Biotechnol. 2003;21(1):71-6. [CrossRef]
- Petruzzella A, Bruand M, Santamaria-Martinez A, Katanayeva N, Reymond L, Wehrle S, et al. Antibody-peptide conjugates deliver covalent inhibitors blocking oncogenic cathepsins. Nat Chem Biol. 2024. [CrossRef]
- Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:61-79.
- Yu H, Kumar S, Frederiksen JW, Kolyadko VN, Pitoc G, Layzer J, et al. Aptameric hirudins as selective and reversible EXosite-ACTive site (EXACT) inhibitors. Nat Commun. 2024;15(1):3977. [CrossRef]
Figure 1.
The Current Model of The Blood Coagulation Cascade, Depicting NOACs Mechanism of Action. There are 2 pathways, the intrinsic pathway and the extrinsic pathway. These multicomponent processes are illustrated as enzymes, inhibitors, zymogens, or complexes. On injury to the vessel wall, tissue factor, the cofactor for the extrinsic tenase complex, is exposed to circulating FVIIa and forms the the extrinsic tenase. FIX and FX are converted to their serine proteases FIXa and FXa, which then form the intrinsic tenase and the prothrombinase complexes, respectively. The combined actions of the intrinsic and extrinsic tenase and the prothrombinase complexes lead to an explosive burst of the enzyme thrombin (IIa). In addition to its multiple procoagulant roles, thrombin also acts in an anticoagulant capacity when combined with the cofactor thrombomodulin in the protein Case complex. The product of the protein Case reaction, APC, inactivates the cofactors FVa and FVIIIa. The cleaved species, FVai and FVIIIai, no longer support the respective procoagulant activities. Once thrombin is generated through procoagulant mechanisms, thrombin cleaves fibrinogen (releasing fibrinopeptide A and B [FPA and FPB]) as well as activates FXIII to form a cross-linked fibrin clot. Thrombin–thrombomodulin also activates thrombin activate-able fibrinolysis inhibitor that slows fibrin degradation by plasmin. The procoagulant response is downregulated by the stoichiometric inhibitor tissue factor pathway inhibitor (TFPI) and antithrombin III (AT-III). TFPI serves to attenuate the activity of the extrinsic tenase trigger of coagulation. AT-III directly inhibits thrombin, FIXa, and FXa. The accessory pathway provides an alternate route for the generation of FIXa. Thrombin has also been shown to activate FXI. The fibrin clot is eventually degraded by plasmin yielding soluble fibrin peptides. Factor Xa inhibitors (apixaban, edoxaban, and rivaroxaban) act by binding to the active site of factor Xa, inhibiting the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade. Dabigatran, conversely, functions as a direct thrombin inhibitor. It binds with high affinity to the active site of thrombin, inhibiting its ability to convert fibrinogen to fibrin, thereby preventing clot formation.
Figure 1.
The Current Model of The Blood Coagulation Cascade, Depicting NOACs Mechanism of Action. There are 2 pathways, the intrinsic pathway and the extrinsic pathway. These multicomponent processes are illustrated as enzymes, inhibitors, zymogens, or complexes. On injury to the vessel wall, tissue factor, the cofactor for the extrinsic tenase complex, is exposed to circulating FVIIa and forms the the extrinsic tenase. FIX and FX are converted to their serine proteases FIXa and FXa, which then form the intrinsic tenase and the prothrombinase complexes, respectively. The combined actions of the intrinsic and extrinsic tenase and the prothrombinase complexes lead to an explosive burst of the enzyme thrombin (IIa). In addition to its multiple procoagulant roles, thrombin also acts in an anticoagulant capacity when combined with the cofactor thrombomodulin in the protein Case complex. The product of the protein Case reaction, APC, inactivates the cofactors FVa and FVIIIa. The cleaved species, FVai and FVIIIai, no longer support the respective procoagulant activities. Once thrombin is generated through procoagulant mechanisms, thrombin cleaves fibrinogen (releasing fibrinopeptide A and B [FPA and FPB]) as well as activates FXIII to form a cross-linked fibrin clot. Thrombin–thrombomodulin also activates thrombin activate-able fibrinolysis inhibitor that slows fibrin degradation by plasmin. The procoagulant response is downregulated by the stoichiometric inhibitor tissue factor pathway inhibitor (TFPI) and antithrombin III (AT-III). TFPI serves to attenuate the activity of the extrinsic tenase trigger of coagulation. AT-III directly inhibits thrombin, FIXa, and FXa. The accessory pathway provides an alternate route for the generation of FIXa. Thrombin has also been shown to activate FXI. The fibrin clot is eventually degraded by plasmin yielding soluble fibrin peptides. Factor Xa inhibitors (apixaban, edoxaban, and rivaroxaban) act by binding to the active site of factor Xa, inhibiting the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade. Dabigatran, conversely, functions as a direct thrombin inhibitor. It binds with high affinity to the active site of thrombin, inhibiting its ability to convert fibrinogen to fibrin, thereby preventing clot formation.
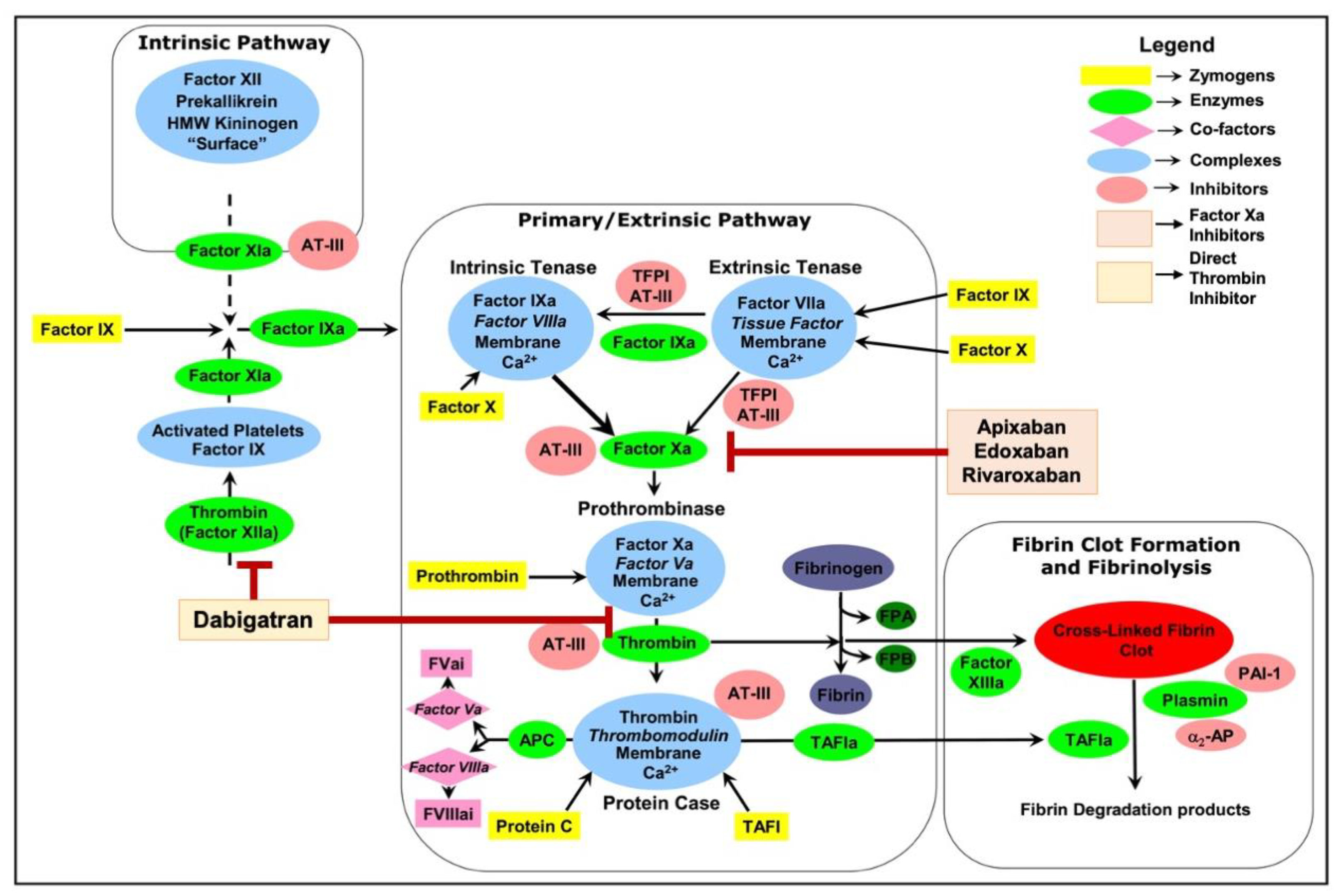
Figure 2.
Schematic Representation of PAR1- & PAR2-mediated Signal Transduction. PAR1 and PAR2 are G protein-coupled receptors that can be activated by thrombin and FXa, initiating a cascade of cellular responses. Upon cleavage, PAR1/2 interact with different G proteins like Gαi, Gα12/13, Gαs and Gαq. Gα12/13 leads to RhoA activation, via RhoGEFs influencing cell hypertrophy. Gαq activates phospholipase C-beta, generating second messengers that trigger calcium release and PKC activation. PKC can further activate the NF-κB signaling pathway to upregulate production of SOX4 and ADAMTS5. Gαi can inhibit AC to regulate downstream cAMP, whereas Gαs can increase cAMP. β-arrestin can activate ERK1/2 signalling pathway however exhibits inhibitory effects on PKC and calcium release. Anticoagulants like dabigatran (thrombin inhibitor) and apixaban, edoxaban and rivaroxaban, (FXa inhibitors) can potentially disrupt this signaling by preventing PAR activation. Ultimately, these signal transduction pathways can trigger physiological changes like inflammatory and immune responses, cell hypertrophy, and cell migration.
Figure 2.
Schematic Representation of PAR1- & PAR2-mediated Signal Transduction. PAR1 and PAR2 are G protein-coupled receptors that can be activated by thrombin and FXa, initiating a cascade of cellular responses. Upon cleavage, PAR1/2 interact with different G proteins like Gαi, Gα12/13, Gαs and Gαq. Gα12/13 leads to RhoA activation, via RhoGEFs influencing cell hypertrophy. Gαq activates phospholipase C-beta, generating second messengers that trigger calcium release and PKC activation. PKC can further activate the NF-κB signaling pathway to upregulate production of SOX4 and ADAMTS5. Gαi can inhibit AC to regulate downstream cAMP, whereas Gαs can increase cAMP. β-arrestin can activate ERK1/2 signalling pathway however exhibits inhibitory effects on PKC and calcium release. Anticoagulants like dabigatran (thrombin inhibitor) and apixaban, edoxaban and rivaroxaban, (FXa inhibitors) can potentially disrupt this signaling by preventing PAR activation. Ultimately, these signal transduction pathways can trigger physiological changes like inflammatory and immune responses, cell hypertrophy, and cell migration.
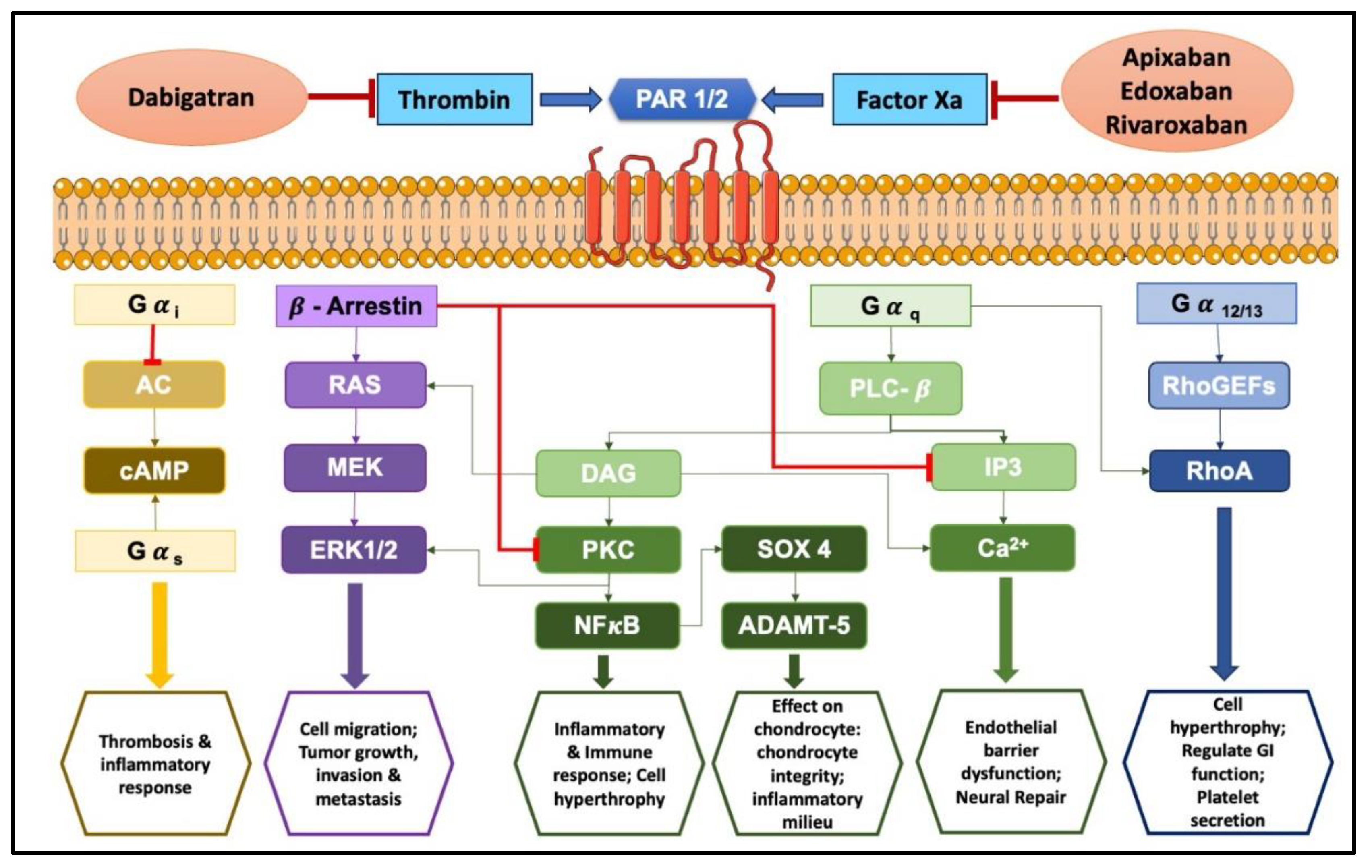
Figure 3.
Schematic Representation of PAR4-mediated Signal Transduction. PAR4 can activate signaling pathways involving Gα12/13 and Gαq. Gα12/13 prompts RhoGEFs to activate RhoA, while Gαq -PLC-b leads to downstream effects such as upregulation of IP3 and DAG, resulting in calcium alterations and PKC upregulation, which ultimately leads to activation of NF-κB signaling pathway. Additionally, β-arrestin can facilitate ERK1/2 phosphorylation but will have inhibitory effects on PKC. Thrombin, known for its ability to cleave PAR4, can influence these pathways, therefore dabigatran (thrombin inhibitor) can modulate signal transduction by attenuating thrombin’s effects. As seen, PAR4 activation can cause physiological alterations such as inflammatory and immune response, endothelial barrier dysfunction, and platelet activation.
Figure 3.
Schematic Representation of PAR4-mediated Signal Transduction. PAR4 can activate signaling pathways involving Gα12/13 and Gαq. Gα12/13 prompts RhoGEFs to activate RhoA, while Gαq -PLC-b leads to downstream effects such as upregulation of IP3 and DAG, resulting in calcium alterations and PKC upregulation, which ultimately leads to activation of NF-κB signaling pathway. Additionally, β-arrestin can facilitate ERK1/2 phosphorylation but will have inhibitory effects on PKC. Thrombin, known for its ability to cleave PAR4, can influence these pathways, therefore dabigatran (thrombin inhibitor) can modulate signal transduction by attenuating thrombin’s effects. As seen, PAR4 activation can cause physiological alterations such as inflammatory and immune response, endothelial barrier dysfunction, and platelet activation.
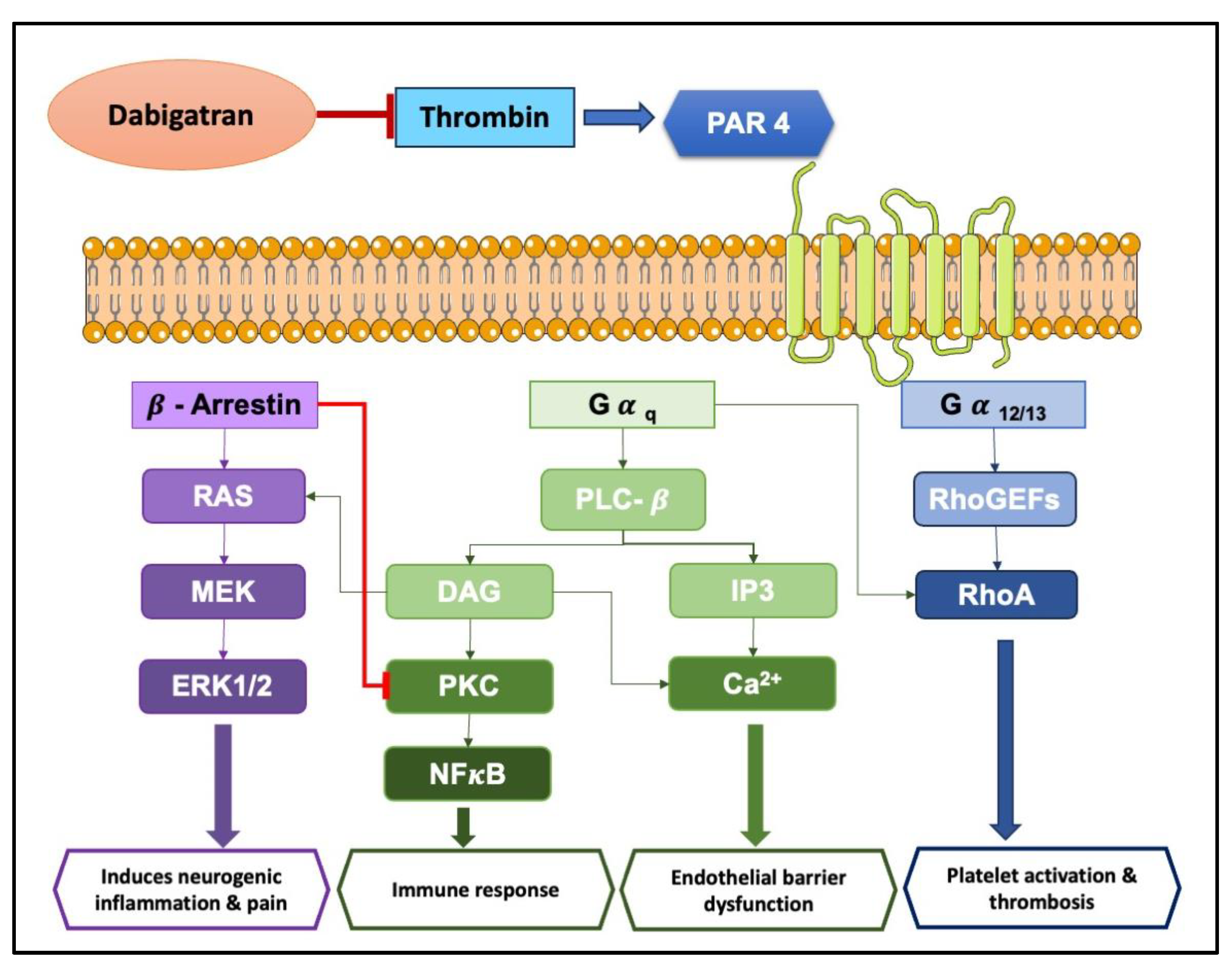
Figure 4.
Proposed Mechanism of Apixaban’s Modulatory Effects on Factor Xa and Associated Inflammatory Signalling Pathways in an Osteoarthritic Chondrocyte Model. This illustration delineates the pathways through which apixaban may exert anti-inflammation in the context of osteoarthritis. Apixaban targets FXa, inhibiting its ability to bind and activate PAR2, which is represented by the red inhibitory line. This intervention most likely attenuates the downstream signalling cascades involved in OA pathophysiology.
Figure 4.
Proposed Mechanism of Apixaban’s Modulatory Effects on Factor Xa and Associated Inflammatory Signalling Pathways in an Osteoarthritic Chondrocyte Model. This illustration delineates the pathways through which apixaban may exert anti-inflammation in the context of osteoarthritis. Apixaban targets FXa, inhibiting its ability to bind and activate PAR2, which is represented by the red inhibitory line. This intervention most likely attenuates the downstream signalling cascades involved in OA pathophysiology.

Table 1.
Pharmacokinetic and clinical profiles of NOACs.
| ANTICOAGULANT | APIXABAN | EDOXABAN | RIVAROXABAN | DABIGATRAN |
| Mechanism | Direct FXa Inhibitor | Direct FXa Inhibitor | Direct FXa Inhibitor | Direct Thrombin Inhibitor |
| Prodrug/absorption | No/3-4h | No/Rapid | No/Rapid | Yes/Rapid |
| Bioavailability/half-life | 50%/12h | 62%/9-11h | 66% w/o food up to 100% with food/5-9h (young) 11-13h (elderly) |
6%/12-17h |
| Vd | 21L | 107L | 50L |
50-70L |
| Time to reach max. Plasma conc./protein binding | 1-4h/87% |
1-2h/55% | 2-4h/92-95% | 0.5-2h/35% |
| Liver metabolism | Yes | Minimal | Yes |
No |
| Renal excretion | 25% | 50% | 35% |
80% |
| Effect of diet | No effect on exposure | No effect on exposure | Peak levels reached at 3h on fasting and 4h with food. |
Delayed absorption |
| Effect of age | Exposure is 32% greater in patients above 65 years | Exposure is 32% greater in patients above 65 years | Bioavailability greater in elderly with no difference in concentration |
2 times higher Bioavailability in elders |
| Effect of body weight | Weight < 50 kg have 20-30% increased exposure & weight >120 kg has 20-30% reduced exposure | Weight < 50 kg have 20-30% increased exposure & weight >120 kg has 20-30% reduced exposure | Weight < 50 kg have 20-30% increased exposure & weight >120 kg has 20-30% reduced exposure |
None |
| Effect of renal impairment | Peak concentration unaffected. However, there is a rise in exposure by 16%, 29%, and 44% corresponding to creatinine clearances of 51–80, 30–50, and 15–29 ml/min, respectively. | Peak concentration unaffected. However, there is a rise in exposure by 16%, 29%, and 44% corresponding to creatinine clearances of 51–80, 30–50, and 15–29 ml/min, respectively. |
Rise in exposure with moderate or severe renal impairment | 6 times higher exposure with severe renal impairment, half-life extended to 28h |
| Effect of hepatic impairment | No change in exposure with Child-Pugh classification A/B | No change in exposure with Child-Pugh classification A/B |
Increased on exposure with Child-Pugh classification B | No change in exposure with Child-Pugh classification B |
| Doses | 2.5 mg, 5 mg | 15 mg, 30 mg, 60mg | 2.5 mg, 10 mg, 15 mg, 20 mg |
75 mg, 110 mg, 150 mg |
| Dosing/dosing form | Two times a day/Tablet | One time a day/Tablet |
One time a day/Tablet | Two time a day/Capsule |
| ADR | >10% haematologic and oncologic haemorrhage; <10% haematuria, epistaxis; <1% hyper-sensitivity reactions | >10% haematologic and oncologic haemorrhage; <10% skin rash, anaemia; <1% intra cranial haemorrhage, interstitial pulmonary disease |
>10% haematologic and oncologic haemorrhage; <10% pruritus, abdominal pain; <1% angioedema cholestasis | >10% gastro-intestinal symptoms; <10% gastritis, esophagitis; <1% allergic oedema, thrombocytopenia |
| Pre- & post-operative care for minor surgical procedures | Suspend treatment for 2 days before the surgical procedure (meaning, skip 1 dose), and recommence 24 hours after the surgery | Suspend treatment for 2 days before the surgical procedure (meaning, skip 1 dose), and recommence 24 hours after the surgery |
Suspend treatment for 2 days before the surgical procedure (meaning, skip 1 dose), and recommence 24 hours after the surgery |
Creatine clearance greater than 50 ml/min suspend treatment for 2 days before the surgical procedure (meaning, skip 1 dose), and recommence 24 hours after the surgery |
| Pre- & post-operative care for major surgical procedures | Suspend treatment for 3 days before the surgical procedure (meaning, skip 4 doses), and recommence 48 hours after the surgery |
Suspend treatment for 3 days before the surgical procedure (meaning, skip 4 doses), and recommence 48 hours after the surgery |
Suspend treatment for 3 days before the surgical procedure (meaning, skip 2 doses), and recommence 48 hours after the surgery |
Creatine clearance greater than 50 ml/min suspend treatment for 3 days before the surgical procedure (meaning, skip 4 doses), and recommence 48 hours after the surgery |
| Laboratory monitoring (optimal method) | Anti-FXa assay | Anti-FXa assay | Anti-FXa assay | Ecarin clotting time; Dilute thrombin time |
| Laboratory monitoring (emergency) | Dilute Prothrombin Time | Limited substantial data | Prothrombin Time (preferably with specific calibrated reagents) | Activated Partial Thromboplastin Time (preferably with specific calibrated reagents) |
Table 2.
Baseline characteristics of the populations studied in atrial fibrillation NOACs trials, adopted from ref Ruff et al.
Table 2.
Baseline characteristics of the populations studied in atrial fibrillation NOACs trials, adopted from ref Ruff et al.
| ARISTOTLE | ENGAGE AF-TIMI 48 |
ROCKET-AF |
RE-LY | |||||||
| Apixaban | Warfarin | Edoxaban 60 mg | Edoxaban 30 mg | Warfarin | Rivaroxaban | Warfarin | Dabigatran 150 mg | Dabigatran 110 mg | Warfarin | |
| Population | 9120 | 9081 | 7035 | 7034 | 7036 | 7131 | 7133 | 6076 | 6015 | 6022 |
|
Age (years) >75 years |
70 31% |
70 31% |
72 41% |
72 40% |
72 40% |
73 43% |
73 43% |
71.5 40% |
71.4 38% |
71.6 39% |
| Women | 36% | 35% | 39% | 39% | 38% | 40% | 40% | 37% | 38% | 39% |
| Persistent AF | 85% | 84% | 75% | 74% | 75% | 81% | 81% | 67% | 68% | 66% |
| Previous stroke/TIA | 19% | 18% | 28% | 29% | 28% | 55% | 55% | 20% | 20% | 20% |
| Previous VKA use | 57% | 57% | 59% | 59% | 59% | 62% | 63% | 50% | 50% | 49% |
| Previous Aspirin use | 31% | 31% | 29% | 29% | 30% | 36% | 37% | 39% | 40% | 41% |
| Median follow-up (years) | 1.8 | 1.8 | 2.8 | 2.8 | 2.8 | 1.9 | 1.9 | 2.0 | 2.0 | 2.0 |
AF= Atrial Fibrillation. TIA= transient ischemic attack. VKA= vitamin K antagonist.
Table 3.
The molecular characteristics of different PAR members.
| Receptor | Amino acids | Tethered ligands | Cleavage type | Classical proteases | Cleavage site | Activating synthetic ligands |
| PAR1 | 425 | h: SFFLR m:SFLLR |
Canonical | Thrombin | ||
| Factor Xa | 38 LDPR*SFLL 45 | SFLLRN-NH2 | ||||
| Plasmin | ||||||
| MMP1 | 36 ATLD*PRSF 43 | PRSFLLRN-NH2 | ||||
| MMP13 | 39DPRS*FLLR46 | |||||
| Non-canonical | Elastase | 42SFLL*RNPN49 | RNPNDKYEPF-NH2 | |||
| Proteinase-3 | 33 ATNA*TLDp40 | TLDPRSF-NH2 | ||||
| Activated Protein C | 43FLLR*NPND50 | NPNDKYEPF-NH2 | ||||
| PAR2 | 395 | h: SLIGKV m: SLIGRL |
Canonical | Trypsin | Isox-Cha-Chg-AR-NH2 | |
| Mast cell Tryptase | 33SKGR*SLIG40 | SLIGKV-NH2 | ||||
| Factor Xa | ||||||
| Thrombin | SLIGRL-NH2 | |||||
| Elastase | 64FSAS*VLTG71 | |||||
| Non-Canonical | Proteinase-3 | 57VFSV*DEFS64 | ||||
| Cathepsin G | 61VDEF*SASV68 | |||||
| Cathepsin S | 53VTVE*TVFS60 | TVFSVDEFSA-NH2 | ||||
| PAR3 | 483 | h: TFRGAP m: SFNGGP |
Canonical | Thrombin | 35LPIK*TFRG42 | TFRGAP-NH2 |
| Non-Canonical | Activated protein C | 38 KTFR*GAPp45 | SFNGGP-NH2 |
|||
| PAR4 | 385 | h: GYPGQV m: GYPGKF |
Canonical | Thrombin | ||
| Trypsin | 44 PAPRIGYPG51 | GYPGQV-NH2 | ||||
| Cathepsin G |
Table 4.
The pro-inflammatory and anti-inflammatory role of PAR members in different inflammatory conditions.
Table 4.
The pro-inflammatory and anti-inflammatory role of PAR members in different inflammatory conditions.
| Inflammatory condition | Anti-inflammatory properties | Pro-inflammatory properties |
| Neuroinflammation |
|
|
| Pruritus and pain |
|
|
| Inflammatory bowel disease (IBS) |
|
|
| Asthma |
|
|
| Pneumonia |
|
|
| Myocarditis |
|
|
| Cystitis |
|
|
| Nephritis |
|
|
| Arthritis |
|
|
Table 5.
Anti-Inflammatory Profile of Apixaban.
| Model or Population | Mono/Combination therapy | Condition Investigated | Inflammatory Markers Examined | Effect observed | Reference |
|---|---|---|---|---|---|
| Inflammatory marker was measured at study entry, and at 2 months in 4830 patients in the ARISTOTLE trial with 1.8 years median follow-up. | Monotherapy | Atrial fibrillation | IL-6 | Repeated measurements of IL-6 suggest that persistent systemic inflammation is independently associated with increased mortality in apixaban treated AF patients, even after considering established clinical risk factors and other strong cardiovascular biomarkers. | Circulation. 2014 Nov 18;130(21):1847-58. |
| 18,201 patients with AF from the ARISTOTLE trial, out of which biomarkers were measured for 14,798 patients. | Monotherapy | Atrial fibrillation | GDF-15 | The effects of apixaban in reducing stroke, mortality, and bleeding were consistent irrespective of GDF-15 levels, but no data is shown if apixaban had any influence on the levels of GDF-15 to mitigate inflammation. | Heart. 2016 Apr;102(7):508-17.(***) |
| 18,201 patients with AF who were randomized to receive either apixaban or warfarin in the ARISTOTLE trial. | Monotherapy | Atrial fibrillation | hs-CRP and IL-6 | No Significant interactions were observed with respect to apixaban and IL-6 or CRP levels and their outcomes. | Heart. 2016 Apr;102(7):508-17. (***) |
| 14,753 patients from the Apixaban for Reduction In STroke and Other ThromboemboLic Events in Atrial Fibrillation (ARISTOTLE) trial. | Monotherapy | Atrial fibrillation | Hs-CRP and IL-6 | Obesity was found to be linked with better survival outcomes in apixaban treated patients. Given that obesity is often associated with chronic inflammation, it is possible that apixaban may have a modulatory role in dampening inflammation in obese, anticoagulated patients with AF. | Open Heart. 2018 Nov 1;5(2):e000908. |
| In vitro model: Glia cell lines (specifically microglial and astrocytic cell lines).In vivo animal model: Adult male mice, of similar genetic lineage as C57BL/6, developed by Institute of Cancer Research.Human model: Human plasma and cerebrospinal fluid (CSF), specifically, the CSF taken from viral meningoencephalitis patients and controls was analysed. | Combination therapy where apixaban individually and in combination with alpha-naphthylsulphonylglycyl-4-amidinophenylalanine piperidine (NAPAP) was used. | Measurement of activated protein C (APC) activity in the context of neural inflammation:
|
Conventional inflammatory markers were not assessed but APC titres were measured. APC modulates inflammation by:
|
Apixaban increases the specificity of APC activity, which in turn alludes to the possibility that APC’s anti-inflammatory effects are advantageously affected in the presence of apixaban. | Int J Mol Sci. 2020 Mar 31;21(7):2422. |
| Venovenous extracorporeal membrane oxygenation (n = 10) due to acute respiratory distress syndrome; and patients treated with venoarterial extracorporeal membrane oxygenation (n = 8) due to cardiocirculatory failure in the ICU of a university hospital. | Monotherapy | Coagulopathy associated with extracorporeal membrane oxygenation (ECMO) in patients with severe cardiocirculatory and/or respiratory failure. | Plasmatic coagulation and platelet aggregation. | Plasmatic coagulation and platelet aggregation were impaired before ECMO due to apixaban. | Crit Care Med. 2020 May;48(5):e400-e408. |
| Macrovascular endothelial cells: HUVEC (Human Umbilical Vein Endothelial Cells)Microvascular endothelial cells: HMEC (Human Microvascular Endothelial Cells) | Monotherapy | Protective role of apixaban in uremia caused by chronic kidney disease (CKD). | VCAM-1, ICAM-1, p38MAPK and p42/44 (also known as ERK1/2) | Apixaban reduced VCAM-1, ICAM-1, and VWF expression, normalized ROS levels and eNOS, and inhibited p38MAPK and p42/44 activation in endothelial cells exposed to uremic serum. | Cardiovasc Drugs Ther. 2021 Jun;35(3):521-532. |
Table 6.
Anti-Inflammatory Profile of Edoxaban.
| Model or Population | Mono/Combination Therapy | Condition Investigated | Inflammatory Markers Examined | Effect Observed | Reference |
|---|---|---|---|---|---|
| Male diabetic mice models, specifically eNOS+/+DM, eNOS+/-DM, and eNOS-/-DM, to investigate the effects of FXa inhibition. Effects of PAR2 absence on DN were assessed using F2rl1-/-; Ins2Akita/+; eNOS+/- mice and compared to F2rl1+/+; Ins2Akita/+; eNOS+/- mice. |
Monotherapy | Diabetic nephropathy | TGFbeta, PAI-1, Collagen Type I, Collagen Type IV, TNF-alpha, MCP-1, IL- 8 and Prostaglandin-endoperoxide Synthase 2 or COX-2 | Edoxaban ameliorated diabetic nephropathy by reducing the expression of key proinflammatory and profibrotic genes, as well as the expression of PAR1 and PAR2. | Arterioscler Thromb Vasc Biol. 2016;36(8):1525-1533. |
| Unilateral ureteral obstruction (UUO) mice (a renal tubulointerstitial fibrosis model) and data from the Food and Drug Administration Adverse Events Reporting System (FAERS) database | Monotherapy | Renal tubulointerstitial injury, which is associated with inflammation and is a major cause of chronic kidney disease (CKD). | PAR1, PAR2, Collagen type 1 and 3, Fibronectin, F4/80 (macrophage marker), TNF-alpha, IL-10, MCP1, IL1beta, TGFbeta, Alpha-smooth muscle actin. | Edoxaban suppressed the elevated levels of FX, PAR-1, and PAR-2, reduced fibrosis and extracellular matrix expression, and attenuated the upregulation of inflammatory molecules and macrophage infiltration in the UUO mouse model of renal tubulointerstitial injury. | Sci Rep. 2018;8(1):10858. |
| Immortalized proximal tubule epithelial cell line (HK-2) derived from normal adult human kidneys | Monotherapy | CKD | The study investigated markers of oxidative stress which is closely interrelated with inflammation. (Intracellular ROS, superoxide anion, peroxynitrite, radical scavenging activity for hydroxyl radicals and hydrogen peroxide. | Edoxaban through its FXa inhibitory and direct radical scavenging activity alleviates oxidative stress induced by FXa, thereby breaking the cycle between oxidative stress and inflammation. | Int J Mol Sci. 2019;20(17):4140. |
| Apolipoprotein E knockout (ApoE -/-) mice fed with cholesterol-rich diet categorized into three groups: a control (Co) group on the diet alone, a Warf group treated with warfarin and vitamin K1, and an Edo group treated with edoxaban. | Monotherapy and combination therapy approaches. Co group received cholesterol-rich diet only, Warf group received warfarin and vitamin K1, and the Edo group received edoxaban. |
Vascular remodelling, atherosclerosis and arteriogenesis. | Histological assessment of smooth muscle cells per collateral artery in both hind limbs; Frequency of perivascular macrophages in the ligated and non-ligated hind limb; Expression of inflammatory cytokines – IL6, MCP-1, IL1b and TNF-alpha. | Edoxaban did not exhibit significant effect on inflammation, specifically the mRNA expression of IL6, MCP-1, IL1b and TNF-alpha in the murine hindlimbs or the spleen remained unaltered. | Vascul Pharmacol. 2020;127:106661. (***) |
| Individuals aged ≥18 years, on continuous ART with maintained HIV RNA <200 copies/mL for 2+ years, having plasma D-dimer level ≥100 ng/mL, creatinine clearance ≥50 mL/min, and weight ≥60 kg, excluding those with recent VTE, contraindications to anticoagulant therapy, certain health conditions like prior stroke, and invasive cancer within the past year, among other criteria. | Monotherapy | Effects of edoxaban in relation to elevated risk of thrombotic events and pro-inflammatory state in the HIV-positive population. | Systemic inflammation – IL6 (high-sensitivity), IL1b and TNFr-1; Monocyte activation – sCD14 and sCD163; Vascular injury – sVCAM; Thrombin generation – D-dimer, TAT; Circulating microparticle procoagulant activity – MPTF, WBTF, functional phospholipid surface assay; immunophenotyping of cryopreserved PBMCs | Edoxaban did not significantly impact most inflammatory or immune activation markers, but it reduced specific coagulation markers like D-dimer and TAT and was associated with a decrease in effector memory T cells. | Open Forum Infect Dis. 2020;7(2):ofaa026. (***) |
| Male C57Bl/6 N mice aged 6-8 weeks subjected to a transient middle cerebral artery occlusion (tMCAO) procedure. Edoxaban was administered in two different doses across three dosing regimens, with a specific dose of 3.3 mg/kg used for tMCAO experiments. Additionally, the vitamin K antagonist (VKA) phenprocoumon was given at 0.3 mg/kg, three times prior to tMCAO, aiming to achieve INR between 2 and 3 during the tMCAO procedure. | Monotherapy where both drugs (edoxaban and phenprocoumon) were given via oral gavage using a gastric tube and were dissolved in 0.5% (w/v) methyl cellulose. | Effect of edoxaban on thrombin mediated inflammatory processes in ischemic stroke. | The inflammatory markers and pathways assessed included IL-1b, IL-6, TNF-alpha gene expression, invasion of immune cells (T cells, neutrophils, macrophages/microglia), and the stabilization of the blood-brain barrier (BBB) through tight junction protein expression and reduced Evans Blue extravasation. | Edoxaban reduced infarct volumes, improved neurological outcomes, enhanced blood-brain barrier function, and attenuated brain tissue inflammation, suggesting its potential protective effects in ischemic stroke. | Int J Mol Sci. 2021;22(18):9893 |
| Male Slc:Wistar rats were used in an LPS-induced microvascular thrombosis model. | Monotherapy | Effect of edoxaban on risk of thrombosis during infection, specifically focusing on lipopolysaccharide (LPS)-induced coagulopathy in rats, where the effect focused on microvascular thrombus formation in the context of this coagulopathy. | The study investigated microvascular thrombus formation in the liver and kidneys, focusing on inflammatory markers IL-6, TNF-alpha and MCP-1. | Edoxaban did not affect the levels of inflammatory cytokines, | J Thromb Thrombolysis. 2021;52(1):9-17. (***) |
| Patients with atrial fibrillation (AF) from the ENGAGE AF-TIMI 48 trial. This randomized trial compared edoxaban versus warfarin in these patients and followed them for a median of 2.8 years. | Monotherapy with either edoxaban or warfarin | Relationship between neutrophil-to-lymphocyte ratio (NLR) and clinical outcomes in AF patients. | NLR | Systemic inflammation (as reflected by NLR) is associated with adverse outcomes in AF patients, and that edoxaban offers protection against some of these outcomes regardless of the inflammatory state | Int J Cardiol. 2023;386:118-124. |
Table 7.
Anti-Inflammatory Profile of Rivaroxaban.
| Human Model or Population | Mono/Combination Therapy | Condition Investigated | Inflammatory Markers Examined | Effect Observed | Reference |
| Japanese subjects with non-valvular chronic AF undergoing anti-coagulation therapy, analysed using unbiased liquid chromatography/tandem mass spectroscopy and candidate multiplexed protein immunoassays. | Monotherapy either with rivaroxaban or warfarin. | Modulation of biologically-relevant plasma proteins in AF |
|
Compared with warfarin, rivaroxaban was associated with a greater increase in thrombomodulin and a trend towards a reduction in MMP-9 over 24 weeks. | Thromb Haemost. 2012;108(6):1180-1191. |
| Ex vivo samples of abdominal aortic aneurysms (AAA) with intraluminal thrombus from six patients. These samples were treated with and without rivaroxaban to assess its effects on inflammation and oxidative stress. Abdominal aortic samples from six organ donors were used as controls. | Monotherapy | AAA with intraluminal thrombus | IL-6, IL-10, MMP-9, nitric oxide synthase 2 and NADPH oxidase subunits (gp67-phox, gp91-phox, and gp47-phox) | Rivaroxaban reduced key inflammatory and oxidative stress markers in human AAA sites. | Br J Clin Pharmacol. 2017;83(12):2661-2670. |
| Ancillary analysis of the X-TRA study with AF patients having left atrial/left atrial appendage (LA/LAA) thrombus. | Monotherapy | Relationship between plasma biomarkers and left atrial thrombus resolution in AF patients using rivaroxaban. |
|
|
Ann Med. 2018 Sep;50(6):511-518 |
| 127 patients with non-valvular atrial fibrillation (NVAF) | Monotherapy (in comparison with warfarin) | AF and its association with inflammation | IL-2, IL-4, IL-10, TNF, IFN-γ, CCL5 (also known as RANTES), CXCL9 (also known as MIG), CCL2 (also known as MCP-1), CXCL10 (also known as IP-10), TGF-β1, ADAMTS13 GDF-15, sICAM-1, p-selectin lipocalin-2 (also known as NGAL), sVCAM-1, SAA |
|
Front Cardiovasc Med. 2020;7:114. |
| Male Wistar rats | Combination therapy of Sunitinib and Rivaroxaban. | Cardiotoxicity induced by Sunitinib. | Serum levels of Ca2+, Mg2+, Fe3+/Fe2+, lipid profiles, and cardiac enzymes. Additionally, measurements of oxidant/antioxidant balance gene and protein expressions in cardiac tissues. |
|
Cardiovasc Toxicol. 2020;20(3):281-290. |
| Patients with atrial fibrillation (AF) scheduled for cardioversion without adequate anticoagulation at baseline. | Rivaroxaban (monotherapy) vs. Vitamin-K antagonist (VKA) (monotherapy). | Effects of anticoagulation in patients with atrial fibrillation (AF) scheduled for cardioversion. |
|
|
TH Open. 2020 Jan 23;4(1):e20-e32. |
| Multi-centre, prospective, randomized, open-label trial involving 179 participants with type 2 diabetes and subclinical inflammation. | Monotherapy | Type 2 diabetes mellitus patients who had subclinical inflammation and were exhibiting stimulated coagulation, activated platelets, and endothelial dysfunction | hsCRP, IL-6, MCP-1, MMP-9 and sCD40L | Rivaroxaban displayed anti-inflammatory effect and improvement of endothelial function. | Diabetologia. 2021;64(12):2701-2712. |
| Observational, multicenter, prospective study involving newly diagnosed AF patients with CKD stages 3b - 4. | Monotherapy comparison of Rivaroxaban and Warfarin. | Effects on inflammation, progression of heart valve calcification, and kidney function in AF patients with CKD stages 3b - 4. | Plasma inflammatory mediators (measured via ELISA) and cytokine (IL-1b, IL-6, TGF-b, TNF-a) levels. |
|
Int J Cardiol. 2021 Dec 15;345:90-97. |
| Non-valvular atrial fibrillation (NVAF) patients treated with rivaroxaban or warfarin. | Monotherapy (rivaroxaban vs. warfarin). | Inflammation and endothelial activation in patients with AF. | Circulating pro-inflammatory extracellular vesicles (EVs) profiles, proteomics of enriched plasma EVs, and levels of soluble P-selectin. |
|
J Thromb Haemost. 2021;19(10):2583-2595. |
| Prospective, randomized study on 228 patients with venous thromboembolism (VTE) |
Combination therapy. Control group received conventional treatment (warfarin or rivaroxaban), whereas the rosuvastatin-intervention group received rosuvastatin 10 mg daily in addition to their conventional treatment. |
The impact of rosuvastatin on D-dimer and other inflammatory serum markers in VTE patients. | D-dimer, mean platelet volume (MPV), neutrophil-to-lymphocyte ratio, and platelet-to-lymphocyte ratio. |
|
Clin Cardiol. 2022;45(7):717-722. |
| Real-world patients with coronary artery disease (CAD) and/or peripheral artery disease (PAD). | Dual pathway inhibition (DPI) using low-dose rivaroxaban and aspirin. | Effect of DPI on plasma inflammation and coagulation markers among patients with CAD and/or PAD. | IL-6, CRP, lipoprotein-associated phospholipase A2, copeptin, and GDF-15. | At the 24-week follow-up, there was a significant reduction in IL-6 and fibrinogen levels and a significant increase in GDF-15. | J Cardiovasc Pharmacol. 2023 Feb 1;81(2):129-133. |
| Cell, Mice or Combined models | Mono/Combination therapy | Condition Investigated | Inflammatory Markers Examined | Effect observed | Reference |
| KK-A(y) mouse model of type 2 diabetes mellitus. | Monotherapy (Rivaroxaban at doses of 5 or 10mg/kg) | Leukocyte-endothelial interaction and microthrombus formation in the context of type 2 diabetes mellitus. | Leukocyte-endothelial interaction and microthrombus formation in the context of type 2 diabetes mellitus. |
|
Thromb Res. 2014 Feb;133(2):276-80 |
| LPS-activated monocytes and THP-1 cells (a human monocytic cell line). | Monotherapy (either rivaroxaban or fondaparinux) | Tissue factor (TF) exposure on activated monocytes and macrophages involved in thrombosis through activation of factor X and cytokine release. | TF expression, prothrombinase activity, cytokine release in cell supernatants (with specific focus on IL-8 and TNFα). |
|
Exp Hematol Oncol. 2014 Dec 17;3(1):30. |
| Apolipoprotein E-deficient (ApoE-/-) mice | Monotherapy | Atherogenesis | PAR-1 and PAR-2 receptors, MMP-9, MMP-13, COX-2, TNF-a, and in vitro experiments that evaluated mRNA expression of IL-1b and TNF-a in mouse macrophages. |
|
Atherosclerosis. 2015;242(2):639-646. |
| Human umbilical vein endothelial cells (in vitro) An ischemic hind limb mouse model (in vivo) |
Monotherapy | The role of FXa in inducing cell senescence and its effect on tissue inflammation and regeneration. | Senescence-associated β-galactosidase, IGFBP-5, EGR-1, p53, and p16INK4a (via RT-qPCR array) and expression of cytokines ((IL-1b, IL-6, MCP-1 and ICAM-1) |
|
Sci Rep. 2016 Oct 18;6:35580. |
| Mouse model with polyurethane catheters placed unilaterally into the external jugular vein (EJV). | Monotherapy (either rivaroxaban or vehicle). | Dysfunction of indwelling central venous catheters (CVC) due to tissue ingrowth or clotting. | Plasma MCP-1 levels, EJV MMP-9 levels, cell proliferation (anti-Ki67), macrophage infiltration (anti-MAC387). |
|
Thromb Res. 2016 Aug;144:106-12 |
| Rat model of brain ischaemia/reperfusion injury using male Wistar rats. | Monotherapy (prestroke anticoagulation with rivaroxaban). | Influence of prestroke anticoagulation with rivaroxaban on stroke severity and associated effects on thrombo-inflammation. | Thrombin/antithrombin complex, intracerebral thrombus formation, CD68-immunoreactivity, expression of cytokines (IL-1b, TNF-alpha, INF-gamma), and adhesion molecules (specifically ICAM-1 and VCAM-1). |
|
Thromb Haemost. 2016 Apr;115(4):835-43. |
| Female SJL/J mice immunized with PLP139-151 to induce autoimmune experimental encephalomyelitis (EAE). | Monotherapy (either warfarin or rivaroxaban). | Effects of anticoagulants (warfarin and rivaroxaban) on autoimmune experimental encephalomyelitis (EAE) as a model for multiple sclerosis. | Neurological deficit scores, histopathological analyses of inflammatory lesions in the spinal cord. |
|
J Neuroinflammation. 2017 Jul 28;14(1):152. |
| Human umbilical vein endothelial cells that natively express protease-activated receptor-1 and -2 | Monotherapy | Function of rivaroxaban in the inactivated coagulation cascade and its role in altering gene transcripts, especially those of pro-inflammatory genes, upon FXa stimulation. |
|
|
J Pharmacol Sci. 2017;133(3):156-161. |
| In vitro study using human atherosclerotic plaques from carotid endarterectomy and vascular smooth muscle cells (VSMC) for experimentation. | Monotherapy | Progression and mechanisms of atherosclerotic plaques, specifically the role of coagulation factor Xa (FXa) in inducing endothelial cell senescence. | PARs, IGFBP-5, p53, and other inflammatory cytokines (IL-1b, IL-6, MCP-1, IGFBP-5). |
|
Sci Rep. 2017 Dec 7;7(1):17172 |
|
Monotherapy | Role of coagulation in acute lung injury (ALI) and the effect of rivaroxaban on it. |
|
|
Am J Transl Res. 2018;10(8):2335-2349. |
| Transluminal femoral artery injury in C57BL/6 mice induced by a straight wire. | Monotherapy | The role of pharmacological blockade of FXa in attenuating neointima formation following wire-mediated vascular injury. | IL-1β, IL-6, TNF-α, stromal cell-derived factor (SDF)-1, TGF-β1, granulocyte-macrophage colony stimulating factor (GM-CSF) |
|
Eur J Pharmacol. 2018;820:222-228. |
| Ten-week-old male CL57/B6 mice subjected to transverse aortic constriction (TAC) surgery | Monotherapy | Atrial fibrillation and inflammatory atrial fibrosis | mRNA levels of TNF-α, IL-6, IL-1β, MCP-1, cardiac PAR-2 expression |
|
J Cardiol. 2018;71(3):310-319. |
| Wild-type (WT) and PAR-2-/- mice subjected to left anterior descending artery (LAD) ligation. | Monotherapy | Cardiac injury and heart failure post-LAD ligation. | IL-6, IL-1β, and MPO(marker for neutrophil infliltration) |
|
Thromb Res. 2018;167:128-134. (***) |
| Male Albino rats | Monotherapy | Liver fibrosis induced by carbon tetrachloride | TNF-α, IL-1β and hydroxyproline | Rivaroxaban restored the inflammatory markers associated with liver fibrosis. | J Biochem Mol Toxicol. 2019;33(5):e22287. |
| Murine model of ischemic cardiomyopathy (ICM) using SR-BI KO/ApoeR61h/h mice (Hypo E mice). | Monotherapy | Effects of FXa inhibitors on atherosclerosis and cardiac remodelling post-MI in Hypo E mice. |
|
|
J Atheroscler Thromb. 2019;26(10):915-930. |
| MI (myocardial infarction) induced in wild-type mice through permanent ligation of the left anterior descending coronary artery. | Monotherapy (rivaroxaban added to regular chow diet). | Protective effects of rivaroxaban against cardiac remodeling after MI. | mRNA expression levels of TNF-α, TGF-β, PAR-1, IL-1 β, IL-6, MCP-1, MMP-2 MMP-9, PAR-2, A-type natriuretic peptides, B-type natriuretic peptides, and phosphorylation of extracellular signal-regulated kinase. |
|
Circ Rep. 2020 Mar 4;2(3):158-166. |
| Wistar rats weighing 200-250 g. | Combination therapy (Rivaroxaban with Sunitinib). | Nephrotoxicity induced by Sunitinib. | TNF-α/NFk-B signaling pathways, Malondialdehyde, Catalase, Glutathione, Glutathione reductase, Caspase-3, IL-17, MCP-1, IKBα. | Rivaroxaban treatment restored altered levels of inflammatory markers and exhibited nephroprotective effects against Sunitinib-induced nephrotoxicity by inhibiting oxidative stress-induced apoptosis and inflammation. | J Thromb Thrombolysis. 2020 Aug;50(2):361-370. |
| Male rats where inflammation was induced post-rivaroxaban therapy using LPS. | Monotherapy | LPS-induced acute vascular inflammatory response. |
|
|
PLoS One. 2020;15(12):e0240669. |
| In vitro study using the tissue factor-expressing prostate carcinoma cell line, 22Rv1. Whole blood was also stimulated with lipopolysaccharide (LPS) or phorbol-myristate-acetate (PMA). |
Monotherapy comparisons of rivaroxaban, dalteparin, and tinzaparin. | Cancer-associated thrombosis (CAT) and the influence of myeloperoxidase (MPO) on anticoagulant activity. | Tumor cell-induced procoagulant activity, platelet aggregation, and the impact of the cationic leukocyte-derived enzyme, MPO. Thrombin generation in plasma supernatants from LPS- or PMA-stimulated whole blood was also measured. |
|
J Thromb Haemost. 2020 Dec;18(12):3267-3279. |
| Ang II-infused ApoE-/- mice and calcium chloride-induced AAA models, as well as human aortic endothelial cells. | Monotherapy | AAA | IL-6, IL-8, IL-1β, MCP-1, MMP-2 as well as adhesive molecules were examined in relation to FXa stimulation and rivaroxaban treatment. |
|
Vascul Pharmacol. 2021;136:106818. |
| Young adult male Wistar Albino type rats with surgically induced Achilles tendon injury followed by primary repair. | Monotherapy (rivaroxaban vs. nadroparin calcium vs. no medication) | Effects of antithrombotic-adjusted prophylactic doses on Achilles tendon healing | Inflammatory cells, capillary vessels, fibroblasts, degrees of inflammation, neovascularization, fibroblastic activity, and collagen fibre sequencing for histopathological evaluation. |
|
J Hand Microsurg. 2021;15(2):133-140. |
| The investigation consisted of two studies: Study 1: PAR2 deficient (PAR2-/-) and wild-type mice infused with angiotensin II (Ang II) or a vehicle. Study 2: Spontaneously hypertensive rats (SHRs) treated after 8h of right atrial rapid pacing. |
Monotherapy (either rivaroxaban, warfarin, or vehicle). | Role of PAR2 signalling in AF arrhythmogenesis and the potential ameliorating effect of rivaroxaban on atrial inflammation and AF prevention. | mRNA expression of collagen1 and collagen3, gene expression of inflammatory (TNF-α, MCP-1, TGF-β, Col1a1, Col31, F2R and F2l1) and fibrosis-related biomarkers in the atrium. |
|
Circ J. 2021;85(8):1383-1391. |
| Pilot, single-centre, randomized, double-blind, placebo-controlled, crossover study with subjects having sickle cell anaemia. | Monotherapy (either rivaroxaban or placebo). | Sickle cell disease (SCD) and its association with coagulation activation. | Thrombin-antithrombin complex, D-dimer, inflammatory (hs-CRP, IL-6, IL-2 and IL-8) and endothelial activation markers, measures of microvascular blood flow. |
|
Transfusion. 2021 Jun;61(6):1694-1698. (***) |
| Male Ldlr-/- mice fed a western-type diet to induce atherosclerosis. | Combination of aspirin (given in water) and rivaroxaban (given in the diet) compared to each agent alone. | Atherosclerosis in Ldlr-/- mice. | Expression of 55 proteins in the aorta and plasma (specific proteins not listed in provided information). |
|
Atherosclerosis. 2022 Mar;345:7-14 |
| LPS stimulation of PBMCs or citrate-anticoagulated whole blood. | Monotherapy with PACMA-31 or DMSO vehicle. | Regulation of TF in monocytes by Protein disulphide isomerase (PDI) and the effect of PACMA-31, a specific PDI inhibitor. | TF expression (antigen, procoagulant activity, mRNA), release of IL-6 and TNFα, and LPS-induced signaling pathways. |
|
Thromb Res. 2022;220:48-59. (***) |
| In vivo: Various strains of mice (wild-type and NLRP3 knockout) fed with standard chow or a high-fat diet. In vitro: examination with mice aortic endothelial cells (MAECs) and smooth muscle cells (MOVASs). |
Monotherapy | Therapeutic role of rivaroxaban in attenuating vascular lesions and dysfunction in type 2 diabetes mellitus mice | NLRP3 inflammasome activation, vascular tension, intima-media thickness, collagen deposition, PAR-1, PAR-2, mitogen-activated protein kinase (MAPK), NF-κB. |
|
J Cell Physiol. 2022 Aug;237(8):3369-3380. |
| ICH induced by collagenase injection into the striatum of wild type (C57BL/6J) anticoagulated mice (warfarin or rivaroxaban) and Mmp10 -/- mice. | Monotherapy using either prothrombin complex concentrate (PCC) or CM-352 (MMP-fibrinolysis inhibitor). | Intracranial hemorrhage (ICH) associated with oral anticoagulants (specifically rivaroxaban). | PAI-1, IL-6, neutrophil infiltration, thrombin-activatable fibrinolysis inhibitor (TAFI) activation. |
|
Thromb Haemost. 2022 Aug;122(8):1314-1325 |
Table 8.
Anti-Inflammatory Profile of Dabigatran.
| Human Model or Population | Mono/Combination therapy | Condition Investigated | Inflammatory Markers Examined | Effect observed | Reference |
| Patients who had elective orthopaedic surgical procedures, specifically total hip or knee arthroplasty. | Monotherapy (Dabigatran vs. low-molecular weight heparin) | Thromboembolic complications post-total hip or knee arthroplasty |
|
No significant difference in the incidence of local or systemic inflammatory complications between dabigatran and low-molecular weight heparin | Pol Orthop Traumatol. 2012 (***) |
|
Monotherapy | Impact of dabigatran on pro-inflammatory cytokines, growth factors, and chemokines; exploring its potential anti-inflammatory effects. | Angiogenin, CXCL1/GRO alpha, IL-1 beta/IL-1F2, human IL-6, IL-8/CXCL8, CCL2/MCP-1, TGF- beta 1, TNF-alpha, VEGF. |
In summary, the study suggests that dabigatran could have anti-inflammatory effects by reducing the expression of pro-inflammatory cytokines, growth factors, and chemokines
|
Life Sci. 2020 Dec 1;262:118474. |
| Cell, Mice or Combined models | Mono/Combination therapy | Condition Investigated | Inflammatory Markers Examined | Effect observed | Reference |
| Female C57BL/6 mice | Mono therapy |
|
|
|
Arthritis Rheum. 2011 May;63(5):1416-25. |
| Transgenic atherosclerosis-prone mice with diminished coagulant or hypercoagulable phenotype (FII(-/WT):ApoE(-/-) and TM(Pro/Pro):ApoE(-/-)) | Monotherapy (Dabigatran etexilate or recombinant activated protein C (APC)) |
|
|
|
PLoS One. 2013;8(2):e55784. |
|
Monotherapy (Dabigatran) |
|
|
|
Front Aging Neurosci. 2013 May 9;5:19. |
|
Monotherapy |
|
|
|
J Pharmacol Exp Ther. 2014 Nov;351(2):288-97. |
|
Monotherapy with the thrombin inhibitor dabigatran at a dosage of 1.2 g/kg/day. |
|
|
Conclusion: The study concludes that dabigatran, a thrombin inhibitor, effectively reduces vascular oxidative stress and inflammation.
|
Arch Med Sci. 2014 Feb 24;10(1):154-60. |
|
Monotherapy |
|
The mean lesion area in the aortic sinus and the innominate artery.
|
The study concluded that inhibition of thrombin with dabigatran etexilate retards the initiation and progression of atherosclerotic lesions in ApoE-/- mice. The treatment also improved endothelial function and reduced the accumulation of macrophages within the vascular wall.
|
Drug Des Devel Ther. 2015 Sep 10;9:5203-11. |
|
Monotherapy using the oral thrombin inhibitor dabigatran (10 mg/g) |
|
|
The study found that while dabigatran was successful in achieving systemic thrombin inhibitory activity similar to human trials, it did not significantly mitigate allergic lung inflammation triggered by HDM in a murine asthma model. Given the limited effects of dabigatran on the evaluated markers of inflammation and asthma pathology, the study suggests that dabigatran might not be a suitable candidate for clinical evaluation in asthma treatment.
|
Am J Physiol Lung Cell Mol Physiol. 2015 Oct 15;309(8):L768-75. (***) |
|
Monotherapy (Dabigatran etexilate (DE) or Warfarin) |
|
|
|
J Neuroimmunol. 2016 Aug 15;297:159-68. |
|
Monotherapy with dabigatran etexilate (DE) at a dosage of 15 mg/kg. |
|
|
Conclusion: The study concluded that prophylactic anticoagulation with dabigatran etexilate improves outcomes following ischemic stroke in rats by reducing thrombin-induced inflammation and thrombus formation.
|
Curr Neurovasc Res. 2016;13(3):199-206. |
| Vascular leak-dependent mice model for pulmonary fibrosis | Monotherapy |
|
|
In summary, dabigatran exhibited significant anti-inflammatory effects in a vascular leak-dependent mouse model for fibrotic lung disease.
|
JCI Insight. 2017 May 4;2(9):e86608. |
|
Monotherapy |
|
|
In summary, dabigatran treatment showed anti-inflammatory effects in a high-fat diet-induced obesity model in mice. It limited weight gain, reduced systemic inflammation, and decreased macrophage counts in adipose tissue, thereby potentially suppressing the progression of obesity-associated diseases.
|
J Clin Invest. 2017 Aug 1;127(8):3152-3166. |
|
Mono therapy (dabigatran administered either during the onset of fibrotic phase (late treatment) or inflammatory phase (early treatment)). |
|
|
|
Clin Exp Rheumatol. 2017 Sep-Oct;35 Suppl 106(4):35-39. (***) |
|
Monotherapy (Dabigatran) |
|
|
|
Acta Ophthalmol. 2018 Aug;96(5):452-458. |
|
MonotherapyGroup 1: Control (No treatment)Group 2: Dabigatran etexilate (10 mg/kg orally for 7 days)Group 3: Bemiparin sodium (250 IU/kg subcutaneously for 7 days) |
|
|
|
World Neurosurg. 2019 Jun;126:e731-e735. (***) |
|
Monotherapy |
|
|
|
Atherosclerosis. 2019 Aug;287:81-88. |
|
Combination therapy (Dabigatran with gentamicin) |
|
|
|
PLoS One. 2019 Apr 19;14(4):e0215333. |
|
Monotherapy (Dabigatran) |
|
|
|
J Thromb Haemost. 2019 Mar;17(3):538-550. |
|
Monotherapy (Dabigatran) |
|
|
|
Biochem Biophys Rep. 2020 Nov 19;24:100862. |
|
Monotherapy (Dabigatran) |
|
|
|
Front Mol Neurosci. 2020 Jun 30;13:114. |
|
- Monotherapy (Dabigatran etexilate) |
|
|
|
Vascul Pharmacol. 2020 |
|
Monotherapy |
|
|
|
Eur J Pharmacol. 2021 Feb 15;893:173838. |
|
Monotherapy |
|
|
|
Liver Transpl. 2021 Feb;27(3):363-384. |
|
Mono therapy (warfarin, dabigatran, and heparin are each used independently). |
|
|
|
Theranostics. 2021 Feb 20;11(9):4251-4261 |
|
Monotherapy Control (Western-type diet only) Dabigatran etexilate (duration: 6 or 18 weeks) Warfarin (duration: 6 or 18 weeks) |
|
|
|
J Thromb Haemost. 2021 May;19(5):1348-1363. |
|
Monotherapy (Dabigatran etexilate or Warfarin) |
|
|
|
J Thromb Haemost. 2021 May;19(5):1348-1363. |
| Retinoic acid (RA)-differentiated human neuroblastoma cell line SH-SY5Y | Monotherapy (250 nM Dabigatran with/without 10-100 nM Thrombin) |
|
|
|
Cereb Circ Cogn Behav. 2021 May 6;2:100014 |
|
Monotherapy |
|
|
In summary, elevated thrombin activity appears to contribute to tissue damage and dysfunction in Crohn’s disease. The study highlights the potential therapeutic benefits of inhibiting thrombin and protease-activated receptor-1 as a strategy for treating inflammatory bowel disease.
|
J Crohns Colitis. 2021 May 4;15(5):787-799. |
| Pig coronary artery stenting model | Combination therapy (dabigatran, aspirin, and clopidogrel) |
|
|
In summary, the short-term peri-interventional triple therapy with dabigatran, aspirin, and clopidogrel enhanced endothelium-dependent vasodilation and reduced thrombin generation and tissue burden, showing anti-inflammatory effects in a porcine model of coronary artery stenting.
|
Front Cardiovasc Med. 2021 Jul 2;8:690476. |
|
Monotherapy |
|
|
|
Front Pharmacol. 2022 Feb 28;13:834472(***) |
| Rat model of complete Freund’s adjuvant (CFA)-induced arthritis. | Monotherapy |
|
|
|
Int J Mol Sci. 2022 Sep 7;23(18):10297. |
| Mouse model (T7K24R trypsinogen mutant mouse and T7D23A mouse) | Monotherapy |
|
|
|
JCI Insight. 2022 Nov 8;7(21):e161145. |
| Pig stenting coronary artery model | Combination therapy: Dabigatran with dual antiplatelet therapy (DAPT) (clopidogrel 75 mg + aspirin 100 mg) |
|
|
|
J Pers Med. 2023 Jan 31;13(2):280 |
Table 9.
Comparative Anti-Inflammatory Profile of NOACs.
| Model or Population | NOACs used | Condition Investigated | Inflammatory Markers Examined | Effect observed | Reference |
|---|---|---|---|---|---|
| The study utilized the Berkeley (BERK) mouse model of Sickle Cell Disease (SCD) which has specific genetic modifications involving human and murine globins. For certain experiments, wild-type (WT) mice and PAR-1 and PAR-2 deficient mice were used. Additionally, bone marrow (BM) transplantation was conducted where irradiated PAR-1 and PAR-2 mice received bone marrow cells from either the BERK or WT control mice. |
Dabigatran and Rivaroxaban | Sickle Cell Disease (SCD) | IL-6, sVCAM-1 was assessed in relation to the activation of TF in sickle mice, MPO which is an enzyme primarily found in neutrophils and is released during the activation of neutrophils. |
|
Blood. 2014;123(11):1747-1756. |
| Human umbilical vascular endothelial cells (HUVECs) | Dabigatran and Rivaroxaban | Transcriptional changes in HUVECs when exposed to thrombin, focusing on the expression levels of preselected pro-inflammatory genes. | ELAM-1, VCAM-1, ICAM-1, MCP-1, IL-8, CXCL1, CXCL2 and TF |
|
Thromb Res. 2016 Jun;142:44-51 |
| 44 patients with acute ischemic stroke patients who were newly prescribed anti-thrombotic agents. | Dabigatran (n =12) and Apixaban (n =14) with antiplatelet agents (n-18) used as control. | Acute ischemic stroke | Hs-CRP, IL-6, pentraxin – 3 |
|
Clin Transl Med. 2018 Jan 12;7(1):2. |
| Polymorphonuclear leukocytes (PMNL) isolated from heparinized venous blood taken from non-smoking, healthy adults. | Dabigatran and Rivaroxaban | Inflammatory response by NOACs | reactive oxygen species (rOS), elastase release, cytosolic calcium flux, neutrophil extracellular trap (NET) formation, cell viability | No significant pro-inflammatory effects of dabigatran or rivaroxaban at concentrations of up to 10 µM was observed on the indicated PMNL markers. | Activation. Pharmaceuticals (Basel). 2018 May 14;11(2):46. |
| 187 patients with non-valvular atrial fibrillation (NVAF) | Dabigatran (n = 96) and Rivaroxaban (n= 91) | NVAF | Hs-CRP, pentraxin-3, IL-1β, IL-6, IL-18, TNF-α, MCP-1,GDF-15, and soluble thrombomodulin. |
|
Heart Vessels. 2019 Jun;34(6):1002-1013 |
| Male Wistar rats weighing between 250 to 350 g | Apixaban, Dabigatran and Rivaroxaban | Ischemia-reperfusion injury in the kidneys | IL-1β and TNF-α. |
|
Turk Gogus Kalp Damar Cerrahisi Derg. 2022 Apr 27;30(2):184-191. |
| Human umbilical vascular endothelial cells (HUVECs) | Dabigatran and Rivaroxaban | Inflammatory activation in endothelial cells caused by 25-hydroxycholesterol (25-OHC), which is associated with atherosclerosis. | Anti-inflammatory cytokines: TGF-β IL-37 IL-35 (specifically its subunits EBI3 and p35) Pro-inflammatory cytokines: IL-18 IL-23 |
25-OHC Effects:
|
Clin Exp Pharmacol Physiol. 2022 Aug;49(8):805-812. |
|
Animal model: 60 C57B/6J mice divided into groups: control (CON) group, atrial fibrillation (AF) group, AF + edoxaban group, and AF + rivaroxaban group. Human Model: Erythrocytes of patients with atrial fibrillation |
Edoxaban and Rivaroxaban | Atrial fibrillation | TNF-α, IL-1β, IL-6, and IL-10. | Both edoxaban and rivaroxaban reduced inflammation in atrial fibrillation, where the effect of edoxaban was superior to that of rivaroxaban. | Front Pharmacol. 2022;13:904317. |
| A syngeneic mouse model comprising male BALB/c mice that were inoculated with colon cancer Colon26 cells. | Dabigatran, Edoxaban and Rivaroxaban | Effect of NOACs on tumour progression in colorectal cancer. | Appraisal of tissue-factor, plasminogen activator inhibitor – 1, IL-6 and MMP-2 in the plasma of Colon26-inoculated mice. | All NOACs displayed anti-inflammatory effect, edoxaban displayed a superior ability not just to suppress tumor growth but also to mitigate elevated levels of inflammatory markers. | TH Open. 2023;7(1):e1-e13. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



