
Submitted:
20 June 2024
Posted:
21 June 2024
You are already at the latest version
Abstract
Visible-light photocatalysis, particularly in its application to organic synthesis, has experienced a big renaissance in the past decade. The rational design of new photocatalysts with designed and improved photocatalytic properties, that at the same time circumvents the necessity of using scarce and toxic transition metals, will predictably play a key role in photocatalysis in the next few years. Although free-base porphyrins were identified some years ago as a promising, easily accessible and easily tunable class of organic photoredox catalysts, a systematic study on the effect of the electronic nature and of the position of the substituents on both the ground-state and the excited-state redox potentials of these compounds is still lacking. We have prepared a set of known functionalized derivatives of meso-tetraphenyl porphyrin containing different substituents either in one of the meso-positions or at a beta-pyrrole carbon and we have determined their ground- and (singlet) excited-state redox potentials. This survey has led us to conclude that the introduction of a carbonyl group at the beta-pyrrolic position strongly increases the facility of the monoelectronic reduction of the porphyrin both in the ground state and in the singlet excited state. We have also prepared a second set of four previously unknown benzoyl derivatives of meso-tetraphenylporphyrin, and we have confirmed the validity of our conclusions.
Keywords:
photosensitizers
; photoredox catalysis
; porphyrins
; redox potential
; substituent effects.
1. Introduction
Visible-light photocatalysis has become an important and well-established synthetic method for the construction of complex molecular architectures, thanks to the renaissance of the field brought about by the influential work of MacMillan [1], Yoon [2], and Stephenson [3], among others [4]. Visible-light photoreactions are made possible by the existence of photocatalysts (PCs), compounds that are promoted to their electronic excited states through irradiation with light; the resulting highly energetic species can exchange energy with surrounding molecules that do not absorb visible radiation (photosensititization) [4d] or engage in single-electron transfer (SET), leading to photoredox catalysis [4a,4b,4e-f]. Most molecular PCs are inorganic or organometallic complexes of Ru(II), Ir(III) or Cu(I), but metal-free organic dyes have emerged as low-cost and green alternatives.[5] Notwithstanding the widespread use of porphyrins as photosensitizers for singlet oxygen generation [6], this important class of organic dyes had been not considered as a source of photoredox catalysts until the seminal report by Zawada, Kadish and Gryko [7], published in 2016. Subsequently, other photocatalytic applications of porphyrins have emerged [8], but the number of porphyrin-based photoredox catalysts is still very limited, particularly when only free-base porphyrins are considered.
In the context of our research in the use of porphyrins as macromolecular catalysts [9] and as switchable organocatalysts [10], we have been recently interested in the photocatalytic activity of functionalized porphyrins [11]. While several studies have been performed concerning the redox electrochemical behaviour of metalloporphyrins [12,13], much less quantitative data are available on the photophysical and electrochemical properties of free-base porphyrins [7,8,14,15], and a systematic study on the effect of the electronic nature and of the position of the substituents on both the ground-state and the excited-state redox potentials of these compounds is still lacking. This is surprising, especially taking into account that, as already recognized by Gryko et al., [7] porphyrins are accessible by a variety of synthetic methods [16] and that their photophysical and electrochemical properties should be readily tuned by the introduction of adequate substituents in the different positions of the porphyrin core.
We decided therefore to prepare a set of known functionalized derivatives (1-5) of meso-tetraphenyl porphyrin (TPPH2), containing different substituents either in one of the meso-positions (1-3) or at a β-pyrrole carbon (4,5), and to determine their ground- and (singlet) excited state redox potentials (Figure 1A). This first survey allowed us to conclude that the introduction of a formyl group at the β-pyrrolic position strongly increases the facility of the monoelectronic reduction of the porphyrin both in the ground state and in the singlet excited state. At the light of these results, we prepared a second set of four previously unknown TPPH2 benzoyl derivatives (6-9, Figure 1B), that confirmed the validity of our conclusions.
2. Results and Discussion
We proceeded on the first place to record the cyclic voltammograms (CVs) of the known porphyrins TPPH2 and 1-5, in order to evaluate their ground-state redox potentials. After checking the solubility of our porphyrin set in various solvents, we selected dichloromethane (DCM) as the solvent, and tetrabutylammonium perchlorate (TBAP, 0.1 M) as the supporting electrolyte. The results obtained are gathered in Table 1, after correction of the obtained values (Ag/AgCl reference electrode) to the standard calomel electrode (SCE, +0.05 V correction). The CVs of TPPH2 (in DMSO) [7] and of 5-(p-nitrophenyl)-10,15,20-triphenylporphyrin 1 [15] had been already reported, and we were pleased to find that our results were essentially the same.
Subsequently, the UV-VIS and the fluorescence spectra of the porphyrins were measured in DCM, to estimate the singlet excited state energies (ES10,0). We determined the wavelength at the intersection between the normalized less energetic UV-VIS absorption (the most red-shifted Q band) and the more energetic emission band (Q0,0) [4d,17]. Contrary to what happens with the redox potentials, this wavelength does not depend on the presence and nature of the substituents and was essentially the same for all of the studied porphyrins (648 ± 1 nm), so that a common value of 1.91 eV can be used for the energy of the first singlet excited state in the whole set.
Combining this value with the ground-state redox potentials extracted for the first SET processes, and making use of the commonly used approximations [4c], we obtain the excited-state potentials. The combined data set is summarized in Table 2.
An inspection of the results gathered in Table 2 shows that the introduction of substituents having non-bonding electron pairs in a nitrogen atom (compounds 2 and 3) increases the facility of oxidation of the excited state (column 5), while the presence of a nitro (1) or a β-formyl group (compounds 4 and 5), that are electron-withdrawing substituents with less oxidable oxygen-centered lone electron pairs has a small effect in the opposite direction. The stronger substituent effects are seen, however, when one compares the relative facility for the reduction of the excited state (column 4), that can be directly related to the electron-donating or electron-accepting nature of the substituents. Thus, the presence of a p-amino group in one of the meso phenyls (compound 2) practically does not change the reduction potential with respect to TPPH2 (+0.69 V vs. +0.70 V), probably due to the compensation between the electron-retrieving inductive effect and the electron-donating resonance effect of this group. The presence of a p-nitro (compound 1) or of a β-formyl group (compound 4) substantially increases the facility of reduction (by +0.15 V and by +0.26 V, respectively). As expected, when the Cu(II) complex 4 is examined, the effect of the formyl substituent is diminished (+0.14 V with respect to TPPH2), probably due to the increase of the formal negative charge (by 2 units) of the porphine core. The replacement of a meso-phenyl group by a 4’-pyridyl moiety (compound 3) brings about a smaller increase (+0.07 V) of the reduction potential of the excited photocatalyst. We concluded therefore that the introduction of a carbonyl substituent strongly increases the facility of reduction of the excited state of the photocatalyst, tuning its photoredox properties towards a reductive quenching cycle.
In the case of TPPH2, the approximate triplet excited-state redox potentials can be readily evaluated, since a -0.50 V energy gap between the singlet and the triplet has been experimentally determined [18]. Although the singlet-triplet difference energy is not known for the other porphyrins, given that the singlet energy value appears to be independent of the substituents, we can assume a similar energy gap for porphyrins 1-5, so that the corresponding triplet excited-state potentials can also be readily estimated. In any case, the observed substituent effects will hold also for the triplet excited states.
In the light of these results, we decided to perform a similar study in a set of previously unknown meso-arylporphyrin derivatives (6-9, Figure 1B), having a benzoyl group at different positions. In this way, we wanted to confirm that the presence of carbonyl substituent increased the facility of reduction in the excited state, and to explore the effect of the position of this substituent. The change of the formyl to the benzoyl substituent was motivated by the diminished chemical reactivity of the latter, on the first place, and by the possibility of observing the photocatalytic-hydrogen atom transfer (photoHAT) activity of diaryl ketones [19], on the second place.
The 5-(4’-benzoyl)-10,15,20-triphenylporphyrin 6 was easily obtained in two steps from the known [11] 5-(4’-formyl) derivative 10 (Scheme 1). Treatment of this compound with an excess of phenylmagnesium bromide in anhydrous THF produced the benzhydryl alcohol 11, that was subsequently oxidized by pyridinium chlorochromate to provide 6 in 63% overall yield, after chromatographic purification.
On the other hand, phenyl(5,10,15,20-tetraphenylporphyrin-7-yl)methanone 7 could be accessed either from the β-formyl derivative 4 [11,20] or from the corresponding Cu-complex 5 [11,21] (Scheme 2).
As in the case of 6 above, the β-benzoyl derivative 7 was synthesized through addition of the phenyl Grignard reagent, and the resulting alcohol 12 was oxidized with PCC. Starting from the Cu complex 5, the same reaction sequence afforded the Cu complex 8, albeit in somewhat lower overall yield. Acid-catalyzed demetallation of 8 to give 7 took place with good yield.
Finally, 5-(benzoyl)-10,15,20-triphenylporphyrin 9 was obtained by the mixed condensation of phenylglyoxal monohydrate 14 with benzaldehyde and pyrrole (Scheme 3), following the conditions previously described by Lindsey for the preparation of 5-(benzoyl)-10,15,20-tris(p-tolyl)porphyrin [16c]. In a single step, after chromatographic purification, pure 9 was isolated in 9% yield.
We recorded next the CV’s of these benzoyl-substituted porphyrins, in DCM solution. The results are summarized in Table 3; the half-wave potentials of TPPH2 are shown for comparison.
We were pleased to find that, according to our prediction, the introduction of an electron-withdrawing benzoyl group resulted in a greater facility of reduction of the neutral species with respect to the unsubstituted meso-tetraphenylporphyrin (compare for instance the values for the first monoelectronic reduction in column 2 of the Table), and at the same time making more difficult the first monoelectronic oxidation (column 3 of the Table). It should also be noted that these effects increased with the proximity of the benzoyl group to the porphine core, so that 5-(benzoyl)-10,15,20-triphenylporphyrin 9 exhibits the less negative value for the monoelectronic reduction potential of the neutral molecule (-0.97 V vs. -1.21 V for TPPH2) and the more positive value for the reduction of the radical cation (+1.32 V vs. +1.09 V for TPPH2).
The photophysical properties of porphyrins 6-9, derived from their UV-VIS and fluorescence spectra, are summarized in Table 4. As it can be seen, the benzoyl-substituted porphyrins 6 and 9 have the same normalized intersection wavelength (648 nm) and therefore the same singlet excited state energy (1.91 V) than the previously studied set of porphyrins (TPPH2, 1-5), together with very low vales for the Stokes shift (3-6 nm). The pyrrolyl-substituted porphyrin 7, on the other hand, has a slightly higher intersection wavelength (657 nm), showing therefore a correspondingly lower (by 0.02 V) singlet excited sate energy. Finally, the Cu(II) complex 8 behaved rather differently than the other members of the set: it presents a low intersection wavelength (618 nm), a large Stokes shift (63 nm) and a high singlet excited state energy (2.00 V).
We measured also the fluorescence quantum yields for these compounds (column 4 in Table 4). The free-base porphyrins 6, 7 and 9 presented values only slightly inferior to that of TPPH2 (that was used as the reference compound), because of the extended π-system. The well-known “heavy atom effect” [22] of porphyrin Ni(II) and Cu(II) complexes was readily apparent in complex 8, with a very low fluorescence quantum yield.
With the measured values of the singlet excited states in our hands, we could finally evaluate both the ground- and the excited-state redox potentials of the benzoyl-substituted porphyrins 6-9, that are shown in Table 5.
By inspection of the values gathered in Table 5, we can see that the introduction of a benzoyl group in the meso-arylporphyrin structure brings about a strong increment of the facility of reduction of the excited state, especially in the case of the meso-benzoyl derivative 9. This compound exhibits a half-wave reduction potential in the excited state of +0.94 V ([*PC/PC.–]), and should therefore behave as a good photooxidant in a reductive quenching cycle.
3. Materials and Methods
3.1. General Methods
Commercially available reagents, catalysts, and solvents were used as received from the supplier. Dichloromethane for porphyrin synthesis was distilled from CaH2 prior to use, and THF was dried by distillation from LiAlH4. Deuterated solvents were supplied by Merck Life Science.
Thin-layer chromatography was carried out on silica gel plates Merck 60 F254, and compounds were visualized by irradiation with UV light and/or and chemical developers (KMnO4, p-anisaldehyde, and phosphomolybdic acid). Chromatographic purifications were performed under pressurized air in a column with silica gel Merck 60 (particle size: 0.040–0.063 mm, Merck Life Science S.L.U., Spain) as stationary phase and solvent mixtures (hexane, ethyl acetate, dichloromethane, and methanol) as eluents.
1H (400 MHz) NMR spectra were recorded with a Varian Mercury 400 spectrometer (Agilent Technologies, Santa Clara, Cal, USA). Chemical shifts (δ) are given in ppm relative to the peak of tetramethylsilane (δ = 0.00 ppm) and coupling constants (J) are provided in Hz. The spectra were recorded at room temperature. Data are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad signal. IR spectra were obtained with a Nicolet 6700 FTIR instrument (Thermo Fisher Scientific, Waltham, Mass., USA), using ATR techniques.
3.2. Photophysical Methods
UV–vis spectra were recorded on a double-beam Cary 500-scan spectrophotometer (Varian); cuvettes (quartz QS Suprasil, Hellma, Hellma GmbH & Co. KG, Mülheim, Germany) of 1 cm were used for measuring the absorption spectra. All spectra were carried out in DCM freshly filtered over basic Al2O3, to eliminate possible traces of hydrochloric acid. Fluorescence spectra measurements were carried out in a PTI Felix GX spectrofluorimeter. All spectra were recorded in an open window of 1.5 mm. The optical path of the cell was 1 cm.
3.3. Cyclic Voltammetry
Cyclic voltammetry measurements were carried out with a computer-controlled potentiostat Model Epsilon EClipse (BASi). The setup comprised an undivided cell, a glassy carbon working electrode (3 mm diameter), a platinum wire counter electrode, and a saturated Ag/AgCl reference electrode. Each cyclic voltammetry was acquired at a sweep rate of 100 mV/s. The examined solvents were initially DCM, DMSO, DMF and MeCN using TBAP (0.1M) as the supporting electrolyte in 50 mL of solution. Best results for the whole set of porphyrins, due to solubility reasons, were obtained for DCM. For the CVs, the reaction mixtures were previously purged under argon to avoid the presence of oxygen during the measurements. To perform the experiments and detect the presence of possible catalytic current, 5 mM stock solutions of the synthetized porphyrins were prepared
3.4. Synthetic Procedures and Product Characterization
5-(4’-Benzoyl)-10,15,20-triphenylporphyrin (6)
A 1 M THF solution of phenylmagnesium bromide (0.62 mL, 0.62 mmol, 4 equiv.) was added dropwise to a flask containing a cold (-78ºC, dry ice-acetone bath) solution of the known aldehyde 10 [11] (100 g, 0.16 mmol) in anhydrous THF (30 mL) under an argon atmosphere. When the addition was finished, the reaction mixture was allowed to warm up to room temperature and stirred for 4 h, at which time it was carefully quenched with aqueous sat. NH4Cl (2 mL). The solution was distilled under vacuum, evaporating most of the THF. In a separatory funnel, the organic phase was separated, and the aqueous phase was washed with ethyl acetate (3x10 mL). The combined organic phases were washed with brine (5 mL) and dried over anhydrous MgSO4. The solvent was evaporated under vacuum. The product was then purified via column chromatography on silica gel (1:1 hexane:DCM) to afford alcohol 11 (77 mg, 63% yield).
1H NMR (400 MHz, CDCl3) δ 8.84 (s, 8H), 8.26 – 8.16 (m, 8H), 7.81 – 7.70 (m, 11H), 7.71 – 7.64 (m, 2H), 7.55 – 7.47 (m, 2H), 7.43 – 7.38 (m, 1H), 6.21 (s, 1H), 3.60 (s, 1H OH), -2.78 (s, 2H NH) ppm. 13C NMR (101 MHz, CDCl3) δ 144.1, 143.3, 142.3, 141.6, 134.9, 134.7, 128.9, 128.1, 127.9, 127.0, 126.8, 125.1, 120.3, 119.9, 76.6 ppm.
Without further characterization, the porphyrin alcohol 11 (166 mg, 0.23 mmol) was directly diluted with DCM (20 mL) and then slowly added to a well-stirred mixture of pyridinium chlorochromate (173 mg, 0.80 mmol, 3.5 eq) and silica gel (0.4 g) in DCM (10 mL). After the addition, the reaction was monitored by TLC. Once completion of the reaction was observed (12 h stirring at rt), the reaction mixture was filtered through a short pad of silica gel, that was washed several times with DCM, to afford the ketone 6 (163 mg, quantitative yield).
1H NMR (400 MHz, CDCl3) δ 8.95 – 8.85 (m, 8H), 8.38 (ddd, J = 8.4, 4.3, 2.0 Hz, 2H), 8.28 – 8.21 (m, 8H), 8.12 (ddt, J = 7.0, 3.3, 1.5 Hz, 2H), 7.85 – 7.74 (m, 9H), 7.70 (d, J = 7.1 Hz, 1H), 7.64 (td, J = 7.6, 7.0, 1.8 Hz, 2H), -2.72 (bs, 1H NH), -2.74 (bs, 1H NH) ppm.
13C NMR (101 MHz, CDCl3) δ 197.0, 146.8, 142.2, 137.9, 136.9, 134.7, 134.7, 132.8, 130.4, 128.7, 128.6, 127.9, 126.9, 120.8, 120.6, 118.6 ppm.
HRMS (ESI) m/z calculated for C51H35N4O [M+H]+, 719.2805; found , 719.2803. m/z calculated for C51H36N4O [M+2H]2+, 360.1439; found, 360.1448.
UV-vis [DCM, λmax (logε), C = 7.2x10-5 M]: 419 (4.78), 515 (3.44), 550 (3.05), 590 (2.83), 645 (2.66).
FTIR (solid, = 1653 (CO)
Phenyl(5,10,15,20-tetraphenylporphyrin-7-yl)methanone (7)
A 1M THF solution of phenylmagnesium bromide (1.4 mL, 1.4 mmol, 5 equiv.) was added dropwise to a flask containing a cold (-78ºC., dry-ice/acetone bath) solution of the known [11,20] 2-formyl-5,10,15,20-tetraphenylporphyrin 4 (183 mg, 0.28 mmol) in anhydrous THF (60 mL) under an argon atmosphere. The reaction was allowed to warm up to room temperature and stirred for 4 h, at which time it was carefully quenched with 5 mL of aq. sat. NH4Cl solution. The organic phase was separated, the aqueous phase was extracted with ethyl acetate (3x15 mL) and the combined organic phases were washed with brine (10 mL) and dried over anhydrous MgSO4. After rotary evaporation, the crude product was then purified via column chromatography in silica gel, eluting with 1:1 hexane:DCM, to afford the alcohol 12 (192 mg, 94% yield).
1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 8.86 (dq, J = 12.1, 4.8 Hz, 4H), 8.74 (d, J = 4.8 Hz, 1H), 8.52 (d, J = 4.8 Hz, 1H), 8.32 – 8.11 (m, 7H), 7.82 – 7.30 (m, 13H), 7.20 – 7.12 (m, 1H), 7.09 (tt, J = 8.2, 6.7, 1.3 Hz, 2H), 6.97 – 6.92 (m, 2H), 6.21 (s, 1H), 3.09 (s, 1H OH), -2.65 (s, 2H NH) ppm. 13C NMR (101 MHz, CDCl3) δ 144.0, 142.6, 142.3, 142.0, 141.6, 134.8, 134.7, 134.3, 133.7, 132.3, 128.4, 128.0, 127x9 (x2), 127.7, 126.9 (x3), 126.8 (x2), 120.6, 120.4, 120.1, 119.8, 72.0 ppm.
Without further characterization, porphyrin alcohol 12 (133 mg, 0.18 mmol) was diluted with DCM (20 mL) and then slowly added to a stirred mixture of pyridinium chlorochromate (139 mg, 0.65 mmol, 3.5 equiv.) and silica gel (0.10 g) in DCM (15 mL). After the addition, the reaction was monitored by TLC. Once completion of the reaction was observed (12 h stirring at rt), the reaction mixture was filtered through a short pad of silica gel, that was washed several times with DCM, to afford the desired ketone 7 (131 mg, quantitative yield).
1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 8.94 (q, J = 4.9 Hz, 2H), 8.87 – 8.76 (m, 4H), 8.30 – 8.19 (m, 6H), 7.87 – 7.69 (m, 11H), 7.50 (tt, J = 7.5, 1.2 Hz, 1H), 7.42 – 7.35 (m, 3H), 7.28 (t, J = 7.9 Hz, 2H), 7.16 – 7.06 (m, 2H), -2.62 (s, 2H) ppm.
13C NMR (101 MHz, CDCl3) δ 195.0, 142.2, 141.9, 141.9, 141.0, 138.6, 136.7, 134.8, 134.8, 132.7, 129.5, 128.1, 128.0, 127.7, 127.0, 126.9, 126.9, 126.6, 121.2, 121.1, 120.6, 120.5 ppm.
HRMS (ESI) m/z calculated for C51H35N4O [M+H]+, 719.2805; found , 719.2801. m/z calculated for C51H36N4O [M+2H]2+, 360.1439; found, 360.1446.
UV-vis [DCM, λmax (logε), C = 7.2x10-6 M]: 422 (4.92), 519 (3.50), 555 (3.02), 596 (2.92), 652 (2.96).
FTIR (solid, = 1655 (CO)
Phenyl(5,10,15,20-tetraphenylporphyrin-7-yl)methanone copper(II) complex (8)
A 1M THF solution of phenylmagnesium bromide (1.9 mL, 1.9 mmol, 3.0 equiv.) was added dropwise to a round-bottom flask containing a cold (-78ºC, dry-ice/acetone bath) solution of the known [11,21] copper(II) 2-formyl-5,10,15,20-tetraphenylporphyrin 5 (455 mg, 0.65 mmol) in anhydrous THF (60 mL) under an argon atmosphere. The flask was allowed to warm up to room temperature and stirred overnight. Then, the reaction mixture was carefully quenched with aq. sat. NH4Cl solution (5 mL). The aqueous phase was extracted with ethyl acetate (3x10 mL), the combined organic phase was washed with brine (10 mL), dried over anhydrous MgSO4 and concentrated under vacuum. The crude product was then purified by column chromatography (silica gel, 1:1 hexane:DCM) to afford 372 mg (74% yield) of the desired alcohol 13, that was not further characterized.
A solution of porphyrin alcohol 13 (372 mg, 0.47 mmol) in DCM (60 mL) was slowly added to a well-stirred suspension of pyridinium chlorochromate (361 mg, 1.7 mmol, 3.6 equiv.) and silica gel (0.4 g) in DCM (10 mL). After the addition, the reaction was monitored by TLC. Once completion of the reaction was observed (12 h stirring at rt), the reaction mixture was filtered through a short pad of silica gel, that was washed several times with DCM, to afford the desired ketone 8 (264 mg, 71% yield).
HRMS (ESI) m/z calculated for C51H33N4OCu [M+H]+, 780.1945; found , 780.1944. m/z calculated for C102H65N8O2Cu2 [2M+H]+, 1559.3817; found, 1559.3824.
UV-vis [DCM, λmax (logε), C = 7.4x10-5 M]: 420 (4.75), 545 (3.48), 587 (2.99).
FTIR (solid, = 1653 (CO)
The identity of 8 was further established by acid-promoted demetallation to 7: In a 50 mL round-bottom flask, equipped with magnetic stirring, the Cu(II) porphyrin complex 8 (26.3 mg, 0.034 mmol) was dissolved in 5 mL of DCM. Concentrated H2SO4 (1 mL, 98%) was added, and the green mixture was vigorously stirred for 30 min. Then the two-phase mixture was carefully poured into a cold (0ºC) aqueous NaOH solution (1 g in 20 mL), transferred into a separatory funnel, and shaken until no green color was observed. The aqueous phase was extracted with DCM (3 x 10 mL), the combined organic layers were washed with an aqueous saturated solution of NaHCO3 (2 x 10 mL), dried over MgSO4 and the solvent was evaporated under reduced pressure. Finally, the obtained crude product was purified via flash chromatography through Et3N-pretreated silica gel (2.5% v/v NEt3) using DCM/AcOEt (1/1) as eluent to obtain the free-base porphyrin 7 (21.5 mg), in 89% yield.
5-(Benzoyl)-10,15,20-triphenylporphyrin (9)
A 1 L round-bottom reaction flask was successively charged with a DCM solution (800 mL) of phenylglyoxal monohydrate 14 (0.302 g, 2.0 mmol), and was purged with argon for 15 min. Then, freshly distilled pyrrole (0.56 mL, 8.0 mmol) and benzaldehyde (0.64 mL, 6.0 mmol) were added. The resulting mixture was stirred for 5 min and trifluoroacetic acid (1.22 mL, 16 mmol) was added dropwise. At this point a change in color was observed. The flask was covered with aluminum foil to protect the reaction from light. After 3 h of stirring at rt, DDQ (1.36 g, 6.0 mmol) was added, and the resulting solution was stirred under reflux for 1 h. After cooling to rt, triethylamine (2.2 mL, 16 mmol) was added dropwise. The solvents were evaporated in vacuo, and the resulting residue was submitted to column chromatographic purification (silica gel, DCM gradient with ethyl acetate). A middle-polarity fraction containing a mixture of monosubstituted and disubstituted porphyrins was separated. A second chromatographic purification (silica gel, DCM/hexanes 1:1) furnished the desired porphyrin 9 (115 mg, 9% yield).
1H NMR (400 MHz, CDCl3) δ 9.03 (d, J = 4.8 Hz, 2H), 8.92 – 8.84 (m, 6H), 8.27 – 8.16 (m, 6H), 7.96 (d, J = 7.6 Hz, 2H), 7.85 – 7.72 (m, 9H), 7.59 (tt, J = 7.6, 1.3 Hz, 1H), 7.41 (ddd, J = 9.1, 7.5, 1.5 Hz, 2H), -2.69 (s, 2H) ppm.
13C NMR (101 MHz, CDCl3) δ 199.5, 142.1, 142.0, 141.8, 134.7, 134.6, 133.6, 131.5, 128.6, 128.1, 128.0, 126.9, 126.9, 122.1, 121.0, 115.8 ppm.
HRMS (ESI) m/z calculated for C45H31N4O [M+H]+, 643.2492; found , 643.2495. m/z C45H32N4O [M+2H]2+, 322.1283; found, 322.1289.
UV-vis [DCM, λmax (logε), C = 5.3x10-5 M]: 418 (4.89), 515 (3.53), 548 (3.06), 589 (3.02), 647 (2.85).
FTIR (solid, ) = 1655 (CO)
4. Conclusions
We have prepared a set of both previously known and of unknown meso-arylporphyrin derivatives, and we have conducted a preliminary study of their photophysical properties and redox potentials. This has allowed us to conclude that the introduction of carbonyl (either formyl or benzoyl) groups can affect in a rational and predictable way the redox behavior of this promising class of organic photosensitizers. Thus, the presence of electron-withdrawing resonance effects, especially at the meso-position results in an enhancement of the facility of reduction of the photocatalyst, both in the ground and in the excited state, so that the redox properties of meso-tetrarylporphyrins can be easily tuned towards a reductive quenching pathway in a photoredox catalytic cycle. The study of the both the photosensitizing ability (olefin isomerization, photocatalyzed HAT) and the photoredox catalytic activity of the newly synthesized porphyrins is underway in our laboratories, and the results will be reported in due course.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: 1H and 13C NMR spectra for porphyrins 6, 7, 9, 11 and 12 (ESI2-ESI6); cyclic voltammetries for compounds TPPH2, 1-9 (ESI7-ESI10)
Author Contributions
Conceptualization, A.M.; methodology, A.M.; validation, A.C.M. and A.M.; investigation, E.P. and A.C.M.; resources, A.M.; data curation, writing—original draft preparation, A.M.; writing—review and editing, A.C.M. and A.M.; supervision, A.M.; project administration, A.M.; funding acquisition, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FEDER, Ministerio de Ciencia e Innovación (MCIN), Agencia Estatal de Investigación (AEI): MCIN/AEI/10.13039/501100011033, and ERDF funds (grant number PID2020-116846GB-C21, to A.M.).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created in addition to those reported here and in the Supplementary Materials.
Acknowledgments
We are grateful to Paula Rodríguez and to Martí Gisbert (UB) for their help in the recording of cyclic voltammograms, and to Drs. Jaume García-Amorós and Alessandro Sorrenti (UB) for their advice with fluorescence experiments. The administrative and technical support of the CCTiUB is also gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nicewicz, D.A.; MacMillan, D.W.C. Merging photoredox catalysis with organocatalysis. Science 2008, 322, 77–80. [Google Scholar] [CrossRef]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef]
- Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R. Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- a) Narayanam, J.W.; Stephenson, C.R. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. https://doi.org/10.1039/b913880n. b) Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. https://doi.org/10.1021/cr300503r. c) Tucker, J.W.; Stephenson, C.R.J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. https://doi.org/10.1021/jo202538x. d) Striet-Kalthoff, F.; James, M.J.; Teders, M.; Pitzer, L.; Glorius, F. Energy transfer catalysis mediated by visible-light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. https://doi.org/10.1039/c8cs00054a. e) Rigotti, Th.; Alemán, J. Visible light photocatalysis – from racemic to asymmetric activation strategies. Chem. Commun. 2020, 56, 11169–11190. https://doi.org/10.1039/d0cc03738a. f) Cauwenbergh, R.; Das, S. Photocatalysis. A Green Tool for Redox Reactions. Synlett 2022, 33, 129–149. https://doi.org/10.1055/s-0040-1706042.
- Wu, Y.; Kim, D.; Teets, T.S. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2022, 33, 1154–1179. [Google Scholar] [CrossRef]
- a) Campestrini, S.; Tonellato, U. Photoinitiated Olefin Epoxidation with Molecular Oxygen, Sensitized by Free Base Porphyrins and Promoted by Hexacarbonylmolybdenum in Homogeneous Solution. Eur. J. Org. Chem. 2002, 3827–3832. https://doi.org/10.1002/1099-0690. b) Clennan, E. L.; Pace, A. Advances in singlet oxygen chemistry. Tetrahedron 2005, 61, 6665–6691. https://doi.org/10.1016/j.tet.2005.04.017. c) Chen, J.-J.; Hong, G.; Gao, L.-J.; Liu, T.-J.; Cao, W.-J. In vitro and in vivo antitumor activity of a novel porphyrin-based photosensitizer for photodynamic therapy, J. Cancer Res. Clin. Oncol. 2015, 141, 1553–1561. https://doi.org/10.1007/s00432-015-1918-1. d) Rostami, M.; Rafiee, L.; Hassanzadeh, F.; Dadrass, A.R.; Khodarahmi, G.A. Synthesis of some new porphyrins and their metalloderivatives as potential sensitizers in photo-dynamic therapy. Res. Pharm. Sci. 2015, 10, 504–513. PMID: 26779270; PMCID: PMC4698861.
- Rybicka-Jasinska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451−15458. [Google Scholar] [CrossRef]
- Costa e Silva, R.; da Silva, L.O.; Bartolomeu, A. A.; Brocksom, T. J.; de Oliveira, K. T. Recent applications of porphyrins as photocatalysts in organic synthesis: batch and continuous flow approaches. Beilstein J. Org. Chem. 2020, 16, 917–955. [Google Scholar] [CrossRef]
- a) Arlegui, A.; El-Hachemi, Z.; Crusats, J.; Moyano, A. 5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies. Molecules 2018, 23, 3363. https://doi.org/10.3390/molecules23123363. b) Arlegui, A.; Soler, B.; Galindo, A.; Orteaga, O.; Canillas, A.; Ribó, J.M.; El-Hachemi, Z.; Crusats, J.; Moyano, A. Spontaneous mirror-symmetry breaking coupled to top-bottom chirality transfer: From porphyrin self-assembly to scalemic Diels–Alder adducts. Chem. Commun. 2019, 55, 12219–12222. https://doi.org/10.1039/c9cc05946f.
- a) Arlegui, A.; Torres, P.; Cuesta, V.; Crusats, J.; Moyano, A. A pH-Switchable Aqueous Organocatalysis with Amphiphilic Secondary Amine–Porphyrin Hybrids. Eur. J. Org. Chem. 2020, 4399-4407. https://doi.org/10.1002/ejoc.202000648. b) Arlegui, A.; Torres, P.; Cuesta, V.; Crusats, J.; Moyano, A. Chiral Amphiphilic Secondary Amine-Porphyrin Hybrids for Aqueous Organocatalysis. Molecules 2020, 25, 3420. https://doi.org/10.3390/molecules25153420.
- Torres, P.; Guillén, M.; Escribà, M.; Crusats, J.; Moyano, A. Synthesis of New Amino-Functionalized Porphyrins: Preliminary Study of Their Organophotocatalytic Activity. Molecules 2023, 28, 1997. [Google Scholar] [CrossRef]
- Kadish, K.M.; Cornillon, J.-L.; Yao, C.-L.; Malinski, T.; Gritzner, G. Solvent effects on electrode potentials of metalloporphyrins. Reduction of 5,10,15,20-tetraphenylporphinato complexes in non-aqueous media. J. Electroanal. Chem. 1987, 235, 189–207. [Google Scholar] [CrossRef]
- Mandal, T.; Das, S.; De Sarkar, S. Nickel(II) Tetraphenylporphyrin as an Efficient Photocatalyst Featuring Visible Light Promoted Dual Redox Reactions. Adv. Synth. Catal. 2019, 361, 3200–3209. [Google Scholar] [CrossRef]
- Bhyrappa, P.; Sankar, M.; Varghese, B. Mixed Substituted Porphyrins: Structural and Electrochemical Redox Properties. Inorg. Chem. 2006, 45, 4136–4149. [Google Scholar] [CrossRef]
- Fang, Y.; Jiang, X.; Ou, Zh.; Michelin, C.; Desbois, N.; Gros, C.P.; Kadish, K.M. Redox properties of nitrophenylporphyrins and electrosynthesis of nitrophenyl-linked Zn porphyrin dimers or arrays. J. Porphyr. Phthalocyanines 2014, 18, 832–841. [Google Scholar] [CrossRef]
- a) Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A simplified synthesis for meso-tetraphenylporphine. J. Org. Chem. 1967, 32, 476–476. https://doi.org/10.1021/jo01288a053. b) Wagner, R.W.; Lawrence, D.S.; Lindsey, J.S. An improved synthesis of tetramesitylporphyrin. Tetrahedron Lett. 1987, 28, 3069–3070. https://doi.org/10.1016/S0040-4039(00)96287-7. c) Lindsey, J.S.; Prathapan, S.; Johnson, T.E.; Wagner, R. W. Porphyrin Building Blocks for Modular Construction of Bioorganic Model Systems. Tetrahedron 1994, 50, 8941–8968. doi: 10.106/S0040-40200185364-3.
- Gadde, K.; De Vos, D.; Maes, B.U.W. Basic Concepts and Activation Modes in Visible-Light Photocatalyzed Organic Synthesis. Synthesis 2023, 55, 164–192. [Google Scholar] [CrossRef]
- a) Seely, G.R. The energetics of electron-transfer reactions of chlorophyll and other compounds. Photochem. Photobiol. 1978, 27, 639–654. https://doi.org/10.1111/j.1751-1097.1978.tb07658.x. b) Rillema, D.P.; Nagle, J.K.; Barringer, L.F., Jr.; Meyer, T.J. Redox Properties of Metalloporphyrin Excited States, Lifetimes, and Related Properties of a Series of Para-Substituted Tetraphenylporphine Carbonyl Complexes of Rutenium(II). J. Am. Chem. Soc. 1981, 103, 56–62. https://doi.org/10.1021/ja00391a013.
- Capaldo, C.; Ravelli, D.; Fagnoni, M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. [Google Scholar] [CrossRef]
- Bonfantini, E.E.; Burrell, A.K.; Campbell, W.M.; Crossley, M.J.; Gosper, J.J.; Harding, M.M.; Officer, D.L.; Reid, D.C.W. Efficient synthesis of free-base 2-formyl-5,10,15,20-tetraarylporphyrins, their reduction and conversion to [(porphyrin-2-yl)methyl]phosphonium salts. J. Porphyr. Phthalocyanines 2002, 6, 708–719. [Google Scholar] [CrossRef]
- Richeter, S.; Jeandon, C.; Gisselbrecht, J.-P.; Ruppert, R.; Callot, H. Synthesis and Optical and Electrochemical Properties of Porphyrin Dimers Linked by Metal Ions. J. Am. Chem. Soc. 2002, 124, 6168–6179. [Google Scholar] [CrossRef]
- Lower, S.K.; El-Sayed, M.A. The triplet state and molecular electronic processes in organic molecules, Chem. Rev. 1966, 66, 199–241. [Google Scholar] [CrossRef]
Figure 1.
Porphyrin derivatives studied in this work. A: previously known compounds. B: new benzoyl-porphyrin derivatives.
Figure 1.
Porphyrin derivatives studied in this work. A: previously known compounds. B: new benzoyl-porphyrin derivatives.
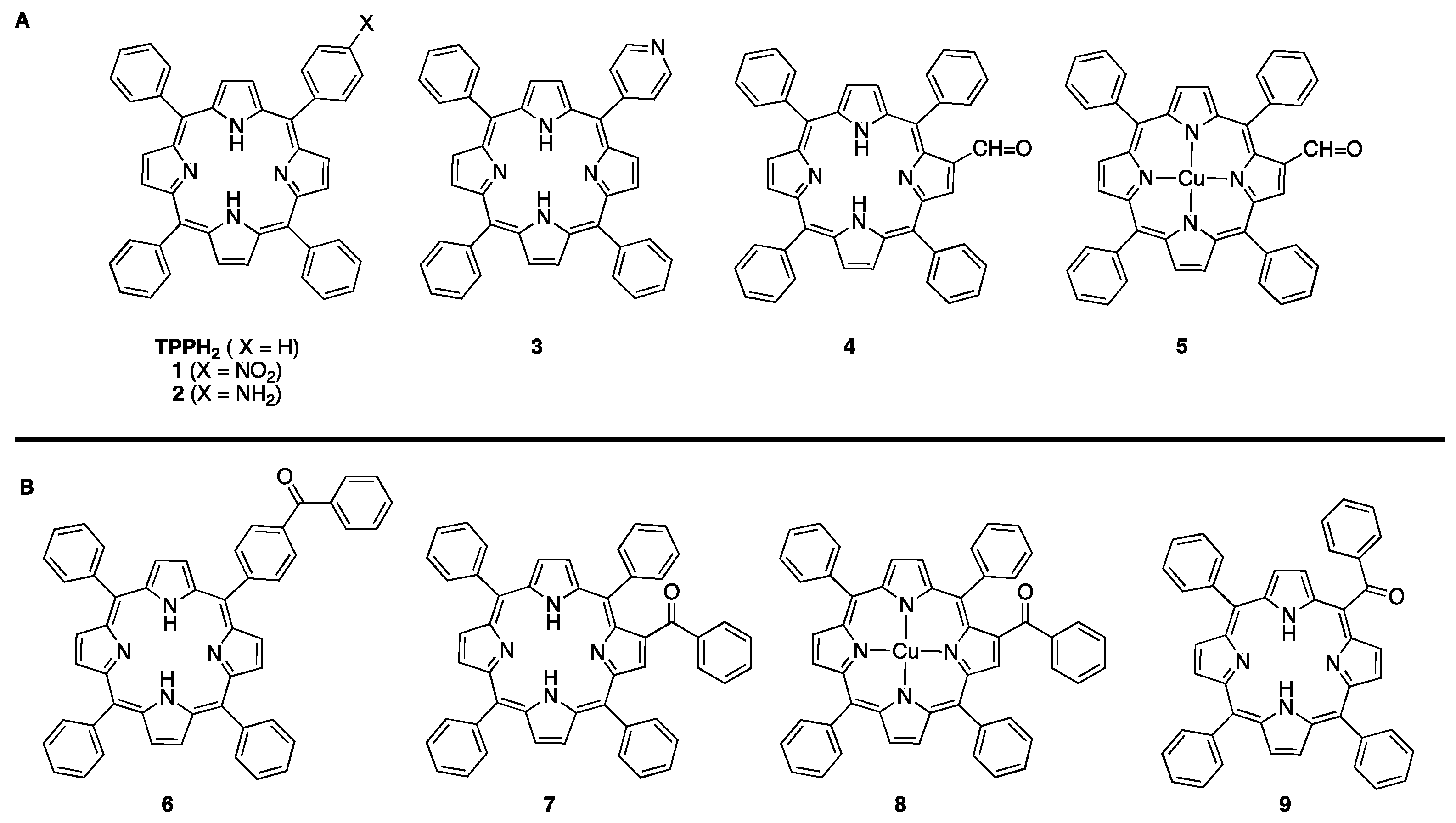
Scheme 1.
Synthesis of 5-(4’-benzoyl)-10,15,20-triphenylporphyrin 6.
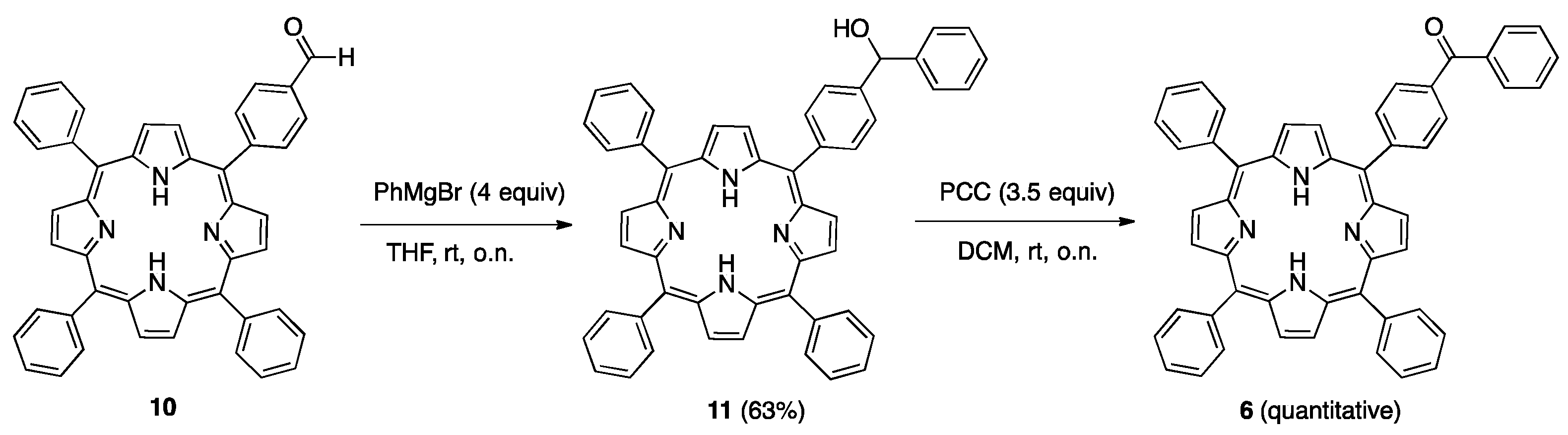
Scheme 2.
Synthesis of benzoyl-porphyrin derivatives 7 and 8.
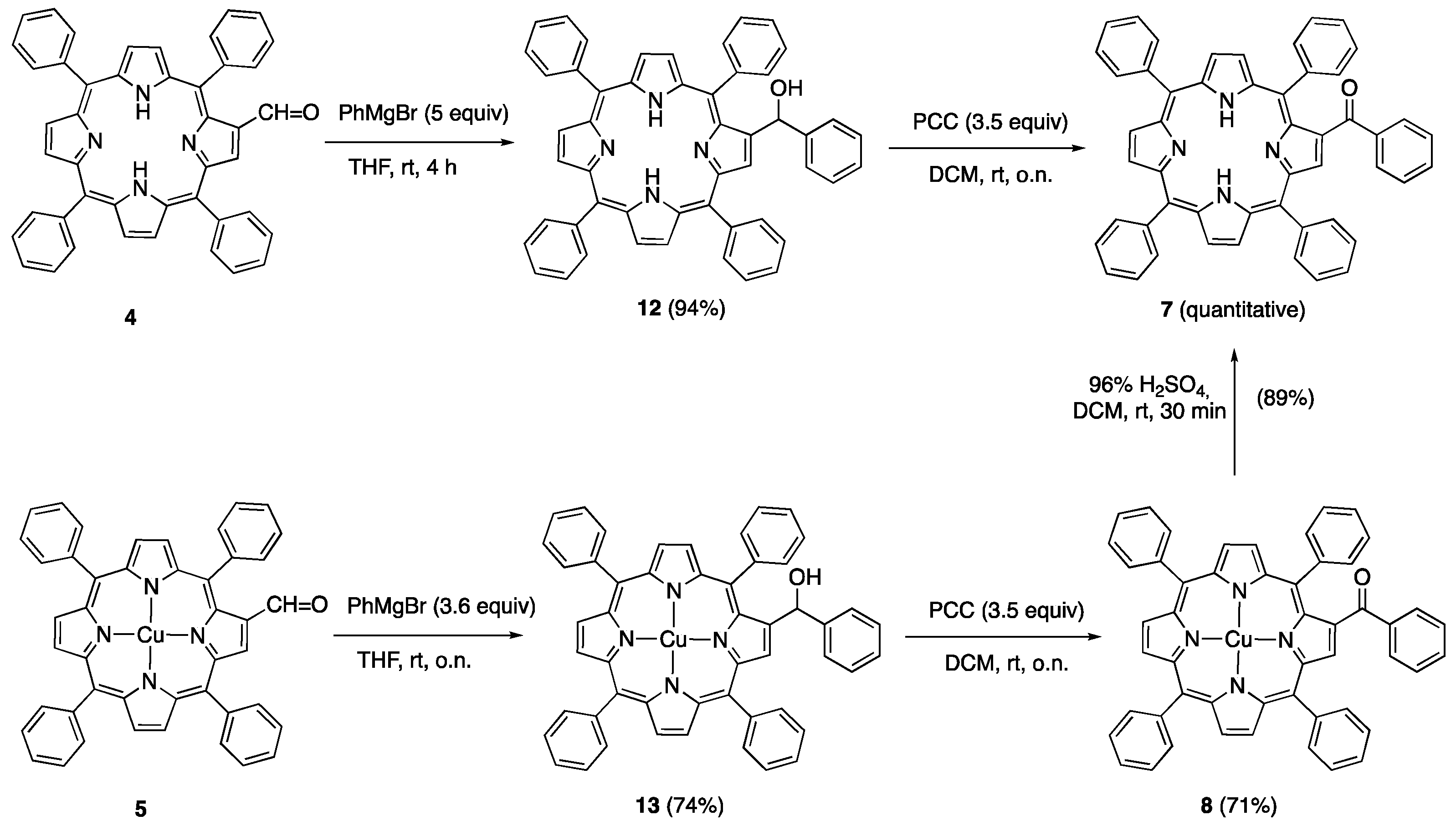
Scheme 3.
Synthesis of 5-(benzoyl)-10,15,20-triphenylporphyrin 9.

Table 1.
Ground-state half-wave potentials of porphyrins TPPH2 and 1-5 in dichloromethane.
| Porphyrin | Experimental ground-state redox half-wave potentials (V vs. SCE) | |||
| [PC.–/PC2–] | [PC/PC.–] | [PC.+/PC] | [PC2+/ PC.+] | |
| TPPH2a | -1.63 | -1.21 | +1.09 | +1.28 |
| 1b | -1.66c | -1.21d/-1.06e | +1.10f | +1.20f |
| 2 | -1.56 | -1.22 | +1.00f,g/+1.10f | +1.70f |
| 3 | -1.49 | -1.14 | +1.05f,h/+1.15 | +1.60f |
| 4 | -1.23 | -0.95 | +1.11 | +1.45 |
| 5 | -1.43 | -1.07 | +1.11 | +1.48 |
a See ref.[7] for the literature values in DMSO. b See ref.[15] for the literature values. c Process overlapped with reduction of the NO2Ph anion. d Reduction of the porphyrin ring. e Reduction of the neutral NO2Ph group. f Irreversible. g Oxidation of the phenylamino group. h Oxidation of the 4’-pyridyl group.
Table 2.
Ground- and (singlet) excited state redox potentials of porphyrins TPPH2 and 1-5.
| Porphyrin | Ground-state redox potentials (V vs. SCE) | (Singlet) excited-state redox potentials (V vs. SCE) | ||
| [PC/PC.–] | [PC.+/PC] | [*PC/PC.–] | [PC.+/*PC] | |
| TPPH2a | -1.21 | +1.09 | +0.70 | -0.82 |
| 1 | -1.06 | +1.10 | +0.85 | -0.81 |
| 2 | -1.22 | +1.00 | +0.69 | -0.91 |
| 3 | -1.14 | +1.05 | +0.77 | -0.86 |
| 4 | -0.95 | +1.11 | +0.96 | -0.80 |
| 5 | -1.07 | +1.11 | +0.84 | -0.80 |
a See ref.[7] for the literature values in DMSO.
Table 3.
Ground-state half-wave potentials of porphyrins TPPH2 and 6-9 in dichloromethane.
| Porphyrin | Experimental ground-state redox half-wave potentials (V vs. SCE) | |||
| [PC.–/PC2–] | [PC/PC.–] | [PC.+/PC] | [PC2+/ PC.+] | |
| TPPH2 | -1.63 | -1.21 | +1.09 | +1.28 |
| 6 | -1.37 | -1.10 | +1.24 | +1.56 |
| 7 | -1.28 | -1.00 | +1.22 | +1.43 |
| 8 | -1.31 | -1.09 | +1.23 | +1.60 |
| 9 | -1.17 | -0.97 | +1.32 | +1.62 |
Table 4.
Selected photophysical data for porphyrins 6-9 in dichloromethane.
| Porphyrin | Less energetic absorption Q band (nm) | More energetic emission Q(0,0) band (nm) | Relative fluorescence quantum yield (φf)a | Singlet excited state energies (ES10,0), V |
| 6 | 645 | 651 | 0.09 | 1.91 |
| 7 | 651 | 662 | 0.09 | 1.89 |
| 8 | 587 | 650 | 0.002 | 2.00 |
| 9 | 647 | 650 | 0.08 | 1.91 |
aH2TPP [(φf) = 0.11] was used as the reference compound.
Table 5.
Ground- and (singlet) excited state redox potentials of porphyrins TPPH2 and 6-9.
| Porphyrin | Ground-state redox potentials (V vs. SCE) | (Singlet) excited-state redox potentials (V vs. SCE) | ||
| [PC/PC.–] | [PC.+/PC] | [*PC/PC.–] | [PC.+/*PC] | |
| TPPH2a | -1.21 | +1.09 | +0.70 | -0.82 |
| 6 | -1.10 | +1.24 | +0.81 | -0.71 |
| 7 | -1.00 | +1.22 | +0.89 | -0.67 |
| 8 | -1.09 | +1.23 | +0.91 | -0.77 |
| 9 | -0.97 | +1.32 | +0.94 | -0.59 |
a See ref.[7] for the literature values in DMSO.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



