
Submitted:
24 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
The gastrointestinal (GI) tract harbors trillions of microorganisms known to influence human health and disease, and next-generation sequencing (NGS) now enables the in-depth analysis of their diversity and functions. Although a significant amount of research has been conducted on the GI microbiome, comprehensive metagenomic datasets covering the entire tract are scarce due to cost and technical challenges. Despite the widespread use of fecal samples, integrated datasets encompassing the entire digestive process, beginning at the mouth and ending with feces, are lacking. With this study, we aimed to fill this gap by analyzing the complete metagenome of the GI tract, providing insights into the dynamics of the microbiota and potential therapeutic avenues. In this study, we delved into the complex world of the GI microbiota, which we examined in five healthy Japanese subjects. While samples from the whole GI flora and fecal samples provided sufficient bacteria, samples obtained from the stomach and duodenum posed a challenge. Using a principal coordinate analysis (PCoA), clear clustering patterns were identified; these revealed significant diversity in the duodenum. Although this study was limited by its small sample size, the flora in the overall GI tract showed unwavering consistency, while the duodenum exhibited unprecedented phylogenetic diversity. A visual heat map illustrates the discrepancy in abundance, with Fusobacteria and Bacilli dominating the upper GI tract and Clostridia and Bacterioidia dominating the fecal samples. Negativicutes and Actinobacteria were found throughout the digestive tract. This study demonstrates that it is possible to continuously collect microbiome samples throughout the human digestive tract. These findings not only shed light on the complexity of GI microbiota but also provide a basis for future research.
Keywords:
Next generation sequence
; microbiome
; whole gastrointestinal
; EGD
; GI tract
1. Introduction
The gastrointestinal (GI) tract contains trillions of microorganisms, the composition of which microorganisms is known to affect human health and disease [1,2,3]. With the development of next-generation sequencing (NGS) technology, we are now able to decipher the genetic information of microorganisms and analyze their diversity, functions, and gene expression patterns [4,5,6]. This has encouraged research on the microbiomes present in the GI tract [7,8]. Fecal samples are widely used as representative samples of the GI tract, comprising an important source of information regarding the composition and function of microbiota; however, the bacteria found in these samples generally only represent those found in the colon. Because GI microbiome studies focus mainly on the colon, there is a lack of research using complete GI metagenomic datasets that encompass the entire GI tract [9,10,11].
The bacteria present in the GI tract are very complex. For example, environments with different pH levels contain different densities and types of microflora [12]. It is therefore important to collect complete metagenomic data from all sites. however, this task is very difficult due to the invasiveness of the sampling procedures, technical issues, and cost. Even when considering studies which focused on a specific disease and accessed metagenomic data for specific sites, the number of sets that cover the entire GI tract remains limited [13,14]. For instance, in the literature concerning the oral microbiota, oral metagenomic data are collected to analyze the diversity and function of the oral microbiota only [15,16,17]. Similarly, data collected from the esophagus, stomach, and duodenum are used to study the characteristics and functions of the microbiota [18,19,20,21,22,23,24]. Few studies have collected mouth, esophagus, stomach, duodenum, and fecal samples [25] Thus, there an abundance of isolated metagenomic data concerning each part of the gastrointestinal tract but a lack of continuous data [26]. We believe that the accumulation of metagenomic data from the entire digestive tract will contribute to our understanding of the metagenomic environment and provide a more comprehensive picture of the microbiota. Therefore, we planned to analyze the metagenome of the entire GI tract in each patient.
2. Materials and Methods
2.1. Sample Preparation
With the approval of the Tohoku University Ethics Committee (Approval No. 2022-1-1066 from the Ethics Committee of the Tohoku University Graduate School of Medicine) and the consent of the research subjects, five healthy Japanese adults underwent upper gastrointestinal endoscopy, and mucus samples were carefully collected from the oral cavity to the duodenum using a brush (ECB-5-180-3-S, Cook Medical). Fecal samples were also collected on the same day.
The technique of brush sampling and preservation of bacterial flora by Esophagogastroduodenoscopy (EGD) was established by us. Details of the collection of gastrointestinal flora by the brush method are as follows.
Before the EGD procedure, the oral cavity was lightly rubbed with a sterile cotton swab to collect the bacterial flora; during the EGD examination, the mucus layer of the esophagus, stomach, and duodenum was lightly rubbed to collect the flora. This brush was originally developed for bronchoscopy and is covered by insurance when used in the intestinal tract. This brush is also compatible with the forceps hole of a small caliber endoscope, and since it does not cut the mucosa, it is non-invasive and carries a low risk of complications. The collected bacteria were frozen at -70°C and analyzed en bloc.
2.2. Extraction of Genomic Bacterial DNA
DNA was extracted from the samples using a UCP kit (a QIAamp UCP Pathogen Mini Kit).
Next, 50 ng of genomic DNA was used in a PCR. The first PCR primer was as follows: 5′ -ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNCCTACGGGNGGCWGCAG-3′ (forward) and 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNGACTACHVGGGTATCTAATCC-3′ (reverse). The details of the PCR process were as follows: 94℃ for 3 min, 30 cycles of 94℃ for 30 sec, 55℃ for 30 sec and 72℃ for 30 sec, 72℃ for 5 min, and 4℃ ad infinitum. Then, 3 ul of the 20 ul amplification product and 2% agarose gel were used to check for PCR bands, which were identified at 540bps.
2.3. 16S rRNA Gene Amplification and Sequencing
The composition and diversity of the fecal microbiome were assessed via the high-throughput sequencing of 16S rRNA gene amplicons using the Illumina MiSeq platform (Illumina Inc., San Diego, USA). The samples were subjected to DNA extraction using a UCP kit (The QIAamp UCP Pathogen Mini Kit, QIAGEN Inc., Germany), according to the manufacturer’s protocol. Sequencing libraries were prepared using a two-step polymerase chain reaction (PCR) method that targeted the V3-V4 hypervariable region of the 16S rRNA gene. The first PCR was conducted using the following gene-specific primers: 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNCCTACGGGNGGCWGCAG-3′ (forward) and 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNGACTACHVGGGTATCTAATCC-3′ (reverse). Subsequently, a second PCR was performed using separate indexing primers that fused the Illumina sequencing adaptors and dual barcodes to the sample amplicons. KOD DNA Polymerase (TOYOBO Bio Inc., Japan) was used to perform the PCR amplification. The pooled library was then quantified using a Qubit 2.0 Fluorometer and a dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA) and diluted to a final concentration of 12 pM using 50% PhiX. Sequencing was performed using a MiSeq Reagent Kit v3 (Illumina, Inc.) with a 300 bp paired-end sequencing protocol, according to the manufacturer’s instructions. In total, 1.87 million paired-end reads were obtained. The samples had a mean read pair count of 7,356 and a maximum read pair count of 12,296.
2.4. Amplicon Sequence Variants
Sequence data for the 16S rRNA gene amplicons were analyzed using the QIIME2 platform, version 2019.10. [27]. For all paired reads, the first 20 bases of both sequences were trimmed to remove primer sequences, the bases after position 200 were truncated to remove low-quality sequence data, and potential amplicon sequencing errors were corrected by using DADA2 to produce an amplicon sequence variant (ASV) dataset. The resulting ASV results were aligned using MAFFT and then used to construct a phylogenetic tree via FastTree2.
α- and ꞵ-diversity metrics were estimated from a subsampled ASV dataset, using 1,000 sequences per sample. Each ASV was identified using a Naïve Bayes classifier trained on 16S rRNA gene sequences from the SILVA dataset (Quast et al., 2013). All reads were assigned to a total of 8 ASVs at the phylum level, 48 ASVs at the family level, 108 ASVs at the genus level, and 168 ASVs at the species level.
α-diversity indices, including the observed ASVs, Shannon index, and Faith’s phylogenetic distance (Faith-pd), were calculated. ꞵ-diversity was evaluated using UniFrac principal PCoA plotting. The microbiome data were represented by the relative abundance at each level of taxonomy, and comparisons between groups were performed for all observed bacteria. At the phylum level, the Firmicutes/Bacteroidetes (F/B) ratio was calculated.
2.5. Statistics
GraphPad Prism and JMP 17.0 were used for statistical analysis. For comparisons between three or more groups, an analysis of variance (ANOVA) was used. A P value of less than 0.05 was considered statistically significant.
3. Results
Using the methods described above, we collected a total of 25 samples from five healthy Japanese individuals (Table 1, Figure 1a). All samples collected contained bacterial DNA and could be amplified via a 16s PCR. However, the gastric and duodenal samples from all patients were relatively difficult to amplify, requiring at least two brushings, whereas the oral and fecal samples contained sufficient bacterial DNA and showed clear bands (Table 1, Figure 1b,c).
We assessed the validity of the bacterial flora analysis conducted using our method and confirmed the amount of oral mucus obtained via swabbing the patient’s mouth and tongue. The number of bacteria in the esophageal, gastric, and duodenal mucus samples obtained via endoscopic brush scraping was sufficient for evaluation (Figure 2). The relationship between the site of specimen acquisition and the number of reads had a higher value in the oral and fecal samples (Figure 2). However, there was a large variation in the number of reads in samples obtained from the stomach, where bacteria are inhibited by high levels of acidity in these samples, some specimens showed a reduction of up to one-tenth.
We then performed a principal component analysis (PCoA) (Figure 3). In this plot, each point represents a sample, and the data are projected onto a two-dimensional space by Principal Component 1 (PC1) and Principal Component 2 (PC2). Clusters in different colors represent different groups, making it possible to visualize similarities between the samples and the separation between groups.
In the unweighted PCoA (Figure 3a), the stomach samples were diverse but formed clusters. In the weighted PCoA, the duodenum samples were varied, but the oral cavity, esophagus, and stomach samples formed almost identical clusters (Figure 3b).
The combined results from the five subjects show that the richness (the number of characteristics observed) of the oral flora was concentrated in a narrow range; meanwhile, the results for the gastric mucus varied significantly among samples (Figure 4a). Statistically, there were no significant differences among the five sites due to the small number of patients. Interestingly, when we plotted Faith-pd, we found that the duodenum was phylogenetically diverse compared to the other sites (Figure 4b).
A heat map was created to depict the abundance of each bacterial species at the class level (Figure 5). The darker the red, the more abundant the species, and the darker the blue, the less abundant the species. The rows show the samples sorted according to collection sites, while the columns contain the assigned bacterial species. When a tree is drawn on the top of the heat map, regardless of the collection site, it can be seen that species with biological similarity generally cluster according to site. It was also visually confirmed that the distribution of bacterial flora in the fecal samples (the bottom five rows) was different from the distribution of the intestinal flora in the upper gastrointestinal tract, which extends from the oral cavity to the duodenum.
We also found that the distribution of microorganisms differed extensively by site. Fusobacteria and Bacilli were abundant in the upper GI tract, while Clostridia and Bacteroidia were abundant in the fecal samples. Negativicutes and Actinobacteria were present throughout the digestive tract (Figure 6).
4. Discussion
Although a significant volume of NGS data on the GI tract has been reported, the bacterial flora of the entire digestive tract have remained largely unknown [9,28]. In this study, we collected specimens from sites ranging from the oral cavity to the large intestine in order to observe the flora of the whole GI in a continuous manner. According to previous reports, we think that fecal samples representing the colon may differ from small intestine [26,29]. In this study, the analysis of the bacterial flora in the small intestines was, indeed, not fully differentiated because obtaining samples from the small intestine requires the use of highly invasive techniques, such as double-balloon endoscopy, which necessitate careful consideration. However, the bacterial flora of the duodenum may show similarities to the flora in intestinal samples. In this study, we identified several bacterial flora that do not vary throughout the intestinal tract. Nevertheless, it would be ideal if a data set representing the small intestinal flora could be collected easily. Future research should focus on determining whether the bacterial flora of the small intestine can be estimated accurately using less invasive methods, which would reduce the need for invasive sampling techniques. We developed a method using a sterilized cytology brush with a sheath that can avoid contamination during EGD (Figure 1a). Bacterial DNA can be easily obtained from the oral cavity and feces, but it is difficult to obtain bacterial DNA from the esophagus, duodenum, and stomach. Even if the stomach contains a significant amount of gastric juice, the acidic environment makes it difficult to collect DNA, so care must be taken to increase the number of samples at the time of collection. In fact, brush scraping had to be performed 2-3 times for two-fifths of the patients. In addition, we used UCP kits for DNA extraction and the KOD enzyme for PCR amplification in order to obtain the necessary amount of high-quality DNA. However, considering the burden this procedure places on the patient, it is necessary to find a method to obtain a complete sample in one step. In the future, we plan to devise ways of making specimen collection easier.
The number of reads obtained from the NGS was approximately 1000, which is considered good. In fact, the bacterial flora at the class level and the bacterial flora between the upper and lower GI tracts could be observed clearly. The upper GI tract was dominated by Fusobacteria and Bacillus, while the lower GI tract was dominated by Actinobacteria and Coriobacteriia. Although there were similarities between the oral and colonic microbiomes, significant differences existed between them. This suggests that the microbial communities of the esophagus and stomach cannot be accurately predicted from fecal samples alone. Therefore, it is crucial to collect samples from different sites, including the upper GI tract, to comprehensively evaluate the GI microbiota. Future research should focus on assessing all gastrointestinal flora, encompassing various diseases and including the upper GI tract, in order to gain a complete understanding of microbial diversity and its implications for health [30,31,32]. It is important to study normal biomes in healthy individuals to provide a reference value for studies of diseased states.
In this study, we discovered that nearly 20% of the bacteria present in the duodenum remain unidentified at the class level. This percentage is significantly higher compared to other regions, with much fewer unidentified bacteria found in the stomach and none observed in the oral cavity or feces. This finding challenges the prevailing understanding that most bacterial populations in the human body have already been identified and classified [33]. Our findings suggest that the duodenum may host a diverse and largely unexplored bacterial ecosystem.
Given that other gastrointestinal regions, such as the stomach, oral cavity, and feces, have well-characterized microbiomes [2,4,12,17,32,34], the high percentage of unidentified bacteria in the duodenum indicates the presence of potentially novel bacterial species. Identifying these unknown bacterial populations is crucial for several reasons. First, the duodenum plays a pivotal role in digestion and nutrient absorption, and its microbial inhabitants likely contribute to these processes. Second, recent research has increasingly recognized the critical role that bacteria play in human health, including their involvement in metabolic functions, immune system modulation, and protection against pathogens. Therefore, understanding the composition of the duodenal microbiome could provide new insights into the complex symbiotic relationships between humans and their microbiota.
Future research should focus on uncovering the identities of these mysterious bacterial populations using advanced genomic and metagenomic techniques, understanding their functions, and determining how they contribute to the overall health of the human host. Such knowledge could lead to advances in science, including the development of new treatments and therapies that leverage the beneficial properties of these bacteria. Overall, this discovery opens up new avenues of research in microbiology and emphasizes the need for a more comprehensive exploration of the human microbiome. By deepening our understanding of the complex interactions between humans and their bacterial counterparts, we can better appreciate the intricate balance that sustains health and well-being.
Finally, we found that it is possible to observe the bacterial communities present in the GI tract in a continuous manner, though it is difficult to statistically characterize them using a small number of samples. However, we proved that this task is technically possible. In the future, we plan to include more patients in our cohort in order to decipher the impact of the microbiome on disease or vice versa.
5. Conclusions
Using sequential sampling and metagenomic analysis, we unlocked the secrets of the entire GI tract. Using advanced UCP technology alongside NGS, we successfully detected abundant bacterial DNA throughout the GI tract. Therefore, the bacterial flora of the entire digestive tract was elucidated, although the sample size used was modest. This study demonstrates the possibility of performing a comprehensive gastrointestinal analysis with limited samples, marking a significant advancement in microbiome research.
Author Contributions
Conceptualization: KI and TKanno; sample collection: SS, YH, BL, MB, RA, NS MO, SK, RO, MS, and KK; data curation: KI; funding acquisition: KI and TKanno; investigation: KI, TT, RS, and TKanno; supervision: ST, MS, KK, JM, KO, RS, TI, TKoike, AM and RS; visualization: TT, YA, and KI; writing original draft: KI, TKanno, and TT; writing review and editing: KI, TKanno, TT, MS, KK, TI, and RS. KI and TKanno contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research (23K05737) from the Japan Society for the Promotion of Science. This work was also supported by the Suzuken Memorial Foundation, Nagoya, Japan; Research grant from Gonryo foundation for the promotion of Medical Science; and the Foundation of Integrative and Kampo Medicine, Tohoku University Graduate School of Medicine, which is a joint research course held with TSUMURA & Co., Tokyo, Japan.
Acknowledgments
We are grateful to Y. Ishii, a Clinical Research Coordinator, for her great help, and. to Yoki M. Ishizawa for reading the paper during the manuscript preparation stage. Finally, I would like to thank the Tohoku Medical Megabank Organization for providing the research environment.
Disclosure/Conflicts of Interest: The authors declare no conflicts of interest.
References
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An Integrated Catalog of Reference Genes in the Human Gut Microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A New Genomic Blueprint of the Human Gut Microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-Analysis of Gut Microbiome Studies Identifies Disease-Specific and Shared Responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.F.; Abubucker, S.; Badger, J.H.; et al. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The Human Microbiome: At the Interface of Health and Disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Guarner, F.; Malagelada, J.-R. Gut Flora in Health and Disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Mindrescu, N.M.; Guja, C.; Jinga, V.; Ispas, S.; Curici, A.; Twakor, A.N.; Stoian, A.M.P. Interactions between Gut Microbiota and Oral Antihyperglycemic Drugs: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 3540. [Google Scholar] [CrossRef]
- Saito, S.; Aoki, Y.; Tamahara, T.; Goto, M.; Matsui, H.; Kawashima, J.; Danjoh, I.; Hozawa, A.; Kuriyama, S.; Suzuki, Y.; et al. Oral Microbiome Analysis in Prospective Genome Cohort Studies of the Tohoku Medical Megabank Project. Front. Cell. Infect. Microbiol. 2021, 10, 604596. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated With Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular Analysis of the Bacterial Microbiota in the Human Stomach. Proc. Natl. Acad. Sci. 2006, 103, 732–737. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host Remodeling of the Gut Microbiome and Metabolic Changes during Pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the Effects of the Environment and Host Genotype on the Gut Microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Group, N.H.W.; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Kawana, T.; Imoto, H.; Tanaka, N.; Tsuchiya, T.; Yamamura, A.; Saijo, F.; Maekawa, M.; Tamahara, T.; Shimizu, R.; Nakagawa, K.; et al. The Significance of Bile in the Biliopancreatic Limb on Metabolic Improvement After Duodenal-Jejunal Bypass. Obes. Surg. 2024, 34, 1665–1673. [Google Scholar] [CrossRef]
- Ikeda, H.; Ihara, E.; Takeya, K.; Mukai, K.; Onimaru, M.; Ouchida, K.; Hata, Y.; Bai, X.; Tanaka, Y.; Sasaki, T.; et al. The Interplay between Alterations in Esophageal Microbiota Associated with Th17 Immune Response and Impaired LC20 Phosphorylation in Achalasia. J. Gastroenterol. 2024, 59, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Mendoza-Ladd, A.; Zeng, M.; Diaz-Arévalo, D.; Morales, E.; Fagan, B.M.; Ordoñez, J.; Velez, P.; Antony, N.; McCallum, R.W. The Human Jejunum Has an Endogenous Microbiota That Differs from Those in the Oral Cavity and Colon. BMC Microbiol. 2017, 17, 160. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Paslier, D.L.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Rajilić-Stojanović, M.; Vos, W.M. de High-Throughput Diversity and Functionality Analysis of the Gastrointestinal Tract Microbiota. Gut 2008, 57, 1605. [Google Scholar] [CrossRef] [PubMed]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E.; Nalin, R.; et al. Towards the Human Intestinal Microbiota Phylogenetic Core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Verdu, E.F.; Collins, S.M. Microbial-Gut Interactions in Health and Disease. Irritable Bowel Syndrome. Best Pr. Res. Clin. Gastroenterol. 2004, 18, 315–321. [Google Scholar] [CrossRef]
- Rieder, R.; Wisniewski, P.J.; Alderman, B.L.; Campbell, S.C. Microbes and Mental Health: A Review. Brain, Behav., Immun. 2017, 66, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial Ecology: Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- NIH Human Microbiome Project Defines Normal Bacterial Makeup of the Body | National Institutes of Health (NIH) Available online:. Available online: https://www.nih.gov/news-events/news-releases/nih-human-microbiome-project-defines-normal-bacterial-makeup-body (accessed on 20 June 2024).
- Yamaki, K.; Tamahara, T.; Washio, J.; Sato, T.; Shimizu, R.; Yamada, S. Intracanal Microbiome Profiles of Two Apical Periodontitis Cases in One Patient: A Comparison with Saliva and Plaque Profiles. Clin. Exp. Dent. Res. 2024, 10, e862. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
a. Sampling brush and bacterial flora collection. The top row of the figure shows the sampling brush used to collect bacterial samples, manufactured by Cook Medical. This brush was used to ensure consistent and efficient sample collection across multiple sites. The bottom row shows photographs of each organ at the time of sampling. These images include the oral cavity, esophagus, stomach, duodenum, capturing the exact conditions under which the samples were collected. Figure 1b. Summary of DNA concentration at each site. Genomic DNA was present in all samples. The oral, esophagus, and fecal samples had sufficient DNA concentrations, whereas the stomach and duodenum samples had relatively low concentrations, making collection challenging. Figure 1c. Testing bacterial DNA via 16S PCR. Bacterial DNA was successfully amplified from all collected samples using 16s PCR, with PCR products identifiable at 550bp. Although amplification was difficult for gastric and duodenal samples (indicated by red and black arrows), oral and fecal samples showed clear bands due to sufficient bacterial DNA.
Figure 1.
a. Sampling brush and bacterial flora collection. The top row of the figure shows the sampling brush used to collect bacterial samples, manufactured by Cook Medical. This brush was used to ensure consistent and efficient sample collection across multiple sites. The bottom row shows photographs of each organ at the time of sampling. These images include the oral cavity, esophagus, stomach, duodenum, capturing the exact conditions under which the samples were collected. Figure 1b. Summary of DNA concentration at each site. Genomic DNA was present in all samples. The oral, esophagus, and fecal samples had sufficient DNA concentrations, whereas the stomach and duodenum samples had relatively low concentrations, making collection challenging. Figure 1c. Testing bacterial DNA via 16S PCR. Bacterial DNA was successfully amplified from all collected samples using 16s PCR, with PCR products identifiable at 550bp. Although amplification was difficult for gastric and duodenal samples (indicated by red and black arrows), oral and fecal samples showed clear bands due to sufficient bacterial DNA.
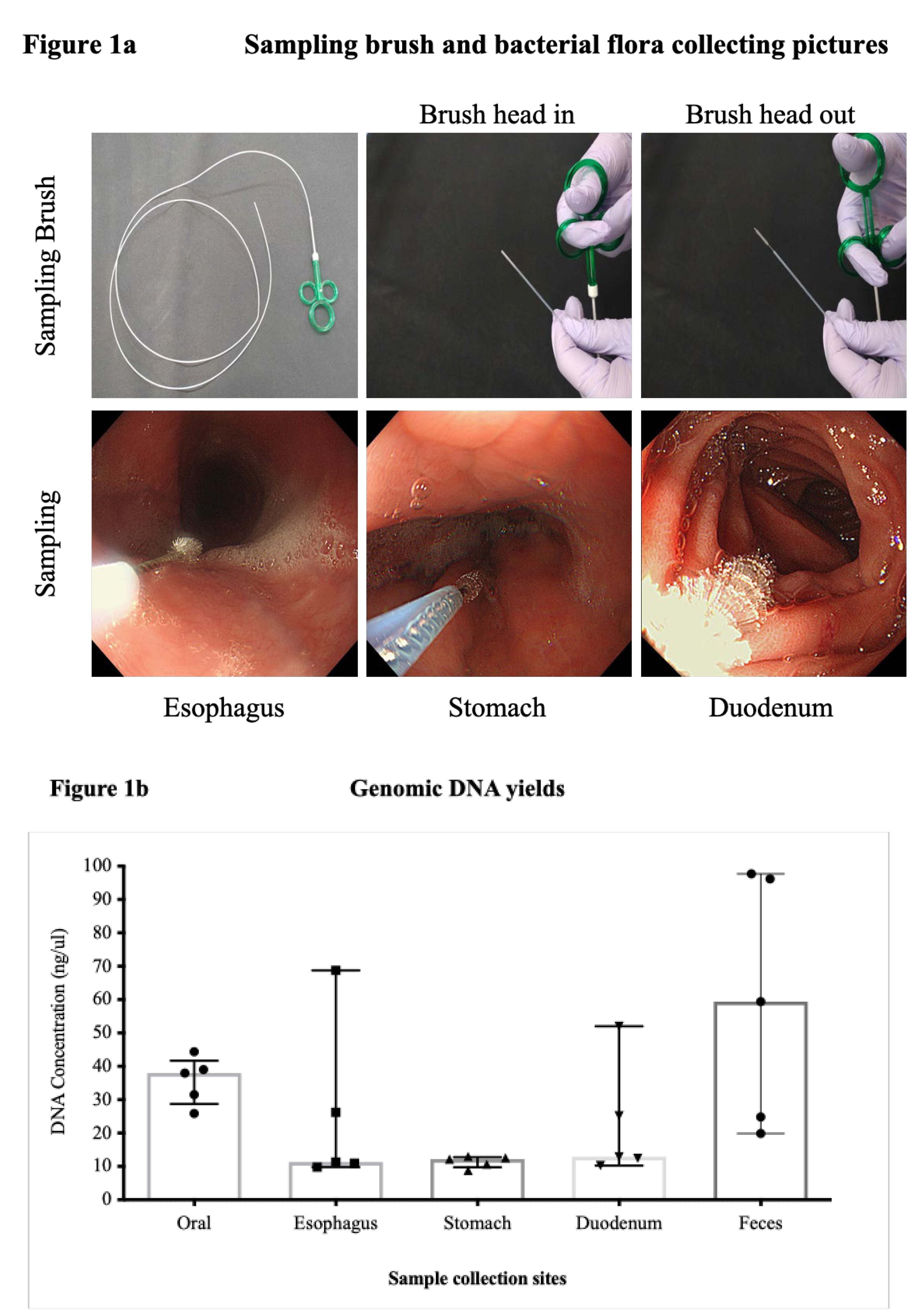
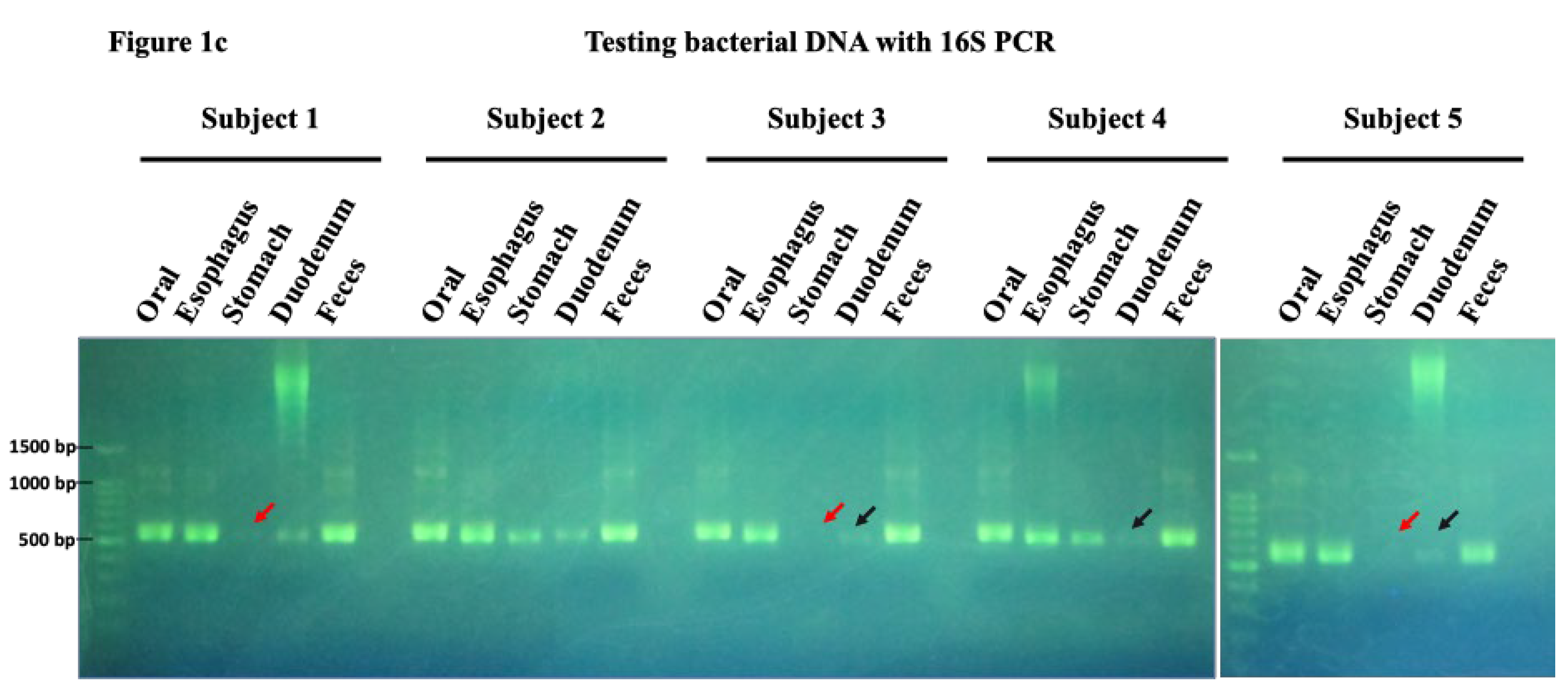
Figure 2.
Number of reads are sufficient for NGS analysis. The graph shows that the number of reads obtained was sufficient for NGS analysis. On the y-axis, the graph represents the number of reads, which indicates the amount of bacterial DNA sequences obtained from each sample. Samples from the esophagus, stomach and duodenum provided enough DNA to effectively assess their bacterial flora. However, in the stomach and duodenum, where conditions are characterized by high acidity or alkalinity, significant variations in read counts were observed. Some samples from these regions yielded only about a tenth of the expected reads, highlighting the challenging conditions for bacterial survival and DNA extraction in these areas.
Figure 2.
Number of reads are sufficient for NGS analysis. The graph shows that the number of reads obtained was sufficient for NGS analysis. On the y-axis, the graph represents the number of reads, which indicates the amount of bacterial DNA sequences obtained from each sample. Samples from the esophagus, stomach and duodenum provided enough DNA to effectively assess their bacterial flora. However, in the stomach and duodenum, where conditions are characterized by high acidity or alkalinity, significant variations in read counts were observed. Some samples from these regions yielded only about a tenth of the expected reads, highlighting the challenging conditions for bacterial survival and DNA extraction in these areas.
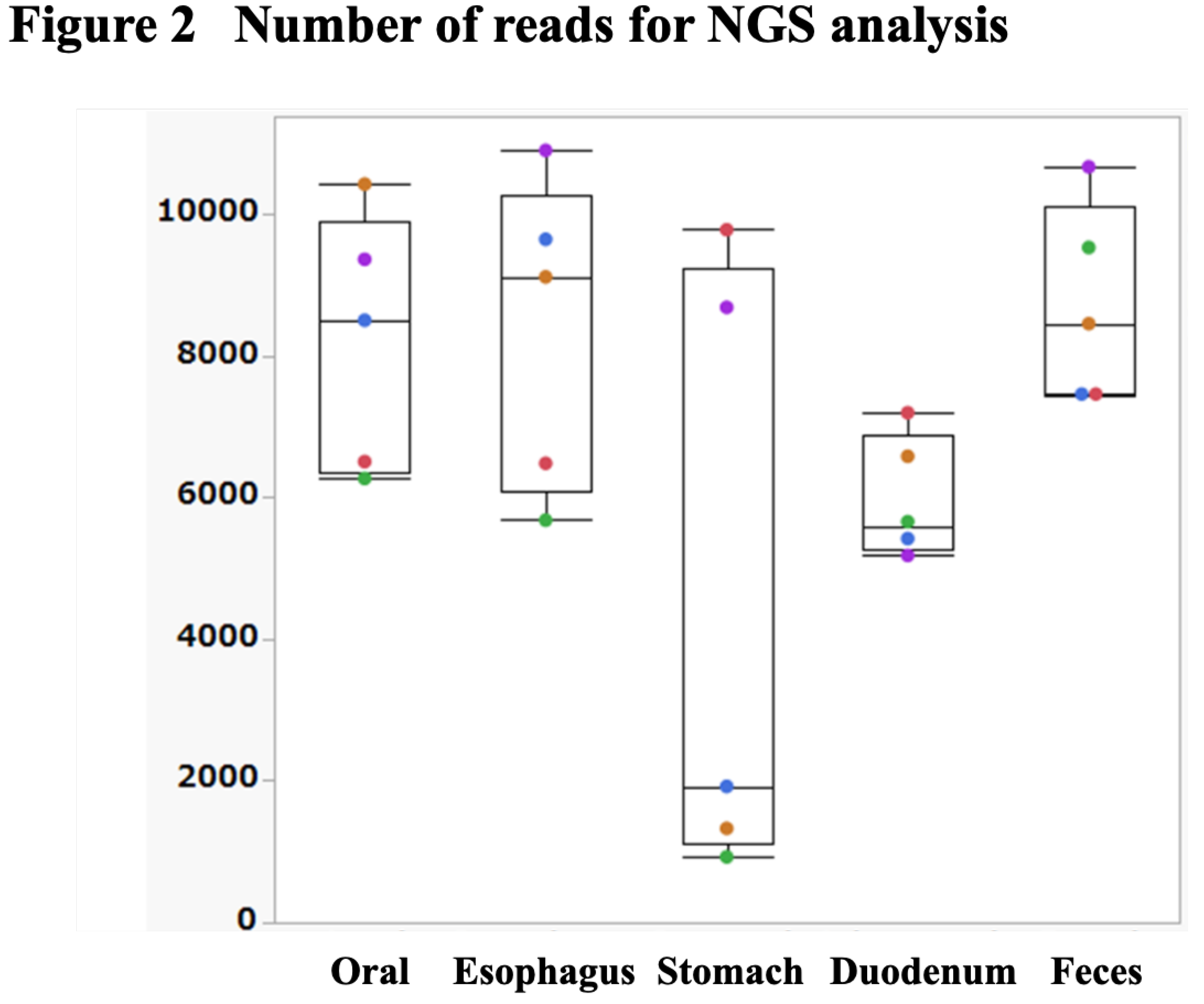
Figure 3.
PCoA analysis. A principal component analysis (PCoA) plot is shown. In this plot, each point represents a sample, and the data are projected onto a two-dimensional space by Principal Component 1 (PC1) and Principal Component 2 (PC2). Clusters in different colors represent different groups, making it possible to visualize similarities between samples and the separation between groups. In the unweighted PCoA (3a), the stomach samples are diverse but form clusters in the other groups. In the weighted PCoA (3b), the duodenal samples are varied, but the oral cavity, esophagus, and stomach samples form almost identical clusters.
Figure 3.
PCoA analysis. A principal component analysis (PCoA) plot is shown. In this plot, each point represents a sample, and the data are projected onto a two-dimensional space by Principal Component 1 (PC1) and Principal Component 2 (PC2). Clusters in different colors represent different groups, making it possible to visualize similarities between samples and the separation between groups. In the unweighted PCoA (3a), the stomach samples are diverse but form clusters in the other groups. In the weighted PCoA (3b), the duodenal samples are varied, but the oral cavity, esophagus, and stomach samples form almost identical clusters.
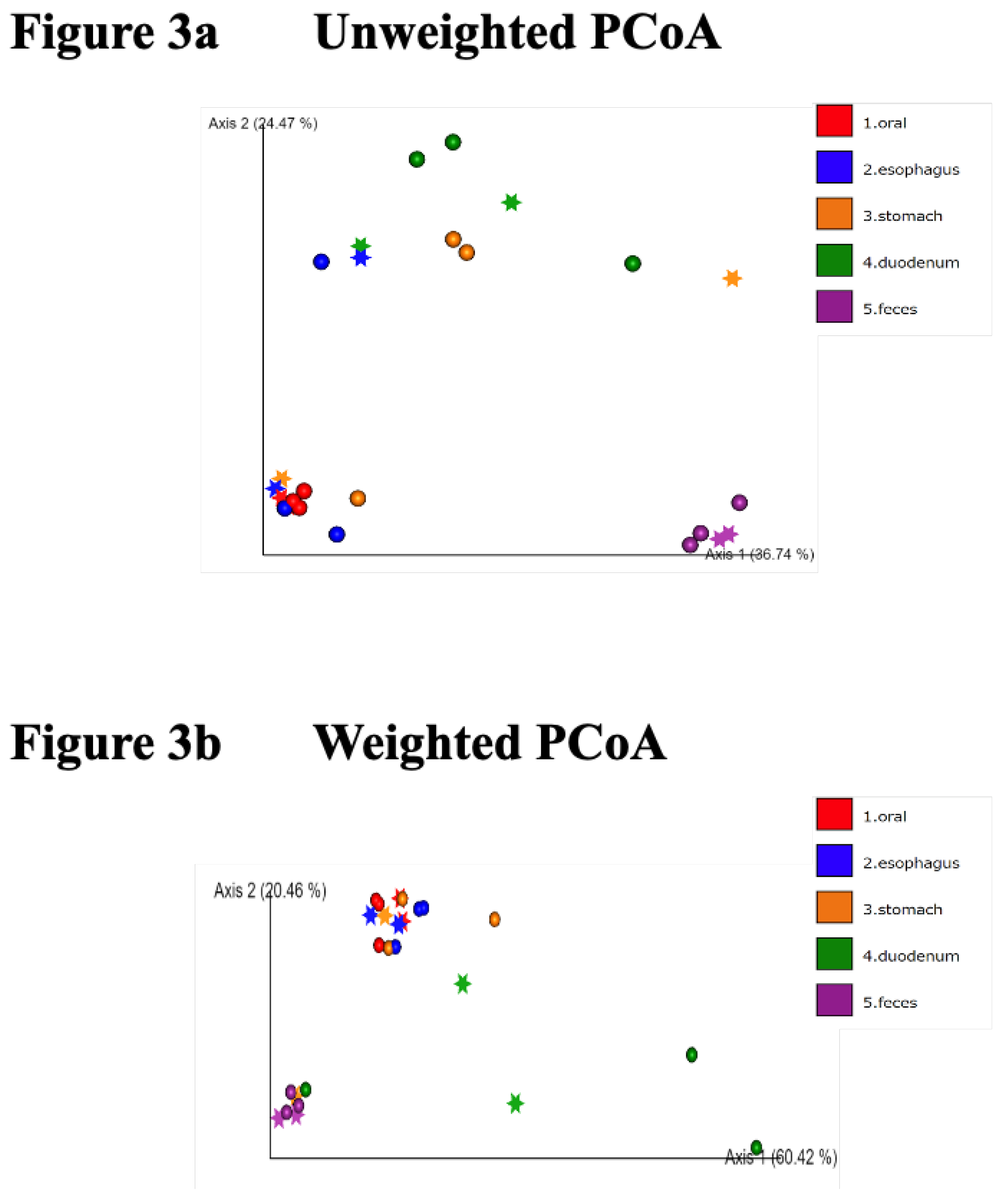
Figure 4.
Comparative analysis of microbial richness and diversity across GI sites.The combined results of the five subjects show that the richness (the number of characteristics observed) of the oral flora is concentrated in a narrow range; meanwhile, the results for gastric mucus vary significantly between samples. Statistically, there are no significant differences among the five groups due to the small number of patients (Figure 4a). The duodenum was found to be phylogenetically diverse (Figure 4b, p=0.049 Willcoxon with Bonfferroni correction).
Figure 4.
Comparative analysis of microbial richness and diversity across GI sites.The combined results of the five subjects show that the richness (the number of characteristics observed) of the oral flora is concentrated in a narrow range; meanwhile, the results for gastric mucus vary significantly between samples. Statistically, there are no significant differences among the five groups due to the small number of patients (Figure 4a). The duodenum was found to be phylogenetically diverse (Figure 4b, p=0.049 Willcoxon with Bonfferroni correction).
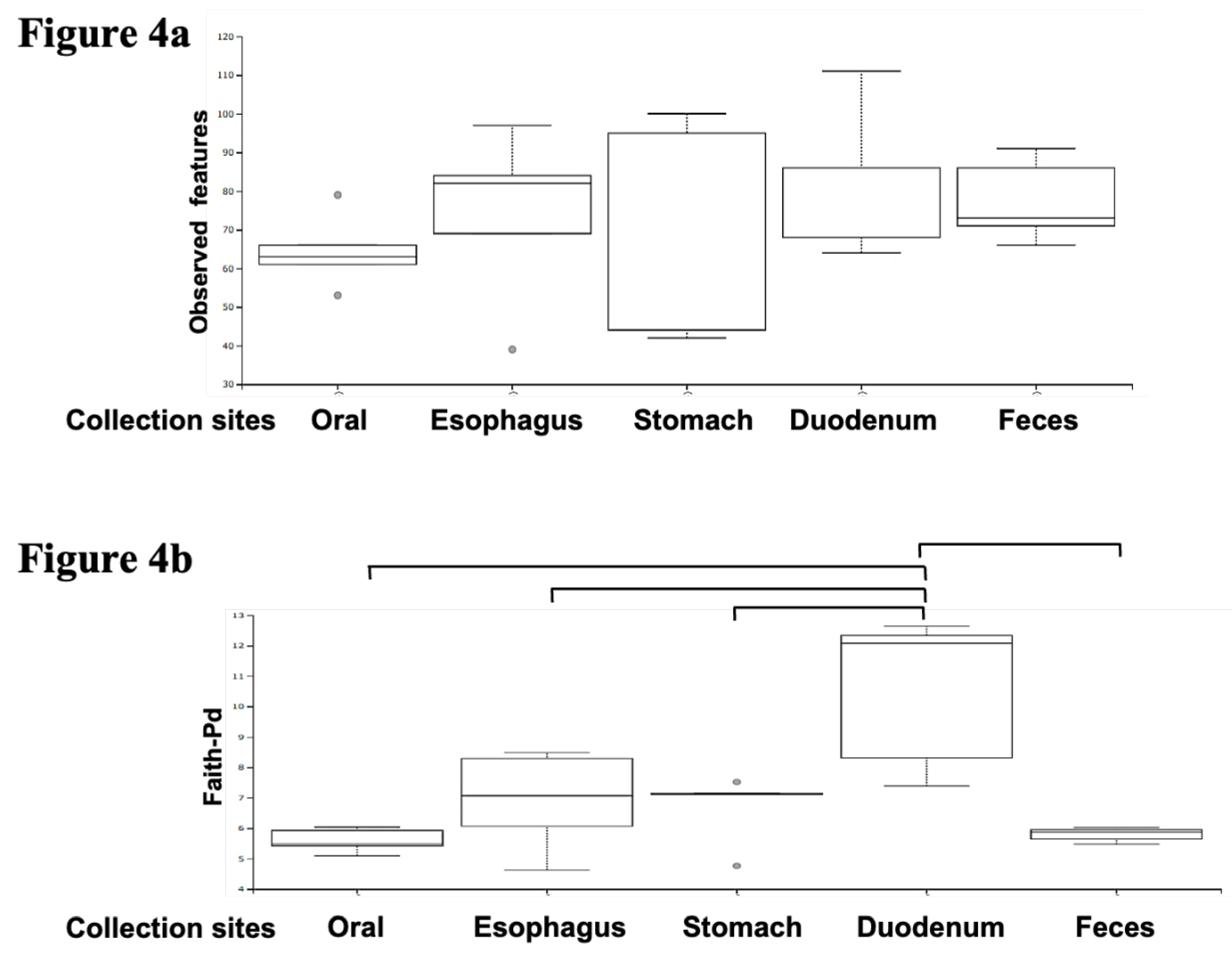
Figure 5.
Visualizing bacterial abundance and distribution across gastrointestinal sites. The darker the red, the more abundant the species, and the darker the blue, the less abundant the species. The rows show samples sorted according to collection sites, while the columns contain assigned bacterial species. When a tree is drawn on the top of the heat map, regardless of the collection site, it can be seen that species with biological similarity are generally clustered according to site. It can also be visually confirmed that the distribution of the bacterial flora in the fecal samples (the bottom five rows) differs from the distribution of the intestinal flora in the upper gastrointestinal tract, which extends from the oral cavity to the duodenum.
Figure 5.
Visualizing bacterial abundance and distribution across gastrointestinal sites. The darker the red, the more abundant the species, and the darker the blue, the less abundant the species. The rows show samples sorted according to collection sites, while the columns contain assigned bacterial species. When a tree is drawn on the top of the heat map, regardless of the collection site, it can be seen that species with biological similarity are generally clustered according to site. It can also be visually confirmed that the distribution of the bacterial flora in the fecal samples (the bottom five rows) differs from the distribution of the intestinal flora in the upper gastrointestinal tract, which extends from the oral cavity to the duodenum.
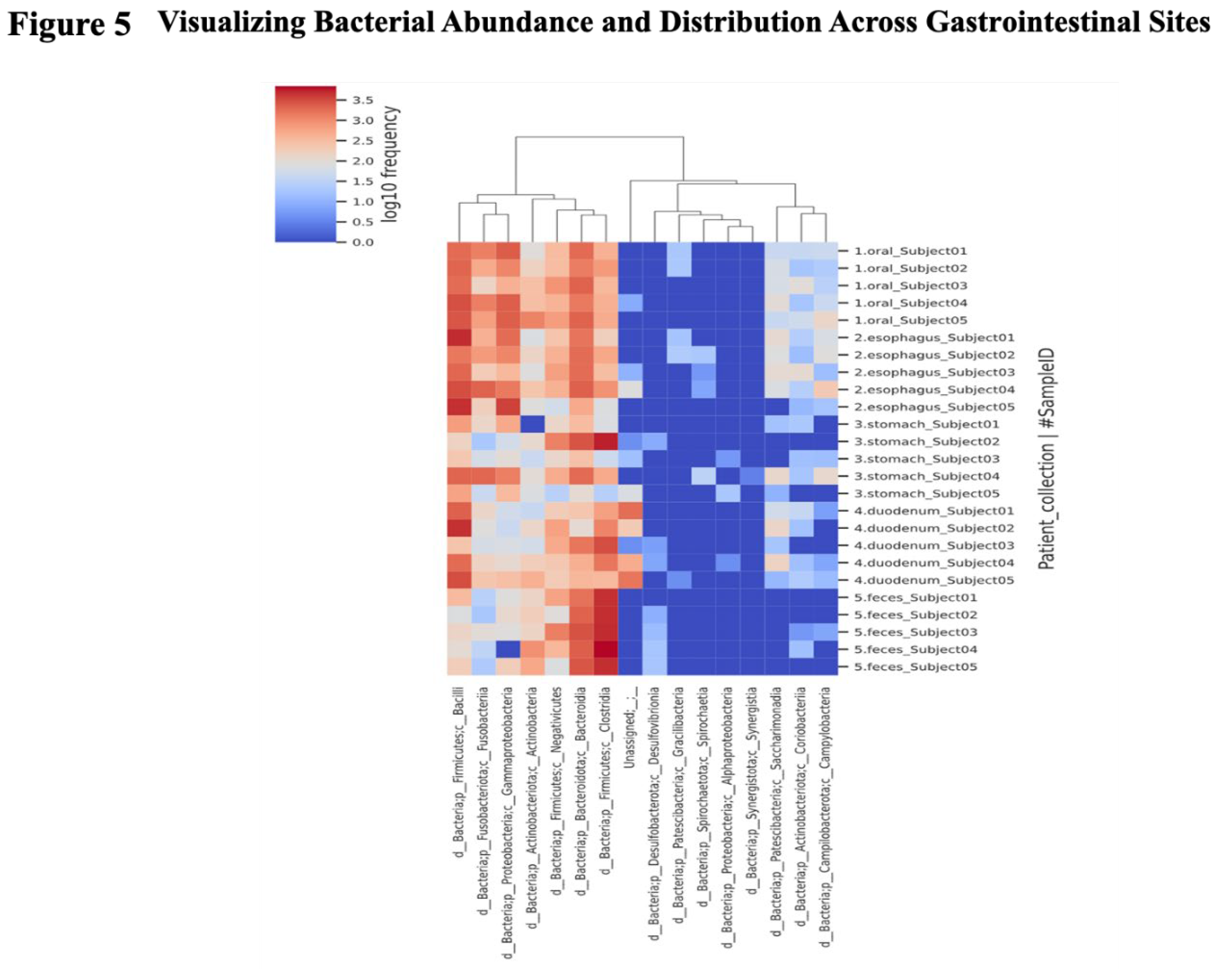
Figure 6.
Class-level microbiome. The charts provided include a stacked bar chart and an area chart. Stacked Bar Chart: The left graph illustrates the relative abundance of different bacterial taxa in different gastrointestinal sites, such as the oral cavity, esophagus, stomach, duodenum, and feces. Each color corresponds to a distinct bacterial group, with the height of each colored section representing the proportion of that group in the sample from each site. Area Chart: The right graph similarly shows the relative abundance of bacterial taxa, but uses continuous areas to show how the proportions of different bacterial groups change across sites. The distribution of microorganisms varies throughout the GI tract: Fusobacteria and Bacilli are dominant in the upper GI tract, while Clostridia and Bacteroidia are dominant in the feces. Negativicutes and Actinobacteria are present throughout the digestive tract.
Figure 6.
Class-level microbiome. The charts provided include a stacked bar chart and an area chart. Stacked Bar Chart: The left graph illustrates the relative abundance of different bacterial taxa in different gastrointestinal sites, such as the oral cavity, esophagus, stomach, duodenum, and feces. Each color corresponds to a distinct bacterial group, with the height of each colored section representing the proportion of that group in the sample from each site. Area Chart: The right graph similarly shows the relative abundance of bacterial taxa, but uses continuous areas to show how the proportions of different bacterial groups change across sites. The distribution of microorganisms varies throughout the GI tract: Fusobacteria and Bacilli are dominant in the upper GI tract, while Clostridia and Bacteroidia are dominant in the feces. Negativicutes and Actinobacteria are present throughout the digestive tract.
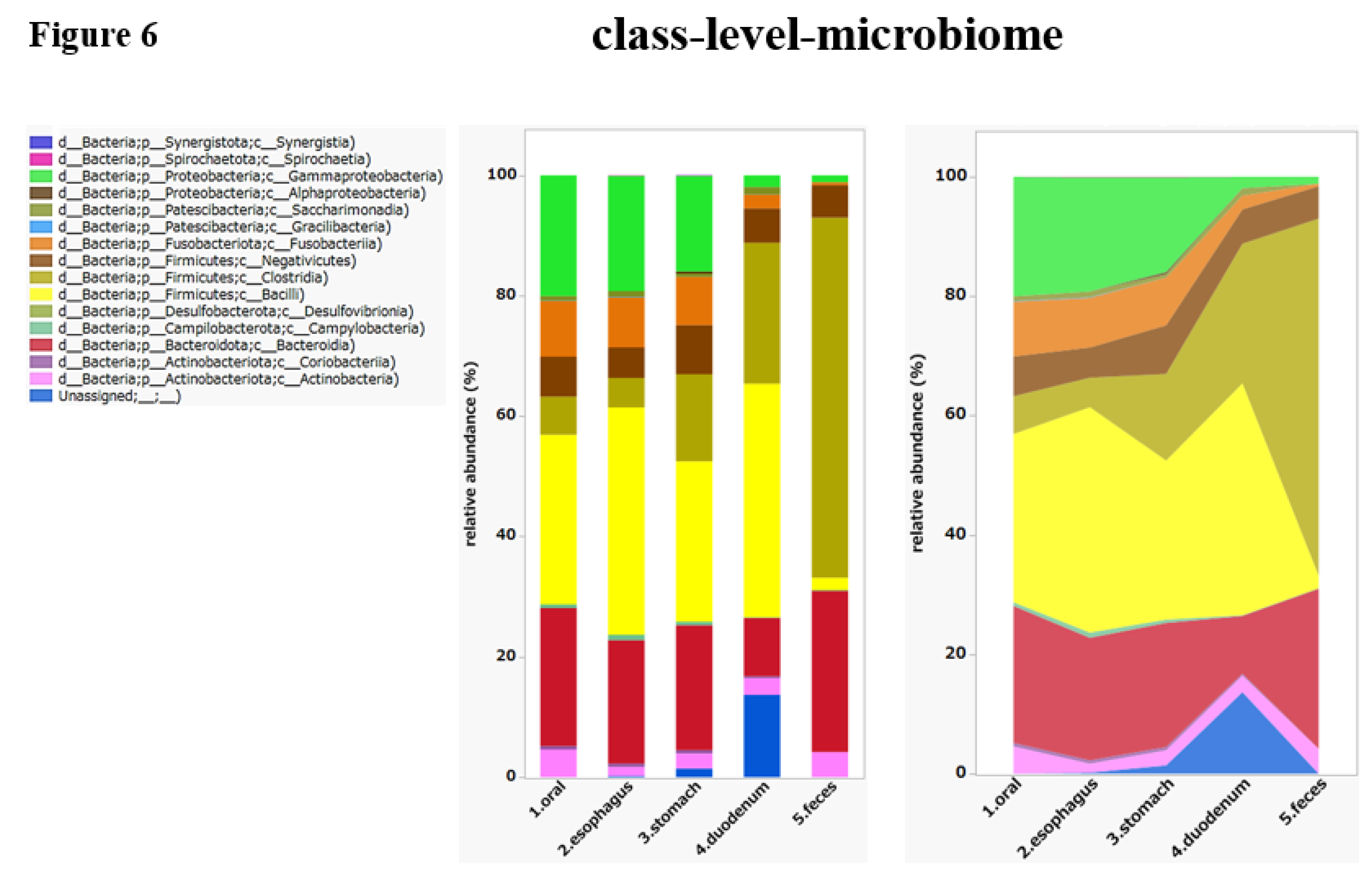

Table 1.
A total of 25 samples were collected from five healthy Japanese individuals. All collected samples contained bacterial DNA with 260/280 ratio as shown in Table 1. However, the DNA concentration of gastric and duodenal samples was relatively low, esophageal and duodenal samples varied with wide range.
Table 1.
A total of 25 samples were collected from five healthy Japanese individuals. All collected samples contained bacterial DNA with 260/280 ratio as shown in Table 1. However, the DNA concentration of gastric and duodenal samples was relatively low, esophageal and duodenal samples varied with wide range.
| Sample ID | Age | Gender | Disease stats | Collection site | DNA concentration (ng/ul) | DNA 260/280 ratio |
|---|---|---|---|---|---|---|
| Subject 1 | 42 | Male | health | oral | 31.5 | 2.45 |
| esophagus | 26.2 | 2.49 | ||||
| stomach | 10.7 | 2.7 | ||||
| duodenum | 52 | 1.99 | ||||
| feces | 24.8 | 2.3 | ||||
| Subject 2 | 52 | Male | health | oral | 38 | 2.14 |
| esophagus | 11 | 2.57 | ||||
| stomach | 12.6 | 2.3 | ||||
| duodenum | 12.4 | 2.29 | ||||
| feces | 97.7 | 2.01 | ||||
| Subject 3 | 54 | Male | health | oral | 44.4 | 2.11 |
| esophagus | 11.3 | 2.54 | ||||
| stomach | 12.2 | 2.28 | ||||
| duodenum | 10.3 | 2.6 | ||||
| feces | 96.2 | 2.1 | ||||
| Subject 4 | 50 | Male | health | oral | 25.9 | 2.23 |
| esophagus | 68.8 | 2.1 | ||||
| stomach | 13 | 2.67 | ||||
| duodenum | 12.9 | 2.58 | ||||
| feces | 59.4 | 2.01 | ||||
| Subject 5 | 41 | Female | health | oral | 39 | 2.12 |
| esophagus | 9.8 | 2.43 | ||||
| stomach | 8.8 | 2.29 | ||||
| duodenum | 25.2 | 2.05 | ||||
| feces | 19.9 | 1.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



