
You are currently viewing a beta version of our website. If you spot anything unusual, kindly let us know.
Preprint
Case Report
Experience of Hereditary Amyloidosis with Rare Variant in Ecuador: A Series of Cases
Altmetrics
Downloads
80
Views
41
Comments
0


This version is not peer-reviewed
Abstract
Approximately more than 120 transthyretin mutations are known whose variation in clinical presentation is heterogeneous, as the course of disease onset depends on genetic variation and level of penetrance. It is little known in Ecuador, and some of the reported cases; suggest to the family tree analysis that they come from a province that is possibly considered endemic. The main objective is to carry out a descriptive cross-sectional analysis on the presentation of transthyretin amyloidosis in families carrying the p.Ser43Asn gene from the index case identified.
Keywords:
Subject: Medicine and Pharmacology - Cardiac and Cardiovascular Systems
1. Introduction
Transthyretin amyloidosis is a rare, multisystem, underdiagnosed disease that causes a great impact on quality of life, with increased mortality when there is cardiac involvement; that deserves to be diagnosed in time and seek measures for multidisciplinary follow-up.[1]There are two forms of transthyretin amyloidosis: the wild form and the genetic form. Some mutations are more frequent and others rare.
Transthyretin amyloidosis is described as having a heterogeneous natural history, and therefore a poor prognosis; with a mean survival from diagnosis of 3.6 years for wild-type amyloidosis and 2.5 years for transthyretin amyloidosis of the Vall 122lle variant.
In our first case, the time elapsed to establish a diagnosis of transthyretin amyloidosis from the onset of symptoms was 7 years, through the retrospective analysis of the clinical history of a patient treated at the Manuel Ygnacio Monteros-IESS Loja General Hospital with a mixed phenotype. This demonstrates a reality in Ecuador about the existing gaps in our health system and the need to carry out studies on the presentation of transthyretin amyloidosis.
The identification of the first case of transthyretin amyloidosis diagnosed at the Manuel Ygnacio Monteros Hospital with the pSer43Asn variant has motivated us to carry out a descriptive study of those index cases identified as carriers of the mutant transthyretin gene . The review of this work will allow establishing inter-institutional behaviors and protocols in the future for the management of this rare pathology not very well known by our Ecuadorian society.
The delay in diagnosis and the rapid progression after the appearance of symptoms led to the death of our first patient, with manifestations of hypertrophic cardiomyopathy, heart failure, and sensory-motor polyneuropathy.[1]
It is considered necessary to make a timely and early diagnosis; surveillance and monitoring of manifestations, their time of presentation, supported by multidisciplinary management, generating comprehensive treatment for each of the patients; which will allow us to avoid systemic damage, or fatal outcomes, higher costs due to hospitalizations, and more complex therapies that increase hospital costs.
In Ecuador, in both the comprehensive public, complementary and private health network, there are no laboratories that perform the molecular study for Familial Amyloidosis; The execution of the genetic study is carried out abroad through private intermediary companies that are responsible for processing the delivery of biological material to international genetics laboratories.
The burden of heart failure disease in Ecuador is continuously increasing. During the period 2014-2018, 90,242 years of healthy life (DALY) were lost, of which 46.72% are represented by years of life lost due to premature death (YLL) and 53.28% by years lived with disability (YLD).[2] The prevalence of Amyloidosis in Ecuador is unknown, and the question remains as to how many of our deceased heart failure patients had a rare underlying disease such as Amyloidosis. It is difficult to carry out a post-mortem study to answer the question; However, monitoring those identified as carriers will allow us to know what the first manifestations of cardiac amyloidosis caused by this p.Ser43Asn variant are, which will allow us to put the country on alert.
The objective of this article is to perform a cross-sectional descriptive analysis on the presentation of transthyretin amyloidosis in families carrying the p.Ser43Asn gene based on the index case identified in the period from January 2020 to January 2022.
2. Case Report
2.1. Case Report Number One
A 59-year-old male patient, born in the city of Loja, is seen in the private outpatient service on September 30, 2020, for presenting episodes of lipothymia, dyspnea at rest, generalized weakness, limb edema for 1 month. lower bowels, hyporexia, abundant diarrheal stools 4 to 6 times a day, the same condition that was reported to be chronic for more than 4 years and that was initially classified as Irritable Bowel Syndrome and when consulted it was mentioned to be more frequent in the last 2 years. months, with significant weight loss. Additionally, as with other pathological history: ICD device placed in 2018, due to diagnosis of Hypertrophic Cardiomyopathy and with diagnosis of ALS (Amyotrophic Lateral Sclerosis). He has received treatment for intestinal tuberculosis 1 year ago without clinical improvement. On physical examination: pale facies, cachectic. BP: 81/52mmHg, HR: 80bpm, SatO2:90%. Cardiac auscultation with norm phonetic sounds. Lungs: ventilated. Lower limbs with pretibial edema +2/4. Usual medication: Propranolol 10mg every 48 hours, Amiodarone 200mg daily, furosemide 10mg daily. The electrocardiogram study revealed: sinus rhythm and inferior Pseudo Q. An Echocardiogram study was performed in the consultation, observing left ventricular hypertrophy (SIV: 19mm), right ventricular hypertrophy (Figure 1), thickening of the interatrial septum, global hypokinesia of the left ventricle with LVEF: 24%, decreased velocities on tissue Doppler, diastolic dysfunction grade III, global strain around – 5% and mild pericardial effusion (Figure 2A). Laboratory studies: elevation of NTproBNP: 13430pg/ml, troponin T: 186pg/ml, urea: 60, creatinine: 1.2mg/dl. Chronic iron deficiency anemia. It was necessary to perform new evaluations, including neurology-neurophysiology, which determined the presence of severe sensory-motor polyneuropathy. A skin biopsy study was requested, which was negative: Kappa and Lambda light chain studies were negative. By the Hematology service, the presence of systemic Lupus Erythematosus was determined as an additional finding to the suspicion of Heredofamilial Amyloidosis due to transthyretin, which was the first presumptive diagnosis after the first evaluation. The patient was evaluated in a hospital unit of the Social Security Institute-Manuel Ygnacio Monteros Hospital, where he continued with the evaluations and follow-up; Scintigraphy studies with Tc99m Pyrophosphate are only available in the city of Quito, and Magnetic Resonance studies were not carried out due to lack of availability in the sector. A genetic study was requested for transthyretin amyloidosis, resulting in the presence of heterozygosity around 3 months later, position: chr18:31,592,954, variation p.Ser43Asn.
The patient was admitted to another hospital unit privately in February 2021 due to an episode of syncope and trauma to the spine and died on 03-08-2021.
When performing a retrospective analysis of his clinical history, initial medical attention since 2013 was found with gastrointestinal manifestations, diarrheal stools alternating with constipation, classified as Irritable Bowel Syndrome. He registered his first attention in cardiology in 2016 due to the presence of dizziness, isolated ventricular extrasystoles, with a delay in diagnosis of approximately 5 years.
Family Tree Analysis
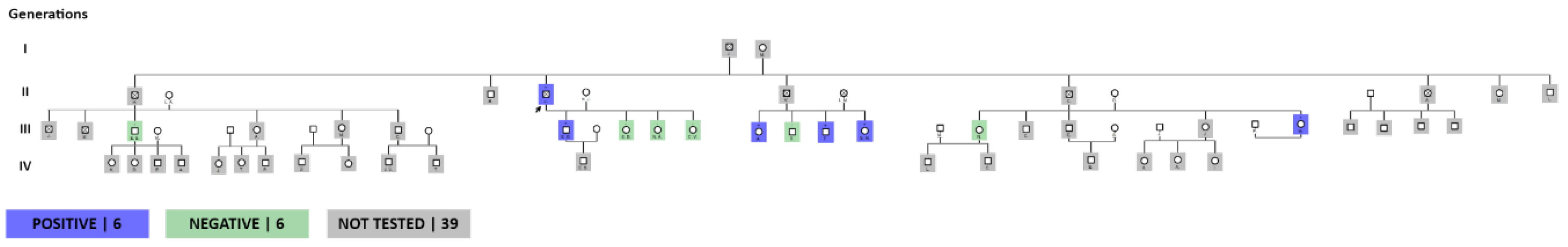
The completion of the family group allowed us to observe a total of 11 siblings of the index patient, with a total of 33 people that make up this family group, of which 19 were applied the test, with a total of 5 people with a positive result, 11 not carried out on the test. test, of these 5 died: 1 from SARS-COV2 pneumonia, 1 from acute myocardial infarction (died in the Province of Pichincha due to a diagnosis of Hypertrophic Cardiomyopathy plus acute myocardial infarction) and 3 of unknown cause with probable death from acute myocardial infarction myocardium (Figure 2). When inquiring about the origin of their parents, they claim to be from the Quilanga canton in the province of Loja. This family group has members residing in the Province of Loja.
2.2. Case Report Number Two
A 49-year-old patient from Loja came to the outpatient clinic at the Manuel Ygnacio Monteros-IESS Loja Hospital due to significant edema in the lower limbs. He denied dyspnea or precordial pain. Other reported symptoms: abdominal distention, diffuse abdominal pain and nausea. History of heart failure with reduced LVEF, with suspicion of cardiac amyloidosis according to a report from the Guayaquil hospital 6 months ago. On physical examination: BP:90/50mmHg, HR:60bpm, SatO2:93%. Rhythmic heart, without murmurs. Lungs: MV preserved. Predominantly cardiac clinical presentation with symptoms of heart and gastrointestinal failure (abdominal distension and diarrhea). Electrocardiogram: First degree AV block, complete right bundle branch block, inferior pseudoinfarction (Figure 3A). Transthoracic echocardiogram: predominantly septal left ventricular hypertrophy (SIV: 24mm), right ventricular hypertrophy, thickening of the interatrial septum and atrioventricular valves. Global hypokinesia, LVEF: 35%, Global Longitudinal Strain: -8%. Mild pericardial effusion (Figure 3B). Cardiac Magnetic Resonance Study: Subendocardial pattern suggesting amyloidosis. Skin and adipose tissue biopsy negative for Congo red stain. Kappa and Lambda light chain tests were negative. Gastric and colon biopsy study negative for Congo red stain. The requested genetic study demonstrates heterozygosity, with the p.Ser43Asn variant. The patient was discharged with a heart transplant referral to a level III hospital.
Family tree analysis
Family tree analyses
The analysis of the family tree allows us to observe a total of approximately 68 members, of which 13 people have been tested, with a positive result in 3, the remaining population to be tested is a total of 55. There are members who died before the research, a number of 4 people from the second generation, with myocardial infarction and heart failure (Figure 4). Origin of the family line from the Quilanga canton, in the province of Loja. This family group has members residing in the Province of Loja, Zamora, Guayas and members residing in Spain.
2.3. Case Report Number Three
Patient, 62 years old, male, from the province of Loja, resident in Quilanga, transferred from another hospital unit for pacemaker placement due to a diagnosis of third-degree AV block, hypertrophic cardiomyopathy, and heart failure, reporting a 5-month clinical condition. with general malaise, dyspnea with medium efforts that progresses to small efforts, accompanied by adynamic, and generalized edema, 72 hours ago he presented oppressive precordial pain with nausea and dizziness. Vital signs upon admission: BP:99/79mmHg, HR:36bpm, RR:16 per min, T:36.2 C, Pulse rate: 93% with FiO2 at 24%.
Electrocardiogram study showed complete AV block, he was subjected to pacemaker placement, evaluated with an echocardiogram study after pacemaker implantation: LVEF: 24% significant hypertrophy of the left ventricle with measurement of the interventricular septum around 33mm, LV diastolic diameter of 26mm, 28mm posterior wall, right ventricular systolic dysfunction, TAPSE:13mm, CAF:25%. Global Longitudinal Strain: -9% (Figure 5).
His neurological evaluation demonstrates the presence of stage III sensory-motor polyneuropathy.
Genetic study result on July 7, 2022, of complete sequencing with a positive result for the p.Ser43Asn variant, with heterozygosity.
Patient died at home on March 17, 2023.
Family tree analysis
Figure 6.
Source: Own elaboration, 2021.
Family tree analyses
The family chart graph indicates a total of 51 members, a total of 8 siblings of the index patient, of whom 7 have died, place of death in the province of Loja. Within their family history, the cause of death is without an established diagnosis, however close relatives express that the deceased relatives presented symptoms of generalized weakness, cachexia, dyspnea with an outcome in sudden death. There is a history of consanguinity in the ancestors of the family. Origin of the family from the Quilanga canton-Province of Loja.
Of the patients tested, one is in the Province of Oro, the rest is in the Malacatos Canton, Province of Loja.
The following table shows the baseline demographic characteristics and the clinical characteristics of ECG and imaging of the 3 clinical cases (Table 1).
3. Discussion
Transthyretin amyloidosis is a systemic disease, of variable incidence and still unknown in various parts of the world, with an incidence of 1 per 100,000 inhabitants worldwide.[3] There are some variants considered more frequent in certain countries where they are stipulated as endemic variants in Spain (Mallorca), Portugal, Norway, Japan, Brazil. The p.Ser43Asn variant is a point mutation in exon 2, codon 43 of the TTR gene that leads to a single amino acid substitution of serine for asparagine; It is considered a rare variant, with little information in the literature;[4] There are few case reports on this particular variant. According to the work published by Maria Papahanasiou and colb, published in December 2020, it documents a report of 2 opportunely diagnosed patients from a family in Italy whose identified variant is the same as our cases and which is considered to have a clinically aggressive course. due to its exclusively cardiac phenotype.[5]
There are at least five studies, in addition to the one already mentioned above, as clinical case reports based on the Ser23Asn variant, which corresponds to the same variant that we mention in our work.
This is due to a change in nomeclature. At the moment, the amino acid sequence from 1 to 20 is the signal peptide and the final protein structure is expressed from amino acids 21 to 147, varying the numbering but corresponding to the same amino acid. The variant causes hereditary transthyretin amyloidosis; this protein has a molecular weight of 14 kDa in monomeric form and exists in tetrameric form (55 kDa) in plasma. As mentioned, this is synthesized in the liver and in the choroid plexus as a homotetrameric structure with a dimer of dimers of quaternary structure.[6,7,8]
One of the first studies published with this variant corresponds to Lawreen Helter Connors and collaborators, published in 1999, in which myocardial amyloidosis was identified in a 44-year-old patient from the north of Portugal with heart block requiring a pacemaker and a heart transplant performed by its involvement exclusively at the cardiac level. Another work that talks about this variant is the publication carried out in 2010 by the Daoko et al group, which mentions a 41-year-old Peruvian patient with symptoms of heart failure, with the same variant of amyloidosis limited to the heart.
Similarly, in another review of clinical cases published by Colombian authors who highlight the relevance of the use of the Technetium 99 pyrophosphate scintigraphy technique for the diagnosis of hereditary transthyretinal amyloidosis in a 41-year-old Ecuadorian patient with symptoms of fatigue, difficulty breathing and peripheral neuropathy, whose identified variant corresponds to Ser23ASn that possibly has Spanish origin. Different researchers mention a possible origin of the variant in Portugal, Spain and Italy[9]; It is difficult to know precisely the origin of the variant and its presence is attributed to migration and colonization processes; with a possible founding effect in the province of Loja, particularly in the Quilanga canton.
In Ecuador, some clinical cases of amyloidosis without specification of the genetic type have been documented since 2018, which is explained by the limitations of the public health system in accessing imaging studies and other studies not available in public hospitals.
In our city of Loja, capital of the province of Loja in the South of the country, our first case of Transthyretin amyloidosis was diagnosed in 2020 with genetic determination of the variant; who debuted with hypertrophic cardiomyopathy, sensory-motor polyneuropathy. as the main manifestations of amyloidosis that led to death. In the retrospective analysis of the first symptoms, gastrointestinal discomfort was found as the first manifestation: diarrhea and constipation, added to the presence of arterial hypotension and dizziness, which confers a mixed phenotype presentation with a delay time of 7 years.
The second case shows gastrointestinal discomfort as first manifestations; There is no neurological involvement, and it was considered to have an exclusively cardiac phenotype. And in the third case, the mixed phenotypic presentation is repeated, with initial gastrointestinal manifestations, with sensory-motor polyneuropathy and cardiac involvement.
According to the families studied, it is possible that there is an approximation of consanguinity between the first and third cases. However, this information cannot be validated by those who would present said consanguinity due to their death.
In case 1, his relatives mention having ancestors from Spain who arrived in Ecuador and traveled to Colombia and Peru, finally deciding to settle in Ecuador in the Province of Loja.
In case number 2, they mention having ancestors from Spain and in the third case they do not know their origins.
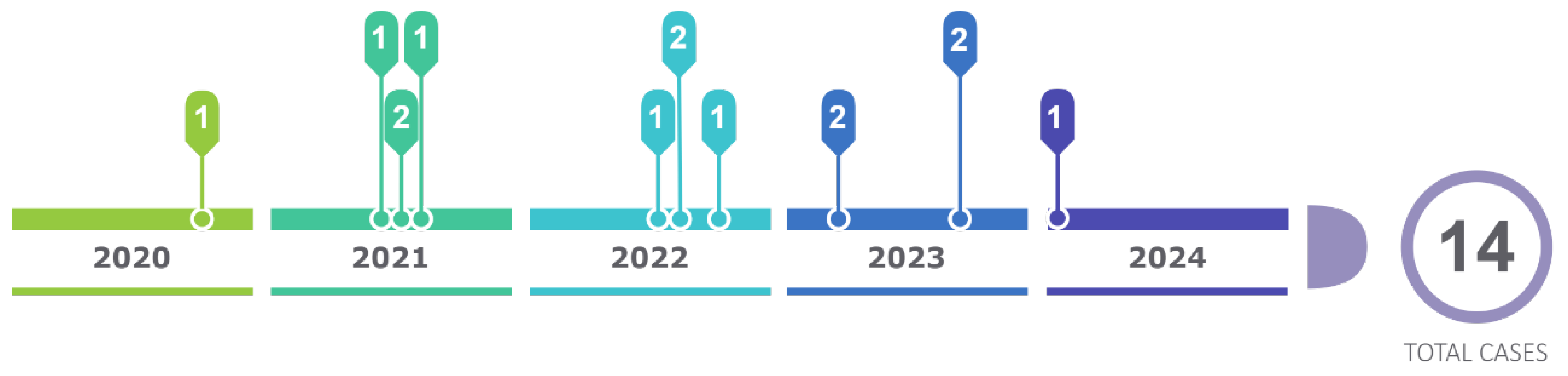
The present cascade study, which consists of carrying out the genetic study on family members, allowed us to discover a total of 14 positive patients in the period from January 2020 to. January 2024, constituting an effective method to carry out research on relatives of I, II, III degree of consanguinity (Figure 7).
Tests were carried out on a total of 44 people from these family trees, of which 14 are carriers of the gene with the p.Ser43Asn variant with an average age of 39 years since their diagnosis through genetic study, with a history of origin in the Loja canton since its first generation, and with current residence in the province and city of Loja, province of Zamora and in the city of Guayaquil. Chronology figure 1
The observation of the first cases allowed us to identify more families different from those mentioned in the article. Currently, from the first families studied, there are a total of 22 cases carrying the variant until January 2024, coming from the same sector.
The so-called “founder effect” occurs when a population is established from a small number of individuals extracted from an ancestral population”.[10]
It is known that the phenotypic expression of hTTR amyloidosis is variable according to the type of mutation; Its geographical origin may not be a predictor of the expression of the disease, however it provides information about the associated variant.[11,12]
Current pharmacological therapies seek to attack different points of amyloidogenesis: production, deposit and elimination.
There are so-called genetic RNA silencers (siRNAs) that cause selective degradation of the TTR mRNA, among these are: Patisiran, Revusiran, Vutrisiran.
Among the antisense oligonucleotides (ASOs) are Inotersen and Eplontersen, which reduces the expression of TTR by introducing nonsense mutations in the TTR gene.
ASOs and siRNAs are designed to alter the disease phenotype of ATTR amyloidosis by degrading both wild-type and mutant TTR RNA transcripts, reducing the synthesis of TTR protein.[13] The two main gene silencing compounds that have undergone extensive clinical assessment in ATTR include inotersen (ASO) and patisiran (siRNA) and have been approved for treating stage 1 and 2 ATTRv.[14]
Patisiran (siRNA) with a reduction [15] of 85% after the second dose to a maximum of 96% with adverse reactions related to infusion and inotersen which was shown through phase III clinical trials to improve the course of polyneuropathy in stages I and II as well as the quality of life of patients, estimated by results in the score on the Norfolk Quality questionnaire reported by the patient.[16]
Life-Diabetic Neuropathy (QOL-DN) questionnaire. The adverse events found in a study of 117 patients were glomerulonephritis in 3%, thrombocytopenia in 2%, and important adverse effects that require safety monitoring for early detection and treatment.[16]
Among the main transthyretin stabilizing drugs are Tafamidis and diflunisal.
Tafamidis or 2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid that acts selectively by bind to TTR and stabilize both wild-type TTR and mutant TTR. There are several clinical trials and extension studies that demonstrate its good efficacy and adequate tolerability. During the first double-blind placebo-controlled trial (Fx-005), four adverse events were identified: diarrhea, urinary tract infection,abdominal pain and vaginal infection.[17] These events were related to disease progression in subsequent studies.
Diflunisal, a non-steroidal anti-inflammatory drug, has also been shown to stabilize transthyretin, however, due to the presence of toxicity in patients with cardiomyopathy, it is not recommended.[18]
Removing amyloid deposits is another important mechanism that is being investigated. The drugs considered to act within cardiac clearance are doxycycline and tauroursodeoxycholic bile acid (TUDCA) with an acceptable profile and safety.[19] EGCG (epigallocatechin-3 gallate)—the most abundant polyphenol in green tea, which has shown a reduction in ventricular mass from 6% to 12.5%.[20,21]
Liver transplantation (LT) was the first approved treatment by eliminating the production of Amyloidogenic mutated TTR produced by the liver.[22]
However, although effective, there are some limitations:a major surgical procedure inevitably associated with its inherent risks; there is a limited number of compatible grafts and some patients still have disease progression due to continued formation of TTR amyloid fibrils.[22]
In a systematic review on the results of isolated heart transplantation, a total of 123 patients were analyzed, finding favorable results with a mean survival of 4.33 years.[23]
With current advances, a multidisciplinary approach, new therapies and adequate patient selection, there is an encouraging future.
In Ecuador, pharmacological therapies are not yet available and there are certain limitations to access to the availability of certain imaging studies and genetic tests that generate delays in the diagnosis of this disease.
It is important to know the possible epigenetic factors that establish the relationship between genetic determinants and external environmental factors such as: diet, toxins, environmental polluting factors, physical activity and mental state that allow establishing the phenotypic expression of this variant.[24]
The real incidence of amyloidosis in Ecuador, and the existing variants, are unknown, so continuing with this type of studies will allow us to know the real data and the implementation of programs that allow improving care and providing support for what is necessary for its diagnosis and treatment.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies with humans were approved by Ethics Committee of the Manuel Ygnacio Monteros Hospital of Loja, Ecuador. The studies were carried out in accordance with local legislation. Participants provided written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for publication of any potentially identifiable images or data included in this article.
Author Contributions
DL: Investigation, Writing – original draft, Visualization, Conceptualization, Methodology, Software, Data curation, Funding, Resources. BA: Investigation, Writing – original draft, Visualization, Conceptualization, Methodology, Software, Data curation, Funding, Resources. HV: Conceptualization, Methodology, Software, Data curation.
Funding
The author(s) declared that they did not receive financial support for the research, authorship, and/or publication of this article from any institution.
Conflict Of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Note And Acknowledgments
Scientific research in medicine in Ecuador faces various challenges that affect its development and progression. Lack of funding is one of the main obstacles to research, the resources allocated to research projects are usually limited, making it difficult to carry out comprehensive studies and acquire advanced equipment. Among other points, the infrastructure dedicated to medical research may be insufficient in terms of equipped laboratories, cutting-edge facilities, and access to specialized technology. Likewise, all this limits the ability of researchers to carry out high-quality research, therefore we have seen many cases where our patients go to other medical entities located in developed countries where medical studies have been carried out and scientific advances have been determined. It is a shame to know that the beginning of the investigation of that patient arose in our country Ecuador and due to lack of resources and support these relevant investigations cannot be developed. The authors would like to thank in the most cordial way the Hospital Manuel Ygnacio Monteros of the city of Loja, for the opening of the project presented on May 04, 2021, in force. The collaboration of the Gastroenterology services of the Manuel Ygnacio Monteros Hospital, Dr. Lenin Alban, and other service colleagues. Collaboration of Drs. Jessenia Bravo, cardiology specialist at Manuel Ygnacio Monteros Hospital. Collaboration of Drs. Ximena Cardenas, neurology specialist at Manuel Ygnacio Monteros Hospital. We thank Dr. Miguel David Alvarez Saltos, Research Physician with Higher Education registration No. REG-INV-23-06967, for his help in correcting and translating this article.
References
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T. , et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016, 29, S27–35. [Google Scholar] [CrossRef] [PubMed]
- Guamán Charco, E.D.; Rodrigo Henríquez, A. Carga de enfermedad por insuficiencia cardiaca en Ecuador durante el periodo 2014-2018. Metro Ciencia. 2021, 29, 83–85. [Google Scholar] [CrossRef]
- González-López, E.; López-Sainz, Á.; Garcia-Pavia, P. Diagnóstico y tratamiento de la amiloidosis cardiaca por transtiretina. Progreso y esperanza. Vol. 70, Revista Espanola de Cardiologia. Ediciones Doyma, S.L.; 2017. p. 991–1004.
- Papathanasiou, M.; Carpinteiro, A.; Kersting, D.; Jakstaite, A.M.; Hagenacker, T.; Schlosser, T.W. , et al. Rare variant (p.Ser43Asn) of familial transthyretin amyloidosis associated with isolated cardiac phenotype: A case series with literature review. Mol Genet Genomic Med. 2021; 9. [Google Scholar]
- Castaño, A.; Bokhari, S.; Brannagan, T.H.; Wynn, J.; Maurer, M.S. Technetium pyrophosphate myocardial uptake and peripheral neuropathy in a rare variant of familial transthyretin (TTR) amyloidosis (Ser23Asn): A case report and literature review. Amyloid 2012, 19, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and treatment approaches involving transthyretin in amyloidogenic diseases. International Journal of Molecular Sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Dra. Dorota Rowczenio, Dr. Dra. Dorota Rowczenio, Dr. Ashutosh Wechalekar. Mutations in Hereditary Amyloidosis. 2024. [Google Scholar]
- Porres-López, E.; de Frutos, F.; Silva-Hernández, L.; Galán, L.; González-López, E.; García-Pavía, P. Hereditary transthyretin amyloidosis caused by p.Ser43Asn variant. A new endemic variant in Ecuador. Revista Española de Cardiología (English Edition) [Internet]. 2017;70(11):564–6. Available from: https://www.revespcardiol.org/en-hereditary-transthyretin-amyloidosis-caused-by-articulo-S1885585723000452. [CrossRef] [PubMed]
- Daoko, J.; Elnahar, Y.; Kersh KEl Mohammad, N.; Shamoon, F. Cardiac mri detection of a rare case of familial cardiac amyloidosis (Ser23asn): Case report with literature review. Reports in Medical Imaging 2010, 3, 123–127. [Google Scholar]
- Kirov, A.; Sarafov, S.; Pavlova, Z.; Todorov, T.; Chamova, T.; Gospodinova, M. , et al. Founder effect of the Glu89Gln TTR mutation in the Bulgarian population. Amyloid. 2019, 26, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y. , et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020, 27, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R. The reality and importance of founder speciation in evolution. BioEssays 2008, 30, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Jono, H. Review Recent Advances in Oligonucleotide-Based Therapy for Transthyretin Amyloidosis: Clinical Impact and Future Prospects [Internet]. Vol. 41, Biol. Pharm. Bull. 1737. Available from: http://www.
- Brannagan, T.H.; Berk, J.L.; Gillmore, J.D.; Maurer, M.S.; Waddington-Cruz, M.; Fontana, M. , et al. Liver-directed drugs for transthyretin-mediated amyloidosis. Journal of the Peripheral Nervous System. 2022, 27, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J. , et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis.
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K. , et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. New England Journal of Medicine. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- de Campos, C.F.; Conceição, I. Updated Evaluation of the Safety, Efficacy and Tolerability of Tafamidis in the Treatment of Hereditary Transthyretin Amyloid Polyneuropathy. Drug, Healthcare and Patient Safety 2023, 15, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Dyck, P.J.; Obici, L.; Zeldenrust, S.R.; Sekijima, Y.; Yamashita, T. , et al. The diflunisal trial: update on study drug tolerance and disease progression. Amyloid [Internet]. 2011 Jun 18;18(sup1):196–7. Available from: http://www.tandfonline.com/doi/full/10.3109/13506129.2011.574354073. [CrossRef] [PubMed]
- Castaño, A.; Bokhari, S.; Brannagan, T.H.; Wynn, J.; Maurer, M.S. Technetium pyrophosphate myocardial uptake and peripheral neuropathy in a rare variant of familial transthyretin (TTR) amyloidosis (Ser23Asn): A case report and literature review. Amyloid 2012, 19, 41–46. [Google Scholar] [CrossRef]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P. , et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clinical Research in Cardiology 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Vol. 15, Frontiers in Neuroscience. Frontiers Media S.A.; 2021.
- Ericzon, B.G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R. , et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Moriguchi, J.; Levine, R.; Chan, J.; Dimbil, S.; Patel, J. , et al. Outcomes of Heart Transplantation in Cardiac Amyloidosis Patients: A Single Center Experience. Transplant Proc. 2021, 53, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Azad, P.; Stobdan, T.; Zhou, D.; Hartley, I.; Akbari, A.; Bafna, V. , et al. High-altitude adaptation in humans: from genomics to integrative physiology. Journal of Molecular Medicine 2017, 95, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Transthoracic Doppler Echocardiogram Study.Source: Own elaboration, 2021. (A) 4-chamber apical section demonstrating septal thickening, interatrial septum, and atrioventricular leaflets. (B) 2-chamber apical section demonstrating thickening of its inferior and anterior walls. (C) 5-chamber apical section denoting ventricular thickening and mitral valve leaflets, and visualization of the aortic valve. (D) Polar map with a total Longitudinal Global Strain: -5%. Doppler ultrasound. (E) significant drop in tissue velocities is observed, e' wave of 0.03m/s at the septal level.
Figure 1.
Transthoracic Doppler Echocardiogram Study.Source: Own elaboration, 2021. (A) 4-chamber apical section demonstrating septal thickening, interatrial septum, and atrioventricular leaflets. (B) 2-chamber apical section demonstrating thickening of its inferior and anterior walls. (C) 5-chamber apical section denoting ventricular thickening and mitral valve leaflets, and visualization of the aortic valve. (D) Polar map with a total Longitudinal Global Strain: -5%. Doppler ultrasound. (E) significant drop in tissue velocities is observed, e' wave of 0.03m/s at the septal level.
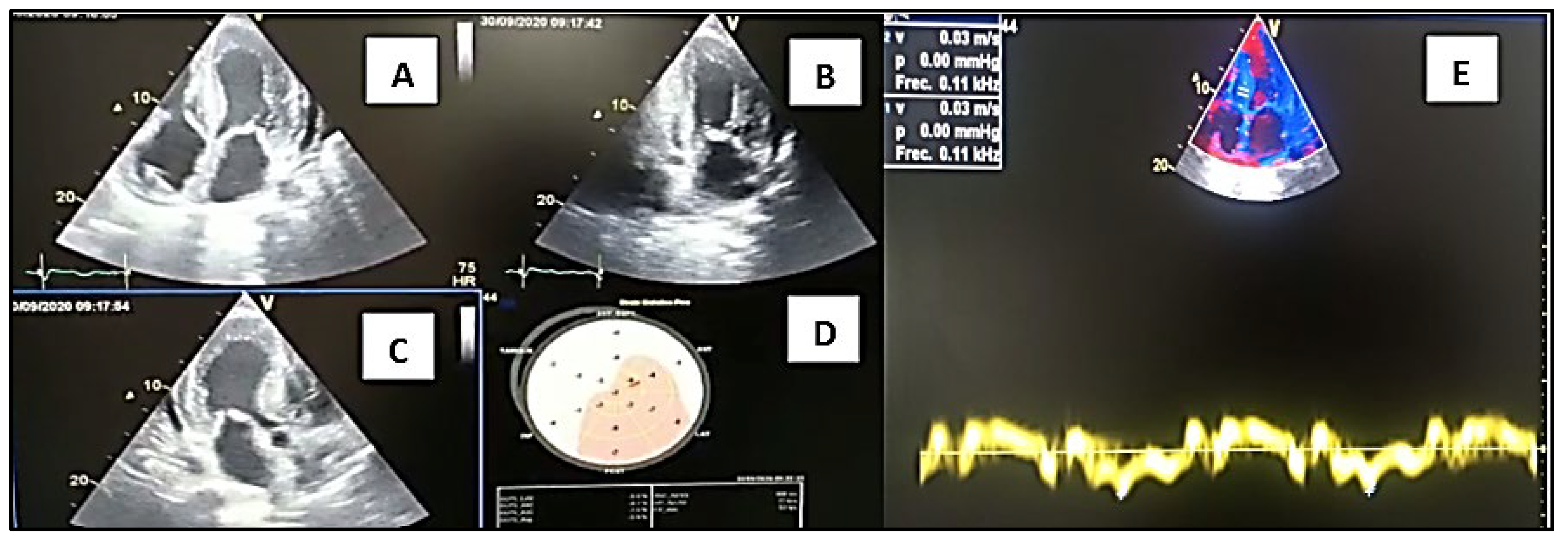
Figure 2.
Source: Own elaboration, 2021.Family tree analyses.
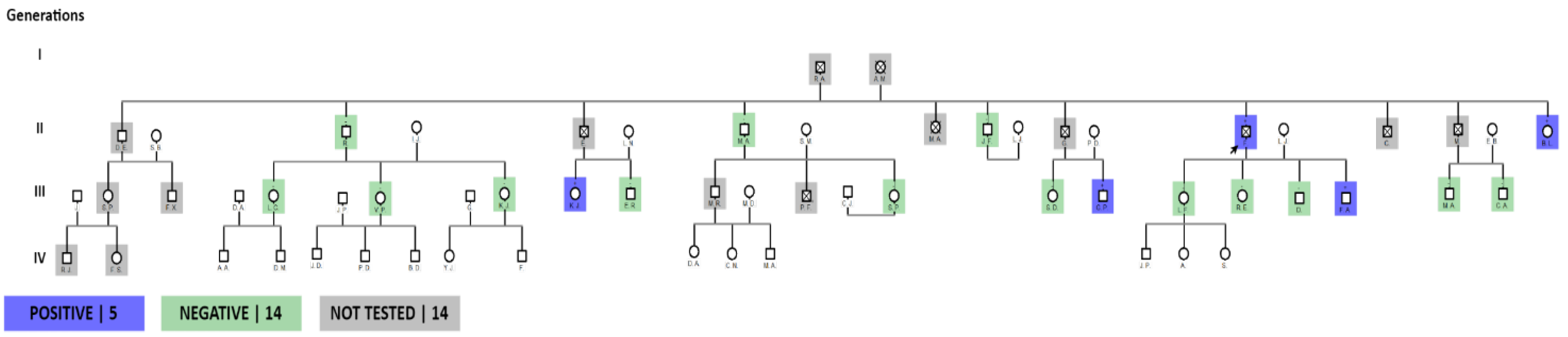
Figure 3.
Source: Own elaboration, 2021. (A) Electrocardiogram demonstrating first degree AV block, complete right bundle branch block, inferior pseudo infarction. (B) Apical 4-chamber image shows a very thickened 24mm interventricular septum, thickened interatrial septum, as well as the mitral valve leaflets and mild pericardial effusion.
Figure 3.
Source: Own elaboration, 2021. (A) Electrocardiogram demonstrating first degree AV block, complete right bundle branch block, inferior pseudo infarction. (B) Apical 4-chamber image shows a very thickened 24mm interventricular septum, thickened interatrial septum, as well as the mitral valve leaflets and mild pericardial effusion.
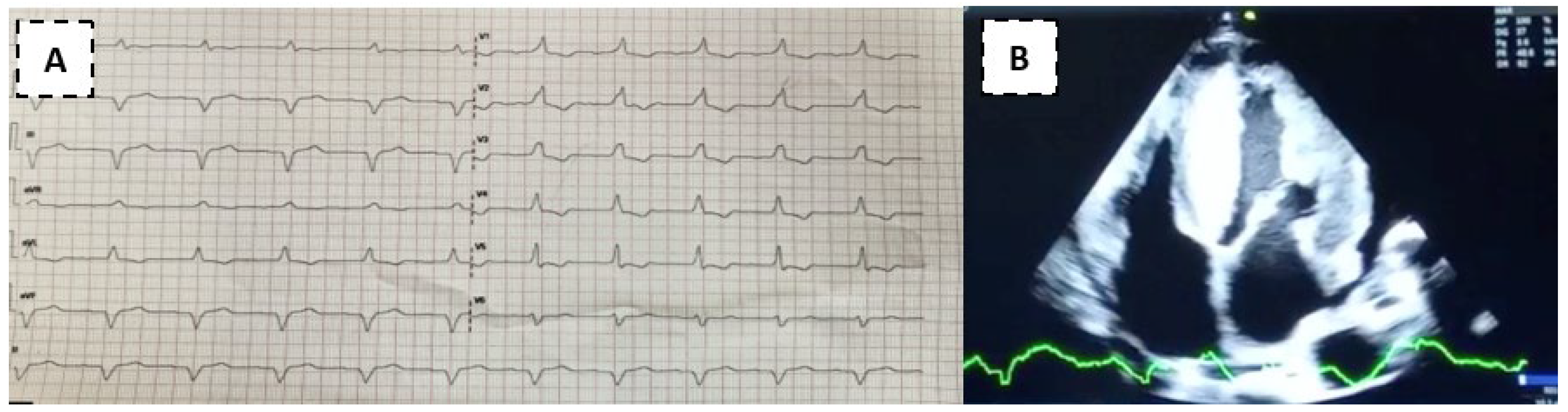
Figure 4.
Source: Own elaboration, 2021.
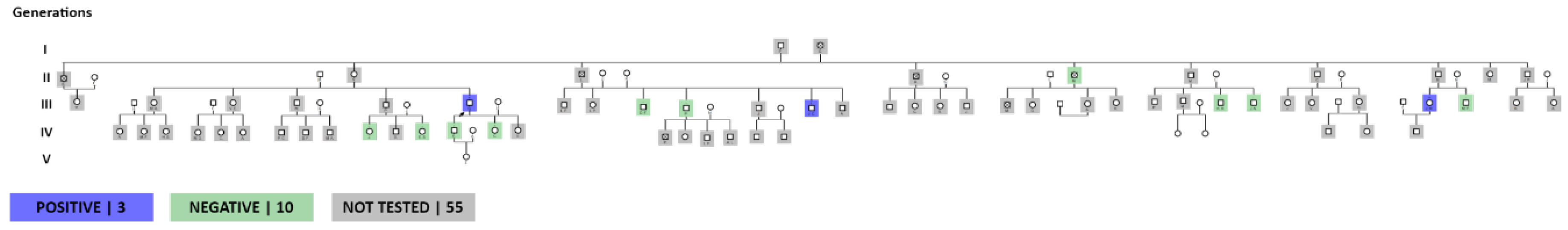
Figure 5.
Source: Own elaboration, 2021. (A) Apical 4-chamber image shows a very thickened 26mm interventricular septum, thickened interatrial septum, and atrioventricular leaflets and pericardial effusion.
Figure 5.
Source: Own elaboration, 2021. (A) Apical 4-chamber image shows a very thickened 26mm interventricular septum, thickened interatrial septum, and atrioventricular leaflets and pericardial effusion.
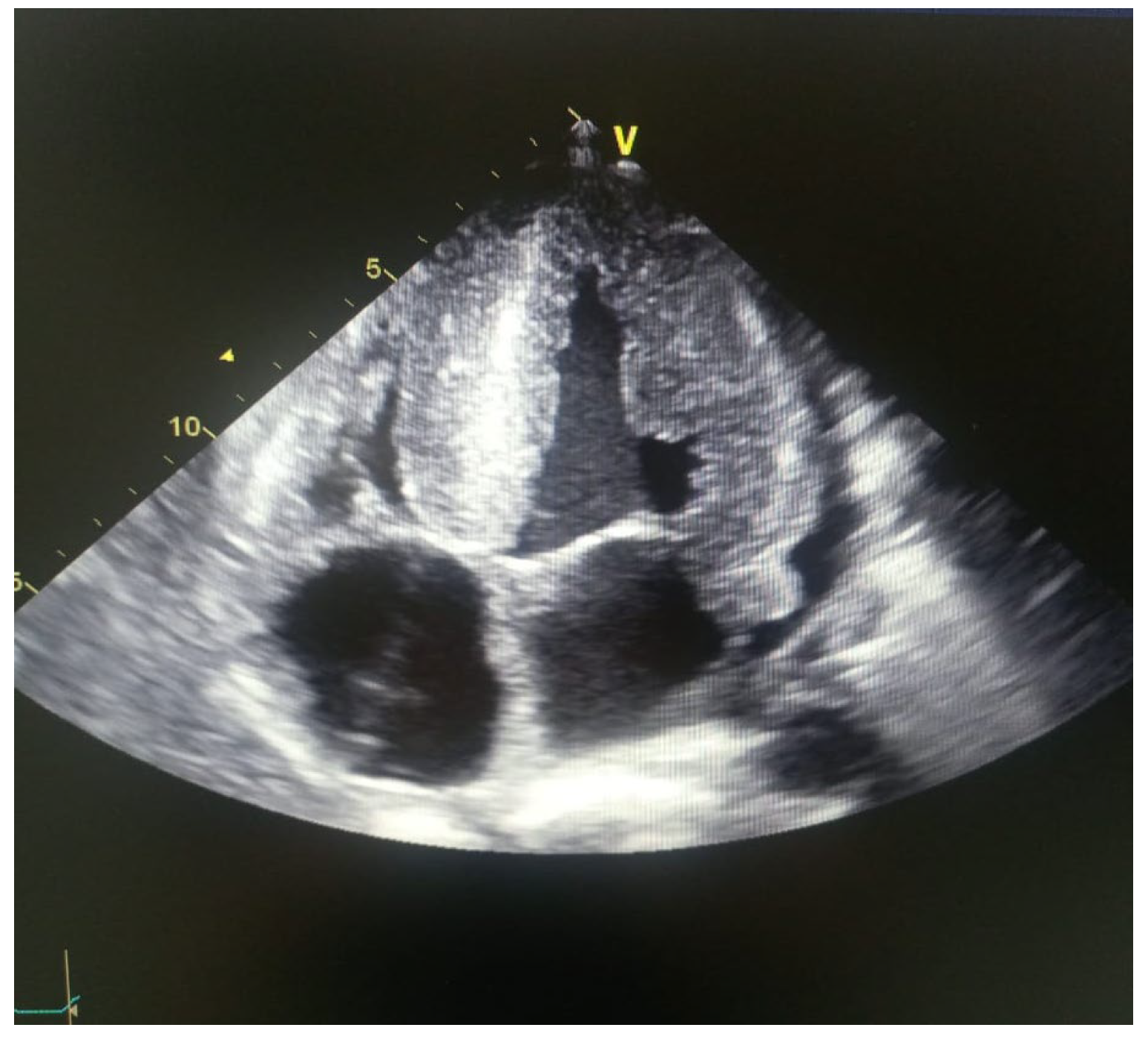
Figure 7.
chronological order.

| DEMOGRAPHIC CHARACTERISTICS. | |||
|---|---|---|---|
| Case 1 | Case 2 | Case 3 | |
| AGE | 59 | 49 | 62 |
| SEX | Male | male | Male |
| PLACE OF ORIGIN | Quilanga | Loja | Quilanga |
| PLACE OF RESIDENCE | Loja | Loja | Quilanga |
| PARENTS' PLACE OF ORIGIN | Quilanga | Quilanga | Quilanga |
| OCCUPATION | Administrator | Businessman | Farmer |
| CLINICAL AND IMAGING CHARACTERISTICS | |||
| ECG FINDINGS | P Inferior pseudo infarction, ventricular extrasystoles | Bifascicular block: RBBB, BAV first degree. Inferior pseudoinfarction | Complete AV block |
| NYHA FUNCTIONAL CLASS | III | II | III |
| MYOCARDIOPATHY ON ECHOCARDIOGRAM | Yes | yes | Yes |
| NEUROLOGICAL EVALUATION/POLYNEUROPATHY NEUROPATHY | Yes | no | Yes |
| CARPAL TUNNEL SYNDROME | No | no | No |
| GASTROINTESTINAL MANIFESTATIONS | Yes | yes | Yes |
| DEVICE PLACEMENT | Yes (CDI) | no | No |
| OUTCOMES | Syncope-TBI/death | Referred to heart transplant | Cardiogenic shock/death |
Source: Own elaboration, 2021s.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Submitted:
20 June 2024
Posted:
21 June 2024
You are already at the latest version
Alerts
This version is not peer-reviewed
Submitted:
20 June 2024
Posted:
21 June 2024
You are already at the latest version
Alerts
Abstract
Approximately more than 120 transthyretin mutations are known whose variation in clinical presentation is heterogeneous, as the course of disease onset depends on genetic variation and level of penetrance. It is little known in Ecuador, and some of the reported cases; suggest to the family tree analysis that they come from a province that is possibly considered endemic. The main objective is to carry out a descriptive cross-sectional analysis on the presentation of transthyretin amyloidosis in families carrying the p.Ser43Asn gene from the index case identified.
Keywords:
Subject: Medicine and Pharmacology - Cardiac and Cardiovascular Systems
1. Introduction
Transthyretin amyloidosis is a rare, multisystem, underdiagnosed disease that causes a great impact on quality of life, with increased mortality when there is cardiac involvement; that deserves to be diagnosed in time and seek measures for multidisciplinary follow-up.[1]There are two forms of transthyretin amyloidosis: the wild form and the genetic form. Some mutations are more frequent and others rare.
Transthyretin amyloidosis is described as having a heterogeneous natural history, and therefore a poor prognosis; with a mean survival from diagnosis of 3.6 years for wild-type amyloidosis and 2.5 years for transthyretin amyloidosis of the Vall 122lle variant.
In our first case, the time elapsed to establish a diagnosis of transthyretin amyloidosis from the onset of symptoms was 7 years, through the retrospective analysis of the clinical history of a patient treated at the Manuel Ygnacio Monteros-IESS Loja General Hospital with a mixed phenotype. This demonstrates a reality in Ecuador about the existing gaps in our health system and the need to carry out studies on the presentation of transthyretin amyloidosis.
The identification of the first case of transthyretin amyloidosis diagnosed at the Manuel Ygnacio Monteros Hospital with the pSer43Asn variant has motivated us to carry out a descriptive study of those index cases identified as carriers of the mutant transthyretin gene . The review of this work will allow establishing inter-institutional behaviors and protocols in the future for the management of this rare pathology not very well known by our Ecuadorian society.
The delay in diagnosis and the rapid progression after the appearance of symptoms led to the death of our first patient, with manifestations of hypertrophic cardiomyopathy, heart failure, and sensory-motor polyneuropathy.[1]
It is considered necessary to make a timely and early diagnosis; surveillance and monitoring of manifestations, their time of presentation, supported by multidisciplinary management, generating comprehensive treatment for each of the patients; which will allow us to avoid systemic damage, or fatal outcomes, higher costs due to hospitalizations, and more complex therapies that increase hospital costs.
In Ecuador, in both the comprehensive public, complementary and private health network, there are no laboratories that perform the molecular study for Familial Amyloidosis; The execution of the genetic study is carried out abroad through private intermediary companies that are responsible for processing the delivery of biological material to international genetics laboratories.
The burden of heart failure disease in Ecuador is continuously increasing. During the period 2014-2018, 90,242 years of healthy life (DALY) were lost, of which 46.72% are represented by years of life lost due to premature death (YLL) and 53.28% by years lived with disability (YLD).[2] The prevalence of Amyloidosis in Ecuador is unknown, and the question remains as to how many of our deceased heart failure patients had a rare underlying disease such as Amyloidosis. It is difficult to carry out a post-mortem study to answer the question; However, monitoring those identified as carriers will allow us to know what the first manifestations of cardiac amyloidosis caused by this p.Ser43Asn variant are, which will allow us to put the country on alert.
The objective of this article is to perform a cross-sectional descriptive analysis on the presentation of transthyretin amyloidosis in families carrying the p.Ser43Asn gene based on the index case identified in the period from January 2020 to January 2022.
2. Case Report
2.1. Case Report Number One
A 59-year-old male patient, born in the city of Loja, is seen in the private outpatient service on September 30, 2020, for presenting episodes of lipothymia, dyspnea at rest, generalized weakness, limb edema for 1 month. lower bowels, hyporexia, abundant diarrheal stools 4 to 6 times a day, the same condition that was reported to be chronic for more than 4 years and that was initially classified as Irritable Bowel Syndrome and when consulted it was mentioned to be more frequent in the last 2 years. months, with significant weight loss. Additionally, as with other pathological history: ICD device placed in 2018, due to diagnosis of Hypertrophic Cardiomyopathy and with diagnosis of ALS (Amyotrophic Lateral Sclerosis). He has received treatment for intestinal tuberculosis 1 year ago without clinical improvement. On physical examination: pale facies, cachectic. BP: 81/52mmHg, HR: 80bpm, SatO2:90%. Cardiac auscultation with norm phonetic sounds. Lungs: ventilated. Lower limbs with pretibial edema +2/4. Usual medication: Propranolol 10mg every 48 hours, Amiodarone 200mg daily, furosemide 10mg daily. The electrocardiogram study revealed: sinus rhythm and inferior Pseudo Q. An Echocardiogram study was performed in the consultation, observing left ventricular hypertrophy (SIV: 19mm), right ventricular hypertrophy (Figure 1), thickening of the interatrial septum, global hypokinesia of the left ventricle with LVEF: 24%, decreased velocities on tissue Doppler, diastolic dysfunction grade III, global strain around – 5% and mild pericardial effusion (Figure 2A). Laboratory studies: elevation of NTproBNP: 13430pg/ml, troponin T: 186pg/ml, urea: 60, creatinine: 1.2mg/dl. Chronic iron deficiency anemia. It was necessary to perform new evaluations, including neurology-neurophysiology, which determined the presence of severe sensory-motor polyneuropathy. A skin biopsy study was requested, which was negative: Kappa and Lambda light chain studies were negative. By the Hematology service, the presence of systemic Lupus Erythematosus was determined as an additional finding to the suspicion of Heredofamilial Amyloidosis due to transthyretin, which was the first presumptive diagnosis after the first evaluation. The patient was evaluated in a hospital unit of the Social Security Institute-Manuel Ygnacio Monteros Hospital, where he continued with the evaluations and follow-up; Scintigraphy studies with Tc99m Pyrophosphate are only available in the city of Quito, and Magnetic Resonance studies were not carried out due to lack of availability in the sector. A genetic study was requested for transthyretin amyloidosis, resulting in the presence of heterozygosity around 3 months later, position: chr18:31,592,954, variation p.Ser43Asn.
The patient was admitted to another hospital unit privately in February 2021 due to an episode of syncope and trauma to the spine and died on 03-08-2021.
When performing a retrospective analysis of his clinical history, initial medical attention since 2013 was found with gastrointestinal manifestations, diarrheal stools alternating with constipation, classified as Irritable Bowel Syndrome. He registered his first attention in cardiology in 2016 due to the presence of dizziness, isolated ventricular extrasystoles, with a delay in diagnosis of approximately 5 years.
Family Tree Analysis
The completion of the family group allowed us to observe a total of 11 siblings of the index patient, with a total of 33 people that make up this family group, of which 19 were applied the test, with a total of 5 people with a positive result, 11 not carried out on the test. test, of these 5 died: 1 from SARS-COV2 pneumonia, 1 from acute myocardial infarction (died in the Province of Pichincha due to a diagnosis of Hypertrophic Cardiomyopathy plus acute myocardial infarction) and 3 of unknown cause with probable death from acute myocardial infarction myocardium (Figure 2). When inquiring about the origin of their parents, they claim to be from the Quilanga canton in the province of Loja. This family group has members residing in the Province of Loja.
2.2. Case Report Number Two
A 49-year-old patient from Loja came to the outpatient clinic at the Manuel Ygnacio Monteros-IESS Loja Hospital due to significant edema in the lower limbs. He denied dyspnea or precordial pain. Other reported symptoms: abdominal distention, diffuse abdominal pain and nausea. History of heart failure with reduced LVEF, with suspicion of cardiac amyloidosis according to a report from the Guayaquil hospital 6 months ago. On physical examination: BP:90/50mmHg, HR:60bpm, SatO2:93%. Rhythmic heart, without murmurs. Lungs: MV preserved. Predominantly cardiac clinical presentation with symptoms of heart and gastrointestinal failure (abdominal distension and diarrhea). Electrocardiogram: First degree AV block, complete right bundle branch block, inferior pseudoinfarction (Figure 3A). Transthoracic echocardiogram: predominantly septal left ventricular hypertrophy (SIV: 24mm), right ventricular hypertrophy, thickening of the interatrial septum and atrioventricular valves. Global hypokinesia, LVEF: 35%, Global Longitudinal Strain: -8%. Mild pericardial effusion (Figure 3B). Cardiac Magnetic Resonance Study: Subendocardial pattern suggesting amyloidosis. Skin and adipose tissue biopsy negative for Congo red stain. Kappa and Lambda light chain tests were negative. Gastric and colon biopsy study negative for Congo red stain. The requested genetic study demonstrates heterozygosity, with the p.Ser43Asn variant. The patient was discharged with a heart transplant referral to a level III hospital.
Family tree analysis
Family tree analyses
The analysis of the family tree allows us to observe a total of approximately 68 members, of which 13 people have been tested, with a positive result in 3, the remaining population to be tested is a total of 55. There are members who died before the research, a number of 4 people from the second generation, with myocardial infarction and heart failure (Figure 4). Origin of the family line from the Quilanga canton, in the province of Loja. This family group has members residing in the Province of Loja, Zamora, Guayas and members residing in Spain.
2.3. Case Report Number Three
Patient, 62 years old, male, from the province of Loja, resident in Quilanga, transferred from another hospital unit for pacemaker placement due to a diagnosis of third-degree AV block, hypertrophic cardiomyopathy, and heart failure, reporting a 5-month clinical condition. with general malaise, dyspnea with medium efforts that progresses to small efforts, accompanied by adynamic, and generalized edema, 72 hours ago he presented oppressive precordial pain with nausea and dizziness. Vital signs upon admission: BP:99/79mmHg, HR:36bpm, RR:16 per min, T:36.2 C, Pulse rate: 93% with FiO2 at 24%.
Electrocardiogram study showed complete AV block, he was subjected to pacemaker placement, evaluated with an echocardiogram study after pacemaker implantation: LVEF: 24% significant hypertrophy of the left ventricle with measurement of the interventricular septum around 33mm, LV diastolic diameter of 26mm, 28mm posterior wall, right ventricular systolic dysfunction, TAPSE:13mm, CAF:25%. Global Longitudinal Strain: -9% (Figure 5).
His neurological evaluation demonstrates the presence of stage III sensory-motor polyneuropathy.
Genetic study result on July 7, 2022, of complete sequencing with a positive result for the p.Ser43Asn variant, with heterozygosity.
Patient died at home on March 17, 2023.
Family tree analysis
Figure 6.
Source: Own elaboration, 2021.
Family tree analyses
The family chart graph indicates a total of 51 members, a total of 8 siblings of the index patient, of whom 7 have died, place of death in the province of Loja. Within their family history, the cause of death is without an established diagnosis, however close relatives express that the deceased relatives presented symptoms of generalized weakness, cachexia, dyspnea with an outcome in sudden death. There is a history of consanguinity in the ancestors of the family. Origin of the family from the Quilanga canton-Province of Loja.
Of the patients tested, one is in the Province of Oro, the rest is in the Malacatos Canton, Province of Loja.
The following table shows the baseline demographic characteristics and the clinical characteristics of ECG and imaging of the 3 clinical cases (Table 1).
3. Discussion
Transthyretin amyloidosis is a systemic disease, of variable incidence and still unknown in various parts of the world, with an incidence of 1 per 100,000 inhabitants worldwide.[3] There are some variants considered more frequent in certain countries where they are stipulated as endemic variants in Spain (Mallorca), Portugal, Norway, Japan, Brazil. The p.Ser43Asn variant is a point mutation in exon 2, codon 43 of the TTR gene that leads to a single amino acid substitution of serine for asparagine; It is considered a rare variant, with little information in the literature;[4] There are few case reports on this particular variant. According to the work published by Maria Papahanasiou and colb, published in December 2020, it documents a report of 2 opportunely diagnosed patients from a family in Italy whose identified variant is the same as our cases and which is considered to have a clinically aggressive course. due to its exclusively cardiac phenotype.[5]
There are at least five studies, in addition to the one already mentioned above, as clinical case reports based on the Ser23Asn variant, which corresponds to the same variant that we mention in our work.
This is due to a change in nomeclature. At the moment, the amino acid sequence from 1 to 20 is the signal peptide and the final protein structure is expressed from amino acids 21 to 147, varying the numbering but corresponding to the same amino acid. The variant causes hereditary transthyretin amyloidosis; this protein has a molecular weight of 14 kDa in monomeric form and exists in tetrameric form (55 kDa) in plasma. As mentioned, this is synthesized in the liver and in the choroid plexus as a homotetrameric structure with a dimer of dimers of quaternary structure.[6,7,8]
One of the first studies published with this variant corresponds to Lawreen Helter Connors and collaborators, published in 1999, in which myocardial amyloidosis was identified in a 44-year-old patient from the north of Portugal with heart block requiring a pacemaker and a heart transplant performed by its involvement exclusively at the cardiac level. Another work that talks about this variant is the publication carried out in 2010 by the Daoko et al group, which mentions a 41-year-old Peruvian patient with symptoms of heart failure, with the same variant of amyloidosis limited to the heart.
Similarly, in another review of clinical cases published by Colombian authors who highlight the relevance of the use of the Technetium 99 pyrophosphate scintigraphy technique for the diagnosis of hereditary transthyretinal amyloidosis in a 41-year-old Ecuadorian patient with symptoms of fatigue, difficulty breathing and peripheral neuropathy, whose identified variant corresponds to Ser23ASn that possibly has Spanish origin. Different researchers mention a possible origin of the variant in Portugal, Spain and Italy[9]; It is difficult to know precisely the origin of the variant and its presence is attributed to migration and colonization processes; with a possible founding effect in the province of Loja, particularly in the Quilanga canton.
In Ecuador, some clinical cases of amyloidosis without specification of the genetic type have been documented since 2018, which is explained by the limitations of the public health system in accessing imaging studies and other studies not available in public hospitals.
In our city of Loja, capital of the province of Loja in the South of the country, our first case of Transthyretin amyloidosis was diagnosed in 2020 with genetic determination of the variant; who debuted with hypertrophic cardiomyopathy, sensory-motor polyneuropathy. as the main manifestations of amyloidosis that led to death. In the retrospective analysis of the first symptoms, gastrointestinal discomfort was found as the first manifestation: diarrhea and constipation, added to the presence of arterial hypotension and dizziness, which confers a mixed phenotype presentation with a delay time of 7 years.
The second case shows gastrointestinal discomfort as first manifestations; There is no neurological involvement, and it was considered to have an exclusively cardiac phenotype. And in the third case, the mixed phenotypic presentation is repeated, with initial gastrointestinal manifestations, with sensory-motor polyneuropathy and cardiac involvement.
According to the families studied, it is possible that there is an approximation of consanguinity between the first and third cases. However, this information cannot be validated by those who would present said consanguinity due to their death.
In case 1, his relatives mention having ancestors from Spain who arrived in Ecuador and traveled to Colombia and Peru, finally deciding to settle in Ecuador in the Province of Loja.
In case number 2, they mention having ancestors from Spain and in the third case they do not know their origins.
The present cascade study, which consists of carrying out the genetic study on family members, allowed us to discover a total of 14 positive patients in the period from January 2020 to. January 2024, constituting an effective method to carry out research on relatives of I, II, III degree of consanguinity (Figure 7).
Tests were carried out on a total of 44 people from these family trees, of which 14 are carriers of the gene with the p.Ser43Asn variant with an average age of 39 years since their diagnosis through genetic study, with a history of origin in the Loja canton since its first generation, and with current residence in the province and city of Loja, province of Zamora and in the city of Guayaquil. Chronology figure 1
The observation of the first cases allowed us to identify more families different from those mentioned in the article. Currently, from the first families studied, there are a total of 22 cases carrying the variant until January 2024, coming from the same sector.
The so-called “founder effect” occurs when a population is established from a small number of individuals extracted from an ancestral population”.[10]
It is known that the phenotypic expression of hTTR amyloidosis is variable according to the type of mutation; Its geographical origin may not be a predictor of the expression of the disease, however it provides information about the associated variant.[11,12]
Current pharmacological therapies seek to attack different points of amyloidogenesis: production, deposit and elimination.
There are so-called genetic RNA silencers (siRNAs) that cause selective degradation of the TTR mRNA, among these are: Patisiran, Revusiran, Vutrisiran.
Among the antisense oligonucleotides (ASOs) are Inotersen and Eplontersen, which reduces the expression of TTR by introducing nonsense mutations in the TTR gene.
ASOs and siRNAs are designed to alter the disease phenotype of ATTR amyloidosis by degrading both wild-type and mutant TTR RNA transcripts, reducing the synthesis of TTR protein.[13] The two main gene silencing compounds that have undergone extensive clinical assessment in ATTR include inotersen (ASO) and patisiran (siRNA) and have been approved for treating stage 1 and 2 ATTRv.[14]
Patisiran (siRNA) with a reduction [15] of 85% after the second dose to a maximum of 96% with adverse reactions related to infusion and inotersen which was shown through phase III clinical trials to improve the course of polyneuropathy in stages I and II as well as the quality of life of patients, estimated by results in the score on the Norfolk Quality questionnaire reported by the patient.[16]
Life-Diabetic Neuropathy (QOL-DN) questionnaire. The adverse events found in a study of 117 patients were glomerulonephritis in 3%, thrombocytopenia in 2%, and important adverse effects that require safety monitoring for early detection and treatment.[16]
Among the main transthyretin stabilizing drugs are Tafamidis and diflunisal.
Tafamidis or 2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid that acts selectively by bind to TTR and stabilize both wild-type TTR and mutant TTR. There are several clinical trials and extension studies that demonstrate its good efficacy and adequate tolerability. During the first double-blind placebo-controlled trial (Fx-005), four adverse events were identified: diarrhea, urinary tract infection,abdominal pain and vaginal infection.[17] These events were related to disease progression in subsequent studies.
Diflunisal, a non-steroidal anti-inflammatory drug, has also been shown to stabilize transthyretin, however, due to the presence of toxicity in patients with cardiomyopathy, it is not recommended.[18]
Removing amyloid deposits is another important mechanism that is being investigated. The drugs considered to act within cardiac clearance are doxycycline and tauroursodeoxycholic bile acid (TUDCA) with an acceptable profile and safety.[19] EGCG (epigallocatechin-3 gallate)—the most abundant polyphenol in green tea, which has shown a reduction in ventricular mass from 6% to 12.5%.[20,21]
Liver transplantation (LT) was the first approved treatment by eliminating the production of Amyloidogenic mutated TTR produced by the liver.[22]
However, although effective, there are some limitations:a major surgical procedure inevitably associated with its inherent risks; there is a limited number of compatible grafts and some patients still have disease progression due to continued formation of TTR amyloid fibrils.[22]
In a systematic review on the results of isolated heart transplantation, a total of 123 patients were analyzed, finding favorable results with a mean survival of 4.33 years.[23]
With current advances, a multidisciplinary approach, new therapies and adequate patient selection, there is an encouraging future.
In Ecuador, pharmacological therapies are not yet available and there are certain limitations to access to the availability of certain imaging studies and genetic tests that generate delays in the diagnosis of this disease.
It is important to know the possible epigenetic factors that establish the relationship between genetic determinants and external environmental factors such as: diet, toxins, environmental polluting factors, physical activity and mental state that allow establishing the phenotypic expression of this variant.[24]
The real incidence of amyloidosis in Ecuador, and the existing variants, are unknown, so continuing with this type of studies will allow us to know the real data and the implementation of programs that allow improving care and providing support for what is necessary for its diagnosis and treatment.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies with humans were approved by Ethics Committee of the Manuel Ygnacio Monteros Hospital of Loja, Ecuador. The studies were carried out in accordance with local legislation. Participants provided written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for publication of any potentially identifiable images or data included in this article.
Author Contributions
DL: Investigation, Writing – original draft, Visualization, Conceptualization, Methodology, Software, Data curation, Funding, Resources. BA: Investigation, Writing – original draft, Visualization, Conceptualization, Methodology, Software, Data curation, Funding, Resources. HV: Conceptualization, Methodology, Software, Data curation.
Funding
The author(s) declared that they did not receive financial support for the research, authorship, and/or publication of this article from any institution.
Conflict Of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Note And Acknowledgments
Scientific research in medicine in Ecuador faces various challenges that affect its development and progression. Lack of funding is one of the main obstacles to research, the resources allocated to research projects are usually limited, making it difficult to carry out comprehensive studies and acquire advanced equipment. Among other points, the infrastructure dedicated to medical research may be insufficient in terms of equipped laboratories, cutting-edge facilities, and access to specialized technology. Likewise, all this limits the ability of researchers to carry out high-quality research, therefore we have seen many cases where our patients go to other medical entities located in developed countries where medical studies have been carried out and scientific advances have been determined. It is a shame to know that the beginning of the investigation of that patient arose in our country Ecuador and due to lack of resources and support these relevant investigations cannot be developed. The authors would like to thank in the most cordial way the Hospital Manuel Ygnacio Monteros of the city of Loja, for the opening of the project presented on May 04, 2021, in force. The collaboration of the Gastroenterology services of the Manuel Ygnacio Monteros Hospital, Dr. Lenin Alban, and other service colleagues. Collaboration of Drs. Jessenia Bravo, cardiology specialist at Manuel Ygnacio Monteros Hospital. Collaboration of Drs. Ximena Cardenas, neurology specialist at Manuel Ygnacio Monteros Hospital. We thank Dr. Miguel David Alvarez Saltos, Research Physician with Higher Education registration No. REG-INV-23-06967, for his help in correcting and translating this article.
References
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T. , et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016, 29, S27–35. [Google Scholar] [CrossRef] [PubMed]
- Guamán Charco, E.D.; Rodrigo Henríquez, A. Carga de enfermedad por insuficiencia cardiaca en Ecuador durante el periodo 2014-2018. Metro Ciencia. 2021, 29, 83–85. [Google Scholar] [CrossRef]
- González-López, E.; López-Sainz, Á.; Garcia-Pavia, P. Diagnóstico y tratamiento de la amiloidosis cardiaca por transtiretina. Progreso y esperanza. Vol. 70, Revista Espanola de Cardiologia. Ediciones Doyma, S.L.; 2017. p. 991–1004.
- Papathanasiou, M.; Carpinteiro, A.; Kersting, D.; Jakstaite, A.M.; Hagenacker, T.; Schlosser, T.W. , et al. Rare variant (p.Ser43Asn) of familial transthyretin amyloidosis associated with isolated cardiac phenotype: A case series with literature review. Mol Genet Genomic Med. 2021; 9. [Google Scholar]
- Castaño, A.; Bokhari, S.; Brannagan, T.H.; Wynn, J.; Maurer, M.S. Technetium pyrophosphate myocardial uptake and peripheral neuropathy in a rare variant of familial transthyretin (TTR) amyloidosis (Ser23Asn): A case report and literature review. Amyloid 2012, 19, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and treatment approaches involving transthyretin in amyloidogenic diseases. International Journal of Molecular Sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Dra. Dorota Rowczenio, Dr. Dra. Dorota Rowczenio, Dr. Ashutosh Wechalekar. Mutations in Hereditary Amyloidosis. 2024. [Google Scholar]
- Porres-López, E.; de Frutos, F.; Silva-Hernández, L.; Galán, L.; González-López, E.; García-Pavía, P. Hereditary transthyretin amyloidosis caused by p.Ser43Asn variant. A new endemic variant in Ecuador. Revista Española de Cardiología (English Edition) [Internet]. 2017;70(11):564–6. Available from: https://www.revespcardiol.org/en-hereditary-transthyretin-amyloidosis-caused-by-articulo-S1885585723000452. [CrossRef] [PubMed]
- Daoko, J.; Elnahar, Y.; Kersh KEl Mohammad, N.; Shamoon, F. Cardiac mri detection of a rare case of familial cardiac amyloidosis (Ser23asn): Case report with literature review. Reports in Medical Imaging 2010, 3, 123–127. [Google Scholar]
- Kirov, A.; Sarafov, S.; Pavlova, Z.; Todorov, T.; Chamova, T.; Gospodinova, M. , et al. Founder effect of the Glu89Gln TTR mutation in the Bulgarian population. Amyloid. 2019, 26, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y. , et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020, 27, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R. The reality and importance of founder speciation in evolution. BioEssays 2008, 30, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Jono, H. Review Recent Advances in Oligonucleotide-Based Therapy for Transthyretin Amyloidosis: Clinical Impact and Future Prospects [Internet]. Vol. 41, Biol. Pharm. Bull. 1737. Available from: http://www.
- Brannagan, T.H.; Berk, J.L.; Gillmore, J.D.; Maurer, M.S.; Waddington-Cruz, M.; Fontana, M. , et al. Liver-directed drugs for transthyretin-mediated amyloidosis. Journal of the Peripheral Nervous System. 2022, 27, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J. , et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis.
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K. , et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. New England Journal of Medicine. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- de Campos, C.F.; Conceição, I. Updated Evaluation of the Safety, Efficacy and Tolerability of Tafamidis in the Treatment of Hereditary Transthyretin Amyloid Polyneuropathy. Drug, Healthcare and Patient Safety 2023, 15, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Dyck, P.J.; Obici, L.; Zeldenrust, S.R.; Sekijima, Y.; Yamashita, T. , et al. The diflunisal trial: update on study drug tolerance and disease progression. Amyloid [Internet]. 2011 Jun 18;18(sup1):196–7. Available from: http://www.tandfonline.com/doi/full/10.3109/13506129.2011.574354073. [CrossRef] [PubMed]
- Castaño, A.; Bokhari, S.; Brannagan, T.H.; Wynn, J.; Maurer, M.S. Technetium pyrophosphate myocardial uptake and peripheral neuropathy in a rare variant of familial transthyretin (TTR) amyloidosis (Ser23Asn): A case report and literature review. Amyloid 2012, 19, 41–46. [Google Scholar] [CrossRef]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P. , et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clinical Research in Cardiology 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Vol. 15, Frontiers in Neuroscience. Frontiers Media S.A.; 2021.
- Ericzon, B.G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R. , et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Moriguchi, J.; Levine, R.; Chan, J.; Dimbil, S.; Patel, J. , et al. Outcomes of Heart Transplantation in Cardiac Amyloidosis Patients: A Single Center Experience. Transplant Proc. 2021, 53, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Azad, P.; Stobdan, T.; Zhou, D.; Hartley, I.; Akbari, A.; Bafna, V. , et al. High-altitude adaptation in humans: from genomics to integrative physiology. Journal of Molecular Medicine 2017, 95, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Transthoracic Doppler Echocardiogram Study.Source: Own elaboration, 2021. (A) 4-chamber apical section demonstrating septal thickening, interatrial septum, and atrioventricular leaflets. (B) 2-chamber apical section demonstrating thickening of its inferior and anterior walls. (C) 5-chamber apical section denoting ventricular thickening and mitral valve leaflets, and visualization of the aortic valve. (D) Polar map with a total Longitudinal Global Strain: -5%. Doppler ultrasound. (E) significant drop in tissue velocities is observed, e' wave of 0.03m/s at the septal level.
Figure 1.
Transthoracic Doppler Echocardiogram Study.Source: Own elaboration, 2021. (A) 4-chamber apical section demonstrating septal thickening, interatrial septum, and atrioventricular leaflets. (B) 2-chamber apical section demonstrating thickening of its inferior and anterior walls. (C) 5-chamber apical section denoting ventricular thickening and mitral valve leaflets, and visualization of the aortic valve. (D) Polar map with a total Longitudinal Global Strain: -5%. Doppler ultrasound. (E) significant drop in tissue velocities is observed, e' wave of 0.03m/s at the septal level.
Figure 2.
Source: Own elaboration, 2021.Family tree analyses.
Figure 3.
Source: Own elaboration, 2021. (A) Electrocardiogram demonstrating first degree AV block, complete right bundle branch block, inferior pseudo infarction. (B) Apical 4-chamber image shows a very thickened 24mm interventricular septum, thickened interatrial septum, as well as the mitral valve leaflets and mild pericardial effusion.
Figure 3.
Source: Own elaboration, 2021. (A) Electrocardiogram demonstrating first degree AV block, complete right bundle branch block, inferior pseudo infarction. (B) Apical 4-chamber image shows a very thickened 24mm interventricular septum, thickened interatrial septum, as well as the mitral valve leaflets and mild pericardial effusion.
Figure 4.
Source: Own elaboration, 2021.
Figure 5.
Source: Own elaboration, 2021. (A) Apical 4-chamber image shows a very thickened 26mm interventricular septum, thickened interatrial septum, and atrioventricular leaflets and pericardial effusion.
Figure 5.
Source: Own elaboration, 2021. (A) Apical 4-chamber image shows a very thickened 26mm interventricular septum, thickened interatrial septum, and atrioventricular leaflets and pericardial effusion.
Figure 7.
chronological order.
| DEMOGRAPHIC CHARACTERISTICS. | |||
|---|---|---|---|
| Case 1 | Case 2 | Case 3 | |
| AGE | 59 | 49 | 62 |
| SEX | Male | male | Male |
| PLACE OF ORIGIN | Quilanga | Loja | Quilanga |
| PLACE OF RESIDENCE | Loja | Loja | Quilanga |
| PARENTS' PLACE OF ORIGIN | Quilanga | Quilanga | Quilanga |
| OCCUPATION | Administrator | Businessman | Farmer |
| CLINICAL AND IMAGING CHARACTERISTICS | |||
| ECG FINDINGS | P Inferior pseudo infarction, ventricular extrasystoles | Bifascicular block: RBBB, BAV first degree. Inferior pseudoinfarction | Complete AV block |
| NYHA FUNCTIONAL CLASS | III | II | III |
| MYOCARDIOPATHY ON ECHOCARDIOGRAM | Yes | yes | Yes |
| NEUROLOGICAL EVALUATION/POLYNEUROPATHY NEUROPATHY | Yes | no | Yes |
| CARPAL TUNNEL SYNDROME | No | no | No |
| GASTROINTESTINAL MANIFESTATIONS | Yes | yes | Yes |
| DEVICE PLACEMENT | Yes (CDI) | no | No |
| OUTCOMES | Syncope-TBI/death | Referred to heart transplant | Cardiogenic shock/death |
Source: Own elaboration, 2021s.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Variant Transthyretin Amyloidosis (ATTRv) in Hungary: First Data on Epidemiology and Clinical Features
Zoltán Pozsonyi
et al.
Genes,
2021
Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy
Torsten Rasmussen
et al.
Cardiogenetics,
2022
Renal Involvement in Hereditary Transthyretin Amyloidosis: An Italian Single-Centre Experience
Pietro Ferraro
et al.
Brain Sciences,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated