
Submitted:
24 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is an age-associated neurodegenerative disorder. A wealth of evidence indicates that the amyloid β (Aβ) aggregates result from dyshomeostasis between Aβ production and clearance, which plays a pivotal role in the pathogenesis of AD. Consequently, therapies targeting Aβ reduction represent a promising strategy of AD intervention. Tetramethylpyrazine nitrone (TBN) is a novel tetramethylpyrazine derivative with the potential for the treatment of AD. Previously, we demonstrated that TBN markedly improved cognitive functions and reduced Aβ, APP, BACE 1 and hyperphosphorylated tau levels in 3 × Tg-AD mice. However, the mechanism by which TBN inhibits Aβ deposition still unclear. In this study, we employed the APP/PS1 mice treated with TBN (60 mg/kg, ig, bid) for six months and N2a/APP695swe cells treated with TBN (300 μM) to explore the mechanism of TBN on the Aβ reduction. Our results indicated that TBN significantly alleviated cognitive impairment and reduced Aβ depositions in APP/PS1 mice. Further investigation of the underlying mechanisms revealed that TBN decreased the expression of APP and BACE1, activated the AMPK/mTOR/ULK1 autophagy pathway and inhibited the PI3K/AKT/mTOR/ULK1 autophagy pathway, as well as decreased the phosphorylation levels of JNK and ERK in APP/PS1 mice. Moreover, TBN was found to significantly reduce the mRNA levels of APP and BACE1, as well as those of SP1, CTCF, TGF-β, and NF-κB, transcription factors involved in regulating gene expression. Additionally, TBN was observed to decrease the level of miR-346 and increase the levels of miR-147 and miR-106a in the N2a/APP695swe cells. These findings indicate that TBN may reduce Aβ levels likely via reducing APP expression by regulating APP gene transcriptional factors and miRNAs, reducing BACE1 expression, and promoting autophagy activities.
Keywords:
Amyloid β
; Amyloid precursor protein
; MicroRNA
1. Introduction
Alzheimer’s disease (AD) is an irreversible, progressive brain disorder that affects more than millions of individuals worldwide, slowly destroying memory and thinking skills and eventually the ability to carry out simple tasks. Although the specific pathogenesis of AD is not fully understood, it is characterized by changes in the brain, including amyloid beta (Aβ) plaques, neurofibrillary tangles (NFTs), neuroinflammation and synaptic loss. Among these, Aβ accumulation is thought to play a central role in the AD pathology, and appears to be an upstream trigger for the downstream alterations, including the NFTs, inflammation and synaptic loss [1]. Aβ is generated from amyloid precursor protein (APP) through two subsequent proteolytic cleavages by β-secretase and γ-secretase, which is known as the amyloidogenic pathway [2]. Conversely, the majority of APP undergo non-amyloidogenic pathway cleavage by α-secretase, resulting in the production of sAPPα, which plays numerous neuroprotective roles in the maintenance of normal physiological function in neurons [3]. Normally, Aβ is degraded by proteasomes and Aβ-degrading enzymes (ADE) in order to maintain equilibrium between Aβ generation and clearance. However, in the pathological cases of AD, the level of APP expression is higher in the entorhinal cortex neurons of AD patients than in normal individuals [3]. Furthermore, the substrate affinity of β- and γ-secretase with APP is increased, while the level of Aβ degradation is decreased, which leads to Aβ dyshomeostasis and the development of AD pathology [3]. Consequently, the adequate reduction of Aβ is considered to be the pivotal therapeutic objective of AD.
The strategies targeting the reduction of Aβ mainly include: the inhibition of amyloidogenic pathway, the promotion of non-amyloidogenic pathway, the promotion of Aβ clearance and the reduction of APP production [4]. In recent decades, the majority of pharmaceutical research into anti-amyloid therapy has focused on the development of secretase inhibitors and enhancers, as well as Aβ clearance agents, including BACE1 inhibitor elenbecestat, α-secretase enzyme enhancers APH1105, Aβ aggregation inhibitor PBT1 and Aβ monoclonal antibody lecanemab [5]. Lecanemab is the second FDA-approved immunotherapies for AD, this represents a significant advance in the ongoing effort to develop effective treatments for AD. Apart from Aβ monoclonal antibody, the autophagy is one of the important pathways involved in cellular response for the removal of protein aggregates, including Aβ [6, 7]. A substantial body of evidence indicates that autophagy enhancers protect against cognitive decline and inhibit Aβ aggregates in AD mouse models, such as rapamycin, lithium, carbamazepine and latrepirdine [6]. Nevertheless, the efficacy of autophagy stimulators for the treatment of AD patients requires further investigation through comprehensive clinical trials [6, 8].Therefore, it is important for researchers to explore additional strategies to combat Aβ.
In recent years, some researchers have attempted to identify a solution by inhibiting Aβ production from the perspective of APP gene expression levels [9]. It has been demonstrated that the number of copies of the APP gene, the level of APP mRNA and the level of APP protein in single neuronal cells of sporadic AD are increased [10, 11]. The process of APP gene transcription to APP mRNA was complicated, involving transcriptional and post-transcriptional regulation. Some of the APP translational modulators include 4-(5-methyl-1H-benzimidazol-2-yl) aniline (JTR-009), 2-[(pyridine-2-ylmethyl)-amino]-phenol (2-PMAP), and posiphen, which have been developed for the treatment of AD [12]. Posiphen, an inhibitor of the transcription of APP mRNA by targeting the iron-response element (IRE), has been demonstrated to reduce APP mRNA at the transcriptional level, having completed the phase III clinical trials with mild to moderate AD [13]. Although the clinical data for posiphen have not yet been published, the inhibition of APP mRNA represents a promising avenue for the treatment of AD [4]. Recent advances in molecular biology have revealed that several microRNAs (miRNAs), mall non-coding RNAs that regulate gene expression post-transcriptionally by binding mRNA targets and inhibiting translation or mRNA degradation [14], are identified to regulate the expression of the APP gene, which is thought to be a causal factor in Aβ formation [9]. The miR-106a, miR-135a, miR-153 and miR-147 have been demonstrated to bind directly to the 3´-UTR of APP mRNA, which has been shown to downregulate APP expression at the post-transcriptional level. Conversely, miR-346 has been demonstrated to target the APP mRNA 5´-UTR, promoting APP translation and Aβ production [15]. Nevertheless, growing numbers of miRNAs have been investigated for their potential role as the inhibitors of mRNA expression related to Aβ clearance pathways. While there are some obstacles to overcome for clinical application, the potential of miRNAs as therapeutic agents for AD is an area of active research [16, 17]. In light of these considerations, it seems unlikely that a single pathway will prove effective in inhibiting Aβ generation or deposition. A promising strategy for reducing Aβ plaque generation may therefore be to combine multiple pathways, such as inhibition of amyloidogenic secretases, promotion of Aβ clearance, and inhibition of APP production.
Tetramethylpyrazine nitrone
TBN (2-[[(1,1-dimethylethyl)oxidoimino]-methyl]-3,5,6-trimethylpyrazine) is a novel tetramethylpyrazine derivative armed with a potent free radical scavenging nitrone moiety[18]. The therapeutic effects of TBN have been demonstrated in various animal models, including those of ischemic stroke, Parkinson’s disease, amyotrophic lateral sclerosis and Alzheimer’s disease[18-21]. Phase 2 clinical trials of TBN for treatment of ischemic stroke, diabetic kidney disease and ALS have been completed and have demonstrated promising results (CTR20190094, CTR20202126, CTR20210206, ChiCTR1900022848, ChiCTR2000039689 and ChiCTR2100043019). The results of the previous study in the 3 × Tg-AD mouse model showed that TBN markedly improved cognitive function by reduction in Aβ levels and plaque deposition, tau hyperphosphorylation, and the restoration of synaptic function, which was likely associated with the inhibition of APP, BACE1, and PS1 protein expression [18]. Furthermore, TBN significantly inhibited the amyloidogenic processing pathway by inhibiting APP, BACE1 and PS1 expression and promoted the non-amyloidogenic processing pathway by increasing ADAM10 expression in the N2a/APP695swe cells [18]. Additionally, the proteomic analysis of hippocampal neurons showed that TBN modulated the MAPK and mTOR pathways, both of which are involved in the regulation of autophagy. In order to provide further evidence for the efficacy of TBN for treatment of AD, we conducted further evaluations of the anti-AD effects and the potential mechanisms of action in Aβ reduction of TBN in APP/PS1 mice in vivo and N2a/APP695swe cells in vitro.
2. Results
2.1. TBN Improves Behavioral Performance in APP/PS1 Mice
Cognitive improvement is an intuitive evaluation indicator of AD. In a series of behavioral experiments that can reflect long-term, short-term memory and learning ability, we tested whether TBN is capable of reestablishing cognitive function in APP/PS1 mice (Figure 1). TBN treatment resulted in a notable increase in latency in the SDA test and a significant elevation in the DI in the NOR test when compared with the vehicle-treated APP/PS1 mice (Figure 1 A, B). The results of the MWM test showed that, in comparison to the APP/PS1 group, TBN treated mice exhibited a significantly shorter latency to locate the platform (Figure 1 D), which had been removed, and a higher frequency of crossing the platform (Figure 1 C). These findings are consistent with those previously reported in 3 × Tg-AD mice [18]. Collectively, these results indicate that TBN is capable of restoring normal cognitive function in AD transgenic mouse models.
2.2. TBN Treatment Reduces the Aβ Plaques in APP/PS1 Mice and Attenuates APP and BACE1 Expression in the Hippocampus of APP/PS1 Mice
The deposit of Aβ plaques was identified by immunohistochemical staining. As illustrated in Figure 2 A, the level of Aβ plaques significantly increased in the hippocampus and cortex of APP/PS1 mice, which was greatly reduced after TBN treatment. It is known that Aβ is generated from APP via the amyloidogenic pathway. Here, we measured the protein expression of APP, BACE1 and PS1 in hippocampus. The results showed in figure 2 B indicated that the expression of APP and BACE1 was significantly decreased in TBN-treated APP/PS1 mice compared to saline-treated APP/PS1 mice. These results are consistent with those of a previous study in 3×Tg-AD mice [18]. However, the expression of PS1 was slightly decreased by TBN treatment and showed no significant change, which differs from the result observed in 3×Tg-AD mice [18]. Moreover, the level of α-secretase, the principal secretase in the non-amyloidogenic pathway, was evaluated. The result showed that TBN increased the expression of ADAM10 without a significant difference. These results demonstrate that TBN can reduce the expression of APP and BACE 1 in both the APP/PS 1 mice and the 3×Tg-AD mice.
2.3. TBN Treatment Restores the Levels of Synapse Proteins in the Hippocampus of APP/PS 1 Mice
The levels of PSD 95, synapsin I and synapsin II were measured by Western blotting. As compared with saline-treated group, the expression of synapsin I and synapsin II was significantly increased after TBN treatment (Figure 3), indicating the protective effects of TBN on synaptic function. The results are consistent with those of a previous study in 3×Tg-AD mice [18].
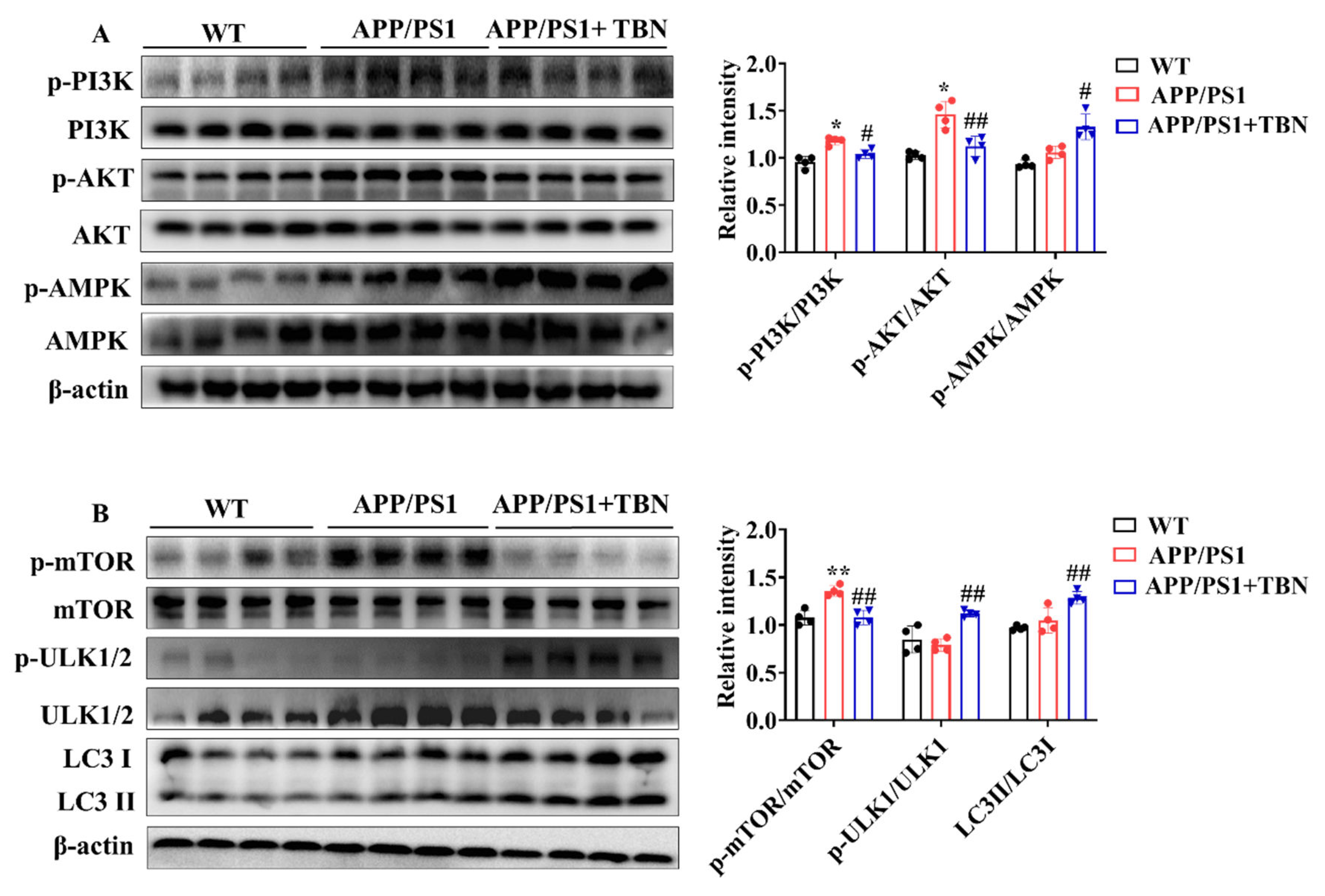
2.4. TBN Enhances Aβ Clearance by Regulating the Expression of Autophagy-Related Proteins in APP/PS1 Mice
The above results indicated that TBN can decrease Aβ production by inhibiting the expression of APP and BACE 1. However, it remains unclear whether TBN has other ways to reduce Aβ aggregation. It was previously established that the metabolism of Aβ is crucially influenced by autopahgy. Consequently, the regulatory effects of TBN on autophagy were evaluated by detecting of the expression of the key regulators involved in the autophagy pathway. AMPK activation has been demonstrated to suppress the activation of mTOR, thereby promoting the process of the autophagy pathway. Furthermore, AMPK activation has also been shown to stimulate ULK-mediated autophagy pathway initiation. In addition to AMPK, the upstream kinase of mTOR, PI3K/AKT kinase suppression can also stimulate the autophagy pathway. The results from Figure 4A showed that TBN treatment significantly increased the ratio of p-AMPK/AMPK and decreased the ratio of p-PI3K/PI3K and p-AKT/AKT compared with saline-treated APP/PS1 mice. Further investigation revealed that TBN treatment resulted in a reduction in the ratio of p-mTOR/mTOR and an increase in the ratio of p-ULK1/ULK1 and LC3II/LC3I. This suggests that the enhancement effect of TBN on Aβ reduction is also mediated by autophagy via activating the AMPK/mTOR/ULK1 pathway and inhibiting PI3K/AKT/mTOR/ULK 1 pathway.
2.5. TBN Reduced the Transcription of the APP gene By Regulating the Transcription Factors and miRNA Levels
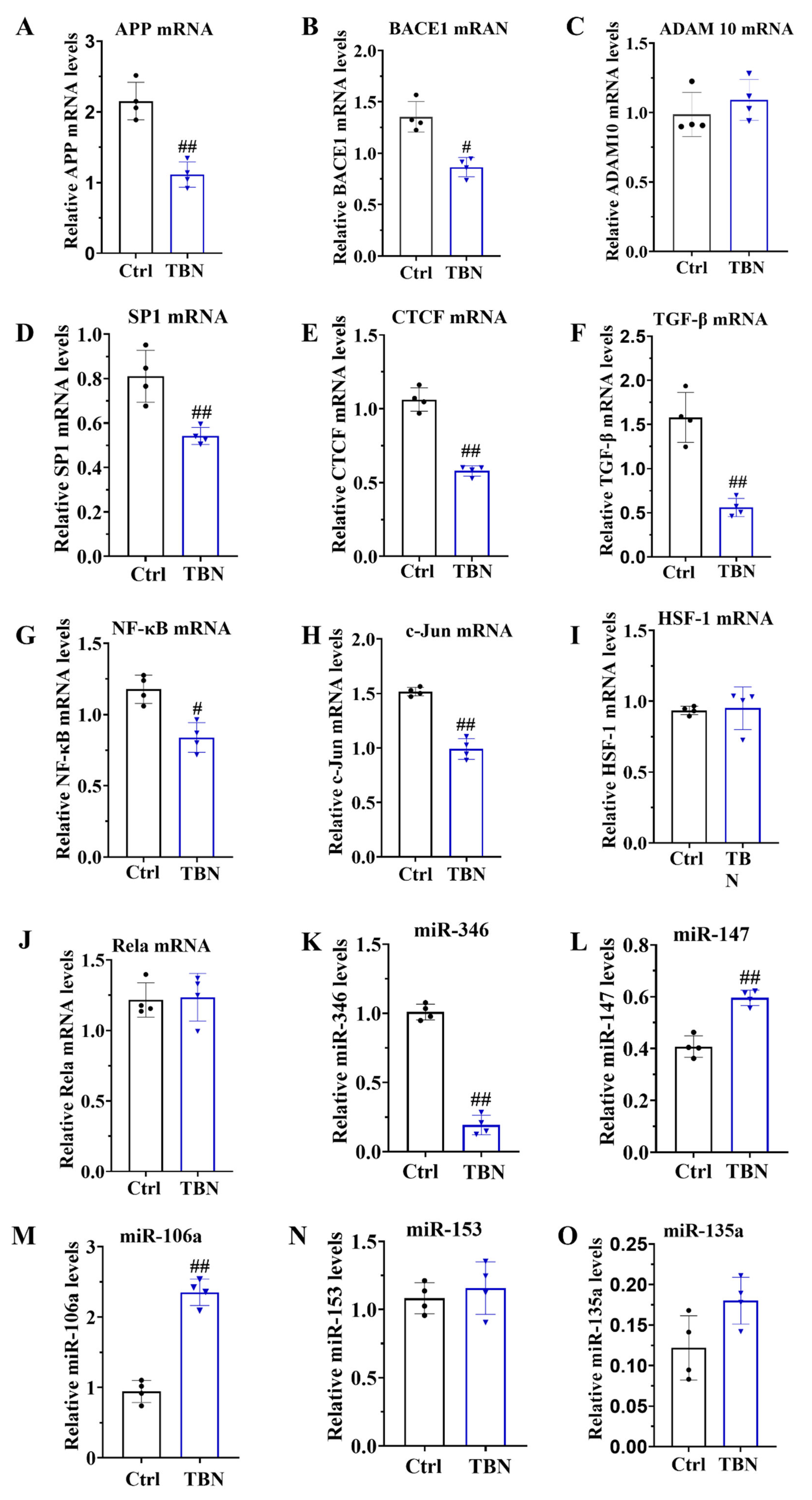
As demonstrated in Figure 1B, TBN can reduce the level of APP expression. However, the mechanism by which TBN affects APP reduction remains unclear. In order to gain further insight, we quantified the levels of APP mRNA, BACE1 mRNA and ADAM 10 mRNA by qRT-PCR in the N2a/APP695swe cells. The results, presented in Figure 5 A-B, indicated that TBN treatment significantly decreased the APP mRNA and BACE 1 mRNA transcription levels when compared with the control group. TBN treatment also exhibited a slight increase in ADAM 10 mRNA levels, although this was not statistically significant when compared with the control group (Figure 5 C).
In order to gain further insight into the underlying mechanisms of TBN on the responsible for the reduction in APP mRNA expression, the transcription factors that promote APP transcription were quantified in the N2a/APP695swe cells using qRT-PCR. These included the following transcription factors: stimulating protein 1 (SP-1), CCCTC-binding factor (CTCF), transforming growth factor-β (TGF-β), heat shock transcription factor 1 (HSF-1), c-Jun and nuclear factor (NF)-κB/Rela. The results, presented in Figure 5 D-J, indicated that the mRNA levels of SP1, CTCF, TGF-β, NF-κB and c-Jun were significantly reduced by TBN treatment, while the levels of HSF-1 and Rela mRNA remained unaltered. These findings indicate that TBN can downregulate the APP mRNA level by inhibiting the expression of transcriptional factors. Furthermore, the levels of miR-346, miR-147, miR-106a, miR-153 and miR-135a were quantified using qRT-PCR in the N2a/APP695swe cells. The results presented in the Figure 5 K-O indicated that TBN treatment significantly decreased the level of miR-346, while simultaneously increasing the levels of miR-147 and miR-106a compared with control cells. TBN treatment also increased the levels of miR-153 and miR-135a, although these changes were not statistically significant. These findings suggest that TBN can inhibit the APP translation by regulating the miRNA levels.
Figure 5.
TBN affects the modulation of APP mRNA in the N2a/APPswe cells. N2a/APPswe cells were treated with or without TBN (300 μm) for 24 h, and the levels of APP mRNA, BACE1 mRNA, ADAM 10 mRNA, SP1 mRNA, CTCF mRNA, TGF-β mRNA, NF-κB mRNA, HSF-1 mRNA, Rela mRNA, miR-346, miR-147, miR-106a, miR-153 and miR-135a were detected by Quantitative reverse transcriptase PCR. Data are presented as mean ± SD, n=4, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, unpaired, two-tailed t test.
Figure 5.
TBN affects the modulation of APP mRNA in the N2a/APPswe cells. N2a/APPswe cells were treated with or without TBN (300 μm) for 24 h, and the levels of APP mRNA, BACE1 mRNA, ADAM 10 mRNA, SP1 mRNA, CTCF mRNA, TGF-β mRNA, NF-κB mRNA, HSF-1 mRNA, Rela mRNA, miR-346, miR-147, miR-106a, miR-153 and miR-135a were detected by Quantitative reverse transcriptase PCR. Data are presented as mean ± SD, n=4, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, unpaired, two-tailed t test.
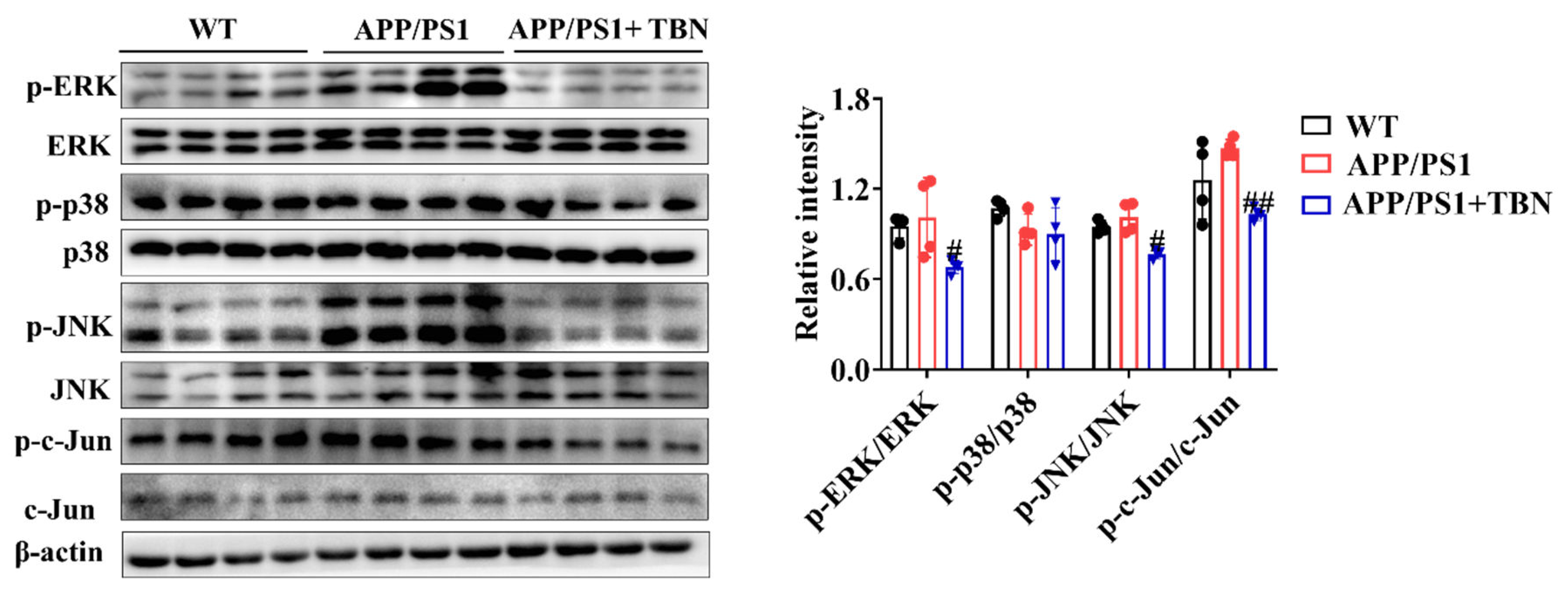
In addition, the results of the investigation displayed in Figure 6, indicated that TBN treatment can reduce the ratio of p-ERK/ERK, p-p38/p38, p-JNK/JNK and p-c-Jun/c-Jun in the hippocampus of APP/PS 1 mice when compared with the saline-treated mice. It is known that c-Jun is a component of activator protein-1 (AP-1), a transcription factor that directly affects APP promoter activity. And AP-1 is activated and regulated by MAP kinases (MAPKs). These results indicate that TBN may lower the AP-1 activity by inhibiting the phosphorylation level of MAPKs, resulting in the downregulation of APP mRNA.
In conclusion, TBN was found to reduce APP expression by regulating the transcriptional factors involved in APP mRNA transcription, including AP-1, SP1, CTCF, TGF-β, and NF-κB. Concurrently, TBN acts on post-transcriptional regulation of APP by decreasing miRNA levels.
Figure 6.
The effects of TBN on the MAPK signaling pathway. Representative images and quantitative analyses of proteins p-ERK, ERK, p-p38, p38, p-JNK, JNK, p-c-Jun, and c-Jun. Data are presented as mean ± SD, n=4, #p < 0.05 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests. .
Figure 6.
The effects of TBN on the MAPK signaling pathway. Representative images and quantitative analyses of proteins p-ERK, ERK, p-p38, p38, p-JNK, JNK, p-c-Jun, and c-Jun. Data are presented as mean ± SD, n=4, #p < 0.05 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests. .
3. Discussion
In our previous study, we have demonstrated that TBN significantly reduced Aβ production and deposition in 3×Tg-AD mice [18]. In the present study, we aimed to further evaluate the therapeutic effects of TBN in the APP/PS1 mice model. Our findings demonstrated that TBN also significantly reduced Aβ production and deposition in APP/PS1 mice. The mechanism by which TBN reduces Aβ levels is likely to be through the inhibition of APP expression via the modulation of APP gene transcriptional factors and miRNAs, the inhibition of BACE 1 expression, and the promotion of autophagy activities.
APP is a type I single-pass transmembrane protein that is the direct source of Aβ generation. As Aβ production can be effectively suppressed by lowering the steady state level of its precursor protein, there has been growing evidence that targeting APP mRNA with a small molecule compound could be a way to treat AD[12]. The regulation of APP gene expression is a complex process involving a number of different factors. These include the gene promoter, which contains a high GC region with five GGGCGC boxes, as well as transcription factors such as SP-1, AP-1, CTCF, TGF-β, HSF-1 and NF-κB, growth factors such as nerve growth factor, cytokines such as interleukin-1 and post-transcriptional regulators such as miRNA. SP-1, AP-1, CTCF, TGF-β, HSF-1 and NF-κB facilitate the transcription in promotor of APP gene [9]. Our studies have demonstrated that TBN significantly decreases the levels of SP-1 mRNA, c-Jun mRNA, TGF-β mRNA and NF-κB mRNA in N2a/APP695swe cells. This suggests that TBN reduces APP mRNA by modulating the levels of related transcription factors. Of note, TBN also decreases p-c-Jun expression in the hippocampus of APP/PS1 mice. C-JUN is the subunit of AP-1. AP-1 is a dimeric protein composed of c-Fos, c-Jun, and related proteins, which effectively promote APP gene transcription to mRNA. This process is stimulated by phosphorylation of MAPKs, which are known to regulate key cellular processes such as gene expression, proliferation, apoptosis, and inflammation, which are involved in the pathophysiology and pathogenesis of AD [22]. Three main distinct subgroups of MAPKs have been identified in mammalian cells, including extracellular-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38. These three pathways are activated in vulnerable neurons in patients with AD [22, 23]. ERK regulates the expression of c-Fos, JNK tightly regulates the levels of c-Jun, and p38 modulates the activity of p53 and ATF2 [24]. A substantial body of research has demonstrated that inhibition of ERK, p38 or JNK restores spatial memory and short-term and long-term memory, and reduces the amount of Aβ deposits [22, 25-27]. Furthermore, MAPKs not only regulate APP mRNA transcription, but also regulate tau phosphorylation [22]. These viewpoints are consistent with the results of our study, which can explain the mechanism of TBN on APP mRNA reduction. It can be postulated that TBN inhibits the phosphorylation levels of JNK and ERK, thereby reducing the level of p-c-jun, which in turn leads to a reduction in AP-1, and finally to a reduction in APP mRNA. Furthermore, our previous study indicated that TBN can reduce tau hyperphosphorylation in 3×Tg-AD mice. This may be related to the inhibition of the MAPKs signaling pathway by TBN.
Another significant finding is that TBN can reduce the level of miR-346, and increase the levels of miR-147 and miR-106a in N2a/APP695swe cells. miRNAs are small, non-coding RNA molecules that play a crucial role in regulating gene expression or mRNA degradation at the post-transcriptional level. They influence various processes associated with AD, including Aβ aggregation, tau pathology, mitochondrial dysfunction, neuroinflammation and synaptic dysfunction [28]. Recently, miRNAs have emerged as promising, cost-effective and non-invasive biomarkers for AD [29]. Several miRNAs have been identified as regulators of Aβ metabolism by targeting the expression of APP and the secretases involved in its processing. For example, miR-106a, miR-135, miR-147, miR-101, miR-16 and miR-520c bind to their predicted target sequences in the APP 3´-UTR and negatively regulate reporter gene expression. Over-expression of these miRNAs results in translational repression of APP mRNA and significantly reduces APP protein levels [14]. Liang et al. have demonstrated that miR-153 suppresses the expression of APP by specifically target site within the APP 3´-UTR [30]. In contrast, miR-346 targets the APP mRNA 5´-UTR to upregulate APP translation and Aβ production [31]. miRNAs can also affect AD pathology in other ways. It has been demonstrated that the expression of the BACE 1 can be increased by miR-29a/b-1, miR-107, miR-298, miR-124, miR-135b, miR-188 and miR-338 [29]. Both miR-34a and miR-26b have been demonstrated to inhibit tau expression and affect NFTs formation [32]. Furthermore, miR-499 and miR-30 have been implicated in the regulation of mitochondrial dynamics, targeting proteins that influence both fission and fusion processes [28]. Additionally, miR-26b, miR-132, and miR-206 have been shown to target neurotrophic factors to regulate synaptic function [32]. Moreover, recent studies have demonstrated that miRNAs modulate autophagic activity. For instance, the gene of mTOR was regulated by several miRNAs, including miR-100, miR-96, miR-338-3p, miR-7 and miR-128. Additionally, ULK 1 expression can be suppressed by miR-290/295 [33]. Our finding also indicated that TBN significantly reduced the BACE 1 mRNA levels, and activated autophagy by decreasing mTOR and increasing ULK expression. These effects may be related to the regulation of miRNAs by TBN, which requires further investigation.
In conclusion, this study represents a preliminary investigation into the potential mechanism of TBN in reducing APP levels. It has been demonstrated that TBN exerts a novel regulatory effect on miRNAs. However, there is still a significant amount of work to be done before these findings can be validated. These includes the verification of the impact of TBN on miRNAs in the hippocampus of APP/PS1 mice, as well as the investigation of whether TBN affects other miRNAs, such as BACE 1, autophagy and synaptic function. Although two antibody-based drugs, aducanumab and lecanemab, have been approved by the FDA, there is still a need for safe and effective pharmacological treatments capable of stopping or at least delaying the course of AD. TBN treatment has been shown to reverse the learning and memory deficits in AD mice, and therefore TBN is a promising drug candidate for AD treatment.
4. Materials and Methods
4.1. Chemicals and Reagents
TBN was provided by Guangzhou Magpie Pharmaceuticals CO., LTD (Guangzhou, China). DMEM, FBS, G418, and Penicillin-Streptomycin solution (100 ×) were purchased from GibcoTM. RIPA buffer and protease/phosphatase inhibitor cocktail were purchased from ThermoFisher Scientific. Antibody APP, BACE1, PS1, ADAM10, synapsin I, synapsin II, PSD 95 and Goat anti-mouse IgG H + L (Alexa Fluor® 488) were purchased from Abcam. Antibody D3D2N, p-PI3K, PI3K, p-AKT, AKT, p-AMPK, AMPK, p-mTOR, mTOR, p-ULK1/2, ULK1/2, LC3, p-ERK, ERK, p-p38, p38, p-JNK, JNK, p-c-Jun, c-Jun, β-actin, anti-rabbit IgG, HRP-linked antibody and anti-mouse IgG antibody were bought from Cell Signaling Technology. Pure Nitrocellulose Blotting Membrane (66485) was obtained from PALL Corporation. TRleasyTM LS Total RNA Extraction Reagent (19201ES60), Hieff qPCR SYBR Green Master Mix (no ROX) (11201ES08) and Hifair III 1st Strand cDNA Synthesis SuperMix for qPCR (11141ES10) were purchased from YEASEN Biotechnology. Step-down avoidance (SDA), novel object recognition (NOR) and mirror water maze (MWM) devices and software were bought from Huaibei Zhenghua Biological instrument equipment Co., LTD.
4.2. Cell Culture
N2a/APP695swe cells were kindly donated by Professor Xifei Yang’ lab (Key Laboratory of Modern Toxicology of Shenzhen, Shenzhen Center for Disease Control and Prevention, Shenzhen, China). N2a/APP695swe cells were cultured in DMDM with 10% FBS, penicillin-streptomycin (1×), and with 200 μg/mL G418 at 37 ℃ with 95% air and 5% CO2 in a humidified incubator. The cells were inoculated in 25 cm2 culture flasks at a specified density for 24 hours, after which they were treated with 300 μM TBN. After a further 24 hours, the cells were collected for RT-PCR, which was measured according to the instructions of the kits.
4.3. Animal Care and Treatment
All experiments were approved by the Animal Ethics Committee of the Shenzhen Center for Disease Control and Prevention (Shenzhen CDC, China) in accordance with the requirements of the Guide for the Care and Use of Laboratory Animals. The APP/PS1 mice were purchased from the Jackson Laboratory and subsequently inbred in the laboratory animal central of the Shenzhen CDC. The mice were housed under controlled conditions, with a temperature range of 20 ℃~26℃, humidity of 40% ~ 70%, and 12-h light/12-h dark cycles. APP/PS1 mice are hemizygous mice expressing human APP and a mutant human PS1-dE9, and their genotype was confirmed by the polymerase chain reaction on a sample of the tail. The littermates of wild-type mice served as the negative control group (WT). Two-month-old APP/PS1 mice were randomly allocated into the vehicle group (APP/PS1) and TBN group (n=16, respectively). The mice in the TBN group were treated with TBN at a dose of 60 mg/kg via intragastric administration twice daily. WT and APP/PS1 group mice were received with parallel volume of saline. Following a six-month period of TBN or saline administration, all mice underwent a behavioural assessment, after which they were sacrificed via the injection of anaesthetics (Zoletil 50/xylazine).
4.3. Behavioral Tests
Behavioral tests commenced at eight months of age and continued for approximately four weeks, throughout which the treatment was maintained. The SDA test, NOR test and MWM test were performed in accordance with the methodology as previously described [18, 34, 35]. In brief, the SDA test was performed by placing the mouse in a separate compartment with a grid floor underneath and an insulated wooden platform. During the training period, the mouse was placed on the grid floor and the electrical shock was delivered for a period of 5 min. In the testing period, the mouse was placed on the wooden platform while the electricity was applied. The time taken for the mouse to touch the grid floor for the first time was recorded as the step-down latency, and the number of times the grid floor was touched was recorded as the error number. The NOR test was conducted on the fourth day following the SDA test. For the NOR test, the mouse was placed in an open field with two identical objects, and allowed to explore freely for 5 min once daily for 3 days during the training phase. On the fourth day, the mouse was placed in the open field with an old object was replaced by a new object, and its exploration trajectory was recorded by the video-tracking software. The discrimination index (DI), which represents the ability to recognize new objects, was calculated using the following equation: (Time exploring new object - Time exploring old object) / (Time exploring new object + Time exploring old object) × 100. Similarly, MWM was performed on the fourth day following the NOR test. The NOR test was carried out in a circular tank with a platform in the third quadrant and filled with opaque water. During the training phase, the mouse was trained to find the platform four times a day for 5 days. A probe trial was conducted on the sixth day following the training session, during which the platform was removed and the mice were placed in the tank to explore the platform site. The mouse’s swimming trajectory was recorded by a movement-tracking system (Super Maze+ software). The latency to cross platform location was defined as the time taken for the mouse to cross the platform site for the first time.
4.4. Immunohistochemistry
The Aβ plaques in the hippocampus and cerebral cortex were quantified using the D3D2N antibody by immunohistochemical staining, in accordance with the methodology as previously described [18, 34]. In brief, the brain tissues were subjected to a series of processes, including fixation in 4% paraformaldehyde, embedding in paraffin, sectioning, dewaxing with xylene, gradual rehydration with ethanol, antigen retrieval, peroxidase removal by 3% hydrogen peroxide, blocking with 10% FBS, primary antibody and secondary antibody incubation, diaminobenzidine colour development, and polyvinylpyrrolidone mounting. To obtain digital images of the sections, an Olympus BX41 microscope was employed.
4.5. Western Blotting
The western blotting procedure was conducted in accordance with the methodology as previously described in [18, 34]. Briefly, hippocampus tissues were homogenized in RIPA lysis buffer containing 1 mM PMSF, and then centrifuged at 12,000 rpm for 20 min at 4 ℃ to obtain the supernatant liquid. Subsequently, the protein concentration was determined, after which the proteins were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) using either 4% or 12% gels. Following electrophoresis, the proteins were transferred to a nitrocellulose (NC) blotting membrane. The NC membrane was then washed for 5 min with Tris-buffered saline with Tween 20 (TBST) and blocked with TBST buffer containing 5% non-fat dry milk for 1 hour at room temperature. The membrane was washed with TBST for 3 times and incubated with the primary antibody overnight at 4 ℃. Following the incubation with the primary antibody, the membrane was incubated with the secondary antibody for 1 hour at room temperature. Finally, the blot was scanned by a chemiluminescent imaging system (Tanon 5200 Multi), and the bands were quantified by densitometry using Image J.
4.6. qRT-PCR
The total RNA was extracted from N2a/APP695swe cells using the TRleasyTM LS Total RNA Extraction Reagent, in according to the manufacturer’s instructions. Quantitative PCR was conducted by Real-Time system (KUBO TECH, q225) using Hifair III 1st Strand cDNA Synthesis SuperMix for qPCR kit and Hieff qPCR SYBR Green Master Mix kit (no ROX) in accordance with the manufacturer’s instructions. The results of the qPCR were normalised using GAPDH for mRNA and U6 for miRNA. The primers used for qRT-PCR are listed below: APP: Forward 5´- CCAGAACTGGTGCAAGCG -3´, reverse 5´- TTTGGCGACGGTGTGCCA -3´. BACE1: forward 5´- TGTGGAGATGGTGGACAACC -3´, reverse 5´- GGACAGCTGCCTCTGGTAG -3´. ADAM 10: forward 5´- AGTAGAGGAAGGAGCCCGG -3´, reverse 5´- GCAATCACAGCTTCTCGTGT -3´. SP1: forward 5´-ATGCTGCTCAACTCTCCTCCATG-3´, reverse 5´-CCTTCTCCACCTGCTGTCTCATC-3´; CTCF: forward 5´- TTGAGGAAGAACAGCAGGAAGGAC-3´, reverse 5´- TGGCTTCGGAGGCTTCATATTACC -3´; TGF-β: forward 5´-GGGCATCGCTCATCTCCACAG-3´, reverse 5´-GCAACAGGTCAAGTCGTTCTTCAC -3´; NF-κB: forward 5´- ATGGGAAACCGTATGAGCCTGTG -3´, reverse 5´- AGTTGTAGCCTCGTGTCTTCTGTC -3´; c-Jun: forward 5′-GTTGCGGCCGCGAAACTT-3′, reverse 5′-CATTGCCCTCGAGCCCTG-3′; HSF-1: forward 5´- CCAAGGAGGTGCTGCCCAAG -3´, reverse 5′- ATGCTGGAACTCGGTGTCATCTC -3′; Rela: forward 5′- AGAGAAGCACAGATACCACCAAGAC -3′, reverse 5′- GGTCAGCCTCATAGTAGCCATCC -3′; GAPDH: forward 5´- TCCACTCACGGCAAATTCAAC -3´, reverse 5´- GTAGACTCCACGACATACTCAGC -3´; miR-346: forward 5´- ACACTCCAGCTGGGTGTCTGCCCGAGTGC -3´, forward 5´-CTCAACTGGTGTCGTGGA-3´; miR-147: forward 5´-ACACTCCAGCTGGGTGGAAACTTTCTGCA-3´, forward 5´-CTCAACTGGTGTCGTGGA -3´; miR-106a: forward 5´-ACACTCCAGCTGGGCAAAGTGCTAACAGT -3´, forward 5´-CTCAACTGGTGTCGTGGA-3´; miR-153: forward 5´-ACACTCCAGCTGGGTTGCATAGTCACAA -3´, forward 5´-CTCAACTGGTGTCGTGGA -3´; miR-135a: forward 5´-ACACTCCAGCTGGGTATAGGGATTGGAGCC-3´, forward 5´-CTCAACTGGTGTCGTGGA-3´.
5. Statistical Analysis
Quantitative data are expressed as mean ± SD. Statistical significance between any two groups was determined by the one-way ANOVA followed by Tukey’s multiple comparison tests or unpaired, two-tailed t test using GraphPad Prism 8. P-value less than 0.05 was considered statistically significant.
Author Contributions
Conceptualization, X.Z., Z.Z. and S.K.; methodology, B.X. and H.C.; data curation, X.Z. and Z.Z.; writing—original draft preparation, X.Z.; writing—review and editing, Z.Z., Y.L. and Y.W.; funding acquisition, X.Z., M.H., Z.Z., X.Y. and K.H.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (NSFC) (Project No. 82304465, 82073821), and Guangzhou Science and Technology Programme (SL2023A03J01103). This work was also funded by Science and Technology Development Fund, Macau SAR (File no. 0023/2020/AFJ), the University of Macau Research Grant (Project No. MYRG2022-00248-ICMS), the Natural Science Foundation of Guangdong Province, China (NO. 2022A1515011729) and Key Program of Shenzhen Basic Research (JCYJ20200109150717745).
Institutional Review Board Statement
All procedures were approved and supervised by the Institutional Animal Care and Use Committee of Shenzhen Center for Disease Control and Prevention.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Yang, H., et al., Based on molecular structures: Amyloid-β generation, clearance, toxicity and therapeutic strategies. Frontiers in Molecular Neuroscience, 2022. 15. [CrossRef]
- Manczak, M., et al., Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Human Molecular Genetics, 2018. 27(8): p. 1332-1342. [CrossRef]
- Hampel, H., et al., The Amyloid-β Pathway in Alzheimer’s Disease. Molecular Psychiatry, 2021. 26(10): p. 5481-5503. [CrossRef]
- Zhu, C.-c., et al., Advances in Drug Therapy for Alzheimer’s Disease. Current Medical Science, 2021. 40(6): p. 999-1008. [CrossRef]
- Yu, T.-W., H.-Y. Lane, and C.-H. Lin, Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review. International Journal of Molecular Sciences, 2021. 22(15). [CrossRef]
- Uddin, M.S., et al., Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Frontiers in Aging Neuroscience, 2018. 10. [CrossRef]
- Liu, J. and L. Li, Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Frontiers in Molecular Neuroscience, 2019. 12. [CrossRef]
- Zhang, Z., et al., Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Research Reviews, 2021. 72. [CrossRef]
- Sato, K., et al., Transcriptional and Post-Transcriptional Regulations of Amyloid-β Precursor Protein (APP) mRNA. Frontiers in Aging, 2021. 2. [CrossRef]
- Delport, A. and R. Hewer, The amyloid precursor protein: a converging point in Alzheimer’s disease. Molecular Neurobiology, 2022. 59(7): p. 4501-4516. [CrossRef]
- Amyloid Precursor Protein: A Regulatory Hub in Alzheimer's Disease. aging and disease, 2023.
- Pankiewicz, J.E. and M.J. Sadowski, Editorial: Translational Control of APP Expression for Alzheimer Disease Therapy. Ann Pharmacol Pharm, 2017. 2(15).
- Chen, X.-Q., et al., Posiphen Reduces the Levels of Huntingtin Protein through Translation Suppression. Pharmaceutics, 2021. 13(12). [CrossRef]
- Patel, N., et al., MicroRNAs can regulate human APP levels. Molecular Neurodegeneration, 2008. 3(1). [CrossRef]
- Amakiri, N., et al., Amyloid Beta and MicroRNAs in Alzheimer’s Disease. Frontiers in Neuroscience, 2019. 13. [CrossRef]
- Madadi, S., et al., Downregulation of serum miR-106b: a potential biomarker for Alzheimer disease. Archives of Physiology and Biochemistry, 2020. 128(4): p. 875-879. [CrossRef]
- Walgrave, H., et al., The promise of microRNA-based therapies in Alzheimer’s disease: challenges and perspectives. Molecular Neurodegeneration, 2021. 16(1). [CrossRef]
- Zhou, X., et al., Evaluation of therapeutic effects of tetramethylpyrazine nitrone in Alzheimer’s disease mouse model and proteomics analysis. Frontiers in Pharmacology, 2023. 14. [CrossRef]
- Guo, B., et al., Therapeutic effects of multifunctional tetramethylpyrazine nitrone on models of Parkinson's disease in vitro and in vivo. Biol Pharm Bull, 2014. 37(2): p. 274-85. [CrossRef]
- Zhang, Z., et al., Tetramethylpyrazine nitrone, a multifunctional neuroprotective agent for ischemic stroke therapy. Scientific Reports, 2016. 6(1). [CrossRef]
- Wen, J., et al., Tetramethylpyrazine nitrone improves motor dysfunction and pathological manifestations by activating the PGC-1α/Nrf2/HO-1 pathway in ALS mice. Neuropharmacology, 2021. 182. [CrossRef]
- Zhu, X., et al., The Role of Mitogen-Activated Protein Kinase Pathways in Alzheimer’s Disease. Neurosignals, 2002. 11(5): p. 270-281. [CrossRef]
- Whitmarsh, A.J. and R.J. Davis, Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med (Berl), 1996. 74(10): p. 589-607. [CrossRef]
- Nagesh, R., et al., Regulation of Jun and Fos AP-1 transcription factors by JNK MAPKs signaling cascade in areca nut extract treated KB cells. Biochemistry and Biophysics Reports, 2021. 27. [CrossRef]
- Gee, M.S., et al., A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimer's Research & Therapy, 2020. 12(1).
- Chang, K.-W., et al., Modulation of the MAPKs pathways affects Aβ-induced cognitive deficits in Alzheimer’s disease via activation of α7nAChR. Neurobiology of Learning and Memory, 2020. 168. [CrossRef]
- Muraleva, N.A., N.G. Kolosova, and N.A. Stefanova, MEK1/2-ERK Pathway Alterations as a Therapeutic Target in Sporadic Alzheimer’s Disease: A Study in Senescence-Accelerated OXYS Rats. Antioxidants, 2021. 10(7). [CrossRef]
- Sharma, M., et al., Regulatory roles of microRNAs in modulating mitochondrial dynamics, amyloid beta fibrillation, microglial activation, and cholinergic signaling: Implications for alzheimer's disease pathogenesis. Neuroscience & Biobehavioral Reviews, 2024. 161. 16/j.neubiorev.2024.105685.
- Siedlecki-Wullich, D., A.J. Miñano-Molina, and J. Rodríguez-Álvarez, microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells, 2021. 10(1). [CrossRef]
- Liang, C., et al., MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Research, 2012. 1455: p. 103-113. [CrossRef]
- Long, J.M., et al., Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Molecular Psychiatry, 2018. 24(3): p. 345-363. [CrossRef]
- Lee, C.Y., et al., miRNAs as Therapeutic Tools in Alzheimer’s Disease. International Journal of Molecular Sciences, 2021. 22(23). [CrossRef]
- Akkoc, Y. and D. Gozuacik, MicroRNAs as major regulators of the autophagy pathway. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2020. 1867(5).
- Zhou, X., et al., Hippocampal proteomic alteration in triple transgenic mouse model of Alzheimer's disease and implication of PINK 1 regulation in donepezil treatment. J Proteome Res, 2018. [CrossRef]
- Zhou, X., et al., Memantine Improves Cognitive Function and Alters Hippocampal and Cortical Proteome in Triple Transgenic Mouse Model of Alzheimer's Disease. Experimental Neurobiology, 2019. 28(3): p. 390-403. [CrossRef]
Figure 1.
TBN improves cognitive abilities in APP/PS1 mouse. (A) Latency in the step-down avoidance (SDA) test. (B) The ability to recognize a new object in the novel object recognition (NOR) test. (C) The swim traces of mice in mirror water maze (MWM) during probe test. (D) The time for the mice to reach the platform at first time during probe test. Data are presented as mean ± SD, n=16, **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
Figure 1.
TBN improves cognitive abilities in APP/PS1 mouse. (A) Latency in the step-down avoidance (SDA) test. (B) The ability to recognize a new object in the novel object recognition (NOR) test. (C) The swim traces of mice in mirror water maze (MWM) during probe test. (D) The time for the mice to reach the platform at first time during probe test. Data are presented as mean ± SD, n=16, **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
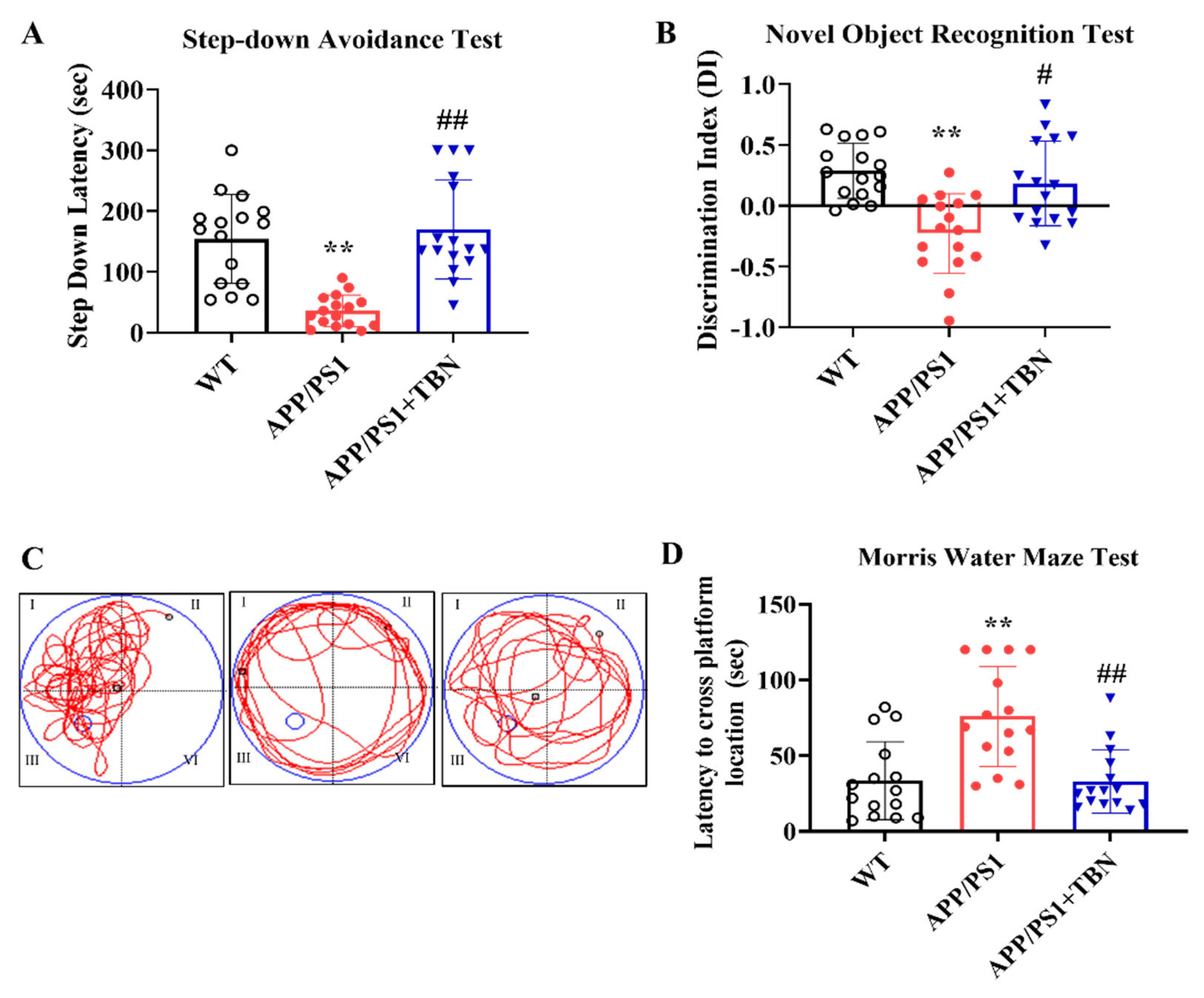
Figure 2.
TBN treatment decreases the Aβ plaques in APP/PS1 mice. (A) Aβ depositions were measured using immunohistochemical staining with β-amyloid (D3D2N) antibody. (B) Western blots and quantitative analyses of APP, BAEC1, ADAM 10 and PS1 in the hippocampal tissues of APP/PS 1 mice. Data are presented as mean ± SD, n=4, *p < 0.05 or **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
Figure 2.
TBN treatment decreases the Aβ plaques in APP/PS1 mice. (A) Aβ depositions were measured using immunohistochemical staining with β-amyloid (D3D2N) antibody. (B) Western blots and quantitative analyses of APP, BAEC1, ADAM 10 and PS1 in the hippocampal tissues of APP/PS 1 mice. Data are presented as mean ± SD, n=4, *p < 0.05 or **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
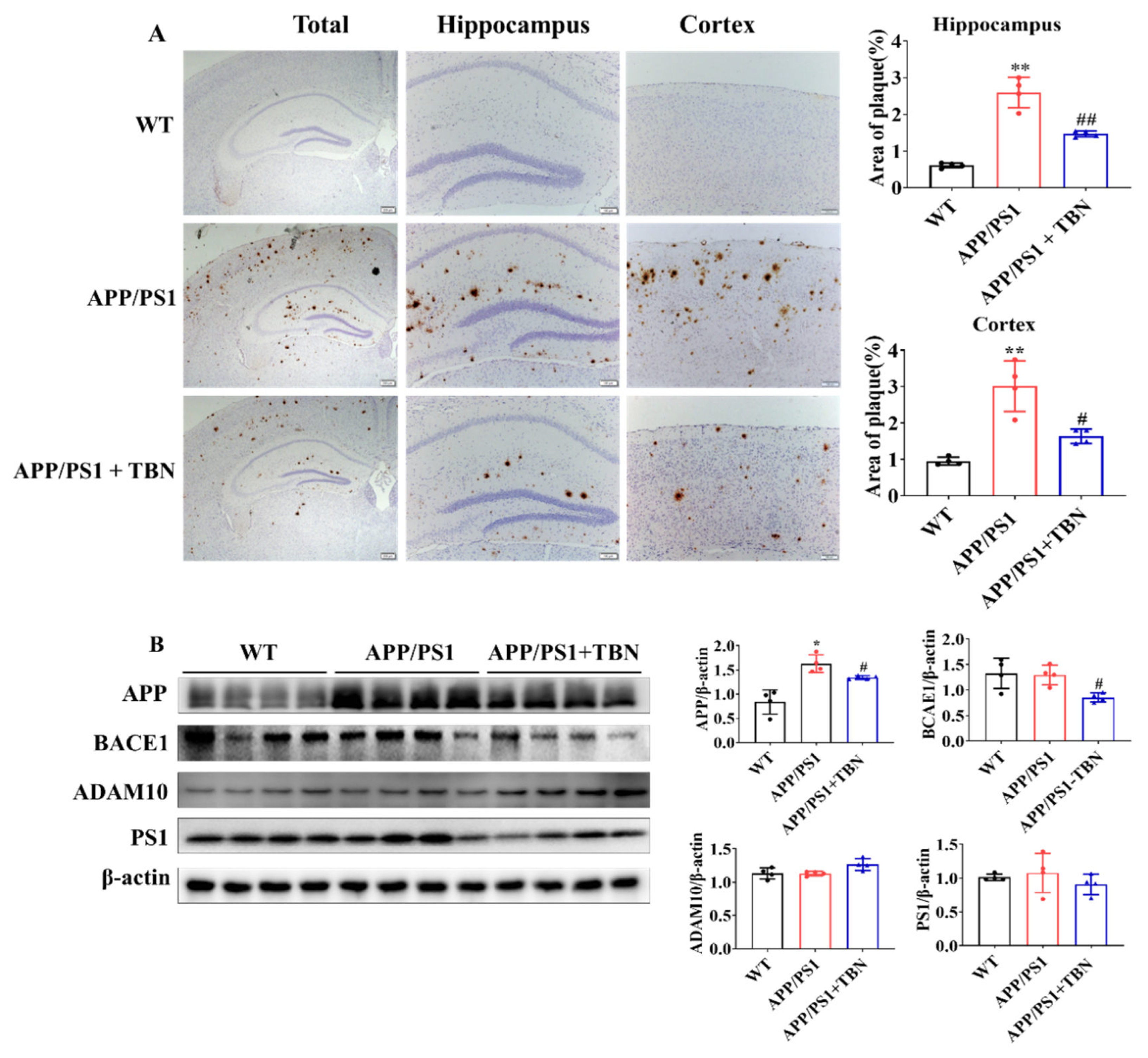
Figure 3.
Effects of TBN on expression of synapse protein in the hippocampal tissues of APP/PS 1 mice. The levels of PSD 95, synapsin I and synapsin II were measured by Western blotting. Data are presented as mean ± SD, n=4, *p < 0.05 vs. WT group, #p < 0.05 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests. .
Figure 3.
Effects of TBN on expression of synapse protein in the hippocampal tissues of APP/PS 1 mice. The levels of PSD 95, synapsin I and synapsin II were measured by Western blotting. Data are presented as mean ± SD, n=4, *p < 0.05 vs. WT group, #p < 0.05 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests. .
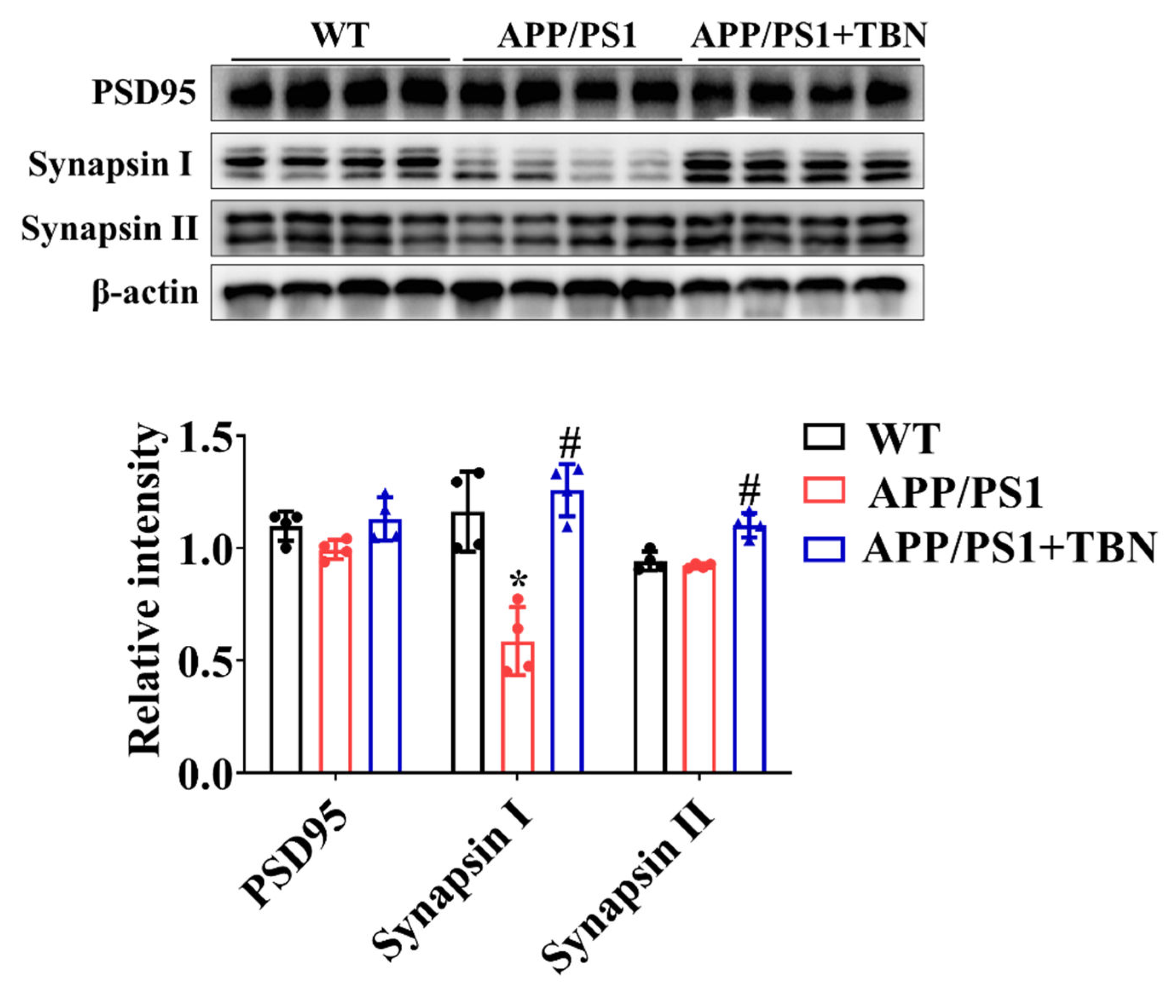
Figure 4.
TBN enhances the expression of autophagy-related proteins in the hippocampal tissues of APP/PS 1 mice. (A and B) Representative images and quantitative analyses of proteins p-PI3K, PI3K, p-AKT, AKT, p-AMPK, AMPK, p-mTOR, mTOR, p-ULK1/2, ULK1/2, LC3 I and LC3 II. Data are presented as mean ± SD, n=4, **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
Figure 4.
TBN enhances the expression of autophagy-related proteins in the hippocampal tissues of APP/PS 1 mice. (A and B) Representative images and quantitative analyses of proteins p-PI3K, PI3K, p-AKT, AKT, p-AMPK, AMPK, p-mTOR, mTOR, p-ULK1/2, ULK1/2, LC3 I and LC3 II. Data are presented as mean ± SD, n=4, **p < 0.01 vs. WT group, #p < 0.05 or ##p < 0.01 vs. APP/PS1 group, one-way ANOVA followed by Tukey’s multiple comparison tests.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



