
Submitted:
21 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
The superfamily of Acid proteases has two catalytic aspartates for proteolysis of their peptide substrates. Here, we show a minimal structural scaffold, the Structural Catalytic Core (SCC), which packs all conserved substructure elements around the catalytic machinery, and which can be found in all enzymes of the Acid proteases superfamily. The SCC is a dimer of several structural blocks, such as the DD-link, the D-loop and the G-loop, around two catalytic aspartates in each protease subunit or within an individual chain. The dimer of two D-loop/DD-link substructures makes a DD-zone, and the dimer of two D-loop/G-loop substructures makes a psi-loop. These structural markers are useful for protein comparison, structure identification, protein family separation and protein engineering.
Keywords:
Pepsin
; Retropepsin
; Ddi1
; Lpg0085
; Acid protease
; three-dimensional structure
; active site
; catalytic aspartate
1. Introduction
Earlier, we have described structural catalytic cores in many serine and cysteine proteases and showed the presence of unique structure/functional environments, “zones”, around the catalytic sites in these proteins [1,2,3,4]. Each zone incorporated a segment of the catalytic core, connected to their respective element of protein functional machinery through a network of conserved hydrogen bonds and other interactions.
Each of the four protease superfamilies studied earlier: (1) alpha/beta-Hydrolases, (2) Trypsin-like serine proteases, (3) Cysteine proteinases and (4) SGNH hydrolase-like proteins, SCOP (Structural Classification of Proteins, https://scop.mrc-lmb.cam.ac.uk/ [5]) IDs: 3000102, 3000114, 3001808 and 3001315, respectively, had only rare, structural exceptions, where aspartic acid could be found in place of the canonical catalytic serine or cysteine residues. At the same time, most of the proteases that predominantly use aspartic acid as a catalytic residue are grouped into the “Acid proteases” superfamily (SCOP ID: 3001059). This superfamily belongs to the “All beta proteins” class (SCOP ID: 1000001) and includes four families, including the “Pepsin-like” family (SCOP ID: 4002301). The 3D structure of a protein from the Pepsin-like family consists of two similar beta barrel domains (N- and C-terminal) with one catalytic aspartate residue in each domain [6,7,8]. Aspartic proteases of this family use an activated water molecule bound to two conserved aspartate residues for catalysis of their peptide substrates. Enzymes of the Pepsin-like family are synthesized as inactive zymogens (pro-enzymes), and later they are subsequently activated by cleavage of the N-terminal pro-peptide, which separate upon activation [9]. The protease 3D structures of the other three families resemble that of one of the structural domains of the peptidase from the “Pepsin-like” family, and they become active when two monomers assemble to form the catalytically active dimer [10].
Here, we propose a general model of the conserved Structural Catalytic Core (SCC) of aspartate proteases. Based on the “key” features of this model, we present a comparative structural analysis of 3D structures of superfamily representative domains in their zymogenic, free and ligand-bound forms found in the Protein Data Bank (PDB [11,12]). In addition, we show a comparative structural analysis of SCC models obtained after dimerization of two identical amino acid chains of proteases or duplication of corresponding amino acid fragments within the same chain.
2. Results and Discussion
2.1. Creating the Dataset of the Acid Proteases Superfamily Fold Proteins
Currently, according to the SCOP, the Acid proteases superfamily consists of 4 families: 1) Lpg0085-like (SCOP ID: 4001811), 2) Retroviral protease (retropepsin) (SCOP ID: 4002288), 3) Pepsin-like (SCOP ID: 4002301) and 4) Dimeric aspartyl proteases (SCOP ID: 4004443) with more than 146 representative domains [5]. Representative 3D structures of this superfamily are tabulated in Table 1. Of the four families, only the Pepsin-like family contains 3D structures of the zymogenic form of aspartic proteases. In addition to the SCOP database, we used data from the Proteopedia and the Uniprot databases (http://proteopedia.org/ wiki/ index.php/ Main_Page [13,14] and https://www.uniprot.org/ [15], respectively). Ten pro-enzyme structures were identified, and they are indicated with a “p” in Table 1. Since each 3D structure of the Pepsin-like pro-enzymes contained two similar domains, both domains were separately analyzed at their catalytic regions, and thus Table 1 contains two lines for each PDB ID of a pro-enzyme labeled as “a” and “b”. For four proteins out of ten, in addition to coordinates of the zymogenic form, there were also available coordinates for both the ligand-free and ligand-bound forms, labeled in Table 1 with letters “c/d” and “e/f”, respectively. For three out of ten proteins, in addition to the coordinates of the zymogenic form, there were coordinates of only the ligand-bound form (i.e., “a”, “b”, “e” and “f” only; rows N: 4, 6 and 7). And for the remaining three proteins there were coordinates available only for the zymogenic form (i.e., “a” and “b” only; rows N: 8-10). In addition to these ten proteases from the Pepsin-like family, three proteolytically nonfunctional proteins in one or two forms were also analyzed (rows N: 11-13). The proteolytic inactivity of the last three proteins is caused by the replacement of their catalytic aspartic acids in the C-domains with serine.
In SCOP, the Retroviral protease (retropepsin) family is represented by the 3D structures of proteases from ten different organisms: HIV-1, HIV-2, HTLV-1, M-PMV, FIV, XMRV, SIV, RSV, MAV and EIAV [5]. Of the ten proteases listed, only the 3D structure of the XMRV protease differs from that of the other retropepsins [16,17]. Therefore, only the 3D structures of HIV-1 and XMRV proteases in the free and ligand-bound forms were chosen for analysis (Table 1, rows 14 and 15).
The Dimeric aspartyl proteases family contains seven representative protein 3D structures [5]. Six of the seven representative proteins are homologues of the DNA damage-inducible protein 1 (Ddi1) protease (PDB ID: 4Z2Z) [18]. The fold of the seventh representative protein, RC1339/APRc from Rickettsia conorii (PDB ID: 5C9F), does not form the mandatory homodimer like all other proteins in the Dimeric aspartyl proteases family [19]. Therefore, two 3D structures from this family, Ddi1 and APRc, were taken for conformational analysis. Finally, the Lpg0085-like family contains only one representative 3D structure (PDB ID: 2PMA) [20] and it was included in the analysis.
2.2. Structural Catalytic Core Around the Catalytic Aspartates in Pepsin
Let us consider three variants of the pepsin 3D structure: the zymogenic propepsin (PDB ID: 3PSG), free pepsin (PDB ID: 4PEP) and ligand-bound pepsin (PDB ID: 6XCZ), which structurally define the Pepsin-like family (SCOP ID: 4000470) (Table 1, rows 1a-1f).

Table 1.
Structural amino acid alignment of the structural catalytic core (SCC) in the Acid proteases superfamily proteins.
Table 1.
Structural amino acid alignment of the structural catalytic core (SCC) in the Acid proteases superfamily proteins.
| N | PDB ID & chain |
R(Å) | Protein | EC: number | Propept. or N-term pept. | DD-link | D-loop | G-loop | Mediator | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Superfamily: Acid proteases | ||||||||||
| Family: Pepsin-like | ||||||||||
| 1a | 3PSG_A,p | 1.65 | Propepsin | EC:3.4.23.1 | 7p VRK 9p | 11 DTEY 14 | 31 FDTGSS 36 | 121 LGLA 124 | Y125 | [21] |
| 1b | 3PSG_A | 1.65 | Propepsin | -׀׀- | 188 GYW 190 | 214 VDTGTS 219 | 301 LGDV 304 | |||
| 1c | 4PEP_A | 1.80 | Pepsin | -׀׀- | 7 ENY 9 | 12 TEY 14 | 31 FDTGSS 36 | 121 LGLA 124 | Y125 | [22] |
| 1d | 4PEP_A | 1.80 | Pepsin | -׀׀- | 188 GYW 190 | 214 VDTGTS 219 | 301 LGDV 304 | |||
| 1e | 6XCZ_A | 1.89 | Pepsin | -׀׀- | 7 ENY 9 | 12 TEY 14 | 31 FDTGSS 36 | 121 LGLA 124 | Y125 | [23] |
| 1f | 6XCZ_A | 1.89 | Pepsin | -׀׀- | 188 GYW 190 | 214 VDTGTS 219 | 301 LGDV 304 | |||
| 2a | 3VCM_A,p | 2.93 | Prorenin | EC:3.4.23.15 | 14p KRM 16p | 11 DTQY 14 | 31 FDTGSS 36 | 121 VGMG 124 | F125 | [24] |
| 2b | 3VCM_A | 2.93 | Prorenin | -׀׀- | 188 GVW 190 | 214 VDTGAS 219 | 301 LGAT 304 | |||
| 2c | 2REN_A | 2.50 | Renin | -׀׀- | 13 TNY 15 | 18 TQY 20 | 37 FDTGSS 42 | 128 VGMG 131 | F132 | [25] |
| 2d | 2REN_A | 2.50 | Renin | -׀׀- | 199 GVW 201 | 225 VDTGAS 230 | 315 LGAT 318 | |||
| 2e | 3K1W_A | 1.50 | Renin | -׀׀- | 13 TNY 15 | 18 TQY 20 | 37 FDTGSS 42 | 128 VGMG 131 | F132 | [26] |
| 2f | 3K1W_A | 1.50 | Renin | -׀׀- | 199 GVW 201 | 225 VDTGAS 230 | 315 LGAT 318 | |||
| 3a | 1PFZ_A,p | 1.85 | Proplasmepsin 2 | EC:3.4.23.39 | 85p KVE 87p | 12 QNIM 15 | 33 LDTGSA 38 | 124 LGLG 127 | W128 | [27] |
| 3b | 1PFZ_A | 1.85 | Proplasmepsin 2 | -׀׀- | 191 LYW 193 | 213 VDSGTS 218 | 301 LGDP 304 | |||
| 3c | 1LF4_A | 1.90 | Plasmepsin 2 | -׀׀- | 9 VDF 11 | 14 IMF 16 | 33 LDTGSA 38 | 124 LGLG 127 | W128 | [28] |
| 3d | 1LF4_A | 1.90 | Plasmepsin 2 | -׀׀- | 191 LYW 193 | 213 VDSGTS 218 | 301 LGDP 304 | |||
| 3e | 2BJU_A | 1.56 | Plasmepsin 2 | -׀׀- | 9 VDF 11 | 14 IMF 16 | 33 LDTGSA 38 | 124 LGLG 127 | W128 | [29] |
| 3f | 2BJU_A | 1.56 | Plasmepsin 2 | -׀׀- | 191 LYW 193 | 213 VDSGTS 218 | 301 LGDP 304 | |||
| 4a | 3QVC_A,p | 2.10 | HAP zymogen | EC:3.4.23.39 | 84p NIE 86p | 9 LANVL 13 | 31 FHTASS 36 | 121 FGLG 124 | W125 | [30] |
| 4b | 3QVC_A | 2.10 | HAP zymogen | -׀׀- | 188 LMW 190 | 214 LDSATS 219 | 301 LGDP 304 | |||
| 4e | 3QVI_A,B | 2.50 | HAP protein | -׀׀- | 7_B K | 12 VLS 14 | 31 FHTASS 36 | 121 FGLG 124 | W125 | [30] |
| 4f | 3QVI_A | 2.50 | HAP protein | -׀׀- | 188 LMW 190 | 214 LDSATS 219 | 301 LGDP 304 | |||
| 5a | 5N7N_A,p | 2.30 | Procathepsin D | N/A | 7p TRF 9p | 37 DVVY 40 | 57 FDTGSA 62 | 147 LGLA 150 | Y151 | [31] |
| 5b | 5N7N_A | 2.30 | Procathepsin D | -׀׀- | 217 GYW 219 | 248 ANTGTS 253 | 336 LGDV 339 | |||
| 5c | 5N71_A | 1.88 | Cathepsin D | -׀׀- | 33 VNL 35 | 38 VVY 40 | 57 FDTGSA 62 | 147 LGLA 150 | Y151 | [31] |
| 5d | 5N71_A | 1.88 | Cathepsin D | -׀׀- | 217 GYW 219 | 248 ANTGTS 253 | 336 LGDV 339 | |||
| 5e | 5N7Q_A | 1.45 | Cathepsin D | -׀׀- | 11 VNL 13 | 16 VVY 18 | 35 FDTGSA 40 | 125 LGLA 128 | Y129 | [31] |
| 5f | 5N7Q_A | 1.45 | Cathepsin D | -׀׀- | 195 GYW 197 | 226 ADTGTS 231 | 314 LGDV 317 | |||
| 6a | 1MIQ_A,p | 2.50 | Proplasmepsin | N/A | 84p KVE 86p | 13 NIM 15 | 33 FDTGSA 38 | 124 LGLG 127 | W128 | [32] |
| 6b | 1MIQ_A | 2.50 | Proplasmepsin | -׀׀- | 191 LYW 193 | 213 VDSGTT 218 | 301 LGDP 304 | |||
| 6e | 1QS8_A | 2.50 | Plasmepsin | -׀׀- | 9 DDV 11 | 14 IMF 16 | 33 FDTGSA 38 | 124 LGLG 127 | W128 | [32] |
| 6f | 1QS8_A | 2.50 | Plasmepsin | -׀׀- | 191 LYW 193 | 213 VDSGTT 218 | 301 LGDP 304 | |||
| 7a | 5JOD_A,p | 1.53 | Proplasmepsin 4 | EC:3.4.23.39 | 85p KID 87p | 13 NLM 15 | 33 FDTGSA 38 | 124 LGLG 127 | W128 | 55 |
| 7b | 5JOD_A | 1.53 | Proplasmepsin 4 | -׀׀- | 191 LYW 193 | 213 VDSGTS 218 | 301 LGDP 304 | |||
| 7e | 1LS5_A | 2.80 | Plasmepsin 4 | -׀׀- | 9 DDV 11 | 14 LMF 16 | 33 FDTGSA 38 | 124 LGLG 127 | W128 | [28] |
| 7f | 1LS5_A | 2.80 | Plasmepsin 4 | -׀׀- | 191 LYW 193 | 213 VDSGTS 218 | 301 LGDP 304 | |||
| 8a | 1QDM_A,p | 2.30 | Prophytepsin | EC:3.4.23.40 | 11p KKR 13p | 15 NAQY 18 | 35 FDTGSS 40 | 126 LGLG 129 | F130 | [33] |
| 8b | 1QDM_A | 2.30 | Prophytepsin | -׀׀- | 195 GYW 197 | 222 ADSGTS 227 | 313 LGDV 316 | |||
| 9a | 1HTR_B,p | 1.62 | Progastricsin | EC:3.4.23.3 | 8p KKF 10p | 11 DAAY 14 | 31 FDTGSS 36 | 121 MGLA 124 | Y125 | [34] |
| 9b | 1HTR_B | 1.62 | Progastricsin | -׀׀- | 189 LYW 191 | 216 VDTGTS 221 | 304 LGDV 307 | |||
| 10a | 1TZS_A,p | 2.35 | Procathepsin E | EC:3.4.23.34 | 9p R | 22 DMEY 25 | 42 FDTGSS 47 | 132 LGLG 135 | Y136 | [35] |
| 10b | 1TZS_A | 2.35 | Procathepsin E | -׀׀- | 201 AYW 203 | 227 VDTGTS 232 | 317 LGDV 320 | |||
| 11c | 1T6E_X | 1.70 | Xylanase inhib. | EC:3.2.1.8 | 8 TKD 10 | 14 SLY 16 | 28 LDVAGP 33 | 141 AGLA 144 | NS146 | [36] |
| 11d | 1T6E_X | 1.70 | Xylanase inhib. | -׀׀- | 204 PAH 206 | 234 LSTRLP 239 | 348 LGGA 351 | |||
| 11e | 1T6G_A | 1.80 | Xylanase inhib. | -׀׀- | 8 TKD 10 | 14 SLY 16 | 28 LDVAGP 33 | 141 AGLA 144 | NS146 | [36] |
| 11f | 1T6G_A | 1.80 | Xylanase inhib. | -׀׀- | 204 PAH 206 | 234 LSTRLP 239 | 348 LGGA 351 | |||
| 12c | 3AUP_A | 1.91 | Basic 7S globulin | N/A | 15 QND 17 | 21 GLH 23 | 40 VDLNGN 45 | 159 AGLG 162 | HA164 | [37] |
| 12d | 3AUP_A | 1.91 | Basic 7S globulin | -׀׀- | 228 GEY 230 | 264 ISTSTP 269 | 361 LGAR 364 | |||
| 13c | 3VLA_A | 0.95 | EDGP (Fragment) | N/A | 14 KKD 16 | 20 LQY 22 | 39 VDLGGR 44 | 155 AGLG 158 | RT160 | [38] |
| 13d | 3VLA_A | 0.95 | EDGP (Fragment) | -׀׀- | 235 VEY 237 | 270 ISTINP 275 | 374 IGGH 377 | |||
| 13e | 3VLB_A | 2.70 | EDGP (Fragment) | -׀׀- | 14 KKD 16 | 20 LQY 22 | 39 VDLGGR 44 | 155 AGLG 158 | RT160 | [38] |
| 13f | 3VLB_A | 2.70 | EDGP (Fragment) | -׀׀- | 235 VEY 237 | 270 ISTINP 275 | 374 IGGH 377 | |||
| Family: Retroviral protease (retropepsin) | ||||||||||
| 14c | 3IXO_A | 1.70 | HIV-1 protease | N/A | N/A | 8 R-P 9 | 24 LDTGAD 29 | 85 IGRN 88 | N/A | [39] |
| 14d | 3IXO_B | 1.70 | HIV-1 protease | -׀׀- | N/A | 8 R-P 9 | 24 LDTGAD 29 | 85 IGRN 88 | N/A | |
| 14e | 5YOK_A | 0.85 | HIV-1 protease | -׀׀- | N/A | 8 R-P 9 | 24 LDTGAD 29 | 85 IGRN 88 | N/A | [40] |
| 14f | 5YOK_B | 0.85 | HIV-1 protease | -׀׀- | N/A | 8 R-P 9 | 24 LDTGAD 29 | 85 IGRN 88 | N/A | |
| 15c | 3NR6_A | 1.97 | XMRV protease | EC:3.4.23.- | N/A | 15 E-P 16 | 31 VDTGAQ 36 | 93 LGRD 96 | R95 | [16] |
| 15d | 3NR6_B | 1.97 | XMRV protease | -׀׀- | N/A | 15 E-P 16 | 31 VDTGAQ 36 | 93 LGRD 96 | R95 | |
| 15e | 3SLZ_A | 1.40 | XMRV protease | N/A | N/A | 15 E-P 16 | 31 VDTGAQ 36 | 93 LGRD 96 | R95 | [41] |
| 15f | 3SLZ_B | 1.40 | XMRV protease | -׀׀- | N/A | 15 E-P 16 | 31 VDTGAQ 36 | 93 LGRD 96 | R95 | |
| Family: Dimeric aspartyl proteases | ||||||||||
| 16c | 4Z2Z_A | 1.80 | Ddi1 protease | EC:3.4.23.- | N/A | 201 VPML 204 | 219 VDTGAQ 224 | 289 IGLD 292 | N/A | [42] |
| 16d | 4Z2Z_B | 1.80 | Ddi1 protease | -׀׀- | N/A | 201 VPML 204 | 219 VDTGAQ 224 | 289 IGLD 292 | N/A | |
| 17c | 5C9F_A | 2.00 | ApRick protease | EC:3.-.-.- | N/A | 121 DGHF 124 | 139 VDTGAS 144 | 209 LGMS 212 | N/A | [19] |
| Family: LPG0085-like | ||||||||||
| 18c | 2PMA_A | 1.89 | Protein Lpg0085 | N/A | N/A | 29 Y | 46 LDTGAK 51 | 145 LGRD 148 | RD148 | [20] |
| 18d | 2PMA_I | 1.89 | Protein Lpg0085 | -׀׀- | N/A | 29 Y | 46 LDTGAK 51 | 145 LGRD 148 | RD148 | |
N/A—Not Available.
The boundary between the N- and C-domains of the 3D structure of pepsinogen is in the vicinity of Gly169 [9]. Asp32 (N-domain) and Asp215 (C-domain) are the two catalytically important aspartate residues. Each aspartate residue is positioned within the hallmark Asp-Thr/Ser-Gly (Asp32-Thr33-Gly34 in 3PSG) motif which, together with a further Hydrophobic-Hydrophobic-Gly sequence motif, forms an essential structural feature known as a psi-loop motif [22,43,44,45,46]. Let us designate two fragments of the protease amino acid sequence involved in formation of the psi-loop motif as the D(Asp)-loop and G(Gly)-loop. In this section, the atomic structure of the D- and G-loops in the N- and C-domains and their position relative to each other in the 3D structures of pepsin will be analyzed in detail.
2.2.1. Propepsin
DD-Zone of Propepsin: A D-loopN—DD-linkN—D-loopC—DD-linkC Circular Motif
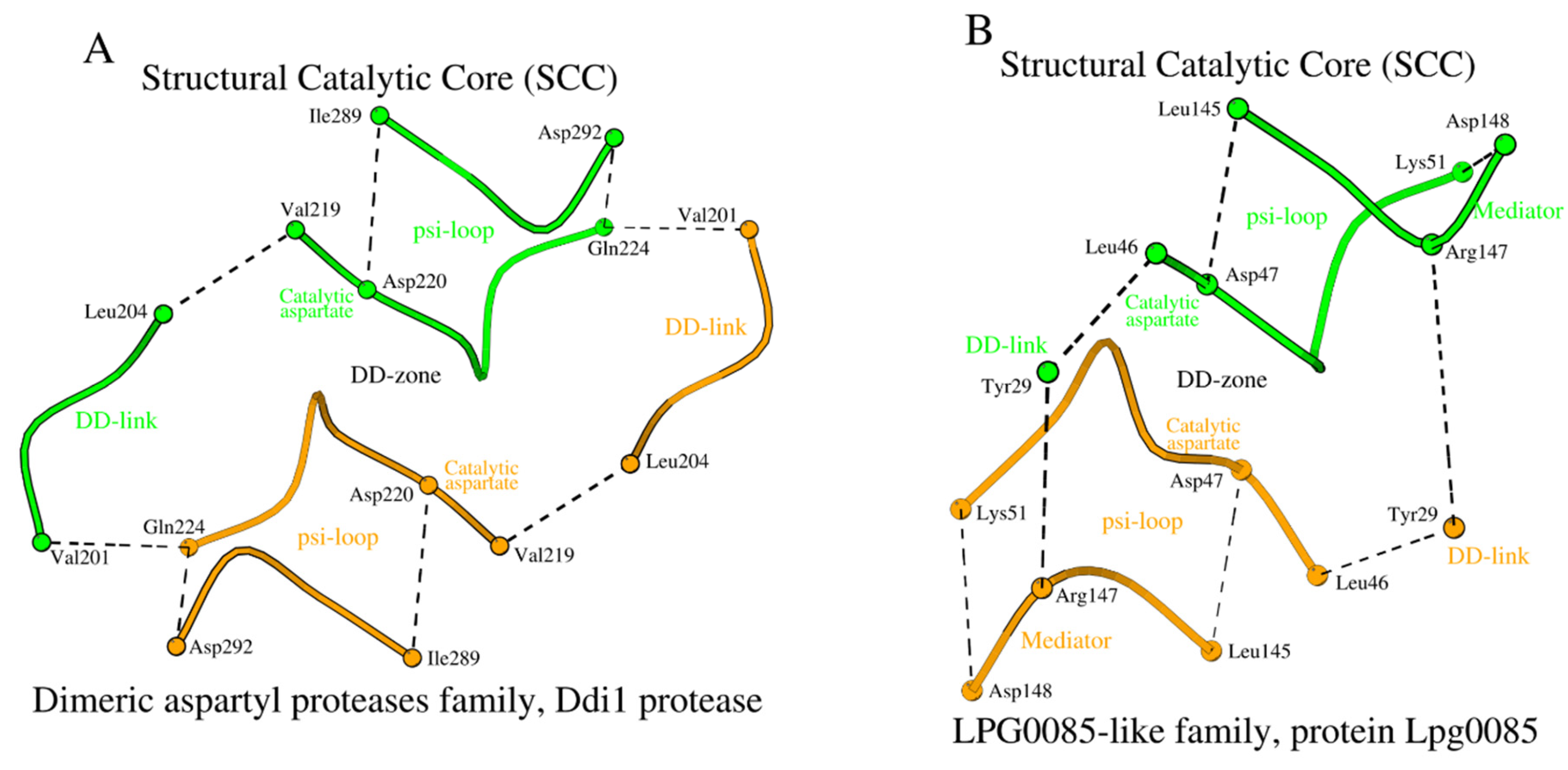
As noted above, the functional activity of pepsin is carried out simultaneously by both of the catalytic residues, Asp32 and Asp215. Therefore, two D-loops, D-loopN for the N-terminal domain and D-loopC for the C-terminal domain, were analyzed in detail (Table 1 and Table S1). It turned out that the two domains of propepsin also contain structurally equivalent short peptides, which we call DD-linkN (Asp11-...-Tyr14) and DD-linkC (Gly188-Tyr189-Trp190), where N and C also stand for the N-terminal domain and C-terminal domain, respectively (Table 1). These two special DD-link peptides “lock” the ends of the D-loopN and D-loopC to form a “circular” structure, which altogether we call the “DD-zone” (Figure 1A).
The DD-zone of propepsin consists of 19 amino acids in total from both D-loops and both DD-links and an additional residue Tyr125. Tyr125 serves as a structural mediator between the C-terminus of the D-loopN and the N-terminus of the DD-linkC (Figure 1A); this residue directly follows Ala124 from G-loopN (Table 1).
Independently, in propepsin, residues Thr33 and Thr216 are located next to the two catalytic aspartates. Their side chain OG1 atoms each make two hydrogen bonds with main-chain nitrogen and oxygen atoms of the opposite D-loop (Figure 1A, Table S1, last column). These interactions are known as the “fireman’s grip” motif [47,48].
The pro-enzyme segment in propepsin is Leu1p-...-Leu44p, where “p” indicates the pro-enzyme sequence region. The pepsin portion in 3PSG starts from Ile1. Glu13 and Phe15 form a short β-sheet-like interaction with Lys9p and Val7p (Figure 1A, Table S2, last column). The residues of this β-sheet undergo a conformational change during the activation process [9].
The Psi-loopN and Psi-loopC Motifs: Interactions between the D-loop and G-loop in the N-and C-domains
In 3PSG, the D-loopN tetrapeptide, Asp32 -...- Ser35, contains a frequently occurring Asx-motif [49], where an aspartate (here, catalytic Asp32) or an asparagine residue within a tetra- or pentapeptide forms two short-range (in terms of sequence location) main-chain and side-chain hydrogen bonds with the sequentially adjacent amino acids (Figure 1B). We observe a similar Asx-motif involving the catalytic Asp215 from the D-loopC tetrapeptide (Figure 1C). Additionally, there are four conserved long-range hydrogen bonds between the D- and G-loops in both N- and C-domains (Figure 1B,C). We will refer to the substructures shown in Figure 1B,C as the psi-loopN and psi-loopC motifs. Each psi motif is an eight-residue 3D structure consisting of D- and G-loop residues that are held together by six hydrogen bonds. The geometric characteristics of these six hydrogen bonds are given in Table S2 (row 1a, columns 4-6).
Comparison of the Psi-loopN and Psi-loopC
Despite the apparent similarity, the psi-loopN and psi-loopC motifs are not identical. While making similar interactions, the D-loopC is five amino acids long (Asp215-...-Ser219) and the D-loopN is only four residues (Figure 1B,C). Moreover, the conformations of the two respective G-loops differ. The G-loopC at its C-terminus contains a β-turn, which is stabilized by the hydrogen bond between O/Gly302 and N/Phe305 (not shown in Figure 1C), while the G-loopN does not have a similar substructure. As a result, there is conformational difference between Phe305 and its structural counterpart in the N-domain, Tyr125, where Phe305 takes part in the conformational arrangement of its respective psi-loop, while Tyr125 does not. Still, the two psi-loop motifs are bound by a set of equivalent interactions, where the O/Asp32-N/Leu123 hydrogen bond in psi-loopN is substituted by the O/Thr218-N/Asp303 hydrogen bond in psi-loopC, and where the O/Ser35-N/Ala124 hydrogen bond in psi-loopN is substituted by the O/Ser219-N/Val304 hydrogen bond in psi-loopC (Figure 1B,C).
The structural changes described above appear to result in tighter binding of Asp32 to the G-loopN than of Asp215 to G-loopC, since the distance from Asp32 to G-loopN is shorter than that from Asp215 to G-loopC. It is possible that this structural fact is the main reason for the differences in functional activity between Asp32 and Asp215 in the proposed models of catalytic hydrolysis of peptide bonds by acid proteases [50,51,52]. If Asp32 is more tightly bound with more potential hydrogen bonds as compared to Asp215, then its nucleophilicity must be somewhat decreased. Thus, Asp215 of the C-domain would play a more prominent role in the proteolytic cleavage of dipeptide substrates than Asp32 of the N-domain.
2.2.2. Activation of Free Pepsin
The conversion of propepsin to active pepsin is achieved through proteolytic cleavage and subsequent removal of the N-terminal amino acid fragment. Here, we are mostly interested in changes that occur in the propepsin structural core, SCC. A structural comparison of propepsin (PDB ID: 3PSG) and mature pepsin (PDB ID: 4PEP) showed that rearrangements occur only in DD-linkN and its immediate environment. First, as described above, the length of the tetrapeptide Asp11-...-Tyr14 was reduced by one residue at its N-terminus (Table 1 and Table S1). Then, the two-stranded β-sheet (Glu13-...-Phe15)/(Val7p-...-Lys9p) is replaced with a structurally similar two-stranded β-sheet (Glu13-...-Phe15)/(Glu7-...-Tyr9) (Table 1 and Table S2). Thus, upon pepsin activation the architecture of the SCC remains largely unchanged.
2.2.3. Pepsin/ligand complex
During activation, the propepsin structure transforms into the active pepsin structure, ligand-free form. How does interaction with the ligand affect the SCC? Let us consider the 3D structure of the pepsin-saquinavir complex (PDB ID: 6XCZ). The key contacts between pepsin and the small-molecule ligand (saquinavir, ROC401) are four hydrogen bonds (Figure 2B; Table S3, rows 1e and 1f). Two pairs of conserved residues from the D-loops of the N- and C-domains, Asp32/Gly34 and Asp215/Gly217, donate four oxygen atoms as part of the four hydrogen bonds. Each of the two aspartates forms an Asx-motif [49], and in addition to the four hydrogen bonds above, there are two additional hydrogen bonds via the mediator-waters HOH527 and HOH645 (Figure 2B), and also there is a hydrogen bond that involves the OH atom of Tyr189, the central residue of the tripeptide DD-linkC. Thus, DD-linkC interacts with the inhibitor. Aside from the extensive hydrogen bonding inventory described above, binding of a ligand does not introduce any visible structural changes to the ligand-free form of the SCC of pepsin (Tables S1 and S2, rows 1c-1f).
2.3. Structural Core in Proteins of the Pepsin-Like Family
2.3.1. DD-Zones
Earlier, we have shown that in propepsin the segment Asp11-Phe15, which includes the DD-linkN, interacts with the pro-tripeptide Val7p-Lys9p (Figure 1A) by means of interactions listed in Table S2. During the transition from the inactive zymogenic form to the enzymatically active form, the DD-linkN is slightly structurally modified as described above, and the pro-tripeptide is spatially substituted by the N-terminal tripeptide (Glu7-Tyr9; Table 1). Interactions between DD-linkN and the N-terminal tripeptide are shown in Table S2. We also observed similar structural rearrangements in the other members of Pepsin-like family although there are variations from the rule: with the histo-aspartic protease (HAP), DD-linkN is one amino acid longer, and with procathepsin E, only one amino acid, R9P, of the pro-peptide contacts DD-linkN (Table 1). However, the general structural trend for the Pepsin-like family is the same.
In propepsin and pepsin, the contact between DD-linkN and D-loopN involves a water molecule as an intermediary (Figure 1; Table S1). In the structure of ligand-bound pepsin, a water molecule does not participate in interactions as an intermediary. A similar water presence and functionality is observed for all of the remaining proteins of the Pepsin-like family. However, considering differences in resolution of structures (Table 1) and the associated difficulties in localization of the bound water molecules, it is not always possible to unambiguously correlate the presence or absence of a water molecule with any form of protein, and thus exceptions are possible.
In pepsin, the contact between D-loopN and DD-linkC involves the amino acid Tyr125 as a structural mediator (Figure 1; Table S1). In a number of proteins, there is also a mediating water molecule in addition to the aromatic amino acid (Table S1, column 5). In three proteins, xylanase inhibitor, basic 7S globulin and EDGP, there are two mediator residues instead of a single Tyr125. A hydrogen bond between the ends of DD-linkC and D-loopC is, however, conserved and contains no mediator insertions in any of the analyzed structures (Table S1, column 6). The contact between D-loopC and DD-linkN does not contain mediators, but can be variable in its nature, being a hydrogen bond, a weak hydrogen bond or a hydrophobic interaction (Table S1, column 7).
2.3.1.1. Fireman’s Grip Motif Reflects Open/Close-Conformation Structural Change
In the Pepsin-like family proteins, the open/close-conformation structural change during the transition from the inactive zymogen to the enzymatically active form can either lead to conformational changes in the DD-zone or not. In proteins, where the hallmark Asp-Thr/Ser-Gly sequence (see Section 2.2) in the C-terminal domain contains serine, the conformational change in the DD-zone does take place, and it is reflected by the change of the Fireman’s grip motif (Table S1, column 8). In proteins, where the hallmark Asp-Thr/Ser-Gly sequence in the C-terminal domain contains threonine, the open/close conformational change in the DD-zone does not take place.
2.3.2. Psi-loops
As noted above, the psi-loop motif includes amino acids from the D- and G-loops. In pepsin, both D-loops contain a catalytic aspartate. Of the thirteen proteins studied, eight are active hydrolases, they have both catalytic aspartates (Table 1). In the HAP protein, an evolutionary Asp32His mutation did occur that, however, did not lead to a loss of catalytic activity because the other Asp215 was still present [30]. The remaining four proteins, cathepsin D, xylanase inhibitor, basic 7S globulin and EDGP, have lost their enzymatic activity due to the replacement of the catalytic aspartate with another amino acid in the C-terminal domain [31,36,37,38]. Loss of catalytic activity in these proteins versus the HAP protein is strong evidence that proteolytic activity requires the aspartate of the C-terminal domain whereas the aspartate of the N-terminal domain maybe dispensable.
Both psi-loopN and psi-loopC motifs are structurally identical among the thirteen proteins of the Pepsin-like family in three different forms (pro-enzyme, mature enzyme and enzyme-ligand complex) (Table S2, columns 4 and 5). That is, replacing the catalytic aspartate with another amino acid either does not affect the conformation of the psi-loop motifs or affects it insignificantly. Structural conservation of the psi-loop conformation also occurs despite structural rearrangement in the tetrapeptides forming the Asx-motif in some proteins (Table S2, column 6). For example, six proteins in one or several forms show a structural transition from the Asx-motif to a Asx-turn [53], which lacks the hydrogen bond between the atoms of the first and fourth residues of the tetrapeptide unlike the Asx-motif. The structures of these six proteins, the HAP protein, plasmepsin 4, phytepsin, xylanase inhibitor, basic 7S globulin and EDGP, have geometrical parameters that formally exceed those of a canonical hydrogen bond [54].
2.3.3. Ligand Bound Pepsin-Like Proteins
Section 2.2.3 identifies seven amino acids of the pepsin’s SCC that are responsible for ligand recognition. These are (1, 2, 3 and 4) catalytic Asp/Gly pairs of (Asp-Thr/Ser-Gly)N and (Asp-Thr/Ser-Gly)C, C-terminal and N-terminal Asp-Thr/Ser-Gly motifs; (5 and 6) two C-terminal serine residues of D-loopN and D-loopC; and (7) the Tyr189, the central residue of the tripeptide DD-linkC. Of the thirteen Pepsin-like representative structures listed in Table 1, only seven had a complex with a ligand close or within the SCC. Six of these seven structures had similar D-loop/ligand contacts (Table S3). And, again, the HAP protein was unique, by lacking the expected contacts of Ala217 and Ser219 with the K95 inhibitor as seen in all of the other structures. With the HAP protein, instead of those contacts, Ala217 and Ser219 of the chain_A formed hydrogen bonds with Asn279 of the chain_B, i.e O/Ala217_A—N/Asn279_B at 2.9 Å and OG/S219-ND2/N279_B at 3.1 Å, respectively, and a weak hydrogen bond with Glu278A of the chain_B (designated as Glu278A_B in the PDB file of 3QVI), O/Ala217_A—CA/Glu278A_B at 3.4 (2.6) 127O (for the definition of parameters of weak hydrogen bonds see [55]). The changes in contact partners for Ala217 and Ser219 is due to the fact that in the inhibitor complex the enzyme forms a tight domain-swapped dimer, not previously seen in any aspartic protease [30]. As a result of such domain-swapped dimerization, Glu278A of chain_B forms contacts with the inhibitor instead of Ala217 and Ser219 of chain_A (Table S3, row 4f and column 5).
Taking together, the Pepsin-like family proteins from Table 1 have their SCC constructed from the same set of conserved amino acids in all three forms, i.e., pro-enzyme, ligand-free enzyme and ligand-bound enzyme, while the most noticeable structural changes concern the transition of the DD-links and fireman’s grips from the zymogenic form to the enzymatic form. The DD-zones include the N-terminal and C-terminal D-loops, D-loopN and D-loopC, with their ends linked by the longer DD-linkN and a water molecule, and a shorter DD-linkC plus a Mediator molecule (Figure 1A).
2.4. SCC in Hydrolases of the Retroviral Protease (Retropepsin) Family
2.4.1. DD-Zones
The Retroviral protease (retropepsin) family is the second family of Acid proteases listed in Table 1. Hydrolases of this family do not have zymogenic form, and the enzyme is a dimer of two identical amino acid chains. Figure 3A shows a DD-zone of HIV-1 protease (PDB ID: 3IXO). The main differences between the DD-zones of pepsin and HIV-1 are the number of residues forming DD-links and an absence of mediators.
A change in the number of residues in the DD-links is usually associated with the presence or absence of the need to form a β-structural contact with either the propeptide or the N-terminal fragment (Figure 3A vs. Figure 1A). However, a decrease in the length of the DD-link by one amino acid does not necessarily lead to a change in the relative position of the D-loops relative to each other. Such is the case for the HIV-1 protease, where atoms of the long side chain of Arg8 (DD-link in HIV-1) interact with Asp29 (D-loop in HIV-1) instead of the oxygen atoms of the shorter side chains of Asp11 (DD-link in pepsin) and Ser219 (D-loop in pepsin) (Figure 3A vs. Figure 1A, Table S1).
In the XMRV protease (PDB ID: 3NR6), there is glutamate (DD-link in XMRV) in place of Arg8 (DD-link in HIV-1) and glutamine (D-loop in XMRV) instead of Asp29 (D-loop in HIV-1) (Table 1), which results in some changes in the architecture of the DD-zone in the XMRV protease compared to HIV-1 (Figure 3B, Table S1). In XMRV, there is an increase in the distance between the ends of the DD-link and the D-loop, which results in the absence of a direct contact between them. However, in XMRV, the D-loop/DD-link contact happens through the Mediator residue Arg95, which also participates in the formation of the psi-loop (Figure 3B).
Thus, the distinctive feature of the Retroviral protease (retropepsin) family hydrolases is within the DD-zones where the D-loops are bound by short DD-links of 2 residues plus a Mediator residue. Additionally, in HIV-1 and XMRV, there is a separate residue Arg87 (in HIV-1; it is not shown in Figure 3A)/Arg95 (in XMRV), which interacts with Asp29 (in HIV-1)/Gln36 (in XMRV) via a conventional hydrogen bond: NH2/R87-OD1/D29 (Table S1, column 5), and stabilizes the conformation of the D-loop. The function of this residue in HIV-1 and XMRV is unknown.
2.4.2. Psi-Loops in HIV-1 and XMRV
As noted above, a homo-dimer of two identical amino acid chains is the active form of a HIV-1 protease. Therefore, one can expect the conformation of the psi-loop motif in chains A and B to be identical. It turned out that HIV-1 and XMRV not only have similar psi-loop motifs, but they are also similar to that observed in the C-domain of pepsin (Figure 1C and Figure 3C). That is, the identical psi-loops in HIV-1 and XMRV have chosen a conformation that provides a catalytic aspartate with higher proteolytic efficiency in both subunits (Table S2). In Table S2 homodimer chains _A and B in HIV-1 (and other retroproteases) are listed as the respective counterparts of the of N- and C-domains in pepsin, but this is an arbitrary assignment.)
2.4.3. Ligand-Bound Forms of Retroviral Proteases
The DD-zones of ligand-bound pepsin and HIV-1 are very similar to each other (Figure 2B and Figure 3D). The main interactions are made by the three amino acids from each of the two D-loops totaling six interacting residues (Table S3). In HIV-1 these residues are Asp25, Gly27 and Asp29 from D-loop-Chain_A and, of course, identical residues are in D-loop-Chain_B of the HIV-1 homodimer (Figure 3D). For comparison, in pepsin those amino acids are Asp32, Gly34 and Ser36 from D-loopN and Asp215, Gly217 and Ser219 D-loopC (Table S3). In addition, with pepsin, Section 2.2.3 describes the additional Tyr189 from the DD-linkC that is involved in contacts with the ligand. In the ligand-bound HIV-1 protease (PDB ID: 5YOK), a combination of Arg8 (DD-link)/Asp29 (D-loop) performs an analogous role. Similar to HIV-1, in the ligand-bound XMRV (PDB ID: 3SLZ) the C-terminal position of the D-loop, Gln36, also participates in ligand binding (Table S3, last column). Replacing Asp29 (in HIV-1) with Gln36 (in XMRV) also results in additional hydrogen bonds formed between XMRV and the inhibitor. Interaction with the ligand does not seem to affect the architecture of the DD-zone in the HIV-1 and XMRV proteases (Table S1). The SCCs of the HIV-1 and XMRV proteases are shown in Figure 4A,B.
2.5. SCCs of the Dimeric Aspartyl Proteases and Lpg0085-Like Family Proteins
In HIV-1 and XMRV we have shown how amino acid changes at the N-terminus of the DD-link and the C-terminus of the D-loop affect the structure of the DD-zone. The Ddi1 protease, like the XMRV protease, has glutamine as the C-terminal amino acid of the D-loop (Table 1 and Table S1, rows 16c and 16d). However, the DD-links of the Ddi1 and XMRV proteases differ in length. In Ddi1, the number of amino acids in the DD-link increases twofold (from 2 to 4 residues) compared to XMRV protease, while in Lpg0085 the DD-link is a single residue (Figure 5A,B; Table 1 and Table S1, rows 18c and 18d). To compensate for such a reduction in the DD-link length in Lpg0085, a Mediator dipeptide Arg147-Asp148 is additionally present for DD-zone formation. Thus, the DD-zones of the Dimeric aspartyl proteases and the Lpg0085-like proteins are characterized by the presence of either a longer DD-link of four residues or a shorter DD-link of one residue plus a separate two-residue Mediator.
As in the case of Retroviral proteases, Ddi1 and Lpg0085 use the psi-loopC motif, which is equivalent to the C-terminal version of the psi-loop motif in Pepsin-like family proteins (Table 1 and Table S2, rows 16c, 16d, 18c and 18d). The ApRick protease does not form a canonical dimer, as do Ddi1 and Lpg0085 [19]. However, the psi-loop in the ApRick protease monomer is still identical to that in Ddi1 and Lpg0085 (Figure 5C; Table 1 and Table S2, row 17c). Li et al. suggested that the ApRick protease “may represent a putative common ancestor of monomeric and dimeric aspartic proteases” [19]. The SCCs in Ddi1 and Lpg0085 are shown in Figure 6A,B.
3. Materials and Methods
The SCOP classification database [5] and the Protein Data Bank (PDB, http://www.rcsb.org/ [11,12]) were used to identify and retrieve 33 representative structures of proteins from the Acid proteases superfamily (SCOP ID: 3001059). Detailed descriptions of the protein structural information contained within this set of PDB files are given in Section 2.1.
Structure visualization and structural analysis of interactions between amino acids in proteins (hydrogen bonds, hydrophobic, other types of weak interactions) were made using Maestro (Schrödinger Release 2023-1: Schrödinger, LLC, New York, NY, 2021; https://www.schrodinger.com/user-announcement/announcing-schrodinger-software-release-2023-4); and software [56] to determine interatomic contacts i.e., of ligand-protein contacts (LPC) and contacts of structural units (CSU).
Pairwise superpositions of representative structures were done using the Dali server (http://ekhidna2.biocenter.helsinki.fi/dali/) [57]. Weak hydrogen bonds from C-H•••O contacts were identified, based on the criteria described in [55]. The π-π stacking and similar contacts were analyzed using the Residue Interaction Network Generator (RING, https://ring. biocomputingup.it/ submit) [58]. Dimers were built using the “Protein interfaces, surfaces and assemblies” service PISA at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) [59]. Figures were drawn with MOLSCRIPT [60].
4. Conclusions
Here, we have outlined the minimal conserved structural arrangement common to the Acid proteases superfamily of proteins, which we refer to as the Structural Catalytic Core (SCC). We began with the Pepsin-like family proteases, where we defined the DD-zone (Figure 1A). The DD-zone is a circular structural motif defined by substructures around the catalytic aspartates in the N- and C-terminal domains, D-loopN and D-loopC, and their interactions with the peptides DD-linkN and DD-linkC that join the ends of D-loopN and D-loopC. Then, we increased the common substructure by defined the psi-loopN and psi-loopC motifs, where the DD-zone interacts through their D-loops with two external tetrapeptides, G-loopN and G-loopC, the residues of which intersect with the Hydrophobic-Hydrophobic-Gly sequence motif [44] (Figure 1B,C). While the two psi-loop motifs use the same logic in their formation, they differ in the environment around the catalytic aspartates, which may determine their different functional roles. Taken together, the psi-loops and the DD-zone define structural boundaries of the SCC in Pepsin-like proteins.
The other families of Acid proteases, Retroviral proteases (retropepsin), Dimeric aspartyl proteases and Lpg0085-like proteins, also have the DD-zone and psi-loop substructures similar to pepsin. However, unlike pepsin, which can be very roughly described as a “hetero psi-loop” protein, where psi-loopN and psi-loopC are not structurally identical unlike the homodimer enzymes. with the psi-loopC to be more functionally active, the The Retroviral proteases, Dimeric aspartyl proteases and Lpg0085-like proteins can be described as having a “homo psi-loop” since they have two identical chains. The homo psi-loops are both structurally similar to psi-loopC of pepsin. As with the Pepsin-like proteases, the other three protein families use DD-links to form a DD-zone (Table 1). If a DD-link is equal or shorter than two amino acids, then there are additional Mediator residues or water molecules filling the gap. Some Mediator residues are located in sequence either at the C-terminus of the G-loop or immediately after it. Based on the structures seen so far, we can argue that a specific “long DD-link” or “DD-link + Mediator”, or “DD-link + water” combination is the same for a structural family within an Acid proteases superfamily, and may distinguish that family from the other proteins.
In summary, we can say that SCC of the Acid proteases superfamily proteins consists of a dimer composed of a DD-link, D-loop and G-loop blocks, where the D-loop plus DD-link forms a DD-zone, and the dimer of D- and G-loops form two psi-loops. Defining the SCC in this way allows us to outline a minimal common sub-structure for the entire superfamily of proteins, such as Acid proteases, which combines amino acid conservation and protein functionality that altogether can be used for protein comparison, structure identification, protein family separation and protein engineering.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org. Table S1. Conserved geometric parameters (distance and angle) of contacts in 33 DD-zones of the acid proteases superfamily proteins. Table S2. Conserved geometric parameters (distance and angle) of contacts in 65 psi-loops of the acid proteases superfamily proteins and contacts between DD-linkN and the propeptide/N-terminal peptide in 13 pepsin-like family proteins. Table S3. Conserved geometric parameters (distance and angle) of contacts between hydrolase and ligand in 9 acid proteases pepsin-like and retroviral protease (retropepsin) families.
Author Contributions
Alexander I. Denesyuk: Study design, Formal analysis, Methodology, Visualization, Writing—Original Draft, Writing—Review & Editing; Konstantin Denessiouk: Formal analysis, Methodology, Visualization, Writing—Original Draft, Writing—Review & Editing; Mark S. Johnson: Formal analysis, Methodology, Writing—Original Draft; Vladimir N. Uversky: Study design, Formal analysis, Methodology, Visualization, Investigation, Writing—Original Draft, Writing—Review & Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank the Biocenter Finland Bioinformatics Network (Dr. Jukka Lehtonen) and CSC IT Center for Science for computational support for the project. The Structural Bioinformatics Laboratory is part of the Solutions for Health strategic area of Åbo Akademi University and within the InFLAMES Flagship program on inflammation and infection, Åbo Akademi University and the University of Turku, funded by the Academy of Finland.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Denessiouk, K.; Denesyuk, A.I.; Permyakov, S.E.; Permyakov, E.A.; Johnson, M.S.; Uversky, V.N. The active site of the SGNH hydrolase-like fold proteins: Nucleophile-oxyanion (Nuc-Oxy) and Acid-Base zones. Curr Res Struct Biol 2024, 7, 100123. [Google Scholar] [CrossRef] [PubMed]
- Denessiouk, K.; Uversky, V.N.; Permyakov, S.E.; Permyakov, E.A.; Johnson, M.S.; Denesyuk, A.I. Papain-like cysteine proteinase zone (PCP-zone) and PCP structural catalytic core (PCP-SCC) of enzymes with cysteine proteinase fold. Int J Biol Macromol 2020, 165, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Denesyuk, A.; Dimitriou, P.S.; Johnson, M.S.; Nakayama, T.; Denessiouk, K. The acid-base-nucleophile catalytic triad in ABH-fold enzymes is coordinated by a set of structural elements. PLoS One 2020, 15, e0229376. [Google Scholar] [CrossRef] [PubMed]
- Denesyuk, A.I.; Johnson, M.S.; Salo-Ahen, O.M.H.; Uversky, V.N.; Denessiouk, K. NBCZone: Universal three-dimensional construction of eleven amino acids near the catalytic nucleophile and base in the superfamily of (chymo)trypsin-like serine fold proteases. Int J Biol Macromol 2020, 153, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Kulesha, E.; Gough, J.; Murzin, A.G. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res 2020, 48, D376–D382. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R. The structure and function of the aspartic proteinases. Annu Rev Biophys Biophys Chem 1990, 19, 189–215. [Google Scholar] [CrossRef]
- Polgar, L. The mechanism of action of aspartic proteases involves ‘push-pull’ catalysis. FEBS Lett 1987, 219, 1–4. [Google Scholar] [CrossRef] [PubMed]
- James, M.N. Catalytic pathway of aspartic peptidases. In Handbook of proteolytic enzymes; Elsevier: 2004; pp. 12–19.
- Sielecki, A.R.; Fujinaga, M.; Read, R.J.; James, M.N. Refined structure of porcine pepsinogen at 1.8 A resolution. J Mol Biol 1991, 219, 671–692. [Google Scholar] [CrossRef] [PubMed]
- Ingr, M.; Uhlikova, T.; Strisovsky, K.; Majerova, E.; Konvalinka, J. Kinetics of the dimerization of retroviral proteases: the “fireman’s grip” and dimerization. Protein Sci 2003, 12, 2173–2182. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Hodis, E.; Prilusky, J.; Martz, E.; Silman, I.; Moult, J.; Sussman, J.L. Proteopedia—a scientific ‘wiki’ bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biol 2008, 9, R121. [Google Scholar] [CrossRef]
- Prilusky, J.; Hodis, E.; Canner, D.; Decatur, W.A.; Oberholser, K.; Martz, E.; Berchanski, A.; Harel, M.; Sussman, J.L. Proteopedia: a status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. J Struct Biol 2011, 175, 244–252. [Google Scholar] [CrossRef]
- UniProt_Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Dimaio, F.; Zhou, D.; Gustchina, A.; Lubkowski, J.; Dauter, Z.; Baker, D.; Wlodawer, A. Crystal structure of XMRV protease differs from the structures of other retropepsins. Nat Struct Mol Biol 2011, 18, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.M.; Goodenow, M.M.; Gustchina, A.; Wlodawer, A. Retroviral proteases. Genome Biol 2002, 3, REVIEWS3006. [Google Scholar] [CrossRef]
- Sirkis, R.; Gerst, J.E.; Fass, D. Ddi1, a eukaryotic protein with the retroviral protease fold. J Mol Biol 2006, 364, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gustchina, A.; Cruz, R.; Simoes, M.; Curto, P.; Martinez, J.; Faro, C.; Simoes, I.; Wlodawer, A. Structure of RC1339/APRc from Rickettsia conorii, a retropepsin-like aspartic protease. Acta Crystallogr D Biol Crystallogr 2015, 71, 2109–2118. [Google Scholar] [CrossRef]
- Tan, K.; Mulligan, R.; Moy, S. ; A., J. The crystal structure of a protein Lpg0085 with unknown function (DUF785) from Legionella pneumophila subsp. pneumophila str. Philadelphia 1. Available online: (accessed on.
- Hartsuck, J.A.; Koelsch, G.; Remington, S.J. The high-resolution crystal structure of porcine pepsinogen. Proteins 1992, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, A.R.; Fedorov, A.A.; Boodhoo, A.; Andreeva, N.S.; James, M.N. Molecular and crystal structures of monoclinic porcine pepsin refined at 1.8 A resolution. J Mol Biol 1990, 214, 143–170. [Google Scholar] [CrossRef]
- Vuksanovic, N.; Silvaggi, N.R. Porcine pepsin in complex with saquinavir. Available online: (accessed on.
- Morales, R.; Watier, Y.; Bocskei, Z. Human prorenin structure sheds light on a novel mechanism of its autoinhibition and on its non-proteolytic activation by the (pro)renin receptor. J Mol Biol 2012, 421, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, A.R.; Hayakawa, K.; Fujinaga, M.; Murphy, M.E.; Fraser, M.; Muir, A.K.; Carilli, C.T.; Lewicki, J.A.; Baxter, J.D.; James, M.N. Structure of recombinant human renin, a target for cardiovascular-active drugs, at 2.5 A resolution. Science 1989, 243, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Remen, L.; Bezencon, O.; Richard-Bildstein, S.; Bur, D.; Prade, L.; Corminboeuf, O.; Boss, C.; Grisostomi, C.; Sifferlen, T.; Strickner, P.; et al. New classes of potent and bioavailable human renin inhibitors. Bioorg Med Chem Lett 2009, 19, 6762–6765. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, N.K.; Cherney, M.M.; Loetscher, H.; Ridley, R.G.; James, M.N. Crystal structure of the novel aspartic proteinase zymogen proplasmepsin II from plasmodium falciparum. Nat Struct Biol 1999, 6, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A.; Gulnik, S.V.; Afonina, E.; Yu, B.; Ellman, J.A.; Haque, T.S.; Silva, A.M. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J Mol Biol 2003, 327, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Prade, L.; Jones, A.F.; Boss, C.; Richard-Bildstein, S.; Meyer, S.; Binkert, C.; Bur, D. X-ray structure of plasmepsin II complexed with a potent achiral inhibitor. J Biol Chem 2005, 280, 23837–23843. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, P.; Xiao, H.; Hidaka, K.; Gustchina, A.; Kiso, Y.; Yada, R.Y.; Wlodawer, A. Structural insights into the activation and inhibition of histo-aspartic protease from Plasmodium falciparum. Biochemistry 2011, 50, 8862–8879. [Google Scholar] [CrossRef]
- Hanova, I.; Brynda, J.; Houstecka, R.; Alam, N.; Sojka, D.; Kopacek, P.; Maresova, L.; Vondrasek, J.; Horn, M.; Schueler-Furman, O.; et al. Novel Structural Mechanism of Allosteric Regulation of Aspartic Peptidases via an Evolutionarily Conserved Exosite. Cell Chem Biol 2018, 25, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, N.K.; Cherney, M.M.; Yowell, C.A.; Dame, J.B.; James, M.N. Structural insights into the activation of P. vivax plasmepsin. J Mol Biol 2003, 329, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Recacha, R.; Jaudzems, K.; Akopjana, I.; Jirgensons, A.; Tars, K. Crystal structure of Plasmodium falciparum proplasmepsin IV: the plasticity of proplasmepsins. Acta Crystallogr F Struct Biol Commun 2016, 72, 659–666. [Google Scholar] [CrossRef]
- Moore, S.A.; Sielecki, A.R.; Chernaia, M.M.; Tarasova, N.I.; James, M.N. Crystal and molecular structures of human progastricsin at 1.62 A resolution. J Mol Biol 1995, 247, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, N.; Gerhartz, B.; Worpenberg, S.; Trappe, J.; Eder, J. Crystal structure of an activation intermediate of cathepsin E. J Mol Biol 2004, 342, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; De Ranter, C.J.; Gebruers, K.; Brijs, K.; Courtin, C.M.; Delcour, J.A.; Rabijns, A. Structural basis for inhibition of Aspergillus niger xylanase by triticum aestivum xylanase inhibitor-I. J Biol Chem 2004, 279, 36022–36028. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Shimizu, T.; Yamabe, M.; Taichi, M.; Nishiuchi, Y.; Shichijo, N.; Unzai, S.; Hirano, H.; Sato, M.; Hashimoto, H. Crystal structure of basic 7S globulin, a xyloglucan-specific endo-beta-1,4-glucanase inhibitor protein-like protein from soybean lacking inhibitory activity against endo-beta-glucanase. FEBS J 2011, 278, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Shimizu, T.; Hirano, H.; Sato, M.; Hashimoto, H. Structural basis for inhibition of xyloglucan-specific endo-beta-1,4-glucanase (XEG) by XEG-protein inhibitor. J Biol Chem 2012, 287, 18710–18716. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.H.; Coman, R.M.; Bracho-Sanchez, E.; Fernandez, M.A.; Gilliland, C.T.; Li, M.; Agbandje-McKenna, M.; Wlodawer, A.; Dunn, B.M.; McKenna, R. Structure of the unbound form of HIV-1 subtype A protease: comparison with unbound forms of proteases from other HIV subtypes. Acta Crystallogr D Biol Crystallogr 2010, 66, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, K.; Kimura, T.; Sankaranarayanan, R.; Wang, J.; McDaniel, K.F.; Kempf, D.J.; Kameoka, M.; Adachi, M.; Kuroki, R.; Nguyen, J.T.; et al. Identification of Highly Potent Human Immunodeficiency Virus Type-1 Protease Inhibitors against Lopinavir and Darunavir Resistant Viruses from Allophenylnorstatine-Based Peptidomimetics with P2 Tetrahydrofuranylglycine. J Med Chem 2018, 61, 5138–5153. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gustchina, A.; Matuz, K.; Tozser, J.; Namwong, S.; Goldfarb, N.E.; Dunn, B.M.; Wlodawer, A. Structural and biochemical characterization of the inhibitor complexes of xenotropic murine leukemia virus-related virus protease. FEBS J 2011, 278, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Trempe, J.F.; Saskova, K.G.; Siva, M.; Ratcliffe, C.D.; Veverka, V.; Hoegl, A.; Menade, M.; Feng, X.; Shenker, S.; Svoboda, M.; et al. Structural studies of the yeast DNA damage-inducible protein Ddi1 reveal domain architecture of this eukaryotic protein family. Sci Rep 2016, 6, 33671. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Taylor, W.R. A structural model for the retroviral proteases. Nature 1987, 329, 351–354. [Google Scholar] [CrossRef]
- Hill, J.; Phylip, L.H. Bacterial aspartic proteinases. FEBS Lett 1997, 409, 357–360. [Google Scholar] [CrossRef]
- Castillo, R.M.; Mizuguchi, K.; Dhanaraj, V.; Albert, A.; Blundell, T.L.; Murzin, A.G. A six-stranded double-psi beta barrel is shared by several protein superfamilies. Structure 1999, 7, 227–236. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Bateman, A. Pepsin homologues in bacteria. BMC Genomics 2009, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.; Blundell, T. The active site of aspartic proteinases. FEBS Lett 1984, 174, 96–101. [Google Scholar] [CrossRef]
- Blundell, T.L.; Jenkins, J.A.; Sewell, B.T.; Pearl, L.H.; Cooper, J.B.; Tickle, I.J.; Veerapandian, B.; Wood, S.P. X-ray analyses of aspartic proteinases. The three-dimensional structure at 2.1 A resolution of endothiapepsin. J Mol Biol 1990, 211, 919–941. [Google Scholar] [CrossRef]
- Wan, W.Y.; Milner-White, E.J. A natural grouping of motifs with an aspartate or asparagine residue forming two hydrogen bonds to residues ahead in sequence: their occurrence at alpha-helical N termini and in other situations. J Mol Biol 1999, 286, 1633–1649. [Google Scholar] [CrossRef]
- James, M.N.; Hsu, I.N.; Delbaere, L.T. Mechanism of acid protease catalysis based on the crystal structure of penicillopepsin. Nature 1977, 267, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Blundell, T.L.; Jones, H.B.; Khan, G.; Taylor, G.; Sewell, B.T.; Pearl, L.H.; Wood, S.P. The Active Site of Acid Proteinases. In Enzyme Regulation and Mechanism of Action, Mildner, P., Ries, B., Eds.; Pergamon: Oxford, 1980; pp. 281–288. [Google Scholar]
- Andreeva, N.S.; Rumsh, L.D. Analysis of crystal structures of aspartic proteinases: on the role of amino acid residues adjacent to the catalytic site of pepsin-like enzymes. Protein Sci 2001, 10, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.J.; Nissink, J.W.; Allen, F.H.; Milner-White, E.J. Mimicry by asx- and ST-turns of the four main types of beta-turn in proteins. Protein Sci 2004, 13, 3051–3055. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An introduction to hydrogen bonding; Oxford university press New York: 1997; Volume 12.
- Derewenda, Z.S.; Derewenda, U.; Kobos, P.M. (His)C epsilon-H...O=C < hydrogen bond in the active sites of serine hydrolases. J Mol Biol 1994, 241, 83–93. [Google Scholar] [CrossRef]
- Sobolev, V.; Sorokine, A.; Prilusky, J.; Abola, E.E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Dali: a network tool for protein structure comparison. Trends Biochem Sci 1995, 20, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res 2022, 50, W651–W656. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Kraulis, P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. Journal of applied crystallography 1991, 24, 946–950. [Google Scholar] [CrossRef]
Figure 1.
Three building blocks of the structural catalytic core (SCC) in propepsin (PDB ID: 3PSG), as a representative member of the Pepsin-like family of the Acid proteases superfamily. (A) DD-zone, (B) psi-loopN and (C) psi-loopC. The dashed lines show long-range hydrogen bonds between the bordering amino acids of fragments of the primary structure of the protein: D-loops, DD-link, Mediator and G-loops, thus determining the cyclic nature and composition of the residues of each block separately. A dimer of dipeptides, Asp32-Thr33 and Asp215-Thr216, from two D-loops form the Fireman’s grip in the DD-zone, which is characterized by four long-range hydrogen bonds, while tetrapeptides, Asp32-...-Ser35 and Asp215-...-Thr218, from two D-loops form the Asx-motif in psi-loopN and psi-loopC, which is characterized by two short-range hydrogen bonds. Structural differences in two long-range hydrogen bonds located within psi-loopN (O/Asp32-N/Leu123 and (O/Ser35-N/Ala124) and psi-loopC (O/Thr218-N/Asp303 and O/Ser219-N/Val304) influence the functional differences between the catalytic aspartates.
Figure 1.
Three building blocks of the structural catalytic core (SCC) in propepsin (PDB ID: 3PSG), as a representative member of the Pepsin-like family of the Acid proteases superfamily. (A) DD-zone, (B) psi-loopN and (C) psi-loopC. The dashed lines show long-range hydrogen bonds between the bordering amino acids of fragments of the primary structure of the protein: D-loops, DD-link, Mediator and G-loops, thus determining the cyclic nature and composition of the residues of each block separately. A dimer of dipeptides, Asp32-Thr33 and Asp215-Thr216, from two D-loops form the Fireman’s grip in the DD-zone, which is characterized by four long-range hydrogen bonds, while tetrapeptides, Asp32-...-Ser35 and Asp215-...-Thr218, from two D-loops form the Asx-motif in psi-loopN and psi-loopC, which is characterized by two short-range hydrogen bonds. Structural differences in two long-range hydrogen bonds located within psi-loopN (O/Asp32-N/Leu123 and (O/Ser35-N/Ala124) and psi-loopC (O/Thr218-N/Asp303 and O/Ser219-N/Val304) influence the functional differences between the catalytic aspartates.
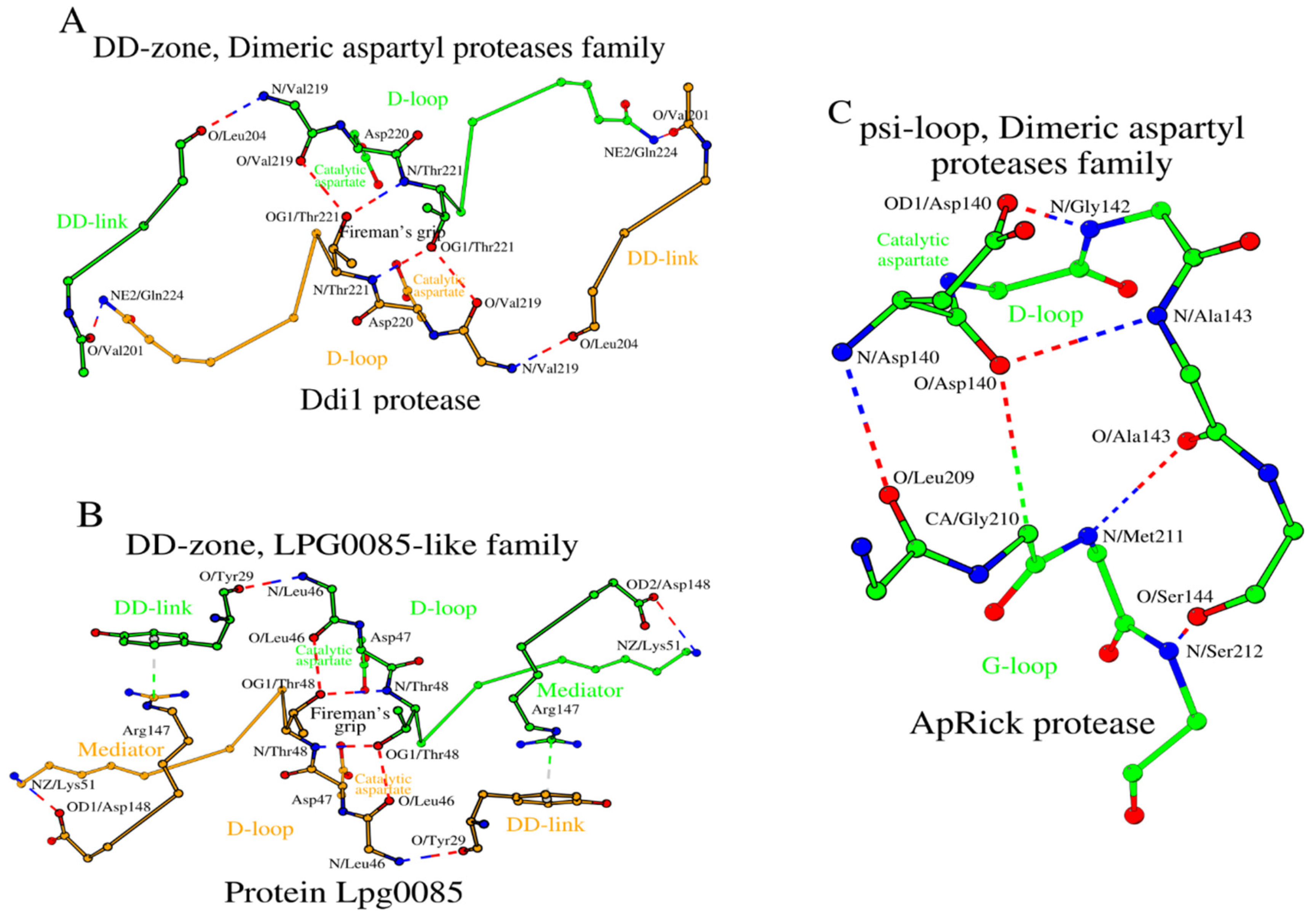
Figure 2.
Interface organization of interactions between the SCC of pepsin and the ligand saquinavir. (A) A smooth coil representation is shown that passes through the CA atom positions of the pepsin’s SCC. The dashed lines show the complete set of long-range hydrogen bonds between the bordering residues of the six amino-acid sequence fragments. (B) The potential hydrogen bonding interactions between the D-loops of the DD-zone and saquinavir are shown with dashed lines.
Figure 2.
Interface organization of interactions between the SCC of pepsin and the ligand saquinavir. (A) A smooth coil representation is shown that passes through the CA atom positions of the pepsin’s SCC. The dashed lines show the complete set of long-range hydrogen bonds between the bordering residues of the six amino-acid sequence fragments. (B) The potential hydrogen bonding interactions between the D-loops of the DD-zone and saquinavir are shown with dashed lines.
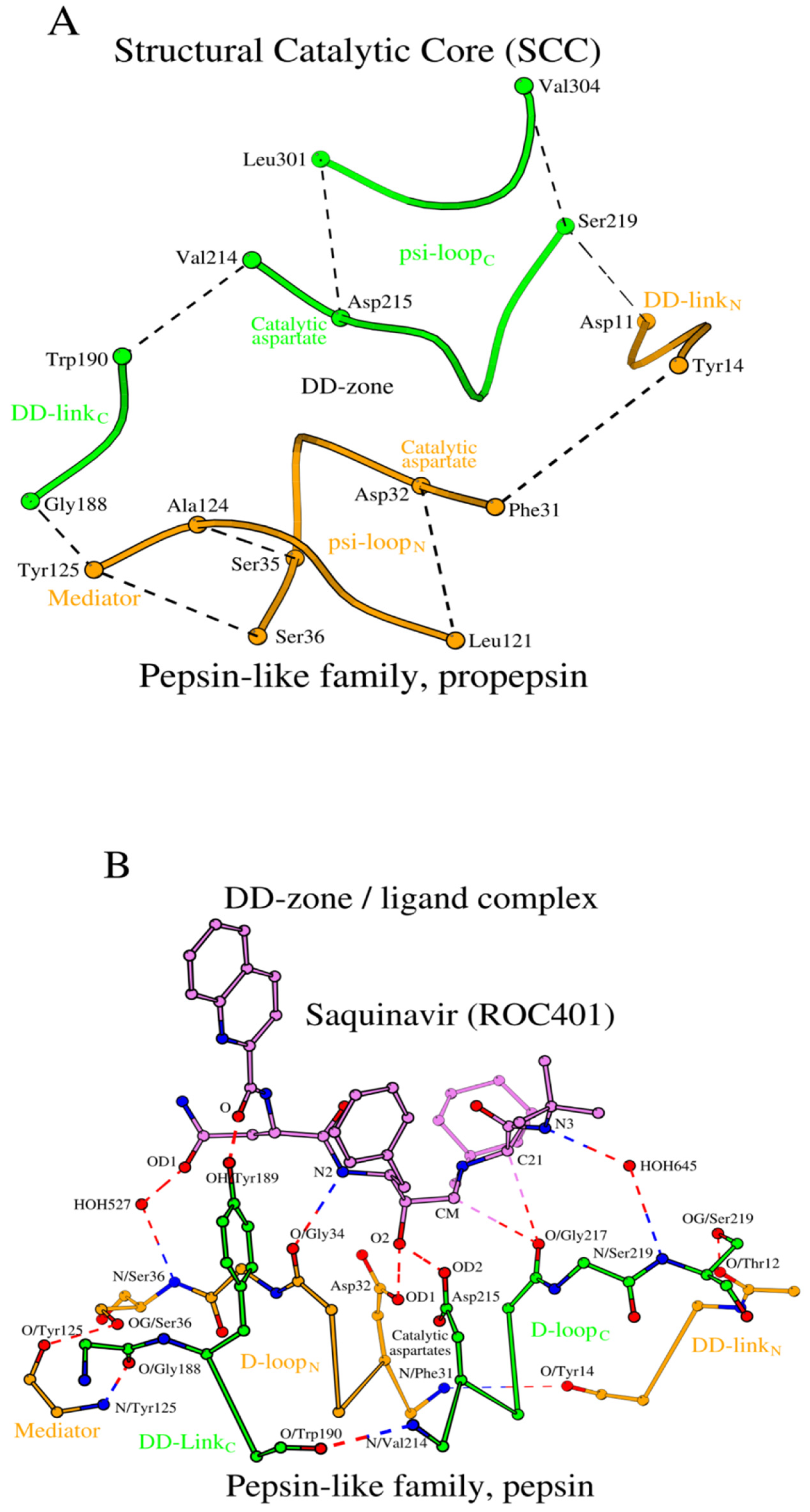
Figure 3.
The building blocks of the SCC in the HIV-1 and XMRV homodimer proteases (PDB IDs: 3IXO and 3NR6, correspondingly), as the representative members of Retroviral protease (retropepsin) family of the Acid proteases superfamily. (A) DD-zone of HIV-1 protease, (B) DD-zone of XMRV protease and (C) psi-loop of HIV-1 protease. (D) The potential hydrogen bonding interactions (dashed lines) between two identical D-loops of the DD-zone and the ligand in the HIV-1 protease with inhibitor KNI-1657 complex (PDB ID: 5YOK).
Figure 3.
The building blocks of the SCC in the HIV-1 and XMRV homodimer proteases (PDB IDs: 3IXO and 3NR6, correspondingly), as the representative members of Retroviral protease (retropepsin) family of the Acid proteases superfamily. (A) DD-zone of HIV-1 protease, (B) DD-zone of XMRV protease and (C) psi-loop of HIV-1 protease. (D) The potential hydrogen bonding interactions (dashed lines) between two identical D-loops of the DD-zone and the ligand in the HIV-1 protease with inhibitor KNI-1657 complex (PDB ID: 5YOK).
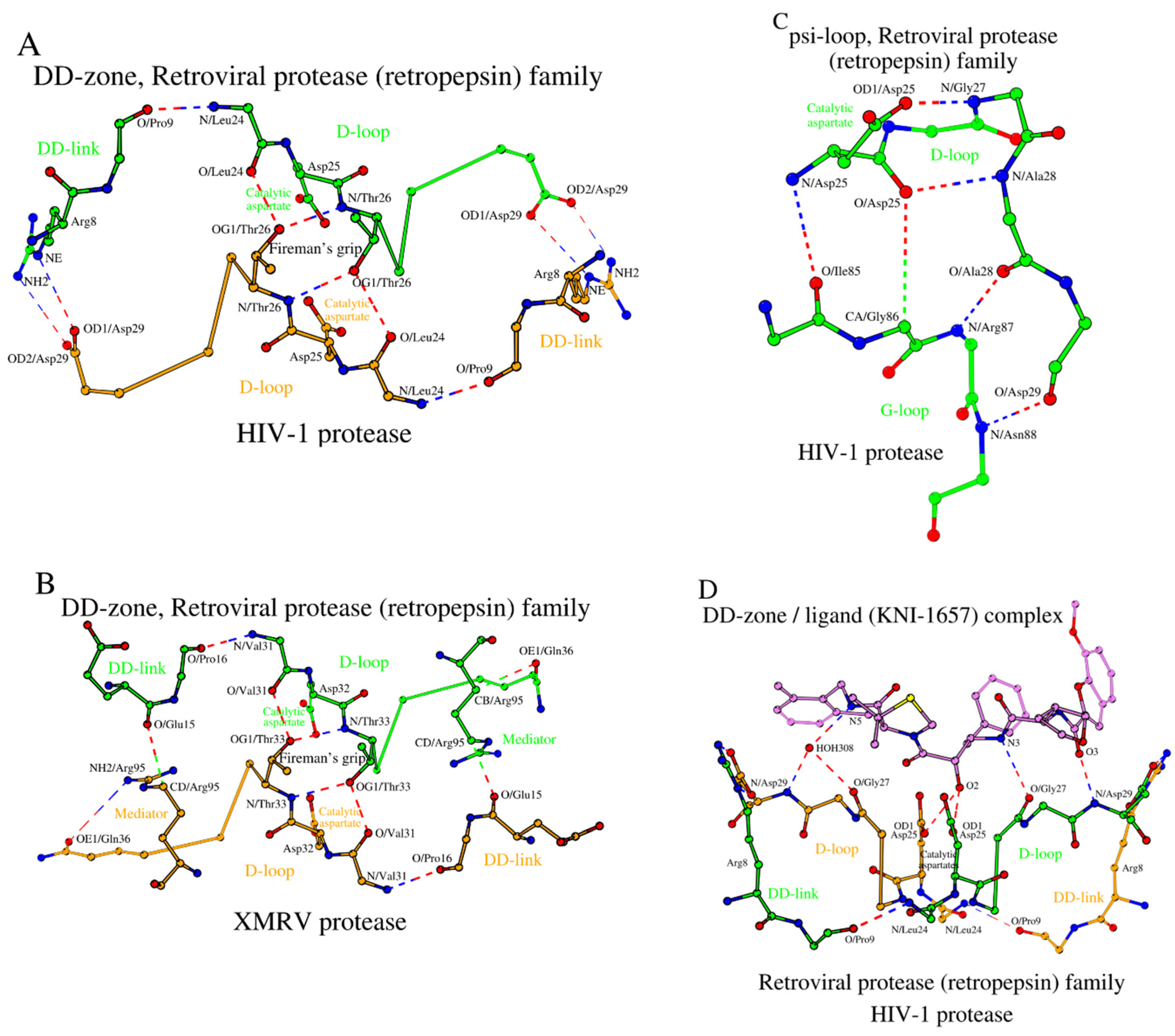
Figure 4.
SCC of (A) HIV-1 and (B) XMRV proteases. A smooth coil representation is used in the figures that passes through the CA atom of SCC positions of the corresponding retroviral proteases. The SCC of the XMRV protease differs from the SCC of the HIV-1 protease by the inclusion of the Mediator residue Arg95 from the G-loop in each monomer.
Figure 4.
SCC of (A) HIV-1 and (B) XMRV proteases. A smooth coil representation is used in the figures that passes through the CA atom of SCC positions of the corresponding retroviral proteases. The SCC of the XMRV protease differs from the SCC of the HIV-1 protease by the inclusion of the Mediator residue Arg95 from the G-loop in each monomer.
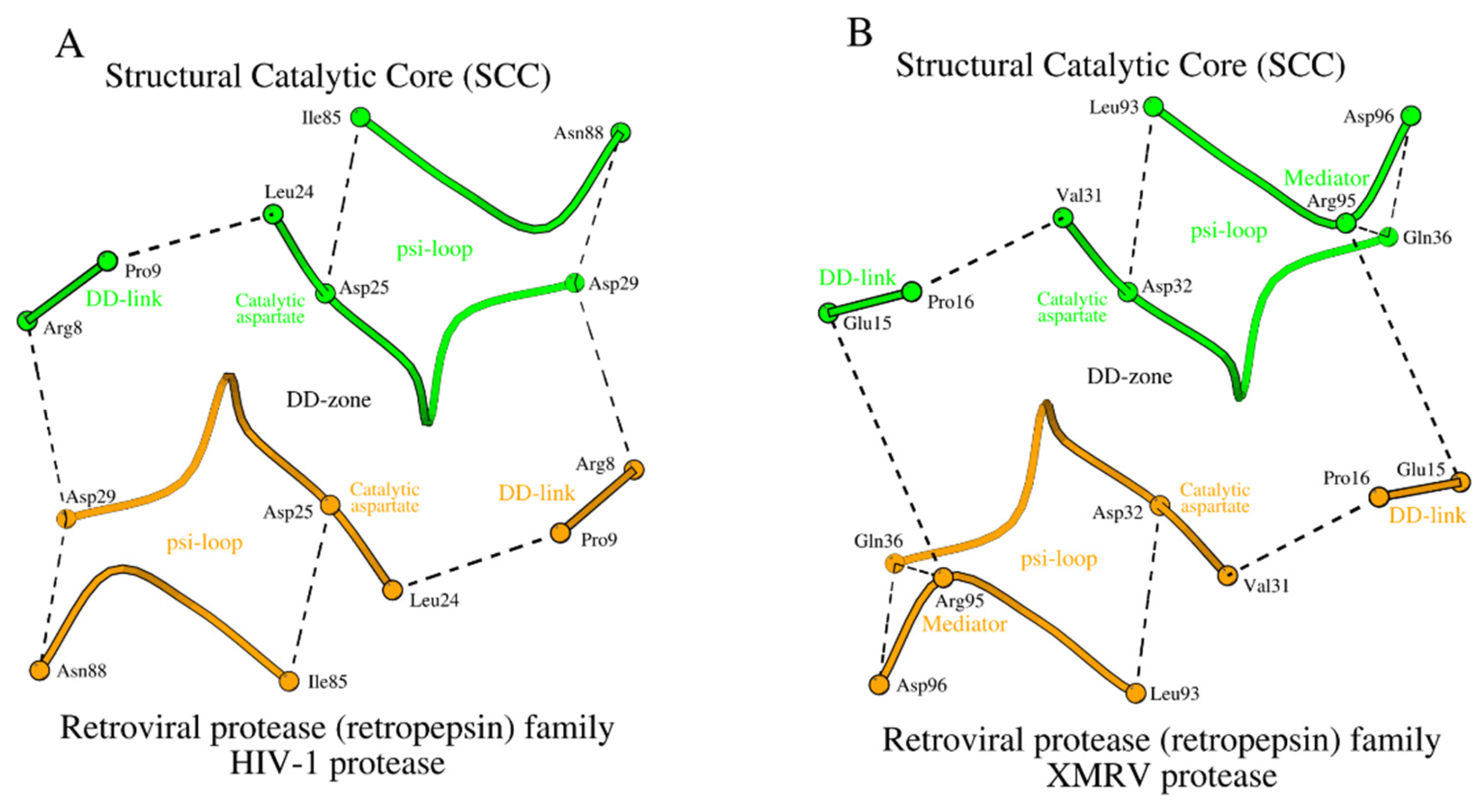
Figure 5.
The building blocks of the SCC in the Ddi1 protease, Lpg0085 protein and ApRick protease (PDB IDs: 4Z2Z, 2PMA and 5C9F, correspondingly), as the representative members of the Dimeric aspartyl proteases and LPG0085-like families of Acid proteases superfamily. (A) DD-zone of Ddi1 protease, (B) DD-zone of protein Lpg0085 and (C) psi-loop of ApRick protease.
Figure 5.
The building blocks of the SCC in the Ddi1 protease, Lpg0085 protein and ApRick protease (PDB IDs: 4Z2Z, 2PMA and 5C9F, correspondingly), as the representative members of the Dimeric aspartyl proteases and LPG0085-like families of Acid proteases superfamily. (A) DD-zone of Ddi1 protease, (B) DD-zone of protein Lpg0085 and (C) psi-loop of ApRick protease.
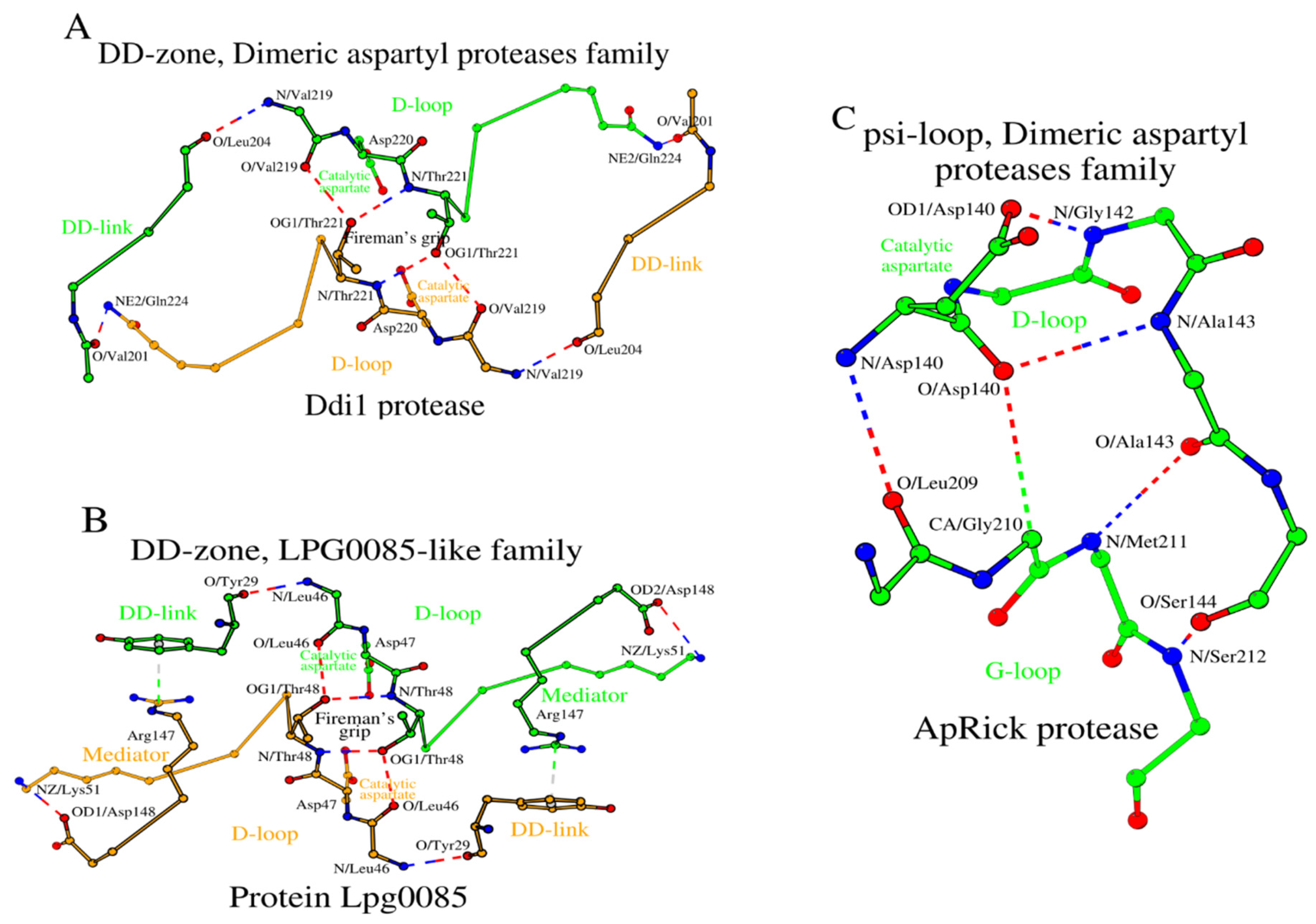
Figure 6.
SCC of (A) Ddi1 protease and (B) protein Lpg0085. The main differences between the SCCs of the two proteins are the amino acid composition of the DD-links and the use of a mediator-dipeptide in the structural formation of the DD-zone in the protein Lpg0085.
Figure 6.
SCC of (A) Ddi1 protease and (B) protein Lpg0085. The main differences between the SCCs of the two proteins are the amino acid composition of the DD-links and the use of a mediator-dipeptide in the structural formation of the DD-zone in the protein Lpg0085.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



