
Submitted:
24 June 2024
Posted:
25 June 2024
You are already at the latest version
Abstract
How hematopoietic stem and progenitor cell (HSPC) fate decisions are affected by genetic alterations acquired during AML leukemogenesis is poorly understood and mainly explored in animal models. Here, we studied isocitrate dehydrogenases (IDH) genes mutations in the human model of HSPC and provide a review of the literature on this topic. IDH1/2 mutations occur in ~20% of AML, are recognized among the mutations earliest acquired during leukemogenesis, and are target of specific inhibitors (ivosidenib and enasidenib, respectively). In order to investigate the direct effects of these mutations on the HSPC we expressed by lentiviral transduction either IDH1-R132H or IDH2-R140Q mutants into human CD34+ healthy donor cells and analyzed the colony-forming unit (CFU) ability. CFU ability was dramatically compromised with a complete trilineage block of differentiation. Strikingly, the block was reversed by the specific inhibitors, confirming it was a specific effect induced by the mutants. In line with this observation, the CD34+ leukemic precursors isolated from a patient with IDH2-mutated AML at baseline and during enasidenib treatment showed over time a progressive and marked improvement of their fitness in terms of CFU ability and propensity to differentiate, attaining clonal trilinear reconstitution of hematopoiesis and complete hematological remission.
Keywords:
acute myeloid leukemia
; IDH1/2 mutation
; human HSPC modeling
1. Introduction
Acute myeloid leukemia (AML) arises from genetic abnormalities in hematopoietic stem or progenitor cells (HSPC), responsible for uncontrolled growth and accumulation of neoplastic blasts in the bone marrow (BM). AML is an aggressive, clinically and biologically heterogeneous disease, in which a large number of recurrently mutated genes have been identified [1]. A stepwise acquisition of more than one alteration is required for leukemia development although it is not always clear how single mutations contribute to leukemogenesis [2,3].
Mutations in Isocitrate Dehydrogenase (IDH) 1 and 2 were originally identified in glioma [4,5], AML, myeloproliferative neoplasm, myelodysplastic syndrome patients [6,7,8] and in other solid tumors [9,10]. In AML approximately 20% of patients carry these mutations, especially in the setting of normal karyotype [1,11]. IDH1 mutations occur thereabout in 7%-10% of patients, while 10%-15% are IDH2 mutated and their frequency increases with age [6,12,13,14].
Isocitrate dehydrogenases are a group of homodimeric enzymes that play an important role in cellular metabolism and regulation of the response to hypoxia [15]. Physiologically the isoform IDH1, localized in the cytoplasm and peroxisomes, is involved in lipid and carbohydrate metabolism, providing NADPH to cells and protecting them from reactive oxygen species. IDH2, on the other hand, is localized in the mitochondrial matrix and participates in the tricarboxylic acid cycle to protect cells from oxidative damage. Both isoforms catalyze the oxidative decarboxylation of isocitrate to generate α-ketoglutarate (αKG) and carbon dioxide (CO2) and produce reduced NADPH from NADP+, a process essential in preserving cellular homeostasis [16].
In AML, IDH mutations are heterozygous and missense. Aminoacidic substitution in IDH1 most often involves arginine 132 with cysteine or histidine (R132C or R132H). In IDH2 arginine is replaced by glutamine at residue 140 (R140Q) and by lysine at residue 172 (R172K) [16,17,18,19]. These mutations result in gain-of-function neomorphic activity that allows the IDH enzymes to catalyze the reduction of αKG to the oncometabolite (R)-2-hydroxyglutarate (2-HG) [20,21,22]. Abnormal accumulation of 2-HG has been shown to inhibit αKG-dependent dioxygenases as the ten-eleven-translocation methylcytosine dioxygenase 2 (TET2), to dysregulate the epigenetic machinery of hematopoietic progenitors; this may explain why IDH1 and IDH2 mutations are mutually exclusive with those affecting TET2 [20,23,24]. On the other hand, IDH1/2 mutations are frequently associated with nucleophosmin (NPM1) gene mutations and internal tandem duplication in FMS-like tyrosine kinase 3 (FLT3-ITD) [12,25].
The bona fide correlation between mutations and leukemogenic mechanisms has prompted intense drug discovery to target the mutants IDH. In particular, two first-in class small molecule inhibitors have been approved by the FDA: enasidenib (AG-221) and ivosidenib (AG-120) [26,27]. Enasidenib is a selective allosteric inhibitor of IDH2 mutation: its function involves the binding and stabilization of the open conformation of the IDH mutated enzyme, and primarily the inhibition of the conversion of αKG to 2-HG2 [19,28]. Ivosidenib is a reversible, allosteric competitive inhibitor of mutated IDH1. Ivosidenib competes for binding with magnesium ion, IDH1 mutant essential cofactor, thereby preventing the formation of a catalytically active site [19,28].
IDH1/2 mutations and the other genetic lesions in genes encoding for epigenetic modifiers (DNMT3A, ASXL1 and TET2) are early events in the development of AML, like observed in deep sequencing and single-cell studies determining clonal evolution in myeloid malignancies [11,29]. These mutations are typically in the founding clone of AML and are almost never found alone [11], suggesting that they are not sufficient to drive leukemia. Effectively, numerous studies in mouse models demonstrated that IDH mutations alone cannot cause overt leukemia in vivo [30,31], but co-operate with additional genetic lesions to initiate cancer [32,33,34]. The direct effects of the single IDH AML-associated mutations on human HSPC are poorly studied, so that it remains unclear how they affect cell fate decision, promotion of leukemogenesis, maintenance and progression of the malignant phenotype. Although some mouse models studies have somehow elucidated how IDH mutations affect mouse HSPC, specific studies in humans are lacking to bridge the gap of species-specific differences in basic biology and hematology.
Here we report our experimental findings on how IDH1/2 mutations and their pharmacological inhibition affect human hematopoietic stem cells fitness and provide a review of the literature on modeling leukemogenesis by IDH1/2 AML-associated mutations (Table 1).
2. Materials and Methods
2.1. Human CD34+ Hematopoietic Stem and Progenitor Cells from Healthy Donors
Human CD34+ HSPC (hCD34+) derived from mobilized peripheral blood of n = 13 healthy donors (HD) (Supplementary Table S1) were obtained upon written informed consent. The study was conducted according to the Declaration of Helsinki and approved by the local ethic committee. After mobilization with rHuG-CSF (Lenograstim), peripheral blood cells were selected for CD34 marker expression with CliniMACS® technology (Miltenyi Biotec Inc, Auburn, CA, USA), according to standard procedures followed at our institute [39]. Leftover cells after infusion to patient were recovered for experiments. Cells were stained with APC/Cyanine7 anti-human CD34 antibody and FITC anti-human CD38 antibody (BioLegend®, San Diego, CA, USA) and analyzed by Fluorescence Activated Cell Sorting (FACS) with BD FACSCanto II™ (Becton Dikinson, NJ, USA) to assess immunophenotype. For each donor up to 10 × 106 CD34+ were cultured in StemSpan™ SFEM II serum free medium (STEMCELL™ Technologies, Vancouver, BC, Canada) supplemented with penicillin/streptomycin (1:100), Recombinant human Fms-related tyrosine kinase 3 ligand/FLT-3 Ligand (100 ng/ml), Stem cell factor/SCF (100 ng/ml) and Recombinant Human Thrombopoietin/TPO (50 ng/ml) (R&D SYSTEMS, Minneapolis, MN, USA) for 48h before cell transduction (hereafter named “HSPC-retention medium”).
2.2. Sequences and Expression Vector
Wild-type coding sequences used for lentiviral transduction are:
- -
- Homo sapiens isocitrate dehydrogenase (NADP(+)) 1, cytosolic (IDH1), transcript variant 1, mRNA (NCBI Reference Sequence: NM_005896.3);
- -
- Homo sapiens isocitrate dehydrogenase (NADP(+)) 2, mitochondrial (IDH2), transcript variant 1, mRNA (NCBI Reference Sequence: NM_002168.3).
- -
- Mutated sequences were virtually designed according to the most common nucleotide change in cytogenetically normal – AML [17]:
- -
- IDH1-R132H (nucleotide change c.395G>A)
- -
- IDH2-R140Q (nucleotide change c.419G>A)
All sequences have been synthesized and provided by Genscript® (Piscataway, NJ, USA) into the pLVX-EF1α-IRES-ZsGreen1 (TakaraBio, Shiga, Japan) vector in which a bicistronic RNA is transcribed by the same promoter and the internal ribosome entry (IRES) site allows for internal/cap-independent translation initiation, obtaining the coordinated expression of GOIs and reporter gene (ZsGreen1).
2.3. Lentivirus Production
HEK 293T packaging cell line was cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin plus 1% glutamine at 37 °C, 5% CO2 atmosphere. For lentiviral particles production, HEK 293T cells were co-transfected by Lipofectamine™3000 (Invitrogen™-Fisher Scientific Inc., Waltham, MA, USA), according to the manufacturer’s instructions, using a second-generation lentiviral system. This system consists of the expression vector pLVX-EF1α-IRES-ZsGreen1 containing the single genes of interest (GOIs) IDH1 wild type or R132H and IDH2 wild type or R140Q, in combination with packaging plasmids, psPAX2, a gift from Didier Trono (Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID: Addgene_12260), and BaEV-TR345, kindly granted by Els Verhoeyen (Université de Lyon, France) [40]. The viral supernatants were collected 48 h after transfection and concentrated 100× by centrifugation at 3000× g for 16 h at +4 °C. Viral titre was estimated by transducing HEK 293T cells with serially diluted vectors and quantifying the proportion of ZsGreen1+ cells by FACS.
2.4. hCD34+ Lentiviral Infection
Lentiviral infections of CD34+ primary cells were performed according to the RetroNectin® (Takara Bio Inc.) protocol guidelines but optimized for our needs. For each well of a 6-well plate, 2 ml of growth HSPC-retention medium were added with lentiviral 100x concentrate supernatant of IDH1 wild type/R132H, or IDH2 wild type/R140Q, or empty vector at Multiplicity of Infection = 30. The experimental conditions are illustrated in Supplementary Table S1. The plate was centrifuged at 2000× g for 2 hours at 32 °C for virus pre-loading. The targets cells CD34+ were counted, resuspended in HSPC-retention medium at 2 × 106 /ml and 1 ml per well was added. The plate was then centrifuged at 1800 RPM for 45 minutes at 32 °C and then incubated at 37 °C, 5% CO2 for 72 hours.
2.5. hCD34+ Cells Staining and Sorting Strategies
After incubation, cells were harvested, washed with PBS and stained for 20 min in dark at RT with APC/Cyanine7 anti-human CD34 antibody. Cells were re-suspended in PBS, HSA (0.5%) and EDTA (2 mM) before sorting. An unstained control was made to set up forward and side scatter on the cell sorter and to set positive gates. The cells were sorted by FACS using a BD FACSAria™ III cell sorter (Becton Dickinson, Franklin Lakes, New Jersey). Transduced bright ZsGreen1+/CD34+ double positive cells expressing either empty vector as control, or IDH1/2 WT, or IDH1-R132H / IDH2-R140Q were used for CFU assays in methylcellulose medium. In our 13 human CD34+ cells samples transduced with GOIs we obtained a ZsGreen1 reporter gene cell positivity and sorting strategy is depicted in Supplementary Figure S1.
2.6. hCD34+ Cells Colony Forming Unit Assays
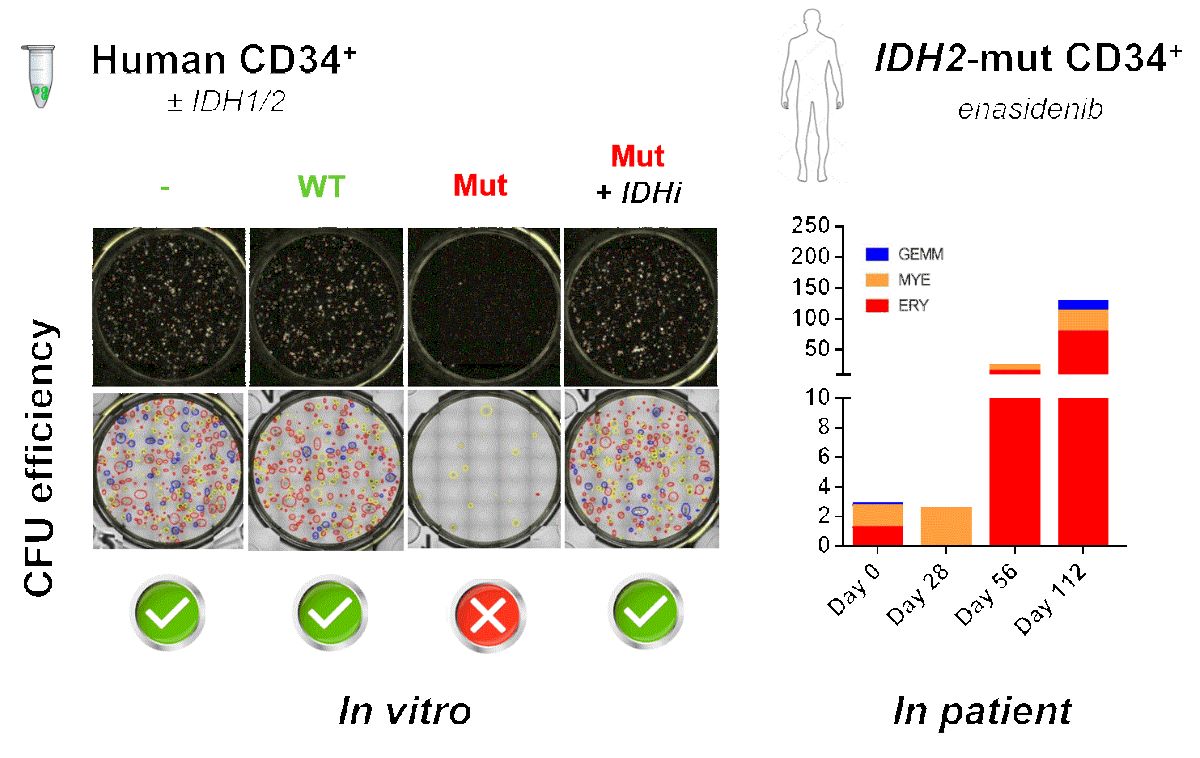
CFU assays were performed by culturing hematopoietic cells in MethoCult™ H4435 Enriched (STEMCELL™ Technologies, Vancouver, BC, Canada) in accordance with the manufacturer’s instructions. After sorting, cells were re-suspended in 300 μl of SFEM II and added to MethoCult™ H4435 Enriched in presence or absence of the specific inhibitor ivosidenib (AG-120, 5 μM) or enasidenib (AG-221, 5 μM) (Selleckchem, Houston, TX, USA) (Supplementary Table S1). Cells were then plated in meniscus-free SmartDish™ cultureware at a density of 500 cells/well in different wells and cultured for 14 days at 37 °C and 5% CO2; 4 wells for each sample condition have been foreseen, for subsequent statistical analyses. Colonies were counted and characterized by subtype [i.e., colony forming unit - Erythroid (CFU-E), burst forming unit - Erythroid (BFU-E), colony forming unit - Granulocyte, Macrophage, Granulocyte/Macrophage (CFU-G/M/GM) and colony forming unit - Granulocyte, Erythrocyte, Macrophage and Megakaryocyte (CFU-GEMM)] after 14 days of culture using STEMvision™ instrument and its analysis software STEMvision Colony Marker (STEMCELL™ Tech, Vancouver, BC, Canada). The experimental workflow is illustrated in Figure 1.
2.7. CD34+ Cells from a IDH2-Mutated AML Patient
Primary cells from a patient with IDH2-mutated R140Q AML were obtained, upon written informed consent, at the Hematology Institute of Perugia (Italy). The study was conducted according to the Declaration of Helsinki and following a protocol approved by local Ethics Committee. Samples were obtained from BM aspirates performed at diagnosis and under therapy with enasidenib (AG-221) 100 mg/die (for 28 days = 1 cycle), as third line chemotherapy, on day 0 (before starting the treatment) and after the I, II and IV cycle of therapy. Mononuclear cells were separated by Lymphoprep™ (Serumwerk Bernburg AG for Alere Technologies AS, Oslo, Norway) according to the manufacturer’s protocol. APC/Cyanine7 anti-human CD34 antibody staining was performed and CD34+ cells were isolated by cell sorter. Sorted CD34+ AML cells were plated in MethoCult™ enriched medium for CFU assays, incubated for 14 days at 37 °C, 5% CO2 and images captured by STEMvision™ as described above.
2.8. Next Generation Targeted-DNA Sequencing (NGS)
Determination of IDH2-mutant allele frequency was carried out by extracting DNA from primary IDH2-R140Q AML BM sample (bulk, myeloid, erythroid, and CD34+ cells) at IV cycle of enasidenib therapy of patient. The DNA sample collected for diagnostic procedures was used for DNA sequencing analysis, in order to obtain a detailed overview about DNA mutations in our AML patient. The DNA concentration was assessed using a fluorometric quantitation using the Qubit 3.0 Fluorometer (Thermo Fisher Scientific cat. no. Q33216), while the DNA Integrity Numbers (DIN) was evaluated by microfluidic electrophoresis on Agilent TapeStation using Agilent High Sensitivity D1000 ScreenTape System (Agilent, USA). The Qiagen Myeloid Neoplasm sequencing panel (Qiagen, USA, cat. Nr. 333502) was used to prepare libraries using 40 ng of DNA from each sample according to the manufacturer instructions. The sequencing process was carried out on MiSeq Illumina platform, generating 150 base-paired end reads. Variant calling was performed using the Geneglobe online data analysis platform (Qiagen, USA).
2.9. Statistical Analysis
For transduced hCD34+ CFU assay, technical replicates of 4 wells for each condition have been foreseen and subsequent statistical analysis was performed on the mean of minimum 3 wells per sample. Graphical representation of data was performed using Graph Pad Prism [v7]. Statistical analyses were performed by One-Way ANOVA test (**** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05).
3. Results
3.1. Either IDH1-R132H or IDH2-R140Q Mutation Blocks CFU Ability Of Human CD34+ HSPC
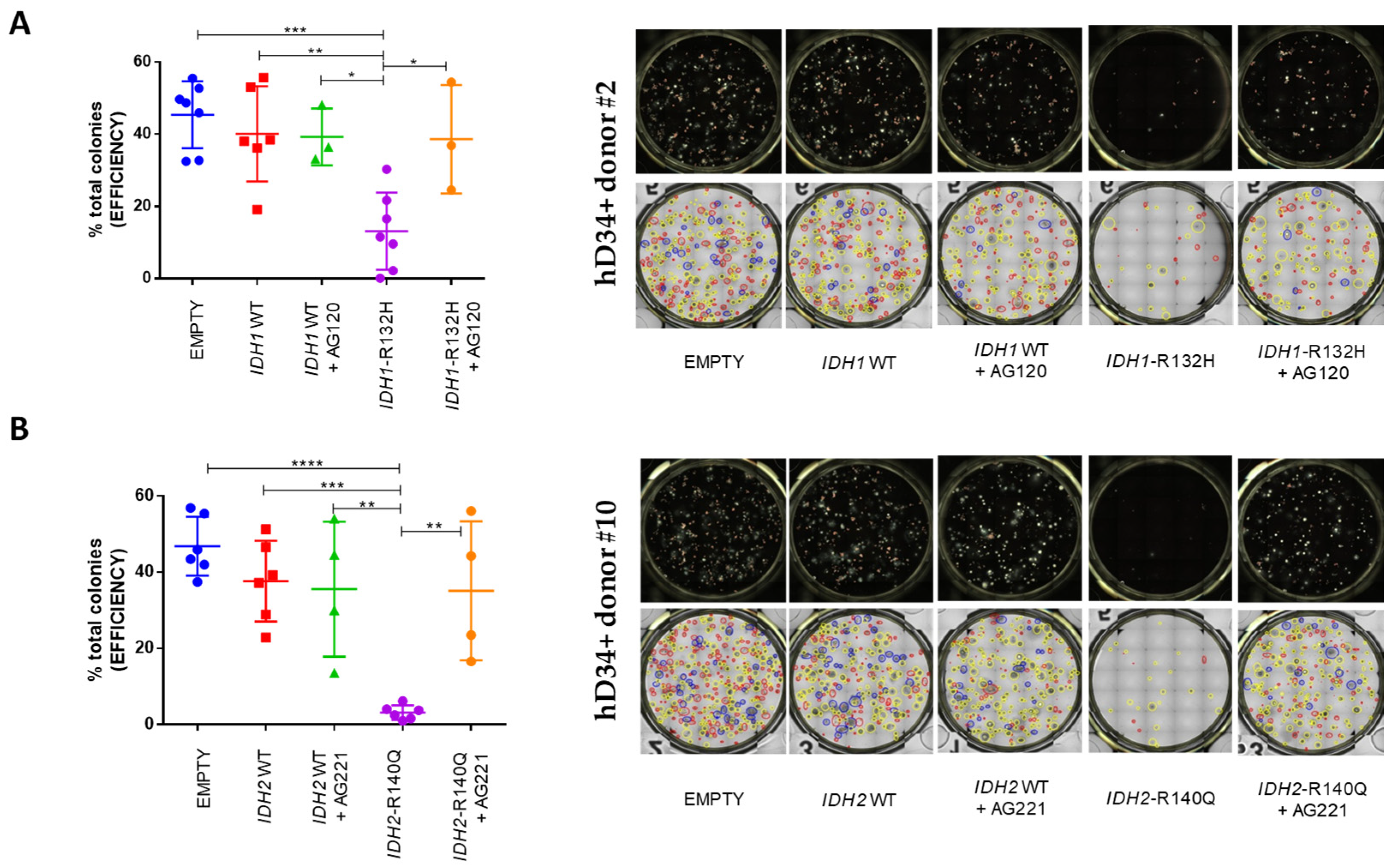
To investigate the direct effect of AML recurring IDH1/2 mutations, hCD34+ from HD were transduced by lentiviral expression vector containing either IDH1 wild-type or IDH1-R132H and ZsGreen1 reporter gene (n = 7 donors) or, alternatively, containing either IDH2 wild-type or IDH2-R140Q and ZsGreen1 reporter gene (n = 6 donors) (Supplementary Table S1) and sorted as described in Materials and Methods.
ZsGreen1-positive sorted cells were used for CFU assays, either in presence or absence of the selective inhibitor AG-120 (n = 3 out of 7 donors for IDH1 experimental set) or AG-221 (n = 4 out of 6 donors in IDH2 set) (Figure 2).
Colonies were assessed at 14 days. To note, in our control groups cloning efficiency from hCD34+ expressing the empty vector was comparable to hCD34+ bearing IDH1 wild-type or IDH2 wild-type and was not significantly influenced by treatment with ivosidenib or enasidenib. Instead, hCD34+ with IDH1-R132H mutation showed an evident block of differentiation compared to hCD34+ transduced with empty vector (p = 0,0002 ***) or wild-type gene (p = 0,0025 **) counterparts. This block was even more pronounced with the IDH2-R140Q mutation when compared with empty vector (p < 0,0001 ****) or with wild-type (p = 0,0004 ***) exogenous expression. In line with being a specific effect of the IDH1 or IDH2 mutations, the block was released, in both cases, by AG-120 (p = 0,0257 *) or AG-221 (p = 0028 **), respectively (Figure 2).
Figure 2.
IDH1-R132H and IDH2-R140Q mutations drive a block of CFU ability in human CD34+ HSPC which is released by the specific inhibitors. CFU assay of transduced hCD34+ cells with empty vector, IDH1 wild-type or IDH1-R132H (n = 7), ± AG-120 inhibitor (n = 3 of 7) (A); empty vector, IDH2 wild-type or IDH2-R140 (n = 6), ± AG-221 inhibitor (n = 4 of 6) (B). Graphs on the left represent the percentage of total colonies rised after 14 days. Panels on the right are representative images of wells (top row) and type of colonies (bottom row) obtained using the STEMvision™ Colony Marker Application: red circles identify BFU-E-derived colonies; yellow circles identify CFU-G/M/GM-derived colonies; blue circles identify CFU-GEMM-derived colonies; and orange circles identify CFU-E-derived colonies. (One-Way ANOVA analyses **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05).
Figure 2.
IDH1-R132H and IDH2-R140Q mutations drive a block of CFU ability in human CD34+ HSPC which is released by the specific inhibitors. CFU assay of transduced hCD34+ cells with empty vector, IDH1 wild-type or IDH1-R132H (n = 7), ± AG-120 inhibitor (n = 3 of 7) (A); empty vector, IDH2 wild-type or IDH2-R140 (n = 6), ± AG-221 inhibitor (n = 4 of 6) (B). Graphs on the left represent the percentage of total colonies rised after 14 days. Panels on the right are representative images of wells (top row) and type of colonies (bottom row) obtained using the STEMvision™ Colony Marker Application: red circles identify BFU-E-derived colonies; yellow circles identify CFU-G/M/GM-derived colonies; blue circles identify CFU-GEMM-derived colonies; and orange circles identify CFU-E-derived colonies. (One-Way ANOVA analyses **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05).
Interestingly, no differences in the proportions of erythroid (BFU/CFU-E), myeloid (CFU-G/M/GM) and mixed (CFU-GEMM) colonies were observed in the different conditions for both IDH analyzed mutations, suggesting that the block of differentiation and release induced by the specific inhibitors affect all three lineages (Figure 3).
3.2. Enasidenib (AG-221) Treatment Induces a Progressive Improvement of CFU Ability of Primary CD34+ Cells in a Patient with IDH2-Mutated AML
In order to evaluate the dynamic of fitness of HSPC in terms of their ability to form colonies, we isolated and studied CD34+ cells from the bone marrow of a patient with IDH2-R140Q mutated AML we had the opportunity to follow under treatment with enasidenib. Specifically, we performed CFU assays with AML patient-derived CD34+ cells either at baseline or at the end of cycle I, II and IV. Moreover, with cells obtained after the IV cycle, we explored also the effect of adding pharmacological AG-221 inhibition in vitro during culture. Colonies were evaluated at 14 days as described above (Figure 4).
Strikingly, we observed that in vivo treatment with enasidenib induced a progressive increase, over time, in the CFU capacity of patient-derived CD34+ cells, whose fitness appeared to progressively improve also in terms of developing mature hematopoietic precursors across the different lineages, a phenomenon that was further strengthened when drug exposure was maintained also in vitro on semi-solid culture (Figure 4). To note, this occurred in parallel with the achievement of hematological complete remission of the patient.
Remarkably, NGS analysis performed after cycle IV, showed with respect to the diagnosis, the persistence of the IDH2-R140Q mutation, on one side in the bulk as well as in isolated myeloid and erythroid cells (Variant Allele Frequency, VAF = 51% for both subpopulations) and on the other side in the CD34+ subpopulation (VAF = 52%) used for the CFU assay (Figure 4).
The VAF values found in the different cell subpopulations suggest that the entire hemopoiesis including the pool of CD34+ cells were clonal (IDH2-R140Q positive) and that the improvement in the fitness of HSPC CD34+ relies on the protracted inhibitory action of enasidenib, that unleashes the block of differentiation mediated by IDH2-R140Q mutation.
These findings are in keeping with the clinical observation underlying that complete remission in patient treated with IDH2 inhibitor is achieved after more cycles of therapy (mean n=5) without undergoing bone marrow aplasia and is associated to cell differentiation of bone marrow cells in absence of clearance of the IDH2 mutation [26,41].
4. Discussion
Our model represents a first example of the AML-associated IDH1/2 mutations modeling in the human system. Indeed, from the review of the models available so far, it emerges that early effects of IDH1/2 mutations particularly in human hematopoiesis are mostly unexplored. Either in vitro or in vivo models from the literature (Table 1) are reviewed and discussed below.
4.1. In Vitro Models of IDH Mutations
Figueroa et al. [24] developed several in vitro cell models to explore the role of IDH mutations in leukemogenesis: transiently transfected 293T human cell line expressing IDH1-R132H or IDH2-R172K mutant, mouse myeloid progenitor 32D cells stably expressing IDH2-R172K mutant and murine primary bone marrow cells bearing IDH1-R132H or IDH2-R172K mutations. All of these models showed a significant increase in DNA methylation compared to control cells expressing IDH wild-type genes; this result was in keeping with the observed association of IDH1/2 mutations with hypermethylation signature in a large cohort of de novo AML patients. Stable expression of mutant IDH2 resulted in an important increase in c-Kit stemness marker expression both in 32D cells and in the murine primary bone marrow cells; in the latter, a reduced expression of the mature myeloid markers (Gr-1 and Mac-1) was also observed. Ultimately, the Figueroa et al. [24] study establishes that IDH1/2 mutant expression may specifically alter DNA methylation in AML cells and this can correlate with their role in impairing myeloid differentiation as observed in the established in vitro models.
Losman et al. [35] chose the TF-1 human erythroleukemia cell line as a model; these cells are GM-CSF-dependent and retain the ability to differentiate in response to erythropoietin (EPO). When stably infected by lentiviral vectors encoding IDH1-R132H, TF-1 cells became cytokine-independent and no longer differentiated upon EPO at the first steps after infection. In this study the inhibitory effect of mutant IDH on hematopoietic differentiation was confirmed on the SCF ER-Hoxb8 cell model, immortalized granulocyte-macrophage progenitor cells derived from primary murine bone marrow cells. In the presence of estrogen, these cells express a functional ER-Hoxb8 fusion protein that promotes their survival and proliferation. Upon estrogen withdrawal, the cells differentiate and up-regulate expression of the monocytic markers CD11b/Mac1 and Gr1. Stable expression of IDH1-R132H transgene in SCF ER-Hoxb8 cells, however, blunted their differentiation in response to estrogen withdrawal. Therefore, growth factor/hormone independence and impaired differentiation seem to be the two fundamental steps in leukemic transformation promoted by IDH mutant in Losman et al. models.
Wang et al. [36] developed a small molecule IDH2-R140Q selective inhibitor (AGI-6780) and used human TF-1 erythroleukemia cell line expressing IDH2-R140Q to study its biological effect. Expression of the mutant protein induced a large production of 2HG, a morphological change in TF-1 cells concomitantly to increase in vimentin expression, and decreased CD38; this evidence suggested that IDH2 mutant expression could induce a more immature phenotype, shifting TF-1 cells to an earlier stage, blocking hematopoietic differentiation. Furthermore, EPO treatment of TF-1 cells bearing IDH2-R140Q failed to induce the differentiation and the expression of genes upregulated during erythropoiesis (i.e., HBG and KLF1); importantly, pharmacological inhibition by AGI-6780 could revert both these phenomena. These results were in keeping with studies of Losman et al. representing the counterpart of the IDH1-R132H mutation in the same in vitro model.
4.2. In Vivo Models of IDH Mutations
Sasaki et al. [30] developed and characterized a conditional knock-in (KI) mice with IDH1-R132H mutation expressed in all hematopoietic cells (Vav-KI mice) or specifically in cells of the myeloid lineage (LysM-KI mice). The LysM-KI mouse model showed increased numbers of early hematopoietic progenitors and developed splenomegaly, signals of dysfunctional bone marrow niche and, main, hypermethylated histones and changes in DNA methylation similar to those observed in human IDH1/2 mutant AML [24]. The evaluation of HPSC compartments in older LysM-KI mice showed a bone marrow with fewer mature cells and more immature elements. In CFU assays, mice showed statistically normal production of CFU-GEMM, CFU-GM, CFU-G and CFU-M colonies. Serial plating experiments showed that, whereas control BM cells stopped proliferating, LysM-KI BM cells continued to grow at an exponential rate. The DNA-methylation analysis of sorted LSK (Lineage−Sca-1+cKit+) cells from young LysM-KI showed a significantly greater proportion of highly methylated CpG sites compared to control mice. DNA-methylation involved several pathways concerning hematopoietic cell proliferation and differentiation, leukemogenesis and leukemic stem cell maintenance. Sasaki et al. [30] therefore did not observe a block of differentiation and of CFU ability in mouse model with the IDH1-R132H mutation; neverthless they detected a significant increase in the pool of hematopoietic precursors and self-renewal capacity associated to the typical hypermethylation profile.
Kats et al. [31] generated transgenic mice expressing IDH2-R140Q in an on/off- and tissue-specific manner using a tetracycline-inducible system, in order to establish the impact of IDH2 mutation on normal HSPC as well as lineage development in vivo. Short-term analyses of hematopoiesis in transgenic IDH2-R140Q animals showed no differences in peripheral blood cell counts and, likewise, there were no alterations in the numbers of hematopoietic stem cells, myeloid progenitors, mature B, T, or myeloid cells in the bone marrow. The effect of this mutation evaluated at longer-term showed an increase in the number of LSK cells in the BM. Evaluating differentiation of primary hematopoietic cells by methylcellulose differentiation assay on KSL cells from IDH2-R140Q mice a potent block of differentiation was observed. Especially erythroid colonies were severely reduced, while the number of myeloid colonies was unaffected. Moreover, in serial plating experiments IDH2-R140Q cells formed more colonies in the second plating when compared with controls. The block of differentiation induced by IDH2 mutation in Kats’s model could be reversed and differentiation of mutant cells was restored either upon IDH2-R140Q doxycycline de-induction or upon AGI-6780 treatment. In conclusion, the expression of the IDH2 mutation in this mouse model is sufficient to induce defects in differentiation of primary hematopoietic cells with restriction to the erythroid lineage and is reversible by the specific pharmacological inhibition. Like IDH1, also IDH2 mutation is associated with an expansion of hematopoietic stem cells with a consequent increase in their self-renewal capacity.
4.3. In Vivo Models of IDH Mutations Combined with Other Genetic Lesions
Chen et al. [32] and Shih et al. [37] demonstrated that IDH2-R140Q and IDH2-R172K mutants can be potent oncogenes in mice, acting as driver mutations that cooperate with the FLT3-ITD or the NRAS-G12D mutation to promote aggressive AML. They showed that IDH2 mutants are required for sustained 2-HG production and leukemia maintenance; moreover, suppression of IDH2 mutant levels and its neo-morphic activity confirmed to trigger myeloid differentiation.
Marshall et al. [33] with their work demonstrated that IDH mutation can cooperate with other “not classic” alterations, creating a mouse model with Mir142 loss-of- function in which IDH2-R140Q mutation synergizes to promote leukemogenesis.
Gruber et al. [34], on their side, developed a multigenic AML mouse model with inducible IDH1-R132H mutation in its prevalent combination with DNMT3A-R882H and NRAS-G12D. Inhibition of mutant IDH1 in this model promoted AML differentiation and prolonged survival of mice. Similarly, Kats et al. [38] created a combinatorial mouse model with IDH2-R140Q, DNMT3A-R882H and NRAS-G12D that develops AML. Silencing of IDH2-R140Q expression again led to terminal myeloid differentiation.
This work and our previous preliminary reports [42,43] show for the first time that the expression of IDH1-R132H or IDH2-R140Q mutant is sufficient to drive in vitro a complete block of hematopoietic cell differentiation in primary human HSPC that is efficiently released by the specific inhibitor with recovery of differentiation ability across all lineages.
Data emerging from our CD34+ HSPC models of IDH mutations are partially in contrast with previous studies in murine models. Indeed, expression of the mutated IDH1 transgene in our human CD34+ HSPC induces a significant block of differentiation that was not observed in Sasaki et al. mouse model, where a significant increase in the hematopoietic precursor pool was described [30]. The marked block of differentiation we show following the expression of IDH2 mutation in human CD34+ HSPC is also different from the in vivo observations of Kats et al. who, unlike the trilinear involvement we report, show only the involvement of erythroid lineage in the block [31].
Instead, strikingly, our findings are in keeping with the recent report from Landberg et al. [44,45] obtained always in human CD34+ HSPC but edited by CRISPR/Cas9 and AAV6-mediated homology directed repair to express IDH1-R132H or IDH2-R140Q mutations: a pronounced reduction in colony formation and differentiation capacity triggered by both IDH mutations and the ability of ivosidenib in restoring block of differentiation driven by IDH1-R132H were shown. The report from Landberg et al. [44] and our findings highlight the direct involvement of the AML-associated IDH1-R132H and IDH2-R140Q mutations in the hematopoietic differentiation process.
Finally, our study on fitness dynamics of primary HSPC from a IDH2-mutated AML patient under enasidenib treatment represents a unique proof-of-concept report, confirming in vivo, in patients, the effects of IDH mutation and of its pharmacological inhibition, providing further explanation to the clinical observation.
5. Conclusions
Our report highligths how the IDH1/2 mutations frequently found in acute myeloid leukemia act at the level of the hematopoietic precursors in the human system by inducing a block of differentiation. The block involves all the hematopoietic myeloid lineages and is specifically reversed by the targeted therapy. Furthermore, we provide evidence that the specific inhibitor act progressively on the HSPC in vivo in patients improving their ability to form colonies in vitro and sustain trilinear hematopoiesis overtime in vivo leading to hematological recovery and complete hematological remission. Our model represents also a suitable model for further mechanistic studies to explore the role of IDH1/2 mutations in human leukemogenesis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Flow cytometry-based sorting strategy for human CD34+ transduced with IDH transgenes; Table S1: Experimental conditions of HSPC CD34+ cells from 13 healthy donors.
Author Contributions
Conceptualization, Maria Paola Martelli; Data curation, Sara Pierangeli and Serena Donnini; Formal analysis, Sara Pierangeli and Serena Donnini; Funding acquisition, Maria Paola Martelli; Investigation, Sara Pierangeli, Serena Donnini, Valerio Ciaurro, Francesca Milano, Valeria Cardinali, Sofia Sciabolacci, Gaetano Cimino, Ilaria Gionfriddo, Roberta Ranieri, Sabrina Cipriani, Eleonora Padiglioni, Roberta Iacucci Ostini and Tiziana Zei; Methodology, Sara Pierangeli, Serena Donnini, Valerio Ciaurro and Maria Paola Martelli; Project administration, Maria Paola Martelli; Supervision, Antonio Pierini and Maria Paola Martelli; Validation, Sara Pierangeli, Serena Donnini, Valerio Ciaurro, Francesca Milano, Ilaria Gionfriddo, Roberta Ranieri, Sabrina Cipriani and Eleonora Padiglioni; Writing—original draft, Sara Pierangeli and Serena Donnini; Writing—review & editing, Maria Paola Martelli. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Research Council (ERC), consolidator grant 2016 n. 725725 (to Maria Paola Martelli). Valerio Ciaurro, Roberta Ranieri and Eleonora Padiglioni were supported by an AIRC/FIRC fellowship for Italy.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the University of Perugia (protocol code: 2018-07; date of approval: 10 may 2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Acknowledgments
We acknowledge Dr. Marcella Sabino for technical help in NGS analyses, and Dr. Laura Pinacoli for the administrative support.
Conflicts of Interest
Maria Paola Martelli declares honoraria/consultancy at scientific advisory board for Abbvie, Amgen, BMS, Delbert, Janssen, Novartis, Pfizer, and Jazz Pharmaceuticals. The other authors declare no conflicts of interest.
References
- Ley, T. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia (vol 368, pg 2059, 2013). New England Journal of Medicine 2013, 369, 98–98. [Google Scholar]
- Gilliland, D. Molecular genetics of human leukemias: New insights into therapy. Seminars in Hematology 2002, 39, 6–11. [Google Scholar] [CrossRef]
- McMurry, H.; Fletcher, L.; Traer, E. Idh inhibitors in aml-promise and pitfalls. Current Hematologic Malignancy Reports 2021, 16, 207–217. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. Idh1 and idh2 mutations in gliomas. N Engl J Med 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Beer, P. Somatic mutations of idh1 and idh2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med USA 2010, 362, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Weissinger, E.M.; Krauter, J.; Wagner, K.; Damm, F.; Wichmann, M.; Göhring, G.; Schumann, C.; Bug, G.; Ottmann, O.; et al. Idh1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica 2010, 95, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. Idh1 and idh2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (idh1) mutations in thyroid cancer. Biochem Biophys Res Commun 2010, 393, 555–559. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of aml in adults: 2022 recommendations from an international expert panel on behalf of the eln. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Neumann, M.; Schroeder, M.P.; Vosberg, S.; Schlee, C.; Isaakidis, K.; Ortiz-Tanchez, J.; Fransecky, L.R.; Hartung, T.; Türkmen, S.; et al. Acute myeloid leukemia in the elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia 2017, 31, 1640–1644. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. Idh2 and npm1 mutations cooperate to activate hoxa9/meis1 and hypoxia pathways in acute myeloid leukemia. Cancer Res 2015, 75, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated idh1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. Idh1 and idh2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A cancer and leukemia group b study. J Clin Oncol 2010, 28, 2348–2355. [Google Scholar] [CrossRef]
- Im, A.P.; Sehgal, A.R.; Carroll, M.P.; Smith, B.D.; Tefferi, A.; Johnson, D.E.; Boyiadzis, M. Dnmt3a and idh mutations in acute myeloid leukemia and other myeloid malignancies: Associations with prognosis and potential treatment strategies. Leukemia 2014, 28, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Martino, G.; Cardinali, V.; Falini, B.; Martinelli, G.; Cerchione, C. Enasidenib and ivosidenib in aml. Minerva Med 2020, 111, 411–426. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated idh1 and idh2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Dang, L.; Su, S.M. Isocitrate dehydrogenase mutation and (r)-2-hydroxyglutarate: From basic discovery to therapeutics development. Annu Rev Biochem 2017, 86, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated idh1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. Idh mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic idh1 and idh2 mutations result in a hypermethylation phenotype, disrupt tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. Idh1-r132 changes vary according to npm1 and other mutations status in aml. Leukemia 2019, 33, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant idh2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivosidenib in idh1-mutated relapsed or refractory aml. N Engl J Med 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol 2019, 9, 417. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. Idh1(r132h) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant idh2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Lu, C.; Cross, J.R.; Morris, J.P.T.; Shroff, A.S.; Ward, P.S.; Bradner, J.E.; Thompson, C.; Lowe, S.W. Cancer-associated idh2 mutants drive an acute myeloid leukemia that is susceptible to brd4 inhibition. Genes Dev 2013, 27, 1974–1985. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Kasturiarachchi, J.; Datta, P.; Guo, Y.; Deltcheva, E.; James, C.; Brown, J.; May, G.; Anandagoda, N.; Jackson, I.; et al. Mir142 loss unlocks idh2(r140)-dependent leukemogenesis through antagonistic regulation of hox genes. Sci Rep 2020, 10, 19390. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.; So, J.; Lewis, A.C.; Franich, R.; Cole, R.; Martelotto, L.G.; Rogers, A.J.; Vidacs, E.; Fraser, P.; Stanley, K.; et al. Inhibition of mutant idh1 promotes cycling of acute myeloid leukemia stem cells. Cell Rep 2022, 40, 111182. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (r)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant idh2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Shih, A.H.; Meydan, C.; Shank, K.; Garrett-Bakelman, F.E.; Ward, P.S.; Intlekofer, A.M.; Nazir, A.; Stein, E.M.; Knapp, K.; Glass, J.; et al. Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in idh2- and tet2-mutant acute myeloid leukemia. Cancer Discov 2017, 7, 494–505. [Google Scholar] [CrossRef]
- Kats, L.M.; Vervoort, S.J.; Cole, R.; Rogers, A.J.; Gregory, G.P.; Vidacs, E.; Li, J.; Nagaraja, R.; Yen, K.E.; Johnstone, R.W. A pharmacogenomic approach validates ag-221 as an effective and on-target therapy in idh2 mutant aml. Leukemia 2017, 31, 1466–1470. [Google Scholar] [CrossRef]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Pierini, A.; Falzetti, F.; Carotti, A.; Terenzi, A.; Reisner, Y.; Aversa, F.; Falini, B.; et al. “Designed” Grafts for hla-haploidentical stem cell transplantation. Blood 2014, 123, 967–973. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.L.; Verhoeyen, E. Baboon envelope pseudotyped lvs outperform vsv-g-lvs for gene transfer into early-cytokine-stimulated and resting hscs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-idh2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Ciaurro, V.; Pierangeli, S.; Falini, B.; Martelli, M.P. Idh1-r132h expression drives in human normal cd34+ hematopoietic cells a block of differentiation released by the specific inhibitor ivosidenib. In Proceedings of the XVI Congress of the Italian Society of Experimental Hematology, Napoli, Italy, 15–17 October 2020. Haematologica 2020, 105, C006. [Google Scholar] [CrossRef]
- Pierangeli, S.; Ciaurro, V.; Donnini, S.; Milano, F.; Sabino, M.; Gionfriddo, I.; Ranieri, R.; Silvestri, S.; Tini, V.; Spinozzi, G.; Falini, B.; Martelli, M.P. Isocitrate dehydrogenases aml-associated point mutations drive a block of differentiation in human normal cd34+hematopoietic cells that is released by specific inhibitors. In Proceedings of the XVII Congress of the Italian Society of Experimental Hematology, Roma, Italy, 31 March–2 April 2022. Haematologica 2022, 107, C60. [Google Scholar] [CrossRef]
- Landberg, N.; Koehnke, T.; Nakauchi, Y.; Fan, A.; Karigane, D.; Thomas, D.; Majeti, R. Targeting idh1-mutated pre-leukemic hematopoietic stem cells in myeloid disease, including ccus and aml. In Proceedings of the 64th Ash Annual Meeting and Exposition, New Orleans, Louisiana, 10–13 December 2022. Blood 2022, 140 (Suppl. 1), 2234–2235. [Google Scholar] [CrossRef]
- Landberg, N.; Köhnke, T.; Feng, Y.; Nakauchi, Y.; Fan, A.C.; Linde, M.H.; Karigane, D.; Lim, K.; Sinha, R.; Malcovati, L.; et al. Idh1-mutant preleukemic hematopoietic stem cells can be eliminated by inhibition of oxidative phosphorylation. Blood Cancer Discov 2024, of1–of18. [Google Scholar] [CrossRef]
Figure 1.
Experimental workflow.
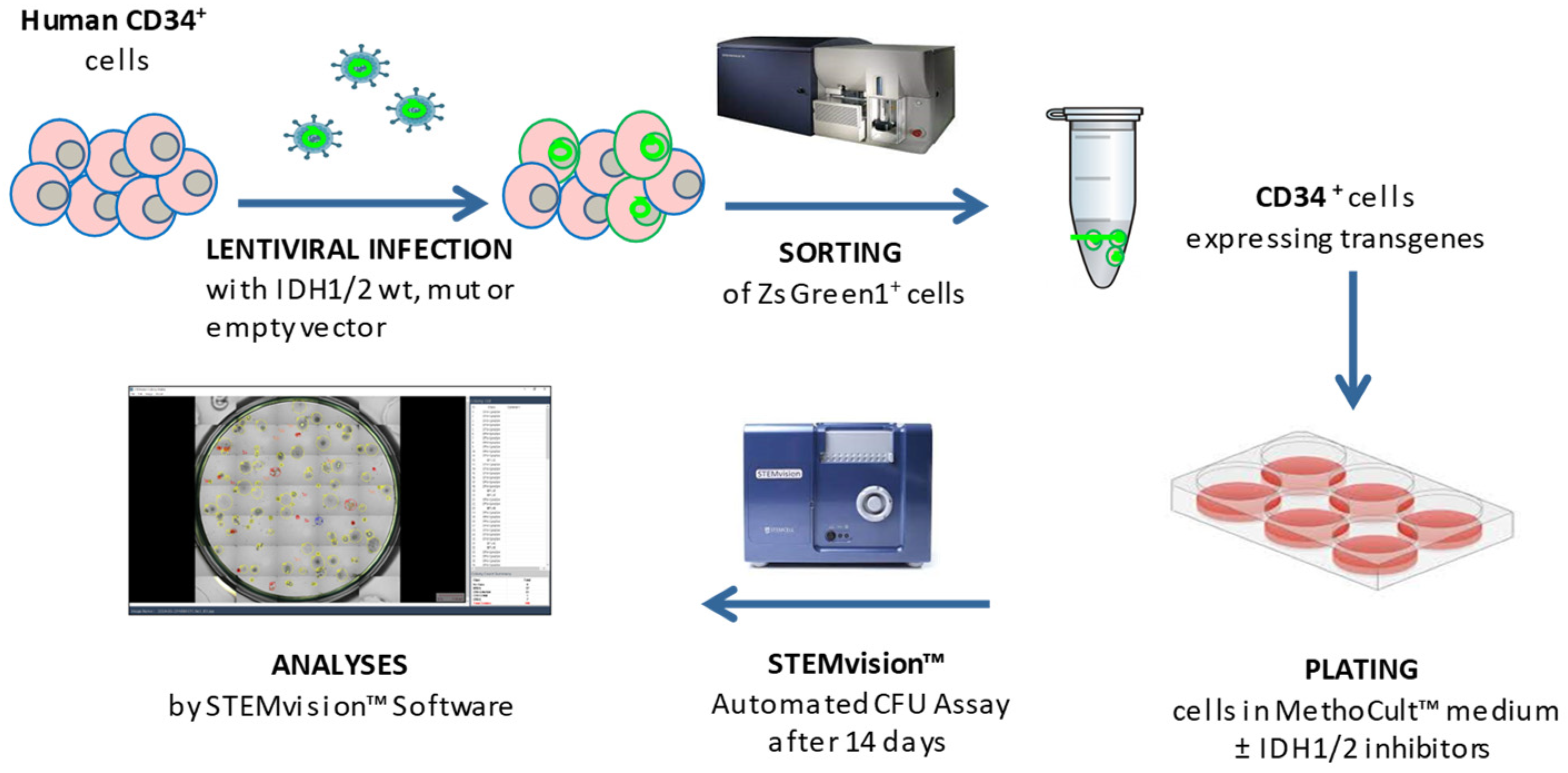
Figure 3.
Block of differentiation induced in human CD34+ by either IDH1 (A) or IDH2 (B) mutations and its release by the specific inhibitors affect all lineages: erythroid (left), myeloid (middle) or GEMM (right) precursors.
Figure 3.
Block of differentiation induced in human CD34+ by either IDH1 (A) or IDH2 (B) mutations and its release by the specific inhibitors affect all lineages: erythroid (left), myeloid (middle) or GEMM (right) precursors.
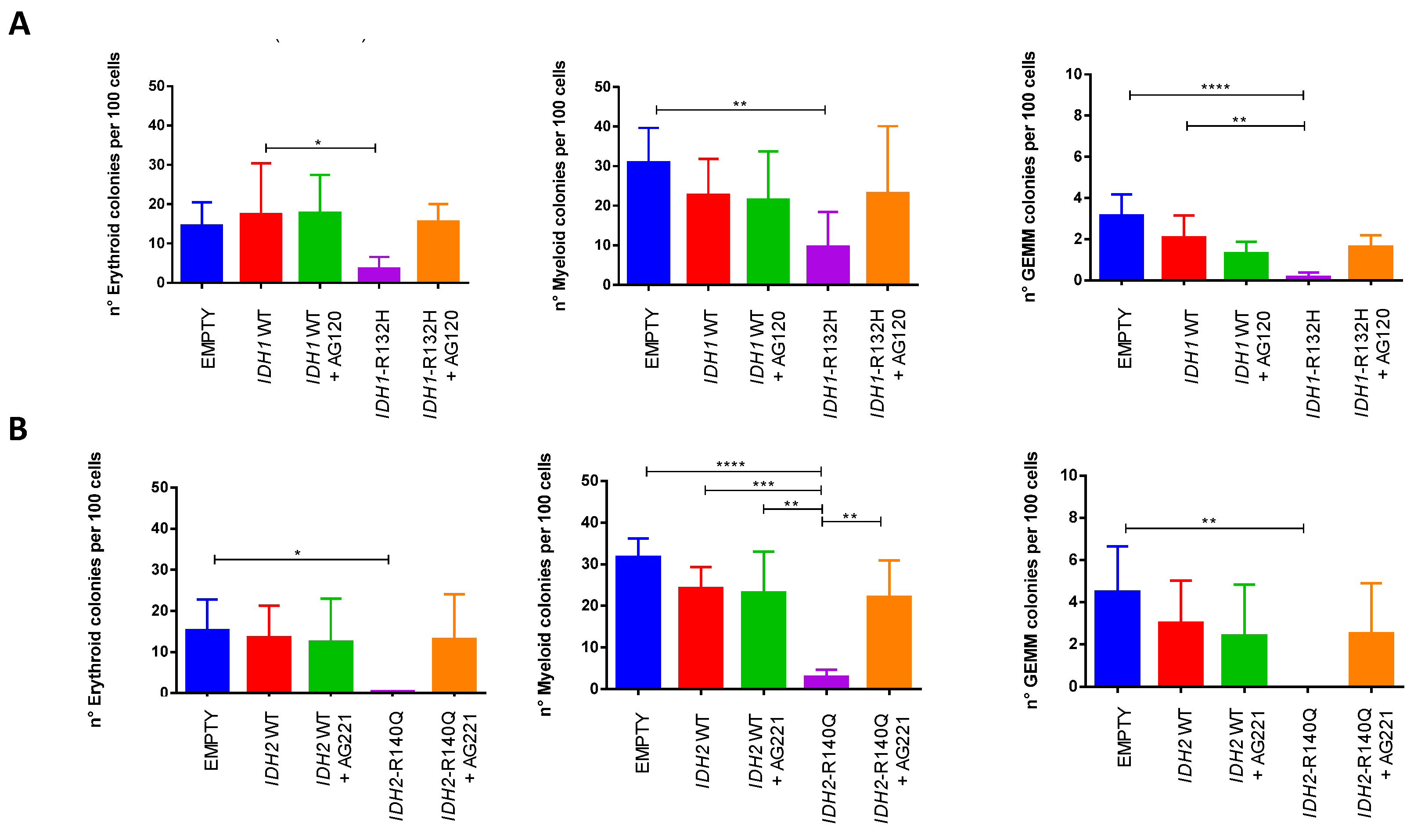
Figure 4.
Enasidenib treatment induces a progressive improvement of CFU ability in primary CD34+ cells carrying IDH2-R140Q mutation. Experimental scheme (left) and stacked columns chart (right) of colonies rised after 14 days from CD34+ cells isolated from patient BM before the start of treatment (Day 0) and at I, II and IV cycle of enasidenib therapy (Day 28, 56, 112 respectively). The table in the middle shows IDH2-R140Q VAF obtained by targeted NGS on DNA extracted from either bulk BM or isolated cell subpopulations at IV cycle/Day 112 of treatment.
Figure 4.
Enasidenib treatment induces a progressive improvement of CFU ability in primary CD34+ cells carrying IDH2-R140Q mutation. Experimental scheme (left) and stacked columns chart (right) of colonies rised after 14 days from CD34+ cells isolated from patient BM before the start of treatment (Day 0) and at I, II and IV cycle of enasidenib therapy (Day 28, 56, 112 respectively). The table in the middle shows IDH2-R140Q VAF obtained by targeted NGS on DNA extracted from either bulk BM or isolated cell subpopulations at IV cycle/Day 112 of treatment.
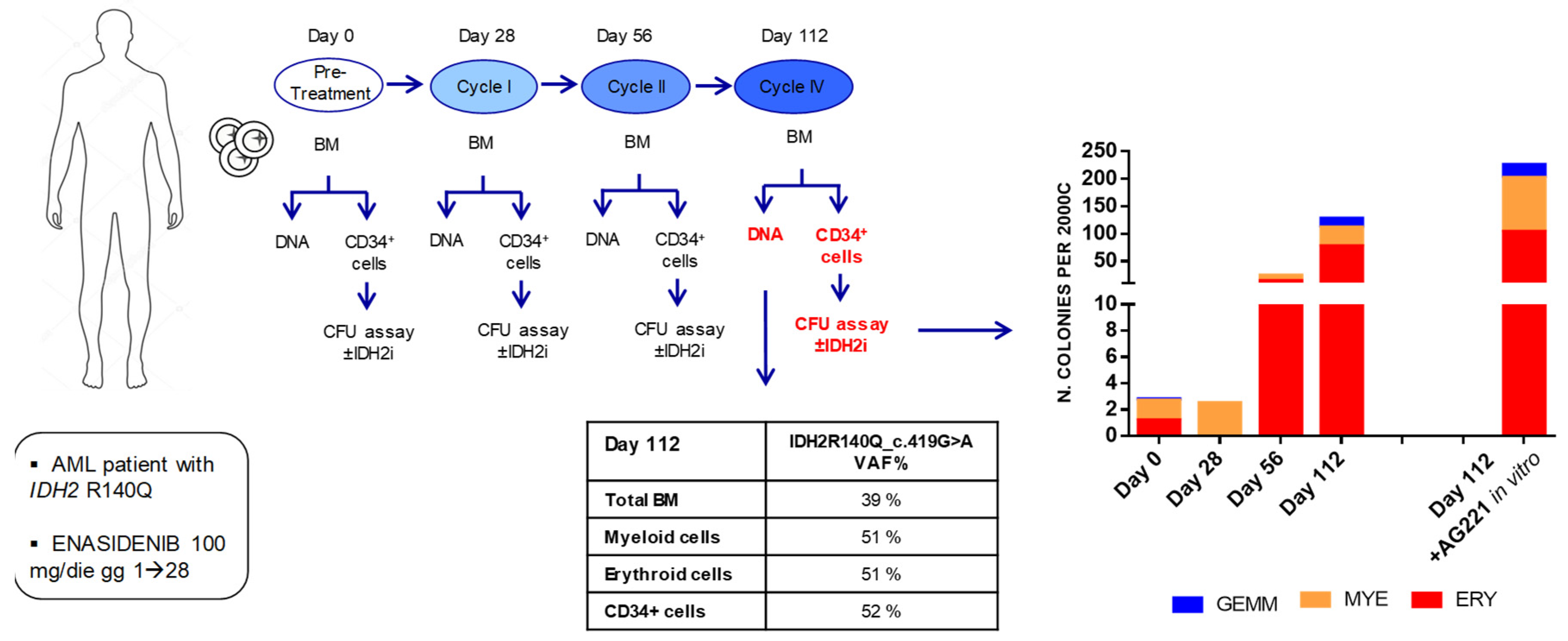

Table 1.
Hematopoiesis models of IDH1/2 AML-associated mutations.
| REFERENCE | IDH mutations | MODELS | OBSERVATIONS |
|---|---|---|---|
| in vitro | |||
| Figueroa et al. [24] | IDH1-R132H or IDH2-R172K/R140Q | 293T human embryo kidney cell line transiently expressing IDH1/2 mutations | IDH1/2 mutants alter DNA methylation and impair myeloid differentiation |
| 32D mouse myeloid progenitor cell line stably expressing IDH2 mutations | |||
| Murine primary bone marrow cells with IDH1/2 mutations | |||
| Losman et al. [35] | IDH1-R132H | TF-1 human erythroleukemia cell line stably expressing IDH1-R132H | IDH1 mutant promote leukemogenesis by growth factor/hormone independence and impaired differentiation |
| SCF ER-Hoxb8 murine granulocyte/macrophage progenitor cells stably expressing IDH1-R132H | |||
| Wang et al. [36] | IDH2‐R140Q | TF-1 human erythroleukemia cell line expressing IDH2-R140Q | IDH2 mutant induce an immature phenotype blocking differentiation |
| in vivo | |||
| Sasaki et al. [30] | IDH1-R132H | Knock-in (KI) mice expressing IDH1-R132H mutation in all hematopoietic cells (Vav-KI mice) or in cells of myeloid lineage (LysM-KI mice) | IDH1 mutant doesn’t block differentiation and increases hematopoietic precursors |
| Kats et al. [31] | IDH2-R140Q | Transgenic mice expressing IDH2-R140Q in an on/off- and tissue-specific manner by a tetracycline- inducible system | IDH2 mutant blocks only erythroid lineage differentiation and increases self-renewal capacity |
| Chen et al. [32] Shih et al. [37] |
IDH2-R140Q /R172K |
Mosaic mouse model FLT3-ITD or NRAS-G12D mutated and transduced with IDH2 mutant | IDH2 mutants sustain leukemia maintenance and their suppression restores differentiation |
| Marshall et al. [33] |
IDH2-R140Q /R172K |
Mouse model with Mir142 loss-of- function and IDH2-R140Q mutation | IDH2 mutation cooperates with peculiar genetic alteration for leukemogenesis |
| Gruber et al. [34] | IDH1-R132H | Multigenic mouse model with inducible IDH1-R132 mutation in combination with DNMT3A-R882H and NRAS-G12D | IDH1 mutant, together with other alterations, if inhibited promote AML differentiation |
| Kats et al. [38] | IDH2-R140Q | Combinatorial mouse model with IDH2-R140Q, DNMT3A-R882H and NRAS-G12D mutations | IDH2 mutant silencing leads to terminal myeloid differentiation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



