
Submitted:
07 June 2024
Posted:
21 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since the first reported case of COVID-19 in December 2019, several SARS-CoV-2 variants has evolved and some of them have shown higher transmissibility becoming the prevalent strains. Genomic epidemiological investigations into strains from different time points including the early stages of the pandemic are very crucial for understanding the evolution and transmission patterns. Using whole genome sequences, our study describes the early landscape of SARS-CoV-2 variants in central India retrospectively (including the first known occurrence of SARS-CoV-2 in Madhya Pradesh). We performed amplicon based whole genome sequencing of randomly selected SARS-CoV-2 isolates (n=38) collected between 2020-2022 at state level VRDL, ICMR-NIRTH, Jabalpur from 11899 RT-qPCR positive samples. We observed the presence of five lineages namely B.1, B.1.1, B.1.36.8, B.1.195 and B.6 in 19 genomes from the first wave cases and variants of concern (VOC) lineages i.e. B.1.617.2 (Delta), and BA.2.10 (Omicron) in the second wave cases. There was a shift in mutational pattern of SRAS-CoV-2 strains from the second wave in contrast to the first wave. We have identified five immune escape variants in the S gene: P681R, P681H, L452R, Q57H, and N501Y in the isolates collected during the second wave. Furthermore, these genomes were compared with 2160 complete genome sequences reported from central India that encompass 109 different SARS-CoV-2 lineages. Among them, VOC lineages Delta (28.93%), and Omicron (56.11%) were circulating predominantly in this region. This study provides useful insights into the genetic diversity of SARS-CoV-2 strains over the initial course of the COVID-19 pandemic in central India.
Keywords:
SARS-CoV-2
; Genetic diversity
; Whole genome sequencing
; Phylogenetic tree
; COVID-19
; Transmission
Introduction
Coronavirus Disease 2019 (COVID-19) is caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The first incidence of SARS-CoV-2 was reported from Wuhan City, China in late December 2019 and the first genome sequence was submitted on 7th January 2020 (NCBI GenBank ID: MN908947.1) [1]. World Health Organisation declared COVID-19 a public health emergency on 30th January 2020 and declared it a pandemic on 11th March 2020 [2]. India too had witnessed the increased incidence of distinct SARS-CoV-2 strains in different states since the beginning of the pandemic. By the end of 31st October 2023, it has been responsible for over 45 million cases and more than 0.53 million deaths in India [3]. SARS-CoV-2 was also found to show higher transmissibility as compared to previously reported coronavirus outbreaks i.e., SARS-CoV (2002) (WHO, 2003) and MERS-CoV (2012) (Zaki et al., 2012). On January 27, 2020, reports of the first COVID-19 case verified in India came from Kerala [4]. There are documented reports of various viral introductions brought about by international travelers. India, a country with a large population and densely populated cities, saw an enormous rise of COVID-19 cases in March 2020. It expanded to rural areas and among all age groups because of airborne viral particles and droplets transmission through aerosol, increased viral multiplication, and superspreading episodes caused by the migration of workers from larger cities. In the state of Karnataka, the B.1.1.7 (Alpha) lineage was found to be predominant among many other lineages [5], and symptomatic cases have been linked to the pandemic’s early spread in the state. Three primary introductions of SARS-CoV-2 were identified in the state of Kerala by genomic sequencing and haplotype analysis. These were followed by many outbreaks that caused the virus to spread locally [6]. In Gujarat state, lineages 20A and 20D were shown to be predominant among deceased patients with respiratory complications [7]. These lineages carry nucleotide changes C28854T in the N-gene and G25563T in Orf3a [15]. A collection of 200 genomes from the southern state of Telangana were sampled between March and July 2020 in order to assess the mutational landscape of SARS-CoV-2 isolates. This analysis helped to detect mutations in non-structural proteins and demonstrated the preponderance of the 20B lineage [8]. Central India is a loosely defined geographical region of India, sometimes called Madhya Bharat, that consists of two Indian states: Madhya Pradesh (MP) and Chhattisgarh. Over 1.05 million cases and approximately 10,786 deaths have been documented in this region till 30th October 2023 [9]. Since the first full genome of SARS-CoV-2 from Wuhan was reported in early January 2020 [4], millions of whole genome sequences from around the world have been added to public databases such as GISAID and GENBANK which helped in identifying the phylodynamics of specific variants and their genetic diversity. However, there has been a paucity of genomic data for SARS-CoV-2 from Central India that needed to be assessed to identify the epidemiology and the prevalence of different lineages of SARS-CoV-2 in this region. Thus, more such studies representing different time frames and geographic regions are needed to understand the evolving landscape of genetic diversity of the virus over the course of pandemic. Therefore, to better understand the spread of SARS-CoV-2 lineages outcomes in the first and second waves, we have sequenced the SARS-CoV-2 strains from the first reported cases in VRDL, ICMR-NIRTH Jabalpur and compared them with the genome sequences submitted in the GISAID database.
Materials and Methods
Sample Acquisition
A total of 11,899 samples tested positive from 2.61 lakh tested cases for SARS-CoV-2 using real-time (RT-PCR) and 38 samples among them underwent whole genome sequencing and mutational analysis for the SARS-CoV-2 variants.
Virus Research and Diagnostic Laboratory (VRDL) under ICMR-National Institute of Research in Tribal Health, Jabalpur is a designated State Level (Grade - II) laboratory under the VRDL network established by the Department of Health Research, Ministry of Health & Family Welfare, Government of India. It was the first lab in Madhya Pradesh (Central India) to start testing for suspected COVID-19 cases from Jabalpur and its surrounding districts of MP and Chhattisgarh. SARS-CoV-2 suspicious samples obtained from several quarantine camps and hospitals in Jabalpur area were processed for diagnostic testing in accordance with WHO recommendations.
RNA Extraction and Real-Time Reverse Transcriptase PCR (RT-PCR)
During the period (March 2020 to May 2022), nasopharyngeal/oropharyngeal swabs (NPS/OPS) (n = 263124) were collected for routine SARS-CoV-2 diagnosis. A total of 140 μL sample of the Virus Transport Medium (VTM) was used for viral RNA extraction using the QIAMP VIRAL RNA mini kit (Qiagen, Hilden, Germany). The SARS-CoV-2 was confirmed using ICMR-NIV multiplex single tube SARS-CoV-2 RT-PCR assay kit by real time RT-PCR. A total of 11899 samples tested positive by real-time RT-PCR. Samples that displayed a cycle threshold (Ct) between 20-28 were considered eligible for sequencing to ensure maximum sequence coverage. Among positive samples, n=19 samples from the very first reported cases of SARS-CoV-2 (Late March-April 2020) and had international travel history were selected. Subsequently, n=17 samples were randomly selected from the second wave and n=2 samples from the third wave were selected to see the presence of Variants of Concern (VOCs). The clinical and epidemiological details of the patients enrolled in the current study are shown in Table 1.
Library Preparation and Whole Genome Sequencing
The cDNA was subjected to a multiplex PCR using the QIAseq SARS-CoV-2 primer panel (Qiagen GmbH, Germany). Subsequently, amplified products were purified using Agencourt AMPure XP beads (Beckman Coulter Inc., TX USA) and quantified using QubitTM dsDNA HS Assay Kit (Invitrogen). The amplified cDNA sample was used for library preparation using the QIASeq FX DNA Library kit (Qiagen GmbH, Germany). The quality-checked normalized libraries were pooled and diluted to a final concentration of 10 pM, spiked with 5% PhiX. Whole genome sequencing of first wave (n=19) samples was performed on the Illumina iSeq platform using iSeq 100 i1 Reagent v2 (300-cycle) kit and subsequent samples of second and third waves were sequenced on the Ion GeneStudio S5 System platform using Ion 540™ Chip Kit.
Raw Read Processing, Genome Assembly and Lineage Assignment
The quality of the whole genome sequences raw (fastq) data was assessed by FastQC (version 0.11.5) [10]. Low-quality sequences were trimmed and filtered using Prinseq-lite (version 0.20.4) with a minimum read length of 30 base pairs [11]. The whole genome assembly of -1 SARS-CoV-2 sequences was obtained by mapping with the Wuhan-Hu genome (GenBank: NC_045512.3) using Burrows-Wheeler Aligner’s (BWA) tool (version 0.7.17). Samtools (version 1.6) and Seqtk (version 1.3) tools were used for generating the consensus sequences. The resultant SARS-CoV-2 genome sequences were compared with the reference genome (Wuhan-Hu-1/2019) to identify Nextstrain clade and Pangolin lineages and genomic variants [12,13].
Phylogenetic Analysis
We have downloaded all complete SARS-Cov-2 genomes (n=2160) (Table S1) from central India for phylogenetic analysis of SARS-CoV-2 genome sequences generated in this study were aligned with the strains reported from central India using Augur Nextstrain’s phylodynamic pipeline toolkit (https://github.com/nextstrain/ncov) [12,14]. Afterward, the maximum likelihood phylogenetic tree was constructed using the Augur tree implementation pipeline with the IQ-TREE v1.6.1 [15]with the default parameters. The selected metadata information plotted in the time-resolved phylogenetic tree was constructed using TreeTime v0.81[16], which was then annotated and visualized in the iTOL website [17]. The effect of mutation on the structure of the protein was predicted using iStable [18].
Furthermore, the metadata of the genomes belonging to the seven lineages identified in this study were downloaded from GISAID database till 31st March 2023 to check the transmission and circulation on a global scale.
Results
Genome Assembly, Lineage Assignment and Mutational Analysis
All these SARS-CoV-2 whole genome sequences mapped to the reference strain (Wuhan/Hu-1/2019, NC_045512.3) with 99.1-99.8% identity. The consensus assembly of 38 samples has been deposited in the BioProject ID: PRJNA759056 (Table S2). To analyse the mutation pattern, these 38 genomes were analysed with the Auspice visualization tool of Nextclade [19](Figure S1). Nextstrain clades and Pangolin classification system was used for classifying the phylogenetic lineages and their corresponding GISAID clade is shown in Table 2. These 38 SARS-CoV-2 of strains belong to seven lineages, namely: B.1.617 (Delta) (n=17), B.1.195 (n=8), B.1 (n=6), B.1.36.8 (n=2), BA.2.10 (Omicron) (n=2), B.1.1 (n=2) and B.6 (n=1). This revealed n=186 single nucleotide polymorphisms (n=129 non-synonymous and n=57 synonymous changes) shown in Table S3. Even though approx. half of these samples are from the early stage of the pandemic, it is interesting to note that only one strain (ICMR-NIRTH-S14, lineage B.6, clade O (19A), date of collection: 08 April 2020) didn’t show a well-characterized D614G mutation in spike protein (Table 2).
In total, 29 novel nucleotide variations in the sequences of SARS-CoV-2 genome have been identified from 38 patient isolates which were unique to the sample and not found in the genomes reported from the central India state as per the data retrieved from GISAID database.
Effect of Novel Mutations on Protein Stability
Using an integrated sequence and structure predictor, iStable [18], the effects of the novel 29 mutations identified were determined. Among 29 mutations, 20 mutations were only identified in the ORF1ab region which was known to encompasses mutational spectra. Out of these 29 mutations, 17 mutations were non-synonymous, 11 were synonymous and one mutation was found in the non-coding region. A total of 15 non-synonymous mutations were found to confer deleterious effects on protein function as they scored in a range between 0 and 1 (Table S4). As most of the mutations were found in ORF1ab region, it was interesting to note that the mutational spectra of this region should be considered while designing new antiviral therapeutics targeting viral ORF1ab.
Phylogenetic Analysis
A neighbor-joining tree was constructed using n=38 strains sequenced in this study (Figure 1) and their phylogenetic relationship along with 2160 SARS-CoV-2 is shown in Figure 2, where the genomes sequenced in this study are shown in beige highlight. All SARS-CoV-2 genomes reported so far from central India comprise 109 different SARS-CoV-2 Pangolin lineages. Importantly, these include B.1.1.7 (Alpha) (64/2160=2.96%); B.1.351 (Beta) (2/2160=0.09%), B.1.617.2+AY.* (Delta) (625/2160=28.93%) and BA.*+BM.1.*+XBB.* (Omicron) (1212/2160=56.11%) VOCs as per the data submitted in GISAID till 31st March 2023 from central India.
A cluster of 8 samples belonging to B.1.195 lineage (Figure 1) comprises three of the first four cases of Jabalpur (a family of 3 patients who had visited Dubai and tested positive on 20 March 2020) and their one contact (ICMR-NIRTH-S7, tested positive on 21 March 2020). The other three individuals in this cluster tested positive between 11-17 April 2020 and were also linked to each other. Another sample with a foreign travel history was ICMR-NIRTH-S3 (lineage B.1.1) which had returned from Germany and tested positive on 20 March 2020. We have made the analysis of the cluster but, this sample didn’t group with other strains reported from central India (Figure 2).
Another cluster representing B.1 lineage strains (n=6 cases: three each from Chhindwara and Indore districts which were epidemiologically linked) (Table 1). Two cases (ICMR-NIRTH-S1 and ICMR-NIRTH-S2) belonged to B.1.36.8 and had tested positive on the same day (27 March 2020). Sample ICMR-NIRTH-S14 (lineage B.6) belongs to a person who had traveled to Delhi and clusters together with other strains sequenced by the Defense Research and Development Establishment (DRDE), Gwalior (Figure 2).
The overall lineages distribution highlighted the dominant occurrence of B.1, B.1.36 and B.6 during the first wave, B.1.617.2 in the second and omicron in the third wave in central India.
Distribution of Circulating Lineages
We have investigated the global prevalence of the seven lineages by analyzing the genomes from GISAID and found that, together, North America and Europe shared nearly 86% load of B.1 and 83% of B.1.1; Europe and Asia shared nearly 82% load of Delta and Omicron VOCs. Though only 6.43% of genomes of B.1 were observed in Asia but among them, nearly half of (3.7%) of the genomes were submitted from India indicating the transmission of B.1 in this region. Interestingly, strains belonging to lineage B.1.195 have not been observed after 2021 whereas the lineages B1 and B.1.1 have been reported until very recently in the year 2022. In fact, in central India, B.1.1 was present in the year 2022 indicating that these strains were in circulation after the first report. B.1.36 was present in a small percentage during the first wave but surged during the second wave. Lineages B.6 and B.1.195, however, were prevalent in the first wave and an almost negligible number of genomes were reported after the first wave. Together, South America and Asia submitted about 85.3% of Lineage B.1.195 so far. Furthermore, Asia alone submitted 62.9% of lineage B.1.36.8 and 79.7% of B.6 SARS-CoV-2 sequences with most of the sequences coming from India (Figure 3).
Discussion
Since the first case of SARS-CoV-2 was discovered in the state of Kerala, India has been on alert for the spread of airborne viruses. To contain and stop the spread of viral transmission, the Ministry of Home Affairs (MHA) subsequently declared a nationwide lockdown that would last from March 25, 2020, until April 14, 2020. A single case regarding lineage B.1.195 (associated with migration) identified in this study may indicate that the initial period (March-April 2020) had effective containment measures, especially for those who had returned from abroad. This strain was associated with travel history to Dubai and was also closely related to another patients from Gujarat and Brazil. All these individuals had a travel history to Dubai around a similar time. However, the subsequent introduction of several lineages via interstate travel might have been the major driver of SARS-CoV-2 transmission in central India. This is supported by the fact that a significant genomic diversity was found during the initial WGS analyses of SARS-CoV-2 patients who travelled to India from other countries.
Another observation from the genomic analysis carried out in this study was that the earliest documented evidence of Delta variants in MP (GISAID ID: EPI_ISL_2461258, Lineage: AY.122) was in September 2020. However, the proportion of this variant among the newly detected strains after May 2021 has increased to over 90% in a short span of time. This indicates its selective advantage over the other circulating lineages [20]. Delta and Omicron variants have been reported to be more transmissible than the other variants and strong evidence is available which states that they were behind the emergence of second and third wave of COVID-19 in central India [21,22]. These observations suggested that the diverse genotypes of SARS-CoV-2 may have emerged as a process of convergent evolution. In addition, certain mutations may lead to changes in their surface antigenic structure whereby the antibodies due to previous infection/vaccination might not bind to them as effectively, exerting an ‘immune evasion’ advantage to them which can serve as a selection pressure and might associate with ‘breakthrough infections’
The D614G (37/38 strains) mutation has been shown to enhance the replication rate and transmissibility of SARS-CoV-2 by more efficient binding of SARS-CoV-2 spike protein to human angiotensin converting enzyme-2 (ACE2) receptors and subsequently increases viral entrance into host cells. Mutations in the spike can enhance viral transmission, disease severity, and the virus’s capacity to elude immune defensive responses [23].
Five immune escape mutations in the S gene at codon position other than D614G were L452R, P681R/H, Q57H, E484A, and N501Y during second and third waves. Substitution at positions P681 and E484 has become increasingly common among clinical isolates. Previous studies showed that the virulence and pathogenesis of the Delta variant could be impacted by D614G and P681R mutations [24]. Aside from D614G, the B.1.617.2 lineage, which has the spike protein mutations L452R and N501Y, may explain the transmission and surge in cases in central India in March 2021. Although the D614G mutation occurs in the Omicron variant, the additional presence of the P681H mutation may result in slow cleavage. Moreover, this may limit the Omicron virus replication to the upper respiratory tract resulting in less fusion and infectivity as compared to the Delta and D614G + P681R double mutants. A previous study reported that, 90.5% of the samples had the D614G mutation, which has recently been linked to higher viral loads and accelerated replication on human lung epithelial cells [25].
Among 29 mutations, 20 mutations were only identified in the ORF1ab region which was known to encompasses mutational spectra. ORF1ab is a conserved area in the SARS-CoV-2 genome[26]. Out of these 29 mutations, 17 mutations were nonsynonymous, 11 were synonymous and one mutation found in the non-coding region. By improving the virus’s adaptability to the utilization of human codons, synonymous SARS-CoV-2 mutations associated with the action of various mutational mechanisms may have a positive effect on viral evolution [26]. The non-synonymous mutation causes epitope loss which might be connected to immune evasion, which would increase viral pathogenicity and propagation [27].
However, after the emergence of new Omicron sub-lineage BA.5, it is now unknown if SARS-CoV-2 is shifting to a more gradual adaptive mechanism or if it will continue to evolve in this beneficial fashion with the frequent emergence of highly divergent lineages [28,29].
In 2022, multiple lineages emerging within BA.2, BA.5, Omicron were observed, in a more step-wise fashion, with several amino acid changes and moderate transmission advantages, which could indicate a shift to a more gradual stepwise evolution. The formation of VOCs and possible future antigenically different lineages may be viewed as ‘shift-like events,’ which are unanticipated, major alterations in the virus’s genetic make-up and, possibly, therapeutically relevant features [28,29]. It is impossible to foresee where future significant lineages will emerge from in viral genetic diversity and whether they will arise from ‘shift-like’ or more gradual, ‘drift-like’ progression similar to that seen in the Omicron clade during 2022 [29,30].
However, these limitations do not affect our conclusions about the genetic diversity and phylogenetic relatedness of the strains from central India. Overall, our study provides useful insights into the extant of genetic diversity of SARS-CoV-2 strains over the course of COVID-19 pandemic right from the very beginning to know its origin, dissemination and evolving pattern of genetic diversity.
Limitations of This Study
This study has few limitations as well: (i) we have sequenced only a limited number of samples from central India. (ii) Although we identified new mutations in SARS-CoV-2 genomes, we were unable to link them with phenotypic effects. Despite these limitations, our study provides valuable insights into the genetic diversity of SARS-CoV-2 infections in central India and its trajectory through the two waves of SARS-CoV-2 pandemic.
Conclusions
To our knowledge, this is the among the very few studies in which the molecular surveillance-based phylogenetic trends based on whole genome sequencing of SARS CoV-2 has been undertaken from central India. In this study we have performed WGS of 38 strains from Jabalpur and adjoining districts and compared them with over 2000 strains from various districts from central India representing different lineages. This study shows that the primary sources of COVID-19 introduction were those with a recent history of foreign travel in the affected countries. This was later on further amplified by inter-state migration, which subsequently led to emergence of newer strains.
The phylogenetic analysis of the SARS-CoV-2 strains in this study points to multiple introductions of the SARS-CoV-2 virus in central India. The genetic diversity, transmission, and evolution of SARS-CoV-2 exhibited a consistent pattern of increasing divergence within major lineages. Continued genomic surveillance strategies are needed for improved understanding of the SARS-CoV-2 pandemic. Furthermore, increased sequencing capacity is necessary for contact tracing and quicker identification of the transmission hotspots. This can be helpful for better preparedness, effective interventions and surveillance activity.
The COVID-19 phenomena show that continuous and sustained monitoring of pathogens using next generation sequencing is an useful tool for monitoring disease transmission and evolution patterns. The pandemic accelerated the capacity building at district level to enable molecular diagnostic with the capability of novel pathogen identification.
Author Contributions
PD performed the NGS experiments: MS, AA and AG and wrote the manuscript which was later updated and revised by SKR, KS and MK. AA and AG analysed the NGS data, whose interpretation was done by AA, MS and AG using bioinformatics tools. SCB, LKS, MU collected the samples and processed them. PVB, AD and PS conceptualised and supervised the study.
Funding
The study has been carried out with the support of Indian Council of Medical Research (ICMR). Lab of P.S. is supported by LRI Netherlands, R2STOP Canada, DBT (Ramalingaswami Fellowship Grants) and ICMR. Infrastructure and funding support from ICMR are also gratefully acknowledged. PD and MS are recipients of Senior Research Fellowship and Research Associateship, respectively from ICMR.
Data Availability Statement
All data generated or analysed during this study are included in this article. The raw data used to support the findings of this study have been deposited in the NCBI having BioProject ID: PRJNA759056
Acknowledgments
The contribution of entire team of Division of Virology and Zoonoses and all other staff of VRDL including Lab technicians, Data Entry Operators and other support staff is sincerely acknowledged for handling and processing of the patient samples and contributing to the diagnostic services amid the challenging times. The authors are thankful to: Dr SK Raghav for their helpful inputs in this manuscript; Sri Krishna, Sanchita Pacholi for their valuable technical assistance. We are also thankful to the patients and the state health au-thorities (clinicians and healthcare workers) for collecting patient samples and their associated epidemiological details. We gratefully acknowledge the GISAID database and all researchers who have submitted information and genomes to this valuable database.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. The Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Ho, Y.-C. SARS-CoV-2: A Storm Is Raging. J Clin Invest 2020, 130. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Cases | WHO COVID-19 Dashboard Available online:. Available online: https://data.who.int/dashboards/covid19/cases?n=c (accessed on 4 June 2024).
- Andrews, M.A.; Areekal, B.; Rajesh, K.R.; Krishnan, J.; Suryakala, R.; Krishnan, B.; Muraly, C.P.; Santhosh, P. V First Confirmed Case of COVID-19 Infection in India: A Case Report. Indian J Med Res 2020, 151, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, C.; Habib, F.; P, K.H.; Rasheed, R.; Prasad, P.; Reddy, V.; Dinesh, P.; Damodar, T.; Hosallimath, K.; George, A.K.; et al. Genomic Epidemiology Reveals Multiple Introductions and Spread of SARS-CoV-2 in the Indian State of Karnataka. PLoS One 2020, 15, e0243412. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, C.; Divakar, M.K.; Jain, A.; Viswanathan, P.; Bhoyar, R.C.; Jolly, B.; Imran, M.; Sharma, D.; Rophina, M.; Ranjan, G.; et al. Initial Insights Into the Genetic Epidemiology of SARS-CoV-2 Isolates From Kerala Suggest Local Spread From Limited Introductions. Front Genet 2021, 12, 630542. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Puvar, A.; Kumar, D.; Ansari, A.; Pandya, M.; Raval, J.; Patel, Z.; Trivedi, P.; Gandhi, M.; Pandya, L.; et al. Genomic Variations in SARS-CoV-2 Genomes From Gujarat: Underlying Role of Variants in Disease Epidemiology. Front Genet 2021, 12, 586569. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Sabarinathan, R.; Bala, P.; Donipadi, V.; Vashisht, D.; Katika, M.R.; Kandakatla, M.; Mitra, D.; Dalal, A.; Bashyam, M.D. A Comprehensive Profile of Genomic Variations in the SARS-CoV-2 Isolates from the State of Telangana, India. J Gen Virol 2021, 102. [Google Scholar] [CrossRef]
- MoHFW | Home Available online:. Available online: https://www.mohfw.gov.in/#latest-update (accessed on 6 June 2024).
- Andrew, S. %J F. Q.C. A Quality Control Tool for High Throughput Sequence Data. 2010, 532, 1. [Google Scholar]
- Schmieder, R.; Edwards, R. %J B. Quality Control and Preprocessing of Metagenomic Datasets. 2011, 27, 863–864. [Google Scholar]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. %J B. Nextstrain: Real-Time Tracking of Pathogen Evolution. 2018, 34, 4121–4123. [Google Scholar]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B. %J V. E. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. 2021, 30. [Google Scholar]
- Hadfield, J.; Megill, C.; Bell, S.M. Genomic Epidemiology of Novel Coronavirus: Global Subsampling. Nextstrain: real-time tracking of pathogen evolution.
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. %J M. biology; evolution IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. 2020, 37, 1530–1534. [Google Scholar]
- Kumar, S.; Stecher, G.; Suleski, M.; Blair Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. %J N. acids research Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. 2021, 49, W293–W296. [Google Scholar]
- Chen, C.W.; Lin, M.H.; Liao, C.C.; Chang, H.P.; Chu, Y.W. IStable 2.0: Predicting Protein Thermal Stability Changes by Integrating Various Characteristic Modules. Comput Struct Biotechnol J 2020, 18, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J Open Source Softw 2021, 6, 3773. [Google Scholar] [CrossRef]
- Brown, K.A.; Gubbay, J.; Buchan, S.A.; Daneman, N.; Mishra, S.; Patel, S.; Day, T. %J medRxiv Inflection in Prevalence of SARS-CoV-2 Infections Missing the N501Y Mutation as a Marker of Rapid Delta (B. 1.617. 2) Lineage Expansion in Ontario, Canada. 2021.
- Mohapatra, R.K.; Tiwari, R.; Sarangi, A.K.; Sharma, S.K.; Khandia, R.; Saikumar, G.; Dhama, K. Twin Combination of Omicron and Delta Variants Triggering a Tsunami Wave of Ever High Surges in COVID-19 Cases: A Challenging Global Threat with a Special Focus on the Indian Subcontinent. J Med Virol 2022, 94, 1761–1765. [Google Scholar] [CrossRef]
- Ray, S.K.; Mukherjee, S. Divulging Incipient SARS-CoV-2 Delta (B.1.617.2) Variant: Possible with Global Scenario. Infect Disord Drug Targets 2022, 23, 35–38. [Google Scholar] [CrossRef]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional Importance of the D614G Mutation in the SARS-CoV-2 Spike Protein. Biochem Biophys Res Commun 2021, 538, 108–115. [Google Scholar] [CrossRef]
- Khatri, R.; Siddqui, G.; Sadhu, S.; Maithil, V.; Vishwakarma, P.; Lohiya, B.; Goswami, A.; Ahmed, S.; Awasthi, A.; Samal, S. Intrinsic D614G and P681R/H Mutations in SARS-CoV-2 VoCs Alpha, Delta, Omicron and Viruses with D614G plus Key Signature Mutations in Spike Protein Alters Fusogenicity and Infectivity. Med Microbiol Immunol 2023, 212, 103–122. [Google Scholar] [CrossRef]
- Franceschi, V.B.; Caldana, G.D.; de Menezes Mayer, A.; Cybis, G.B.; Neves, C.A.M.; Ferrareze, P.A.G.; Demoliner, M.; de Almeida, P.R.; Gularte, J.S.; Hansen, A.W.; et al. Genomic Epidemiology of SARS-CoV-2 in Esteio, Rio Grande Do Sul, Brazil. BMC Genomics 2021, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E. V Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc Natl Acad Sci USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.M.; Chakrabarti, J.; Mandal, S. Non-Synonymous Mutations of SARS-CoV-2 Leads Epitope Loss and Segregates Its Variants. Microbes Infect 2020, 22, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Markov, P. V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nature Reviews Microbiology 2023 21:6 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.D.; et al. The Origins and Potential Future of SARS-CoV-2 Variants of Concern in the Evolving COVID-19 Pandemic. Current Biology 2021, 31, R918–R929. [Google Scholar] [CrossRef]
- Bano, I.; Sharif, M.; Alam, S. Genetic Drift in the Genome of SARS COV-2 and Its Global Health Concern. J Med Virol 2022, 94, 88–98. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic analysis of 38 SARS-COV-2 genomes sequenced at ICMR-NIRTH with the reference genome (NC_045512) hCoV19/Wuhan/WH01/2019. The lineage distribution is depicted in different colours.
Figure 1.
Phylogenetic analysis of 38 SARS-COV-2 genomes sequenced at ICMR-NIRTH with the reference genome (NC_045512) hCoV19/Wuhan/WH01/2019. The lineage distribution is depicted in different colours.
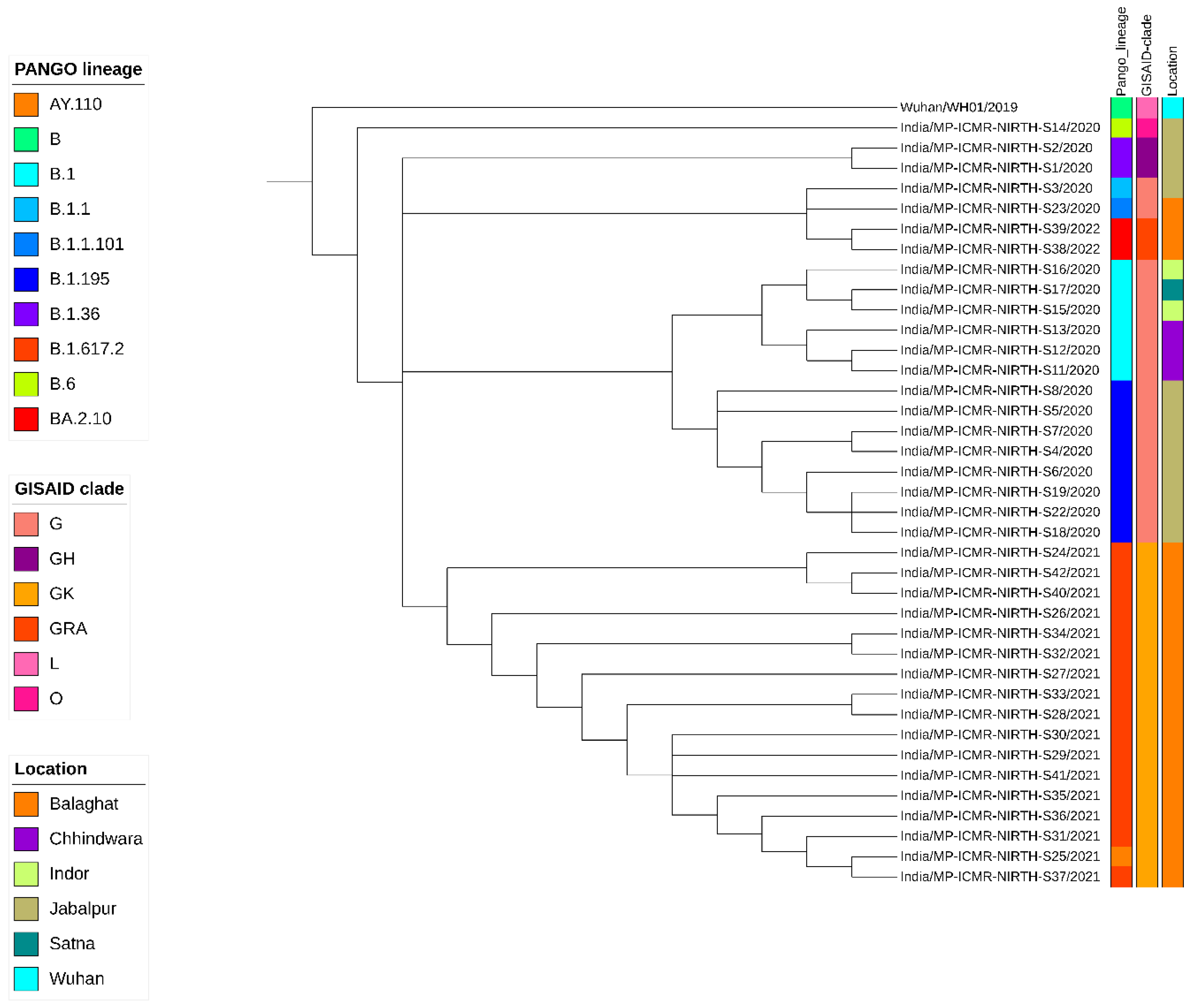
Figure 2.
Phylogenetic analysis of 38 SARS-COV-2 genomes sequenced at ICMR-NIRTH with the 2160 genomes obtained from GISAID. Classification of the genome sequences according to the Pangolin lineages, Nextstrain, and GISAID clade are shown in colour. The labels corresponding to the ICMR-NIRTH genome sequences generated during this study are highlighted and have been marked with light yellow colour.
Figure 2.
Phylogenetic analysis of 38 SARS-COV-2 genomes sequenced at ICMR-NIRTH with the 2160 genomes obtained from GISAID. Classification of the genome sequences according to the Pangolin lineages, Nextstrain, and GISAID clade are shown in colour. The labels corresponding to the ICMR-NIRTH genome sequences generated during this study are highlighted and have been marked with light yellow colour.
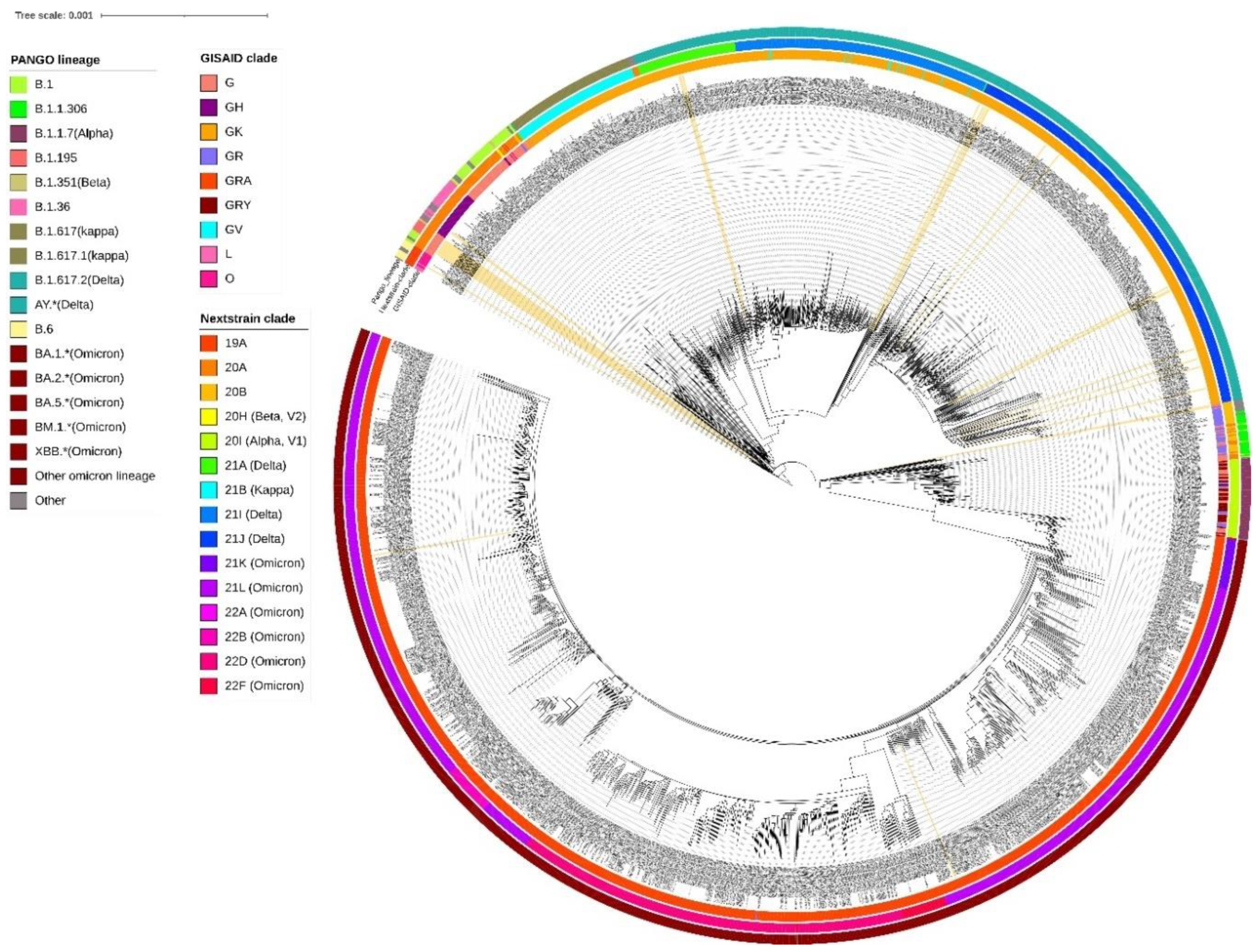
Figure 3.
Global distribution of 5 lineages of SARS-CoV-2 from GISAID database till August 2022. A) Month-wise distribution of lineages from worldwide submitted data. B) Distribution of lineages across different continents regions of the world. C) Comparison of lineages present in Central Indian states with other Indian states and Asian countries.
Figure 3.
Global distribution of 5 lineages of SARS-CoV-2 from GISAID database till August 2022. A) Month-wise distribution of lineages from worldwide submitted data. B) Distribution of lineages across different continents regions of the world. C) Comparison of lineages present in Central Indian states with other Indian states and Asian countries.
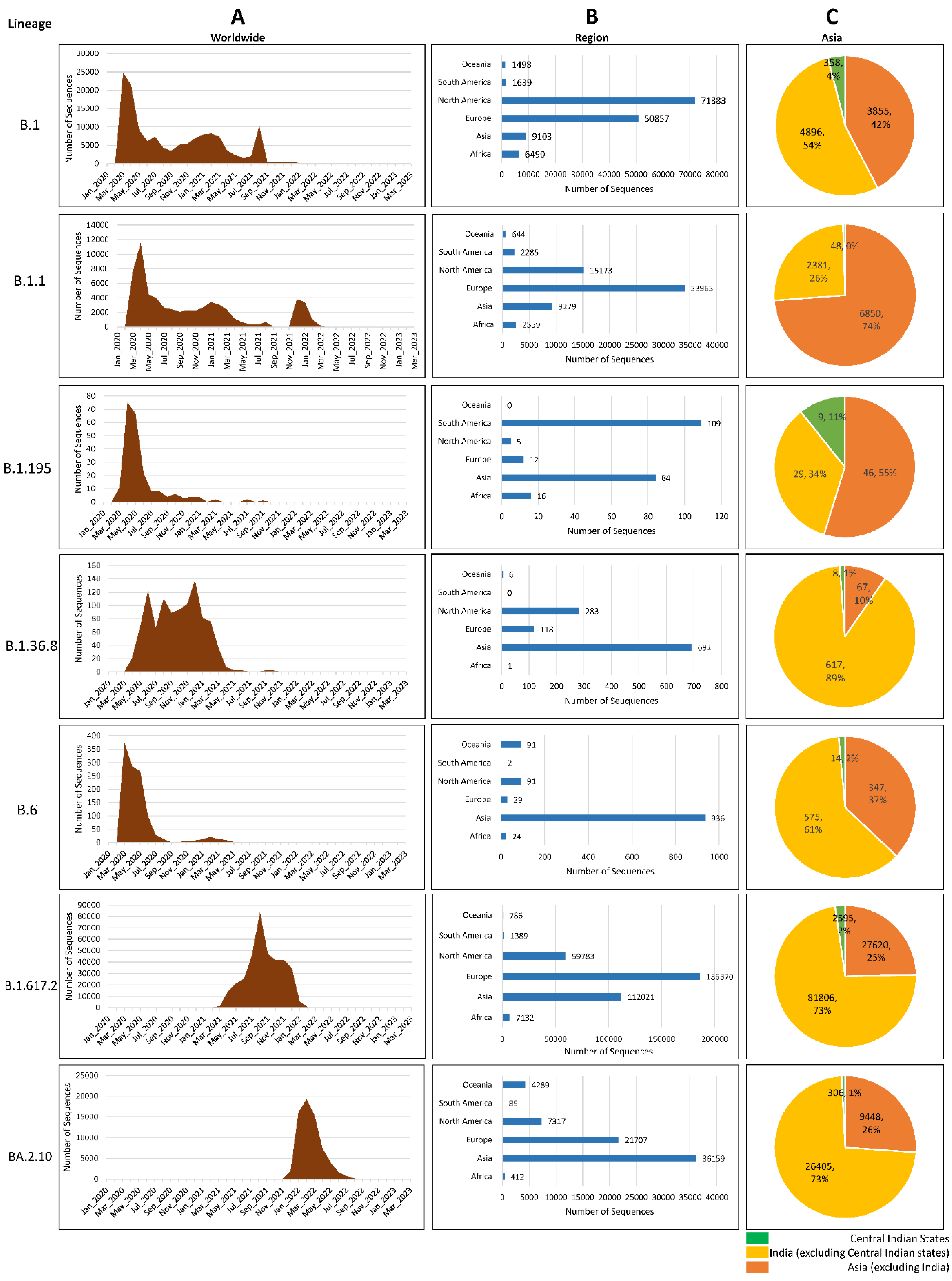

Table 1.
Epidemiological features of laboratory confirmed COVID-19 cases.
| Sample-ID | Age (in years) | Gender | District of Residence | Travel History/ Contact | Date of Sample Collection | Sample Type | Symptoms |
|---|---|---|---|---|---|---|---|
| ICMR-NIRTH-S1 | 46 | M | JABALPUR | NA | 2020-03-27 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S2 | 48 | M | JABALPUR | NA | 2020-03-27 | Oropharyngeal swab | fever |
| ICMR-NIRTH-S3 | 24 | M | JABALPUR | Germany | 2020-03-20 | Nasopharyngeal swab | fever cough |
| ICMR-NIRTH-S4 | 59 | M | JABALPUR | Dubai,UAE | 2020-03-20 | Nasopharyngeal swab | fever cough |
| ICMR-NIRTH-S5 | 45 | F | JABALPUR | Dubai,UAE | 2020-03-20 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S6 | 22 | F | JABALPUR | Dubai,UAE | 2020-03-20 | Nasopharyngeal swab | cough |
| ICMR-NIRTH-S7 | 53 | M | JABALPUR | Dubai(Contact of ICMR-NIRTH-S4) | 2020-03-21 | Nasopharyngeal swab | cough |
| ICMR-NIRTH-S8 | 40 | M | JABALPUR | NA | 2020-03-22 | Nasopharyngeal swab | cough bodyache |
| ICMR-NIRTH-S11 | 33 | M | CHHINDWARA | Indore travel history | 2020-04-01 | Nasopharyngeal swab | fever cough breathlessness sore throat body ach |
| ICMR-NIRTH-S12 | 26 | F | CHHINDWARA | Delhi travel history | 2020-04-07 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S13 | 27 | M | CHHINDWARA | Contact of ICMR-NIRTH-S12 | 2020-04-07 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S14 | 61 | M | JABALPUR | Delhi travel history | 2020-04-08 | Nasopharyngeal swab | Fever cough breathlessness body ach sputum |
| ICMR-NIRTH-S15 | 30 | M | INDORE | Indore travel history | 2020-04-10 | Nasopharyngeal swab | fever cough sore throat |
| ICMR-NIRTH-S16 | 22 | M | INDORE | Indore travel history | 2020-04-10 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S17 | 24 | M | SATNA | Indore travel history | 2020-04-10 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S18 | 70 | M | JABALPUR | NA | 2020-04-11 | Nasopharyngeal swab | fever cough sore throat |
| ICMR-NIRTH-S19 | 50 | M | JABALPUR | Contact of ICMR-NIRTH-S18 | 2020-04-14 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S22 | 44 | F | JABALPUR | Contact of ICMR-NIRTH-S19 | 2020-04-17 | Throat swab | cough sore throat |
| ICMR-NIRTH-S23 | 35 | M | BALAGHAT | NA | 2020-07-27 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S24 | 28 | F | BALAGHAT | NA | 2021-04-16 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S25 | 28 | M | BALAGHAT | NA | 2021-04-16 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S26 | 24 | M | BALAGHAT | NA | 2021-04-17 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S27 | 24 | M | BALAGHAT | NA | 2021-04-18 | Throat swab | Symptomatic |
| ICMR-NIRTH-S28 | 38 | M | BALAGHAT | NA | 2021-04-19 | Throat swab | Symptomatic |
| ICMR-NIRTH-S29 | 25 | M | BALAGHAT | NA | 2021-04-19 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S30 | 19 | M | BALAGHAT | NA | 2021-04-20 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S31 | 14 | M | BALAGHAT | NA | 2021-04-09 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S32 | 45 | M | BALAGHAT | NA | 2021-04-09 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S33 | 70 | F | BALAGHAT | NA | 2021-04-12 | Throat swab | Symptomatic |
| ICMR-NIRTH-S34 | 54 | M | BALAGHAT | NA | 2021-04-13 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S35 | 41 | M | BALAGHAT | NA | 2021-04-18 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S36 | 18 | M | BALAGHAT | NA | 2021-04-21 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S37 | 45 | F | BALAGHAT | NA | 2021-04-24 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S38 | 26 | F | BALAGHAT | NA | 2022-02-03 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S39 | 40 | F | BALAGHAT | NA | 2022-02-04 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S40 | 40 | M | BALAGHAT | NA | 2021-04-20 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S41 | 25 | M | BALAGHAT | NA | 2021-04-21 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S42 | 16 | F | BALAGHAT | NA | 2021-04-27 | Throat swab | Asymptomatic |
| NA= Contact history Not Available | |||||||
Table 2.
Pangolin lineage, GISAID clade and Nextstrain clade information of 38 SARS-COV-2 genomes sequenced in this study.
Table 2.
Pangolin lineage, GISAID clade and Nextstrain clade information of 38 SARS-COV-2 genomes sequenced in this study.
| Sr No | Sample ID | Date of Sample Collection | GISAID Clade | Nextstrain Clade | Pangolin lineage | WHO name |
|---|---|---|---|---|---|---|
| 1 | ICMR-NIRTH-S1 | 2020-03-27 | GH | 20A | B.1.36.8 | unassigned |
| 2 | ICMR-NIRTH-S2 | 2020-03-27 | GH | 20A | B.1.36.8 | unassigned |
| 3 | ICMR-NIRTH-S3 | 2020-03-20 | G | 20B | B.1.1 | unassigned |
| 4 | ICMR-NIRTH-S4 | 2020-03-20 | G | 20A | B.1.195 | unassigned |
| 5 | ICMR-NIRTH-S5 | 2020-03-20 | G | 20A | B.1.195 | unassigned |
| 6 | ICMR-NIRTH-S6 | 2020-03-20 | G | 20A | B.1.195 | unassigned |
| 7 | ICMR-NIRTH-S7 | 2020-03-21 | G | 20A | B.1.195 | unassigned |
| 8 | ICMR-NIRTH-S8 | 2020-03-22 | G | 20A | B.1.195 | unassigned |
| 9 | ICMR-NIRTH-S11 | 2020-04-01 | G | 20A | B.1 | unassigned |
| 10 | ICMR-NIRTH-S12 | 2020-04-07 | G | 20A | B.1 | unassigned |
| 11 | ICMR-NIRTH-S13 | 2020-04-07 | G | 20A | B.1 | unassigned |
| 12 | ICMR-NIRTH-S14 | 2020-04-08 | O | 19A | B.6 | unassigned |
| 13 | ICMR-NIRTH-S15 | 2020-04-10 | G | 20A | B.1 | unassigned |
| 14 | ICMR-NIRTH-S16 | 2020-04-10 | G | 20A | B.1 | unassigned |
| 15 | ICMR-NIRTH-S17 | 2020-04-10 | G | 20A | B.1 | unassigned |
| 16 | ICMR-NIRTH-S18 | 2020-04-11 | G | 20A | B.1.195 | unassigned |
| 17 | ICMR-NIRTH-S19 | 2020-04-14 | G | 20A | B.1.195 | unassigned |
| 18 | ICMR-NIRTH-S22 | 2020-04-17 | G | 20A | B.1.195 | unassigned |
| 19 | ICMR-NIRTH-S23 | 2020-07-27 | G | 20B | B.1.1.101 | unassigned |
| 20 | ICMR-NIRTH-S24 | 2021-04-16 | GK | 21A | B.1.617.2 | Delta |
| 21 | ICMR-NIRTH-S25 | 2021-04-16 | GK | 21J | B.1.617.2 | Delta |
| 22 | ICMR-NIRTH-S26 | 2021-04-17 | GK | 21J | B.1.617.2 | Delta |
| 23 | ICMR-NIRTH-S27 | 2021-04-18 | GK | 21J | B.1.617.2 | Delta |
| 24 | ICMR-NIRTH-S28 | 2021-04-19 | GK | 21J | B.1.617.2 | Delta |
| 25 | ICMR-NIRTH-S29 | 2021-04-19 | GK | 21J | B.1.617.2 | Delta |
| 26 | ICMR-NIRTH-S30 | 2021-04-20 | GK | 21J | B.1.617.2 | Delta |
| 27 | ICMR-NIRTH-S31 | 2021-04-09 | GK | 21J | B.1.617.2 | Delta |
| 28 | ICMR-NIRTH-S32 | 2021-04-09 | GK | 21J | B.1.617.2 | Delta |
| 29 | ICMR-NIRTH-S33 | 2021-04-12 | GK | 21J | B.1.617.2 | Delta |
| 30 | ICMR-NIRTH-S34 | 2021-04-13 | GK | 21J | B.1.617.2 | Delta |
| 31 | ICMR-NIRTH-S35 | 2021-04-18 | GK | 21J | B.1.617.2 | Delta |
| 32 | ICMR-NIRTH-S36 | 2021-04-21 | GK | 21J | B.1.617.2 | Delta |
| 33 | ICMR-NIRTH-S37 | 2021-04-24 | GK | 21J | B.1.617.2 | Delta |
| 34 | ICMR-NIRTH-S38 | 2022-02-03 | GRA | 21L | BA.2.10 | Omicron |
| 35 | ICMR-NIRTH-S39 | 2022-02-04 | GRA | 21L | BA.2.10 | Omicron |
| 36 | ICMR-NIRTH-S40 | 2021-04-20 | GK | 21A | B.1.617.2 | Delta |
| 37 | ICMR-NIRTH-S41 | 2021-04-21 | GK | 21J | B.1.617.2 | Delta |
| 38 | ICMR-NIRTH-S42 | 2021-04-27 | GK | 21A | B.1.617.2 | Delta |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



