
Submitted:
21 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
L-gulonolactone oxidase enzyme (GULO) catalyzes the last step of L-ascorbic acid (vitamin C) biosynthesis. This enzymatic activity is lost in primates. The full-length rat GULO has been previously produced in plants and demonstrated to be active. In this study, we compare activity of two variants of GULO produced in Escheriachia coli cells, full-length rat GULO (fGULO) and its C-terminal catalytic domain (cGULO). The expression and purification of the recombinant proteins were optimised and their biological activity was confirmed by two methods, the GULO activity assay in the protein extracts and the ‘in-gel’ staining for GULO activity. Both variants of recombinant GULO were biologically active in both assays. However, cGULO is more promising than fGULO for ascorbic acid production because it is more efficiently produced by bacteria.
Keywords:
Gulonolactone oxidase
; recombinant protein
; GULO activity
; His-tag
1. Introduction
L-gulonolactone oxidase (GULO) is required for the final step of L-ascorbic acid biosynthesis. It catalyzes the reaction of L-gulono-1,4-lactone with oxygen to produce L-ascorbate and hydrogen peroxide and it uses L-flavin adenine dinucleotide (FAD) as a cofactor [1,2]. Although several species are capable of synthesizing vitamin C, this enzymatic activity is lost in humans, guinea pigs, bats and other primates [3,4]. The inability of these species to synthesize L-ascorbic acid is due to the absence of a functional GULO gene. Analysis of the nucleotide sequence of human genome reveals that the GULO gene has accumulated a numerous mutations as long as it stopped being active so that it is now present as a pseudo gene [5,6]. The GULO enzyme belongs to aldonolactone oxidoreductases family. The enzymes from this family contain two conserved domains: FAD-binding domain (Pfam Id: 01565) at the N-terminus and an arabinono-1,4-lactone oxidase (ALO) catalytic domain (Pfam Id: 04030) at the C-terminus.
The full-length rat GULO has been previously produced in plants and proved to elevate the level of ascorbic acid and increase plant tolerance to abiotic stresses [7,8]. However there is no reports on the production of an active recombinant GULO in the bacterial expression system. In this study we focused on the usage of such expression system for the production of enzymatically active GULO enzyme. Assuming that C-terminal cytoplasmic domain of GULO possesses catalytic activity not only the full-length GULO but also the truncated variant composed of only C-terminal domain of GULO was produced in Escherichia coli. Both proteins were purified based on the presence of C-terminally located His-tag, and their biological activity was demonstrated.
2. Materials and Methods
2.1. Plasmids, Bacterial Strains and Chemicals
pET28b(+) expression vector and Escherichia coli strains (DH5α, Rosetta(DE3), Rosetta(DE3)pLysS, BL21(DE3)pRARE, HMS174 and C43(DE3)) were obtained from Novagen (USA). cOmplete His-tag purification columns were from Roche Life Science Products. All the restriction enzymes, DNA and protein markers, PCR purification kit and plasmid miniprep kit were obtained from Thermo-Scientific Co. Ascorbic acid kit was purchased from Abcam Co. Monoclonal Anti-polyHistidine antibody, L-gulonolactone, flavin adenine dinucleotide, reduced glutathione, oxidized glutathione, Triton X-100, phenazine methosulfate and nitro blue tetrazolium were purchased from Sigma-Aldrich Co. This study used commercially available materials of analytical grade.
2.2. Cloning of GULO-Encoding Fragments into Bacterial Expression Plasmids
The sequence encoding rat full-length GULO (UniProt ID: P10867.3, 1-440 aa., (Figure 1) was synthesized and cloned into pUC57 vector by GenScript Company. For further cloning purposes GULO cDNA were amplified (Kapa HIFi hotstart DNA polymerase, Roche) using the 5’-TGATCCATGGTTCATGGCTACAAAGG-3’ and 5’-AGCACCATGGATAACAGATTCTTTTTCTGGA-3’ oligonucleotides as forward primers for fGULO and cGULO, respectively, and the 5’-TATTCTCGAGATAGAACACTTTTTCCAG-3’ oligonucleotide as a reverse primer for both products. Following restriction digestion with NcoI and XhoI, the PCR products were ligated into pET28b in frame with the sequence encoding 6x-His tag resulting in pET28b-fGULO and pET28b-cGULO plasmids.
2.3. Production of Recombinant fGULO-His and cGULO-His and Testing Solubility of Recombinant Proteins
The plasmids were transformed into several different E. coli strains to optimize fGULO and cGULO production. Finally, BL21(DE3)pRARE and Rosetta(DE3)pLysS were chosen for fGULO and cGULO production, respectively. For fGULO, the bacteria were cultured in Terrific broth (TB) with 50 μg/ml kanamycin to OD600=1 and induced with 1.0 mM isopropyl-ß-D-1-thiogalactopyranoside (IPTG) and incubated overnight at 16oC and 25oC. For cGULO, bacteria were grown in lysogeny broth (LB) with 50 μg/ml kanamycin to OD600=0.6 then IPTG was added to a final concentration of 0.5 mM and incubated at 37oC for 3 hours.
To test the expression of fGULO-His and cGULO-His recombinant proteins, total protein extract obtained from 1 ml of bacterial cultures were precipitated, resuspended in 1× Laemmli loading buffer, boiled at 95oC and checked by SDS-PAGE. To determine if GULO-His recombinant proteins were present in a soluble form in bacterial cells, the pellets from 50 ml induced cultures were suspended in 5 ml of phosphate buffered saline (PBS), pH 7.4, supplemented with 0.1% Triton X-100. A Branson SX150 sonifier was used to lyse the resuspended pellets using five cycles of 15 s sonication interspersed with 30 s cooling on ice, followed by centrifugation at 10,000 x g (4oC, 60 min) to separate the soluble and insoluble fractions. Aliquots of the supernatants and pellets (resuspended in PBS with 0.1% Triton X-100, pH 7.4) were mixed with 4X Laemmli loading dye, boiled for 10 min and analysed using SDS-PAGE.
2.4. Protein Purification under Denaturing Conditions
Bacterial cultures (100 ml) were centrifuged and resuspended in 10 ml lysis/equilibration buffer (0.1 M sodium phosphate, pH 8.0, 0.1% Triton X-100 and 8 M urea) and sonicated as described above. The cell lysate was then centrifuged at 10,000 x g for 60 min at 4°C. The supernatant was collected and applied to a poly-histidine affinity tag column previously equilibrated with the same lysis buffer. There were 10 volumes of wash buffer (0.1 M sodium phosphate, pH 6.5, 0.1% Triton X-100 and 8 M urea). Elution was then performed with elution buffer (0.1 M sodium phosphate, pH 5.5, 0.1% Triton X-100, 500 mM imidazole and 8 M urea). SDS-PAGE was used to investigate the eluted fractions.
The fractions containing the maximum concentration of purified GULO-His recombinant proteins were pooled and their buffer exchanged in five consecutive dialysis steps in dialysis buffer (1x PBS, pH 7.4, 2 mM reduced glutathione 1 mM oxidised glutathione, 2 mM EDTA, 10 µM FAD and 0.1% Triton X-100) with a gradual decrease in urea concentration (8M, 6M, 4M, 2M, 1M and 0M). The first two dialysis steps were carried out using a dialysis bag of molecular weight cutoff of 10 kDa with gentle agitation for 4 h at 16oC and the last three steps were performed using PD-10 (Sephadex G-25) column according to manufacturer’s instructions to avoid protein aggregation which started to appear within dialysis bag at the stage of changing urea concentration from 4 M to 2 M. The dialyzed fractions from PD-10 column were collected, concentrated and stored as aliquots at −20◦C for further analyses. The protein content was determined by Bradford's method, using BSA as a standard [9].
2.5. Western Blot Analysis
Approximately 30 µg of recombinant protein was fractionated by SDS-PAGE and transferred to a nitrocellulose membrane using standard transblotting buffer in an electrophoretic transfer cell (Bio-Rad). Following visualisation of the transferred proteins with 0.02% Ponceau-S, the membranes were blocked with 5% (w/v) non-fat dry milk in PBS, pH 7.4 for 1 h and incubated with mouse anti-polyhistidine monoclonal antibody (1:6000). Immunodetection was performed with secondary goat anti-mouse IgG conjugated to alkaline phosphatase (1:10000, 1h) and 1-step NBT/BCIP (ThermoFisher Scientific).
2.6. GULO Activity Assay
The activity of GULO was determined using a technique adapted from Hassan et al. 2004 [3] with a few adjustments. The reaction solution (1.0 ml) included a potassium phosphate buffer (50 mM, pH 7.0), 50 mM sodium citrate, 1 mM dithiothreitol, 10 µM FAD, and the enzyme. The reaction began with the addition of 10 mM L-gulono-γ-lactone as the substrate and was carried out under aerobic conditions at 37oC, with vigorous shaking for 15 minutes, and then halted by the addition of trichloroacetic acid to a final concentration of 5%. Following this, the mixture was centrifuged at 15,000 × g for 15 minutes at 4oC. Ascorbic acid produced in the supernatants was quantified by a colorimetric assay using ascorbic acid assay kit (Abcam Co., Cat. No. AB65346). During the incubation, L-ascorbic acid underwent autoxidation, so the measured value was adjusted to account for this autoxidation that occurred when L-ascorbic acid was added to the reaction without a substrate. A single unit of the enzyme is considered to be the amount that can catalyze the production of 1 nmol of ascorbic acid per minute under the conditions of the assay. The assays were performed in triplicates.
2.7. Polyacrylamide Gel Electrophoresis and Staining Gels for GULO Activity
Polyacrylamide gel electrophoresis (8%) was performed following Lee et al. 1999 [10] protocol. To this end, Triton X-100 (0.1%) was incorporated into the gel and the electrophoresis buffer. The proteins were then stained with Coomassie blue. The activity of GULO was marked using Nishikimi, 1976 [11] technique, with slight adjustments. The gels were then incubated in a solution that included 2.5 mM L-gulono-lactone, 0.33 mM phenazine methosulfate, 0.12 mM nitro blue tetrazolium, 1 mM EDTA, 10 µM FAD, and potassium phosphate buffer (50 mM, pH 7.5). The incubation was conducted in the dark at room temperature until the bands were clearly appeared and the reaction was halted by immersing the gel in 7% acetic acid.
2.8. Statistical Analysis
The data are presented as mean values from (3-5) independent experiments with standard deviation (±SD) indicated. Data analysis was performed using Origin 8.0.
3. Results and Discussion
This study reports on production of two variants of His-tagged recombinant rat GULO in E.coli cells, full-length (fGULO containing 440 amino acids) and C-terminal cytoplasmic domain (cGULO containing 167 amino acids due to deletion of 273 N-terminal amino acids). The low level expression of fGULO-His was successfully augmented by increasing the biomass density (to OD600=1) and using the low cultivation temperature. Such conditions allow for a slower production of correctly folded protein and a reduction of aggregation [12]. We selected the highest producer, which was in this case BL21(DE3)pRARE [13,14]. Using Terrific Broth (TB) instead of Lysogeny broth (LB) resulted in attaining the required cell density for the extended induction time, as previously documented [14,15]. For fGULO, a protein band of expected size, migrating slightly lower than the 50 kDa protein marker, was detected. The expression level of recombinant cGULO protein was similar in different hosts and the protein size was in agreement with the expected size of about 20 kDa.
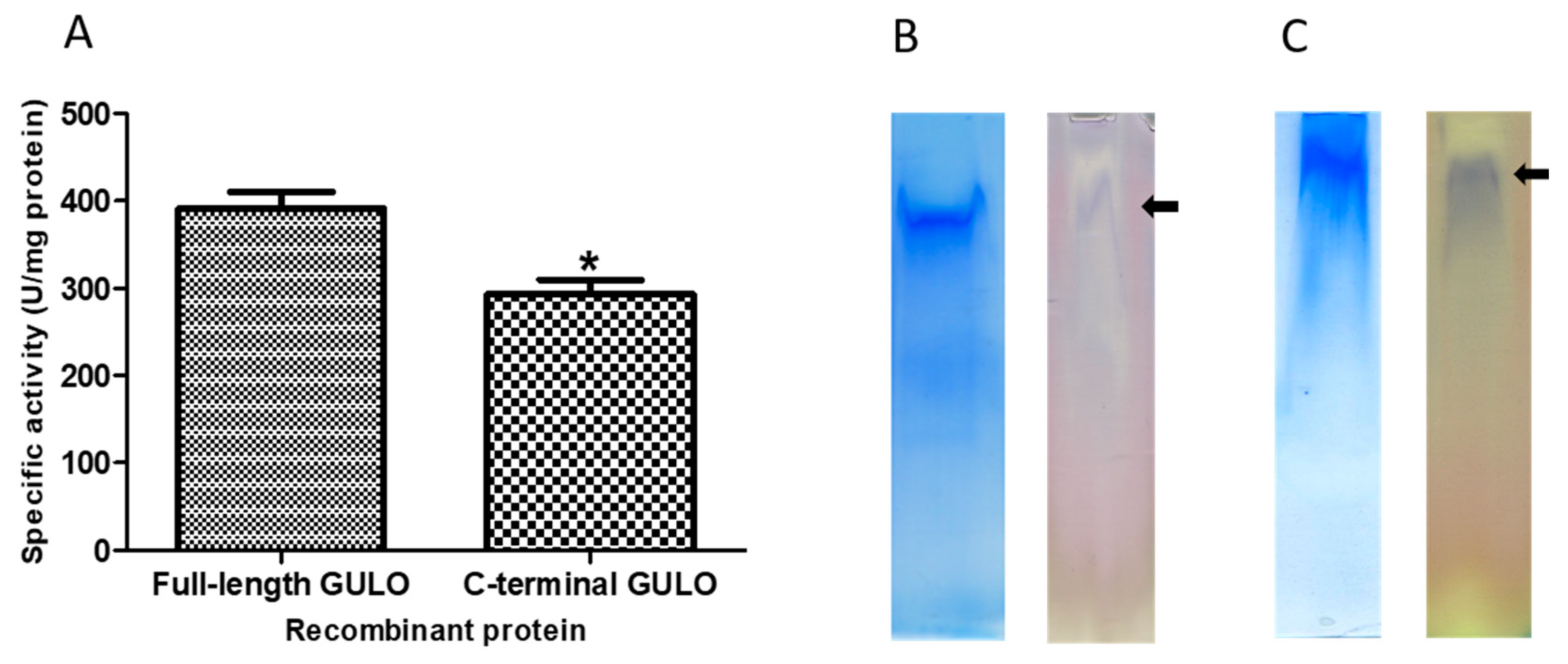
The fGULO and cGULO had different isoelectric points (pI) and net charges (Supplementary Figure S1), however it did not affect their solubility in bacterial cells. Both recombinant proteins were present almost exclusively in the insoluble fraction and only small amount was recovered from the soluble fraction. In such cases the common practice is to solubilize the protein using a chaotropic agent, such as urea, which successfully enhances protein solubility [16]. Therefore, the purification procedures were carried out under denaturing conditions (in the presence of 8M urea in lysis, washing and elution buffers) to allow solubilization of the recombinant proteins, following elution with 0.5 M imidazole. The amounts of recombinant fGULO and cGULO proteins obtained after purification steps were monitored. The specific enzymatic activity of cGULO was lower that the specific enzymatic activity of fGULO. (Figure 2). Enzymatic activity for recombinant proteins was verified either directly in protein extracts (Figure 2) or ‘in-gel’, after proteins separation in a native PAGE (Figure 3). The GULO proteins in insoluble fractions of bacterial extracts were first solubilized, purified, renaturated and assayed for GULO enzymatic activity. The protein extract from non-induced bacterial strains was used as a negative control. Both GULO variants were enzymatically active, however, the specific enzymatic activity of cGULO, consisting of only 167 C-terminal amino acid was significantly lower (about 75%) than that of fGULO, consisting of 440 amino acids. It could be attributed to impaired protein folding, to a decoupling the FAD-binding domain from the catalytic domain or to the higher proteolytic degradation of the truncated variant than the full-length variant in bacterial cells [17]. The native PAGE of the renatured affinity purified recombinant fGULO and of cGULO revealed only one protein band in each preparation (Figure 3). Similarly, in the preparations of fGULO and cGULO a single corresponding band was also detected using the ‘in-gel” assays for GULO activity. The GULO enzymatic activity resulted in formation of insoluble blue purple formazan dye (dark band) due to reduction of nitro blue tetrazolium in the presence of substrate and phenazine methosulphate [18].
GULO enzyme which belongs to ascorbate-synthesizing (aldonolactone oxidoreuctases) family, includes two conserved domains: N-terminal FAD-binding region and C-terminal HWXK motif which is specific for D-arabinono-1,4-lactone oxidase (ALO) involved in the final step of D-ascorbic acid biosynthesis. The Arabidopsis GULO4 lost GULO activity due to the absence of ALO domain [19]. The mutations in HWXK motif led to GULO activity loss, as the positions His-447 and Trp-448, which are part of a C-terminal motif, are essential for FMN formation [20]. The conserved Lys residue in this motif (Lys-450) is not needed for cofactor binding, yet its substitution with Gly makes the protein inactive. This suggests that the absence of activity in these mutations backs the theory that the conserved HWXK motif is vital for creating the active site of the enzyme. In flavoprotein aldonolactone oxidoreductases, a C-terminal histidine residue is covalently bonded to the FAD cofactor, while an N-terminal histidine residue facilitates this bond. This covalent bond enhances the enzyme's ability to carry out redox reactions, increase cofactor binding, improve protein stability, and prevent flavin modification. The significant increase in thermal stability with excess FAD suggests that the cofactor protects the enzyme from irreversible denaturation or aggregation. [22]. Mutation in the FAD-binding region could contribute to the loss of GULO activity in bat Pteropus vampyrus [23]. It could also explain that failure of Biyani and Madhubala, 2011 [24] to detect ALO activity in Leishmania donovani in the absence of FAD.
Rat GULO full-length has been previously produced in transgenic plants and proved to be active in such plants as tobacco and lettuce [7], potato [8] and Arabidopsis [25]. On the other hand, production of vitamin C from D-glucose in E. coli was achieved on one-step fermentation by expression of ten genes from Arabidopsis thaliana included in vitamin C biosynthesis pathway [4].
4. Conclusion and Perspective
It is the first report about heterologous production of enzymatically active C-terminal cytoplasmic fragment of rat GULO. The protein was catalytically active, as demonstrated by its ability to produce ascorbic acid from L-gulonolactone in the presence of FAD (cofactor). The cGULO appears to be a more promising option than fGULO for the synthesis of ascorbic acid as it is produced more efficiently than fGULO in bacteria. Future studies are planned for production of active recombinant GULO in fusion with proteins enabling polymerization in both bacterial cells and transgenic plants. In the further perspective, such approach will permit efficient production of the matrix, which will provide the missing enzymatic activity in human tissues, which could be applied topically to the wounded skin. It is anticipated that the local content of vitamin C would be increased by externally provided active recombinant GULO, while the polymerizing protein would be used to replace decayed tissues in the wound site.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Abdul Aziz M. Gad: Writing – review & editing, Writing – original draft, Methodology, Investigation, Conceptualization, Formal analysis. Anna Góra-Sochacka: Writing – review & editing, Investigation, Methodology. Agnieszka Sirko: Writing – review & editing, Supervision.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Maria Skłodowska-Curie grant agreement No 847639 and from the Ministry of Education and Science.
Data availability statement
Data will be made available on request.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Maria Skłodowska-Curie grant agreement No 847639 and from the Ministry of Education and Science.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Linster, C.L.; Van Schaftingen, E. Vitamin C: biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, N. Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radic. Biol. Med. 2018, 122, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Hasan, L.; Vogeli, P. , Stoll, P.; KramerStranzinger, Š.Š.G.; Neuenschwander, S. Intragenic deletion in the gene encoding L-gulonolactone oxidase causes vitamin C deficiency in pigs. Mamm. Genome. 2004, 15, 323–333. [Google Scholar] [CrossRef]
- Tian, Y.S.; Deng, Y.D.; Zhang, W.H.; Xu, J.; Gao, J.J.; Fu, X.Y.; Han, H.J.; Li, Z.J.; Wang, L.J.; Peng, R.H.; Yao, Q.H. Metabolic engineering of Escherichia coli for direct production of vitamin C from D-glucose. Biotechnol. biofuels bioprod. 2022, 15, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, M.; Yagi, K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J. Clin. Nutr. 1991, 4, l203S–1208S. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.F.; Duque, P.; Lopez-Fernandez, H.; Vázquez, N.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. Multiple independent L-gulonolactone oxidase (GULO) gene losses and vitamin C synthesis reacquisition events in non Deuterostomian animal species. BMC Evol. Biol. 2019, 19, 126–137. [Google Scholar] [CrossRef]
- Jain, A.K.; Nessler, C.L. Metabolic engineering of an alternative pathway for ascorbic acid biosynthesis in plants. Mol. Breed. 2000, 6, 73–78. [Google Scholar] [CrossRef]
- Hemavathi, Upadhyaya, C. P.; Akula, N.; Young, K.E.; Chun, S.C.; Kim, D.H.; Park, S.W. Enhanced ascorbic acid accumulation in transgenic potato confers tolerance to various abiotic stresses. Biotechnol. Lett. 2010, 32, 321–330. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lee, B.H.; Huh, W.K.; Kim, S.T.; Lee, J.S.; Kang, S.O. Bacterial Production of d-Erythroascorbic Acid and L-Ascorbic Acid through Functional Expression of Saccharomyces cerevisiae D-Arabinono-1,4 Lactone Oxidase in Escherichia coli. Appl. Environ Microb. 1999, 65, 4685–4687. [Google Scholar] [CrossRef]
- Nishikimi, M. Purification and characterization of L-gulono-γ-lactone oxidase from rat and goat liver. Arch. Biochem. Biophys. 1976, 175, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014, 5, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Chernikova, T.N.; Yakimov, M.M.; Golyshin, P.N.; Timmis, K.N. Chaperonins govern growth of Escherichia coli at low temperatures. Nat. Biotechnol. 2003, 21, 1266–126. [Google Scholar] [CrossRef] [PubMed]
- Malash, M.N.; Hussein, N.A.; Muawia, S.; Nasr, M.I.; Siam, R. An optimized protocol for high yield expression and purification of an extremophilic protein. Protein Expr. Purif. 2020, 169, 105585. [Google Scholar] [CrossRef] [PubMed]
- Madurawe, R.D.; Chase, T.E.; Tsao, E.I.; Bentley, W.E. A recombinant lipoprotein antigen against Lyme disease expressed in E. coli: fermentor operating strategies for improved yield. Biotechnol. Prog. 2000, 16, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S.E.; Bicknell, A. Detection and prevention of protein aggregation before, during, and after purification. Anal. Biochem. 2003, 316, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wolucka, B.A.; Communi, D. Mycobacterium tuberculosis possesses a functional enzyme for the synthesis of vitamin C, L-gulono-1,4-lactone dehydrogenase. FEBS J. 2006, 273, 4435–4445. [Google Scholar] [CrossRef]
- Okamura, M. Purification and properties of L-gulono-1, 4-lactone oxidase from Grifola frondosa. J. Nutr. Sci. Vitaminol, 2001; 47, 258–262. [Google Scholar] [CrossRef]
- Maruta, T.; Ichikawa, Y.; Mieda, T.; Takeda, T.; Tamoi, M.; Yabuta, Y.; Ishikawa, T.; Shigeoka, S. The contribution of Arabidopsis homologs of L-gulono-1, 4-lactone oxidase to the biosynthesis of ascorbic acid. Biosci. Biotechnol. Biochem. 2010, 74, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Logan, F.J.; Taylor, M.C.; Wilkinson, S.R.; Kaur, H.; Kelly, J.M. The terminal step in vitamin C biosynthesis in Trypanosoma cruzi is mediated by a FMN-dependent galactonolactone oxidase. Biochem. J. 2007, 407, 419–426. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Van Berkel, W.J.; Benen, J.A.; Visser, J.; Mattevi, A. A novel oxidoreductase family sharing a conserved FAD-binding domain. Trends Biochem. Sci. 1998, 23, 206–207. [Google Scholar] [CrossRef]
- Leferink, N.G.H. L-Galactono-γ-lactone dehydrogenase from Arabidopsis thaliana, a flavoprotein involved in vitamin C biosynthesis. FEBS J. 2008, 275, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Pan, Y.H.; Zhang, Y.; Jones, G.; Zhang, S. Progressive pseudogenization: vitamin C synthesis and its loss in bats. Mol. Biol. Evol. 2011, 28, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Biyani, N.; Madhubala, R. Leishmania donovani encodes a functional enzyme involved in vitamin C biosynthesis: Arabino-1,4-lactone oxidase. Mol. Biochem. Parasit. 2011, 180, 76–85. [Google Scholar] [CrossRef]
- Aboobucker, S.I.; Suza, W.P.; Lorence, A. Characterization of two Arabidopsis L-gulono-1, 4-lactone oxidases, AtGulLO3 and AtGulLO5, involved in ascorbate biosynthesis. Reactive oxygen species (Apex, NC) 2017, 40, 389–427. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Nucleotide sequence encoding GULO protein used in this study. The deduced protein sequence is indicated above the nucleotide sequence. The non-cytoplasmic domain (amino acids 1-250), the trans-membrane domain (amino acids 251-273) is highlighted in blue, and the cytoplasmic domain (amino acids 274-440) is highlighted in green. The domains were identified in recombinant rat GULO by a BLAST search.
Figure 1.
Nucleotide sequence encoding GULO protein used in this study. The deduced protein sequence is indicated above the nucleotide sequence. The non-cytoplasmic domain (amino acids 1-250), the trans-membrane domain (amino acids 251-273) is highlighted in blue, and the cytoplasmic domain (amino acids 274-440) is highlighted in green. The domains were identified in recombinant rat GULO by a BLAST search.


Figure 2.
Purification steps of fGULO-His and cGULO-His using the SDS-PAGE (A) and Western blot with anti-His antibody (B). Panel A, left hand: 10% SDS-PAGE of elution fractions from poly-histidine affinity tags column. M, prestained protein marker with size indicated next to the gel; 1, non-induced lysate of E. coli BL21(DE3)pRARE transformed with pET28b-fGULO; 2, induced lysate; 3, soluble fraction of lysate; 4, insoluble fraction of lysate; 5, flow through; 6, wash; 7, an aliquot of the pooled eluted fractions; 8, an aliquot of the pooled renatured eluted fractions. Panel A, right hand: 15% SDS-PAGE of elution fractions from poly-histidine affinity tags column. M, prestained protein marker with size indicated next to the gel, 1, non-induced lysate of E. coli Rosetta(DE3)pLysS transformed with pET28b-cGULO; 2, induced lysate; 3, soluble fraction of lysate; 4,: insoluble fraction of lysate; 5, flow through; 6, wash; 7, an aliquot of the pooled eluted fractions; 8, an aliquot of the pooled renatured eluted fractions. Panel B, left hand: M, prestained protein marker; 1, fGULO crude extract; 2, affinity purified fGULO; 3, renatured affinity purified fGULO. Panel B, right hand: M, prestained protein marker; 1, cGULO crude extract; 2, affinity purified cGULO; 3, renatured affinity purified cGULO. The arrows indicate the presumed fGULO and cGULO.
Figure 2.
Purification steps of fGULO-His and cGULO-His using the SDS-PAGE (A) and Western blot with anti-His antibody (B). Panel A, left hand: 10% SDS-PAGE of elution fractions from poly-histidine affinity tags column. M, prestained protein marker with size indicated next to the gel; 1, non-induced lysate of E. coli BL21(DE3)pRARE transformed with pET28b-fGULO; 2, induced lysate; 3, soluble fraction of lysate; 4, insoluble fraction of lysate; 5, flow through; 6, wash; 7, an aliquot of the pooled eluted fractions; 8, an aliquot of the pooled renatured eluted fractions. Panel A, right hand: 15% SDS-PAGE of elution fractions from poly-histidine affinity tags column. M, prestained protein marker with size indicated next to the gel, 1, non-induced lysate of E. coli Rosetta(DE3)pLysS transformed with pET28b-cGULO; 2, induced lysate; 3, soluble fraction of lysate; 4,: insoluble fraction of lysate; 5, flow through; 6, wash; 7, an aliquot of the pooled eluted fractions; 8, an aliquot of the pooled renatured eluted fractions. Panel B, left hand: M, prestained protein marker; 1, fGULO crude extract; 2, affinity purified fGULO; 3, renatured affinity purified fGULO. Panel B, right hand: M, prestained protein marker; 1, cGULO crude extract; 2, affinity purified cGULO; 3, renatured affinity purified cGULO. The arrows indicate the presumed fGULO and cGULO.
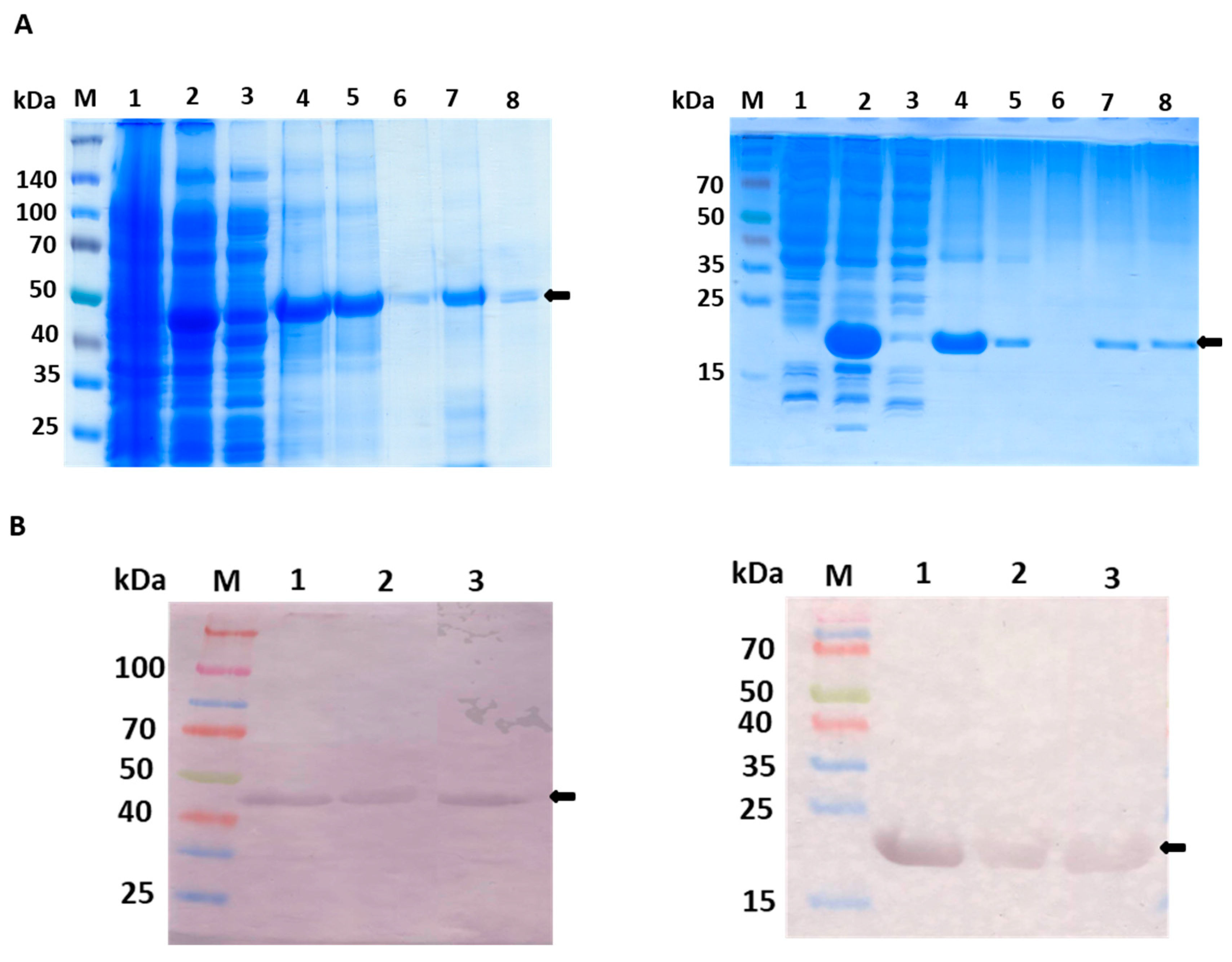
Figure 3.
Verification of enzymatic activity of fGULO-His and cGULO-His recombinant proteins. (A) The GULO activity assays in the protein extracts are presented as means with standard deviation (SD) indicated. One unit of GULO activity is defined as the amount of enzyme capable of production of one nanomole of ascorbic acid/minute/mg of protein. The asterisk indicates the statistically significant differences in the specific GULO activity from fGULO, * for p<0.01. The ‘in-gel’ GULO activity assay of fGULO-His (panel B) and cGULO-His (panel C). The respective gel slices, containing the partially purified recombinant fGULO or cGULO proteins, were either stained with Coomassie blue or assayed ‘in-gel’ for GULO activity. The arrows indicate the position of bands visualised by staining for GULO activity.
Figure 3.
Verification of enzymatic activity of fGULO-His and cGULO-His recombinant proteins. (A) The GULO activity assays in the protein extracts are presented as means with standard deviation (SD) indicated. One unit of GULO activity is defined as the amount of enzyme capable of production of one nanomole of ascorbic acid/minute/mg of protein. The asterisk indicates the statistically significant differences in the specific GULO activity from fGULO, * for p<0.01. The ‘in-gel’ GULO activity assay of fGULO-His (panel B) and cGULO-His (panel C). The respective gel slices, containing the partially purified recombinant fGULO or cGULO proteins, were either stained with Coomassie blue or assayed ‘in-gel’ for GULO activity. The arrows indicate the position of bands visualised by staining for GULO activity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



